

Abstract
Gastrin-releasing peptide (GRP) binds to its receptor (GRP receptor [GRPR]) to regulate multiple biological processes, but the function of GRP/GRPR axis in acute kidney injury (AKI) remains unknown. In the present study, GRPR is highly expressed by tubular epithelial cells (TECs) in patients or mice with AKI, while histone deacetylase 8 may lead to the transcriptional activation of GRPR. Functionally, we uncovered that GRPR was pathogenic in AKI, as genetic deletion of GRPR was able to protect mice from cisplatin- and ischemia-induced AKI. This was further confirmed by specifically deleting the GRPR gene from TECs in GRPRFlox/Flox//KspCre mice. Mechanistically, we uncovered that GRPR was able to interact with Toll-like receptor 4 to activate STAT1 that bound the promoter of MLKL and CCL2 to induce TEC necroptosis, necroinflammation, and macrophages recruitment. This was further confirmed by overexpressing STAT1 to restore renal injury in GRPRFlox/Flox/KspCre mice. Concurrently, STAT1 induced GRP synthesis to enforce the GRP/GRPR/STAT1 positive feedback loop. Importantly, targeting GRPR by lentivirus-packaged small hairpin RNA or by treatment with a novel GRPR antagonist RH-1402 was able to inhibit cisplatin-induced AKI. In conclusion, GRPR is pathogenic in AKI and mediates AKI via the STAT1-dependent mechanism. Thus, targeting GRPR may be a novel therapeutic strategy for AKI.
Keywords: GRP, GRPR, STAT1, acute kidney injury, inflammation, necroptosis
Graphical abstract
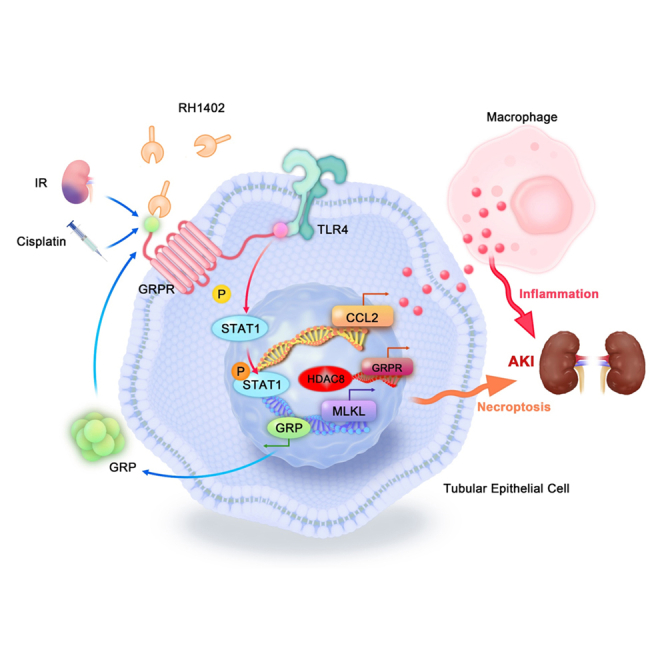
Meng et al. first revealed the important mechanism of GRPR mediating AKI through inflammation and necroptosis, suggested that targeting GRPR may be a novel therapeutic strategy for AKI.
Introduction
Acute kidney injury (AKI) is a serious syndrome characterized by sudden renal dysfunction, which affects millions of people worldwide.1,2 Severe and repeated AKI may cause abnormal cellular repair and fibrosis, leading to chronic diseases and end-stage renal disease.3 AKI is mainly caused by ischemia/reperfusion (I/R) injury, nephrotoxic insult, or sepsis. Sustained inflammation and programmed cell death are the major pathological features of AKI.4,5,6 Unfortunately, specific therapies for AKI are still lacking.7,8,9,10
The gastrin-releasing peptide (GRP) is a mammalian homolog of bombesin (BN), which is a tetradecapeptide originally isolated from the skin of the frog Bombina bombina.11,12 GRP binds with GRP receptor (GRPR) to induce intracellular changes that influence multiple cell behaviors and biological processes, including neuroendocrine regulation,13,14 gastrointestinal system functions, cancer growth,15 cell proliferation, and immune cell regulation.16,17 GRPR is a member of the G-protein coupled receptor family (GPCRs), also known as BN-like peptide binding receptor subtypes 2, and shows the highest affinity for GRP. Notably, GRP/GRPR may serve as a potential therapeutic target in inflammatory diseases,18 such as sepsis,19 asthma,20 rheumatoid arthritis,21 inflammatory bowel disease,22 hepatic I/R injury,23 and periodontitis.24 Although the involvement of GRP/GRPR signaling in the kidney was largely unknown until recently, emerging evidence has demonstrated the possibility of its participation in kidney diseases. GRPR is expressed in murine and sheep kidneys and is highly induced in renal cell carcinoma, while GRP promotes the growth of renal tumors.25,26 Recently, evidence from GRPR-targeted cancer therapy has shown that GRPR-targeted radioconjugates are highly concentrated in the kidney at a very early stage,27 further confirming the existence of GRPR in the kidney. ProGRP, the precursor of GRP, is a classic biomarker for clinical diagnosis of small cell lung carcinoma because it has higher stability compared with GRP.28 It is noteworthy that a key elimination symptom for high circulating ProGRP levels is kidney dysfunction, because ProGRP level is significantly higher in the renal failure group than in the healthy controls. Although accumulation of ProGRP in the kidney and circulation may be partly caused by the impaired renal excretion function, since ProGRP is primarily metabolized by the kidneys,28,29 whether kidney cells could produce GRP or whether the high level of GRP in the kidney could further promote renal dysfunction by binding GRPR and initiating downstream signaling remains to be determined.
In the present study, we observed highly activated GRP/GRPR signaling in biopsies of human patients with AKI, especially in the tubular region, which was further confirmed by animal models of AKI and cisplatin-treated tubular epithelial cells (TECs). Then, we explored the mechanisms by which GRPR was elevated in AKI. Considering the potential role of GRP/GRPR in inflammation regulation, we hypothesized that GRP/GRPR signaling in TECs promotes AKI by enhancing renal inflammation. To verify our hypothesis, we performed in vivo experiments using GRPR global knockout (KO) mice, in addition to GRPRFlox/Flox/KspCre mice in which GRPR was specifically knocked out from TECs, with distinct types of AKI models. In addition, we conducted in vitro studies in cisplatin-treated TECs with GRPR silencing or overexpression and explored the detailed mechanisms using RNA sequencing in GRPR-overexpressed TECs. GRPR-targeted therapy was evaluated in cisplatin-treated mice using two protocols: lentivirus-mediated in vivo GRPR knockdown and treatment with a novel GRPR antagonist termed RH-1402. RH-1402 was administered before and after AKI model establishment to evaluate both preventive and treatment effects.
Results
GRP/GRPR was upregulated in patients and mice with AKI
Serum ProGRP levels were significantly higher in patients with AKI than in healthy controls (Figure 1A). In human patient biopsies, we found that GRPR was highly expressed in AKI kidneys with the positive signal mostly in the tubules (Figure 1B). In the AKI models induced by I/R injury and cisplatin, the circulating ProGRP and GRP mRNA levels in the injured kidneys were both consistently increased (Figures 1C and 1D). To explore the location of GRPR in the setting of cisplatin-induced AKI, immunofluorescence of lotus tetragonolobus lectin (a proximal tubule marker), calbindin D28k (a distal tubule marker), aquaporin-3 (a collecting duct marker), and GRPR was performed. As a result, cisplatin markedly upregulated GRPR expression in proximal tubule epithelial cells and collecting ducts (Figures 1E and S1A, supporting information). Increased GRPR levels were also confirmed by immunohistochemistry (IHC), real-time PCR, western blotting, and quantitative analysis (Figures 1F–1H). Additionally, in the cisplatin-treated TECs, GRPR was induced as shown by immunofluorescence, real-time PCR, western blotting, and quantitative analyses (Figures S1B–S1D, supporting information). There is no significant difference of GRPR mRNA between the sham and I/R groups (Figure S1E, supporting information). The mRNA level of GRPR increased gradually at 0 h, 12 h, 18 h, and 24 h after cisplatin treatment (Figure S1F, supporting information). According to the data, we chose 18 h for the follow-up experiment, sodium butyrate (NaB), a class I histone deacetylase (HDAC) inhibitor, increased the transcription level of GRPR mRNA in cisplatin-treated TECs (Figure 1I). We then detected the protein level of class I HDACs and found that HDAC1 was upregulated, HDAC2 was unchanged, but HDAC3 and HDAC8 were significantly downregulated (Figure 1J). To further determine which HDAC regulates GRPR, we transfected TECs with HDAC3 and HDAC8 overexpression plasmid. The results of real-time PCR showed that HDAC3 and HDAC8 were successfully overexpressed (Figure S1G, supporting information). Real-time PCR and western blotting results showed that the overexpression of HADC8, but not HDAC3, by TECs decreased GRPR production at the mRNA and protein levels in response to cisplatin stimulation (Figures 1K, 1L, S1G, and S1H, supporting information). The chromatin immunoprecipitation (ChIP) assay further showed decreased enrichment of HDAC8 rather than HDAC3 in the GRPR promoter region, indicating that GRPR may be increased by the deacetylation mechanism of HDAC8 in cisplatin-treated TECs (Figure 1M). Compared with cisplatin stimulation, HDAC8 decreased significantly only at 12 h of hypoxia and 3 h of re-oxygenation (hypoxia and re-oxygenation [H/R]) (Figure S1I, supporting information). These data provide a molecular basis for our study of the GRP/GRPR axis in AKI.
Figure 1.

