

Summary
Trauma, vascular events, or neurodegenerative processes can lead to axonal injury and eventual transection (axotomy). Neurons can survive axotomy, yet the underlying mechanisms are not fully understood. Excessive water entry into injured neurons poses a particular risk due to swelling and subsequent death. Using in vitro and in vivo neurotrauma model systems based on laser transection and surgical nerve cut, we demonstrated that axotomy triggers actomyosin contraction coupled with calpain activity. As a consequence, neurons shrink acutely to force water out through aquaporin channels preventing swelling and bursting. Inhibiting shrinkage increased the probability of neuronal cell death by about 3-fold. These studies reveal a previously unrecognized cytoprotective response mechanism to neurotrauma and offer a fresh perspective on pathophysiological processes in the nervous system.
Subject areas: Biological sciences, Physiology, Neuroscience
Graphical abstract
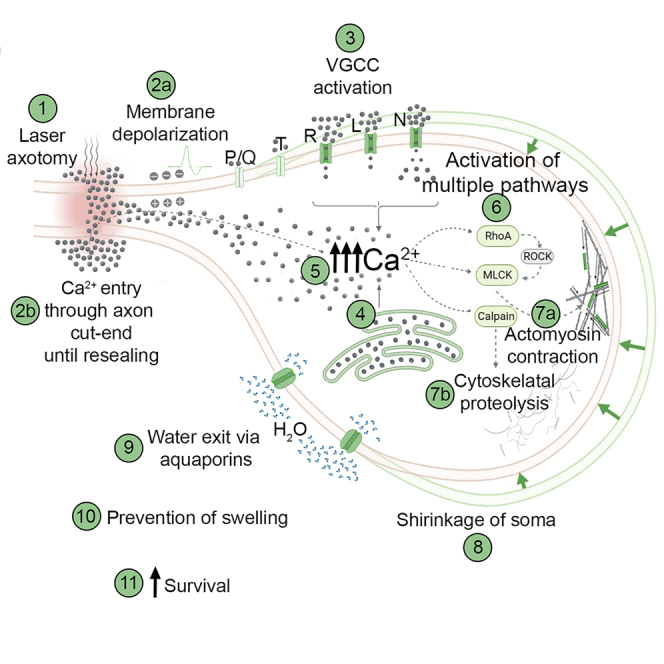
Highlights
-
•
Axonal damage increases cytosolic calcium in neurons
-
•
Increased calcium activates proteolytic enzymes and actomyosin contraction
-
•
Neurons shrink and pump out water through aquaporin channels and avoid deadly swelling
-
•
Axotomy-induced shrinkage helps neurons survive the trauma
Biological sciences; Physiology; Neuroscience
Introduction
Axons of most neurons can extend to several thousand-fold lengths of the cell’s diameter. Such a prominent structure represents a substantial cellular surface area susceptible to intrusions of various kinds, ranging from traumas to autoimmunity. Traumatic brain injuries may physically break axons by extreme stretching or rotating forces or by direct laceration, which lead to primary axotomy. Traumas or neurodegenerative processes may also cause partial axonal damage which can start molecular mechanisms that ultimately result in secondary axotomy.1,2 Many nervous system disorders start or progress by axonal injury, which may result in the death of the affected neuron, acutely or over extended periods.1,3 However, substantial proportion of axotomized neurons can survive especially when the injury is far from the cell body.4,5 Yet, full repertoire of responses neurons employ to survive axotomy has not been characterized.
