

Abstract
Objects: Epidemiologic studies have linked exposure to airborne pollutant particulate matter (PM) with increased rates of chronic cardiopulmonary diseases, including asthma and idiopathic pulmonary fibrosis (IPF). Several investigations have suggested that the epithelial-to-mesenchymal transition (EMT) may contribute to the complex pathobiology of environmental exposure-mediated pulmonary fibrosis. The present study was designed to characterize the mechanisms of PM-mediated EMT in human lung epithelial cells (HBECs). Methods and results: PM induced significant dose (0-100 μg/ml) and time (0-72 h)-dependent increases in transforming growth factor β (TGFβ) and fibronectin (FN) protein levels in HBECs lysates. PM-activated TGFβ and FN protein production in HBECs was prevented by the antioxidant N-acetyl-cysteine (NAC, 5 mM). Furthermore, the NF-κB inhibitor BAY11-7082 (5 μM) abolished PM-induced FN production in HBECs. Biomarkers of EMT (ACTA2, SNAIL1 and SNAIL2) in PM-treated HBECs were significantly increased at the mRNA level compared to control cells. Conclusions: These results demonstrate that PM increases protein levels of TGFβ and FN via reactive oxygen species (ROS)-dependent pathways. In addition, PM exposure induces EMT in human lung epithelial cells, supporting a novel mechanism for PM-induced pulmonary fibrosis.
Keywords: Particulate matter, epithelial-to-mesenchymal transition, reactive oxygen species, lung epithelial cells
Introduction
Inhalation of ambient particulate matter (PM) is known to cause or exacerbate chronic cardiopulmonary disease morbidity and mortality, including asthma and idiopathic pulmonary fibrosis (IPF) [1]. PM is composed of organic and inorganic agents, including transition metals, hydrocarbons, and endotoxins, which are capable of penetrating alveolar epithelial cells (AECs) and causing pulmonary distress, inducing epithelial cell apoptosis and fibrotic remodeling stimulation [2,3]. The epithelial-to-mesenchymal transition (EMT) is a process by which cells lose epithelial apical-basal polarity and acquire mesenchymal characteristics. EMT leads to initiation and progression of tissue differentiation during organ development, as well as to tissue fibrosis and carcinoma progression [4]. EMT has been identified in the alveolar epithelium and airway epithelium in pulmonary fibrosis as a hallmark of fibrosis [5].
Transforming Growth Factor-beta (TGFβ) is a cytokine that plays an essential role in multiple cellular functions and tissue engineering, including cell differentiation, migration, and proliferation [6]. An increasing number of studies have revealed that TGFβ signal transduction is a vital factor in stimulating EMT during tumorigenesis [7] and tissue fibrosis [8]. TGFβ is the primary factor that drives several pulmonary disease processes, particularly those involving pulmonary fibrosis, lung cancer, and inflammatory lesions [9]. Transgenic mice overexpressing the mature form of TGFβ1 in the liver exhibit multiple organ fibrosis and inflammatory lesions [10]. The effects of TGFβ on EMT progression in AECs suggest that AECs are the source of pathogenic mesenchymal cells during lung fibrogenesis [5]. Fibronectin (FN) is a primary component of the extracellular matrix (ECM), which regulates the cellular network between intra- and extracellular environments, controlling cell fibrillogenesis [11,12]. It has been reported that TGFβ mediates lung fibrosis by stimulating ECM production and FN overexpression in human lung fibroblasts [13]. Additionally, several EMT biomarkers are stimulated by TGFβ treatment, such as alpha smooth muscle actin (α-SMA), snail family transcriptional repressor 1 (SNAI1) and snail family transcriptional repressor 2 (SNAI2) [14,15]. In alveolar epithelial cells, PM exposure enhances TGFβ activation based on an increase in reactive oxygen species (ROS) generation [3]. We hypothesize that PM exposure induces EMT by activating TGFβ in human lung epithelial cells (HBECs).
Our group previously revealed that PM exposure induces human lung endothelial cell injury in a ROS-dependent manner. Herein, we report that PM also stimulates EMT in lung epithelial cells upon ROS generation. PM exposure induces TGFβ and fibronectin protein production, which are key effectors that promote mammary mesenchymal formation. Finally, EMT biomarker genes (α-SMA, SNAI1 and SNAI2) are significantly upregulated in PM-exposed cells, confirming EMT remodeling in HBECs, all of which favor a profibrotic phenotype. Taken together, these findings provide an avenue to understand the mechanism of PM-induced pulmonary fibrosis.