GRP/GRPR in patients with AKI, mouse model and in vitro model of AKI
(A) Serum ProGRP level in in healthy control and AKI patients were detected. (B) IHC staining of KIM1 and GRPR in kidneys of healthy control and AKI patients. (C) Serum ProGRP level in two different AKI models induced by I/R and cisplatin. (D) GRP mRNA levels in two different AKI models. (E) IF staining of GRPR and lotus tetragonolobus lectin (LTL) in cisplatin-induced AKI mouse kidneys. (F) IHC staining of GRPR in cisplatin-induced AKI mouse kidneys. (G) GRPR mRNA level in kidneys of AKI mice. (H) GRPR protein level in kidneys of AKI mice. (I) GRPR expression was detected in HK2 by real-time PCR. (J) Western blot and quantification of HDAC1/2/3/8. (K) GRPR expression in vitro were determined by real-time PCR. (M) HDAC8 and GRPR protein level in vitro. (M) The binding of HDAC8 on GRPR promoter regions were detected by ChIP assay. Independent experiments were performed throughout the in vitro studies in triplicate. Data represented the mean ± SEM for at least 6 persons or mice. ∗∗p < 0.01, ∗∗∗p < 0.001 vs. sham/saline/NC/EV. ###p < 0.001 vs. EV + Cis. Cis, cisplatin control; EV, empty vector; NC, normal; OE, overexpression.
Global or tubular-specific knockout of GRPR protected against I/R or cisplatin-induced AKI
We constructed GRPR KO mice to determine the function of GRPR in AKI (Figure 2A). GRPR deficiency was confirmed at the genomic DNA and protein levels (Figures 2B and 2C). periodic acid-Schiff (PAS) staining showed that GRPR deficiency attenuated cisplatin-induced renal injury (Figure 2D) and restored the renal dysfunction as detected by serum creatinine and blood urea nitrogen (BUN) assays (Figures 2E and 2F). The protein and mRNA levels of KIM1, a biomarker for tubular injury, were increased in cisplatin nephropathy and downregulated by GRPR KO (Figures 2G and 2H). Additionally, GRPR KO alleviated the synthesis of pro-inflammatory factors, including tumor necrosis factor (TNF)-α, IL-1β, and CCL2, compared with that in control mice (Figure 2H). The function of GRPR was then evaluated in the second AKI model. GRPR KO protected against I/R-induced renal dysfunction, injury, and inflammation (Figures 2I–2K).
Figure 2.

Global knockout of GRPR protected against renal dysfunction, renal injury, and inflammation in AKI models induced by cisplatin and I/R
(A) Genotyping strategy. (B) GRPR deficiency was confirmed by detecting genomic DNA. (C) Global knockout of GRPR was verified by western blot. (D) Representative PAS staining pictures of kidneys from GRPR WT and KO mice treated with cisplatin and their quantification. Scale bar, 50 μm. (E and F) Serum creatinine and BUN levels. (G) KIM1 protein level detected by western blot and quantification. (H) Relative mRNA levels of KIM1, TNF-α, IL-1β, and CCL2 were determined by real-time PCR. (I) Representative PAS staining of kidneys from GRPR WT and KO mice treated with I/R injury. (J and K) Serum creatinine and BUN levels. Data represented the mean ± SEM for six mice. ∗∗∗p < 0.001 vs. WT + saline/WT + sham. ##p < 0.01, ###p < 0.001 vs. WT + Cis/WT + I/R. Cis, cisplatin; WT, wild type.
To demonstrate the importance of GRPR in TECs, we constructed GRPRFlox/Flox/KspCre mice (Figure 3A). Western blotting and real-time PCR showed that approximately 80% of GRPR was removed from the kidney (Figures 3B and 3C). Conditional knockout of GRPR from kidney attenuated renal damage and restored the decline of renal function (Figures 3D–3F). The KIM1 levels and production of proinflammatory indexes were substantially decreased in GRPRFlox/Flox/KspCre mice (Figures 3G and 3H). Nevertheless, GRPRFlox/Flox mice developed obvious damage after being intraperitoneally injected with cisplatin, as indicated by mitochondrial swelling and loss of cell organelle contents of TECs based on transmission electron microscopy. These pathological changes were attenuated in GRPRFlox/Flox/KspCre mice (Figure 3I). The function of GRPR in TECs was also evaluated in the I/R-induced AKI model. The finding showed that conditional knockout of GRPR from TECs protected against I/R-induced renal dysfunction, injury, and inflammation (Figures 3J–3N).
Figure 3.

Conditional knockout of GRPR from kidney TECs protected against renal dysfunction, renal injury, and inflammation in AKI models induced by cisplatin and I/R
(A) The construction of GRPRFlox/Flox/KspCre mice. (B) Conditional knockout of GRPR from kidney TECs was confirmed by western blot. (C) GRPR expression was determined by real-time PCR. (D) Representative PAS staining pictures of kidneys from GRPRFlox/Flox and GRPRFlox/Flox/KspCre mice treated with cisplatin and their quantification. (E, F) Serum creatinine and BUN levels. (G) KIM1 protein level was detected by Western blot. (H) Relative mRNA levels of KIM1, TNF-α, IL-1β, and CCL2 were determined by real-time PCR. (I) Representative electron micrographs of kidneys are shown. (J) Representative PAS staining pictures of kidneys from GRPRFlox/Flox and GRPRFlox/Flox/KspCre mice treated with I/R injury and their quantification. (K, L) Serum creatinine and BUN levels. (M) Relative mRNA levels of KIM1, TNF-α, IL-1β, and CCL2 were determined by real-time PCR. (N) IF staining of F4/80. Data represented the mean ± SEM for six mice. ∗∗p < 0.01, ∗∗∗p < 0.001 vs. FF/FF + saline/FF + sham. #p < 0.05, ##p < 0.01, ###p < 0.001 vs. FF + Cis/FF + I/R. Cis, cisplatin.
Cisplatin or H/R-induced injury and inflammatory response in TECs was inhibited by silencing GRPR, but promoted by overexpressing GRPR
We validated our in vitro findings in human HK2 cells by transfecting the small hairpin RNA (shRNA) plasmid (Figure S2A, supporting information). Western blotting confirmed the silencing of GRPR at the protein level; we chose sequence 2, which showed the most significant suppressive effect on GRPR, for further experiments (Figure S2B, supporting information). Western blotting and real-time PCR results showed that silencing GRPR could diminish the increase in KIM1 induced by cisplatin at the protein and mRNA levels (Figures S2C and S2D, supporting information). GRPR knockdown decreased the synthesis of cisplatin-induced pro-inflammatory factors, including TNF-α, IL-1β, and CCL2, compared with the control group (Figure S2D, supporting information). For comparison, we constructed a GRPR overexpression plasmid and transfected it into TECs (Figure S2E, supporting information). The GRPR protein level increased 3-fold after transfection (Figure S2F, supporting information). Western blotting and real-time PCR results showed that overexpression of GRPR further increased KIM1 at the protein and mRNA levels in response to cisplatin (Figures S2G and S2H, supporting information). The inflammatory response was also induced in the GRPR overexpression group in the absence or presence of cisplatin (Figure S2H, supporting information). Furthermore, we constructed a GRPR KO cell line in TECs (Figure S2I, supporting information), which was confirmed by Western blot assay (Figure S2J, supporting information). GRPR KO decreased cisplatin-induced increases in KIM1 and pro-inflammatory factors at the protein and mRNA levels (Figures S2K and S2L, supporting information). We also validated in cellular experiments of H/R 3 or 6 h that these data show that GRPR has the same function as cisplatin model in H/R (Figures S3A–S3F, supporting information). These in vitro data confirmed that GRPR promoted cisplatin or H/R-induced cell damage and inflammation in TECs.
GRPR promoted renal injury and inflammation via a STAT1-dependent mechanism
To further explore the mechanisms by which GRPR promotes renal injury and inflammation, we performed RNA sequencing in GRPR overexpression and control medullary thymic epithelial cells (mTECs). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that GRPR overexpression was positively correlated with many inflammation-regulatory pathways, including the NOD-like receptor signaling pathway, TNF signaling pathway, and Toll-like receptor (TLR) signaling pathway (Figure 4A). It is noteworthy that GRPR overexpression was also correlated with the necroptosis-regulatory pathway, as this is a key pathway mediating programmed cell death and necro-inflammation (Figure 4A). Changes in several key genes were clearly evident in the heatmap. The results showed that GRPR was successfully overexpressed. Consequently, the transcription factor STAT1 was largely induced, and the necroptosis-regulatory genes MLKL and CCL2 were also upregulated (Figure 4B). We then confirmed the changes in these pathways in GRPR knockdown HK2 cells and GRPR KO mice. Western blot analysis demonstrated that silencing GRPR suppressed both cisplatin-induced STAT1 and its phosphorylation, and inhibited the RIPK1/RIPK3/MLKL axis and P65 nuclear factor (NF)-κB signaling. These results were confirmed in vivo in GRPR KO mice with AKI (Figures 4C and 4D). Importantly, immunofluorescence and co-IP results showed that GRPR co-localized and interacted with TLR4 (Figures 4E, 4F, and S4A, supporting information), molecular docking showed that GRPR and TLR4 had mutiple potential interaction sites (Figure S4B, supporting information), and western blotting further demonstrated that knockdown of TLR4 decreased GRPR overexpression-induced STAT1 production and phosphorylation, knockdown of GRPR also down-regulated TLR4 expression. These data indicated the importance of TLR4 in GRPR-induced STAT1 upregulation and phosphorylation (Figures 4G and S4C, supporting information). A ChIP assay showed that transcriptional factor STAT1 bound with the promoter regions of MLKL and CCL2 to induce their production, and STAT1 knockdown prevented the upregulation of MLKL and CCL2 (Figures 4H and 4I). Interestingly, we found that GRPR activation induced the synthesis of its ligand GRP, thereby forming a positive feedback loop. Cisplatin-induced GRP was decreased by GRPR knockdown, but further promoted by GRPR overexpression (Figure 4J). Mechanistically, STAT1 could also bind the promoter region of GRP to induce its production, and STAT1 knockdown suppressed cisplatin-induced GRP upregulation (Figures 4K and 4L). Immunofluorescence analyses showed that silence GRPR decreased phosphorylation and membrane translocation of MLKL in cisplatin-induced TECs (Figure 4M).
Figure 4.

