

Abstract
Purpose:
This prospective pharmacokinetic (PK) ancillary study of the TEXCAN phase II GERCOR trial of chemorefractory metastatic colorectal cancer (mCRC) patients treated with regorafenib (REGO) investigated correlations between overall survival (OS) and concentrations (C) of REGO and its active metabolites, M-2 and M-5.
Methods:
55 patients received REGO 160 mg/day for 21 days of a 28-day cycle (NCT02699073). REGO, M-2, M-5 were measured by liquid chromatography-mass spectrometry (LC-MS/MS) assay at day 15 of cycle 1 (C1) and 2 (C2). We studied the association between OS and Cmin of REGO, M-2, and M-5 at C1 and their accumulations between C1 and C2.
Results:
Medians of C2/C1 M-2 and M-5 ratios were 0.82 (Interquartile range [IQR] 0.50–1.78) and 0.75 (IQR 0.41–1.93), respectively. Patients with C2/C1 M-2 ratio ≥ median had improved survival compared to those < median (12.6 vs. 4.0 months, P = 0.023), corresponding to a66% mortality risk reduction in multivariate analysis.
The C2/C1 M-2 ratio correlated with C1 REGO+M-2+M-5 (Csum; P = 0.006). Restricted cubic spline analysis showed an increased OS benefit as the C2/C1 M-2 ratio raises and when C1 Csum ranged between 2.5 and 5.5 mg/L. Patients within the Csum range had a reduced incidence of serious adverse events and improved OS.
Conclusions:
We identified pharmacokinetic parameters associated with a survival benefit in mCRC patients treated by REGO. OS and safety were favorable when C1 REGO+M-2+M-5 Csum ranged between 2.5 and 5.5 mg/L. These results pave the way for individual REGO dose modification strategies based on pharmacokinetic monitoring.
Keywords: Regorafenib; chemorefractory metastatic colorectal cancer; M-2, M-5, survival, pharmacokinetic, pharmacological monitoring
Introduction
Regorafenib (REGO) is a recommended therapeutic option for chemorefractory metastatic colorectal cancer (mCRC) (1,2). It was evaluated in two phase III, multicentric, randomized trials showing a significant overall survival (OS) improvement in patients treated by 160 mg REGO for 21 days of a 28-day cycle (3,4). Patients presented interindividual differences in tolerance to REGO: 54% of patients in the CORRECT trial had high grade (3–4) adverse events, 61% temporary or definitive discontinuation of treatment and to 38% dose reduction. Decrease of dose-intensity may limit the benefit derived from REGO. Therapeutic monitoring seems therefore compelling to optimize REGO exposure while preventing high grade toxicities in clinical practice.
REGO pharmacokinetics (PK) is characterized firstly by hepatic metabolism via cytochrome P450 3A4 leading to the production of two active metabolites (M-2 and M-5) excreted in feces and secondly via glucuronide conjugation by UDP-glucuronosyltransferase 1A9 catalyzing formation of inactive glucuronides, which are mainly excreted in urine (5,6). M-2 and M-5 progressively accumulate over the course of treatment as a result of hepatic metabolism and enterohepatic cycle. Phases I trials have shown an important inter and intra-patient variability in concentrations of REGO and its metabolites (5,7). Moreover, both metabolites, M-2 and M-5, exhibit accumulation after a single daily dose of regorafenib exceeding 80 mg (5,7). In the CORRECT trial, a population PK analysis didn’t identify clinically relevant parameters to adjust the initial dose of treatment (8), indicating the need for further investigations addressing dose adjustment strategies based on individual therapeutic monitoring. As REGO, and its metabolites M-2 and M-5 have similar pharmacological activity and concentrations at steady state, we hypothesized that PK parameters assessing their accumulation and exposure altogether would better predict the pharmacodynamic of REGO than usual PK endpoints.
Here, we present the results of the GERCOR TEXCAN phase II study, which assessed prospectively the correlations between OS, toxicities, and trough concentrations (Cmin) of REGO and its metabolites (M-2 and M-5) and their target concentration range in chemorefractory mCRC patients treated by REGO.
Patients and Methods
Study design and participants
This is a prospective, PK ancillary study of the TEXCAN multicentric phase II GERCOR study in chemorefractory mCRC patients treated by REGO (NCT02699073). The results on the primary objective of the study have been published previously (9). The main objective of the current study was to investigate the correlations between OS and trough concentrations of REGO and its pharmacologically active metabolites M-2 and M-5. Secondary objectives were to assess the correlation between progression-free survival (PFS), overall response rate (ORR), safety, and PK parameters. Patients were included according to the inclusion and exclusion criteria of the CORRECT study (4). Exclusion criteria included the use of drugs potentially interacting with REGO as inductors/inhibitors of CYP450 3A4 or UGT1A9. Patients were treated orally with 160 mg REGO daily for 3 weeks on and 1 week off. All patients a signed specific informed consent for this ancillary study. The PK population included all patients treated by REGO who received treatment at least one day before PK sampling to ensure an adequate Cmin assessment. Compliance was recorded in the electronic case report form based on the number of remaining pills assessed by clinical research assistant. The FAS-CORRECT prognosis groups combining performance status, time since initial diagnosis, number of metastatic sites, and the presence of liver metastasis, was used for prognosis stratification (10).
