

Abstract
Mutant Z alpha-1 antitrypsin (ATZ) accumulates in globules in the liver and is the prototype of proteotoxic hepatic disease. Therapeutic strategies aiming at clearance of polymeric ATZ are needed. Transient receptor potential mucolipin-1 (TRPML1) is a lysosomal Ca2+ channel that maintains lysosomal homeostasis. In this study, we show that by increasing lysosomal exocytosis, TRPML1 gene transfer or small-molecule-mediated activation of TRPML1 reduces hepatic ATZ globules and fibrosis in PiZ transgenic mice that express the human ATZ. ATZ globule clearance induced by TRPML1 occurred without increase in autophagy or nuclear translocation of TFEB. Our results show that targeting TRPML1 and lysosomal exocytosis is a novel approach for treatment of the liver disease due to ATZ and potentially other diseases due to proteotoxic liver storage.
Keywords: alpha-1 antitrypsin deficiency, lysosomal exocytosis, alpha-1 antitrypsin, TRPML1, mucolipin-1
Graphical abstract
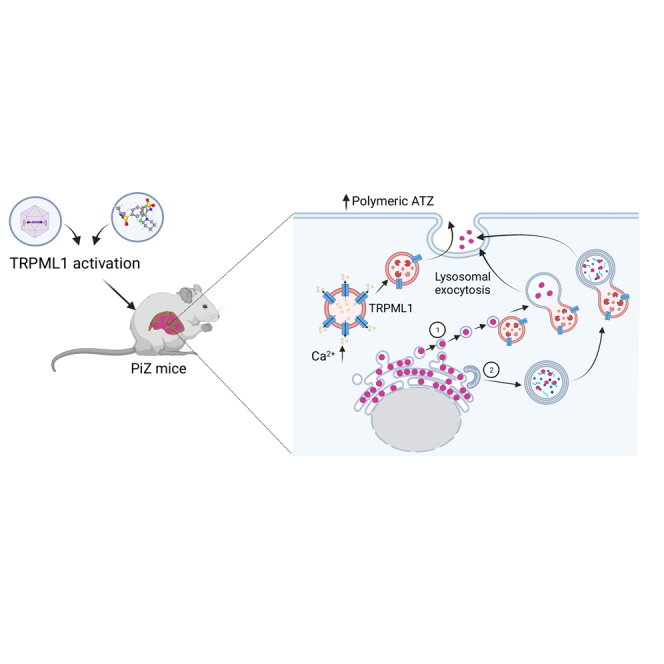
Brunetti-Pierri, Pastore, and colleagues showed that by increasing lysosomal exocytosis, TRPML1 gene transfer or small-molecule-mediated activation of TRPML1 improves the liver disease due to mutant alpha-1 antitrypsin in mice.
Introduction
Alpha-1 antitrypsin (AAT) deficiency affecting 1 in 2,000 to 3,000 live births is caused by pathogenic variants in the SERPINA1 gene that encodes an acute phase plasma glycoprotein and serine-protease inhibitor primarily synthesized in the liver.1,2 The most frequent AAT-deficient allele is the Z variant, a single amino acid substitution (p.Glu342Lys) that alters the protein structure resulting in the formation of polymers of AAT in the endoplasmic reticulum (ER). As a result of hepatic accumulation of the Z variant of AAT (ATZ), serum AAT is reduced, causing lung emphysema by a loss-of-function mechanism.3 In contrast, by a gain-of-function mechanism, ATZ polymers retained in the liver cause a spectrum of hepatic diseases ranging from acute and chronic hepatitis, fibrosis, cirrhosis, and increased risks of hepato-carcinoma.4,5
Although it did not achieve serum concentrations within the therapeutic range, AAV-mediated gene transfer of SERPINA1 into muscles resulted in sustained expression of AAT in patients with AAT deficiency.6 Moreover, muscle-directed gene therapy has limitations related to the vector dose that can be administered and the need of multiple injections. Nevertheless, while delivery of the SERPINA1 gene to muscle or liver is a long-term solution for lung disease, it does not affect liver disease. Treatment of both lung and liver disease indeed requires hepatic knockout or downregulation of mutant SERPINA1 gene to eliminate the expression of toxic ATZ protein, in addition to providing the wild-type gene. To achieve this goal, one strategy that has been investigated is the suppression of the expression of the mutant SERPINA1 gene with concomitant expression of the wild-type SERPINA1. This strategy has been achieved in preclinical studies by adeno-associated virus (AAV) vectors delivering microRNA (miRNA) sequences targeting SERPINA1 gene while also driving the expression of miRNA-resistant wild-type SERPINA1 gene, thus achieving concomitant ATZ knockdown in the liver and increased expression of AAT.7 While this AAV-based approach is still at the preclinical stages of development, a siRNA-based treatment reducing expression of the ATZ might become clinically available soon.8 Meanwhile, only liver transplantation is available for therapy of patients with severe liver disease. The siRNA therapy for liver disease under clinical development is not expected to clear pre-existing polymers, and the development of strategies to increase the degradation of polymers accumulated in the ER is still needed. Various approaches have been investigated in preclinical models to reduce the burden of toxic ATZ, such as inhibition of polymerization, or increased clearance. 9,10. In our previous work, we showed that hepatic gene transfer of the transcription factor EB (TFEB), the master regulator of autophagy and lysosomal biogenesis,11,12 is effective at reducing ATZ polymers by increasing its degradation in autophagolysosomes.13 TFEB-induced autophagy is regulated by the non-selective cation channel transient receptor potential mucolipin 1 (TRPML1), also known as mucolipin-1 (MCOLN1).14,15,16 TRPML1 can also activate autophagy in a TFEB-independent manner.17 TRPML1 is a calcium (Ca2+)-release channel residing on the lysosomal membrane that is involved in a variety of processes, such as lysosome to trans-Golgi network retrograde trafficking, autophagic vesicle (AV)-lysosome fusion, lysosome reformation, and lysosomal exocytosis.18,19 As therapeutic target, TRPML1 has been investigated in various diseases.20,21,22 Of note, drugs acting on TRPML1 are expected to be more targeted compared to TFEB, which regulates a wide variety of functions. In this study, we investigated TRPML1 as candidate target for therapy of the liver disease induced by ATZ by both genetic and pharmacological approaches.