GRPR promoted renal injury and inflammation in STAT1-dependent mechanisms
(A) KEGG pathway analysis of RNA-seq in GRPR overexpression and control mTECs. (B) Heatmap. (C) Correlated signaling pathways in GRPR knockdown and control HK2 cells. (D) Correlated signaling pathways in GRPR WT and KO mice. (E) IF staining of GRPR and TLR4 in TECs. (F) Co-IP of GRPR and TLR4 in cisplatin-treated TECs. (G) Protein levels of P-STAT1/STAT1/TLR4/GRPR and quantification. (H) The binding of P-STAT1 on MLKL or CCL2 promoter regions were detected by ChIP assay. (I) Relative mRNA levels of STAT1, MLKL and CCL2 in STAT1 knockdown mTECs. (J) The mRNA level of GRP in GRPR knockdown or overexpressed mTECs. (K) The binding of P-STAT1 to GRP promoter by ChIP assay. (L) The mRNA level of GRP in STAT1 knockdown mTECs. (M) IF staining of P-MLKL. Independent experiments were performed throughout the in vitro studies in triplicate. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. EV + NC/WT + saline/TLR4-KD/P-STAT1+IgG. #p < 0.05, ###p < 0.001 vs. EV + Cis/WT + Cis/TLR4-KD/EV + Cis. $$p < 0.01, $$$p < 0.001 vs. GRPR-OE. Cis, cisplatin; EV, empty vector; KD, knockdown; NC, normal control; OE, overexpression; WT, wild type.
Overexpression of STAT1 restored AKI in GRPRFlox/Flox/KspCre mice or cells in vivo and in vitro
To further elucidate the key function of STAT1 in the GRP/GRPR axis, we constructed lentivirus-mediated STAT1 overexpression in GRPRFlox/Flox/KspCre mice, which was confirmed by western blotting and real-time PCR (Figures 5A and 5B). We found that the restoration of STAT1 expression and phosphorylation increased renal injury, serum levels of creatinine, and BUN (Figures 5C–5E). Immunofluorescence, Western blot, and real-time PCR analysis demonstrated that overexpression of STAT1 increased production of proinflammatory indexes, activation of P65 NF-κB signaling pathway, as well as KIM1 protein and mRNA in cisplatin-treated GRPRFF/KspCre mice (Figures 5A, 5B, and 5F). We also verified this result in in vitro experiments and came to the same conclusion (Figures 5G–5I). These results suggest that STAT1 plays a key role in the regulation of GRPR in AKI.
Figure 5.

Rescue of STAT1 expression in conditional KO mice or KO cell restored renal damage
(A) P-STAT1/STAT1, PP65/P65, KIM1 protein level was determined by western blot. (B) Relative mRNA levels of STAT1, KIM1 and proinflammatory genes. (C) Representative PAS staining of kidney in lentivirus-mediated STAT1 overexpression in mice treated with cisplatin. (D, E) Serum creatinine and BUN levels. (F) IF staining of KIM1, F4/80, and TNF-α. (G) Protein levels of P-STAT1/STAT1, PP65/P65, and quantification. (H) Relative mRNA levels of STAT1, KIM1, and proinflammatory indexes were determined by real-time PCR. (I) IF staining of P-STAT1 in TECs. Data represented the mean ± SEM for six mice. ∗p < 0.05, ∗∗∗p < 0.001 vs. FF + Cis. #p < 0.05, ##p < 0.01, ###p < 0.001 vs. KspCre+Cis. Independent experiments were performed throughout the in vitro studies in triplicate. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. WT + Cis. #p < 0.05, ###p < 0.001 vs. KO + Cis. Cis, cisplatin; WT, wild type.
Lentivirus-mediated in vivo GRPR knockdown protected against cisplatin-induced AKI
We then analyzed the effects of GRPR-targeted therapy using two methods. First, we used lentivirus-mediated GRPR knockdown in mice (Figure 6A). Western blotting results showed that GRPR expression was decreased in GRPR shRNA-transfected kidneys (Figure 6B). PAS staining and quantitative data demonstrated that GRPR silencing relieved cisplatin-induced kidney damage (Figure 6C). Results of serum creatinine and BUN assays showed that GRPR silencing restored renal dysfunction in response to cisplatin (20 mg/kg) (Figures 6D and 6E). Real-time PCR and western blot results indicated that GRPR silencing decreased the cisplatin-induced increase in KIM1 (Figures 6F and 6G). In addition, disruption of GRPR attenuated cisplatin-induced renal inflammation, as shown by reduced pro-inflammatory factors and F4/80+ macrophage infiltration (Figures 6G and 6H). Mechanistically, GRPR silencing prevented the activation of STAT1 and P65 NF-κB signaling (Figure 6I).
Figure 6.

Lentivirus-mediated in vivo GRPR knockdown protected against cisplatin-induced AKI
(A) Construction of GRPR-KD mice. (B) Lentivirus-mediated GRPR knockdown in mice was confirmed by western blot. (C) Representative PAS staining of kidney in NC, EV, and GRPR KD mice treated with cisplatin. (D and E) Serum creatinine and BUN levels. (F) KIM1 protein level was determined by western blot and quantitative analysis. (G) Relative mRNA levels of KIM1 and proinflammatory genes. (H) IF staining of F4/80 and TNF-α. (I) Protein levels of P-STAT1/STAT1 and PP65/P65. Data represented the mean ± SEM for six mice. ∗p < 0.05, ∗∗∗p < 0.001 vs. EV + saline. #p < 0.05, ##p < 0.01, ###p < 0.001 vs. EV + Cis. Cis, cisplatin; EV, empty vector.
Treatment with a GRPR antagonist RH-1402 attenuated cisplatin-induced AKI in vivo and in vitro
Second, We have synthesized a series of GRPR antagonists. Preliminary observation of their effects on cisplatin-induced cell activity was observed by MTT (Figure S5, supporting information), and we found RH-1402 and evaluated its protective effect both in vitro and in vivo. Molecular docking showed that RH-1402 could bind the key amino acid residues of GRPR (Figure 7A), and the binding affinity between GRPR and RH-1402 was confirmed by radioligand binding assays (Figure 7B). The cellular thermal shift assay (CETSA) assay also indicated the binding of RH-1402 to GRPR, and showed clear differences in the detected soluble GRPR proteins with and without RH-1402 treatment, at denaturation temperatures ranging from 47°C to 59°C. RH-1402-treated HK2 cells showed a significant increase in the thermal stability of GRPR compared with those without RH-1402 treatment (Figure 7C). We then used the MTT assay to analyze the impact of RH-1402 on cell viability in HK2 cells with or without cisplatin. RH-1402 treatment began to show suppressive effects on cell viability at concentrations of >64 μmol/L (Figure 7D). RH-1402 at concentrations of 1, 2, and 4 μmol/L significantly restored cell viability after cisplatin treatment (20 μmol/L) (Figure 7E). Western blotting and real-time PCR showed that RH-1402 decreased KIM1 levels and the production of pro-inflammatory factors, and the suppressive effect of RH-1402 on CCL2 was very impressive (Figures 7F and 7G). In this setting, we co-cultured mTECs with RAW246.7 cells, and transwell analyses showed that RH-1402 treatment in cisplatin-treated mTECs alleviated macrophage migration compared with the control group (Figure 7H). We also found that RH-1402 downregulated the activation of the NF-κB and STAT1 signaling pathways induced by cisplatin (Figure 7I). PI/Hoechst double staining analyses showed that cisplatin significantly increased the portion of PI-positive cells, while pre-treatment of RH-1402 decreased that of PI-positive cells (Figure 7J). Flow cytometry and Western blot results showed that RH-1402 decreased cisplatin-induced programmed cell death and RIPK/MLKL signaling in HK2 cells (Figures 7K and 7L). We treated GRPR-silenced HK2 cells with RH-1402 to further confirm that RH-1402 functioned in a GRPR-dependent manner. NF-κB promoter assay and western blotting results showed that RH-1402 failed to further suppress NF-κB activation in GRPR-deficient HK2 cells (Figures 7M and 7N). This finding was confirmed by real-time PCR to detect mRNA levels of TNF-α, IL-1β, IL-6, and CCL2 (Figure 7O), which indicates that GRPR plays a key role in mediating the protective effect of RH-1402.
Figure 7.

RH-1402 attenuated cisplatin-induced cell injury and inflammation by antagonizing GRPR in HK2 cells
(A) The potential binding of GRPR and RH-1402 was conducted by molecular docking. (B) The binding affinity between GRPR and RH-1402 was confirmed by radioligand binding assays. (C) The thermal stability of RH-1402 to GRPR was indicated by CETSA assay. (D, E) The impacts of RH-1402 on cell viability in HK2 cells with or without cisplatin were analyzed by MTT assay. (F) KIM1 protein level was determined by western blot, while cisplatin-induced HK2 cells were treated with RH-1402. (G) Relative mRNA levels of KIM1, TNF-α, IL-1β, and CCL2 were determined by real-time PCR. (H) The effect of RH-1402 on macrophage migration by transwell assay. (I) Protein levels of P-STAT1/STAT1 and PP65/P65 in cisplatin-induced HK2 with or without RH-1402. (J) PI/Hoechst double staining and IF of p-MLKL in HK2 cells. (K) The effect of RH-1402 on programmed cell death induced by cisplatin was detected by flow cytometric analysis. (L) Protein levels of RIPK1/RIPK3/MLKL axis with quantification in vitro. (M) The effect of RH1402 on NF-κB promoter activity. (N) Protein levels of PP65/P65 in cisplatin-induced HK2 with or without RH-1402 while GRPR was silenced. (O) Relative mRNA levels of KIM1 and proinflammatory indexes. Independent experiments were performed throughout the in vitro studies in triplicate. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. NC/EV + Con. #p < 0.05, ##p < 0.01, ###p < 0.001 vs. Cis/EV + Cis. $p < 0.05, $$p < 0.01, $$$p < 0.001 vs. EV+Cis+RH-1402. Cis, cisplatin; EV, empty vector.
