

Abstract
NEUROD1 is a transcription factor that helps maintain a mature phenotype of pancreatic β cells. Disruption of Neurod1 during pancreatic development causes severe neonatal diabetes; however, the exact role of NEUROD1 in the differentiation programs of endocrine cells is unknown. Here, we report a crucial role of the NEUROD1 regulatory network in endocrine lineage commitment and differentiation. Mechanistically, transcriptome and chromatin landscape analyses demonstrate that Neurod1 inactivation triggers a downregulation of endocrine differentiation transcription factors and upregulation of non-endocrine genes within the Neurod1-deficient endocrine cell population, disturbing endocrine identity acquisition. Neurod1 deficiency altered the H3K27me3 histone modification pattern in promoter regions of differentially expressed genes, which resulted in gene regulatory network changes in the differentiation pathway of endocrine cells, compromising endocrine cell potential, differentiation, and functional properties.
Subject terms: Histone post-translational modifications, Endocrine system and metabolic diseases
Errors during pancreas development and specification of the endocrine lineage can result in severe neonatal diabetes. Here they show that loss of NEUROD1 leads to disturbances in endocrine cell identity acquisition during pancreas development, with cellular reprogramming occurring at the single-cell level within both α- and β-cell populations.
Introduction
The endocrine pancreas is a key regulatory tissue of glucose metabolism1. It consists of the islets of Langerhans, small scarce spherical cell clusters containing five hormone-producing cell types [α cells (glucagon), β cells (insulin), γ cells (pancreatic polypeptide), δ cells (somatostatin) and ε cells (ghrelin)]. In mice, the β-cells are predominately located in the central core of islets, while other hormone-producing cells localized in the periphery, forming a mantle. Dysfunction of these endocrine cells can lead to severe pathophysiology, especially in the case of β-cells failing to produce insulin, resulting in diabetes mellitus. The incidence of diabetes is gradually increasing worldwide, necessitating the development of novel therapies to either compensate for decreased insulin levels or replace dysfunctional β-cells in situ. As the endocrine pancreas bears no signs of postnatal regeneration2, the solution possibly lies in direct cell reprogramming3–6 via gene regulatory networks7,8 driving pancreatic development from multipotent progenitors (MPs) to differentiated endocrine cells9.
Pancreatic organogenesis starts around embryonic day 8.5 (E8.5) in mice and between 29–33 days post-conception in humans, from MPs expressing a characteristic combination of Pdx1, Sox9, and Ptf1a (reviewed in9). As morphogenesis of the pancreas progresses, Ptf1a expression becomes limited to acini located at the tips10, whereas Pdx1, Sox9, and Nkx6.1 define bipotent precursors in the trunk area, establishing both ductal and endocrine progenitors (EPs)11. Sox9 activates the pro-endocrine gene Neurogenin 3 (Neurog3), and thus, initiates endocrine differentiation12. Endocrine differentiation in mice proceeds in two temporal waves13, termed the primary transition starting at E8.5 and secondary transition from E12.5 to E16.514, (humans have one wave only15). The first differentiated endocrine cells are glucagon+ cells, detected in the dorsal pancreas at E9.5. The first insulin+ cells, expressing high levels of PDX1, do not appear until E12.5, but their numbers then increase exponentially beginning at E14.516. The upregulation of Neurog3 is necessary for the induction of cascades of transcription factors orchestrating endocrine cell differentiation, such as NEUROD1, INSM1, RFX6, IRX1, and PAX49,17,18. Many involved transcription factors cooperate with epigenetic regulators19 and together, they direct cell-fate determination. Polycomb group protein complexes play a significant role in this process20, especially the histone demethylase KDM6B21. KDM6B allows the removal of transcriptionally repressive histone modifications from bivalent loci in the genome. Bivalency can be defined as the presence of both activation (H3K4me3) and repression (H3K27me3) marks allowing time-effective changes in the gene expression, and such phenomenon is vital in embryonic development22–24.
In both mice and humans, Neurod1 expression follows Neurog3 directly25–28, indicating a role of NEUROD1 in the gene regulatory network of the basal endocrine lineage commitment. NEUROD1 serves as an early and comprehensive marker for all endocrine cells29. NEUROD1+ EPs leave their proliferative state30 and begin the process of differentiation into specific endocrine cell types. This change corresponds to the Neurog3-low to Neurog3-high transition31. During this 12h-long cellular process31,32, a KDM6B-driven switch from bivalency to a H3K4me3-only state occurs in hundreds of promoters, including Neurod1, Insm1, and Rfx6, resulting in gene expression shifting from a Sox9, Onecut1, and Hes1-enriched Neurog3-low EP profile towards a Neurod1, Insm1, Rfx6, Isl1, and Pax6 elevated expression pattern leading to endocrine cell differentiation. This transition is accompanied by enhancer priming and activation of genes typical for endocrine functions, predominantly with NEUROD1 binding sites31, suggesting NEUROD1’s pro-endocrine pioneering function.
A pioneering potential of NEUROD1 was reported in neuron differentiation33. This pioneering activity involves both activation of cell type-specific genes and repression of genes associated with alternative non-neuronal lineages. NEUROD1 is indispensable for neuronal differentiation, survival, and reprogramming, by reshaping the epigenetic and transcriptional landscape33–39. NEUROD1 promotes neuronal fate and preserves neuronal identity by suppressing non-neuronal fates.
Unlike Neurog340, which exhibits transient expression during endocrine specification, Neurod1 demonstrates a more dynamic and temporally diverse expression profile across various endocrine cell subtypes throughout development and maturation41,42. In mice, Neurod1 expression initiates around E9.5, persists until E12.5 in early glucagon+ cells, dissipates temporarily, and then peaks again around E17.5 in nascent islets41. This spatiotemporal fluctuation is fundamental for the establishment43 and proper differentiation of the endocrine lineage44,45. The significance of NEUROD1 becomes evident when considering the consequences of its deficiency. First, mice lacking Neurod1 during pancreas development die shortly after birth from severe diabetes partly due to a reduced number of endocrine cells, particularly β cells44–47. Second, NEUROD1 is indispensable for β-cell maturation and maintenance44,48,49. Third, rare mutations in Neurod1 in humans manifest as a subtype of maturity-onset diabetes of the young (MODY6)50–52, since NEUROD1 is one of the direct transcription activators of the insulin gene53–55, and is therefore, critical for modulating glucose homeostasis. Fourth, Neurod1 deletion is accompanied by an altered islet architecture43,45,46,56.
In this study, we focused on molecular cues shaping the endocrine differentiation path associated with Neurod1. Using a Neurod1Cre endocrine-specific Neurod1 knockout (Neurod1-self terminating; Neurod1ST), we introduced an early Neurod1 deletion disrupting fetal pancreatic development to provide further insight on the role of NEUROD1 in the early endocrine lineage establishment. We identified changes associated with Neurod1 deletion on a transcriptomic (endocrine specific RNA-seq) and epigenetic level using the CUT&Tag-seq approach, which helps overcome the low-input limits of existing epigenetic-landscape profiling methods such as ChIP-seq57.
Herein, we link an early endocrine deficiency of NEUROD1 to an altered chromatin landscape of numerous genes resulting in transcriptome dysregulation and ambiguous cell fate commitment, eventually leading to critical developmental defects of the endocrine pancreas, which are manifested both functionally and morphologically.
Results
Neurod1 elimination results in a severe diabetic phenotype
To evaluate the role of Neurod1 during the early development of the pancreas, we generated a Neurod1-“self-terminating” conditional knockout mouse model (Neurod1ST) by breeding Neurod1loxP/loxP 58 with Neurod1Cre/+ 59. Using CRE-dependent Ai14-tdTomato reporter mice60, we showed that the expression pattern of tdTomato matched the formation of the first differentiated endocrine cell clusters and NEUROD1 expression in the E10.5 pancreas (Supplementary Fig. 1a). We confirmed that Neurod1-CRE activity in early endocrine cell clusters emerging from the dorsal pancreatic bud is exclusive to the endocrine lineage in the pancreas and corresponds to the expression of Neurod1 gene42,43,61. The CRE-mediated deletion of Neurod1 was efficient, with a more than 65% elimination of NEUROD1 in the E10.5 pancreas of Neurod1ST (Neurod1loxP/loxP; Neurod1Cre/+) compared to Controls (Neurod1loxP/loxP or Neurod1loxP/+) (Supplementary Fig. 1b, c).
We observed neonatal mortality in Neurod1ST (Supplementary Fig. 2) correlating with a trend of reduced body weight (Fig. 1a) and elevated levels of glycemia (Fig. 1b) measured on the first day after birth (P0). Decreased total pancreatic insulin levels confirmed a severe diabetic phenotype of the Neurod1ST (Fig. 1c). No surviving Neurod1ST homozygous mutant mice were detected beyond P3. These findings are in line with observations in newborns with global deletion of Neurod146. We also characterized the phenotype of mice with the heterozygous genotype (Het: Neurod1loxP/+; Neurod1Cre/+) containing one functional Neurod1 allele and Cre allele. Mice with Het genotype with the respect to development, body weight, blood glucose, and survival were undistinguishable from Control mice, confirming no unintended effects of the Cre recombinase and the reduced level of Neurod1 gene (Fig. 1a, b, and Supplementary Fig. 2). These results allowed us to utilize the Cre driver in combination with the Ai14-tdTomato reporter (Control-Ai14) for cell-type-specific molecular and morphological analyses.
Fig. 1. Neurod1ST mice demonstrate a severe diabetic phenotype with reduced endocrine cell mass.

a Genotype-determined differences in the measured values of body weight in newborn mice at postnatal day P0-P2 (One-way ANOVA, Tukey’s multiple comparisons test). b Differences in blood glucose in newborn mice fed ad libitum at P0 (One-way ANOVA, Tukey’s multiple comparisons test). c Total pancreatic insulin levels at embryonic day 17.5, E17.5 (n = 8 pancreases/group). Data are presented as mean ± SEM. d Representative immunostaining for α-amylase (α-AMY, a marker of exocrine tissue), glucagon (GCG), and insulin (INS) in pancreatic tissue sections of Control and Neurod1ST at P0. Boxes indicate the areas magnified in the right panels. e Representative microscopy images of tdTomato+ cells and immunostaining of α-AMY, and GCG in P0 pancreatic tissue sections of Control-Ai14 and Neurod1ST-Ai14. Boxes indicate the areas magnified in the right panels. f The number of GCG+ and INS+ cells counted in the central section with the largest pancreatic footprint of Neurod1ST and Control pancreases per arbitrary unit (arb. units = 0.0003 mm3; n = 24 views of area/Control from 5 pancreases and 22 views of area/Neurod1ST from 5 pancreases). Data are presented as mean ± SEM. g Representative images of immunolabeled somatostatin+ (SST+) cells and quantification of the percentage of SST+ per tdTomato+ endocrine cells at P0 (n = 4 pancreases/genotype). Data are presented as mean ± SD. h, i Quantification of the percentage of proliferating INS+ and GCG+ cells in the Neurod1ST and Control pancreas at E17.5 and P0. Representative immunostainings for proliferation marker, Ki67, β-cells expressing INS, and α cells expressing GCG. GCG+ cells forming the mantle around INS+-cell core in the Control are indicated by arrows (in i). Note a disrupted islet core-mantle organization in Neurod1ST with a missing mantle of GCG+ cells around β-cell core compared to Control. Nuclei were stained by Hoechst (HS). Data are presented as mean ± SD (n = 5 pancreases/genotype/each age). Unpaired two-tailed t test (c, f–i). Source data for (a–c, f–i) are provided as a Source Data file. Scale bars, 50 µm. ns not significant.
Next, we evaluated the histology of islets of Langerhans in Neurod1ST compared to both Control and Control-Ai14. We observed reduced endocrine tissue content in the number of glucagon-producing (GCG) α cells and insulin-producing (INS) β cells at P0 (Fig. 1d–f). The percentage of somatostatin (SST)-secreting cells was not affected (Fig. 1g). Consistent with previous studies44,45, we found a major defect in α- and β-cell proliferation without noticeable increase in apoptotic cells in Neurod1ST, as evaluated by TUNEL (Supplementary Fig. 3). The proliferation of β cells in Neurod1ST was significantly reduced at both E17.5 (Fig. 1h) and P0 (Fig. 1i). Furthermore, we confirmed previous results45 indicating no significant effect of Neurod1 elimination on the α-cell proliferation at E17.5 (Fig. 1h). The β-cell proliferative state in physiological conditions in mice reportedly starts around E17.5 and peaks approximately in P3, preceding a process of maturation62. α cells follow the same trend but peak around P062. Concomitantly, α-cell proliferation was moderately reduced in Neurod1ST at P0 (Fig. 1i; p = 0.0463). Since both β and α cells are a major part of the islet cell mass, a lower proliferation of both β and α cells corresponds to reduced endocrine content of the Neurod1ST. Similar to prior reports44–46, the lack of Neurod1 was accompanied by disrupted architecture of the islets of Langerhans, forming smaller cell clusters with the distinctive α-cell mantle absent in P0 Neurod1ST (Fig. 1i).