Blood sampling and PK analysis
Blood samples for PK analysis were withdrawn into heparinized tubes at day 15 (±2 days) of C1 and C2, in fasting conditions before intake of regorafenib, and/or at progression, and/or during an episode of grade III-IV toxicities. The blood was immediately centrifuged for 15 min (2500 g at +4°C) and plasma were stored at −80°C until analysis. Trough concentrations (Cmin) of REGO, M-2, and M-5 were quantified by a validated liquid chromatography-mass spectrometry (LC-MS/MS) method (11). Csum was defined as the sum of the Cmin of REGO, M-2, and M-5, corresponding to the expected overall active drug exposure. Accumulation ratios are the ratio between concentrations measured at C2 and C1. Chemical structure of REGO, M-2 and M-5 are given in table 1A (6,11).
Table 1.
Pharmacokinetic characteristics of the mITT population
| REGO mg/L |
M-2 mg/L |
M-5 mg/L |
Accumulation ratio | Csum REGO+M-2+M-5 mg/L |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cycle | C1 | C2 | C1 | C2 | C1 | C2 |
REGO
C2/C1 |
M-2
C2/C1 |
M-5
C2/C1 |
C1 | C2 |
| N | 34 | 27 | 34 | 27 | 34 | 27 | 26 | 26 | 26 | 34 | 27 |
| Mean ± SD | 2.10 ± 1.17 | 2.10 ± 1.36 | 1.69 ± 1.01 | 1.52 ± 0.91 | 1.72 ± 1.19 | 1.59 ± 1.28 | 1.20 ± 0.72 | 1.23 ± 0.99 | 1.17 ± 0.90 | 5.50 ± 2.99 | 5.21 ± 2.81 |
| Median concentration (Q1-Q3) | 1.985 (1.03–2.73) |
1.9 (1.10–2.76) |
1.436 (0.89–2.49) | 1.294 (0.77–2.24) | 1.611 (0.73–2.37) | 1.174 (0.45–2.42) | 1.07 (0.65–1.53) |
0.82 (0.50–1.78) |
0.75 (0.41–1.93) |
5.21 (3.17–8.33) | 5.26 (3.01–7.93) |
| Min-Max concentrations,mg/L | 0.54–4.84 | 0.43–6.49 | 0.11–3.86 | 0.31–3.34 | 0.14–4.65 | 0.20–5.10 | 0.28–2.96 | 0.23–4.46 | 0.21–3.21 | 0.89–11.08 | 1.05–10.01 |
|
C1 vs C2, p-value
|
0.69 | 0.74 | 0.65 | - | - | - | 0.89 | ||||
| Coefficient of correlation between C1 and C2, p-value | 0.56 (0.003) | 0.33 (0.09) | 0.38 (0.05) | - | - | - | |||||
Abbreviations: REGO, regorafenib; Csum, Concentration sum of regorafenib, M-2, and M-5 trough concentrations; ITT, Intention-to-treat
Statistical analysis
Pharmacokinetic parameters were described with mean, standard deviation (SD), median, interquartile range (IQR), min and max, and were compared between C1 and C2 using the Wilcoxon signed-rank test. The Pearson’s correlation coefficient was provided. Baseline characteristics were described with frequencies in the population with PK information and compared according to Cmin at C1 with the chi-squared test or Fisher’s exact test, as appropriate.
OS was defined as the time from treatment initiation to death due to any cause. Patients alive were censored at the last date they were known to be alive. PFSwas defined as the time from treatment initiation to progression (RECIST, clinical, or biological) or death whatever occurred first. Patients without documented objective progression at the time of the final analysis were censored at the date of their last objective tumor assessment. Survival curves were estimated by the Kaplan-Meier method, described with median and 95% confidence interval (CI), and compared using the log-rank test. The association between PK parameters and outcomes were estimated using the Cox proportional hazard regression model, with hazard ratios (HR) and their 95% CIs. Proportional hazards assumptions were examined graphically by plotting log-minus-log of survival and cumulative sums of martingale residuals. The restricted cubic spline method was used to evaluate the association between pharmacological parameters and OS to identify a cut-off of interest for Cmin REGO, the M-2 accumulation ratio, and Csum. Radiological response and toxicities were compared according to PK parameters at C1 with the Fisher’s exact test. All analyses were performed using SAS version 9.4 (SAS Institute, Cary NC) and R software version 2.15.2 (R Development Core Team, Vienna, Austria; http://www.r-project.org).