Considering the safety of the compound, we detected body weight (Figure S6A, supporting information), aspartate aminotransferase/alanine aminotransferase (Figures S6B and S6C, supporting information), and hematoxylin and eosin staining of each organ (Figure S6D, supporting information), and the results showed that RH-1402 had no potential toxicity. The protective effect of RH-1402 was verified in a cisplatin-induced AKI model using separate prevention and treatment experiments in which RH-1402 was applied before or after AKI model establishment, respectively. First, we performed the preventative experiments (Figure 8A). PAS and score data showed that RH-1402 application reduced cisplatin-induced tubular necrosis, dilation, and cast formation in a dosage-dependent manner (Figure 8B), and prevented renal dysfunction, as shown by serum creatinine and BUN assays (Figures 8C and 8D). Western blotting, immunofluorescence (IF), IHC, and real-time PCR data consistently showed that RH-1402 application decreased KIM1 levels in a dose-dependent manner, and cisplatin-induced macrophage infiltration and pro-inflammatory factors were also prevented in the RH-1402 application group (Figures 8E and S7A–S7C, Supporting Information). Western blot results showed that RH-1402 inhibited the activation of PP65 NF-κB and STAT1 signaling in cisplatin nephropathy (Figure S7D, supporting information).
Figure 8.

The preventive and therapeutic effects of RH-1402 in three AKI mouse models
(A) Experimental timeline of prevention. (B) Representative PAS staining pictures of kidneys from AKI mice in which RH-1402 was applied before cisplatin injection. (C, D) Serum creatinine and BUN levels. (E) IF and IHC staining of TNF-α and F4/80 and their quantification. (F) Experimental timeline of treatment. (G) Representative PAS staining pictures of kidneys from AKI mice in which RH-1402 was applied after model establishment. (H and I) Serum creatinine and BUN levels. (J) IF staining of KIM1, F4/80, and TNF-α and their quantification. (K) PAS staining and scores in the kidneys of I/R-induced AKI mice subjected to I/R treatment. (L and M) BUN levels and serum creatinine levels. Data represented the mean ± SEM for six mice. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 vs. Saline. #p < 0.05, ##p < 0.01, ###p < 0.001 vs. Cis/Cis D3. Cis, cisplatin; D, day.
In the treatment experiment, mice were intraperitoneally injected with RH-1402 1 day after cisplatin injection to detect therapeutic effects in the established AKI model (Figure 8F). PAS staining, serum creatinine, and BUN assays consistently showed that RH-1402 treatment attenuated renal damage and renal dysfunction (Figures 8G–8I). Real-time PCR results showed that the production of cisplatin-induced KIM1 and pro-inflammatory factors were largely reduced by RH-1402 treatment, and this finding was further confirmed by IF analysis (Figures S8A and S8B, supporting information).
Also, intraperitoneal injection of RH-1402 into mice before I/R surgery (Figure 8J). PAS staining and quantitative analysis showed that RH-1402 attenuated I/R-induced renal injury while decreasing serum BUN and creatinine levels (Figures 8K–8M). IF of F4/80 and TNF-α suggested that RH-1402 significantly decreases the expression of inflammatory factors in the kidney (Figure S8C, supporting information). In vivo experiments confirmed that RH-1402 could significantly improve renal function and alleviate renal injury in three AKI models.
Discussion
In the current study, we found that GRPR was induced and highly expressed in tubules from human biopsy, murine, and TEC models of AKI. GRPR promoted renal damage and dysfunction by inducing CCL2-mediated macrophage infiltration, MLKL-mediated necroptosis, and GRP auto-production in STAT1-dependent mechanisms (Figure S9, supporting information). Lentivirus-mediated in vivo silencing of GRPR or the novel GRPR antagonist RH-1402 showed significant renoprotective effects, indicating that GRPR may be an attractive therapeutic target for AKI.
First, we determined the expression, localization, and function of GRPR in AKI. Previous studies indicated that the concentration of ProGRP in the circulation of normal individuals is low (<60 ng/L), but it was highly increased in patients with renal insufficiency30; however, information regarding its receptor GRPR is very limited. Currently, the antitumor function of receptor-targeted radiopharmaceuticals is a topical research field.31 Previous studies investigating GRPR-targeted therapy found that the kidney is the major non-target organ, since significant uptake of GRPR-targeted radio-conjugates was observed at a very early stage after injection.27 Cell profiling of mouse AKI from single-cell RNA sequencing revealed that GRP/GRPR were expressed in the kidney, and GRP expression in the tubules was increased while the expression of GRPR in I/R-induced AKI model was unchanged.32,33 Our data also showed that the mRNA level of GRPR in I/R group was not significantly increased, and this was inconsistent with the cisplatin model in which GRPR was largely induced. In view of this phenomenon, GRPR may be regulated by different mechanisms in cisplatin and I/R models, and the time of injury and individual differences affect the mRNA level of GRPR, which leads to inconsistent changes in mRNA expression in the two AKI models. In the current study, GRPR was highly induced in the kidneys of patients with AKI and murine AKI model induced by cisplatin. Positive signals for GRPR were located at the renal tubule, and this finding was confirmed using cisplatin-treated TECs as an in vitro model. It is interesting that GRPR was highly expressed in proximal tubule epithelial cells and collecting ducts, but not distal tubules, in response to cisplatin. We will further detect the detailed function in different types of renal TECs in the future. Additionally, it is of note that the mRNA and protein level of GRPR was not significantly induced in kidneys of I/R-induced AKI mice, our data were supported by the single-cell RNA seq data that I/R has limited impact on the level of tubular GRPR.33 This inconsistency of GRPR expression between cisplatin- and I/R-induced AKI mice may lead by the distinct pathophysiologies of these AKI models. However, we confirmed that GRPR also has important functions in I/R-induced AKI. As a ligand of GRPR, the serum level of ProGRP was upregulated in both patients with AKI and the murine AKI models, which is consistent with recent research showing that ProGRP increased in patients with renal dysfunction.30 Additionally, single-cell RNA sequencing (RNA-seq) databases confirmed that GRP was induced in TECs in I/R-induced AKI model.32,33 Importantly, we explored the mechanisms by which GRPR was induced in cisplatin nephropathy. Histone acetylation is one of important epigenetic mechanisms that regulates gene expression involved in AKI, and a set of HDACs are involved in the regulation of kidney injury in several AKI models.34,35,36 NaB, a class I HDAC inhibitor, has been reported to reverse the effects of the GRPR antagonist RC-3095.37 We currently found that the inhibition of class I HDACs by NaB treatment aggravated transcriptional activation of GRPR. HDAC8, but not other class I HDACs, binds to the promoter region of GRPR and may lead to its transcriptional inhibition in cisplatin nephropathy. Interestingly, we could not observe a significant decrease in HDAC8 in H/R-treated TECs; this may be one of the major reasons for the phenomenon that the GRPR mRNA expression was unchanged in an I/R-induced AKI model. To date, little has been known about the function of GRP/GRPR signaling in kidney diseases, except for renal cell carcinoma.38 Another study mentioned that GRPR may promote the formation of calcium oxalate crystals in mice with kidney stones, but its functional role and mechanism of action in kidney injury remain to be determined.39 In fact, we recently found that two analogues of PD176252 decreased cisplatin-induced renal toxicity in both in vivo and in vitro models of AKI, but the detailed function of GRP/GRPR axis in AKI remains obscure.40,41 In the current study, we have answered the important clinical question of whether activation of the GRP/GRPR axis plays a critical role in AKI. Knockout of GRPR protected against renal injury and inflammation in two AKI models induced by I/R and cisplatin; more important, conditional knockout of GRPR from TECs had an almost equivalent suppressive effect on the restoration of renal dysfunction and downregulation of inflammatory factors and chemokines. These results demonstrated that GRPR activation in TECs plays a key role in AKI.
Second, we explored the detailed mechanisms by which GRPR promotes AKI. Previous studies have shown that GRP rapidly signals through GRPR, activating the PLC-β2, PI3K, and MAPK pathways, which promote neutrophil chemotaxis, suggesting that GRPR may mediate inflammatory responses through chemokines.42 In the current study, we performed RNA-seq using GRPR overexpression and control TECs and detected significant correlations between GRPR and signaling pathways regulating inflammation and necroptosis. Within a set of pro-inflammatory indexes, the CCL2 levels were largely induced by GRPR, which is a powerful chemokine in mediating renal inflammation.43,44 The regulatory effect of GRPR on CCL2 was also confirmed by our in vivo and in vitro data; among the indexes studied, this showed one of the most significant changes when GRPR was overexpressed or deleted. Additionally, the recruitment of macrophages was substantially inhibited when we blocked GRPR signaling in TECs with RH-1402 in the transwell assay. Therefore, we presumed that tubule-derived CCL2 may recruit more macrophages into the injured kidney to promote inflammation. We also found that MLKL was highly induced in GRPR-overexpressing cells and suppressed in GRPR-silenced TECs and GRPR KO mice. MLKL is a core factor regulating necroptosis, a novel and important pattern of programmed cell death in AKI, whereby the release of intracellular contents induces severe necro-inflammation and accelerates AKI progression.45,46,47 We found that regulation of MLKL by GRPR may be another mechanism by which GRPR promotes renal inflammation. Furthermore, from the RNA-seq data, we detected a significant increase in STAT1 in direct response to GRPR overexpression. STAT1 is a member of the STAT family and functions as a signal messenger and transcription factor, which regulates the expression of genes related to cell proliferation, apoptosis, and inflammation.48,49,50,51 Using co-IP and IF techniques, we found that GRPR could activate downstream STAT1 signaling by binding to TLR4 and silencing of TLR4 in GRPR-overexpressing TECs inhibited GRPR-induced STAT1 activation. Notably, GRPR signaling induced STAT1 at both transcription and phosphorylation levels, and STAT1 was found to bind the promoter regions of CCL2 and MLKL to induce their transcription, thereby accelerating renal inflammation and programmed cell death. This was confirmed by our finding that silencing STAT1 downregulated cisplatin-induced CCL2 and MLKL production. Interestingly, we found that activation of GRPR could induce the production of GRP, its major ligand, in a STAT1-dependent mechanism, thereby forming a GRP/GRPR/STAT1-positive feedback loop. We found that forced overexpression of STAT1 on conditional KO mice or KO cells restored kidney injury and inflammation. All these data indicate that STAT1 is a key transcriptional factor mediating GRPR function.
