

Abstract
Objectives: Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, a novel class of cholesterol-lowering drugs, can reduce atherosclerosis independent of systemic lipid changes. However, the mechanism by which PCSK9 inhibition protects against arteriosclerosis has not been fully elucidated. Recent evidence has demonstrated a correlation between PCSK9 inhibitors and oxidative stress, which accelerates atherosclerotic development. Moreover, an increasing number of studies have shown that autophagy protects the vasculature against atherosclerosis. Therefore, the aims of this study were to investigate the effect of PCSK9 inhibition on oxidative stress and autophagy in atherosclerosis and determine whether autophagy regulates PCSK9 inhibition-mediated oxidative stress and inflammation in macrophages. Methods: Male apolipoprotein E (ApoE)-/- mice were fed a high-fat diet (HFD) for 8 weeks and then received the PCSK9 inhibitor (evolocumab), vehicle, or evolocumab plus chloroquine (CQ) for another 8 weeks. ApoE-/- mice in the control group were fed a regular (i.e., non-high-fat) diet for 16 weeks. Additional in vitro experiments were performed in oxidized low-density lipoprotein (ox-LDL)-treated human acute monocytic leukemia cell line THP-1-derived macrophages to mimic the pathophysiologic process of atherosclerosis. Results: PCSK9 inhibitor treatment reduced oxidative stress, lipid deposition, and plaque lesion area and induced autophagy in HFD-fed ApoE-/- mice. Most importantly, the administration of chloroquine (CQ), an autophagy inhibitor, significantly reduced the beneficial effects of PCSK9-inhibitor treatment on oxidative stress, lipid accumulation, inflammation, and atherosclerotic lesions in HFD-fed ApoE-/- mice. The in vitro experiments further showed that the PCSK9 inhibitor enhanced autophagic flux in ox-LDL-treated THP-1-derived macrophages, as indicated by increases in the numbers of autophagosomes and autolysosomes. Moreover, the autophagy inhibitor CQ also reduced PCSK9 inhibition-mediated protection against oxidative stress, generation of reactive oxygen species (ROS) and inflammation in ox-LDL-treated THP-1-derived macrophages. Conclusions: This study reveals a novel protective mechanism by which PCSK9 inhibition enhances autophagy and thereby reduces oxidative stress and inflammation in atherosclerosis.
Keywords: Atherosclerosis, proprotein convertase subtilisin/kexin type 9, autophagy, oxidative stress, inflammation
Introduction
Cardiovascular disorders remain the leading cause of morbidity and mortality worldwide. Atherosclerosis, the major cause of cardiovascular disorders, is a chronic inflammatory disease characterized by progressive accumulation of macrophage foam cells, inflammation, and oxidative stress in the vessel wall. Excess oxidative stress promotes endothelial dysfunction, macrophage infiltration, and subsequent inflammation [1], thus amplifying the development of atherosclerosis. Elucidating the regulatory mechanisms underlying oxidative stress in macrophages may facilitate the development of novel strategies to prevent and treat atherosclerosis.
Proprotein convertase subtilisin/kexin type 9 (PCSK9), a serine protease, is pivotal in regulating cholesterol homeostasis; it targets the low-density lipoprotein receptor (LDLR) to enable lysosomal degradation [2,3], resulting in an increase in plasma low-density lipoprotein-cholesterol (LDL-C) levels. Thus, PCSK9 has emerged as a key target for the treatment of hypercholesterolemia. Clinical trials have demonstrated that PCSK9 inhibitors (e.g., alirocumab and evolocumab) not only reduce plasma LDL-C levels, but also decrease the risk of cardiovascular events [4,5], indicating that PCSK9 may be an important target for preventing and treating atherosclerosis. Recent studies suggest that hepatic cholesterol metabolism is involved in regulating PCSK9-induced atherosclerosis. However, other studies suggest that PCSK9 inhibition or absence reduces atherosclerosis without affecting plasma lipid levels [6-9], implying that PCSK9 might promote atherosclerosis through mechanisms beyond its metabolic effect. Especially, the direct effect of PCSK9 on macrophages activation, an important component of the atherosclerotic plaque, cannot be ignored. Thus, PCSK9 attracts intense interest as a target for the treatment of atherosclerosis. Further study is required to understand the effect of PCSK9 inhibition on macrophage biology in the pathogenesis of atherosclerosis.
Evidence has indicated a correlation between PCSK9 inhibition and oxidative stress in various diseases. Cammisotto et al. reported that the PCSK9 inhibitor evolocumab reduces serum oxidized low-density lipoprotein (ox-LDL) concentrations in patients with heterozygous familial hypercholesterolemia [10]. Similarly, alirocumab, another PCSK9 inhibitor, regulates oxidative stress by reducing lipid peroxidation in alcohol-induced liver injury [11], and evolocumab both counteracts hydrogen peroxide-induced damage in human umbilical vein endothelial cells (HUVECs) [12] and alleviates ox-LDL-induced myocardial injury through mitochondrial fission induced by oxidative stress [13]. Thus, the role and mechanism of PCSK9 inhibition in regulating oxidative stress during atherosclerosis is of interest.
Autophagy is an evolutionarily conserved protective system by which cytoplasmic materials, such as defective proteins and organelles, are degraded in lysosomes [14]. A number of studies have shown that autophagy is required to maintain lipid homeostasis and protects against vascular atherosclerosis [15,16]. Impaired autophagy in macrophages during atherosclerotic development promotes lipid accumulation [15], excessive inflammatory responses [17], greater reactive oxygen species (ROS) generation and oxidative stress [18]. Recently, it was shown that PCSK9 deficiency reduces the secretion of apolipoprotein B by increasing autophagic flux in the liver [6]. PCSK9 knockdown by small interfering RNA (siRNA) in vascular smooth muscle cells (SMCs) was also shown to reduce the activation of the mammalian target of rapamycin (mTOR) signaling pathway [19], which is critical for the negative regulation of autophagy.
Thus, the present study aimed (1) to determine the effect of PCSK9 inhibitor treatment on oxidative stress and autophagy in atherosclerosis and (2) to elucidate whether autophagy regulates the effects of PCSK9 inhibition on oxidative stress and inflammation in macrophages.
Materials and methods
Materials
Evolocumab (a PCSK9 inhibitor) was purchased from Amgen (Amgen Inc. USA). Oxidized low-density lipoprotein (ox-LDL) was purchased from Peking Union-Biology (Beijin, China). Phorbol 12-myristate 13-acetate (PMA), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody, 2’,7’-dichlorofluorescein diacetate (DCFDA), and dihydroethidium (DHE) were obtained from Sigma-Aldrich (St. Louis, MO, USA). 8-Hydroxy-2’-deoxyguanosine (8-OHdG) antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-NF-κBp65 (P-NF-κBp65) antibody, anti-NF-κBp65 antibody, anti-sequestosome 1 (p62/SQSTM1) antibody and anti-microtubule-associated protein 1 light chain 3 (LC3) A/B antibody were obtained from Cell Signaling Technology (CST, Beverly, MA, USA). Chloroquine (CQ) was purchased from MedChemExpress (MCE, Monmouth Junction, NJ, USA).
Animals
The study procedures were approved by the Institutional Animal Care and Use Committees of Shanxi Cardiovascular Hospital.