Results
Fifty-five patients were included (the TEXCAN cohort) (9). Of these, 13 hadn’t PK samples at C1, five had PK samples 2 days or more after the last administration of regorafenib, leaving 37 patients in the modified intention-to-treat (miTTb) PK population with at least one interpretable PK sample at C1D14 or C2D14. In total, 34 and 27 patients had a measure of Cmin at C1 and at C2, respectively (Supplementary Figure 1). Patients baseline characteristics are provided in Table S1 (Supplementary data). No difference in OS was observed between the whole TEXCAN cohort and the PK cohort (Supplementary Figure 2).
Table 1 shows Cmin of REGO and its metabolites at days 15 of C1 and C2, the accumulation ratios of each active analyte, and Csum. There was no correlation between Cmin of REGO at C1 and baseline patient and tumor characteristics (Supplementary Table S1).
Figure 1 displays univariate OS analysis according to Cmin of REGO, the C2/C1 accumulation ratio, and Csum. A tendency towards worse survival was observed for Cmin of REGO at C1 ≥ median and Csum at C1 ≥ median. C1 Cmin of M-2 or M-5 were not associated with OS (respectively, P = 0.49 and P = 0.42; Supplementary Figure S3). The C2/C1 M-2 accumulation ratio was significantly associated with better OS (12.6 months if ratio ≥ median vs. 4.0 months if ratio < median, log-rank P = 0.0173) but not the C2/C1 M-5 accumulation ratio. Univariate analysis of PFS showed no statistically significant relationship between PFS and each analyte or the accumulation ratios (Supplementary Figure S4).
Figure 1. Overall survival of mCRC patients receiving REGO in the TEXCAN phase II trial according to predefined pharmacokinetic parameters.
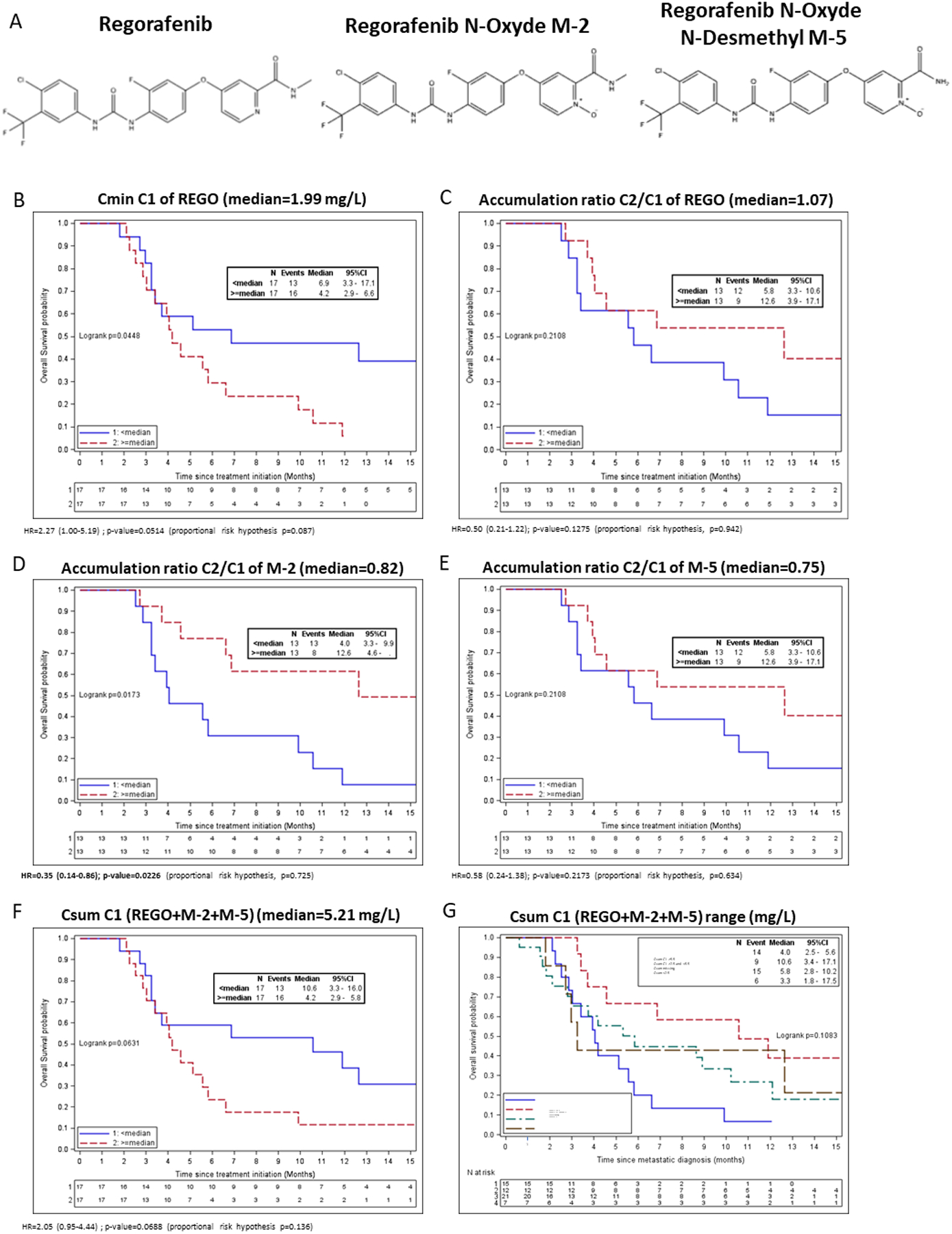
A: Chemical structure of Regorafenib, M-2 and M-5 metabolites; B: Cmin of REGO at Cycle 1 D14; C: REGO accumulation ratio between Cycle 2 and Cycle 1 D14; D: M-2 accumulation ratio between Cycle 2 and Cycle 1 D14; E: M-5 accumulation ratio between Cycle 2 and Cycle 1 D14; F: Csum (REGO+M-2+M-5) at Cycle 1 D14; G: three defined threshold of Csum at Cycle 1 D14 and patients with missing PK data