In the aftermath of an axotomy, the neuron becomes prone to swelling started by influx of Na+, Cl−, and Ca2+ through the cut-end until effective resealing, especially in injuries close to the neuronal body. Ion influx occurs through specific channels in the soma that open due to changes in the plasma membrane’s electrical potential or mechanical stimuli caused by the injury.6 Water accompanies ions into the neuron as the cytosol becomes more and more hyperosmolar.6,7 Two main mechanisms are known to decrease solute load and reduce the risk of swelling after axonal injury. First, during strong depolarization triggered by an axonal injury, the Na+-Ca2+ exchange pump drives out Na+ in exchange for Ca2+.8,9 However, cytosolic Ca2+ overload may damage the plasma membrane, mitochondria, and cytoskeleton.8,10 Second, to remove excessive Cl− from the cytosol, cells use Cl−-K+ cotransporters to release Cl− along with K+ that spontaneously diffuses out due to concentration gradient. To restore cytosolic K+ levels, the ATP-dependent Na+-K+ pump is overactivated, which adds to the impending energy crisis.7,11,12 If the neuron cannot cope with this emergency, uncontrolled water influx swells and ultimately bursts the cell.7
Experimental axotomy in neuron cultures or living animals is a common approach for examining the factors influencing neuronal survival and degenerative or regenerative processes following axonal injury. We observed a hitherto unreported phenomenon during axotomy experiments: shrinkage in neuronal soma. This critical observation aligns with earlier findings that neurons employ local axonal contractions to promote resealing of cut axon ends.13 We hypothesized that actomyosin contraction (AMC) in the neuronal body might be an early response to axotomy, which counteracts the ensuing deadly swelling. Indeed, we have shown that by shrinking, neurons increase survival chance after axotomy both in vitro and in vivo.
Results
Axotomy causes a reduction in cell size, which is preceded by calcium influx and membrane depolarization
The in vitro experimental paradigm employs time-lapse imaging of adult primary sensory neurons during the transection of outgrown axons with a laser beam at physiological temperature and atmosphere. To confidently use all kinds of DRG neuronal subtypes in the experiments without unacceptable deviations, we performed correlation analysis between the relative amount of shrinkage and the initial cell size, which defines various neuronal phenotypes.14 The results indicated that all sizes of neurons shrink with similar ratios (Figure S1). To confirm that changes in sizes measured in two dimensions represent volume changes in three dimensions, we exposed DRG neurons to hypo- and hyperosmotic challenges, which caused swelling and shrinking, respectively. Correlation between cross-sectional area and volume measurements was quite strong, which validated the method (Figure S2). In response to laser axotomy, neurons immediately started to shrink. Cell size change analysis revealed that the neuronal bodies shrank about 12% within the first 15 min (Figures 1A, 1B, and Video S1). Notably, nearly half of the shrinkage occurred during the first 5 min, which we selected as the reference time point for other experiments. Next, we sought to understand this observation’s in vivo relevance and utilized Thy1-GFP line M strain mice with sparse GFP expression in dorsal root ganglia (DRG) neurons,15 where individual axons can be identified, allowing precise in vivo laser transection. An imaging window was surgically formed to expose the second lumbar (L2) DRGs, as described before.16 Cutting single axons with a femtosecond infrared laser of a two-photon microscope confirmed that these neurons in vivo shrank comparable to isolated cells in vitro (Figures 1A and 1B). We next investigated whether this phenomenon is unique to DRG neurons or also occurs in CNS. For this, we axotomized cultured adult cortical and hippocampal neurons, both of which also shrank. Experiments with immature newborn hippocampal neurons also yielded similar results (Figures 1A and 1B). We then continued with in vivo confirmation of these findings by cutting axons of cortical and hippocampal neurons under two-photon microscope as described previously, this time through a cranial window constructed in Thy1-GFP line M mice.17 Both types of CNS neurons had significant size reduction upon axotomy in this in vivo setting.
Figure 1.

Axotomy causes a reduction in cell size, which is preceded by calcium influx and membrane depolarization
(A) Images show adult mouse dorsal root ganglion (DRG), hippocampal (HPC), and cortical (Cor) and newborn (NB) HPC neurons in vitro and adult DRG and HPC neurons of a live Thy1-GFP mouse before and after laser axotomy (scale bar, 15 μm; arrowheads point to the site of axotomy) (see also Video S1).
(B) The scatter dot plot shows changes in the cell size 5 min following axotomy in cultured adult DRG (n = 6/65), cortical (n = 2/6), HPC (n = 2/10), and newborn HPC (n = 3/25) neurons and 15 min after in vivo axotomy of adult DRG (n = 3/9) and central neurons (CNS - Cortical and HPC neurons, (n = 2/4). Inlet shows size changes in DRG neurons during 5 min after axotomy. Paired t test for ratios was used to compare cell sizes before and after axotomy (See also Figures S1 and S2).