Materials and methods
Reagents and chemicals
The PM sample (0.1-0.3 μm aerodynamic diameter) used in this study was collected from the Fort McHenry Tunnel, Baltimore, MD, USA, at a high volume; the chemical components, without any detectable endotoxin contamination in the PM suspension (1 mg/ml) water, were described previously [16]. TGFβ antibody was purchased from cell signal and fibronectin antibody was purchased from Sigma. Cell lysis buffer was purchased from Thermo-Fisher Scientific (Carlsbad, CA). N-acetylcysteine (NAC) and BAY 11-7082 (BAY) were purchased from Sigma (St. Louis, MO). qPCR TaqMan Real-Time PCR Assay was purchased from Applied Biosystems (now a part of Thermo-Fisher): Hs00195591_m1 (SNAIL1); Hs00161904_m1 (SNAIL2); Hs00426835_g1 (ACTA2); Hs02786624_g1 (GAPDH). All other biochemicals were purchased from Sigma unless stated otherwise.
Cell culture
HBECs were cultured in Minimal Essential Medium with Earle’s Minimal Essential Medium (MEM/EBSS, Thermo Fisher) supplemented with 10% FBS and 1% penicillin/streptomycin (P/S) at 37°C in a humidified atmosphere of 5% CO2 and 95% air, with 90% confluency for experimentation. Before PM challenge, HBEC media were changed to serum-free media for 24 h of synchronization. For the chemical blockade experiment, NAC and BAY were used to treat cells 30 min before PM exposure.
Western blotting
Treated or untreated HBECs were scraped into radioimmunoprecipitation assay buffer (RIPA buffer, Cell Signaling, Danvers, MA) with adequate protease and phosphatase inhibitors (Thermo Fisher) after washing with cold PBS three times. The protein concentration was quantified using the Bradford protein assay (Bio-Rad, Hercules, CA); equal amounts of protein samples were separated by Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis (SDS-PAGE), followed by transfer to polyvinylidene difluoride (PVDF) membranes. After incubation with 5% BSA in TBS-Tween 20 buffer (TBST, Thermo Fisher) for 1 h, the PVDF membrane was further incubated in primary antibody in 5% BSA in TBST buffer overnight at 4°C. After washing with TBST three times, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:5,000 in TBST with 5% BSA) at room temperature for 1 h. Western blot images were obtained by using a myECL imager system (Thermo Fisher) and chemiluminescence reagent (Bio-Rad) after three washes with TBST. Immunoblot bands were quantified with intensity using ImageJ software.
Real-time PCR
Total RNA was extracted from HBECs treated with PM exposure for 24 h using TRIzol reagent (Life Technology, Carlsbad, CA). Total RNA was treated with DNase I (Thermo Fisher), and 1 µg total RNA was used for first-strand cDNA synthesis with random primer p(N)6 and Maxima H Minus Reverse Transcriptase (Thermo Fisher). Real-time PCR was conducted by using Taqman Probe (Applied Biosystems, Waltham, MA) and Universal Master Mix II (Life Technology) following the supplier’s protocol. The GAPDH gene was used as an internal control. Real-time PCR assays were performed following the protocol of CFX96 Touch Real-Time PCR Detection System (Bio-Rad). All reactions were performed in triplicate.
Statistical analysis
Data are presented as the mean ± SEM for each experimental group. We performed statistical comparisons among treatment groups by randomized design and one-way ANOVA, followed by the Newman-Keuls post hoc test for more than two groups or by an unpaired Student’s t test for two groups. In all cases, statistical significance was defined as P<0.05. These statistical analyses were performed by using GraphPad Prism software version 9.5.
Results
PM exposure induces TGFβ expression in HBECs
We first confirmed the effects of PM exposure on production of TGFβ, a marker of fibrosis. In HBECs, TGFβ was increased along with PM stimulation (6-48 h) and peaked at 48 h (Figure 1A and 1B). In a dose-dependent study, after exposure to PM for 48 h, TGFβ protein was gradually upregulated with increasing concentration (2 µg/ml, 10 µg/ml and 100 µg/ml) and reached the highest level at 100 µg/ml (Figure 1C and 1D). These results indicate that PM exposure induces TGFβ production in a time- and dose-dependent manner.