In multivariate analyses including the FAS-CORRECT groups (Table 2), an increased OS was observed for the C2/C1 M-2 accumulation ratio ≥ median (HR = 0.36, IC 95% of 0.14–0.88, P = 0.0253) and decreased OS for Cmin REGO and Csum at C1 ≥ median.
Table 2. Multivariate analyses of OS according to PK parameters and the CORRECT prognostic groups.
Each model includes one PK parameter (Cmin REGO C1, Csum C1 or Ratio M-2 C2/C1) and the FAS-CORRECT prognostic group (10)
| N (events) |
HR | 95% CI | p-value | ||
|---|---|---|---|---|---|
| Model 1 : CminREGO C1 | 34 (29) | ||||
| Cmin REGO C1 | <median | 1 | 0.0267 | ||
| ≥median | 2.64 | 1.12–6.23 | |||
| -CORRECT prognostic group | Good | 1 | 0.2193 | ||
| Poor/moderate | 1.68 | 0.73–3.84 | |||
| Model 2 : Csum C1 | 34 (29) | ||||
| Csum (REGO+M-2+M-5) C1 | <median | 1 | 0.0146 | ||
| ≥median | 2.99 | 1.24–7.21 | |||
| CORRECT prognostic group | Good | 1 | 0.0859 | ||
| Poor/moderate | 2.19 | 0.90–5.36 | |||
| Model 3 : Ratio M-2 C2/C1 | 26 (21) | ||||
| Ratio M-2 C2/C1 | <median | 1 | 0.0253 | ||
| ≥median | 0.36 | 0.14–0.88 | |||
| CORRECT prognostic group | Good | 1 | 0.8294 | ||
| Poor/moderate | 1.11 | 0.42–2.94 | |||
Abbreviations: REGO, regorafenib; Csum, Concentration sum of regorafenib, M-2, and M-5 trough concentrations; Cmin, trough concentration
Restricted cubic spline analysis revealed different patterns depending on the PK parameters (Figure 2). An increased risk of death was observed for higher Cmin of REGO at C1, while risk of death decreased when the C2 to C1 M-2 ratio increased, with a plateau effect for M-2 C2/C1 ≥ 0.8. We observed a decreased risk of death for Csum at C1 with a turning point at 5.0 mg/L, which was followed by an increased risk of death for Csum ≥ 7 mg/L.
Figure 2. Cubic spline analyses of overall survival according to selected PK parameters.