To uncover the effects of Neurod1-deficiency on the formation, and spatial distribution of the islets of Langerhans in the 3D tissue microenvironment of the pancreas, we performed light sheet fluorescence microscopy at E18.5 on both Control-Ai14 (Supplementary Movie 1) and Neurod1ST-Ai14 (Supplementary Movie 2), visualizing α- (GLP1; green) and β-cell (INS; white) mass overlap with endocrine tissue (tdTomato; magenta). Surprisingly, apart from a disrupted α-cell islet mantle and increased variability among islet size in Neurod1ST, the endocrine cell mass appeared to be semi-organized into strings/sheets along the invisible lines of pancreatic ducts. In contrast, Control-Ai14 islets shared a more spherical shape, regular size span, and core-mantle organization. Abnormal clustering of islets near ductal structures may lead to a failure in establishing the normal dispersed islet morphology within the exocrine tissue63. This altered organization may disrupt the communication and interactions not only between neighboring islets but also between endocrine and non-endocrine tissues. Consequently, it can disrupt the intricate network that enables intra-islet and inter-islet synchronous hormone secretion64. Such alterations in the spatial architecture and communication could have significant implications for glucose responsiveness and overall endocrine cell function. The 3D endocrine pancreas architecture is a critical factor in maintaining glucose homeostasis and preserving β‑cell function, as its disruption has been associated with diabetes mellitus65,66.
Endocrine cell differentiation is dysregulated in Neurod1ST
The pancreas starts budding from the endodermal foregut around E8.5 and is specified by a multipotent progenitor marker PDX167. Once the endocrine lineage developmentally diverges from its multipotent pro-pancreatic precursors, Pdx1 expression dissipates and is reconstituted later and only in the β-cell lineage68,69. The appearance of the first endocrine cells, GCG-expressing cells, coincides with the onset of Neurod1 expression during the primary transition in the developing dorsal pancreatic bud at E9.543. However, only a subset of GCG+ cells (30%) express NEUROD1 in the E9.5 pancreas43. Consistent with the limited requirements of Neurod1 for the earliest endocrine cells, efficient reduction of NEUROD1 did not have any significant impact on the formation of endocrine clusters in the dorsal pancreatic bud of Neurod1ST at E10.5 (Fig. 2a, b). The relative size of the GCG+ cell clusters compared to the PDX1+ area of the dorsal pancreatic bud was unchanged. (Fig. 2b). We also observed a persistent expression of PDX1 in the differentiating α-cell lineage in Neurod1ST during the primary (E10.5, E11.5) and secondary pancreatic transition (E12.5, E15.5) (Fig. 2c). Although the generation of GCG+ endocrine cells was not affected, the persistent expression of PDX1 in GCG+ cells, indicates that the differentiation of these cells might be altered or delayed in the absence of NEUROD1.
Fig. 2. Neurod1 elimination impairs PDX1 expression in the developing pancreas.

a Representative images of the E10.5 dorsal pancreatic bud during the primary transition visualized by whole-mount immunostaining of PDX1 (a marker of pancreatic progenitors), ISL1 (a marker of pancreatic endocrine cells and pancreas-surrounding mesenchyme), and tdTomato+ cell clusters. b Whole-mount immunolabeling of glucagon (GCG) clusters in the dorsal pancreatic bud delineated by the expression of PDX1 at E10.5. Semi-quantitative measurements of GCG+ area in the PDX1+ domain of Control and Neurod1ST. Data are presented as mean ± SD (n = 5 pancreases/genotype). c Representative whole-mount immunostaining images of the developing pancreas during the primary transition (E10.5, E11.5) and the secondary transition (E12.5, E15.5) show an increased number of cells with PDX1 and GCG co-expression (arrows) in Neurod1ST compared to Control. Note some PDX1+ cells without characteristic insulin (INS) expression within developing islets in Neurod1ST at E15.5 (arrowheads). The percentage of cells co-expressing PDX1+ and GCG+ cells quantified in the Neurod1ST and Control pancreas at E10.5. Data are presented as mean ± SD (n = 4 pancreases/genotype). Unpaired two-tailed t test. Source data are provided as a Source Data file. Scale bars, 50 µm. ns, not significant.
During the secondary transition of pancreas development marked by a major wave of endocrine cell differentiation, the islets of Langerhans emerge, and the endocrine tissue starts producing all related hormones. A significant larger percentage of the endocrine tdTomato+ cells did not produce either GCG or INS hormones at E15.5, suggesting that these tdTomato+ cells might have an altered or disrupted differentiation process (Fig. 3a, b). Noticeably, some endocrine cells expressed PDX1 as a marker of differentiating β cells but without INS co-expression in Neurod1ST at E15.5 (Fig. 2c). In line with expression dysregulation in differentiating α and β cells, the hormone production at the mRNA level was affected in the Neurod1ST pancreas at E15.5 (Fig. 3c). Rodents express two nonallelic insulin genes, Ins1 and Ins2, encoding the proinsulin 1 and proinsulin 2 isoforms70. The relative expression of Ins1 but not Ins2 mRNA was significantly reduced in Neurod1ST compared to Controls at E15.5 (Fig. 3c). These data are consistent with previous studies that reported that deletion of Neurod1 results in a loss of Ins1 expression, whereas Ins2 expression is relatively unaffected45,48. Therefore, we assessed the expression of peptides C1 and C2, which are cleaved from the corresponding inactive prohormones. At P0, peptide C1 was apparently reduced in Neurod1ST-Ai14 compared to the Control-Ai14 in contrast to peptide C2 (Fig. 3d), consonantly to different mouse models with conditional Neurod1 deletions45,48. Our results further confirm that loss of Neurod1 thwarts endocrine lineage differentiation leading to reduced endocrine cell mass and defective hormone production.
Fig. 3. Neurod1 deficiency results in decreased hormone production.
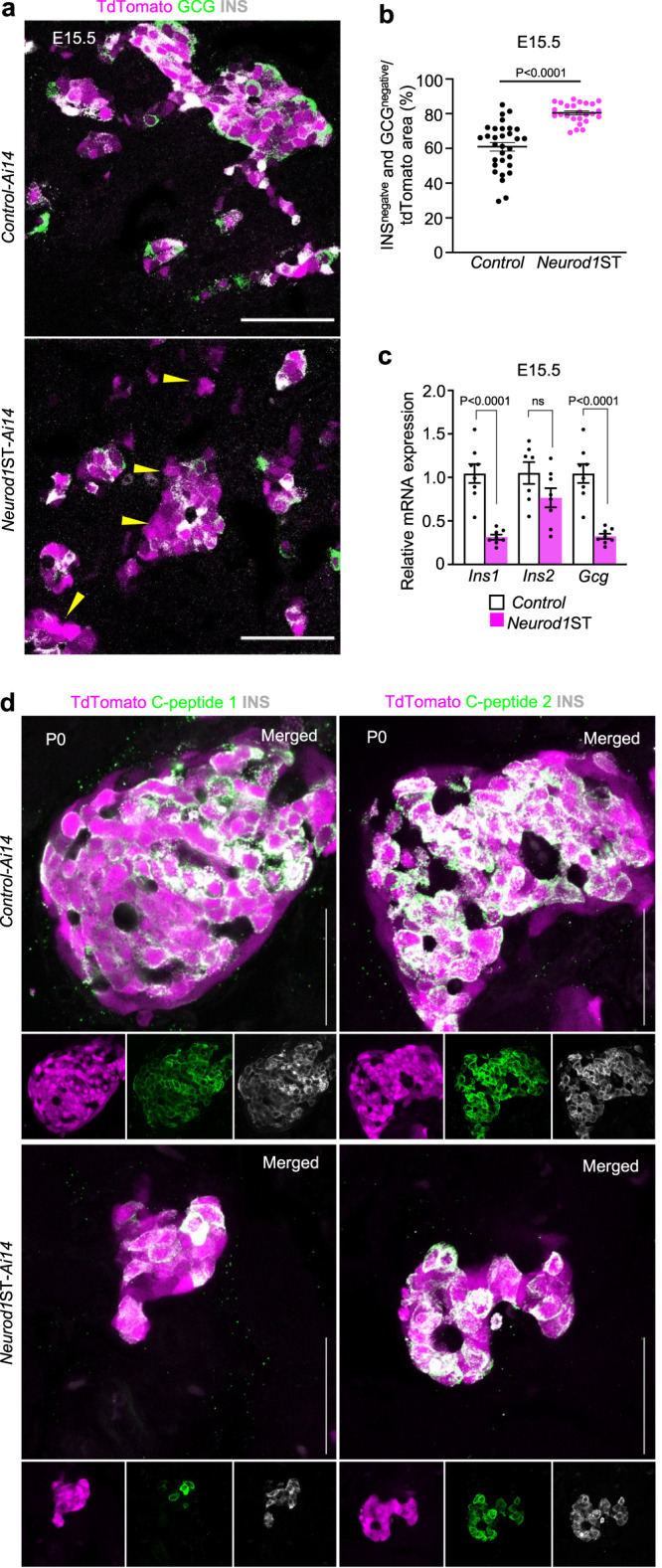
a Representative confocal microscopy images of tdTomato+ cells and immunostaining of glucagon (GCG), and insulin (INS) during the secondary transition at E15.5. Note the increased number of tdTomato+ cells without a detectable expression of INS or GCG (arrowheads). b Quantification of tdTomato+ area without GCG and INS expression in the central section of pancreas with the largest pancreatic footprint. Data are presented as mean ± SEM; (n = 31 tdTomato+ areas/Control from 4 pancreases and 25 tdTomato+ areas/Neurod1ST from 4 pancreases); unpaired two-tailed t test. Source data are provided as a Source Data file. c Relative mRNA expression of glucagon (Gcg), insulin 1 (Ins1), and insulin 2 (Ins2) by RT-qPCR using RNA extracted from E15.5 pancreases. Data are presented as mean ± SEM; (n = 8 pancreases/genotype); unpaired two-tailed t test, Source data are provided as a Source Data file. d Representative confocal microscopy images of tdTomato+ cells and immunostaining of INS, C-peptide 1 and C-peptide 2 in P0 pancreatic tissue sections. Scale bars, 50 µm. ns, not significant.
NEUROD1 reinforces endocrine cell fate
To analyze the molecular changes resulting from the elimination of Neurod1 in developing pancreatic endocrine cells, we performed a bulk-cell-RNA sequencing (bulk-RNA-seq) analysis to evaluate the overall transcriptomic profile in Neurod1ST-Ai14 and validated representative differentially expressed genes using qRT-PCR in independent biological samples (Fig. 4a). Each of four biological replicates per genotype for the bulk-RNA-seq analysis contained 100 tdTomato+ FACS-sorted single cells from the dissociated E15.5 embryonic pancreas. tdTomato-expressing cells represent differentiating pancreatic endocrine cells with an active Neurod1-promoter.
Fig. 4. Neurod1 elimination disrupts gene regulatory network in the developing endocrine cells.

a RNA sequencing experiment design—1: The embryonic pancreases (E15.5) were extracted and dissociated into single cells. Endocrine tdTomato+ cells were collected using FACS (100 cells/sample, 4 samples/genotype). Lysed cells total RNA served as template for RNA-seq library preparation. 2: Whole extracted E15.5 pancreases were lysed and used for RT-qPCR validation of several differentially expressed endocrine-specific genes identified by RNA-seq. Created with BioRender.com. b RNA-seq data analysis identified 422 differentially expressed genes with 112 downregulated and 153 upregulated protein-coding genes (see also Supplementary data). c Downregulated gene functional enrichment (Gene Ontology [GO] biological processes, KEGG pathways, REACTOME). d Upregulated GO enrichment (biological processes). e Profiled differential expression heatmap: All differentially expressed genes with affiliation to endocrine/non-endocrine single cell expression profile. f Transcription regulation heatmap: Differentially expressed genes identified in transcription regulation cluster. g Boxplots show the deconvolved cell type percentage in E15.5 endocrine population from our bulk RNA-seq data (circle marks identified outliers). Centerline of the boxplot indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers represent the rest of the data distribution. h RT-qPCR validation of several identified differentially expressed genes at E15.5 in the whole pancreas. Relative mRNA expression data are presented as mean ± SEM (n = 8 pancreases/Control and 7 pancreases/Neurod1ST), unpaired two-tailed t test, ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05). Source data are provided as a Source Data file. i Quantification of cells co-expressing SOX9 and PDX1. Data are presented as mean ± SD; (n = 4 pancreases/genotype); unpaired two-tailed t test. Source data are provided as a Source Data file. j Representative confocal microscopy images of tdTomato+ cells and immunolabeled SOX9, insulin (INS), and PDX1 cells. A box indicates the area magnified in the right panel. Arrowheads indicate tdTomato+ cells co-expressing SOX9 and PDX1. Scale bars, 50 µm. k Top 10 transcription factors identified by de novo motif enrichment analysis of differentially expressed genes.
Differential expression analysis identified 112 downregulated and 153 upregulated protein-coding genes in the Neurod1ST-Ai14 endocrine-specific cell population compared to the corresponding Control-Ai14 (Fig. 4b, Supplementary Data 1). This analysis confirmed significant reduction of Ins1 mRNA levels but not Ins2 in Neurod1ST, consistent with the necessity of NEUROD1 for Ins1 expression45,48. Functional enrichment analyses71 of the set of 112 downregulated genes showed enrichment of Gene Ontology (GO) biological processes associated with endocrine functions such as regulation of hormone levels, cell-cell signaling, response to monosaccharide, peptide hormone processing, hormone, peptide, and insulin secretion and their transport (Fig. 4c, Supplementary Data 1). For example, this analysis identified an enrichment of genes involved in peptide hormone processing, Cpe72, and insulin secretion regulation, Ffar173, Nnat74, Scg575, Pcsk276, Pcsk677, Nipal178. Furthermore, the analysis uncovered the following KEGG pathways79 and endocrine pathologies: AMPK signaling, Insulin secretion, and Maturity onset diabetes of the young (MODY), Type II diabetes mellitus (T2DM), respectively. Ins1, Mafa80, Cacna1c81, Irs482 included in T2DM, and MODY, confirm the diabetic phenotype, and Rapgef483 is involved in the cAMP signaling pathway crucial for Ca2+-dependent hormone secretion84. Additional genes such as G6pc285, Prkag286, and Hmgcr87 are involved in AMP-activated protein kinase (AMPK) signaling, whose activation enhances extracellular glucose uptake88, and with Npy89 also affects lipid metabolism and cellular physiology90. Interestingly, within the Reactome enrichment of downregulated genes, we found genes associated with Anchoring of the basal body to the plasma membrane, which may affect ciliary functions vital for islet development and glucose homeostasis maintenance91 (Fig. 4c).