Male ApoE-/- mice weighing approximately 18-20 g were purchased from Chauncey Biology (Beijing, China). All mice were kept in standard laboratory cages in a temperature-controlled (22-25°C) room and were fed a regular chow diet for 1 week before the experiment. ApoE-/- mice were exposed to a high-fat diet (HFD) (20% fat and 1.25% cholesterol) for 8 weeks and then subjected to different drug treatments for another 8 weeks. ApoE-/- mice were randomly categorized into three groups and administered the following: saline (equal volume), the PCSK9 inhibitor (evolocumab) (30 mg/kg, subcutaneous injections every 2 weeks), or evolocumab plus chloroquine (CQ) (50 mg/kg, intraperitoneal injections every other day). ApoE-/- mice fed a regular diet (RD; regular chow diet) for 16 weeks served as the control group. In this study, all animals were euthanized by intravenous injection of 10% potassium chloride (KCL). After the hearts were arrested, the aortic root was removed for further experimental analyses.
Culture and treatment of human acute monocytic leukemia cell line THP-1-derived macrophages
Human acute monocytic leukemia cell line THP-1 were purchased from the Cell Bank at Shanghai Institute for Biological Sciences and cultured with Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco, CA, USA) containing 10% fetal bovine serum (Gibco) and 1000 U/mL penicillin and 100 µg/mL streptomycin (Boster, Wuhan, China) at 37°C in a humidified atmosphere with 5% CO2. THP-1-derived macrophages were induced with 100 ng/mL phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, MO, USA) at a density of 0.5-1 × 106 cells per well in six-well plates for 48 h. After overnight serum starvation, the cells were treated with the drugs.
Atherosclerotic lesion analysis
A cross-sectional analysis of the aortic root was performed to evaluate atherosclerotic lesions and lipid deposition. The aortic roots were embedded in optimum cutting temperature (OCT) compound, cut into 6-μm-thick sections and placed on glass slides. Three or four sections were obtained from each mouse, spaced 30 μm apart beginning at the base of the aortic root and were stained with hematoxylin and eosin (H&E) and oil red O (Solarbio, Beijing, China) according to standard laboratory procedures, respectively. Plaque area and lipid deposition size were quantified with Image J software.
Immunohistochemistry
Immunohistochemical analyses for 8-OHdG were used to indicate the presence of DNA damage due to oxidative stress in the aortic root plaques. Briefly, after blocking endogenous peroxidase with 3% hydrogen peroxide solution, frozen cross-sections were blocked in 3% bovine serum albumin (BSA; Servicebio, Wuhan, China) solution for 30 min, and then sections were incubated with mouse 8-OHdG monoclonal antibody (1:100, Santa Cruz Biotechnology, CA, USA) overnight at 4°C. Vascular sections were washed in phosphate-buffered saline (PBS). Anti-mouse secondary antibodies conjugated to horseradish peroxidase (HRP) were used. After washing, the sections were stained with 3,3N-diaminobenzidine tetrahydrochloride (DAB) and counterstained with hematoxylin. Vascular images were observed by a light microscope.
Western blot analysis
Protein samples from THP-1-derived macrophages and vascular tissue protein were lysed and homogenized in radioimmunoprecipitation assay (RIPA) buffer, as previously described. Then, the resulting homogenate was centrifuged at 14,000 r.p.m. for 20 min at 4°C. The concentration of extracted protein lysates was determined by the bicinchoninic acid (BCA) protein assay kit (Beyotime Biotechnology, Beijing, China). Protein samples (30-40 μg/lane) were prepared and subjected to 8% or 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE); they were then transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked in 5% (w/v) dried milk in Tris-buffered saline (TBS)-Tween (TBST) for at least 2 h at room temperature. Then, the blots were incubated with primary antibodies overnight at 4°C: anti-LC3A/B antibody (Cell Signaling), anti-p62 antibody (Cell Signaling), anti-PCSK9 antibody (Abcam), anti-phospho-NF-κBp65 antibody (Cell Signaling), anti-NF-κBp65 antibody (Cell Signaling) and anti-GAPDH antibody (Sigma). After washing, the membranes were exposed to the appropriate peroxidase-conjugated secondary antibodies (1:1,000) for 1 h at 37°C. Western blot bands were developed by enhanced chemiluminescence (ECL) using the Molecular Imager ChemiDoc XRS system (Bio-Rad) and quantified by Image LabTM 5.2. Bands were normalized to the GAPDH or β-actin expression levels.
Foam cell formation analysis
To visualize foam cell formation, oil red O staining (Solarbio) was performed in each treated group. In brief, THP-1-derived macrophages were fixed with 4% paraformaldehyde for 2 hours. After washing, the cells were exposed to 60% isopropanol for 1 min and stained with oil red O for 10 min. After differentiation with 60% isopropanol, the nuclei were stained with hematoxylin solution. For the visualization of oil red O staining, cells were photographed under a microscope.
Autophagy flux analysis
THP-1-derived macrophages on coverslips were transfected with lentivirus-red fluorescent protein (RFP)-green fluorescent protein (GFP)-LC3 (GENECHEM, Shanghai, China) according to the manufacturer’s protocols for 48 hours. Then, the cells were pretreated with a PCSK9 inhibitor for 5 min and treated with ox-LDL for 24 h. The cells were fixed with 4% paraformaldehyde for 2 h and washed with PBS. To determine the fluorescence of red fluorescent protein (RFP) and green fluorescent protein (GFP), the cellular images were examined with a confocal microscopy (FV3000 Leica, Wetzlar, Germany). The mean number of yellow fluorescent puncta (RFP-positive and GFP-positive dots) and red fluorescent puncta (RFP-positive and GFP-negative dots) were analyzed using Image-Pro Plus 6 software. Approximately 30 cells were measured for each treatment analysis.
Immunofluorescent staining
Fluorescent staining was conducted to visualize of LC3 and 8-OHdG. Briefly, cells cultured on cover slides were permeabilized with 0.1% Triton X-100 for 10 min after fixation with 4% paraformaldehyde, and then cells were incubated with primary rabbit anti-LC3 antibody (Cell Signaling) or anti-8-OHdG antibody (Santa Cruz) overnight at 4°C. After washing, the cells were exposed to an anti-rabbit secondary IgG conjugated to Alexa Fluor 488 or tetramethylrhodamine isothiocyanate (TRITC) for 1 h at 37°C. 4’,6-Diamidino-2-phenylindole (DAPI) staining was used to indicate the nuclei. Fluorescent images were captured with a laser confocal microscope (FV3000 Leica). At least 50 cells from 20-50 fields were examined from each treatment for data analysis.
Intracellular reactive oxygen species (ROS) analysis
The production of reactive oxygen species (ROS) was evaluated by analyzing the fluorescence intensity by dihydroethidium (DHE; Sigma, St Louis, MO, USA) and 2’,7’-dichlorofluorescein diacetate (DCFDA) (Sigma). Briefly, OCT-embedded 6-μm-thick sections of the aortic root were stained with DHE solution and DAPI (nuclei) according to standard laboratory procedures. Additionally, THP-1-derived macrophages were seeded in six-well plates. After drug treatment, the cells were washed in PBS and then cultured in RPMI-1640 medium with supplemental DCFDA (Sigma) at a concentration of 20 μM for 20 min. After incubation, the cells were imaged under a confocal microscope (FV3000 Leica).
Macrophage autophagy staining analysis
For the CD68 and LC3 staining analyses, OCT-embedded sections were prepared and incubated with primary antibodies against CD68 (1:100, mouse polyclonal; Servicebio, Wuhan, China) and LC3A/B (1:100, rabbit monoclonal; Cell Signaling, MA, USA) overnight at 4°C. After washing, the sections were exposed to anti-mouse-CY3 and anti-rabbit-fluorescein isothiocyanate (FITC) secondary antibodies for visualization using a microscope (FV3000 Leica). 4’,6’-Diamidino-phenylindole (DAPI) staining was used to indicate the nuclei.