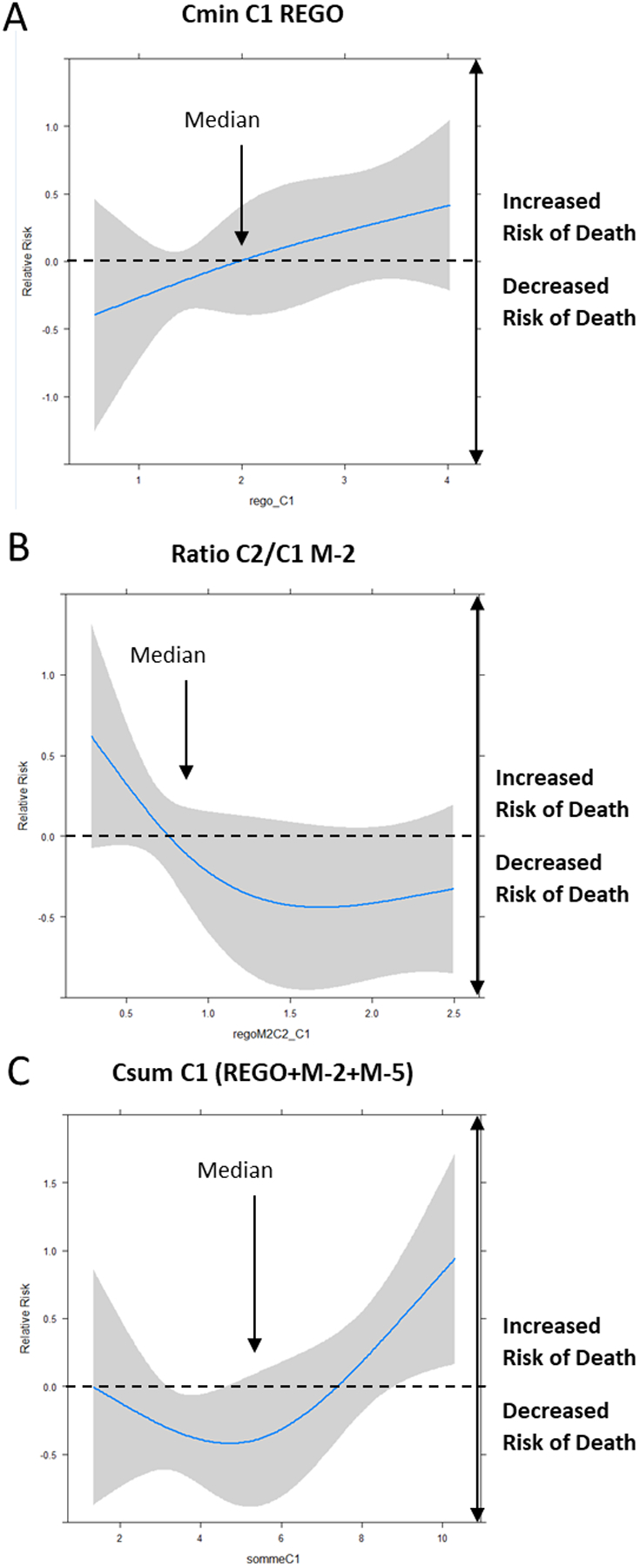
A: Cmin of REGO at Cycle 1 D14; B: Ratio of M-2 between Cycle 2 and Cycle 1 at D14; C: Csum (REGO+M-2+M-5) at Cycle 1 D14
Given that the C2/C1 M-2 ratio is a late biomarker, difficult to implement in clinical practice as requiring C1 and C2 Cmin, we assessed the correlation between the M-2 accumulation ratio and Csum. Csum at C1 correlated with the M-2 accumulation as a continuous variable (Pearson’s correlation = −0.53, P = 0.0058). An inverse correlation between parameters was seen: an increased Csum at C1 correlated with a decreased M-2 accumulation ratio (Supplementary Table S2). This phenomenon was due to subsequent treatment interruptions and/or cumulative dose reductions done in the patients with the highest Csum at day 15 of C1 (Supplementary Table S5). Moreover, a higher rate of interruption or discontinuation and a lower cumulative dose were observed for patients with Csum < 2.5 mg/L, indicating that Csum was low because of temporary interruption and doses adjustment before PK sampling (Supplementary Table S5). We therefore defined ranges of Csum at C1, with a target range of 2.5 to 5.5 mg/L, which was found in 35% of patients from the PK cohort (n = 12/34, Supplementary Table S2). Considering best ORR, patients within the C1 Csum target range achieved a 100% control rate (n = 12/12; Supplementary Table S3). Progressive disease rates were 28.6% (n = 2/7), 26.7% (n = 4/15), and 33.3% (n = 7/21) for Csum < 2.5 mg/L, Csum ≥ 5.5 mg/L, or no PK data, respectively. Higher control rates at week 8 were observed for patients with C1 Csum < 2.5 mg/L (71%, n = 4/7) and C1 Csum 2.5–5.5 mg/L (50%, n = 6/12) compared to those with C1 Csum ≥ 5.5 mg/L (33.3%, n = 5/15) and with missing PK data (23.8%, n = 5/21). Moreover, patients reaching a Csum within 2.5–5.5 mg/L at C2 has a significant higher control rate at 8W of 80% (n = 8/10) compared to 40% (n = 2/5) for C2 Csum < 2.5 mg/L, 50% (n = 6/12) for C2 Csum ≥5.5 mg/L and 14.29% (n = 4/28) for unknow PK (P =0.0006) (Supplementary table 3). These observations translated into a survival advantage within the range [2.5–5.5 [mg/L with an OS of 10.6 months (Figure 1F) compared with 3.3, 4.0 Csum < 2.5 mg/L, Csum ≥ 5.5 mg/L (P = 0.0498).