The GO analysis of the cohort of 153 upregulated genes showed enrichment of biological processes uncommon in the developing pancreatic endocrine cells (Fig. 4d, Supplementary Data 1), which were related to exocrine system and gland development, containing characteristic ductal genes, such as multipotency-maintaining Sox992, the WNT secretion mediator wntless, Wls gene93,94, or Ntn4, a member of the Netrin family, abundantly expressed by pancreatic ductal cells95. Thus, the increased expression of non-endocrine genes suggested a flawed differentiation process in Neurod1ST. We also found an increased abundance of non-endocrine cell adhesion and cell junction organization markers such as ductal Ocln96, Rhoc97, S100a1098, Sparcl199, mesenchymal Vcan100, or acinar Cldn10101, Pcdh1102. Interestingly, upregulated Cdh4 is commonly expressed in both early ductal and endocrine cells but diminishes during embryonic development and remains only in non-endocrine pancreatic tissue103. Besides that, we found few upregulated endocrine-related genes, including a membrane raft component Cd55104, β-cell protective Nid1105, and an immune cell adaptor Skap1106. Notably, 26 genes were linked to programmed cell death, which highly overlapped with the cell adhesion cohort. Many of those genes were related to various kinds of neoplasia progression (Tpx2, Id3, Top2a, Dab2, Mecom, Nrp1107–112), including pancreatic ductal adenocarcinoma, or pancreatitis (Gsn113). Furthermore, we observed an increased expression in genes involved in the MAPK/ERK cascade, which reportedly restrains endocrine specification114. In the context of the affected endocrine functions, we identified a group of genes related to aldosterone metabolic process and renin-angiotensin-mediated blood flow regulation (regulation of systemic arterial blood pressure by mediated by a chemical signal), including Ace2115,116 that has negative effects on glucose tolerance and insulin sensitivity117, or Adrb2 which impairs endocrine development by generating hypervascularized islets118. Additionally, β-cell proliferation and insulin secretion regulator Sgk3119 were upregulated in Neurod1ST.
Mammalian Phenotype Ontology enrichment analysis uncovered the enrichment of differentially expressed genes in Neurod1ST associated with phenotype-matching categories (Supplementary Data 1), such as abnormal islet morphology (downregulated genes) and abnormal homeostasis (upregulated genes). Interestingly, downregulated genes were enriched exclusively in pancreatic endocrine tissues, whereas upregulated genes were found in a wide span of multiple tissues, e.g., lung, renal, neural, dermal, and gastrointestinal, including pancreatic.
Next, using an open-access transcriptomic database of the mouse embryonic (E15.5) single-cell pancreas29, we linked identified differentially expressed genes from our bulk-RNA-seq to their characteristic single-cell transcriptomic signature profiles in the cell subpopulations of the differentiating pancreas (Supplementary Data 1). We categorized differentially expressed genes based on their single-cell transcriptomic signature profiles as endocrine, non-endocrine, and combining both. Strikingly, we observed a clear pattern of non-endocrine genes upregulated in Neurod1ST (Fig. 4e), representing mesenchymal, ductal, trunk, and acinar cell subpopulations. Conversely, genes with an endocrine expression pattern were downregulated in Neurod1ST. Based on the functional characterization of differentially expressed genes using DAVID120, non-endocrine genes were enriched in the cluster of transcription regulators (Fig. 4f), cell differentiation (Supplementary Fig. 4a), and cell cycle (Supplementary Fig. 4b) in Neurod1ST compared to the Control. Prominent endocrine differentiation-driving transcription factors Insm1121,122, Isl1123, Mafa124, and Tox3125, including Neurod1, were downregulated in Neurod1ST as well as chromatin remodelers Kdm1b126, and α-cell enriched Smarca1127. Additionally, the β-cell apoptosis regulator St18128, endocrine β-cell survival regulator Slit1129, and acinar-to-ductal inductor Klf4130 were decreased. In contrast, various transcription factors were upregulated in Neurod1ST such as ductal Sox992 and its cooperator Maf131,132, acinar-reprogramming Mecom111, or transcriptional corepressor Cbfa2t3133, and positive proliferation marker Mki67, cyclin Ccnb2, negative proliferation regulator Zfp36l1134, β-cell protective regulator Rgs2135, or ductal cell cycle-driver Id3108. Two chromatin remodelers were also upregulated; the ubiquitous Smarca4 and EPs-distinct Smarcd2125.
To better understand the ambiguous cell identities in Neurod1ST, we performed the deconvolution of our bulk-RNA-seq data to estimate cell type proportions136. Single-cell transcriptomic signatures of five mostly abundant (at E15.5) and closely cell-fate-related pancreatic cell types (three endocrine and two non-endocrine), namely α cells, β cells, endocrine progenitors, and trunk epithelium, and tip epithelium were used for the deconvolution analysis137. We identified a shift in the transcriptome signatures of β-cell populations between the Neurod1ST and Control (Fig. 4g).
Selected differentially expressed genes were validated by qRT-PCR with RNA isolated from whole E15.5 embryonic pancreas (Fig. 4h). However, validation of differentially expressed non-endocrine genes was not possible due to their abundance in non-endocrine tissue, which would greatly skew the expression differences. Apart from supporting the conclusions of disrupted endocrine functions (Cpe, Resp18, Scg5, Slc2a5) and EP-related transcription dysregulation (Tox3, Smarcd2), we also confirmed that Neurod1 elimination affects its downstream target Nnat138. Furthermore, we used immunohistochemistry to assess the expression of the non-endocrine ductal SOX9 transcription factor (Fig. 4i, j). Consistent with our bulk-RNA-seq data, we observed frequent co-expression of SOX9 and PDX1 in tdTomato+ endocrine cells in Neurod1ST, whereas such cells were rarely detected in the Control pancreas at E15.5.
Next, we searched for transcription factor binding motifs enriched within the promoters of all differentially expressed genes found in the Neurod1ST mutant, using HOMER de novo motif analysis139. This analysis identified significant enrichments for binding sites of NEUROD1 and ISL1 within the extracted top ten hits for the promoters of down-regulated genes (Fig. 4k), confirming direct gene expression network regulation involving both downregulated transcription factors. Interestingly, the motifs of lineage-determining transcription factors, such as MEIS2, NFIA and RBPJ, were also significantly enriched in the promoters of down-regulated genes. RBPJ140,141 is a crucial modifier of endocrine-versus-acinar transcriptional activity and both RBPJ and MEIS2142 modulate PDX1 activity in a lineage-specific manner, while NFIA regulates Notch signaling and promotes pro-endocrine development143. Among motifs enriched in differentially expressed upregulated genes, we identified HNF4α, which is a master regulator of epithelial differentiation in multiple tissues, including the gastrointestinal tract and liver144,145. In the pancreas, HNF4α controls β-cell function and glucose metabolism and is associated with MODY1146,147 but HNF4α also regulates early pancreatic genes in the pancreatic progenitors148. Additionally, significant enrichments for binding sites of EGR1, HES1, ELF5, HIC1, SPDEF, and NR2E1 transcription factors were identified in upregulated gene promotors. EGR1 regulates PDX1 expression in β cells149 and was also described as their stress marker150. HES1 controls cell fate determination between ductal-versus-endocrine fate choice in bipotent progenitors via the Notch signaling axis12,151. ELF5 is specifically expressed in trunk cells, while SPDEF and HIC1 in acinar cells at E15.529. On the other hand, NR2E1 possess β-cell protective traits152.
Taken together, these data identified significant downregulation of endocrine genes on the global transcriptomic level within the Neurod1-deficient endocrine cell population, while the expression of numerous genes characteristic for the non-endocrine tissues is substantially elevated. These deviations fully support the phenotypic pathophysiology of Neurod1ST and emphasize the flawed developmental gene regulatory network derailing endocrine differentiation and cell cycling. Furthermore, these results provide further evidence of the essential role of NEUROD1 in defining basal characteristics of the endocrine lineage commitment.
NEUROD1 shapes the endocrine cell epigenetic landscape
Following the findings of NEUROD1 pioneering potential31,33,34, we decided to further explore its regulatory role in the endocrine identity determination of the developing pancreas using CUT&Tag sequencing (Fig. 5a). To assess NEUROD1-related changes in bivalency, we compared the H3K4me3 and H3K27me3 endocrine profiles of Neurod1ST-Ai14 with Control-Ai14. Both profiles were prepared from FACS-sorted tdTomato+ endocrine cells isolated from several E15.5 embryonic pancreases. To correlate H3K4me3 and H3K27me3 modifications with gene expression changes, we focused only on histone modifications of differentially expressed genes identified by our bulk-RNA-seq. We also included NEUROD1 binding sites identified in silico by HOMER139. As either the presence of the repressive H3K27me3 mark153 or the combination of H3K27me3 with H3K4me3 histone modifications154 result in downregulation of gene expression or silencing, we centered our scope on H3K27me3 peaks that might possibly cause gene downregulation.
Fig. 5. NEUROD1 is essential for shaping the epigenetic landscape distinct to the endocrine cell lineage.

a CUT&Tag-seq experiment design: tdTomato+ nuclei were immobilized on magnetic beads and “Cleavage Under Targets and Tagmentation” was performed with antibodies against H3K4me3 or H3K27me3. Reads from amplified libraries were mapped on reference genome and putative NEUROD1 binding sites were identified in silico. Created with BioRender.com. b Among 112 downregulated genes in Neurod1ST, we found 66 genes containing elevated H3K27me3 peaks (Group 1). In cohort of 153 upregulated genes in Neurod1ST, we identified 28 genes (Group 2) with increased H3K27me3 peaks in Control compared to Neurod1ST. c Representative genes for both Group1 and 2 are presented as an UCSC genome browser output. NEUROD1 binding motifs identified in silico are often adjacent to the detected peak loci of gene promoters.
Based on the H3 methylation pattern, two main gene groups were identified (Fig. 5b). Group 1 contained genes downregulated in Neurod1ST-Ai14 with increased H3K27me3 peaks within the gene promoter, gene-adjacent loci, or both. Many identified genes were directly linked to endocrine functions by our analysis of bulk-RNA-seq data, indicating Neurod1-dependent modulation of the gene regulatory network driving endocrine differentiation, e.g., Isl1, Tox3, Mafa (Fig. 5c). Group 2 consisted of genes containing upregulated H3K27me3 peaks in the Control samples, such as Sox9, Vcan, Ocln, correlating with downregulated expression in endocrine cells of the Control-Ai14 pancreas based on our bulk-RNA-seq data (Fig. 5b, c). In contrast, H3K27me3 modification was undetected in Group 2 genes in Neurod1ST endocrine cells, supporting a putative cause of their upregulated expression in Neurod1ST.
These findings indicate that NEUROD1 is a key player in the gene regulatory networks underlying cell fate commitment, defining endocrine progenitor identity by the remodeling of the chromatin landscape and thus, driving the terminal differentiation of endocrine cells in the pancreas. Furthermore, our results uncover a new point of view on the global scope of yet unclear epigenetic regulations behind such phenomena.
Molecular reprogramming in Neurod1-deficient α and β cells
Although a bulk-cell RNA-seq provides extensive sequencing depth, it represents an average of gene-expression of multiple cells. Therefore, to examine the effects of Neurod1 deletion in a heterogeneous endocrine population, we utilized single-cell RNA-seq (scRNA-seq) analysis. We specifically focused on adult islets of Langerhans to minimize a possible asynchrony of generation of endocrine cells between the control and mutant pancreases. For the scRNA-seq study, we used a conditional Neurod1 deletion model (Neurod1CKO; Neurod1loxP/loxP;Isl1Cre/+ genotype)45. Unlike our Neurod1ST mice that die shortly after birth, 40% of Neurod1CKO mice survive into adulthood. Isolated pancreatic islets were dissociated into single cells, and an estimated 12,000 cells per each genotype were used for scRNA-seq. Unsupervised clustering of cell endocrine profiles revealed six transcriptionally distinct subgroups in the Control (Fig. 6a). Four principal clusters were identified based on endocrine markers such as Ins2 and Ins1 for the β cell population, Gcg for α cells, pancreatic polypeptide-expressing cells (Ppy) for PP/γ-cell population cluster, and Sst-expressing cells (δ cells). Some cells within the δ cell cluster co-expressed Neurog3 at a low level155. Notably, many β cells co-expressed Sst and Ins2 and/or Ppy in Neurod1CKO. In addition, we found a higher proportion of polyhormonal cells in Neurod1CKO compared to the Control clusters (Fig. 6b).
Fig. 6. Molecular identity of the β-cell subpopulation is altered by Neurod1 deletion.

a Two-dimensional embedding and unsupervised clustering of endocrine profiles from Control pancreatic islets revealed six cell clusters, identified by markers shown in a cropped and transformed UMAP plots: β cells (Ins1, Ins2), α cells (glucagon, Gcg), PP cells (pancreatic polypeptide, Ppy), polyhormonal cells (expressing Ins1, Ins2, Gcg, Sst, or/and Ppy), immature/transitioning β cell cluster (Ins1, Ins2), and δ cells (Sst). The detected clusters are indicated by different colors. In Neurod1CKO, endocrine profiles revealed the same six cell clusters with one additional cluster representing a molecularly different β-cell subpopulation. b Cellular composition per sample. c UMAP plot showing the distribution of endocrine cells from the Control and Neurod1CKO. d Heatmaps of differentially expressed genes between Control and Neurod1CKO in α and β cells (cut off: Padj < 0.05, log2FC = 0.6).