Inflammation assay
Whole blood samples from ApoE-/- mice were centrifuged immediately after collection at 3,000 rpm for 10 min at 4°C, and the serum samples were then collected and stored at -80°C. All samples were diluted to a 1:4 ratio in assay buffer before measurement of the inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and interleukin (IL)-10. Cytokine levels were quantified by Luminex assay according to the manufacturer’s protocols, and multiplex panels were performed and measured using a Luminex Assay System (EMD Millipore).
Quantitative reverse transcription-polymerase chain reaction
The total RNA was obtained from cultured THP-1-derived macrophages in vitro using the TRIzol reagent and quantified using a spectrophotometer. cDNA was generated using the Primescript reverse transcriptase kit with a gDNA eraser (TaKaRa, Japan). The TB Green Premix Ex Taq II (Tli RNaseH Plus) kit (TaKaRa) was used to assess the mRNA levels of tumor necrosis factor (TNF)-α and interleukin (IL)-6 in an Applied Biosystems 7300 real-time polymerase chain reaction (PCR) system. The sequence primers of each gene in this study were listed as follows: TNF-α (5’-TGC ACT TTG GAG TGA TCG GC-3’ and 5’-ACT CGG GGT TCG AGA AGA TG-3’); IL-6 (5’-GGC ACT GGC AGA AAA CAA CC-3’ and 5-GCT CTG GCT TGT TCC TCA CT-3’); and GAPDH (5’-ATC ATC CCT GCC TCT ACT GC-3’ and 5’-GTCAGGTCCACCACTGACAC-3’). In this study, GAPDH served as the reference control gene for value normalization. The comparative 2-ΔΔCt method was used for analyzing the relative expression of the target genes.
Statistical analysis
All continuous variables are presented as the mean ± standard error of the mean (SEM). When three or four groups were compared, a one-way analysis of variance (ANOVA) was performed to determine the overall treatment effect, and Tukey’s tests were used for multiple comparisons. Differences between two groups were assessed with Student’s t test. If P < 0.05, the data were considered to have a significant difference.
Results
PCSK9 inhibition decreased oxidative stress in atherosclerosis
Oxidative stress is an important proatherogenic mechanism in atherosclerosis. To evaluate the effect of PCSK9 inhibition on oxidative stress during atherosclerotic development, ApoE-/- mice were fed an HFD for 8 weeks and then exposed to the PCSK9 inhibitor evolocumab or an equal volume of saline for another 8 weeks. As shown in Figure 1A-D, we found that treatment with evolocumab significantly reduced lipid deposition area and plaque lesions compared to saline treatment. Importantly, immunohistochemical analysis indicated that administration of evolocumab attenuated 8-OHdG expression; 8-OHdG is a key biomarker of DNA damage due to oxidative stress [20] (Figure 1E, 1F). Moreover, DHE staining showed that evolocumab treatment reduced reactive oxygen species (ROS) generation (Figure 1G, 1H). Additionally, the PCSK9 inhibitor enhanced antioxidant capacity, as shown by the systematic levels of superoxide dismutase (SOD) (Figure 1I). Furthermore, no significant differences were observed in the plasma lipid levels between the PCSK9 inhibitor-treated and saline-treated groups (Supplementary Figure 1). These findings demonstrate that PCSK9 inhibition alleviates oxidative stress in atherosclerotic progression.
Figure 1.

PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibition decreased oxidative stress in the aortic root plaque of ApoE-/- mice. A-I. Apolipoprotein E (ApoE)-/- mice fed a high-fat diet (HFD) for 8 weeks were treated with saline or a PCSK9 inhibitor evolocumab (Evomab) for another 8 weeks. Regular diet (RD)-fed ApoE-/- mice were used as the control group. A, B. Oil red O staining analysis for plaque lipid deposition size (n = 6 mice per group, scale bar = 500 μm). C, D. Hematoxylin and eosin (H&E) staining of the aortic root sections from ApoE-/- mice in two groups. Quantification of plaque lesion area (n = 8 mice per group; scale bar = 500 μm). E, F. Immunohistochemical analysis of 8-hydroxy-2’-deoxyguanosine (8-OHdG) expression in the aortic root plaques of ApoE-/- mice in two groups. Quantification analysis of 8-OHdG expression (n = 5 mice per group; scale bar = 100 μm). G, H. Dihydroethidium (DHE) staining for reactive oxygen species (ROS) in the aortic root sections of ApoE-/- mice in two groups. Lesions were stained with 4’,6-diamidino-2-phenylindole (DAPI) for nuclei. Quantification of relative DHE fluorescence (n = 5 mice per group; scale bar = 200 μm). I. Quantification of the circulating levels of superoxide dismutase (SOD) by colorimetric assay (n = 7-8 mice per group). All values represent the mean ± standard error of the mean (SEM). **P < 0.01; ***P < 0.001.
PCSK9 inhibition increased autophagy during atherosclerotic progression
A number of studies have demonstrated that autophagy plays a protective role in the development of atherosclerosis [18,21]. To investigate the effect of PCSK9 inhibition on autophagy during atherosclerotic progression, we evaluated the protein expression of an autophagic marker in ApoE-/- mice fed an HFD. Western blot analysis showed that HFD-fed ApoE-/- mice displayed significantly decreased LC3-II and increased P62 protein expression compared with RD-fed ApoE-/- mice. Importantly, treatment with the PCSK9 inhibitor evolocumab obviously increased LC3-II and decreased P62 protein expression in HFD-fed ApoE-/- mice (Figure 2A-D). Furthermore, evolocumab treatment increased macrophage autophagy, as shown by analysis of the percentage of CD68-postive macrophages showing LC3 staining (Figure 2E, 2F). These results demonstrate that PCSK9 inhibition enhances macrophage autophagy during atherosclerotic progression.
Figure 2.