While no significant differences in high grade toxicities were observed according to Csum ranges, patients displaying a C1 Csum between 2.5 to 5.5 mg/L had no serious Adverse Event (SAE, 0%, N=0/12) compared to Csum<2.5 mg/L, Csum≥5.5 mg/L or patients with missing PK data, respectively with 43% (n = 3/7), 20% (n = 3/15) and 24% (n = 5/21; Supplementary Table S4). Considering drug discontinuation or cumulative dose intake (Supplementary Table S5), patients within the C1 Csum range of 2.5 to 5.5 mg/L had fewer treatment interruption or discontinuation (P = 0.0043) and received higher cumulative doses (P = 0.05). Correlation study of demographic parameters with REGO and active metabolite showed that female patients had an increase in M-5 at C1D14 compared to male patients and patients with Body Mass Index >25 kg/m2 had increase REGO exposure at C2D14 (Supplementary table S6). Altogether, these data suggest that overexposure and underexposure at C1D15 are detrimental, in line with toxicity-related early and late temporary interruptions, precluding to obtain satisfactory exposure to REGO and active metabolites.
Discussion
In the TEXCAN phase II trial, we showed that the monitoring of Cmin of REGO, M-2, and M-5 from day 15 is feasible and clinically relevant in chemorefractory mCRC patients. PK analyses showed results in line with phase I trials (7). Three PK parameters, C1 REGO, C1 Csum, and the C1 to C2 accumulation ratio of M-2 were independently associated with survival. While high concentrations of REGO and Csum at C1 were detrimental, M-2 accumulation between C1 and C2 was independently associated with improved OS. However, this biomarker is obtained at the end of C2 and may therefore only reflect the ability of patients to receive an adequate dose intensity of treatment. M-2 accumulation correlated with, C1 Csum of REGO+M-2+M-5 and we identified that C1 Csum ranging from 2.5 to 5.5 mg/L translated into an OS advantage with decreased occurrence of SAE. As an earlier biomarker, this parameter seems implementable in clinical practice.
In this PK analysis of the TEXCAN phase II trial we showed that the approved 160 mg schedule fits only to one third of patients and the others would need a baseline and/or rapid dose adjustment. Indeed, REGO and its active metabolites exposure above defined threshold were detrimental leading ultimately to decreased survival. The observed association of improved survival and M-2 accumulations between C1 and C2, a marker of continuous favorable exposure to REGO, suggests also that complete discontinuation of the drug in case of toxicity should be avoided at the utmost by early dose modifications. Therefore, individual personalization of the REGO dose may help to decrease the onset of SAE, avoid long drug interruptions or discontinuation, and thus guarantee adequate exposure and improve OS (12). Kubota et al. studied the area under curve (AUC) for the REGO, M-2, and M-5 concentrations at day 1 and showed that the relationship between PFS and M-5 AUC, but not for REGO or M-2 (12). AUC measure requires multiple points to perform a 24-h concentration-time curve limiting its applicability. Furthermore, REGO and its active metabolites have long elimination half-lives (REGO: 26–28 hours, M-2: 20–30 hours, M-5: 40–100 hours) (7,13) and the correlation of their concentrations at day 1 with concentrations at steady state remains to be established. Day 15 Cmin samples seem more easily implementable in clinical practice and allow overall active metabolite exposure assessment at steady state. Importantly, as reported by Keunecke et al, we found that female gender and high body max index correlated with increased exposure to REGO and/or metabolites (14), demographic parameters that could be useful to adjust the initial dose. Influence of entero-hepatic cycle and food intake on the PK of REGO and metabolites remains uncertain in our study and should be considered carefully for future applications.
Fukudo et al. recently published significantly longer PFS (112 vs 57 days, p=0.044) and lower cumulative incidence of DLTs in patients with summed trough concentrations of regorafenib and its active metabolites ranging between 2.9 and 4.3 μg/mL in patients treated for CRC, hepatocellular carcinoma and GIST (15). In their population, only nine patients out of 34 started regorafenib treatment at the recommended dose of 160 mg once daily, 3 weeks on and one week off. Nevertheless, the concordance of their findings with ours, despite different population and doses, strengthen the general idea that regorafenib doses needs to be adjusted individually to fit in a concentration range optimized both for safety and efficacy. The identified range of Csum associated with benefit deserves to be validated prospectively in larger cohorts.