Results
Hepatic TRPML1 gene transfer reduces ATZ globules in PiZ mice
To investigate the efficacy of TRPML1 at correcting the liver disease induced by ATZ, 6-week-old PiZ mice, recapitulating the liver pathology of human AAT deficiency,23 were injected intravenously (i.v.) with a serotype 8 adeno-associated virus (AAV8) vector expressing the human TRPML1 under the control of the hepatocyte-specific thyroxin-binding globulin (TBG) promoter (AAV-TRPML1) and sacrificed 12 weeks post injection. Age- and gender-matched PiZ mice were injected i.v. with either AAV-GFP expressing the reporter green fluorescent protein (GFP) as negative control or with AAV-TFEB expressing TFEB as positive control (Figure 1A).13 Hepatic AAV-mediated delivery of TRPML1 gene was confirmed in livers of PiZ mice by real-time PCR that showed expression of the human TRPML1 mRNA in AAV-TRPML1-injected mice (Figure 1B). Liver immunofluorescence showed transgene expression (TRPML1, GFP, or TFEB) in approximately 70% of hepatocytes of PiZ mice (Figure 1C). Compared to AAV-GFP-injected controls, livers of PiZ mice injected with AAV-TRPML1 showed a significant reduction in Periodic acid-Schiff staining with diastase digestion (PAS-D)-positive globules (Figure 1D), the pathology hallmark of liver disease due to ATZ, which are absent in wild-type mice (SI Appendix, Figure S1).23 In line with our previous study,13 AAV-TFEB decreased PAS-D-positive globules, and this reduction was similar to the PiZ mice injected with AAV-TRPML1 (Figure 1D). Consistent with the reduction in globules, Sirius red staining showed decreased liver fibrosis in PiZ mice injected with AAV-TRPML1 compared to control PiZ mice injected with AAV-GFP (Figure 1E), whereas wild-type mice of similar age do not show any liver fibrosis (SI Appendix, Figure S1). Liver immunofluorescence with an antibody recognizing ATZ polymers showed a decrease in number and size of ATZ polymers (Figure 1F), and the immunoblotting on soluble and insoluble fractions confirmed the reduction in ATZ polymers (Figure 1G). By 2 weeks post injection, PiZ mice injected with AAV-TRPML1 also showed an increase of total circulating AAT (Figure 1H), suggesting increased AAT secretion. Notably, by 2 and 4 weeks post injection, an increase of ATZ polymers was detected in PiZ mice injected with AAV-TRPML1 but not in those injected with AAV-TFEB (Figure 1H), suggesting a difference in the mechanisms underlying ATZ clearance between TRPML1 and TFEB. Hepatic TRPML1 gene transfer was not associated with liver toxicity, as suggested by serum ALT and AST activities that were unaffected compared to AAV-GFP-injected PiZ mice (SI Appendix, Figure S2). Altogether, these results suggest that TRPML1 reduces hepatic ATZ accumulation and fibrosis. In contrast to 6-week-old injected mice, PiZ mice injected with AAV-TRPML1 at 16 weeks of age showed a trend in reduction of PAS-D-positive globules, but liver fibrosis was unaffected by 12 weeks post injection (SI Appendix, Figure S3). This suggests that the therapeutic potential of AAV-TRPML1 is reduced at later stages of disease. Although reduced in number,24 larger ATZ globules might be more resistant to clearance.
Figure 1.

TRPML1 gene transfer in PiZ livers reduces ATZ accumulation
(A) Schematic representation of AAV8 vectors injected in PiZ mice. (B) RT-PCR analysis for TRPML1 expression in livers of wild-type mice injected with AAV-GFP or AAV-TRPML1 (AAV-GFP, n = 7; AAV-TRPML1, n = 9). Human TRPML1 expression was normalized on murine Trpml1 mRNA. (C) Representative immunofluorescence staining for GFP, TRPML1, and TFEB in livers of PiZ mice injected with AAV vectors and harvested at 12 weeks post injection (n = 9 per group). Representative PAS-D (D) and Sirius Red (E) staining of livers of PiZ mice harvested 12 weeks after AAV injections and their corresponding quantifications (n = 9 per group). (F) Representative immunofluorescence staining for ATZ polymers in livers of PiZ mice injected with AAV vectors and harvested at 12 weeks post-injection. (G) Immunoblots for soluble and insoluble fractions on livers of PiZ mice injected with AAV-GFP or AAV-TRPML1 and their corresponding quantifications (AAV-GFP, n = 8; AAV-TRPML1, n = 9). GAPDH and H3 were used for normalization. AAV-GFP and AAV-TRPML1 are on the same gel but not contiguous. (H) Changes in percentages of serum total AAT (n = 9 per group) or polymeric ATZ (AAV-GFP, n = 6; AAV-TRPML1, n = 5; AAV-TFEB, n = 6) in PiZ mice after AAV injections. Data are shown as mean ± standard error. Student’s t test: ∗p value < 0.05; ∗∗p value < 0.01; ∗∗∗p value < 0.001. A.U., arbitrary units; p.i., post-injection.
TRPML1 stimulates secretion of ATZ polymers and lysosomal exocytosis in PiZ livers
The increased concentrations of total serum AAT and polymeric ATZ following hepatic TRPML1 gene transfer suggested that the reduction of PAS-D-positive globules in the liver is due to increased lysosomal exocytosis of ATZ polymers induced by TRPML1. TRPML1 was found to co-localize with lysosomal-associated membrane protein 1 (LAMP1) (Figure 2A). Upon lysosomal fusion, LAMP1 is exposed on the plasma membrane, and increased LAMP-1 staining on the plasma membrane was observed in livers of mice injected with AAV-TRPML1 4 weeks post injection compared to controls (Figures 2A and 2B). Translocation of lysosomes to the plasma membrane is the hallmark of lysosomal exocytosis,25 and by electron microscopy, an increased number of lysosomes close to the plasma membrane was detected in liver cells of PiZ mice injected with AAV-TRPML1 compared to controls at 4 and 12 weeks post injection (Figure 2C). Furthermore, triple immunostaining of livers of mice injected with AAV-TRPML1 showed distribution of LAMP1-positive structures in close proximity to the plasma membrane, which is positive for both the basolateral membrane marker Na-K ATPase and the plasma membrane marker laminin 2 (Figure 2D).26,27 Moreover, increased activities of the lysosomal enzymes iduronate sulfatase (IDS) and galactosamine (N-acetyl)-6-sulfatase (GALNS) were detected in sera of AAV-TRPML1-injected PiZ mice compared to controls (Figure 2E). Altogether, these results suggest an increase of lysosomal exocytosis in livers of PiZ mice injected with AAV-TRPML1.