Figure 1.
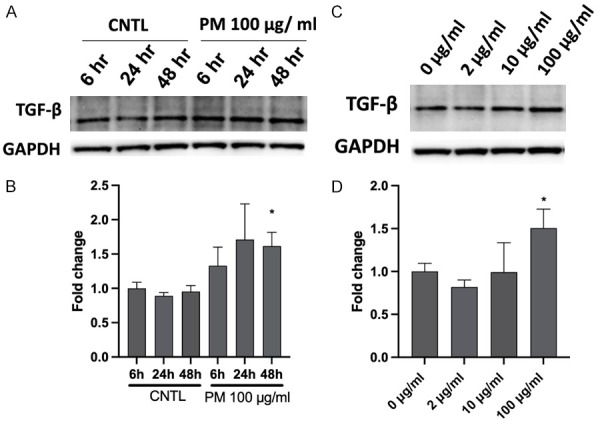
PM challenge induces TGF-β expression in a time- and dose-dependent manner. A. HBECs were challenged with PM (100 μg/ml) for 6 h, 24 h, or 48 h, and cell lysates were analyzed by western blotting for TGF-β protein expression. B. Bar graph of relative TGF-β levels. *, P<0.05 compared to control. C. HBECs were exposed to different concentrations of PM for 48 h (0-100 μg/ml), and TGF-β protein expression levels were assayed by western blotting. D. Bar graph of relative TGF-β levels. *, P<0.05 compared to control. PM, particulate matter; HBECs, human lung epithelial cells.
PM induces TGFβ increase through the ROS pathway
PM exposure of lung cells trigger ROS production via polycyclic aromatic hydrocarbons (PAHs) or transition metals, leading to direct or indirect effects on lipids and proteins through activation of intracellular oxidant pathways [17]. Our laboratory’s previous studies revealed that PM induces oxidative stress in lung cells in vitro and in vivo [16,18,19]. To assess whether cellular oxidative stress mediates the effect of PM treatment on TGFβ production in HBECs, we used the ROS scavenger N-acetylcysteine (NAC), which abolishes PM-induced ROS generation (Supplementary Figure 1). NAC pretreatment (5 mM, 30 min) reduced PM-induced TGFβ production (Figure 2), suggesting that PM-induced TGFβ upregulation is ROS dependent.
Figure 2.
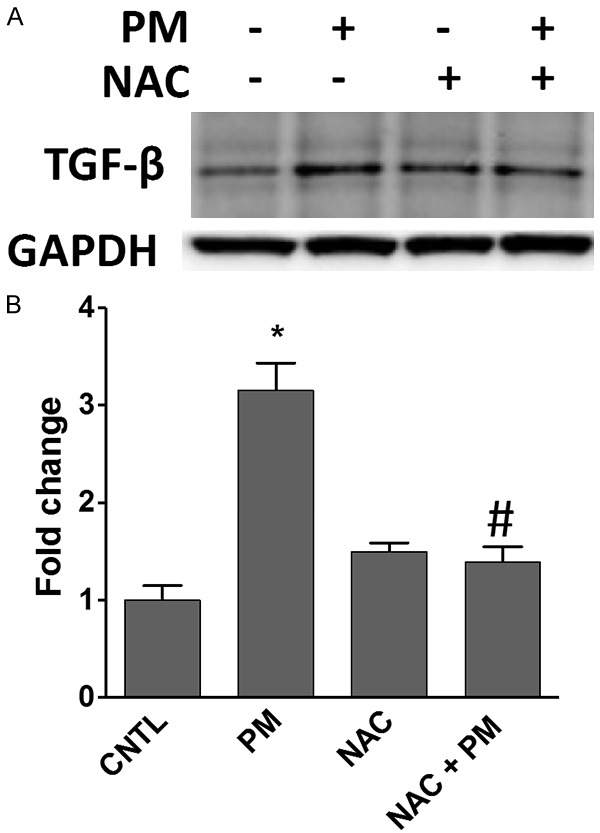
PM-activated TGF-β upregulation is attenuated by N-acetyl cysteine (NAC). HBECs were pretreated with 5 mM NAC for 30 min and then challenged with PM (100 μg/ml) for 48 h. A. TGF-β protein levels were assayed by western blotting. B. Bar graph of relative TGF-β levels. *, P<0.05 compared to control. #, P<0.05 compared to the PM group. PM, particulate matter; HBECs, human lung epithelial cells.