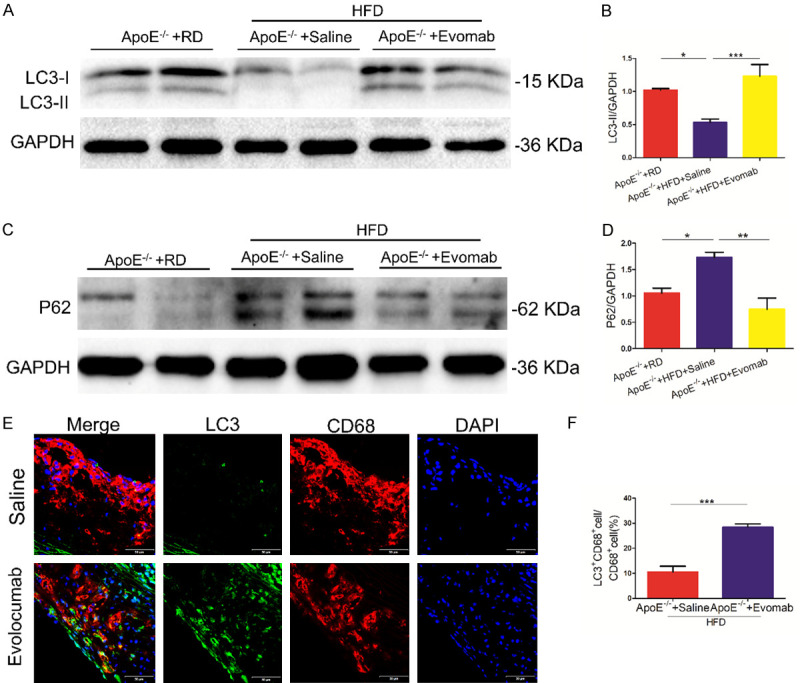
PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibition promoted autophagy during arteriosclerotic progression. A, B. Western blot analysis of microtubule-associated protein 1 light chain 3 (LC3) protein in the whole-aorta lysates of apolipoprotein E (ApoE)-/- mice fed with fed a high-fat diet (HFD) in the saline and evolocumab (Evomab) groups. Regular diet (RD)-fed ApoE-/- mice were used as the control group. Quantification of relative protein levels (n = 5-6 mice per group). C, D. Western blot analysis of sequestosome 1 (P62) protein in ApoE-/- mice in three groups. Quantification of relative protein levels (n = 4-5 mice per group). E, F. Representative fluorescent staining for LC3 (green) and CD68 (red) in the aortic root plaques of ApoE-/- mice fed with HFD in the saline and evolocumab groups. (4’,6-Diamidino-2-phenylindole, DAPI: nuclei; scale bar = 50 μm). Quantification of the percentage of LC3-postive macrophages in two groups. All data are presented as the mean ± standard error of the mean (SEM). *P < 0.05; **P < 0.01; ***P < 0.001.
PCSK9 inhibition promoted autophagic flux in ox-LDL-treated THP-1-derived macrophages
Ox-LDL is the key activator of oxidative stress in atherosclerosis. Previous studies have shown that ox-LDL induces PCSK9 expression in vascular SMCs [22] and macrophages [23]. Moreover, LDL-loaded macrophages secreted PCSK9 into the medium [24]. Therefore, we further investigated the effect of PCKS9 inhibition on autophagy in ox-LDL-induced THP-1-derived macrophages. This study showed that ox-LDL treatment induced an increase in PCSK9 protein expression in THP-1-derived macrophages (Figure 3A, 3B). Most importantly, we found that treatment with the PCSK9 inhibitor evolocumab significantly increased LC3-II protein expression and decreased P62 protein expression in ox-LDL-treated THP-1-derived macrophages (Figure 3C-F). Moreover, immunofluorescence staining demonstrated that treatment with evolocumab increased the number of LC3 puncta in ox-LDL-treated THP-1-derived macrophages (Figure 3G, 3H), suggesting that PCSK9 inhibition increases the level of autophagosomes. To further explore the effect of PCKS9 inhibition on autophagic flux in lipid-accumulating macrophages, lentivirus-RFP-GFP-LC3 was transfected into THP-1-derived macrophages. Representative fluorescent illumination showed that the PCSK9 inhibitor evolocumab enhanced autophagic flux in ox-LDL-treated THP-1-derived macrophages, as indicated by analysis of the autophagosomes (GFP-positive/RFP-positive dots) and autolysosomes (GFP-negative/RFP-positive dots) (Figure 3I, 3J). Hence, these data demonstrate that PCSK9 inhibition enhances autophagic flux in ox-LDL-treated THP-1-derived macrophages.
Figure 3.
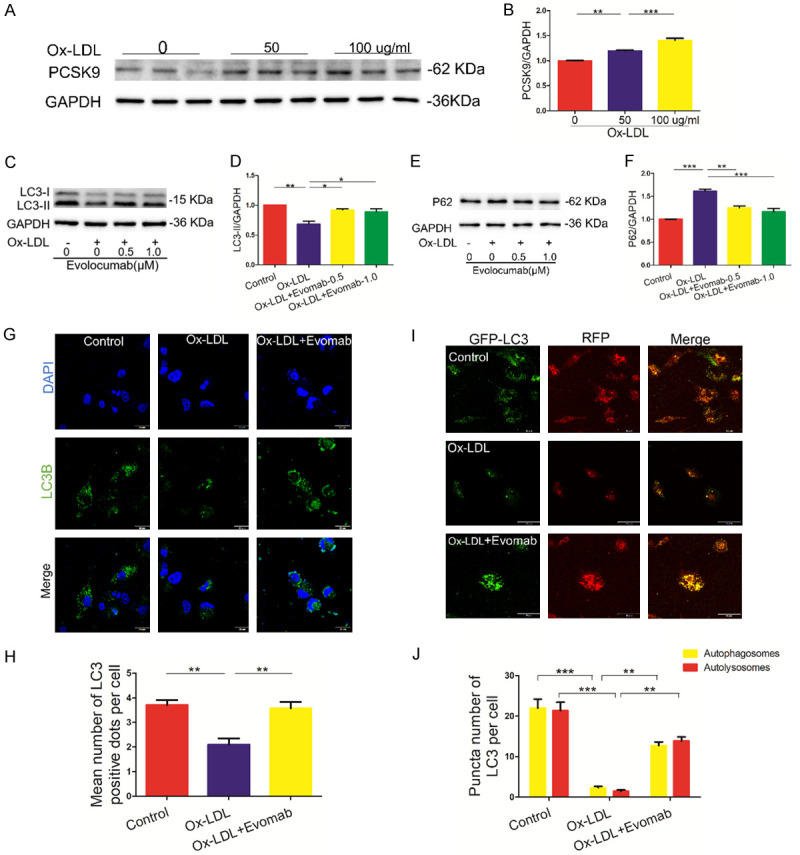
PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibition promoted autophagy in ox-LDL-treated THP-1-derived macrophages. A, B. Western blot analysis of PCSK9 protein in human acute monocytic leukemia cell line THP-1-derived macrophages induced by oxidized low-density lipoprotein (ox-LDL) (50 and 100 µg/mL) for 24 h. Quantification of relative protein levels. C-F. Serum-starved THP-1-derived macrophages were pretreated for 5 min with the indicated concentrations of evolocumab (Evomab), followed by stimulation with ox-LDL (100 µg/mL) for 24 h. Western blot analysis for the protein levels of microtubule-associated protein 1 light chain 3 (LC3) and sequestosome 1 (P62). Quantification of relative protein levels. G, H. Representative fluorescent staining for LC3 (DAPI: nuclei; scale bar = 20 μm). Quantification of the number of LC3 dots. At least 50 cells from 20-50 fields were examined from each treatment for data analysis. I, J. THP-1-derived macrophages were transfected with lentivirus-red fluorescent protein (RFP)-green fluorescent protein (GFP)-LC3 and then treated with ox-LDL (100 µg/ML) in the absence or presence of the PCSK9 inhibitor for 24 h. Representative fluorescent analysis for autophagosomes (GFP-positive/RFP-positive dots) and autolysosomes (GFP-negative/RFP-positive dots). Quantification of the number of autophagosomes (yellow) and autolysosomes (red). At least 50 cells were used for data analysis. Each data analysis was performed thrice, and the data are presented as the mean ± standard error of the mean (SEM). *P < 0.05; **P < 0.01; ***P < 0.001.