Figure 2.

Increased lysosomal exocytosis in PiZ livers after TRPML1 gene transfer
(A) Representative images of immunofluorescence for LAMP1 and TRPML1-Myc in livers of PiZ mice harvested 4 weeks after AAV injections. (B) Representative images of immunofluorescence for LAMP1 and polymeric ATZ in livers of PiZ mice harvested 4 weeks after AAV injections. The dashed lines are superimposed to the margins of the plasma membranes to highlight single cells. (C) Representative electron microscopy images of PiZ livers injected with AAV-GFP or AAV-TRPML1 and harvested either 4 or 12 weeks post injection showing lysosomes (white arrows) close to the plasma membrane shown in red. The relative quantifications of lysosomes close to the plasma membrane is shown (n = 3 per group). (D) Triple immunostaining for the lysosome marker LAMP1, the plasma membrane marker laminin 2, and the basolateral membrane marker Na-K ATPase on livers of PiZ mice injected with AAV-GFP or AAV-TRPML1 and harvested at 4 weeks post injection. Quantification of LAMP1/laminin 2 co-localization is shown (n = 3 per group; n = 4 fields/sample). (E) Serum IDS and GALNS activities in AAV-GFP- and AAV-TRPML1-injected PiZ mice at 2 weeks post injection (IDS, n = 6 per group; GALNS, n = 4 for AAV-GFP; and n = 6 per AAV-TRPML1). Data are shown as mean ± standard error. Student’s t test: ∗p value < 0.05; ∗∗p value < 0.01. Abbreviations are as follows: BC, bile canaliculi; lyso, lysosomes; PM, plasma membrane.
TRPML1 can induce autophagy through TFEB activation,14,15,16 and consistently, TFEB nuclear translocation was detected in wild-type control mice injected with AAV-TRPML1 (Figure 3A). In contrast, very little to absent TFEB-positive nuclei were detected in PiZ liver cells either with or without AAV-TRPML1 injection (Figure 3A). Moreover, compared to AAV-GFP-injected mice, western blots on liver lysates showed increased LAMP1, SQSTM1/p62, and LC3II in wild-type but not in PiZ mice following injections of AAV-TRPML1 (Figure 3B), suggesting that ATZ globule clearance induced by TRPML1 is dependent on lysosomal exocytosis rather than activation of autophagy.
Figure 3.

TFEB nuclear translocation is not induced in PiZ livers by TRPML1 gene transfer
(A) Immunohistochemistry for TFEB in livers of wild-type or PiZ mice injected with AAV-GFP or AAV-TRPML1 and harvested at 4 weeks post injection. Quantification of TFEB-positive nuclei in livers is shown (n = 3 per group). (B) Immunoblots for LAMP1, SQSTM1/p62, and LC3 in livers of wild-type and PiZ mice injected with AAV-TRPML1 or AAV-GFP controls and relative quantifications (n = 3–4 per group). Data are shown as mean ± standard error. Student’s t test: ∗p value < 0.05; ∗∗p value < 0.01. WT, wild-type.
Drug-mediated activation of TRPML1 reduces ATZ globules in PiZ mouse livers
We next investigated the efficacy of a TRPML1 small-molecule agonist at reducing the hepatic accumulation of ATZ. First, we investigated the TRPML1 activator ML-SA528 in HTO/Z cells, a HeLa cell line engineered for doxycycline-regulated expression of ATZ.29 Incubation of HTO/Z cells with ML-SA5 reduced intracellular ATZ and increased the extracellular-to-intracellular ratio of ATZ, suggesting increased secretion of ATZ (SI Appendix, Figure S4A). An increase in the LAMP1 signal on the plasma membrane was detected by fluorescence-activated cell sorting (FACS) in HTO/Z compared to HeLa cells following incubation with ML-SA5 for 30 min (SI Appendix, Figure S4B). The small increase in LC3II in HTO/Z cells incubated with ML-SA5 and bafilomycin A1 compared to HeLa cells suggests that autophagy does not play a major role in the increased disposal of ATZ in HTO/Z mediated by ML-SA5 (SI Appendix, Figure S4C).
6-week-old PiZ mice receiving daily intraperitoneal injections of ML-SA528 for 15 days showed reduced hepatic PAS-D globules and soluble and insoluble AAT fractions (Figures 4A–4D). Moreover, a transient increase of serum ATZ polymers was detected along with a concomitant increase of total serum AAT (Figure 4E). ML-SA5 treatment did not result in liver toxicity, as shown by serum AST and ALT activities (SI Appendix, Figure S5). Consistent with the data with AAV-mediated TRPML1 gene transfer, PiZ mice treated with ML-SA5 showed little to absent TFEB nuclear translocation and no increase in LC3, in contrast to wild-type mice (Figures 4F and 4G). In contrast to AAV-mediated TRPML1 gene transfer, a reduction in SQSTM1/p62 was detected after ML-SA5 treatment, but the reasons for such a difference are unclear. Altogether, these results suggest that small-molecule-mediated activation of TRPML-1 has therapeutic potential for clearance of the hepatic accumulation of toxic ATZ.
Figure 4.