PM induces FN expression
TGFβ directly acts on fibroblast-type cells to induce progression of fibrosis via regulation of profibrotic molecule expression, including fibronectin (FN). Compared to control cells, PM (100 µg/ml)-challenged cells exhibited a higher level of FN in a time-dependent manner (6-72 h, Figure 3A and 3B). Additionally, PM exposure dose-dependently induced FN expression in HBECs (2-100 µg/ml, Figure 3C and 3D). These results indicate that PM exposure induces FN expression in the cell fibrosis process.
Figure 3.
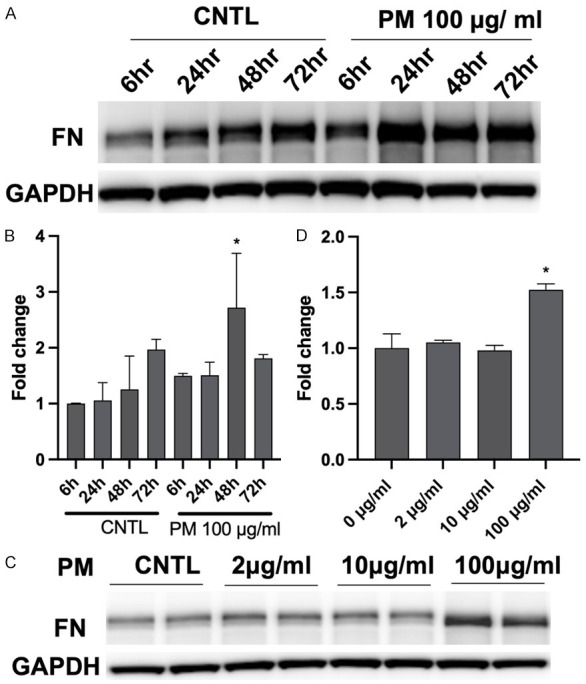
PM challenge induces FN expression in a time- and dose-dependent manner. A. HBECs were challenged with PM (100 μg/ml) for 6 h, 24 h, or 48 h, and cell lysates were analyzed by western blotting for PM-induced FN expression. B. Bar graph of relative FN levels. *, P<0.05 compared to control. C. HBECs were exposed to different concentrations of PM for 48 h (0-100 μg/ml), and FN protein expression levels were assayed by western blotting. D. Bar graph of relative FN levels. *, P<0.05 compared to control. PM, particulate matter; FN, fibronectin; HBECs, human lung epithelial cells.
PM induces FN through ROS and the NFκB pathway
PM exposure induces ROS production to initiate lung cell pathogenesis [16,18,19], and excessive cellular ROS activate fibronectin expression [20]. We used NAC as a ROS scavenger to evaluate the ROS effect on FN increase in HBECs. Similar to TGFβ, PM-induced FN expression was significantly suppressed by NAC pretreatment (5 mM, 30 min) in HBECs exposed to PM for 48 h (Figure 4).
Figure 4.
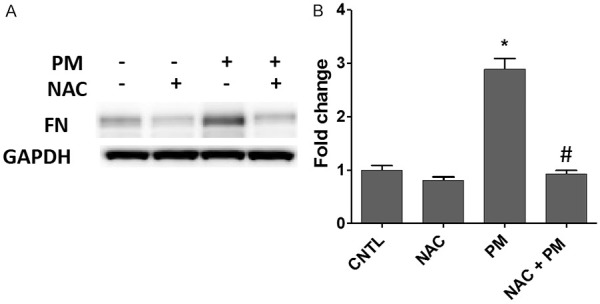
PM-activated FN upregulation is attenuated by N-acetyl cysteine (NAC). HBECs were pretreated with 5 mM NAC and then challenged with PM (100 μg/ml) for 48 h. A. FN protein levels were assayed by western blotting. B. The bar graph of relative FN levels. *, P<0.05 compared to CNTL. #, P<0.05 compared to the PM group. PM, particulate matter; FN, fibronectin; HBECs, human lung epithelial cells.