Our scRNA-seq of islets of Langerhans of Neurod1CKO revealed a remarkable difference in the β-cell population, which segregated into two molecularly distinct subpopulations. One β-cell subpopulation of Neurod1CKO overlapped with the β-cell population in Control animals in the UMAP space, suggesting that this β-cell subpopulation, which shares molecularly similarities with the Control β-cell subpopulation, is responsible for the survival of Neurod1CKO mutants. The second β-cell subpopulation exhibited a loss of Ins1 and Neurod1 and therefore, was further investigated to gain insights into the molecular characteristics of β cells lacking Neurod1. The relative proportions of endocrine subtypes were altered in Neurod1CKO with a reduced proportion of α and PPY cells, and increased proportions of β-cell subpopulations compared to endocrine subpopulations in the Control (Fig. 6b). A noticeable shift in the distribution of α-cell clusters indicates the molecular identities of α cells were also different between Neurod1CKO and Control (Fig. 6c). Other mutant endocrine cells were clustered into the similar subgroups as those observed in the Control in the UMAP space (Fig. 6c).
We next compared the gene profiles of Neurod1-negative cell subtypes in Neurod1CKO with Control endocrine cells. To reveal changes in expression patterns underlying the distinct molecular shifts of α- and β-cell subpopulations in Neurod1CKO, we performed a differential gene expression analysis. By comparing the single-cell transcriptomes of Neurod1CKO Neurod1negative α and β cells with Neurod1positive cells of the Control, we observed clear differences in gene expression (Fig. 6d, Supplementary Data 2), indicating that Neurod1 deletion had a significant impact on the global expression profiles of both endocrine cell subtypes.
The most significant transcriptional differences were observed between β cells of the Control and Neurod1CKO. In the Neurod1CKO β-cell subpopulations, we observed a downregulation of genes associated with the insulin secretion machinery and hormone production, such as Ins1, Ins2, G6pc2, and Ffar1. These findings are consistent with the gene expression changes we detected in our bulk-RNA-seq analysis of E15.5 endocrine cells. Importantly, our scRNA-seq analysis focused on adult β cells, which allowed us to better discern the molecular differences between the altered matured, immature, and healthy β-cell states. Surprisingly, the expression of key maturation markers of β cells such as Slc2a2 and Ucn3156,157 in Neurod1CKO was comparable to Control β-cell subpopulations, except for the absence of Ucn3 in a smaller subcluster of adult β cells in Neurod1CKO (indicated by the arrowhead in Fig. 7). However, there was a notable increase in the expression of an immature β-cell marker, Mafb (Fig. 7b). Mafb is a transcription factor that serves as an immature β-cell marker highly expressed during the development of pancreatic α and β cells, but its expression becomes restricted to α cells in adult mice158,159. Other upregulated markers of immature β cells included retinol binding protein 4 (Rbp4)62 and a pan-endocrine lineage marker gene, Chga125. Rbp4-rich cells in the adult pancreas represent dedifferentiated β cells with reduced functionality157,160. Our analysis revealed an enrichment of Wnt4 in Neurod1CKO, which may indicate an adaptive response by the β cells to enhance their metabolic maturation161 (Fig. 7). Additionally, we observed the upregulation of key transcription factors for insulin gene expression and β-cell survival, Isl1, Nkx6.1, and Pdx161,162–164. These finding suggest a compensatory response by the dysfunctional β cells in Neurod1CKO to overcome the absence of Neurod1 and maintain their survival and functional integrity (Fig. 7 and Supplementary Fig. 5b). It is interesting to note that this contrasts with previous reports where these transcription factors were significantly reduced in diabetic islets, indicating a loss of β-cell identity under glucotoxic conditions157,165,166. Nevertheless, metabolic stress in failing pancreatic β cells in Neurod1CKO was evident by the upregulation of metabolic genes associated with glutathione and lipid metabolism, glycolysis, and protein processing in the ER. These included lactate dehydrogenase A (Ldha), free fatty acid receptor (Ffar4), mitochondrial fatty acid beta-oxidation enzyme (Hadh), the ER-resident sterol O-acyltransferase (Soat1), and glutathione peroxidase 1 and 3 (Gpx1, Gpx3) (Fig. 7, Supplementary Fig. 5b, and Supplementary Data 2).
Fig. 7. Molecular reprograming of Neurod1CKO endocrine cells.

a UMAP visualization (in two dimensions [UMAP_1 and UMAP_2]) of endocrine cells from the adult Control and Neurod1CKO pancreas (different colors mark distinct cell types annotated based on the expression of signature genes; each dot represents one cell). Clusters corresponding to α, β, PP (γ), δ, polyhormonal cells, immature β, and β cells not expressing Neurod1 (in Neurod1CKO) are shown in cropped and transformed UMAP plots that excludes a cluster of β cells expressing Neurod1 in Neurod1CKO plots for easier visualization. b Feature plots showing differentially expressed genes between Control and Neurod1CKO endocrine cells.
Consistent with the findings from our bulk-RNAseq analysis of E15.5 endocrine cells, our single-cell transcriptomic analysis confirmed the upregulation of non-endocrine genes in Neurod1CKO adult endocrine cells. These included the ductal marker Sox992, the WNT secretion mediator Wls93,94, Ocln96, Cdh4103, and the acinar-reprogramming factor Mecom111 (Fig. 7, Supplementary Fig. 5b). These expression changes suggest a shift in the cellular identity and a compromised commitment to the endocrine lineage in the Neurod1CKO adult endocrine cells.
Remarkably, we identified an upregulation of lineage-determining factors and regulators of Notch-signaling regulators, Nfia143 and Rbpj140,141, in Neurod1CKO β cells (Fig. 7). A significant enrichment of NFIA and RBPJ binding sites within the promoters of downregulated genes from the bulk-RNAseq data was revealed in our HOMER analysis (Fig. 4k). These findings further support the involvement of these factors in the transcriptional dysregulation observed in the Neurod1CKO β cells and suggests their potential role in mediating the downstream effects of Neurod1 deficiency.
Furthermore, our scRNA-seq analysis uncovered changes in gene expression patterns in Neurod1CKO that have not yet been investigated in the context of developing endocrine cells, dysfunctional β cells, or diabetic islets. For example, we found the enrichment of the transcriptional corepressor Tle4, which has been implicated in various pathways and transcription factor interactions, including Wnt and Notch signaling167; upregulation of citron rho-interacting kinase (Cit), which is known to play roles in cell division, proliferation, and neuronal development168; and increased levels of the transcription factor Nfix that plays multiple roles in cell fate determination, differentiation, cell migration, and oxidative stress169–171. Conversely, we observed a marked reduction in the expression of the cadherin gene Cadh8 in Neurod1CKO.
Altogether these findings provide additional evidence of cellular reprogramming and dysregulation occurring at the single-cell level within both α- and β-cell populations and offer valuable insights into the molecular mechanisms underlying the deficiency of Neurod1.
Discussion
In this study, we identified a previously unrecognized function of NEUROD1 during the differentiation of pancreatic endocrine cells. Our results suggest that NEUROD1-induced changes in the transcriptome and epigenome reinforce endocrine cell fate commitment and promote endocrine differentiation (Summary in Fig. 8). We demonstrated that NEUROD1 is not required for α-cell generation during the primary transition of pancreas development, as the percentage of generated GCG-producing α cells during the primary transition are comparable between Neurod1ST and Control. Although the “first wave” of α-cell generation seems to be unaffected in Neurod1ST, many of these cells co-expressed PDX1, indicating abnormalities in the pancreatic lineage differentiation pathway. Consistent with previous studies44,45, we confirmed an impaired proliferative expansion of α and β cells lacking NEUROD1 in the Neurod1ST perinatal pancreas. Although previous studies suggested that NEUROD1 is a dominant factor for the maturation of β cells and Ins1 expression44,45,48, there are no mechanistical investigations to date on NEUROD1 requirements for differentiation programs of endocrine cells.
Fig. 8. NEUROD1 function during endocrine cell differentiation.

Our study reveals dysregulated transcriptome and altered histone methylation patterns in Neurod1ST endocrine cells. Compared to the Control islets of Langerhans, the Neurod1ST endocrine cells exhibit reduced expression of endocrine-related genes and enrichment of non-endocrine genes associated with the exocrine system and pancreatic ductal cells. These findings emphasize the critical role of NEUROD1 in governing gene networks that determine the commitment and terminal differentiation of endocrine cells in the pancreas. Created with BioRender.com.
Our transcriptomic analyses of endocrine cells investigated the secondary transition of endocrine differentiation at E15.5, when endocrine progenitors have a higher potential to form β cells125. At E15.5, most endocrine progenitors come from the bipotent trunk epithelium, and it is plausible that the differentiation of endocrine progenitors is reinforced by changes in the transcriptome and epigenome. Accordingly, Neurod1 elimination in Neurod1ST resulted in downregulation of transcription factor networks driving endocrine differentiation, maintaining processes associated with endocrine functions, and insulin expression, including Insm1, Isl1, and Mafa49,122,124,162. In addition, the expression of chromatin remodelers was changed in Neurod1ST endocrine cells, the ubiquitous Smarca4 and EPs-distinct Smarcd2 were upregulated, while Kdm1b126, and α-cell enriched Smarca1127 were downregulated. Strikingly, we identified the upregulation of non-endocrine genes in Neurod1ST, including ductal Sox992 and its cooperator Maf131,132, or acinar-reprogramming Mecom111. The characterization of enriched transcription factor binding motifs within promoters of downregulated genes showed a distinct enrichment for endocrine lineage-determining transcription factors, NFIA and RBPJ140,141,143, and endocrine lineage specific transcription factors, ISL1 and NEUROD1, correlating with Neurod1 deficiency and changes in transcription signatures of Neurod1ST endocrine cells. Among enriched transcription factor binding motifs in the upregulated gene promoters were the Notch effector HES1 that controls cell fate bipotent progenitors151, ELF5 associated with the trunk epithelium of the pancreas29, and HNF4a important for the expression of early pancreatic progenitor genes148. These results suggest that NEUROD1 reinforces the downstream transcriptional network of bipotent progenitor cells toward endocrine lineage differentiation preferentially towards β cells in the secondary transition of the E15.5 pancreas.
Like the transcriptome changes, we found a corresponding shift in the chromatin landscape of H3K4me3 and H3K27me3 modifications in Neurod1ST pancreatic endocrine cells. The silencing H3K27me3 modification was detected in the gene promoter regions of Isl1, Insm1, and Mafa genes corresponding to the downregulation of mRNA of these genes in Neurod1ST. In contrast, the upregulation of non-endocrine mRNA, Sox9, Vcan, and Ocln, in Neurod1ST was associated with a missing H3K27me3 silencing modification in Neurod1ST compared to Control endocrine cells. These data support an epigenetic regulatory role of NEUROD1 in the differentiating endocrine cells in the pancreas and highlight more robust requirements of NEUROD1 for the formation of fully functional endocrine cells.
In line with our bulk RNA-seq and epigenetic analyses, the single-cell profiling of endocrine cells from the adult pancreatic islets demonstrated that β-cell identity and function were dramatically altered by the embryonic deletion of Neurod1. The transcriptional β-cell states of Neurod1CKO represented a mixture of upregulated immature (Mafb, Chga, Wls, Nfia, Rbpj, Ldha) and mature β-cell marker genes (Slc2a2, Ucn3) together with upregulated genes representing non-endocrine fate choices (Sox9, Cdh4, Ocln, Mecom). Interestingly, we found a higher proportion of β cells co-expressing Sst and Ins2 and/or Ppy, as well as a higher proportion of polyhormonal cells within the Neurod1CKO endocrine subpopulations. The ability of NEUROD1 to govern cellular and fate choice identities is not without precedent. Several studies highlighted the significance of NEUROD1 in promoting neuronal fate and preserving neuronal identity by suppressing non-neuronal fates33–35.
In the context of β-cell identities, Neurod1 deficiency affected numerous genes that serve as markers for β-cell dysfunction and the diabetes-induced dedifferentiation of β-cells157,160. These genes play crucial roles in insulin secretion, metabolism, and maturity. For instance, RBP4 is known as a diabetogenic factor157,160 but is also expressed in immature β-cells during embryonic development62. Likewise, LDHA, a glycolytic enzyme, is normally suppressed in mature β-cells since it is critical for coupling glucose metabolism with insulin secretion. In contrast to reports of diabetes-induced dedifferentiated β cells157,166, we found an increased expression of transcription factors associated with β-cell identity and the regulation of insulin genes, such as Pdx1, Isl1, and Nkx6.1, indicating a compensatory regulatory response aimed at overcoming the absence of Neurod1. Similarly, an enrichment of Wnt4 and Ffar4 in Neurod1CKO may indicate an adaptive response by the dysfunctional β cells to enhance their metabolic maturation and stimulate failing insulin secretion161,172.
Recent findings have provided compelling evidence supporting the existence of transcriptional and functional heterogeneity among insulin-secreting β cells within the context of normal islet homeostasis. This heterogeneity encompasses a range of cellular states and subpopulations, highlighting the diverse characteristics of these cells62,136,160. Epigenetic silencing represents an important axis of β-cell heterogeneity173. Importantly, under conditions of metabolic stress, these subpopulations undergo dynamic changes that lead to functional impairments. An intriguing area for future investigation is whether Neurod1-deficient β cells, which exhibit a mixture of mature, immature, and non-endocrine identities, represent a subpopulation that possesses inherent plasticity and adaptive capacity to sustain chronic metabolic stress. Understanding such a heterogeneous transcriptional regulatory landscape, featuring rigidity and flexibility in expression across different gene families, would provide valuable insights into the functional diversity and adaptability of β cells.