Pharmacological activation of TRPML1 reduces ATZ accumulation in PiZ mouse livers
(A) Experimental plan for ML-SA5 treatment in PiZ mice; each arrow indicates an ML-SA5 administration and blood drops indicates timing of blood collections. (B) Liver PAS-D staining of vehicle- and ML-SA5-treated PiZ mice and relative quantifications (n = 7 per group). (C) Immunofluorescence for ATZ polymers on livers of vehicle- and ML-SA5-treated PiZ mice (n = 7 per group). (D) Immunoblot for soluble and insoluble fractions of livers from vehicle- or ML-SA5-treated PiZ mice and their quantifications (n = 3 per group). GAPDH and H3 were used as loading controls. (E) Changes in percentages of serum total AAT (n = 8 per group) and ATZ polymers (vehicle, n = 7; ML-SA5, n = 8) in PiZ mice. (F) Immunohistochemistry for TFEB in livers from wild-type or PiZ mice treated with vehicle or ML-SA5 (n = 3 per group). (G) Immunoblots for autophagy markers SQSTM1/p62 and LC3 in livers of wild-type (n = 3 per group) and PiZ mice (n = 5 per group) treated with vehicle or ML-SA5 and their relative quantifications. Vehicle and MLSA5 samples are on the same gel but are not contiguous. Data are shown as mean ± standard error. Student’s t test: ∗p value < 0.05; ∗∗p value < 0.01. A.U., arbitrary units; p.i., post-injection; WT, wild-type.
Discussion
In this study, using either hepatic gene transfer or pharmacological activation, we showed the efficacy of TRPML1 at reducing the toxic burden of ATZ and fibrosis in a mouse model of liver disease due to AAT deficiency. Among several functions including endocytosis, membrane trafficking, and lysosomal biogenesis, the release of Ca2+ into the cytosol from lysosomes or late endosomal lumen mediated by TRPML114,17 regulates lysosomal exocytosis.30,31 Lysosomal exocytosis entails lysosomal fusion with the plasma membrane for expulsion of their storage materials outside the cells.18 This pathway plays a major role in several processes, including immune responses, bone resorption, and repair of injured plasma membrane.32,33 By increasing intracellular Ca2+, TRPML1 expands the pool of lysosomes close to and their fusion with the plasma membrane. Previous studies showed that lysosomal exocytosis induced by TFEB through the upregulation of TRPML1 promotes lysosomal clearance of pathological storage in models of lysosomal storage disorders (LSDs)31 and Alzheimer disease.34 In the present study, we expand the disease targets of TRPML1-induced lysosomal exocytosis to include the liver disease due to ATZ. Indeed, we found increased lysosomal exocytosis and serum secretion of polymeric ATZ by hepatocytes following TRPML1 gene transfer in a mouse model of AAT deficiency.
TRPML1 upregulates autophagy by multiple mechanisms. First, it activates calcineurin, a phosphatase that promotes TFEB nuclear translocation and activation.14,15,16 Second, it activates calmodulin-dependent protein kinase kinase b and AMP-activated protein kinase, leading to autophagosome formation in a TFEB-independent manner.17 Third, it increases autophagosome-lysosome fusion.35 Fourth, it is required for lysosome tubulation and autophagic lysosome reformation35,36 and lysosomal biogenesis.37
Despite its role in TFEB-mediated activation of autophagy,14,15,16 we did not observe induction of TFEB nuclear translocation in PiZ mice after TRPML1 gene transfer or treatment with a TRPML1 agonist. This observation suggests an impairment of TFEB-mediated autophagy in livers with ATZ storage but also suggests that the ATZ globule clearance induced by TRPML1 is independent from TFEB. The reasons underlying the impaired TFEB nuclear translocation in PiZ mice compared to wild-type mice after TRPML1 gene transfer are unclear and will require further investigations. Interestingly, the lack of TFEB nuclear translocation after TRPML1 gene transfer or activation points to lysosomal exocytosis and not increased autophagy as the main and novel mechanism involved in TRPML1-mediated clearance of hepatic ATZ storage.
Further studies are required to understand delivery of ATZ to lysosomes for lysosomal exocytosis. The mechanisms underlying ATZ polymer delivery from the ER lumen to endolysosomes may require ER capture within autophagosomes or single-membrane, ER-derived, ATZ-containing vesicles releasing their content within endolysosomes upon membrane fusion through ER-to-lysosome-associated degradation pathway (ERLAD), a recently described pathway in ATZ-expressing cells (Figure 5).38
Figure 5.

Genetic or pharmacological activation of TRPML1 ameliorates liver disease of PiZ mice
Graphic representation of therapeutic efficacy of TRPML1-mediated lysosomal exocytosis in PiZ mice. Delivery of ATZ polymers to lysosome for exocytosis may rely on ER-to-lysosome-associated degradation pathway (ERLAD) (1) or autophagosome-lysosome fusion (2).
Molecules that modulate the activity of the TRPML1 channel20,28 have shown therapeutic potential in various disorders, including Nieman Pick disease21 and Parkinson disease.39 By promoting lysosomal exocytosis, these TRPML1 agonists have also been shown to promote sarcolemma repair, thus alleviating the muscle defect in mouse models of Duchenne muscular dystrophy.28 Preclinical and clinical developments in these disorders might facilitate therapeutic applications of these compounds also in AAT deficiency.
In conclusion, we found that TRPML1-mediated lysosomal exocytosis is a druggable pathway for therapy of the liver disease due to ATZ that can be added to therapeutic strategies based on downregulation of ATZ expression by siRNA8 and activation of intracellular degradation pathways, such as the proteasome and autophagy.9,40 Based on the different mode of action, TRPML1 activation might be used in combination with siRNA or drugs increasing autophagy to increase their therapeutic efficacy.
Materials and methods
Adeno-associated viral vectors
PCR amplification of hTFEB3xflag from pAAV-CMV-TFEB-3xflag41 and standard cloning procedures were used to produce the pAAV-TBG-TFEB-3xflag vector. TRPML1-Myc-DDK from pCMV6-TRPML1-Myc-DDK (RC201010 Origene) and standard cloning procedures were used to produce the pAAV-TBG-hTRPML1-Myc-DDK vector. pAAV-TBG-GFP vector was provided by AAV TIGEM Vector Core. AAV8 viral vectors were produced by the AAV Vector Core at TIGEM. pAAV2/8-TBG-TRPML1-Myc-DDK, pAAV2/8-TBG-hTFEB-3xflag, and pAAV2/8-TBG-EGFP were triple-transfected in sub-confluent HEK-293 cells along with pAd-Helper and pack2/1 packaging plasmids as previously described.42 Recombinant vectors were purified by CsCl gradient centrifugation as previously described.42 Vector titers were expressed as genome copies per milliliter (GC/mL) that were determined by both qPCR and dot-blot analysis.