Impaired autophagy attenuated the protective effects of PCSK9 inhibition against oxidative stress and atherosclerotic lesions in vivo
To further determine whether or not autophagy mediates the effects of PCSK9 inhibition on oxidative stress and plaque formation in vivo, the autophagy inhibitor chloroquine (CQ) was employed. ApoE-/- mice were fed an HFD for 8 weeks and then received the PCSK9 inhibitor evolocumab, an equal volume of saline, or evolocumab plus CQ for another 8 weeks. The results showed that the autophagy inhibitor CQ further increased LC3-II protein expression induced by treatment with evolocumab in ApoE-/- mice (Figure 4A, 4B). Furthermore, CQ administration significantly reduced the protective effects of evolocumab against lipid accumulation, plaque formation, and oxidative stress, as indicated by analysis of oil Red O staining, hematoxylin - eosin staining, 8-OHdG expression, ROS production and plasma SOD levels in HFD-fed ApoE-/- mice (Figure 4C-K). These data suggest that impaired autophagy attenuates the protective effects of PCSK9 inhibition against oxidative stress and plaque formation in atherosclerotic development.
Figure 4.
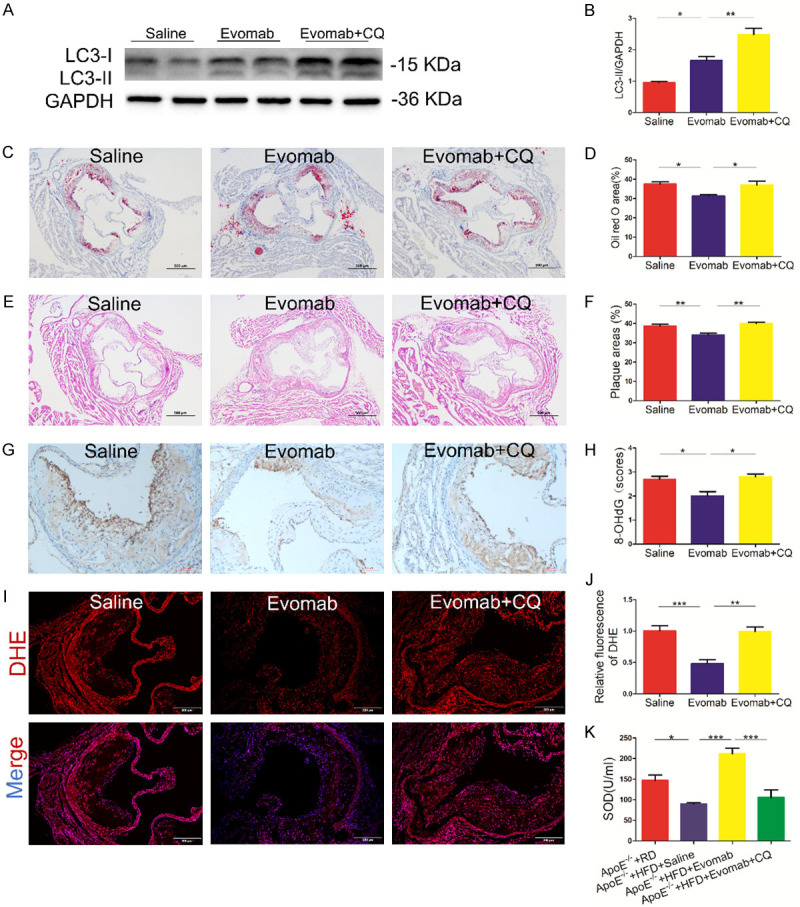
Impaired autophagy attenuated the protective effects of evolocumab against oxidative stress and plaque lesions in vivo. A-J. Apolipoprotein E (ApoE)-/- mice fed a high-fat diet (HFD) for 8 weeks were treated with saline, evolocumab (Evomab), and Evomab plus chloroquine (CQ) for another 8 weeks. Regular diet (RD)-fed ApoE-/- mice were used as the control group. A, B. Western blot analysis of microtubule-associated protein 1 light chain 3 (LC3) in the aortic vascular tissue of ApoE-/- mice in the saline, Evomab, and Evomab plus CQ groups. Quantification of relative protein levels (n = 4 mice per group). C, D. Representative oil red O staining of the aortic root lesions of ApoE-/- mice in each group (scale bar = 500 μm). Quantification of lipid deposition area (n = 5 mice per group). E, F. Hematoxylin and eosin staining of the aortic root sections of ApoE-/- mice in three groups (scale bar = 500 μm). Quantification of plaque lesions (n = 5 mice per group). G, H. Immunohistochemical analysis of 8-hydroxy-2’-deoxyguanosine (8-OHdG) expression in the aortic root plaques of ApoE-/- mice in three groups (scale bar = 100 μm). Quantification of 8-OHdG staining (n = 5 mice per group). I, J. Dihydroethidium (DHE) staining for detecting reactive oxygen species (ROS) in the aortic root plaques of ApoE-/- mice in three groups (DAPI: nuclei; scale bar = 200 μm). Quantification of DHE relative fluorescent intensity (n = 5 mice per group). K. Quantification of the serum levels of superoxide dismutase (SOD) by colorimetric assay (n = 6-8 mice per group). The data are presented as the mean ± standard error of the mean (SEM). *P < 0.05; **P < 0.05; ***P < 0.001.
Autophagy inhibition reduced the effects of PCSK9 inhibition against oxidative stress in ox-LDL-treated THP-1-derived macrophages
We further determined whether or not autophagy mediates the effects of PCSK9 inhibition on oxidative stress in ox-LDL-treated THP-1-derived macrophages. The results indicated that treatment with evolocumab plus CQ caused a marked increase in LC3-II and P62 protein expression in ox-LDL-treated THP-1-derived macrophages compared with treatment with evolocumab alone (Figure 5A-C), suggesting that CQ decreases autophagic flux mediated by the PCSK9 inhibitor evolocumab in ox-LDL-treated THP-1-derived macrophages. Importantly, the effect of PCSK9 inhibition on oxidative stress in ox-LDL-treated THP-1-derived macrophages was attenuated by CQ, as demonstrated by analysis of 8-OHdG expression and ROS generation (Figure 5D-G). Moreover, the effect of PCSK9 inhibition on lipid-droplet accumulation was also attenuated by CQ in ox-LDL-treated THP-1-derived macrophage foam cells (Figure 5H, 5I). Hence, these lines of evidence suggest that autophagy facilitates PCSK9 inhibition-induced protection against oxidative stress in ox-LDL-induced THP-1-derived macrophages.
Figure 5.