Third, since GPCRs are the most productive drug targets for disease treatment,52 we evaluated the potential of GRPR-targeted prevention and therapy in an AKI mouse model. First, lentivirus-mediated GRPR knockdown in mice with cisplatin-induced AKI alleviated renal dysfunction, renal damage, and inflammation. Then we used RH-1402, a novel GRPR antagonist recently discovered by our group, to detect the prevention and treatment effects of the GRPR-targeted strategy. We have previously shown the anti-inflammatory effects of RH-1402 in vitro.40 However, its effect in vivo had not previously been evaluated. In the prevention experiment, RH-1402 showed a good renoprotective effect and suppressed renal injury and inflammation. Considerable protective effects of RH-1402 were also detected in the treatment experiment. Application of RH-1402 at 1 day after model establishment successfully maintained AKI progression while limiting renal inflammation. These data provide valuable evidence that GRPR-targeted therapy against AKI may have a bright future in clinical settings. Of note, a recent study explored the structure of the GRPR by using cryo-electron microscopy; this may accelerate the searching of GRPR inhibitors.53
In conclusion, we found that TEC-generated GRPR accelerated renal damage and dysfunction by activating STAT1-mediated GRP auto-production, necroptosis, and renal inflammation. Lentivirus-mediated GRPR in vivo silencing or the novel GRPR antagonist RH-1402 showed preventive and therapeutic potential against renal inflammation and programmed cell death, indicating that GRPR may serve as a good therapeutic target for AKI. Considering that GRPR is currently a potential target in cancer therapy, the GRPR antagonist RH-1402 may show specific importance as a dual-effect agent by protecting against antitumor drug (e.g., cisplatin)-induced nephrotoxicity.
Materials and methods
Human renal biopsy and blood samples
This clinical study has been approved by the Biomedical Ethics Committee of Anhui Medical University (Anhui, China) (#20190240) and followed the principles of the Declaration of Helsinki. Written informed consent was obtained from all participants. According to the Kidney Disease: Improving Global Outcomes criteria, AKI is diagnosed if serum creatinine increases by 0.3 mg/dL (or ≥26.5 mM) in ≤48 h or increases to ≥1.5-fold from baseline within the prior 7 days and/or by a decrease in urine output of <0.5 mL/kg/h for 6–12 h. Exclusion criteria included small cell lung cancer, prostate cancer, arthritis, enteritis, allergic dermatitis, sepsis, infectious diseases, and so on. After obtaining patient and ethics committee consent, serum from healthy volunteers and patients with AKI were collected and processed within 6 h of collection. We included nine healthy volunteers and seven patients with AKI. Renal biopsies had been performed as part of routine clinical diagnostic investigation. The samples of renal biopsies were obtained from department of nephropathy, the First Affiliated Hospital of Anhui Medical University. Control samples were obtained from the healthy kidney poles of individuals who underwent tumor nephrectomies without renal disease. Fresh renal tissues were fixed with formalin for 24 h and embedded in paraffin until use. Characteristics of healthy volunteers and AKI patients are described in Table S1.
ProGRP detection
To examine the content of ProGRP, we used GRP precursor detection kit (electrochemiluminescence method) from Roche Biotechnology Co Ltd (Mannheim, Germany). The content of ProGRP were tested according to the product protocols and the detection was performed by Laboratory Division, the First Affiliated Hospital of USTC, University of Science and Technology of China.
Mouse models
Male C57BL/6 mice (at 6–8 weeks of age with body weight of 20–22 g) were provided by the Experimental Animal Center, Anhui Medical University. All animal procedures were approved by the Animal Experimentation Ethics Committee of the Anhui Medical University and conducted by the Guide for the Care and Use of Laboratory Animals, eighth edition. Mice were housed with ad libitum access to food and water in light- and temperature-controlled environments. In the cisplatin-induced AKI model, mice were injected with 20 mg/kg cisplatin, while the control group was injected with the same amount of saline. In the I/R-induced AKI model, mice were anesthetized and placed on a thermostat plate to maintain body temperature. Bilateral renal pedicles were clipped for 40 min, and the clamps were released for 24 h reperfusion.
Generation and verification of GRPR KO mice and conditional KO mice
GRPR global KO mice (C57BL/6) was constructed by Cyagen Biosciences (Guangzhou, China), homozygous and wild-type mice were obtained by backcrossing heterozygotes. Mice with conditional deletion of GRPR from the kidney TEC was generated by mating the GRPRFlox/Flox mouse (C57BL/6) with the KspCre mouse (C57BL/6), all were constructed by GemPharmatech Co. Ltd (Jiangsu, China). PCR, quantitative real-time PCR, and western blotting analyses were used to identify the genotype of these mice.
Lentivirus-mediated GRPR Knockdown/STAT1 overexpression in mice
Lentivirus with GRPR knockdown/STAT1 Overexpression vector was purchased from GenePharma (Shanghai, China). First, we constructed three GRPR shRNA sequences to search for the one with the most silencing efficiency in mTECs. Then the lentivirus labeled with GFP was synthesized according to effective sequence. The tails of mice were then wiped with alcohol to expand the tail vein for injection. Mice were slowly injected with 100 μL lentivirus with a concentration of 1 × 109 TU/mL through the tail vein using a 0.5-mL insulin syringe at the speed of approximately 0.2 μL/min. At 72 h after lentivirus infection, mice were intraperitoneally injected with either 20 mg/kg cisplatin or the equal volume of saline for further analysis.
Assessment of renal function
Blood samples collected from mice were used to measure creatinine, BUN, they were harvested from mice with or without intraperitoneal injection of 20 mg/kg cisplatin after 3 days. The levels of creatinine and BUN in blood samples were measured using the Creatinine and BUN Assay Kit (Nanjingjiancheng, China) according to the manufacturer’s instructions.
PAS staining and immunohistochemical analysis
To evaluate the histological damage, PAS staining was performed with the PAS Kit according to the protocol of the manufacturer. Renal tissues were fixed in 4% paraformaldehyde immediately. After dehydration, samples were embedded in parafin. According to the manufacturer’s instruction, PAS staining was performed in parafin sections (3–5 μm) to assess the degree of tubulointerstitial damage and examined by light microscope (Olympus, Tokyo, Japan) at 200× magnification. On PAS-stained kidney sections (n = 6–8), kidney damage in the cortical proximal was scored as the approximate extent of tubules that displayed tubular necrosis, cast formation, and tubular dilation as follows: 0 = normal; 1 = 10%; 2 = 10–25%; 3 = 26%–50%; 4 = 51%–75%; 5 = 75%–95%; 6 = >96%. In addition, IHC was performed in paraffin sections to examine renal damage and inflammation using a microwave-based antigen retrieval technique. These sections were incubated for 12 h at 4°C with a solution of rabbit anti-KIM1 (Bioss, Woburn, MA), anti-TNF-α, anti-F4/80 antibody (Servicebio, Woburn, MA), and the anti-GRPR primary antibody (Abcam, Cambridge, UK). After incubation in secondary antibody and chromagen liquid DAB (3, 30-diaminobenzidine tetrahydrochloride), the slides were counterstained with hematoxylin. The results were analyzed by Image Analysis System (AxioVision 4, Carl Zeiss, Jena, Germany).
IF
Cells were grown on glass coverslips, fixed with acetone at 37°C for 10 min, and then blocked with 10% bovine serum albumin (Beyotime, Jiangsu, China) at 37°C for 10 min. They were incubated with the primary antibody overnight, washed three times with PBS, and finally incubated with goat anti-rabbit IgG-rhodamine (Bioss) antibody for 1 h in the dark at 37°C. The cells were counterstained with DAPI to stain the nuclei and imaged using an inverted fluorescence microscope (Zeiss Spot, Carl Zeiss, Gottingen, Germany) or confocal microscope (Leica, Wetzlar, Germany). For kidney paraffin sections, after dewaxing, rehydration, antigen retrieval, reducing endogenous peroxidase activity, and blocking, sections were then incubated with primary antibodies and fluorescein isothiocyanate (FITC)-conjugated anti-rabbit/anti-mouse secondary antibodies and then observed with fluorescence microscope (Zeiss Spot, Carl Zeiss, Gottingen, Germany).
Western blot analysis
Protein from cultured TECs and mouse kidney tissue were extracted with ice-cold RIPA lysis buffer. A BCA protein quantitative kit (Beyotime) was used to evaluate the protein concentration. Total protein was loaded in 10% SDS-PAGE and transferred onto nitrocellulose membranes. Then membranes were blocked with 5% milk for 2 h. Subsequently, membranes were incubated with the primary antibody against β-actin (Servicebio), KIM1 (Bioss), GRPR/P65/PP65 (Zenbio, Research Triangle Park, NC), HDAC1/3 (Proteintech, Rosemont, IL), HDAC2/8 (Huabio, Woburn, MA) overnight at 4°C. Signals were captured with LiCor/Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE). The intensities of each Western blot band were quantified and analyzed by using the ImageJ software (NIH, Bethesda, MD). The ratio for the quantification of interest was normalized and presented as the mean ± SEM value.
RNA extraction and real-time PCR examination
Total RNA from cultured human TECs and mouse kidney tissue was isolated by using Trizol reagent according to the manufacturer’s instructions, and quantified by using Drop 2000 Spectrophotometer (ThermoFisher Scientific, Waltham, MA). Single-stranded cDNA was synthesized from total RNA using RealMasterMix (Toyobo, Osaka, Japan). Real-time PCR was performed in a total volume of 10 μL, including 2 μL cDNA solution, 5μL Bio-Rad iQ SYBR Green supermix with Opticon 2 (Bio-Rad, Hercules, CA), 2.4 μL nuclease-free water, and 0.6 μL of each primer. The ratio for the mRNA of interest index was normalized to β-actin. The sequences of primers were described in Table S2.