Mouse studies
Animal procedures were approved by the Italian Ministry of Health. PiZ transgenic mice43 were maintained in a C57BL/6J background. C57BL/6J wild-type mice (Charles River) were used as control mice. Injections of AAV8-TBG-GFP, AAV8-TBG-TRPML1-Myc-DDK, and AAV8-TBG-TFEB-3xflag were performed in a volume of 200 μL at the dose of 5 x 1012 GC/kg of body weight in the retro-orbital plexus of 6- or 16-week-old male PiZ or wild-type mice. Mice were sacrificed at 4 or 12 weeks post injection, and liver samples were harvested for analyses. ML-SA5 (kindly provided by CASMA Therapeutics) was dissolved in 1:3 DMSO:PEG400 and injected intraperitoneally daily for 15 days at a dose of 2 mg per kg of body weight. Blood samples were collected by retro-orbital bleeding at the indicated time points.
Analyses on serum samples
Serum samples were analyzed by ELISA for ATZ detection. To detect human ATZ by ELISA, multi-well plates (Nunc Maxisorp) were coated with Cappel goat anti-human AAT (MP Biomedicals) and then blocked in 0.1% phosphate buffered saline (PBS) Tween 20 containing 5% non-fat milk. Serial dilutions of purified human AAT were loaded to build a standard curve. Rabbit anti-human AAT (Dako; 1:4,000) was used as capturing antibody and goat anti-rabbit IgG-horseradish peroxidase (HRP) (Dako; 1:2,000) as secondary antibody. For detection of the polymeric ATZ, the 2C1 anti-polymer monoclonal antibody (Hycultbiotech; 1:400) was used.
For IDS enzyme activity, serum samples were incubated with 1.25 mM 4-methylumbelliferyl-α-L-iduronide-2-sulfate (Carbosynth) for 4 h. The reaction was continued by adding 30 μL of hIDUA solution (1 μg/mL) in Pi/Ci buffer (0.02% Na-azide in 0.2 M sodium-phosphate/0.1 M citrate buffer, pH 4.5) for a 24-h incubation. The reaction was stopped by adding 200 μL of stop buffer (0.025% Triton X-100 in 0.5 M NaHCO3/0.5 M Na2CO3 buffer), and fluorescence was read at excitation 350 nm/emission 460 nm on a fluorometer (GloMax Explorer Multimode Microplate of Reader).
GALNS activity was determined fluorometrically. Briefly, 4 μL of serum was incubated with 22 mM 4-methylumbelliferyl-β-D-galactopyranoside-6-sulfate fluorogenic substrate (Carbosynth) in 0.1 M NaCl/sodium acetate buffer, pH 4.3, for 24 h at 37°C. Next, the reaction mixtures underwent a second incubation with 10 mg/mL β-galactosidase from Aspergillus oryzae (Sigma-Aldrich) in 0.1 M NaCl/sodium acetate buffer, pH 4.3, for an additional 30 min at 37°C. The reactions were stopped with 1 M glycine/NaOH, pH 10.5, solution. Released fluorescence was read at excitation λ = 366 nm and emission λ = 450 nm with a Synergy Neo microplate reader (BioTek). A standard curve was generated using 4-methylumbelliferone sodium salt (Sigma-Aldrich). GALNS activity was expressed as nanomoles of 4-methylumbelliferone released per ml of serum per hour.
Liver stainings
PAS-D staining was performed on 6-μm-thick paraffin sections of livers. Liver specimens were fixed in 4% Paraformaldehyde for 12 h and stored in 70% ethanol, embedded into paraffin blocks, and cut into 6-μm sections. Sections were rehydrated and treated with amylase solution 0.5% (α-amylase type VI-B, Sigma) for 20 min and then stained with PAS-D reagent according to the manufacturer’s instructions (Bio-Optica). Sirius red staining was performed on 6-μm liver sections rehydrated and stained for 1 h in picro-Sirius red solution (0.1% Sirius red in saturated aqueous solution of picric acid). After two changes of acidified water (5 mL glacial acetic acid in 1 L water), sections were dehydrated in three changes of 100% ethanol, cleared in xylene, and mounted in a resinous medium. Quantification was performed using the ImageJ plug-in “Analyze particles” on the segmented images calculating the percentage area staining positively in five to nine randomly selected low power views.
For the immunostaining, 6-μm-thick liver paraffin sections were deparaffinized and rehydrated. Heat-induced epitope retrieval was made by using 10 mM sodium citrate buffer (pH 6.0) and maintaining sections at a sub-boiling temperature for 15 min. Sections were washed in PBS, pH 7.4, for 5 min and then permeabilized with 0.2% Triton in PBS (Sigma) for 20 min. Sections were then incubated for 1 h at room temperature with blocking solution (3% bovine serum albumin [Sigma], 5% donkey serum [Millipore], 0.1% Triton in PBS) and overnight at 4°C with primary antibodies (SI Appendix, Table S1) diluted in the blocking solution. Sections were incubated for 1 h at room temperature with the secondary antibodies. Secondary antibodies made in donkey were Alexa Fluor 488 anti-rabbit (Invitrogen) and Alexa Fluor 594 anti-mouse (Invitrogen). Nuclei were counterstained with DAPI (Invitrogen).
Immunohistochemistry analyses were performed in 6-μm paraffin sections with VENTANA BenchMark Ultra automated staining instrument (Ventana Medical Systems, Roche), using VENTANA reagents except as noted, according to manufacturer’s instructions. Slides were deparaffinized using EZ Prep solution (cat # 950-102) for 16 min at 72°C. Epitope retrieval was accomplished with CC1 solution (cat # 950-224) at a high temperature (95°C) for a period that is suitable for a specific tissue type. Antibodies were titered with a blocking solution into user-fillable dispensers for use on the automated stainer. For bright-field detection, slides were developed using the VENTANA ultraview Universal DAB detection kit (cat #760-500) according to the manufacturer’s instructions. Slides were then counterstained with hematoxylin II (cat # 790-2208) for 8 min, followed by bluing reagent (cat # 760-2037) for 4 min. For fluorescent detection, slides were developed using DISCOVERY FAM Kit (cat # 760-243) and DISCOVERY Cy5 Kit (cat# 760-238) for 8 min. Slides were then counterstained with DISCOVERY QD DAPI (cat # 760-4196) for 8 min. Whole digital slides were viewed by ZEN Blue software. Bright-field sections were scanned with Zeiss Axio Scan.Z1, and fluorescent sections were acquired with a confocal microscope. Antibodies are listed in SI Appendix and Table S1.