To avoid early toxicity and discontinuations of treatment, other regimens have been proposed as in the REDOs trial (16). In this study, a progressive weekly dose escalation schedule from 80 to 160 mg/day based on tolerance was implemented. This modified regimen translated into increased initiation of cycle 3 with decreased occurrence of high-grade toxicities. In the RESET trial with a reduced dose regimen, REGO concentrations were lower (3978 vs 7244 nM) in patients for whom the dose was progressively increased to 160 mg/day compared to those who did not escalated (17). Flexible REGO dosing has also benefited to patients with mCRC in the REARRANGE trial (18). This study compared the approved REGO schedule (160 mg/day at 3 weeks on, 1 week off) with an initially reduced dosing (120 mg/day at 3 weeks on, 1 week off for the first cycle or intermittent regimen (160 mg/day at 1 week on, 1 week off followed by the standard regimen for subsequent cycles), subsequently increased to approved dose if the tolerance was favorable. Even if numerical decreased of high grade toxicities were observed for specific side effects, this trial did not meet its primary endpoint of improved tolerability in the reduced doses arms. The high PK inter-patient variability may explain discrepancies between those trials. Indeed, in the TEXCAN trial only one third of patients with the standard REGO regimen achieved a C1 Csum of 2.5–5.5 mg/L associated with better survival, increased control rate at 8 weeks, and decreased occurrence of SAE while out of range variations of REGO and its metabolites exposure were detrimental.
Our PK analysis suggests that REGO interruption should be avoided by using decreased doses to prevent the onset of SAE, to allow adequate exposure to the drug, and to ultimately offer an individualized REGO dosing and schedule. These results may lead to individual REGO dose modification strategies based on PK monitoring and paving the way for a prospective clinical trial evaluating REGO, M-2, and M-5 concentrations monitoring in mCRC patients.
Supplementary Material
Highlights:
Therapeutic monitoring of regorafenib in metastatic colorectal cancer is feasible.
We define a safe range of residual concentration regorafenib+M-2+M-5.
This range translates into a survival benefit and decreased serious adverse events.
Only one third of the patients are within range at C1 D14.
Therapeutic monitoring of regorafenib may help to optimize its dose.
Acknowledgments
Authors would like to thank Mike Foote (Dr) and Magdalena Benetkiewicz (Sc.D) editorial support. BR received salary support from Nuovo Soldati Swim Across America foundations and MSKCC core lab grant P30 CA008748, and Bayer for financial support of the TEXCAN clinical trial.
Abbreviations
- CI 95%
95% confidence interval
- Cmin
Trough concentration
- Csum
Concentration sum of regorafenib, M-2, and M-5 trough concentrations
- CTCAE
Common Terminology Criteria for Adverse Events
- HR
Hazard ratio
- IQR
Interquartile range
- ITT
Intention-to-treat
- mCRC
Metastatic colorectal cancer
- M-2
Regorafenib M-2 active metabolite
- M-5
Regorafenib M-2 active metabolite
- PK
Pharmacokinetic
- PFS
Progression-free survival
- ORR
Overall response rate
- OS
Overall survival
- RCS
Restricted cubic spline
- REGO
Regorafenib
- SAE
Serious Adverse Event
- SD
Standard deviation
Footnotes
Declaration of interest statement: BR has served in a consulting/advisory role for Bayer, Roche, Novartis, Gilead, and Servier and has received travel, accommodations, and expenses from Bayer, Servier, and Astellas. RC has received honoraria from Amgen, MSD Oncology, Sanofi, and Servier, and research grant from Servier Institute. TA has received consulting/advisory role and or received honoraria from Amgen, Bristol-Myers Squibb, Chugai, Clovis, GlaxoSmithKline, Gristone Oncology, HalioDx, MSD Oncology, Pierre Fabre, Roche/Ventana, Sanofi, Servier, and Tesaro and has received travel, accommodations, and expenses from Roche/Genentech, MSD Oncology, and Bristol-Myers Squibb. OL has received honoraria from Bayer, Roche, Hologic and Bracco. DV has served in a consulting/advisory role for GERCOR, NOVARTIS, INCYTE, INVECTYS, FSK, AC Biotech SAS, CEllprothera and HalioDX. JBB has served in a consulting/advisory role for Amgen, Astra Zeneca, Bayer, Merck Serono, Pierre Fabre, Sanofi, Servier and Viatris, and has received travel, accommodations, and expenses from Amgen, Merck Serono, Sanofi, and Servier. SK has served as consulting/advisory role and received honoriaria from Beigne, Boehringer-Ingelheim, Incyte, Ipsem, MSD, Pfizer, Sanofi, Servier. CT has received consulting/advisory role and or received honoraria from MSD, Roche, Bayer, Sanofi and has received travel accommodations MSD, Bayer. TM has received honoraria from Sandoz, Pierre Fabre, AAA, Bristol Myers Squibb, Merck Serono, Servier and Sanofi/Regeneron Pharmaceuticals; research funding from AMGEN and reimbursement for travel expenses from Amgen, Pierre Fabre and Servier. CL has served in a consulting/advisory role for MSD, Roche, Servier, Amgen, Daichi-Sankyo and has received travel, accommodation and expenses from Roche and MSD. All remaining authors have declared no conflicts of interest.