Based on our transcriptomic and epigenetic analyses, we propose two regulatory roles of NEUROD1. Firstly, NEUROD1 controls the functional differentiation of insulin-producing β cells. Despite the perturbation of the β-cell differentiation program at the endocrine progenitor stage due to Neurod1 loss, adult Neurod1-deficient β cells of Neurod1CKO still express key transcription factors associated with β-cell identity and insulin gene transcription, such as Pdx1, Isl1, and Nkx6.1. Our investigation of the maturation characteristics of adult β cells in Neurod1CKO revealed a combination of immature and mature states. This is evident from the enrichment of glycolytic genes (Ldha and Eno1), the pan-endocrine gene (Chga), and immature β-cell markers (Mafb and Rbp4), which resemble immature fetal or neonatal β cells. Interestingly, the expression of mature β-cell marker genes (Slc2a2 and Ucn3) in Neurod1CKO was similar to the Control, while the upregulation of Wnt4 suggests an attempt to enhance β-cell functional maturation.
Secondly, NEUROD1 plays a role in reinforcing endocrine commitment during the differentiation of endocrine progenitors. This is supported by the upregulation of non-endocrine genes, including Sox9, Cdh4, Ocln, and Mecom, observed in Neurod1ST. Endocrine cells primarily originate from Sox9+ bipotent progenitors in the trunk epithelium174. These progenitors either differentiate into endocrine cells or ducts12. SOX9 triggers endocrine differentiation by inducing Neurog3 expression92,174, and Neurod1 expression follows Neurog325–28. NEUROD1 expression was also found in transitioning Sox9low cells43. Furthermore, ectopic expression of NEUROD1 induces endocrine differentiation in the pancreas, demonstrating its pro-endocrine role25. Similarly, in Neurod1CKO adult β cells, there is an upregulation of non-endocrine genes, indicating permanent changes in β-cell identities. Moreover, the increased proportions of bihormonal and polyhormonal β cells in Neurod1CKO suggest a potential involvement of NEUROD1 in controlling monohormonal β-cell state.
Taken together, our findings provide insights into the regulatory roles of NEUROD1 in promoting the functional differentiation of β cells. Our data suggest that NEUROD1 reprograms the transcription factor and epigenetic landscapes, favoring the differentiation of endocrine cells and facilitating the generation of functional insulin-producing β cells. Our study, combined with the well-established role of NEUROD1 in neuronal differentiation, emphasizes the broader significance of NEUROD1 as an epigenetic and transcriptional regulator in embryonic development.
Limitations of the study
One limitation of our analysis is the lack of spatial resolution. By single-cell dissociation, information regarding intra-islet connectivity, the distribution of β-cell subtypes, and regional functional differences within the pancreas or islets is lost. Additionally, we did not explore the temporal gene expression kinetics, which represents another important aspect that could enhance our understanding of pancreatic development. Investigating the dynamic changes in gene expression patterns over time would provide insights into the molecular events and regulatory mechanisms involved in the formation of the Neurod1-deficient endocrine pancreas. The study also does not identify direct NEUROD1 target genes during the formation of endocrine cells in the pancreas. Finally, the main conclusions of the current study are based on a mouse in vivo model, although the findings should translate to human pancreas development, this has not been confirmed.
Methods
Ethics approval and consent to participate
Experiments were carried out following the animal welfare guidelines 2010/63/EC of the European Communities Council Directive, agreeing with the Guide for the Care and Use of Laboratory Animals (National Research Council. Washington, DC. The National Academies Press, 1996). The design of experiments was approved by the Animal Care and Use Committee of the Institute of Molecular Genetics, Czech Academy of Sciences (protocol # 878/2022).
Experimental model
Animal experiments were conducted according to protocols approved by the Animal Care and Use Committee of the Institute of Molecular Genetics, Czech Academy of Sciences. All experiments were performed with littermates (males and females) cross-bred from two transgenic mouse lines: floxed Neurod1 [Neurod1loxP/loxP, 58], and Neurod1Cre/+ [Tg(Neurod1-Cre)1Able, Stock No: 028364 Jackson Laboratory59]. The lines were maintained on the C57BL/6 background. Neurod1Cre/+ mice do not have any detectable phenotype. Breeding scheme: Female mice Neurod1loxP/loxP were crossed with Neurod1loxP/+; Neurod1Cre/+ males, in which the Neurod1-Cre knock-in allele was inherited paternally to minimize the potential influence of maternal genotype on the developing embryos. Neurod1loxP/+ or Neurod1loxP/loxP without the Neurod1-Cre allele individuals were used as the Control. The Het mice of the Neurod1loxP/+; Neurod1Cre/+ genotype, containing one functional Neurod1 allele, were functionally and phenotypically similar to the Control. The reporter line (Ai14, B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze, Stock No: 7914 Jackson Laboratory) was used, generating Control-Ai14 (Neurod1loxP/+; Neurod1Cre/+; tdTomatoloxP/+) and Neurod1ST-Ai14 (Neurod1loxP/loxP; Neurod1Cre/+; tdTomatoloxP/+). For scRNA-seq, we used mice with a conditional deletion of Neurod1, Neurod1CKO, with Neurod1loxP/loxP;Isl1Cre/+ genotype45. The mice with the genotype Neurod1loxP/+ or Neurod1loxP/loxP without the Isl1-Cre allele were used for the preparation of the Control pancreatic cells. Genotyping was performed by PCR on tail DNA (Supplementary Table 1). Mice were kept under standard experimental conditions with a constant temperature (23–24 °C) and fed the standard diet (#1324, Altromin International, Germany). The females were housed individually during the gestation period, and the litter size was recorded. Blood glucose levels were measured in animals by glucometer (COUNTOUR TS, Bayer); blood glucose levels maintained above 13.9 mmol/L are classified as diabetic. For total pancreatic insulin content, pancreases were excised, weighed, minced, and homogenized in acid-ethanol. Hormone concentration in extracts was measured by ELISA using Mouse Insulin ELISA kit (Mercodia, Sweden).
Reverse transcription-quantitative real-time polymerase chain reaction
RT-qPCR was performed as described previously175. Briefly, total RNA was isolated from the whole pancreas at E15.5 by Trizol RNA extraction. The Hprt1 gene was selected as the best reference gene for our analyses from a panel of 12 control genes (TATAA Biocenter AB, Sweden). Primers were designed using Primer-BLAST176. Primer sequences are presented in Supplementary Table 2.
Immunohistochemical staining and morphological evaluations
For vibratome sections, dissected tissues were fixed in 4% PFA, embedded in 4% agarose gel and sectioned at 80 µm on a Leica VT1000S vibratome. Used primary and secondary antibodies are in Supplementary Table 3. The nuclei were in specific cases counterstained with Hoechst 33342. Apoptotic cells were labeled by TUNEL (#11684795910, Roche). Image acquisition was completed using the Zeiss LSM 880 NLO scanning confocal microscope with ZEN lite program. The expression of NEUROD1 was quantified in the whole pancreases of Control and Neurod1ST at E10.5 using the thresholding tool of the ImageJ, and the results were expressed as a percentage of the NEUROD1+ area per tdTomato+ area. The numbers of GCG+, INS+, and Ki67+ cells were counted in all viewing areas of a vibratome section from the central part of the pancreas with the largest pancreatic footprint from Neurod1ST and Control embryos or pups. The results for GCG proliferating and INS proliferating cells were expressed as a percentage of the total number of GCG+ and INS+ cells per evaluated area of the pancreas. The Cell Counter plugin of ImageJ177 was used for quantification. GCG+ and INS+ cells were counted in a vibratome section from the central part of the pancreas with the largest pancreatic footprint (n = 5 pancreases/genotype in total 24 areas of view/Control and 22/areas of view/Neurod1ST). The expression of SST was quantified in all viewing areas of a vibratome section from the central part of the pancreas with the largest pancreatic footprint in Neurod1ST-Ai14 and Control-Ai14 pups. The thresholding tool in ImageJ was utilized for quantification, and the results were expressed as a percentage of SST+ areas per the total area of tdTomato+ per evaluated area of the pancreas. The evaluation of GCG+ delaminating cells at E10.5 was quantified using the thresholding tool in ImageJ and the results were expressed as a percentage of the total GCG+ area to PDX1+ area. GCG+ cells co-expressing PDX1 were quantified in the entire pancreas at E10.5. Quantification was performed using the Cell Counter plugin of ImageJ, the results were expressed as a percentage of GCG+ PDX1+ cells per total GCG+ cells. The thresholding tool in ImageJ was utilized for the evaluation of tdTomato+ cells, which did not express GCG or INS in the Neurod1ST-Ai14 and Control-Ai14 pancreas at E15.5. The expression of tdTomato was quantified in all viewing areas of a vibratome section from the central part of the pancreas with the largest pancreatic footprint. The results were expressed as a percentage of GCGnegative and INSnegative areas per the tdTomato+ areas.
Light-sheet fluorescent microscopy (LFSM) and analysis of images
The pancreas was microdissected from Control-Ai14 and Neurod1ST-Ai14 embryos (E18.5) and tissue was processed as described previously178. In addition to tdTomato expression, cleared samples were immunolabeled using anti-insulin and anti-GLP1 antibodies (Supplementary Table 4). Zeiss Lightsheet Z.1 microscope with illumination objective Lightsheet Z.1 5x/0.1 and detection objective Dry objective Lightsheet Z.1 5x/0.16 was used for imaging at the Light Microscopy Core Facility of the Institute of Molecular Genetics of the Czech Academy of Sciences. IMARIS software v8.1.1 (Bitplane AG, CA, USA) was used for image processing.
Isolation of pancreatic endocrine cells
The pancreases microdissected from E15.5 embryos were directly trypsinized to generate single cells and prepared for FACS as described61,179. Cell suspensions were filtered through 40-μm nylon mesh and tdTomato+ cells were immediately sorted using a flow cytometer (BD FACSAriaTM Fusion), through a 100 μm nozzle in 20 psi, operated with BD FACSDiva™ Software. For RNA sequencing, 100 cells per sample were collected into the individual wells of 96-well plate containing 5 μl of lysis buffer of NEB Next single-cell low input RNA library prep kit (New England Biolabs #E6420). For CUT&Tag profiling study, ~5000-20000 tdTomato+ cells per sample were sorted. The total time from euthanasia to cell collection was ~3 hrs. Cell sorting was performed in the Imaging Methods Core Facility at BIOCEV.
Bulk-cell RNA sequencing and analyses
Using NEB Next single-cell low input RNA library prep kit for Illumina (New England Biolabs #E6420), RNA-seq libraries were prepared from the pancreases of Neurod1ST-Ai14 (n = 4) and Control-Ai14 (n = 4) by following the manufacturer’s instructions. The libraries were sequenced on an Illumina NextSeq 500 next-generation sequencer. NextSeq 500/550 High Output kit 75 cycles (Illumina #200024906) were processed at the Genomics and Bioinformatics Core Facility (Institute of Molecular Genetics CAS, Czechia). RNA-Seq reads in FASTQ files were mapped to the mouse genome using STAR [version 2.7.0c180] GRCm38 primary assembly and annotation version M8. The raw data of RNA sequencing were processed with a standard pipeline. Using cutadapt v1.18181, the number of reads (minimum, 23.4 million; maximum, 27.5 million) was trimmed by Illumina sequencing adaptor and of bases with reading quality lower than 20, subsequently reads shorter than 20 bp were filtered out. Ribosomal RNA and reads mapping to UniVec database were filtered out using bowtie v1.2.2. with parameters -S -n 1 and SortMeRNA database182. A count table was generated by Rsubread v2.0.1 package using default parameters without counting multi-mapping reads.
DESeq2 [v1.26.0183] default parameters were used to normalize data and compare the different groups. Genes were then filtered using the criteria: adjusted P-value Padj < 0.05, and a base mean ≥ 50, and Fold change > 1.3 and <0.7. Only protein coding genes were kept in the analysis, following Mouse Genome Database184. The functional enrichment analysis was performed using: g:Profiler (ver. e106_eg53_p16_65fcd97) with g:SCS multiple testing correction method applying significance threshold of 0.0571; and MouseMine (http://www.mousemine.org/), using Mammalian Phenotype Ontology185 and Mouse Anatomy and Development Ontology186. De novo motif analysis was performed by HOMER [ver. 4.10139] using default settings; up to top 5 scoring targets were considered per a detected motif.
Deconvolution of endocrine cell subtypes for bulk-cell RNA sequencing data
Deconvolution was performed using the CibersortX algorithm at cibersortx.stanford.edu187. Single-cell transcriptomic profiling dataset of cells in the embryonic pancreas137 was used as reference, including count matrix and metadata labels. The reference matrix was built out of the 2589 cells and gene list of 18565 gene features, as deposited by137. Every cell population counted over 250 cells. The units of reference matrix were UMI counts. Calculation of scRNA-seq signature matrix was done in default mode (quantile normalization disabled, minimal expression of 0.75, replicates of 5, sampling of 0.5). Imputation of cell fractions and high-resolution gene expression imputation were calculated in the default settings, with S-mode batch correction enabled, quantile normalization disabled and n = 100 permutations for significance analysis. Sample mixture file was submitted with unfiltered gene list of 26447 features for Neurod1ST and in UMI counts.