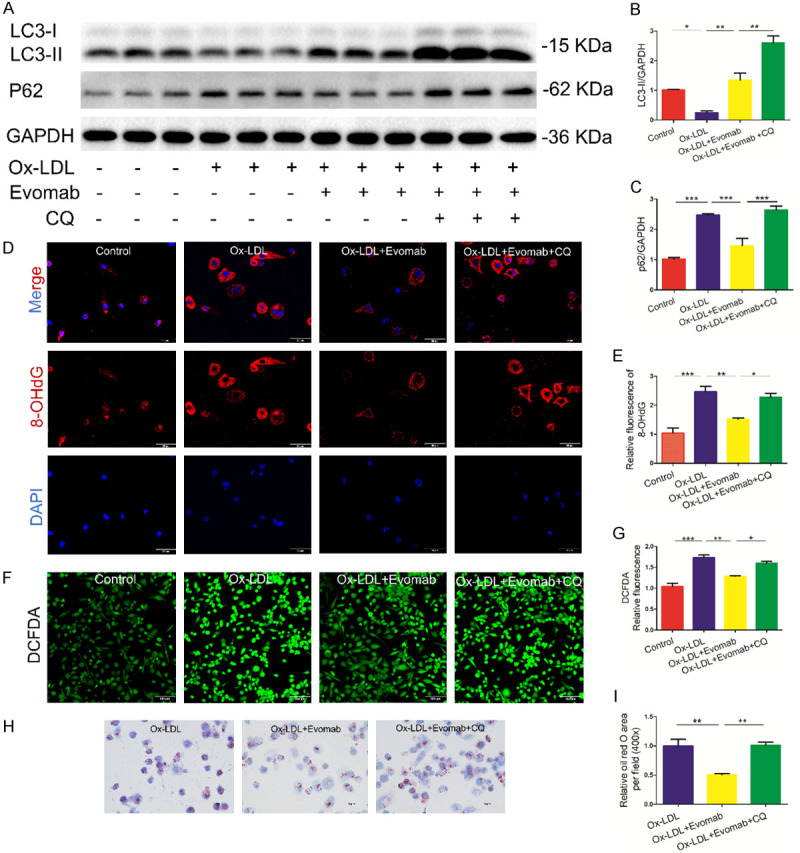
Autophagy inhibition reduced the effects of PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibition against oxidative stress in ox-LDL-treated THP-1-derived macrophages. Serum-starved human acute monocytic leukemia cell line THP-1-derived macrophages were pretreated with 12.5 μΜ autophagy inhibitor chloroquine (CQ) for 1 h and then exposed to oxidized low-density lipoprotein (ox-LDL) (100 µg/mL) in the absence or presence of the proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor evolocumab (Evomab) (1 µΜ) for 24 h. A-C. Western blot analysis of the protein expression of microtubule-associated protein 1 light chain 3 (LC3) and sequestosome 1 (P62) in each group. Quantification of relative protein levels. D, E. Representative fluorescent staining for 8-hydroxy-2’-deoxyguanosine (8-OHdG) expression (DAPI: nuclei; scale bar = 50 μm). Quantification of 8-OHdG relative fluorescent intensity. F, G. 2’,7’-Dichlorofluorescein diacetate (DCFDA) staining for reactive oxygen species (ROS) generation in ox-LDL-induced THP-1-derived macrophages in each group (scale bar = 100 μm). Quantification of DCFDA relative fluorescent intensity. H, I. THP-1-derived macrophages were stained using oil red O staining for indicating lipid accumulation. Quantification of oil red O area per field. Each experiment was performed thrice, and the results are representative ones. All the data are presented as the mean ± standard error of the mean (SEM). *P < 0.05; **P < 0.01; ***P < 0.001.
Impaired autophagy attenuated the anti-inflammatory effects of PCSK9 inhibition in vivo and in vitro
A previous study demonstrated that PCSK9 deficiency by gene inactivation suppresses inflammatory response in atherosclerotic development. We further investigated whether or not autophagy regulates the effects of PCSK9 inhibition against inflammatory response. We found that autophagy inhibition by CQ attenuated the anti-inflammatory effects of PCSK9 inhibition, as illustrated by analysis of the serum levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and interleukin (IL)-10 in HFD-fed ApoE-/- mice (Figure 6A-C). Moreover, autophagy inhibition by CQ also blunted PCSK9 inhibition-mediated the decrease in the levels of inflammatory cytokines and phosphorylation of NF-κBp65 in ox-LDL-induced THP-1-derived macrophages (Figure 6D-G). Hence, these findings demonstrate that impaired autophagy reduces the anti-inflammatory effect of PCSK9 inhibition in vivo and in vitro.
Figure 6.
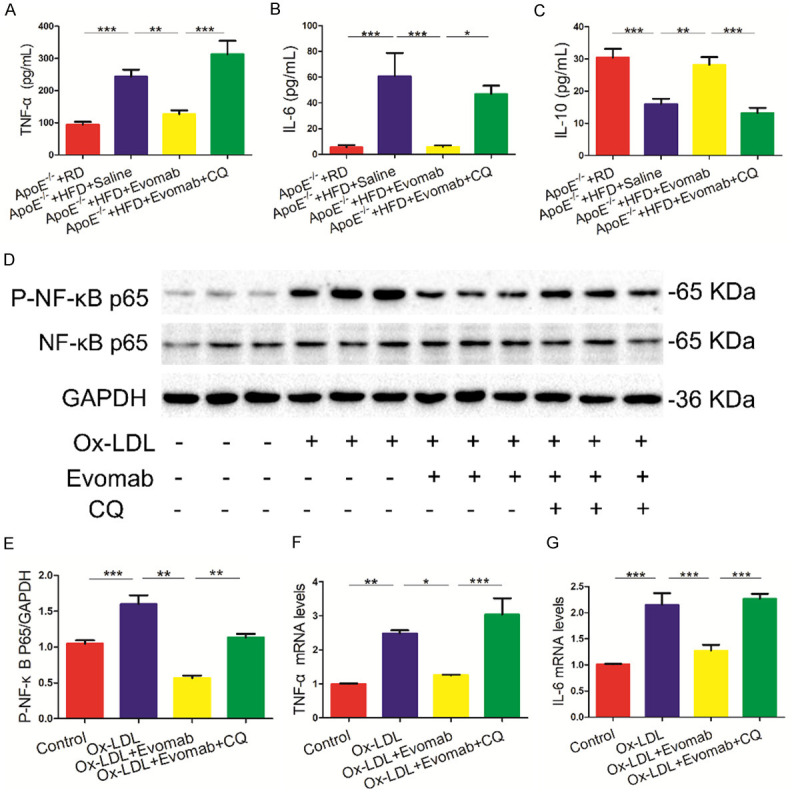
Autophagy inhibition decreased the anti-inflammatory effects of PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibition in vivo and in vitro. A-C. Luminex assay for the serum levels of inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-6 and interleukin (IL)-10 in high-Fat diet (HFD)-fed Apolipoprotein E (ApoE)-/- mice treated with saline, evolocumab (Evomab), or evolocumab plus chloroquine (CQ); Regular diet (RD)-fed ApoE-/- mice served as the control group (n = 7-8 mice per group). D-G. Oxidized low-density lipoprotein (Ox-LDL) (100 µg/mL)-induced human acute monocytic leukemia cell line THP-1-derived macrophages were treated with evolocumab in the absence or presence of CQ (12.5 μΜ) for 24 h. D, E. Western blot analysis of phosphorylated NF-kappaB p65 (P-NF-κB P65) protein expression. F, G. mRNA levels of TNF-α and IL-6 were assessed by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assay. Each data analysis was performed at least thrice. All data are presented as the mean ± standard error of the mean (SEM). *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
This study indicated a novel role of PCSK9 inhibitors in mediating oxidative stress and autophagy in atherosclerotic development. Moreover, we confirmed that autophagy regulates the protective effects of PCSK9 inhibition against oxidative stress and inflammation in THP-1-derived macrophages and in atherosclerotic development. These findings revealed a novel mechanism by which PCSK9 inhibition protects against atherosclerotic progression.
PCSK9, a serine protease, is mainly secreted by the liver and hinders the clearance of LDL-C by promoting the degradation of LDLRs in lysosomes. PCSK9 inhibitors reduce the risk of cardiovascular events [4,25]. Clinical studies have shown that the circulating levels of PCSK9 are associated with carotid intima media thickness [26] and PCSK9 inhibitors in combination with statins result in a greater decrease in atheroma volume in patients [27], implying a role for PCSK9 in atherosclerotic development. Indeed, functional experiments demonstrated that PCSK9 overexpression obviously increased plaque size [28,29], while PCSK9 knockdown by short hairpin RNA reduced plaque size [30]. In this study, we found that treatment with a PCSK9 inhibitor reduced lipid accumulation and plaque lesion size in the aortic roots from HFD-fed ApoE-/- mice, which is consistent with a previous study revealing that a vaccine targeting PCSK9 attenuates atherosclerosis in ApoE-deficient mice [9].