Clinical trial reference: NCT02699073
References
- 1.Chiorean EG, Nandakumar G, Fadelu T, Temin S, Alarcon-Rozas AE, Bejarano S, et al. Treatment of Patients With Late-Stage Colorectal Cancer: ASCO Resource-Stratified Guideline. JCO Global Oncology. 2020;414–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Qin S, Xu R, Yau TCC, Ma B, Pan H, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015;16:619–29. [DOI] [PubMed] [Google Scholar]
- 4.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–12. [DOI] [PubMed] [Google Scholar]
- 5.Strumberg D, Scheulen ME, Schultheis B, Richly H, Frost A, Büchert M, et al. Regorafenib (BAY 73–4506) in advanced colorectal cancer: a phase I study. Br J Cancer. 2012;106:1722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zopf D, Fichtner I, Bhargava A, Steinke W, Thierauch K-H, Diefenbach K, et al. Pharmacologic activity and pharmacokinetics of metabolites of regorafenib in preclinical models. Cancer Med. 2016;5:3176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mross K, Frost A, Steinbild S, Hedbom S, Büchert M, Fasol U, et al. A phase I dose-escalation study of regorafenib (BAY 73–4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012;18:2658–67. [DOI] [PubMed] [Google Scholar]
- 8.Trnkova ZJ, Grothey A, Sobrero A, Siena S, Falcone A, Ychou M, et al. Population Pharmacokinetics Analysis of Regorafenib and Its Active Metabolites From the Phase III Correct Study of Metastatic Colorectal Cancer. Annals of Oncology. 2013;24:iv37. [Google Scholar]
- 9.Lucidarme O, Wagner M, Gillard P, Kim S, Bachet J-B, Rousseau B, et al. RECIST and CHOI criteria in the evaluation of tumor response in patients with metastatic colorectal cancer treated with regorafenib, a prospective multicenter study. Cancer Imaging. 2019;19:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adenis A, de la Fouchardiere C, Paule B, Burtin P, Tougeron D, Wallet J, et al. Survival, safety, and prognostic factors for outcome with Regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: results from a multicenter study (REBECCA) nested within a compassionate use program. BMC Cancer. 2016;16:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allard M, Khoudour N, Rousseau B, Joly C, Costentin C, Blanchet B, et al. Simultaneous analysis of regorafenib and sorafenib and three of their metabolites in human plasma using LC-MS/MS. J Pharm Biomed Anal. 2017;142:42–8. [DOI] [PubMed] [Google Scholar]
- 12.Kubota Y, Fujita K, Takahashi T, Sunakawa Y, Ishida H, Hamada K, et al. Higher Systemic Exposure to Unbound Active Metabolites of Regorafenib Is Associated With Short Progression-Free Survival in Colorectal Cancer Patients. Clinical Pharmacology & Therapeutics. 2020;108:586–95. [DOI] [PubMed] [Google Scholar]
- 13.Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, et al. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet. 2011;50:551–603. [DOI] [PubMed] [Google Scholar]
- 14.Keunecke A, Hoefman S, Drenth H-J, Zisowsky J, Cleton A, Ploeger BA. Population pharmacokinetics of regorafenib in solid tumours: Exposure in clinical practice considering enterohepatic circulation and food intake. British Journal of Clinical Pharmacology. 2020;86:2362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukudo M, Asai K, Tani C, Miyamoto M, Ando K, Ueno N. Pharmacokinetics of the oral multikinase inhibitor regorafenib and its association with real-world treatment outcomes. Invest New Drugs. 2021;39:1422–31. [DOI] [PubMed] [Google Scholar]
- 16.Bekaii-Saab TS, Ou F-S, Ahn DH, Boland PM, Ciombor KK, Heying EN, et al. Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): a randomised, multicentre, open-label, phase 2 study. Lancet Oncol. 2019;20:1070–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki T, Sukawa Y, Imamura CK, Masuishi T, Satake H, Kumekawa Y, et al. A Phase II Study of Regorafenib With a Lower Starting Dose in Patients With Metastatic Colorectal Cancer: Exposure–Toxicity Analysis of Unbound Regorafenib and Its Active Metabolites (RESET Trial). Clinical Colorectal Cancer. 2020;19:13–21.e3. [DOI] [PubMed] [Google Scholar]
- 18.Argiles G, Margalef NM, Valladares-Ayerbes M, de Prado JV, Grávalos C, Alfonso PG, et al. Results of REARRANGE trial: A randomized phase 2 study comparing different dosing approaches for regorafenib (REG) during the first cycle of treatment in patients (pts) with metastatic colorectal cancer (mCRC). Annals of Oncology. 2019;30:iv135. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.