CUT&Tag profiling
Bench top CUT&Tag version 3 was performed as previously described57,188 with minor modifications61. Briefly, nuclei from freshly FACS-sorted tdTomato+ pancreatic endocrine cells were captured by Concanavalin A-coated magnetic beads to facilitate subsequent washing steps. CUT&Tag validated primary antibodies anti-H3K4me3, anti-H3K27me3, normal rabbit IgG negative control, and anti-rabbit secondary antibody were used (Supplementary Table 5). Each purified DNA library for Illumina sequencing was tested on Agilent High Sensitivity DNA Chip. DNA libraries were sequenced at the OMICS Genomics facility (BIOCEV) on the MiSeq (Illumina) using MiSeq Reagent Kit v3 (Illumina), which allows extended read lengths up to 2x 75 bp. Both CUT&Tag-seq. H3K27me3 and H3K4me3 data are from one independent biological replicate, each sample was pooled together from 3 to 7 pancreases.
Data analysis was performed following CUT&Tag Data processing tutorial189. Paired-end sequencing data were mapped using bowtie2 (ver. 2.2.5)190 to mouse genome GRCm38 primary assembly. PCR duplicates were not removed. Reads mapping to SNAP-CUTANA barcodes were counted using bash shell tool grep. For visualization bam files were normalized using scale factor = “% genome reads” / “% spike-in reads” and converted to bigwig format using bedtools (ver. 2.30.0). Factors are available in Supplementary Table 6. After filtering and conversion to bedgraph format, peak calling was performed with usage of relevant IgG controls and stringent mode with tool SEACR (ver. 1.3)191. Peaks were annotated using CHIPseeker (ver. 1.30.3)192 and annotation version M8 of mouse genome. NEUROD1 motifs were identified in mm10 mouse genome by HOMER (ver. 4.10)139 using internal NeuroD1(bHLH)/Islet-NeuroD1-ChIP-Seq (GSE30298) motif file and converted to bedfile. Data were visualized in IGV (ver. 2.12.3)193 and final images were generated with UCSC browser194,195 as mean values.
Single-cell RNA sequencing and analysis
We used adult mice Neurod1CKO (genotype: Neurod1loxP/loxP;Isl1Cre/+) and Control (Neurod1loxP/+ or Neurod1loxP/loxP genotype from the same breeding) to isolate pancreatic islets. Mouse pancreatic islets were isolated (and pooled) from four mice per each genotype by perfusion of the common bile duct with Collagenase P (1 mg/ml; Merck) in HBS (Merck) supplemented with Actinomycin D (25 µg/ ml; Merck) to stop transcription, digestion of the pancreases with 0.8 mM Collagenase P and purification of the islets by Ficoll discontinuous gradient. Single cell dispersion was carried out as follows: islets were settled by centrifugation (140g, 4 °C, 4 min), washed with 2 ml of HBS with Actinomycin D (2.5 µg/ ml). Islets were centrifuged (140g, 4 °C, 4 min) again and Dulbecco’s PBS was used for the final wash. Digestion was performed with 0.6 ml of trypsin/0.53 mM EDTA179 supplemented with Actinomycin D (25 µg/ ml). Samples were incubated at 37 °C for 5 min. Following incubation, trypsinization was stopped by adding of 1.2 ml of PBS with 10 mM EGTA supplemented with 10% fetal bovine serum (FBS; Merck). Cells were gently dispersed mechanically using a P1000 pipette. Cells were then spun down (800 × g, 4 °C, 10 min) and resuspended in PBS with 10 mM EGTA supplemented with 2% FBS. Single-cell suspension was used for library preparation: Chromium Next GEM Single Cell 3’ Reagent Kits v3.1 (10 × Genomics, Pleasanton, CA) was used to prepare the sequencing libraries, according to the manufacturer’s protocols. Briefly, 10 × Chromium platform was used to encapsulate individual cells into droplets along with beads covered in cell-specific 10 × Barcodes, unique molecular identifiers (UMIs) and poly(dT) sequences. After reverse transcription, the cDNA libraries were amplified (13–14 cycles), fragmented and ligated to sequencing adaptors. SPRISelect magnetic beads were used for purification of the cDNA suspension and size selection of the fragments. The concentration and quality of the libraries were measured using Qubit dsDNA HS Assay Kit (Invitrogen) and Fragment Analyzer HS NGS Fragment Kit (#DNF-474, Agilent). The libraries were pooled and sequenced in paired-end mode using Illumina NovaSeq 6000 SP Reagent Kit.
Single-cell sequencing data was aligned to a custom reference mouse genome GRCm38 containing the active transcript sequence of tdTomato. Aligned reads were annotated by STARsolo (STAR version 2.7.3a)180 to the custom GENCODE version M8 annotation containing the transcript information of tdTomato. Seurat R package (version 4.3.0)196 was used to visualize, cluster and annotate clusters on SCTransformed and integrated data. EmptyDrops function (DropletUtils R package)197 was applied to preserve only cell-containing droplets. DoubletFinder198 R package was used for the identification of doublets. Seurat functions FindMarkers and DoHeatmap were used for differential expression analysis and heatmaps production respectively. Proportions of cell types were extracted with Prop.table function.
Experimental design, statistics and reproducibility
All comparisons were made between animals with the same genetic background, typically littermates, and we used male and female mice. The number of samples (n) for each comparison are given in the corresponding figure legends. Phenotyping and data analysis were performed blind to the genotype of the mice. All values are presented either as the mean ± standard deviation (SD) or standard error of the mean (SEM). For statistical analysis, GraphPad Prism (ver. 9.3.1) software and RStudio (ver. 2022.7.1.554) were used. To assess differences in the mean, one-way ANOVA with Tukey’s multiple comparison test, and unpaired two-tailed t tests were employed. A P value < 0.05 denoted the presence of a statistically significant difference. Significance was determined as P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), P < 0.0001 (****). The complete results of the statistical analyses are included in the figure legends. Each experiment presented was independently repeated a minimum of three times, unless otherwise specified.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
This research was supported by the Czech Science Foundation (GA19-07378S and GA22-11516S to GP), and by the institutional support of the Czech Academy of Sciences (RVO: 86652036 to GP). We thank Prof. Lori Sussel (University of Colorado) for reading and commenting of the revised MS, and A. Pavlinek (King’s College London) for editing the MS. We thank M. Anderova for providing tdTomato-Ai14 reporter line. We acknowledge Imaging Methods Core Facility at BIOCEV supported by the MEYS CR (Large RI Project LM2018129 Czech-BioImaging) and ERDF (project No. CZ.02.1.01/0.0/0.0/18_046/0016045) for its support with obtaining imaging data and FACS experiments presented in this paper; the Light Microscopy Core Facility of the Institute of Molecular Genetics of the Czech Academy of Sciences for its support for LFSM generated 3D images; Biocev GeneCore Facility for its support with gene expression/transcriptome analyses; and Biocev Animal facility (LM 2018126 Czech Centre for Phenogenomics by MEYS OP RDE CZ.02.1.01/0.0/0.0/18_046/0015861 CCP Infrastructure Upgrade II by MEYS and ESIF).
Author contributions
G.P., R.B., and F.S. conceived and designed the study; R.B., V.F., O.S., and Z.B., performed experiments; R.B., and V.F. performed 3D visualization; V.F., P.A., and L.L-M performed single-cell RNA-seq analyses; S.B., L.V., O.S., and D.Z. performed bioinformatic analyses of bulk-RNA-seq and CUT&Taq-seq; O.S. wrote the first original draft; R.B., and V.F. contributed to the manuscript review & editing; G.P. wrote the manuscript and obtained research funding.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
All raw sequencing data have been deposited at NIH GEO. The bulk RNA-seq and Cut&Tag-seq data are under accession number GSE212084 and the single-cell RNA-seq data are under accession number GSE236559. Analyzed data are in Supplementary Data 1 and Data 2. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Romana Bohuslavova, Valeria Fabriciova, Ondrej Smolik.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-41306-6.
References
- 1.Röder PV, Wu B, Liu Y, Han W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016;48:e219–e219. doi: 10.1038/emm.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Frances M, et al. Adult pancreatic islet endocrine cells emerge as fetal hormone-expressing cells. Cell Rep. 2022;38:110377. doi: 10.1016/j.celrep.2022.110377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandian GN, Taniguchi J, Sugiyama H. Cellular reprogramming for pancreatic β-cell regeneration: clinical potential of small molecule control. Clin. Transl. Med. 2014;3:6–6. doi: 10.1186/2001-1326-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim H-S, Lee M-K. β-Cell regeneration through the transdifferentiation of pancreatic cells: pancreatic progenitor cells in the pancreas. J. Diabetes Investig. 2016;7:286–296. doi: 10.1111/jdi.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez Fallas ME, et al. Stem/progenitor cells in normal physiology and disease of the pancreas. Mol. Cell Endocrinol. 2021;538:111459–111459. doi: 10.1016/j.mce.2021.111459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isaacson A, Spagnoli FM. Pancreatic cell fate specification: insights into developmental mechanisms and their application for lineage reprogramming. Curr. Opin. Genet. Dev. 2021;70:32–39. doi: 10.1016/j.gde.2021.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Arda HE, Benitez CM, Kim SK. Gene regulatory networks governing pancreas development. Dev. Cell. 2013;25:5–13. doi: 10.1016/j.devcel.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jennings RE, Scharfmann R, Staels W. Transcription factors that shape the mammalian pancreas. Diabetologia. 2020;63:1974–1980. doi: 10.1007/s00125-020-05161-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szlachcic WJ, Ziojla N, Kizewska DK, Kempa M, Borowiak M. Endocrine pancreas development and dysfunction through the lens of single-cell RNA-sequencing. Front Cell Dev. Biol. 2021;9:629212. doi: 10.3389/fcell.2021.629212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Q, et al. A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell. 2007;13:103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Jennings RE, Berry AA, Strutt JP, Gerrard DT, Hanley NA. Human pancreas development. Development. 2015;142:3126–3137. doi: 10.1242/dev.120063. [DOI] [PubMed] [Google Scholar]
- 12.Shih HP, et al. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villasenor A, Chong DC, Cleaver O. Biphasic Ngn3 expression in the developing pancreas. Dev. Dyn. 2008;237:3270–3279. doi: 10.1002/dvdy.21740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dassaye R, Naidoo S, Cerf ME. Transcription factor regulation of pancreatic organogenesis, differentiation and maturation. Islets. 2016;8:13–34. doi: 10.1080/19382014.2015.1075687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jennings RE, et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes. 2013;62:3514–3522. doi: 10.2337/db12-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrera PL, et al. Embryogenesis of the murine endocrine pancreas; early expression of pancreatic polypeptide gene. Development. 1991;113:1257–1265. doi: 10.1242/dev.113.4.1257. [DOI] [PubMed] [Google Scholar]
- 17.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl Acad. Sci. USA. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreiber V, et al. Extensive NEUROG3 occupancy in the human pancreatic endocrine gene regulatory network. Mol. Metab. 2021;53:101313. doi: 10.1016/j.molmet.2021.101313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaimala S, Kumar CA, Allouh MZ, Ansari SA, Emerald BS. Epigenetic modifications in pancreas development, diabetes, and therapeutics. Med. Res. Rev. 2022;42:1343–1371. doi: 10.1002/med.21878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dumasia NP, Pethe PS. Pancreas development and the Polycomb group protein complexes. Mech. Dev. 2020;164:103647. doi: 10.1016/j.mod.2020.103647. [DOI] [PubMed] [Google Scholar]
- 21.Ding Y, et al. JMJD3: a critical epigenetic regulator in stem cell fate. Cell Commun. Signal. 2021;19:72. doi: 10.1186/s12964-021-00753-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voigt P, Tee W-W, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shpargel KB, Starmer J, Yee D, Pohlers M, Magnuson T. KDM6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet. 2014;10:e1004507. doi: 10.1371/journal.pgen.1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harikumar A, Meshorer E. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 2015;16:1609–1619. doi: 10.15252/embr.201541011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwitzgebel VM, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127:3533–3542. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 26.Huang HP, et al. Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol. Cell Biol. 2000;20:3292–3307. doi: 10.1128/mcb.20.9.3292-3307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon J, Correa-Medina M, Ricordi C, Edlund H, Diez JA. Endocrine Cell Clustering During Human Pancreas Development. J. Histochem. Cytochem. 2009;57:811–824. doi: 10.1369/jhc.2009.953307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasa R, et al. Induction of pancreatic islet cell differentiation by the neurogenin-neuroD cascade. Differentiation. 2008;76:381–391. doi: 10.1111/j.1432-0436.2007.00228.x. [DOI] [PubMed] [Google Scholar]
- 29.Krentz NAJ, et al. Single-cell transcriptome profiling of mouse and hESC-derived pancreatic progenitors. Stem Cell Rep. 2018;11:1551–1564. doi: 10.1016/j.stemcr.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen J, et al. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49:163–176. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- 31.Yu X-X et al. Dynamics of chromatin marks and the role of JMJD3 during pancreatic endocrine cell fate commitment. Development145 (2018). [DOI] [PubMed]
- 32.Bankaitis ED, Bechard ME, Wright CVE. Feedback control of growth, differentiation, and morphogenesis of pancreatic endocrine progenitors in an epithelial plexus niche. Genes Dev. 2015;29:2203–2216. doi: 10.1101/gad.267914.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda T, et al. Pioneer factor NeuroD1 rearranges transcriptional and epigenetic profiles to execute microglia-neuron conversion. Neuron. 2019;101:472–485.e477. doi: 10.1016/j.neuron.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 34.Pataskar A, et al. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. 2016;35:24–45. doi: 10.15252/embj.201591206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akol I, et al. Multimodal epigenetic changes and altered NEUROD1 chromatin binding in the mouse hippocampus underlie FOXG1 syndrome. Proc. Natl Acad. Sci. USA. 2023;120:e2122467120. doi: 10.1073/pnas.2122467120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Z, et al. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 2009;12:1090–1092. doi: 10.1038/nn.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filova I, et al. Early deletion of Neurod1 alters neuronal lineage potential and diminishes neurogenesis in the inner ear. Front Cell Dev. Biol. 2022;10:845461. doi: 10.3389/fcell.2022.845461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci. Res. 2006;55:223–233. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 39.Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 41.Chu K, Nemoz-Gaillard E, Tsai MJ. BETA2 and pancreatic islet development. Recent Prog. Horm. Res. 2001;56:23–46. doi: 10.1210/rp.56.1.23. [DOI] [PubMed] [Google Scholar]