Homology modeling and molecular docking study
In recent years, a lot of GPCRs three-dimensional (3D) structures have been solved, such as bovine rhodopsin, adrenergic, dopaminergic, histaminergic, chemokines, opioids, and neurotensin receptors, which provide further insights into the 3D topology of GPCRs. For homology building of GRPR receptor, MODELLER software in the Discovery Studio (Accelrys, San Diego, CA) was used. The results of PSI-BLAST searched showed the crystallographic coordinates of rat NTR1 (4BUO) had a good degree (33%) of similarity with GRPR sequence.54 In the homology modeling, the number of models parameter was set to 20 and the remaining parameters were set to the default values. The best model according to the Discrete Optimized Protein Energy score was added transmembrane region with solvate with Explicit Membrane. Solvate was added to the addition of water system and then carried out molecular dynamics. After the optimization, the model was used to further molecular docking study. The previous study demonstrated that mutation of Phe301, Arg287, Tyr284, Asp104, Arg100, and Asp97 significant decreased the affinity, especially, mutation of Arg287 70-fold decreased in affinity.55 The docking study was carried out with MOE 2019 to understand the ligand-protein interactions. According the previous method to carry out the docking study. Briefly, the target compound was minimized the energy in the MOE, then the optimized model was loaded into MOE. Addition of polar hydrogens, protonation, and energy minimization were carried out to get the stabilized conformation. The active site was found with a ‘Site Finder’ module to define the docking site. Then, the standard docking procedure was implemented. The docking result of RH-1402 is shown in Figure 7A. The results showed that the amino acids residue of Arg287, Cys196, Asp104, and Arg100 formed H-bonds with protein and Phe301 and Tyr101 formed H-Pi-bonds.
Determination of binding affinity
The human prostate cancer (PC-3) cell line was supplied by the Shanghai Cell Bank, Chinese Academy of Sciences. PC-3 cell was cultured in RPMI 1640 (BI) medium with 10% fetal bovine serum (FBS) (ExCell) and 1% penicillin-streptomycin (Gibco, Grand Island, NY). Cells were cultured in humidified incubator with the growing environment (37°C and 5% CO2). In vitro competitive cell binding assays of RH1402 was performed against 125I-Tyr4-BBN for the GRPR according previous method.56 We seeded 105 PC-3 cells at per well in 24-well plates and incubated them for 12 h; then, the medium was aspirated and washed twice with binding buffer. 125I-Tyr4-BBN (20,000 cpm) (PerkinElmer, Waltham, MA; Product Number: NEX258) and different concentrations of RH1402 (0–10,000 nM) were added, and then incubated for 1 h at 37°C. After incubation, the cells were washed twice with binding buffer and solubilized with 2 mL/1 N NaOH. The NaOH solution was measured with γ-counter (Berthold Technologies, Bad Wildbad, Germany), and the median inhibition concentration was calculated by nonlinear regression analysis. Note that the binding buffer comprised (N-2-Hydroxyethylpiperazine- N′-2-ethanesulfonic acid (HEPES) (50 mM), NaCl (125 mM), KCl (7.5 mM), MgCl2 (5.5 mM), ethylene glycol Bis(2-aminoethyl Ether)-N,N,N′,N′-tetra-acetic Acid (1 mM), bovine serum albumin (2 mg/mL), chymostatin (2 mg/L), soybean trypsin inhibitor (100 mg/L), and bacitracin (50 mg/L).
Cell culture and transfection
Kidney TECs of human (HK2) and mouse (mTEC) were provided by Prof. Hui-yao Lan, The Chinese University of Hong Kong. CRISPR-Cas9-edited GRPR-deficient mTEC used guide RNA was 5′-GGGTT TATTA CAGGA CAGCA-3'. In brief, the lentiviral vector contains a pair of targeted mouse GRPR exon 1 and CRISPR related protein 9 (Cas 9; lentivirus Cas 9-GFP). MTECs were infected with GFP lentivirus and classified. Single GFP-labeled MTECs were inoculated in a 96-well plate for monoclonal screening for 8–12 days. Finally, KO verification is performed. TECs were cultured in DMEM/F12 containing 5% FBS (VivaCell, Shanghai, China) under 37°C and 5% CO2 condition. GRPR shRNA plasmid (Genechem, Shanghai, China), HDAC3/HDAC8/GRPR overexpression plasmid/TLR4 shRNA/STAT1 siRNA (Hanbio, Shanghai, China) and their control constructs were transfected using Lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. After starvation in 0.5% FBS for 12 h, cells were incubated with 20 μmol/L cisplatin for 24 h and harvested for further analysis. For the H/R model, hypoxia (0.1% O2) was induced in 0.5% low-glucose medium for 12 h before reoxygenation in normal medium for western blot or real-time PCR analysis. In cell culture we used mycoplasma clearance reagents (SaveIt, Hanbio) according to the manufacturer’s instructions.
CETSA
The CETSA was conducted according to the protocol as previously described.8 Treating HK2 cells with or without RH-1402 for 24 h, and then we extracted cellular proteins. The samples were adjusted to similar concentrations using the BCA kit. Then, it was distributed equally into different PCR tubes and denatured the samples at various temperatures for 8 min on PCR instrument (Eppendorf, Hamburg, Germany), and freeze-thawed the samples three times using liquid nitrogen. Samples were centrifuged and the supernatants were analyzed by western blot.
Transwell migration assay
Co-culture of RAW246.7 and mTEC cells were performed using transwell chambers with 8-μm pores on the membrane (BD, Franklin Lakes, NJ) in a 24-well plate. mTEC cells were cultured in the lower compartment for 12 h with RH-1402 stimulation and 24 h with cisplatin stimulation. RAW246.7 were cultured in upper compartments of the transwell chamber and co-cultured mTEC cells for 24 h. Transwell were removed by cotton swabs and was fixed with methanol for 30 min and then stained with 0.1% crystal violet for 30 min. The migrated cells were counted under a fluorescence microscope (Zeiss Spot, Carl Zeiss, Gottingen, Germany).
Transmission electron microscopy
Kidney tissue was immersed in 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate (pH 7.4) at 4°C, and postfixed in buffered 1% osmium tetroxide on ice. After rinsing and dehydrating, embedded in LR White resin (London Resin Company, Reading, UK). Polymerization was achieved in gelatin capsules at 60°C for 48 h. Specimens were then detected by a transmission electron microscope (H-7700, Tokyo, Japan).
IP assay
Cells were suspended in standard NP-40 IP buffer (Sigma-Aldrich, St. Louis, MO) and the proteins were incubated in NP-40 IP buffer at 4°C. The mixture was combined with anti-TLR4 antibody 3 h later and then precipitated with Protein G Sepharose beads followed by overnight incubation at 4°C. The beads were washed three times with 1 mL IP buffer and finally eluted. The immune complexes were then subjected to Western blot for determination of protein expression.
Dual-luciferase reporter assay
DualLuciferase Reporter Assay System (Promega, Madison, WI) was used for luciferase activity measurement according to the manufacturer’s instructions. Cells were seeded into 24-well plates, cotransfected with plasmid containing NF-κB promoter sequence and empty vector. After Lipofectamine 3000 transfection reagent is transfected for 6 h, add 0.5% FBS with RH-1402 for 12 h. Finally, cells were stimulated with cisplatin for 24 h and then harvested for further analysis.
Flow cytometric analysis
The number of apoptotic and necrotic cells was detected by flow cytometric analysis. The HK2 cells were treated in the presence or absence of 20 μmol/L cisplatin for 24 h after incubation with RH-1402 overnight. Then HK2 cells were digested with trypsin for 2min and centrifuged at 1,500 rpm for 5 min. According to the manufacturer’s instructions, the density of cells is approximately 106 cells/mL with the addition of 400 μL Annexin V binding fluid. Subsequently, the cells were stained with 5 μL FITC for 15 min at 4°C. Cells were re-stained with 10 μL PI for 5 min and lightly placed at 4°C in the dark. Then the cells were immediately measured with a laser eight-color flow cytometer (FACSVerse, BD). The results were quantified using the FlowJo 7.6 software.
RNA-seq
RNA samples isolated from the mTECs were submitted to the BGI (Shenzhen, China), and performed quantitative and qualitative assessments of RNA samples including concentration and contamination. In this project, a total of 6 samples were tested using the BGISEQ platform. The average comparison rate of the sample comparison genome is 96.61%, and the average comparison rate of the comparison gene set is 84.57%; a total of 17,409 genes were detected. According to the KEGG pathway annotation to classify and then perform enrichment analysis (with a p value of <0.05 and a fold change of >2). To reflect the correlation of gene expression between samples, Pearson correlation coefficients of all gene expression levels between every two samples were calculated, and these coefficients were reflected in the form of heatmap.
ChIP assay
Cells were stimulated with Cisplatin. Then ChIP assay was conducted using the SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads) (Cell Signaling Technology, Danvers, MA) according to the manufacturer’s instructions, anti-PSTAT1 (Zenbio), HDAC3 (Proteintech), HDAC8 (Huabio) and IgG were used to immunoprecipitated cross-linked protein-DNA complexes. The immunoprecipitated DNA was identified by real-time PCR, with specific primers designed for the predicted binding sites within the promoter of mouse MLKL, CCL2, GRP and human GRPR. The sequences of primers were described in Table S3.
Cell viability assay
An MTT assay was used to evaluate cell viability according to the manufacturer’s instruction. Human HK2 cells were grown in 96-well plates with treatments by a set of concentrations of RH-1402 (arranged from 0.5 to 64 μmol/L). Twelve hours later, cells in each well of 96-well plates were treated with cisplatin (20 μmol/L) for 24 h in incubator. Cells were treated with a 5 mg/mL MTT solution for 4 h at 37°C. Living cells can reduce exogenous MTT to formazan and formazan crystals were dissolved in 150 μL of DMSO. Finally, the absorbance was measured at 492 nm using a microplate reader (Multiskan MK3, ThermoFisher Scientific). The information of all compounds is shown in Figures S10–S13, Tables S4, and S5.