Several studies have demonstrated that NF-κB plays a critical role in EMT by modulating expression of mesenchymal genes [21,22]. Furthermore, it has been reported that NF-κB directly binds to the FN promoter to regulate FN gene transcription [23,24]. In addition, our previous study confirmed that PM exposure activates NF-κB in human lung epithelial cells [25]. Therefore, to address the role of NF-κB in PM-activated FN expression, an NF-κB pharmacological inhibitor (BAY 5 µM, 30 min pretreatment) was used. BAY significantly abolished PM-induced FN protein expression (Figure 5). Taken together, PM challenge activated FN expression via ROS- and NF-κB-dependent pathways.
Figure 5.
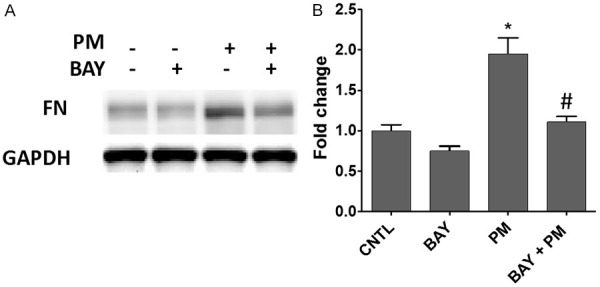
PM-activated FN upregulation is attenuated by the NF-κB inhibitor Bay. HBECs were pretreated with 5 μM Bay for 30 min and then challenged with PM (100 μg/ml) for 48 h. A. FN protein levels were assayed by western blotting. B. The bar graph of relative FN levels. *, P<0.05 compared to CNTL. #, P<0.05 compared to the PM group. PM, particulate matter; FN, fibronectin.
PM upregulates EMT biomarker gene expression
Several studies have demonstrated that EMT progression induces many marker genes, such as cytoskeleton α-SMA, which is encoded by the ACTA2 gene, and the transcription factors Snail1 and Snail2, which are encoded by the SNAIL1 and SNAIL2 genes, respectively. To confirm PM-induced EMT in HBECs, we determined ACTA2, SNAIL1 and SNAIL2 mRNA levels after PM exposure for 24 h. As shown, ACTA2 (Figure 6A), Snail1 (Figure 6B), and Snail2 (Figure 6C) were significantly increased in PM-exposed HBECs compared to control cells, suggesting that PM exposure preferentially induces EMT in HBECs.
Figure 6.
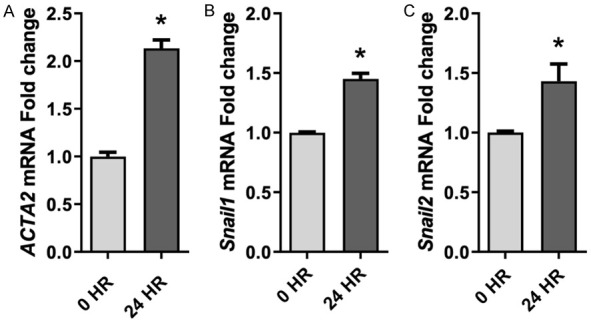
PM exposure induces EMT biomarker gene transcription. ACTA2 (A), SNAIL1 (B) and SNAIL2 (C) mRNA levels were significantly upregulated in PM-treated HBECs (24 h) compared to control cells. All experiments were conducted with at least three biological repeats. *, P<0.05 compared to control. PM, particulate matter; EMT, epithelial-to-mesenchymal transition; HBECs, human lung epithelial cells.
Discussion
In this study, we demonstrated that PM exposure induces EMT and initiates fibrosis in HBECs. In particular, PM stimulates production of TGFβ and fibronectin, two important primary factors that promote myofibrosis and cell migration, via PM-induced oxidative stress. The increase in the biomarkers α-SMA, Snail1 and Snail2 confirmed the EMT phenotype in HBECs after PM stress. As one more descriptive confirmation, PM indeed induced elongated EMT-like cell morphology (Supplementary Figure 2), which was attenuated by blockade of ROS or NF-κB.
Epithelial cells form an adherent layer and intimately communicate with and attach to each other by forming integral adhesion complexes, especially cell-cell contact structures, including tight junctions. The epithelial layer exhibits apical-basal polarity that separates it from other tissues, with the apical side toward the lumen and basolateral localization on the basement membrane [26]. Controversially, mesenchymal cells lack cell polarization and intercellular junctions due to disassembly of the organized actin cytoskeleton [27]. In addition to mediating embryonic development, EMT occurs during organ injury to initiate fibrosis, such as prolonged myofibroblasts and fibrogenesis accumulation in airway remodeling during lung inflammation. In this case, EMT is deemed a key initiation step of lung fibrosis. It has been well elucidated that fibrosis leads to several other lung diseases, such as asthma, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) [28].