- 42.Itkin-Ansari P, et al. NeuroD1 in the endocrine pancreas: localization and dual function as an activator and repressor. Dev. Dyn. 2005;233:946–953. doi: 10.1002/dvdy.20443. [DOI] [PubMed] [Google Scholar]
- 43.Mastracci TL, Anderson KR, Papizan JB, Sussel L. Regulation of Neurod1 contributes to the lineage potential of neurogenin3+ endocrine precursor cells in the pancreas. PLoS Genet. 2013;9:e1003278. doi: 10.1371/journal.pgen.1003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romer AI, Singer RA, Sui L, Egli D, Sussel L. Murine perinatal β-cell proliferation and the differentiation of human stem cell-derived insulin-expressing cells require NEUROD1. Diabetes. 2019;68:2259–2271. doi: 10.2337/db19-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bohuslavova R, et al. NEUROD1 is required for the early α and β endocrine differentiation in the pancreas. Int. J. Mol. Sci. 2021;22:6713. doi: 10.3390/ijms22136713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naya FJ, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dudek K. D., Osipovich A. B., Cartailler J-P, Gu G. & Magnuson M. A. Insm1, Neurod1, and Pax6 promote murine pancreatic endocrine cell development through overlapping yet distinct RNA transcription and splicing programs. G3 Genes|Genomes|Genetics11 (2021). [DOI] [PMC free article] [PubMed]
- 48.Gu C, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010;11:298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia S, et al. Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic β-cell function. EMBO J. 2015;34:1417–1433. doi: 10.15252/embj.201490819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malecki MT, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat. Genet. 1999;23:323–328. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- 51.Rubio-Cabezas O, et al. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes. 2010;59:2326–2331. doi: 10.2337/db10-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horikawa Y, Enya M. Genetic dissection and clinical features of MODY6 (NEUROD1-MODY) Curr. Diab. Rep. 2019;19:12. doi: 10.1007/s11892-019-1130-9. [DOI] [PubMed] [Google Scholar]
- 53.Docherty HM, et al. Relative contribution of PDX-1, MafA and E47/beta2 to the regulation of the human insulin promoter. Biochem J. 2005;389:813–820. doi: 10.1042/BJ20041891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Babu DA, Chakrabarti SK, Garmey JC, Mirmira RG. Pdx1 and BETA2/NeuroD1 participate in a transcriptional complex that mediates short-range DNA looping at the insulin gene. J. Biol. Chem. 2008;283:8164–8172. doi: 10.1074/jbc.M800336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naya FJ, Stellrecht CM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 56.Huang HP, Chu K, Nemoz-Gaillard E, Elberg D, Tsai MJ. Neogenesis of beta-cells in adult BETA2/NeuroD-deficient mice. Mol. Endocrinol. 2002;16:541–551. doi: 10.1210/mend.16.3.0784. [DOI] [PubMed] [Google Scholar]
- 57.Kaya-Okur HS, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goebbels S, et al. Cre/loxP-mediated inactivation of the bHLH transcription factor gene NeuroD/BETA2. Genesis. 2005;42:247–252. doi: 10.1002/gene.20138. [DOI] [PubMed] [Google Scholar]
- 59.Li HJ, Kapoor A, Giel-Moloney M, Rindi G, Leiter AB. Notch signaling differentially regulates the cell fate of early endocrine precursor cells and their maturing descendants in the mouse pancreas and intestine. Dev. Biol. 2012;371:156–169. doi: 10.1016/j.ydbio.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bohuslavova R, et al. ISL1 controls pancreatic alpha cell fate and beta cell maturation. Cell Biosci. 2023;13:53. doi: 10.1186/s13578-023-01003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qiu WL, et al. Deciphering pancreatic islet β cell and α cell maturation pathways and characteristic features at the single-cell level. Cell Metab. 2017;25:1194–1205.e1194. doi: 10.1016/j.cmet.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 63.Pauerstein PT, et al. A radial axis defined by semaphorin-to-neuropilin signaling controls pancreatic islet morphogenesis. Development. 2017;144:3744–3754. doi: 10.1242/dev.148684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roscioni SS, Migliorini A, Gegg M, Lickert H. Impact of islet architecture on beta-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016;12:695–709. doi: 10.1038/nrendo.2016.147. [DOI] [PubMed] [Google Scholar]
- 65.Brereton MF, Vergari E, Zhang Q, Clark A. Alpha-, delta- and pp-cells: are they the architectural cornerstones of islet structure and co-ordination? J. Histochem. Cytochem. 2015;63:575–591. doi: 10.1369/0022155415583535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Migliorini A, Bader E, Lickert H. Islet cell plasticity and regeneration. Mol. Metab. 2014;3:268–274. doi: 10.1016/j.molmet.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burlison JS, Long Q, Fujitani Y, Wright CV, Magnuson MA. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev. Biol. 2008;316:74–86. doi: 10.1016/j.ydbio.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bastidas-Ponce A, et al. Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic beta-cells. Mol. Metab. 2017;6:524–534. doi: 10.1016/j.molmet.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–1768. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duvillie B, et al. Phenotypic alterations in insulin-deficient mutant mice. Proc. Natl Acad. Sci. USA. 1997;94:5137–5140. doi: 10.1073/pnas.94.10.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raudvere U, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liew CW, et al. Insulin regulates carboxypeptidase E by modulating translation initiation scaffolding protein eIF4G1 in pancreatic β cells. Proc. Natl Acad. Sci. USA. 2014;111:E2319–E2328. doi: 10.1073/pnas.1323066111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kohara K, et al. Suppression of free fatty acid receptor 1 expression in pancreatic β-cells in obese type 2 diabetic db/db mice: a potential role of pancreatic and duodenal homeobox factor 1. Endocr. J. 2019;66:43–50. doi: 10.1507/endocrj.EJ18-0203. [DOI] [PubMed] [Google Scholar]
- 74.Millership SJ, et al. Neuronatin regulates pancreatic β cell insulin content and secretion. J. Clin. Invest. 2018;128:3369–3381. doi: 10.1172/JCI120115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Farber CR, et al. Overexpression of Scg5 increases enzymatic activity of PCSK2 and is inversely correlated with body weight in congenic mice. BMC Genet. 2008;9:34. doi: 10.1186/1471-2156-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jonsson A, et al. Effect of a common variant of the PCSK2 gene on reduced insulin secretion. Diabetologia. 2012;55:3245–3251. doi: 10.1007/s00125-012-2728-5. [DOI] [PubMed] [Google Scholar]
- 77.Choi S, Korstanje R. Proprotein convertases in high-density lipoprotein metabolism. Biomark. Res. 2013;1:27. doi: 10.1186/2050-7771-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Manialawy Y, Khan SR, Bhattacharjee A, Wheeler MB. The magnesium transporter NIPAL1 is a pancreatic islet–expressed protein that conditionally impacts insulin secretion. J. Biol. Chem. 2020;295:9879–9892. doi: 10.1074/jbc.RA120.013277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic. Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang C, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005;25:4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schulla V, et al. Impaired insulin secretion and glucose tolerance in beta cell-selective Ca(v)1.2 Ca2+ channel null mice. EMBO J. 2003;22:3844–3854. doi: 10.1093/emboj/cdg389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sadagurski M, Dong XC, Myers MG, Jr., White MF. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol. Metab. 2013;3:55–63. doi: 10.1016/j.molmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sugawara K, Shibasaki T, Takahashi H, Seino S. Structure and functional roles of Epac2 (Rapgef4) Gene. 2016;575:577–583. doi: 10.1016/j.gene.2015.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tengholm A, Gylfe E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metab. 2017;19:42–53. doi: 10.1111/dom.12993. [DOI] [PubMed] [Google Scholar]
- 85.Prentki M, Matschinsky Franz M, Madiraju SRM. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 86.Yavari A, et al. Chronic activation of γ2 AMPK induces obesity and reduces β cell function. Cell Metab. 2016;23:821–836. doi: 10.1016/j.cmet.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Loh K, et al. Inhibition of adenosine monophosphate-activated protein kinase-3-hydroxy-3-methylglutaryl coenzyme a reductase signaling leads to hypercholesterolemia and promotes hepatic steatosis and insulin resistance. Hepatol. Commun. 2019;3:84–98. doi: 10.1002/hep4.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Joshi T, et al. Targeting AMPK signaling pathway by natural products for treatment of diabetes mellitus and its complications. J. Cell Physiol. 2019;234:17212–17231. doi: 10.1002/jcp.28528. [DOI] [PubMed] [Google Scholar]
- 89.Machida Y, et al. Pancreatic islet neuropeptide Y overexpression has minimal effect on islet morphology and β-cell adaptation to high-fat diet. Endocrinology. 2014;155:4634–4640. doi: 10.1210/en.2014-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lodh S. Primary Cilium, An Unsung Hero in Maintaining Functional β-cell Population. Yale J. Biol. Med. 2019;92:471–480. [PMC free article] [PubMed] [Google Scholar]
- 92.Seymour PA, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl Acad. Sci. USA. 2007;104:1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu J, et al. WLS retrograde transport to the endoplasmic reticulum during Wnt secretion. Dev. Cell. 2014;29:277–291. doi: 10.1016/j.devcel.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 94.Baumgartner BK, Cash G, Hansen H, Ostler S, Murtaugh LC. Distinct requirements for beta-catenin in pancreatic epithelial growth and patterning. Dev. Biol. 2014;391:89–98. doi: 10.1016/j.ydbio.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yebra M, et al. Endothelium-derived Netrin-4 supports pancreatic epithelial cell adhesion and differentiation through integrins α2β1 and α3β1. PLoS ONE. 2011;6:e22750. doi: 10.1371/journal.pone.0022750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kojima T, et al. Tight junctions in human pancreatic duct epithelial cells. Tissue Barriers. 2013;1:e24894. doi: 10.4161/tisb.24894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Suwa H, et al. Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br. J. Cancer. 1998;77:147–152. doi: 10.1038/bjc.1998.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bydoun M, et al. S100A10, a novel biomarker in pancreatic ductal adenocarcinoma. Mol. Oncol. 2018;12:1895–1916. doi: 10.1002/1878-0261.12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Esposito I, et al. Tumor-suppressor function of SPARC-like protein 1/Hevin in pancreatic cancer. Neoplasia. 2007;9:8–17. doi: 10.1593/neo.06646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guerriero, I. et al. Exploring the molecular crosstalk between pancreatic bud and mesenchyme in embryogenesis: novel signals involved. Int. J. Mol. Sci.20 (2019). [DOI] [PMC free article] [PubMed]
- 101.Willmann SJ, et al. The global gene expression profile of the secondary transition during pancreatic development. Mech. Dev. 2016;139:51–64. doi: 10.1016/j.mod.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 102.Ogaki S, Harada S, Shiraki N, Kume K, Kume S. An expression profile analysis of ES cell-derived definitive endodermal cells and Pdx1-expressing cells. BMC Dev. Biol. 2011;11:13. doi: 10.1186/1471-213X-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sjödin A, Dahl U, Semb H. Mouse R-cadherin: expression during the organogenesis of pancreas and gastrointestinal tract. Exp. Cell Res. 1995;221:413–425. doi: 10.1006/excr.1995.1392. [DOI] [PubMed] [Google Scholar]
- 104.Nagaraj V, et al. Complement inhibitor CD55 governs the integrity of membrane rafts in pancreatic beta cells, but plays no role in insulin secretion. Biochem. Biophys. Res. Commun. 2015;460:518–524. doi: 10.1016/j.bbrc.2015.03.062. [DOI] [PubMed] [Google Scholar]
- 105.Zbinden A, et al. Nidogen-1 mitigates ischemia and promotes tissue survival and regeneration. Adv. Sci. 2021;8:2002500. doi: 10.1002/advs.202002500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Q, et al. CD8+ effector T cell migration to pancreatic islet grafts is dependent on cognate antigen presentation by donor graft cells. J. Immunol. 2016;197:1471–1476. doi: 10.4049/jimmunol.1600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Warner SL, et al. Validation of TPX2 as a potential therapeutic target in pancreatic cancer cells. Clin. Cancer Res. 2009;15:6519–6528. doi: 10.1158/1078-0432.CCR-09-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee S-H, et al. The Id3/E47 axis mediates cell-cycle control in human pancreatic ducts and adenocarcinoma. Mol. Cancer Res. 2011;9:782–790. doi: 10.1158/1541-7786.MCR-10-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pei YF, Yin XM, Liu XQ. TOP2A induces malignant character of pancreatic cancer through activating β-catenin signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864:197–207. doi: 10.1016/j.bbadis.2017.10.019. [DOI] [PubMed] [Google Scholar]
- 110.Hocevar BA. Loss of disabled-2 expression in pancreatic cancer progression. Sci. Rep. 2019;9:7532. doi: 10.1038/s41598-019-43992-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Backx E, et al. MECOM permits pancreatic acinar cell dedifferentiation avoiding cell death under stress conditions. Cell Death Differ. 2021;28:2601–2615. doi: 10.1038/s41418-021-00771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.He LH, et al. Neuropilin1 silencing impairs the proliferation and migration of cells in pancreatic cancer. J. Clin. Lab Anal. 2020;34:e23394. doi: 10.1002/jcla.23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang HY, et al. Gelsolin impairs barrier function in pancreatic ductal epithelial cells by actin filament depolymerization in hypertriglyceridemia-induced pancreatitis in vitro. Exp. Ther. Med. 2022;23:290. doi: 10.3892/etm.2022.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu X-X, et al. Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J. 2019;38:e100164. doi: 10.15252/embj.2018100164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Memon B, Abdelalim EM. ACE2 function in the pancreatic islet: Implications for relationship between SARS-CoV-2 and diabetes. Acta Physiol. (Oxf.) 2021;233:e13733. doi: 10.1111/apha.13733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Favre GA, Esnault VLM, Obberghen EV. Modulation of glucose metabolism by the renin-angiotensin-aldosterone system. Am. J. Physiol.—Endocrinol. Metab. 2015;308:E435–E449. doi: 10.1152/ajpendo.00391.2014. [DOI] [PubMed] [Google Scholar]