For triple immunostaining, 6-μm cryosections were rehydrated, permeabilized with PBS 0.2% Triton X-100, blocked, and incubated overnight at 4°C with primary antibodies (SI Appendix, Table S1) and then with secondary antibodies (AlexaFluor) for 1 h at room temperature. Nuclei were counterstained with DAPI (Invitrogen). Stained liver sections were mounted, covered with a cover slip, and examined under a Zeiss LSM 710 confocal laser-scanning macroscope and Leica DM500 microscope for bright field.
Electron microscopy
Liver tissue was fixed for 24 h at 4°C in a mixture of 2% paraformaldehyde and 1% glutaraldehyde prepared in 0.2 M HEPES buffer (pH 7.4). Samples were post-fixed as previously described.44 After dehydration, the specimens were embedded in epoxy resin and polymerized at 60°C for 72 h. Thin, 60-nm sections were cut on a Leica EM UC7 microtome. Electron microscopy images were acquired using an FEI Tecnai-12 electron microscope (Thermo Fisher) equipped with a VELETTA CCD digital camera. The number of lysosomes associated with the plasma membrane was counted considering a reference concentric area at radii of 0–4 μm including both the bile canaliculus and basolateral membrane45 using EM images acquired at low magnification (×9900, 12 micrographs for each condition).
Western blot analyses
Liver samples were homogenized in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, pH 8.0, 0.1% SDS) containing complete protease inhibitor cocktail and phosphatase inhibitor (Roche Applied Science). Samples were incubated for 20 min at 4°C and then centrifuged at 16,000 g for 10 min. Pellets were discarded, and cell lysates were used for western blots. 10–20 μg of liver proteins was electrophoresed on a 4%–20% SDS-PAGE. After transfer to PVDF membrane, blots were blocked in TBS-Tween 20 containing 5% non-fat milk for 1 h at room temperature, and the primary antibody was then applied overnight at 4°C. Anti-rabbit IgG, anti-mouse IgG, or anti-rat-IgG conjugated with horseradish peroxidase (GE Healthcare, 1:3,000) and ECL (Pierce) were used for detection. Equal gel loading was confirmed with immunoblot for β-actin (Novus Biological). Primary antibodies are listed in the SI Appendix and Table S1.
Preparation of soluble and insoluble fractions from livers was performed as previously described,46 and immunoblotting was performed using antibodies against the total AAT and the polymeric Z-AAT. GAPDH (Santa Cruz) and H3 (Cell Signaling) were used as markers of cytosolic or nuclear fractions, respectively.
Real-time PCR
Total RNA was extracted from liver tissue in TRIzol reagent (Invitrogen) using the RNeasy kit (Qiagen). RNA was reverse transcribed using a first-strand complementary deoxyribonucleic acid kit with random primers according to the manufacturer’s protocol (Applied Biosystems). The RT-PCR reactions were performed using the SYBR Green Master Mix (Roche) and the Roche Light Cycler 480 system (Roche). PCR conditions for all analyzed genes were as follows: pre-heating, 5 min at 95°C; cycling, 40 cycles of 15 s at 95°C, 15 s at 60°C, and 25 s at 72°C. Results were expressed in terms of cycle threshold (Ct). Ct values were averaged for each duplicate. β2-microglobulin or Ribosomal protein S16 genes were used as endogenous controls (reference markers). Differences between the mean Ct values of the tested genes and those of the reference gene were calculated as ΔCtgene = Ctgene – Ctreference. Relative fold increase in expression was determined as 2–ΔΔCt. Primers used for RT-PCR are listed in SI Appendix and Table S2.
Cell culture studies
HeLa and HTO/Z cells were cultured in Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum, 5% penicillin/streptomycin, and 2 mM L-glutamine. Cells were treated with 10 μM ML-SA5 or DMSO as vehicle. Following incubation for 24 h, cells were incubated with 40 nM bafilomycin A1 (Sigma) or DMSO for 4 h, washed once with cold PBS, and scraped with RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, pH 8.0, 0.1% SDS) containing complete protease and phosphatase inhibitor cocktails (Roche). Samples were incubated for 20 min at 4°C, centrifuged at 16,000 g for 10 min, and cell lysates were used for western blot analysis. For flow cytometry, HeLa and HTO/Z cells were incubated with ML-SA5 for 30 min, harvested with trypsin-EDTA, and washed in PBS. Staining for luminal LAMP1 was performed using a specific LAMP1-PE antibody (Santa Cruz; 1:200). FACS analysis was performed using the cytometer BD FACS ARIA III. Data were analyzed using Flow-Jo software (Flow-Jo, Ashland, OR). In all experiments, at least 10,000 events were acquired. Dead cells and doublets were excluded from analysis based on FSC-A/SSC-A and FSC-A/FSC-H dot-plot, respectively.
Statistical analyses
Data are expressed as mean ± standard error. Statistical significance was calculated using the Student’s two-tailed t test or two-way ANOVA. A p value <0.05 was considered statistically significant.
Acknowledgments
We are grateful to Cathal Wilson for editing and critical reading of the manuscript. We thank TIGEM AAV Vector Core for production of AAV vectors and TIGEM Advance Histopathology Facility for tissue processing and embedding and for setup of protocols for detection of mouse TFEB. We are grateful to CASMA Therapeutics for providing ML-SA5 and Jeff Teckman for HTO/Z cells. This work was supported by the Alpha-1 Foundation (to N.B.-P. and N.P.) and Associazione Italiana sul Cancro (AIRC) (to N.P.).