Oxidative stress is critical in atherosclerotic progression [31], although this process is adaptive under normal conditions. Increased oxidative stress is involved in various key proatherogenic events, such as oxidation of LDL-C, endothelial dysfunction, and macrophage infiltration or activation. This study showed that the PCSK9 inhibitor decreased ROS generation and 8-OHdG expression, a sensitive and specific indicator of damage to mitochondrial DNA due to oxidative stress [20], in aortic root plaques in ApoE-deficient mice, suggesting that the PCSK9 inhibitor plays an essential role in regulating mitochondrial oxidative stress in atherosclerosis. Moreover, we observed that the PCSK9 inhibitor decreased inflammatory response in HFD-fed ApoE-/- mice and in oxidatively stressed THP-1-derived macrophages, which is consistent with previous studies [30] reporting an important role of PCSK9 in regulating inflammation during atherosclerotic progression. Wang et al. showed that mitochondrial oxidative stress in macrophages amplifies atherosclerotic lesion development by promoting the NF-κB-mediated inflammatory response [1] and extracellular neutrophil traps [32]. A previous study showed that the PCSK9 inhibitor evolocumab counteracted damage due to oxidative stress in endothelial cells [12]. Therefore, PCSK9 inhibitors might attenuate atherosclerotic lesions by decreasing oxidative stress relevant to inflammation and endothelial dysfunction in atherosclerosis. Recently, PCSK9 inhibition was reported to suppress platelet activation and in vivo thrombosis by regulating oxidative stress [33]. Moreover, patients with acute coronary syndrome treated with a PCSK9 inhibitor displayed reduced ox-LDL levels, a key indicator of oxidative stress. Given these findings, it is reasonable to conclude that PCSK9 inhibitors might provide a therapeutic strategy for reducing cardiovascular risk related to oxidative stress.
Autophagy plays an essential and protective role against the development of atherosclerosis. Impaired autophagy in macrophages was found to aggravate vascular plaques in HFD-fed LDLR-knockout mice by increasing oxidative stress [18] and inflammatory hyperactivation [17]. In contrast, autophagy facilitates the degradation of lipids and further inhibits atherosclerotic progression [15]. Moreover, autophagy enhancement suppresses the formation and progression of macrophage foam cells and prevents atherosclerosis [34]. In this study, we found that autophagic protein markers were decreased in the vascular tissue of ApoE-/- mice, suggesting that the HFD impairs autophagy in ApoE-/- mice. Importantly, we observed that treatment with a PCSK9 inhibitor increased macrophage autophagy in aortic vascular tissue. The in vitro investigation further demonstrated that PCSK9 inhibition increased autophagic flux in lipid-loaded THP-1-derived macrophages. Furthermore, the inhibition of autophagy attenuated the protective effects of PCKS9 inhibition against oxidative stress and inflammation in both atherosclerosis and ox-LDL-treated macrophages, suggesting that autophagy is, at least in part, required for the protective effects of PCSK9 inhibition against the development of atherosclerosis. A previous study suggested that PCSK9 deficiency regulates ApoB metabolism by increasing autophagic flux in the liver [6], indirectly supporting our findings. However, PCSK9 inhibition has been shown to play a protective role in myocardial ischemia and ischemia-reperfusion injury by suppressing autophagy [35,36]. The discrepancy between our findings and those of the aforementioned studies may be related to the different cell types and experimental conditions. Interestingly, another study reported that PCSK9 deficiency impacts cardiac lipid metabolism and promotes the development of heart failure with preserved ejection fraction [37]. Thus, clarification of the role of PCSK9 in cardiomyocytes (i.e., beneficial or detrimental) is needed.
The present study had some limitations. First, we did not confirm whether PCSK9 inhibition improves mitochondrial function by decreasing oxidative stress; this topic warrants further investigation. Second, our study showed that PCSK9 inhibition reduced phospho-NF-κB p65 expression in ox-LDL-treated macrophages. Thus, PCSK9 inhibition suppresses inflammation at least partially by regulating NF-κB pathway. This result is supported by previous studies revealing that PCSK9 promotes macrophage inflammation by regulating NF-κB pathway [30,38]. However, the results should be interpreted with caution, because PCSK9 inhibition could also regulate inflammation through cholesterol regulation. Further work is needed to sort out how much of the pro-inflammatory response of PCSK9 is cholesterol-independent.
In conclusion, this study demonstrated that PCSK9 inhibition alleviated oxidative stress, inflammation, and plaque lesions by promoting autophagy in HFD-fed ApoE-/- mice. In vitro investigations indicated that PCSK9 inhibition promoted autophagic flux that attenuated oxidative stress and inflammation in ox-LDL-treated THP-1-derived macrophages. This study provides the first evidence of the novel role of PCSK9 inhibition in regulating autophagy and oxidative stress during atherosclerotic progression and indicates that autophagy is a critical mechanism by which PCSK9 inhibition reduces oxidative stress and inflammation in the development of atherosclerosis.
Acknowledgements
We acknowledge the following funding for supporting this research: Key Research and Development Plan Program of Shanxi province (201903D321182), Shanxi Cardiovascular Hospital Incentive Plan Fund (XYS20190107 and XYS20210101), Health Commission of Shanxi Province (2022014), and Natural Science Foundation of China (82070374).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB-mediated inflammation in macrophages. Circ Res. 2014;114:421–433. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Urban D, Pöss J, Böhm M, Laufs U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013;62:1401–1408. doi: 10.1016/j.jacc.2013.07.056. [DOI] [PubMed] [Google Scholar]
- 3.Glerup S, Schulz R, Laufs U, Schlüter KD. Physiological and therapeutic regulation of PCSK9 activity in cardiovascular disease. Basic Res Cardiol. 2017;112:32. doi: 10.1007/s00395-017-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, Jukema JW, Lecorps G, Mahaffey KW, Moryusef A, Pordy R, Quintero K, Roe MT, Sasiela WJ, Tamby JF, Tricoci P, White HD, Zeiher AM ODYSSEY OUTCOMES Committees and Investigators. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–2107. doi: 10.1056/NEJMoa1801174. [DOI] [PubMed] [Google Scholar]
- 6.Sun H, Krauss RM, Chang JT, Teng BB. PCSK9 deficiency reduces atherosclerosis, apolipoprotein B secretion, and endothelial dysfunction. J Lipid Res. 2018;59:207–223. doi: 10.1194/jlr.M078360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kühnast S, van der Hoorn JW, Pieterman EJ, van den Hoek AM, Sasiela WJ, Gusarova V, Peyman A, Schäfer HL, Schwahn U, Jukema JW, Princen HM. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res. 2014;55:2103–2112. doi: 10.1194/jlr.M051326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denis M, Marcinkiewicz J, Zaid A, Gauthier D, Poirier S, Lazure C, Seidah NG, Prat A. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation. 2012;125:894–901. doi: 10.1161/CIRCULATIONAHA.111.057406. [DOI] [PubMed] [Google Scholar]