TGFβ is a central factor that mediates and regulates EMT propagation since TGFβ treatment was recognized as the most important inducer of EMT in various epithelial cells [29]. TGFβ induces EMT development in bronchial epithelial cells, as accompanied by cell morphology changes, including a spindle fibroblast-like cell shape and a reduction in cell-cell contact [30]. The effects of PM exposure on upregulation of TGFβ protein expression in HBECs (Figure 1) provide evidence that PM stress induces EMT. Furthermore, TGFβ has diverse cellular functions and is related to several cellular biological responses, including tissue fibrosis and carcinogenesis, via crosstalk with other pathways, such as mitogen-activated protein kinases (p38, JNK and ERK) and p53 [31].
Our previous study revealed that ambient particulate matter exerts robust cellular injury on lung cells in a ROS-dependent manner [16]. The increase in ROS generation in the lung tissues of animal models after PM exposure contributes to PM-induced lung injury [32,33]. In HBECs, the ROS scavenger NAC attenuated the PM-induced increase in TGFβ expression, as well as fibronectin expression, suggesting that PM exposure initiates EMT after ROS generation (Figures 2 and 4). However, TGFβ might also induce ROS production, which is reported to be involved in induction of EMT in metastatic breast and pancreatic cancer cells [34], suggesting that positive feedback may exist between TGFβ and ROS.
It has been reported that FN is significantly increased in human bronchial epithelial cells and human lung fibroblasts exposed to TGFβ [13,30]. FN is a stromal extracellular matrix (ECM) protein that exerts its function by binding to integrins to mediate intercellular signaling. FN is maintained at low levels in normal cells but shows a robust increase at a trend correlating with the severity of disease, especially in fibrotic disorders [35]. Herein, we show that FN was increased after PM challenge as a downstream effect of NF-κB signaling (Figure 5). The NF-κB pathway is widely considered a major activator of immunity and inflammation, as well as a pivotal regulator of cell proliferation and migration [36]. NF-κB is essential for carcinoma progression and EMT induction [37]. PM exposure stimulates NF-κB subunit p65 activation, which directly binds to the fibronectin promoter and increases binding ability under TGFβ treatment [23,38]. We speculate that PM induces TGFβ production and activates NF-κB transcriptional function to increase fibronectin expression.
Several genes have been recognized as biomarkers to identify EMT initiation, including cytoskeleton genes and transcription factor genes [39]. α-SMA is one of six actin family members and is commonly considered a marker of active myofibroblast formation. Activation of snail transcription factors, particularly Snail1 and Snail2, is associated with EMT progression, such as fibrosis and cancer development [39,40]. In the present study, we show that α-SMA, Snail1, and Snail2 were upregulated by PM exposure, strongly suggesting EMT initiation in HBECs (Figure 6). Interestingly, we found that although PM-increased α-SMA/Snail2 can be attenuated by the ROS scavenger NAC and NFκB inhibitor BAY, as expected (Supplementary Figure 3), increased Snail1 was not prevented by either reagent, suggesting that Snail1 might be activated in a ROS-independent pathway.
Rho activation is also required for TGFβ-induced EMT through stimulation of ROCK activity. Rho-ROCK activity stimulates the formation of actin stress fibers as scaffolds for cellular signaling. Inhibition of Rho/ROCK eliminates the TGF-β-induced EMT effect in vitro and in vivo [41]. Similarly, in our previous study, PM exposure induced Rho/ROCK signaling, resulting in actin rearrangement with stress fiber formation [42] and suggesting that PM-activated TGFβ coordinates with PM-activated Rho to exert EMT in HBECs.
One of the early events in EMT is disassembly of epithelial tight junctions to disrupt normal epithelial cell integrity. For example, the tight junction protein Zona occludens-1 (ZO-1) is distributed during the EMT process [27,43]. Our previous study demonstrated that PM stress induces relocation of ZO-1 from the cell periphery accompanying ZO-1 protein degradation [18]. Additionally, ZO-1 protein inhibition and localization alteration depend on NF-κB activation [44]. In this study, pretreatment with an NF-κB inhibitor attenuated the increase in FN under PM exposure, suggesting that the NF-κB pathway is associated with the EMT process in HBECs (Figure 5). We will evaluate ZO-1 alterations in further studies to determine the role of ZO-1 in PM-induced EMT in HBECs.