Statistical analysis
Data are expressed as the mean ± SEM. The results were analyzed by GraphPad Prism 8.0 software. Nonparametric tests were used to evaluate the statistical differences between the two groups, and 1-way or two-way ANOVA nonparametric analysis of variance was used to analyze the statistical differences between more than two groups. A p value of <0.05 was considered to be significant. Statistical results were labeled in each figure as ns = p ≥ 0.05,∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Acknowledgments
This work was supported, in whole or in part, by the National Natural Science Foundation of China (No. 81970584), the National Key R&D Program (2022YFC2502503), the Major Projects of Science and Technology in Anhui Province (202103a07020013), the Project of Collaborative Innovation for Colleges of Anhui Province (NO. GXXT-2021-070), and the Scientific Research Fund of Anhui Medical University (NO.2019xkj057). The authors thank the Laboratory Animal Center of Anhui Medical University and the Center for Scientific Research of Anhui Medical University for valuable help in our experiment. We thank Professor Yang Kai of Soochow University for his valuable help in radioligand binding assays.
Author contributions
X-M.M, R-S.Y., and J.J designed and supervised the study. C.L. performed most of the experiments. Q-Y.M., X-Q.L., H-D.L. designed the experiments, conducted the experiments, and analyzed the data. M-J.Y. and S-S.D. designed and synthesized compounds, and analyzed spectrum; M-M.L. and J-G.W. checked and identified compounds. S-S.X., W-X.M., J-N.W., Y.H., Y.C., L.G., R-B.H., and H-G.B performed the animal experiments, scored the histology slides, conducted microscopy, and interpreted the results. H-M.Z., J.J., Q.G., C.Y., J.L., H-Y.C., and H-Y.L. performed clinical tissues studies and reviewed the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.06.016.
Contributor Information
Juan Jin, Email: jinjuan@ahmu.edu.cn.
Ri-sheng Yao, Email: rishengyao@163.com.
Xiao-ming Meng, Email: mengxiaoming@ahmu.edu.cn.
Supplemental information
Data and code availability
No publicly available data or shared data are cited. All data needed to evaluate the conclusion of the current study are present in the paper and/or the Supplementary Materials. Additional data are available from the corresponding author on request.
References
- 1.Vijayan A. Tackling AKI: prevention, timing of dialysis and follow-up. Nat. Rev. Nephrol. 2021;17:87–88. doi: 10.1038/s41581-020-00390-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ronco C., Bellomo R., Kellum J.A. Acute kidney injury. Lancet. 2019;394:1949–1964. doi: 10.1016/S0140-6736(19)32563-2. [DOI] [PubMed] [Google Scholar]
- 3.Gao L., Zhong X., Jin J., Li J., Meng X.M. Potential targeted therapy and diagnosis based on novel insight into growth factors, receptors, and downstream effectors in acute kidney injury and acute kidney injury-chronic kidney disease progression. Signal Transduct. Target. Ther. 2020;5:9. doi: 10.1038/s41392-020-0106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linkermann A. Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 2016;89:46–57. doi: 10.1016/j.kint.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Wang J.N., Yang Q., Yang C., Cai Y.T., Xing T., Gao L., Wang F., Chen X., Liu X.Q., He X.Y., et al. Smad3 promotes AKI sensitivity in diabetic mice via interaction with p53 and induction of NOX4-dependent ROS production. Redox Biol. 2020;32:101479. doi: 10.1016/j.redox.2020.101479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Q., Ren G.L., Wei B., Jin J., Huang X.R., Shao W., Li J., Meng X.M., Lan H.Y. Conditional knockout of TGF-betaRII/Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics. 2019;9:8277–8293. doi: 10.7150/thno.35686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allison S.J. Acute kidney injury: AIMing to enhance debris clearance and improve outcomes in AKI. Nat. Rev. Nephrol. 2016;12:123. doi: 10.1038/nrneph.2016.3. [DOI] [PubMed] [Google Scholar]
- 8.Wang J.N., Wang F., Ke J., Li Z., Xu C.H., Yang Q., Chen X., He X.Y., He Y., Suo X.G., et al. Inhibition of METTL3 attenuates renal injury and inflammation by alleviating TAB3 m6A modifications via IGF2BP2-dependent mechanisms. Sci. Transl. Med. 2022;14:eabk2709. doi: 10.1126/scitranslmed.abk2709. [DOI] [PubMed] [Google Scholar]
- 9.Yu J.T., Hu X.W., Yang Q., Shan R.R., Zhang Y., Dong Z.H., Li H.D., Wang J.N., Li C., Xie S.S., et al. Insulin-like growth factor binding protein 7 promotes acute kidney injury by alleviating poly ADP ribose polymerase 1 degradation. Kidney Int. 2022;102:828–844. doi: 10.1016/j.kint.2022.05.026. [DOI] [PubMed] [Google Scholar]
- 10.He X.Y., Wang F., Suo X.G., Gu M.Z., Wang J.N., Xu C.H., Dong Y.H., He Y., Zhang Y., Ji M.L., et al. Cpd-42 Alleviates Acute Kidney Injury via Targeting RIPK3-mediated Necroptosis. Br. J. Pharmacol. 2023 doi: 10.1111/bph.16152. [DOI] [PubMed] [Google Scholar]
- 11.Anastasi A., Erspamer V., Bucci M. Isolation and structure of bombesin and alytesin, 2 analogous active peptides from the skin of the European amphibians Bombina and Alytes. Experientia. 1971;27:166–167. doi: 10.1007/BF02145873. [DOI] [PubMed] [Google Scholar]
- 12.Alshehri S., Fan W., Zhang W., Garrison J.C. In Vitro Evaluation and Biodistribution Studies of HPMA Copolymers Targeting the Gastrin Releasing Peptide Receptor in Prostate Cancer. Pharm. Res. 2020;37:229. doi: 10.1007/s11095-020-02952-3. [DOI] [PubMed] [Google Scholar]
- 13.Chen S., Gao X.F., Zhou Y., Liu B.L., Liu X.Y., Zhang Y., Barry D.M., Liu K., Jiao Y., Bardoni R., et al. A spinal neural circuitry for converting touch to itch sensation. Nat. Commun. 2020;11:5074. doi: 10.1038/s41467-020-18895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melzer S., Newmark E.R., Mizuno G.O., Hyun M., Philson A.C., Quiroli E., Righetti B., Gregory M.R., Huang K.W., Levasseur J., et al. Bombesin-like peptide recruits disinhibitory cortical circuits and enhances fear memories. Cell. 2021;184:5622–5634.e25. doi: 10.1016/j.cell.2021.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W., Wu K.J., Vellaisamy K., Leung C.H., Ma D.L. Peptide-Conjugated Long-Lived Theranostic Imaging for Targeting GRPr in Cancer and Immune Cells. Angew. Chem. Int. Ed. Engl. 2020;59:17897–17902. doi: 10.1002/anie.202007920. [DOI] [PubMed] [Google Scholar]
- 16.Genton L., Kudsk K.A. Interactions between the enteric nervous system and the immune system: role of neuropeptides and nutrition. Am. J. Surg. 2003;186:253–258. doi: 10.1016/s0002-9610(03)00210-1. [DOI] [PubMed] [Google Scholar]
- 17.Tokita K., Katsuno T., Hocart S.J., Coy D.H., Llinares M., Martinez J., Jensen R.T. Molecular basis for selectivity of high affinity peptide antagonists for the gastrin-releasing peptide receptor. J. Biol. Chem. 2001;276:36652–36663. doi: 10.1074/jbc.M104566200. [DOI] [PubMed] [Google Scholar]
- 18.Petronilho F., Danielski L.G., Roesler R., Schwartsmann G., Dal-Pizzol F. Gastrin-releasing peptide as a molecular target for inflammatory diseases: an update. Inflamm. Allergy Drug Targets. 2013;12:172–177. doi: 10.2174/1871528111312030003. [DOI] [PubMed] [Google Scholar]
- 19.Dal-Pizzol F., Di Leone L.P., Ritter C., Martins M.R., Reinke A., Pens Gelain D., Zanotto-Filho A., de Souza L.F., Andrades M., Barbeiro D.F., et al. Gastrin-releasing peptide receptor antagonist effects on an animal model of sepsis. Am. J. Respir. Crit. Care Med. 2006;173:84–90. doi: 10.1164/rccm.200507-1118OC. [DOI] [PubMed] [Google Scholar]
- 20.Zhou S., Potts E.N., Cuttitta F., Foster W.M., Sunday M.E. Gastrin-releasing peptide blockade as a broad-spectrum anti-inflammatory therapy for asthma. Proc. Natl. Acad. Sci. USA. 2011;108:2100–2105. doi: 10.1073/pnas.1014792108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Oliveira P.G., Grespan R., Pinto L.G., Meurer L., Brenol J.C.T., Roesler R., Schwartsmann G., Cunha F.Q., Xavier R.M. Protective effect of RC-3095, an antagonist of the gastrin-releasing peptide receptor experimental arthritis. Arthritis Rheum. 2011;63:2956–2965. doi: 10.1002/art.30486. [DOI] [PubMed] [Google Scholar]
- 22.ter Beek W.P., Muller E.S.M., Van Hogezand R.A., Biemond I., Lamers C.B.H.W. Gastrin releasing peptide receptor expression is decreased in patients with Crohn's disease but not in ulcerative colitis. J. Clin. Pathol. 2004;57:1047–1051. doi: 10.1136/jcp.2003.014993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo L., Wu X., Zhang Y., Wang F., Li J., Zhu J. Protective effects of gastrin-releasing peptide receptor antagonist RC-3095 in an animal model of hepatic ischemia/reperfusion injury. Hepatol. Res. 2019;49:247–255. doi: 10.1111/hepr.13315. [DOI] [PubMed] [Google Scholar]
- 24.Choi Y., Heo S.C., Kim Y.N., Joo J.Y., Hwang J.J., Bae M.K., Kim H.J. Gastrin-Releasing Peptide (GRP) Stimulates Osteoclastogenesis in Periodontitis. Cells. 2020;10:50. doi: 10.3390/cells10010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pansky A., DE Weerth A., Fasler-Kan E., Boulay J.L., Schulz M., Ketterer S., Selck C., Beglinger C., VON Schrenck T., Hildebrand P. Gastrin releasing peptide-preferring bombesin receptors mediate growth of human renal cell carcinoma. J. Am. Soc. Nephrol. 2000;11:1409–1418. doi: 10.1681/ASN.V1181409. [DOI] [PubMed] [Google Scholar]