- 117.Luther JM. Effects of aldosterone on insulin sensitivity and secretion. Steroids. 2014;91:54–60. doi: 10.1016/j.steroids.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ceasrine, A. M., Lin, E. E., Lumelsky, D. N., Iyer, R. & Kuruvilla R. Adrb2 controls glucose homeostasis by developmental regulation of pancreatic islet vasculature. Elife7 (2018). [DOI] [PMC free article] [PubMed]
- 119.Yao LJ, et al. Novel role for SGK3 in glucose homeostasis revealed in SGK3/Akt2 double-null mice. Mol. Endocrinol. 2011;25:2106–2118. doi: 10.1210/me.2010-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sherman BT, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 2022;50:W216–W221. doi: 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mellitzer G, et al. IA1 is NGN3-dependent and essential for differentiation of the endocrine pancreas. EMBO J. 2006;25:1344–1352. doi: 10.1038/sj.emboj.7601011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gierl MS, Karoulias N, Wende H, Strehle M, Birchmeier C. The zinc-finger factor Insm1 (IA-1) is essential for the development of pancreatic beta cells and intestinal endocrine cells. Genes Dev. 2006;20:2465–2478. doi: 10.1101/gad.381806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature. 1997;385:257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- 124.Matsuoka TA, et al. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl Acad. Sci. USA. 2004;101:2930–2933. doi: 10.1073/pnas.0306233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Scavuzzo MA, et al. Endocrine lineage biases arise in temporally distinct endocrine progenitors during pancreatic morphogenesis. Nat. Commun. 2018;9:3356. doi: 10.1038/s41467-018-05740-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Y, Sun L, Luo Y, He S. Knockdown of KDM1B inhibits cell proliferation and induces apoptosis of pancreatic cancer cells. Pathol. Res. Pr. 2019;215:1054–1060. doi: 10.1016/j.prp.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 127.Duvall E, et al. Single-cell transcriptome and accessible chromatin dynamics during endocrine pancreas development. Proc. Natl Acad. Sci. 2022;119:e2201267119. doi: 10.1073/pnas.2201267119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Henry C, Close AF, Buteau J. A critical role for the neural zinc factor ST18 in pancreatic β-cell apoptosis. J. Biol. Chem. 2014;289:8413–8419. doi: 10.1074/jbc.M114.554915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang YH, Manning Fox JE, Zhang KL, MacDonald PE, Johnson JD. Intraislet SLIT-ROBO signaling is required for beta-cell survival and potentiates insulin secretion. Proc. Natl Acad. Sci. USA. 2013;110:16480–16485. doi: 10.1073/pnas.1214312110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wei D, et al. KLF4 is essential for induction of cellular identity change and acinar-to-ductal reprogramming during early pancreatic carcinogenesis. Cancer Cell. 2016;29:324–338. doi: 10.1016/j.ccell.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang C, Guo Z-M. Multiple functions of Maf in the regulation of cellular development and differentiation. Diabetes/Metab. Res. Rev. 2015;31:773–778. doi: 10.1002/dmrr.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang W, Lu N, Eberspaecher H, de Crombrugghe B. A new long form of c-Maf cooperates with Sox9 to activate the type II collagen gene*. J. Biol. Chem. 2002;277:50668–50675. doi: 10.1074/jbc.M206544200. [DOI] [PubMed] [Google Scholar]
- 133.Rossetti S, Hoogeveen AT, Sacchi N. The MTG proteins: chromatin repression players with a passion for networking. Genomics. 2004;84:1–9. doi: 10.1016/j.ygeno.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 134.Suk F-M, et al. ZFP36L1 and ZFP36L2 inhibit cell proliferation in a cyclin D-dependent and p53-independent manner. Sci. Rep. 2018;8:2742. doi: 10.1038/s41598-018-21160-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dong H, et al. Regulator of G protein signaling 2 is a key regulator of pancreatic β-cell mass and function. Cell Death Dis. 2017;8:e2821. doi: 10.1038/cddis.2016.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baron M, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3:346–360.e344. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.van Gurp, L. et al. A transcriptomic roadmap to alpha- and beta-cell differentiation in the embryonic pancreas. Development146 (2019). [DOI] [PubMed]
- 138.Chu K, Tsai MJ. Neuronatin, a downstream target of BETA2/NeuroD1 in the pancreas, is involved in glucose-mediated insulin secretion. Diabetes. 2005;54:1064–1073. doi: 10.2337/diabetes.54.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fujikura J, et al. Rbp-j regulates expansion of pancreatic epithelial cells and their differentiation into exocrine cells during mouse development. Dev. Dyn. 2007;236:2779–2791. doi: 10.1002/dvdy.21310. [DOI] [PubMed] [Google Scholar]
- 141.Fujikura J, et al. Notch/Rbp-j signaling prevents premature endocrine and ductal cell differentiation in the pancreas. Cell Metab. 2006;3:59–65. doi: 10.1016/j.cmet.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 142.Swift GH, et al. An endocrine-exocrine switch in the activity of the pancreatic homeodomain protein PDX1 through formation of a trimeric complex with PBX1b and MRG1 (MEIS2) Mol. Cell Biol. 1998;18:5109–5120. doi: 10.1128/mcb.18.9.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Scavuzzo MA, et al. Pancreatic cell fate determination relies on notch ligand trafficking by NFIA. Cell Rep. 2018;25:3811–3827.e3817. doi: 10.1016/j.celrep.2018.11.078. [DOI] [PubMed] [Google Scholar]
- 144.Santangelo L, et al. The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4alpha. Hepatology. 2011;53:2063–2074. doi: 10.1002/hep.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Saha SK, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stoffel M, Duncan SA. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc. Natl Acad. Sci. USA. 1997;94:13209–13214. doi: 10.1073/pnas.94.24.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang H, Maechler P, Antinozzi PA, Hagenfeldt KA, Wollheim CB. Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic beta-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J. Biol. Chem. 2000;275:35953–35959. doi: 10.1074/jbc.M006612200. [DOI] [PubMed] [Google Scholar]
- 148.Ng NHJ, et al. HNF4A haploinsufficiency in MODY1 abrogates liver and pancreas differentiation from patient-derived induced pluripotent stem cells. iScience. 2019;16:192–205. doi: 10.1016/j.isci.2019.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Eto K, Kaur V, Thomas MK. Regulation of pancreas duodenum homeobox-1 expression by early growth response-1*. J. Biol. Chem. 2007;282:5973–5983. doi: 10.1074/jbc.M607288200. [DOI] [PubMed] [Google Scholar]
- 150.Leu SY, et al. Loss of EGR-1 uncouples compensatory responses of pancreatic β cells. Theranostics. 2020;10:4233–4249. doi: 10.7150/thno.40664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Seymour PA, et al. Jag1 modulates an oscillatory Dll1-Notch-Hes1 signaling module to coordinate growth and fate of pancreatic progenitors. Dev. Cell. 2020;52:731–747.e738. doi: 10.1016/j.devcel.2020.01.015. [DOI] [PubMed] [Google Scholar]
- 152.Shi X, et al. Nr2e1 deficiency augments palmitate-induced oxidative stress in beta cells. Oxid. Med. Cell Longev. 2016;2016:9648769. doi: 10.1155/2016/9648769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kundu S, et al. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol. Cell. 2017;65:432–446.e435. doi: 10.1016/j.molcel.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 155.Gribben C, et al. Ductal Ngn3-expressing progenitors contribute to adult beta cell neogenesis in the pancreas. Cell Stem Cell. 2021;28:2000–2008.e2004. doi: 10.1016/j.stem.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Blum B, et al. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat. Biotechnol. 2012;30:261–264. doi: 10.1038/nbt.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sachs S, et al. Targeted pharmacological therapy restores beta-cell function for diabetes remission. Nat. Metab. 2020;2:192–209. doi: 10.1038/s42255-020-0171-3. [DOI] [PubMed] [Google Scholar]
- 158.Artner I, et al. MafB: an activator of the glucagon gene expressed in developing islet alpha- and beta-cells. Diabetes. 2006;55:297–304. doi: 10.2337/diabetes.55.02.06.db05-0946. [DOI] [PubMed] [Google Scholar]
- 159.Artner I, et al. MafB is required for islet beta cell maturation. Proc. Natl Acad. Sci. USA. 2007;104:3853–3858. doi: 10.1073/pnas.0700013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Camunas-Soler J, et al. Patch-seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metab. 2020;31:1017–1031.e1014. doi: 10.1016/j.cmet.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yoshihara E, et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature. 2020;586:606–611. doi: 10.1038/s41586-020-2631-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang H, et al. The LIM-homeodomain protein ISL1 activates insulin gene promoter directly through synergy with BETA2. J. Mol. Biol. 2009;392:566–577. doi: 10.1016/j.jmb.2009.07.036. [DOI] [PubMed] [Google Scholar]
- 163.Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013;4:1262–1275. doi: 10.1016/j.celrep.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ediger BN, et al. Islet-1 Is essential for pancreatic beta-cell function. Diabetes. 2014;63:4206–4217. doi: 10.2337/db14-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19:872–882. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hoffman BG, Zavaglia B, Beach M, Helgason CD. Expression of Groucho/TLE proteins during pancreas development. BMC Dev. Biol. 2008;8:81. doi: 10.1186/1471-213X-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.D’Avino PP. Citron kinase—renaissance of a neglected mitotic kinase. J. Cell Sci. 2017;130:1701–1708. doi: 10.1242/jcs.200253. [DOI] [PubMed] [Google Scholar]
- 169.Messina G, et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell. 2010;140:554–566. doi: 10.1016/j.cell.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 170.Fraser J, et al. Common regulatory targets of NFIA, NFIX and NFIB during postnatal cerebellar development. Cerebellum. 2020;19:89–101. doi: 10.1007/s12311-019-01089-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ribeiro, V. et al. NFIXing cancer: the role of NFIX in oxidative stress response and cell fate. Int. J. Mol. Sci.24 (2023). [DOI] [PMC free article] [PubMed]
- 172.McCloskey AG, Miskelly MG, Flatt PR, McKillop AM. Pharmacological potential of novel agonists for FFAR4 on islet and enteroendocrine cell function and glucose homeostasis. Eur. J. Pharm. Sci. 2020;142:105104. doi: 10.1016/j.ejps.2019.105104. [DOI] [PubMed] [Google Scholar]
- 173.Dror E, et al. Epigenetic dosage identifies two major and functionally distinct beta cell subtypes. Cell Metab. 2023;35:821–836.e827. doi: 10.1016/j.cmet.2023.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lynn FC, et al. Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc. Natl Acad. Sci. USA. 2007;104:10500–10505. doi: 10.1073/pnas.0704054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cerychova R, et al. Adverse effects of Hif1a mutation and maternal diabetes on the offspring heart. Cardiovasc. Diabetol. 2018;17:68. doi: 10.1186/s12933-018-0713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ye J, et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Filova I, et al. ISL1 is necessary for auditory neuron development and contributes toward tonotopic organization. Proc. Natl Acad. Sci. USA. 2022;119:e2207433119. doi: 10.1073/pnas.2207433119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sugiyama T, Rodriguez RT, McLean GW, Kim SK. Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc. Natl Acad. Sci. USA. 2007;104:175–180. doi: 10.1073/pnas.0609490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:3. [Google Scholar]
- 182.Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 183.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Blake JA, et al. Mouse Genome Database (MGD): knowledgebase for mouse-human comparative biology. Nucleic Acids Res. 2021;49:D981–d987. doi: 10.1093/nar/gkaa1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Smith CL, Eppig JT. The mammalian phenotype ontology: enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009;1:390–399. doi: 10.1002/wsbm.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hayamizu TF, Baldock RA, Ringwald M. Mouse anatomy ontologies: enhancements and tools for exploring and integrating biomedical data. Mamm. Genome. 2015;26:422–430. doi: 10.1007/s00335-015-9584-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Newman AM, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kaya-Okur HS, Janssens DH, Henikoff JG, Ahmad K, Henikoff S. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 2020;15:3264–3283. doi: 10.1038/s41596-020-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zheng, Y., Ahmad, K. & Henikoff, S. CUT&Tag Data Processing and Analysis Tutorial https://yezhengstat.github.io/CUTTag_tutorial/index.html (2020).
- 190.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Meers MP, Tenenbaum D, Henikoff S. Peak calling by sparse enrichment analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin. 2019;12:42. doi: 10.1186/s13072-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 193.Robinson JT, et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010;26:2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kent WJ, et al. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hao Y, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e3529. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lun ATL, et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019;20:63. doi: 10.1186/s13059-019-1662-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e324. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
All raw sequencing data have been deposited at NIH GEO. The bulk RNA-seq and Cut&Tag-seq data are under accession number GSE212084 and the single-cell RNA-seq data are under accession number GSE236559. Analyzed data are in Supplementary Data 1 and Data 2. Source data are provided with this paper.