Author contributions
N.P. performed in vivo experiments, most of the assays included in the study, and interpreted the data; F.A. performed in vitro experiments and immunostainings on liver sections; R.C. handled mouse colonies, performed genotyping, ELISA, and western blot analyses; V.M. performed and analyzed ML-SA5 experiments in vivo; T.G. performed and analyzed TRPML1 gene transfer experiments; B.M.C. performed ELISA assays; B.L. performed triple immunostaining on liver sections; E.P. performed EM studies; V.C. performed TRPML1 and TFEB cloning, IDS activity on mouse serum, immunoblot for validation, and immunohistochemistry for TFEB; L.D.S. performed GALNS activity; E.N. provided technical support with mouse experiments; N.C.S. generated the TRPML1 and TFEB expression cassettes, performed the validation experiments, and contributed to interpretation of the corresponding results; P.P. contributed to interpretation of the results; N.P. and N.B.-P. designed, led, and supervised the experiments and wrote the manuscript.
Declaration of interests
The authors have no conflict of interest to disclose.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.06.018.
Contributor Information
Nunzia Pastore, Email: pastore@tigem.it.
Nicola Brunetti-Pierri, Email: brunetti@tigem.it.
Supplemental information
Data availability
All data presented in this work are available from the authors upon request.
References
- 1.de Serres F.J. Worldwide racial and ethnic distribution of alpha1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveys. Chest. 2002;122:1818–1829. doi: 10.1378/chest.122.5.1818. [DOI] [PubMed] [Google Scholar]
- 2.Silverman G.A., Bird P.I., Carrell R.W., Church F.C., Coughlin P.B., Gettins P.G., Irving J.A., Lomas D.A., Luke C.J., Moyer R.W., et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 3.Carrell R.W., Lomas D.A. Alpha1-antitrypsin deficiency--a model for conformational diseases. N. Engl. J. Med. 2002;346:45–53. doi: 10.1056/NEJMra010772. [DOI] [PubMed] [Google Scholar]
- 4.Perlmutter D.H. Alpha(1)-Antitrypsin Deficiency. Curr. Treat. Options Gastroenterol. 2000;3:451–456. doi: 10.1007/s11938-000-0033-8. [DOI] [PubMed] [Google Scholar]
- 5.Fromme M., Schneider C.V., Trautwein C., Brunetti-Pierri N., Strnad P. Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J. Hepatol. 2022;76:946–958. doi: 10.1016/j.jhep.2021.11.022. [DOI] [PubMed] [Google Scholar]
- 6.Brantly M.L., Chulay J.D., Wang L., Mueller C., Humphries M., Spencer L.T., Rouhani F., Conlon T.J., Calcedo R., Betts M.R., et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc. Natl. Acad. Sci. USA. 2009;106:16363–16368. doi: 10.1073/pnas.0904514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mueller C., Tang Q., Gruntman A., Blomenkamp K., Teckman J., Song L., Zamore P.D., Flotte T.R. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol. Ther. 2012;20:590–600. doi: 10.1038/mt.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strnad P., Mandorfer M., Choudhury G., Griffiths W., Trautwein C., Loomba R., Schluep T., Chang T., Yi M., Given B.D., et al. Fazirsiran for Liver Disease Associated with Alpha(1)-Antitrypsin Deficiency. N. Engl. J. Med. 2022;387:514–524. doi: 10.1056/NEJMoa2205416. [DOI] [PubMed] [Google Scholar]
- 9.Hidvegi T., Ewing M., Hale P., Dippold C., Beckett C., Kemp C., Maurice N., Mukherjee A., Goldbach C., Watkins S., et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 10.Lomas D.A., Irving J.A., Arico-Muendel C., Belyanskaya S., Brewster A., Brown M., Chung C.W., Dave H., Denis A., Dodic N., et al. Development of a small molecule that corrects misfolding and increases secretion of Z α1 -antitrypsin. EMBO Mol. Med. 2021;13:e13167. doi: 10.15252/emmm.202013167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A., Di Malta C., Donaudy F., Embrione V., Polishchuk R.S., et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 12.Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P., et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pastore N., Blomenkamp K., Annunziata F., Piccolo P., Mithbaokar P., Maria Sepe R., Vetrini F., Palmer D., Ng P., Polishchuk E., et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. 2013;5:397–412. doi: 10.1002/emmm.201202046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A., et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X., Cheng X., Yu L., Yang J., Calvo R., Patnaik S., Hu X., Gao Q., Yang M., Lawas M., et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016;7:12109. doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W., Gao Q., Yang M., Zhang X., Yu L., Lawas M., Li X., Bryant-Genevier M., Southall N.T., Marugan J., et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA. 2015;112:E1373–E1381. doi: 10.1073/pnas.1419669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scotto Rosato A., Montefusco S., Soldati C., Di Paola S., Capuozzo A., Monfregola J., Polishchuk E., Amabile A., Grimm C., Lombardo A., et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat. Commun. 2019;10:5630. doi: 10.1038/s41467-019-13572-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H., Ren D. Lysosomal physiology. Annu. Rev. Physiol. 2015;77:57–80. doi: 10.1146/annurev-physiol-021014-071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medina D.L. TRPML1 and TFEB, an Intimate Affair. Handb. Exp. Pharmacol. 2023;278:109–126. doi: 10.1007/164_2022_603. [DOI] [PubMed] [Google Scholar]
- 20.Chen C.C., Keller M., Hess M., Schiffmann R., Urban N., Wolfgardt A., Schaefer M., Bracher F., Biel M., Wahl-Schott C., Grimm C. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat. Commun. 2014;5:4681. doi: 10.1038/ncomms5681. [DOI] [PubMed] [Google Scholar]