- 26.Dumesny C., Whitley J.C., Baldwin G.S., Giraud A.S., Shulkes A. Developmental expression and biological activity of gastrin-releasing peptide and its receptors in the kidney. Am. J. Physiol. Ren. Physiol. 2004;287:F578–F585. doi: 10.1152/ajprenal.00416.2003. [DOI] [PubMed] [Google Scholar]
- 27.Zhang W., Fan W., Ottemann B.M., Alshehri S., Garrison J.C. Development of Improved Tumor-Residualizing, GRPR-Targeted Agents: Preclinical Comparison of an Endolysosomal Trapping Approach in Agonistic and Antagonistic Constructs. J. Nucl. Med. 2020;61:443–450. doi: 10.2967/jnumed.119.231282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang L., Huang Z., Lin Y., Fu J., Liang X., Liu F. Clinical Application of Pro-Gastrin-Releasing Peptide. Clin. Lab. 2018;64:1259–1268. doi: 10.7754/Clin.Lab.2018.180316. [DOI] [PubMed] [Google Scholar]
- 29.Nordlund M.S., Bjerner J., Warren D.J., Nustad K., Paus E. Progastrin-releasing peptide: stability in plasma/serum and upper reference limit. Tumour Biol. 2008;29:204–210. doi: 10.1159/000148188. [DOI] [PubMed] [Google Scholar]
- 30.Dai Z., Zhu J., Huang H., Fang L., Lin Y., Huang S., Xie F., Sheng N., Liang X. Expression and clinical value of gastrin-releasing peptide precursor in nephropathy and chronic kidney disease. Nephrology (Carlton) 2020;25:398–405. doi: 10.1111/nep.13642. [DOI] [PubMed] [Google Scholar]
- 31.Valverde I.E., Vomstein S., Mindt T.L. Toward the Optimization of Bombesin-Based Radiotracers for Tumor Targeting. J. Med. Chem. 2016;59:3867–3877. doi: 10.1021/acs.jmedchem.6b00025. [DOI] [PubMed] [Google Scholar]
- 32.Kirita Y., Wu H., Uchimura K., Wilson P.C., Humphreys B.D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. USA. 2020;117:15874–15883. doi: 10.1073/pnas.2005477117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixon E.E., Wu H., Muto Y., Wilson P.C., Humphreys B.D. Spatially Resolved Transcriptomic Analysis of Acute Kidney Injury in a Female Murine Model. J. Am. Soc. Nephrol. 2022;33:279–289. doi: 10.1681/ASN.2021081150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W., Guan Y., Bayliss G., Zhuang S. Class IIa HDAC inhibitor TMP195 alleviates lipopolysaccharide-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020;319:F1015–F1026. doi: 10.1152/ajprenal.00405.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang J., Shi Y., Liu N., Xu L., Zang X., Li P., Zhang J., Zheng X., Qiu A., Zhuang S. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018;132:339–359. doi: 10.1042/CS20171417. [DOI] [PubMed] [Google Scholar]
- 36.Liu J., Livingston M.J., Dong G., Tang C., Su Y., Wu G., Yin X.M., Dong Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018;9:322. doi: 10.1038/s41419-018-0374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petry F.S., Dornelles A.S., Lichtenfels M., Valiati F.E., de Farias C.B., Schwartsmann G., Parent M.B., Roesler R. Histone deacetylase inhibition prevents the impairing effects of hippocampal gastrin-releasing peptide receptor antagonism on memory consolidation and extinction. Behav. Brain Res. 2016;307:46–53. doi: 10.1016/j.bbr.2016.03.041. [DOI] [PubMed] [Google Scholar]
- 38.Ischia J., Patel O., Sethi K., Nordlund M.S., Bolton D., Shulkes A., Baldwin G.S. Identification of binding sites for C-terminal pro-gastrin-releasing peptide (GRP)-derived peptides in renal cell carcinoma: a potential target for future therapy. BJU Int. 2015;115:829–838. doi: 10.1111/bju.12886. [DOI] [PubMed] [Google Scholar]
- 39.Wang X.F., Zhang B.H., Lu X.Q., Wang R.Q. Gastrin-releasing peptide receptor gene silencing inhibits the development of the epithelial-mesenchymal transition and formation of a calcium oxalate crystal in renal tubular epithelial cells in mice with kidney stones via the PI3K/Akt signaling pathway. J. Cell. Physiol. 2019;234:1567–1577. doi: 10.1002/jcp.27023. [DOI] [PubMed] [Google Scholar]
- 40.Yao S., Wei B., Yu M., Meng X., He M., Yao R. Design, synthesis and evaluation of PD176252 analogues for ameliorating cisplatin-induced nephrotoxicity. Medchemcomm. 2019;10:757–763. doi: 10.1039/c8md00632f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu M.J., Li C., Deng S.S., Meng X.M., Yao R.S. Discovery of a novel GRPR antagonist for protection against cisplatin-induced acute kidney injury. Bioorg. Chem. 2022;124:105794. doi: 10.1016/j.bioorg.2022.105794. [DOI] [PubMed] [Google Scholar]
- 42.Czepielewski R.S., Porto B.N., Rizzo L.B., Roesler R., Abujamra A.L., Pinto L.G., Schwartsmann G., Cunha F.d.Q., Bonorino C. Gastrin-releasing peptide receptor (GRPR) mediates chemotaxis in neutrophils. Proc. Natl. Acad. Sci. USA. 2012;109:547–552. doi: 10.1073/pnas.1110996109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sen P., Helmke A., Liao C.M., Sörensen-Zender I., Rong S., Bräsen J.H., Melk A., Haller H., von Vietinghoff S., Schmitt R. SerpinB2 Regulates Immune Response in Kidney Injury and Aging. J. Am. Soc. Nephrol. 2020;31:983–995. doi: 10.1681/ASN.2019101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilkening A., Krappe J., Mühe A.M., Lindenmeyer M.T., Eltrich N., Luckow B., Vielhauer V. C-C chemokine receptor type 2 mediates glomerular injury and interstitial fibrosis in focal segmental glomerulosclerosis. Nephrol. Dial. Transpl. 2020;35:227–239. doi: 10.1093/ndt/gfy380. [DOI] [PubMed] [Google Scholar]
- 45.Pefanis A., Ierino F.L., Murphy J.M., Cowan P.J. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019;96:291–301. doi: 10.1016/j.kint.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y., Ma H., Shao J., Wu J., Zhou L., Zhang Z., Wang Y., Huang Z., Ren J., Liu S., et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015;26:2647–2658. doi: 10.1681/ASN.2014080741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang L., Liu X.Q., Ma Q., Yang Q., Gao L., Li H.D., Wang J.N., Wei B., Wen J., Li J., et al. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. FASEB J. 2019;33:3523–3535. doi: 10.1096/fj.201801711R. [DOI] [PubMed] [Google Scholar]
- 48.Ying W.Z., Li X., Rangarajan S., Feng W., Curtis L.M., Sanders P.W. Immunoglobulin light chains generate proinflammatory and profibrotic kidney injury. J. Clin. Invest. 2019;129:2792–2806. doi: 10.1172/JCI125517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piaszyk-Borychowska A., Széles L., Csermely A., Chiang H.C., Wesoły J., Lee C.K., Nagy L., Bluyssen H.A.R. Signal Integration of IFN-I and IFN-II With TLR4 Involves Sequential Recruitment of STAT1-Complexes and NFkappaB to Enhance Pro-inflammatory Transcription. Front. Immunol. 2019;10:1253. doi: 10.3389/fimmu.2019.01253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Begitt A., Droescher M., Meyer T., Schmid C.D., Baker M., Antunes F., Knobeloch K.P., Owen M.R., Naumann R., Decker T., Vinkemeier U. STAT1-cooperative DNA binding distinguishes type 1 from type 2 interferon signaling. Nat. Immunol. 2014;15:168–176. doi: 10.1038/ni.2794. [DOI] [PubMed] [Google Scholar]
- 51.Huang F., Wang Q., Guo F., Zhao Y., Ji L., An T., Song Y., Liu Y., He Y., Qin G. FoxO1-mediated inhibition of STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis in diabetic kidney disease. EBioMedicine. 2019;48:491–504. doi: 10.1016/j.ebiom.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marti-Solano M., Crilly S.E., Malinverni D., Munk C., Harris M., Pearce A., Quon T., Mackenzie A.E., Wang X., Peng J., et al. Combinatorial expression of GPCR isoforms affects signalling and drug responses. Nature. 2020;587:650–656. doi: 10.1038/s41586-020-2888-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C., Xu Y., Liu H., Cai H., Jiang Y., Xu H.E., Yin W. Molecular recognition of itch-associated neuropeptides by bombesin receptors. Cell Res. 2023;33:184–187. doi: 10.1038/s41422-022-00743-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carrieri A., Lacivita E., Belviso B.D., Caliandro R., Mastrorilli P., Gallo V., Niso M., Leopoldo M. Structural Determinants in the Binding of BB2 Receptor Ligands: In Silico, X-Ray and NMR Studies in PD176252 Analogues. Curr. Top. Med. Chem. 2017;17:1599–1610. doi: 10.2174/1568026617666161104102459. [DOI] [PubMed] [Google Scholar]
- 55.Uehara H., Hocart S.J., González N., Mantey S.A., Nakagawa T., Katsuno T., Coy D.H., Jensen R.T. The molecular basis for high affinity of a universal ligand for human bombesin receptor (BnR) family members. Biochem. Pharmacol. 2012;84:936–948. doi: 10.1016/j.bcp.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ding H., Kothandaraman S., Gong L., Williams M.M., Dirksen W.P., Rosol T.J., Tweedle M.F. A human GRPr-transfected Ace-1 canine prostate cancer model in mice. Prostate. 2016;76:783–795. doi: 10.1002/pros.23172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No publicly available data or shared data are cited. All data needed to evaluate the conclusion of the current study are present in the paper and/or the Supplementary Materials. Additional data are available from the corresponding author on request.