In conclusion, this study confirms that PM induces EMT and early fibrotic signals in lung epithelial cells in an in vitro model of PM exposure. Therefore, inhibition of ROS/NF-κB may provide useful therapeutic strategies for treatment of PM-induced lung fibrosis.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Brunekreef B, Holgate ST. Air pollution and health. Lancet. 2002;360:1233–1242. doi: 10.1016/S0140-6736(02)11274-8. [DOI] [PubMed] [Google Scholar]
- 2.Upadhyay D, Panduri V, Ghio A, Kamp DW. Particulate matter induces alveolar epithelial cell DNA damage and apoptosis: role of free radicals and the mitochondria. Am J Respir Cell Mol Biol. 2003;29:180–187. doi: 10.1165/rcmb.2002-0269OC. [DOI] [PubMed] [Google Scholar]
- 3.Dysart MM, Galvis BR, Russell AG, Barker TH. Environmental particulate (PM2.5) augments stiffness-induced alveolar epithelial cell mechanoactivation of transforming growth factor beta. PLoS One. 2014;9:e106821. doi: 10.1371/journal.pone.0106821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 6.Ma J, Sanchez-Duffhues G, Goumans MJ, Ten Dijke P. TGF-beta-induced endothelial to mesenchymal transition in disease and tissue engineering. Front Cell Dev Biol. 2020;8:260. doi: 10.3389/fcell.2020.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hao Y, Baker D, Ten Dijke P. TGF-beta-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20:2767. doi: 10.3390/ijms20112767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frangogiannis N. Transforming growth factor-beta in tissue fibrosis. J Exp Med. 2020;217:e20190103. doi: 10.1084/jem.20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito A, Horie M, Nagase T. TGF-beta signaling in lung health and disease. Int J Mol Sci. 2018;19:2460. doi: 10.3390/ijms19082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parisi L, Toffoli A, Ghezzi B, Mozzoni B, Lumetti S, Macaluso GM. A glance on the role of fibronectin in controlling cell response at biomaterial interface. Jpn Dent Sci Rev. 2020;56:50–55. doi: 10.1016/j.jdsr.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradbury P, Nader CP, Cidem A, Rutting S, Sylvester D, He P, Rezcallah MC, O’Neill GM, Ammit AJ. Tropomyosin 2.1 collaborates with fibronectin to promote TGF-beta1-induced contraction of human lung fibroblasts. Respir Res. 2021;22:129. doi: 10.1186/s12931-021-01730-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 15.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Chiang ET, Moreno-Vinasco L, Lang GD, Pendyala S, Samet JM, Geyh AS, Breysse PN, Chillrud SN, Natarajan V, Garcia JG. Particulate matter disrupts human lung endothelial barrier integrity via ROS- and p38 MAPK-dependent pathways. Am J Respir Cell Mol Biol. 2010;42:442–449. doi: 10.1165/rcmb.2008-0402OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lodovici M, Bigagli E. Oxidative stress and air pollution exposure. J Toxicol. 2011;2011:487074. doi: 10.1155/2011/487074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang T, Wang L, Moreno-Vinasco L, Lang GD, Siegler JH, Mathew B, Usatyuk PV, Samet JM, Geyh AS, Breysse PN, Natarajan V, Garcia JG. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part Fibre Toxicol. 2012;9:35. doi: 10.1186/1743-8977-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang T, Moreno-Vinasco L, Huang Y, Lang GD, Linares JD, Goonewardena SN, Grabavoy A, Samet JM, Geyh AS, Breysse PN, Lussier YA, Natarajan V, Garcia JG. Murine lung responses to ambient particulate matter: genomic analysis and influence on airway hyperresponsiveness. Environ Health Perspect. 2008;116:1500–1508. doi: 10.1289/ehp.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HB, Yu MR, Song JS, Ha H. Reactive oxygen species amplify protein kinase C signaling in high glucose-induced fibronectin expression by human peritoneal mesothelial cells. Kidney Int. 2004;65:1170–1179. doi: 10.1111/j.1523-1755.2004.00491.x. [DOI] [PubMed] [Google Scholar]
- 21.Huber MA, Beug H, Wirth T. Epithelial-mesenchymal transition: NF-kappaB takes center stage. Cell Cycle. 2004;3:1477–1480. doi: 10.4161/cc.3.12.1280. [DOI] [PubMed] [Google Scholar]