- 21.Shen D., Wang X., Li X., Zhang X., Yao Z., Dibble S., Dong X.P., Yu T., Lieberman A.P., Showalter H.D., Xu H. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong X.Z., Sun X., Cao Q., Dong G., Schiffmann R., Dong X.P. BK channel agonist represents a potential therapeutic approach for lysosomal storage diseases. Sci. Rep. 2016;6:33684. doi: 10.1038/srep33684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlson J.A., Rogers B.B., Sifers R.N., Finegold M.J., Clift S.M., DeMayo F.J., Bullock D.W., Woo S.L. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J. Clin. Invest. 1989;83:1183–1190. doi: 10.1172/JCI113999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piccolo P., Annunziata P., Soria L.R., Attanasio S., Barbato A., Castello R., Carissimo A., Quagliata L., Terracciano L.M., Brunetti-Pierri N. Down-regulation of hepatocyte nuclear factor-4alpha and defective zonation in livers expressing mutant Z alpha1-antitrypsin. Hepatology. 2017;66:124–135. doi: 10.1002/hep.29160. [DOI] [PubMed] [Google Scholar]
- 25.Reddy A., Caler E.V., Andrews N.W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 26.Omairi S., Hau K.L., Collins-Hooper H., Scott C., Vaiyapuri S., Torelli S., Montanaro F., Matsakas A., Patel K. Regulation of the dystrophin-associated glycoprotein complex composition by the metabolic properties of muscle fibres. Sci. Rep. 2019;9:2770. doi: 10.1038/s41598-019-39532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jokela M., Lehtinen S., Palmio J., Saukkonen A.M., Huovinen S., Vihola A., Udd B. A novel COL6A2 mutation causing late-onset limb-girdle muscular dystrophy. J. Neurol. 2019;266:1649–1654. doi: 10.1007/s00415-019-09307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu L., Zhang X., Yang Y., Li D., Tang K., Zhao Z., He W., Wang C., Sahoo N., Converso-Baran K., et al. Small-molecule activation of lysosomal TRP channels ameliorates Duchenne muscular dystrophy in mouse models. Sci. Adv. 2020;6:eaaz2736. doi: 10.1126/sciadv.aaz2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teckman J.H., Burrows J., Hidvegi T., Schmidt B., Hale P.D., Perlmutter D.H. The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J. Biol. Chem. 2001;276:44865–44872. doi: 10.1074/jbc.M103703200. [DOI] [PubMed] [Google Scholar]
- 30.Medina D.L., Ballabio A. Lysosomal calcium regulates autophagy. Autophagy. 2015;11:970–971. doi: 10.1080/15548627.2015.1047130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medina D.L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J.A., Sardiello M., et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 2011;21:421–430. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Paola S., Medina D.L. TRPML1-/TFEB-Dependent Regulation of Lysosomal Exocytosis. Methods Mol. Biol. 2019;1925:143–144. doi: 10.1007/978-1-4939-9018-4_12. [DOI] [PubMed] [Google Scholar]
- 33.Andrews N.W., Almeida P.E., Corrotte M. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014;24:734–742. doi: 10.1016/j.tcb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bae M., Patel N., Xu H., Lee M., Tominaga-Yamanaka K., Nath A., Geiger J., Gorospe M., Mattson M.P., Haughey N.J. Activation of TRPML1 clears intraneuronal Abeta in preclinical models of HIV infection. J. Neurosci. 2014;34:11485–11503. doi: 10.1523/JNEUROSCI.0210-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X., Rydzewski N., Hider A., Zhang X., Yang J., Wang W., Gao Q., Cheng X., Xu H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016;18:404–417. doi: 10.1038/ncb3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y., Xu M., Zhu X., Yao J., Shen B., Dong X.P. Lysosomal Ca(2+) release channel TRPML1 regulates lysosome size by promoting mTORC1 activity. Eur. J. Cell Biol. 2019;98:116–123. doi: 10.1016/j.ejcb.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Cao Q., Yang Y., Zhong X.Z., Dong X.P. The lysosomal Ca(2+) release channel TRPML1 regulates lysosome size by activating calmodulin. J. Biol. Chem. 2017;292:8424–8435. doi: 10.1074/jbc.M116.772160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fregno I., Fasana E., Bergmann T.J., Raimondi A., Loi M., Soldà T., Galli C., D'Antuono R., Morone D., Danieli A., et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018;37:e99259. doi: 10.15252/embj.201899259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsunemi T., Perez-Rosello T., Ishiguro Y., Yoroisaka A., Jeon S., Hamada K., Rammonhan M., Wong Y.C., Xie Z., Akamatsu W., et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca(2+) Channel Agonists Protects Human Dopaminergic Neurons from alpha-Synuclein Toxicity. J. Neurosci. 2019;39:5760–5772. doi: 10.1523/JNEUROSCI.3085-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaushal S., Annamali M., Blomenkamp K., Rudnick D., Halloran D., Brunt E.M., Teckman J.H. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. 2010;235:700–709. doi: 10.1258/ebm.2010.009297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gatto F., Rossi B., Tarallo A., Polishchuk E., Polishchuk R., Carrella A., Nusco E., Alvino F.G., Iacobellis F., De Leonibus E., et al. AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci. Rep. 2017;7:15089. doi: 10.1038/s41598-017-15352-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao W., Chirmule N., Berta S.C., McCullough B., Gao G., Wilson J.M. Gene therapy vectors based on adeno-associated virus type 1. J. Virol. 1999;73:3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlson J.A., Rogers B.B., Sifers R.N., Hawkins H.K., Finegold M.J., Woo S.L. Multiple tissues express alpha 1-antitrypsin in transgenic mice and man. J. Clin. Invest. 1988;82:26–36. doi: 10.1172/JCI113580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polishchuk E.V., Polishchuk R.S. Pre-embedding labeling for subcellular detection of molecules with electron microscopy. Tissue Cell. 2019;57:103–110. doi: 10.1016/j.tice.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 45.Sewell R.B., Dillon C., Grinpukel S., Yeomans N.D., Smallwood R.A. Pericanalicular location of hepatocyte lysosomes and effects of fasting: a morphometric analysis. Hepatology. 1986;6:305–311. doi: 10.1002/hep.1840060225. [DOI] [PubMed] [Google Scholar]
- 46.Attanasio S., Ferriero R., Gernoux G., De Cegli R., Carissimo A., Nusco E., Campione S., Teckman J., Mueller C., Piccolo P., Brunetti-Pierri N. CHOP and c-JUN up-regulate the mutant Z alpha1-antitrypsin, exacerbating its aggregation and liver proteotoxicity. J. Biol. Chem. 2020;295:13213–13223. doi: 10.1074/jbc.RA120.014307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in this work are available from the authors upon request.