- 9.Wu D, Pan Y, Yang S, Li C, Zhou Y, Wang Y, Chen X, Zhou Z, Liao Y, Qiu Z. PCSK9Qβ-003 vaccine attenuates atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Drugs Ther. 2021;35:141–151. doi: 10.1007/s10557-020-07041-6. [DOI] [PubMed] [Google Scholar]
- 10.Cammisotto V, Baratta F, Castellani V, Bartimoccia S, Nocella C, D’Erasmo L, Cocomello N, Barale C, Scicali R, Di Pino A, Piro S, Del Ben M, Arca M, Russo I, Purrello F, Carnevale R, Violi F, Pastori D, Pignatelli P. Proprotein convertase subtilisin kexin type 9 inhibitors reduce platelet activation modulating ox-LDL pathways. Int J Mol Sci. 2021;22:7193. doi: 10.3390/ijms22137193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JS, Mukhopadhyay P, Matyas C, Trojnar E, Paloczi J, Yang YR, Blank BA, Savage C, Sorokin AV, Mehta NN, Vendruscolo JCM, Koob GF, Vendruscolo LF, Pacher P, Lohoff FW. PCSK9 inhibition as a novel therapeutic target for alcoholic liver disease. Sci Rep. 2019;9:17167. doi: 10.1038/s41598-019-53603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safaeian L, Mirian M, Bahrizadeh S. Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H(2)O(2)-induced oxidative stress. Arch Physiol Biochem. 2020;128:1681–1686. doi: 10.1080/13813455.2020.1788605. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Dai F, Wang H, Wei G, Jiang Q, Yin P, Wang S, Ge J, Yang C, Wu J, Zou Y. PCSK9 participates in oxidized-low density lipoprotein-induced myocardial injury through mitochondrial oxidative stress and Drp1-mediated mitochondrial fission. Clin Transl Med. 2022;12:e729. doi: 10.1002/ctm2.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 15.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang S, Wan X, Zou X, Sun S, Hao X, Liang C, Zhang Z, Zhang F, Sun B, Li H, Yu B. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis. 2021;12:88. doi: 10.1038/s41419-020-03357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Z, Liu S, Wang X, Mathur P, Dai Y, Theus S, Deng X, Fan Y, Mehta JL. Cross-talk between PCSK9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxid Redox Signal. 2016;25:997–1008. doi: 10.1089/ars.2016.6631. [DOI] [PubMed] [Google Scholar]
- 20.Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2’-deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:120–139. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- 21.Robichaud S, Rasheed A, Pietrangelo A, Doyoung Kim A, Boucher DM, Emerton C, Vijithakumar V, Gharibeh L, Fairman G, Mak E, Nguyen MA, Geoffrion M, Wirka R, Rayner KJ, Ouimet M. Autophagy is differentially regulated in leukocyte and nonleukocyte foam cells during atherosclerosis. Circ Res. 2022;130:831–847. doi: 10.1161/CIRCRESAHA.121.320047. [DOI] [PubMed] [Google Scholar]
- 22.Ding Z, Liu S, Wang X, Deng X, Fan Y, Shahanawaz J, Shmookler Reis RJ, Varughese KI, Sawamura T, Mehta JL. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc Res. 2015;107:556–567. doi: 10.1093/cvr/cvv178. [DOI] [PubMed] [Google Scholar]
- 23.Ding Z, Liu S, Wang X, Theus S, Deng X, Fan Y, Zhou S, Mehta JL. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res. 2018;114:1145–1153. doi: 10.1093/cvr/cvy079. [DOI] [PubMed] [Google Scholar]
- 24.Badimon L, Luquero A, Crespo J, Peña E, Borrell-Pages M. PCSK9 and LRP5 in macrophage lipid internalization and inflammation. Cardiovasc Res. 2021;117:2054–2068. doi: 10.1093/cvr/cvaa254. [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM. Mortality differences associated with treatment responses in CANTOS and FOURIER: insights and implications. Circulation. 2018;137:1763–1766. doi: 10.1161/CIRCULATIONAHA.117.033254. [DOI] [PubMed] [Google Scholar]
- 26.Lee CJ, Lee YH, Park SW, Kim KJ, Park S, Youn JC, Lee SH, Kang SM, Jang Y. Association of serum proprotein convertase subtilisin/kexin type 9 with carotid intima media thickness in hypertensive subjects. Metabolism. 2013;62:845–850. doi: 10.1016/j.metabol.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373–2384. doi: 10.1001/jama.2016.16951. [DOI] [PubMed] [Google Scholar]
- 28.Tavori H, Giunzioni I, Predazzi IM, Plubell D, Shivinsky A, Miles J, Devay RM, Liang H, Rashid S, Linton MF, Fazio S. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc Res. 2016;110:268–278. doi: 10.1093/cvr/cvw053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roche-Molina M, Sanz-Rosa D, Cruz FM, Garcia-Prieto J, Lópezl S, Abia R, Muriana FJ, Fuster V, Ibanez B, Bernal JA. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–59. doi: 10.1161/ATVBAHA.114.303617. [DOI] [PubMed] [Google Scholar]
- 30.Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, Wang Z, Wei DH, Liu LS, Zheng XL, Jiang ZS. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-kapaB pathway. Atherosclerosis. 2017;262:113–122. doi: 10.1016/j.atherosclerosis.2017.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, Huang Y, Bernatchez P, Giordano FJ, Shadel G, Sessa WC, Min W. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am J Pathol. 2007;170:1108–1120. doi: 10.2353/ajpath.2007.060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler Thromb Vasc Biol. 2017;37:e99–e107. doi: 10.1161/ATVBAHA.117.309580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi Z, Hu L, Zhang J, Yang W, Liu X, Jia D, Yao Z, Chang L, Pan G, Zhong H, Luo X, Yao K, Sun A, Qian J, Ding Z, Ge J. PCSK9 (proprotein convertase subtilisin/kexin 9) enhances platelet activation, thrombosis, and myocardial infarct expansion by binding to platelet CD36. Circulation. 2021;143:45–61. doi: 10.1161/CIRCULATIONAHA.120.046290. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Tang Y, Cui Y, Zhang H, Zhang D. Autophagy is associated with cell fate in the process of macrophage-derived foam cells formation and progress. J Biomed Sci. 2016;23:57. doi: 10.1186/s12929-016-0274-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding Z, Wang X, Liu S, Shahanawaz J, Theus S, Fan Y, Deng X, Zhou S, Mehta JL. PCSK9 expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc Res. 2018;114:1738–1751. doi: 10.1093/cvr/cvy128. [DOI] [PubMed] [Google Scholar]
- 36.Huang G, Lu X, Zhou H, Li R, Huang Q, Xiong X, Luo Z, Li W. PCSK9 inhibition protects against myocardial ischemia-reperfusion injury via suppressing autophagy. Microvasc Res. 2022;142:104371. doi: 10.1016/j.mvr.2022.104371. [DOI] [PubMed] [Google Scholar]
- 37.Da Dalt L, Castiglioni L, Baragetti A, Audano M, Svecla M, Bonacina F, Pedretti S, Uboldi P, Benzoni P, Giannetti F, Barbuti A, Pellegatta F, Indino S, Donetti E, Sironi L, Mitro N, Catapano AL, Norata GD. PCSK9 deficiency rewires heart metabolism and drives heart failure with preserved ejection fraction. Eur Heart J. 2021;42:3078–3090. doi: 10.1093/eurheartj/ehab431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang Z, Jiang L, Peng J, Ren Z, Wei D, Wu C, Pan L, Jiang Z, Liu L. PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF-κB activation in THP-1-derived macrophages. Int J Mol Med. 2012;30:931–938. doi: 10.3892/ijmm.2012.1072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.