- 22.Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104:733–744. doi: 10.1002/jcb.21695. [DOI] [PubMed] [Google Scholar]
- 23.Stanisavljevic J, Porta-de-la-Riva M, Batlle R, de Herreros AG, Baulida J. The p65 subunit of NF-kappaB and PARP1 assist Snail1 in activating fibronectin transcription. J Cell Sci. 2011;124:4161–4171. doi: 10.1242/jcs.078824. [DOI] [PubMed] [Google Scholar]
- 24.Lee BH, Park SY, Kang KB, Park RW, Kim IS. NF-kappaB activates fibronectin gene expression in rat hepatocytes. Biochem Biophys Res Commun. 2002;297:1218–1224. doi: 10.1016/s0006-291x(02)02356-2. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Usatyuk PV, Gorshkova IA, He D, Wang T, Moreno-Vinasco L, Geyh AS, Breysse PN, Samet JM, Spannhake EW, Garcia JG, Natarajan V. Regulation of COX-2 expression and IL-6 release by particulate matter in airway epithelial cells. Am J Respir Cell Mol Biol. 2009;40:19–30. doi: 10.1165/rcmb.2008-0105OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 28.Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res. 2018;19:136. doi: 10.1186/s12931-018-0834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009;10:100. doi: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 32.Tang W, Du L, Sun W, Yu Z, He F, Chen J, Li X, Li X, Yu L, Chen D. Maternal exposure to fine particulate air pollution induces epithelial-to-mesenchymal transition resulting in postnatal pulmonary dysfunction mediated by transforming growth factor-beta/Smad3 signaling. Toxicol Lett. 2017;267:11–20. doi: 10.1016/j.toxlet.2016.12.016. [DOI] [PubMed] [Google Scholar]
- 33.Kim GD, Oh J, Park HJ, Bae K, Lee SK. Magnolol inhibits angiogenesis by regulating ROS-mediated apoptosis and the PI3K/AKT/mTOR signaling pathway in mES/EB-derived endothelial-like cells. Int J Oncol. 2013;43:600–610. doi: 10.3892/ijo.2013.1959. [DOI] [PubMed] [Google Scholar]
- 34.Boudreau HE, Casterline BW, Rada B, Korzeniowska A, Leto TL. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic Biol Med. 2012;53:1489–1499. doi: 10.1016/j.freeradbiomed.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park J, Schwarzbauer JE. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene. 2014;33:1649–1657. doi: 10.1038/onc.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlowski RZ, Baldwin AS Jr. NF-kappa B as a therapeutic target in cancer. Trends Mol Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- 37.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappa B is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silbajoris R, Osornio-Vargas AR, Simmons SO, Reed W, Bromberg PA, Dailey LA, Samet JM. Ambient particulate matter induces interleukin-8 expression through an alternative NF-kappaB (nuclear factor-kappa B) mechanism in human airway epithelial cells. Environ Health Perspect. 2011;119:1379–1383. doi: 10.1289/ehp.1103594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assani G, Zhou Y. Effect of modulation of epithelial-mesenchymal transition regulators Snail1 and Snail2 on cancer cell radiosensitivity by targeting of the cell cycle, cell apoptosis and cell migration/invasion. Oncol Lett. 2019;17:23–30. doi: 10.3892/ol.2018.9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho HJ, Yoo J. Rho activation is required for transforming growth factor-beta-induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol Int. 2007;31:1225–1230. doi: 10.1016/j.cellbi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Wang T, Shimizu Y, Wu X, Kelly GT, Xu X, Wang L, Qian Z, Chen Y, Garcia JGN. Particulate matter disrupts human lung endothelial cell barrier integrity via Rho-dependent pathways. Pulm Circ. 2017;7:617–623. doi: 10.1086/689906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das V, Bhattacharya S, Chikkaputtaiah C, Hazra S, Pal M. The basics of epithelial-mesenchymal transition (EMT): a study from a structure, dynamics, and functional perspective. J Cell Physiol. 2019;234:14535–14555. doi: 10.1002/jcp.28160. [DOI] [PubMed] [Google Scholar]
- 44.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.