(C) Membrane potential recordings from a control and an axotomized neuron. Images show simultaneous patch-clamp recording and axotomy (arrowhead points to the injury site).
(D) Bars represent levels of the resting membrane potential (RMP) of neurons before (n = 4/29), immediately (n = 3/16)), and 24 h (n = 3/15) after axotomy and following the second axotomy (n = 3/12); Kruskal Wallis and Mann-Whitney U tests were used to compare groups.
(E) Time-lapse images and real-time Ca2+ traces of GCamp6-expressing DRG neurons upon axotomy (See also Videos S2 and S3). Error bars: SEM; n = number of animals used/number of cells analyzed; for all tests: ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001.
For the remainder of the study, we selected DRG neurons as a model system due to their excellent in vitro survival and the ease of intravital imaging.
Axotomy causes membrane depolarization and action potentials in cortical neurons in vitro.6 Since many post-axotomy events can be related to this and concurrent Ca2+ influx, we first characterized these phenomena using patch-clamp recordings. We exploited DRG neurons from genetically engineered mice expressing the genetically encoded Ca2+ biosensor GCamp6 to capture neuronal body shrinkage and subsequent Ca2+ influx in response to laser axotomy. Laser axotomy significantly depolarized the membrane with or without generating action potentials (AP) (Figure 1C). Application of AP blocker tetrodotoxin did not prevent shrinkage suggesting that generation of AP is not essential for this response (Figure 2D). After 24 h, the resting membrane potentials of axotomized neurons were similar to the controls, and when re-axotomized then, the membrane potential depolarized comparable to naive neurons, but AP generation was sporadic (Figure 1D and supplemental Text). Live-cell Ca2+ imaging showed that cytosolic Ca2+ levels plateaued sharply after axotomy and returned to baseline within an hour (Figure 1E). We also observed a persistent Ca2+ influx that led to enhanced cytosolic Ca2+ accumulation when the proximal stump was not promptly resealed (Videos S2 and S3).
Figure 2.

Calcium entry through voltage-gated channels triggers AMC which partially depends on RhoA signaling
(A) Effect of inhibition of voltage-gated calcium channels (VGCC) and internal Ca2+ stores on size change after axotomy. VGCC subtypes L, N, P/Q, R, and T, were blocked with nifedipine (NIF) (n = 4/46), ω-conotoxin GVIA (CNX) (n = 3/30), ω-agatoxin-IVA(AGX) (n = 3/23), SNX-482(SNX) (n = 3/18), and mibefradil (MIB) (n = 3/28), respectively. Thapsigargin (TG) (n = 4/44) was used to deplete internal Ca2+ stores before axotomy.
(B) Effect of inhibition of AMC on size change 5 min after axotomy in Ca2+-free medium (n = 3/38), with myosin inhibitors butanedione monoxime (BDM (n = 3/30)) and blebbistatin (BB) (n = 4/48) and actin inhibitors cytochalasin E (CytE) (n = 3/33) and swinholide A (SWH) (n = 3/41) (See also Figure S3).
(C) Effect of inhibition of RhoA signaling with clostridium botulinum exoenzyme C3 (n = 3/20) or RhoA’s effector ROCK with Y-27632 (n = 3/30) on size change after axotomy.
(D) Effect of inhibition of voltage-gated Na+ channels thus prevention of AP generation with tetrodotoxin (TTX) on size change after axotomy (n = 2/14). Mann-Whitney U test was used to compare size changes in each group to the changes 5 min (B) or at indicated time points (A, D) after control axotomy (n5 = 6/65, n15 = 6/28, n30 = 6/43, n60 = 6/60). Error bars: SEM; n = number of animals used/number of cells analyzed; for all tests: ∗p < 0.05, ∗∗∗∗p < 0.00001.
The neuron expresses Ca++ sensor - GCaMP6s.
The neuron expresses Ca++ sensor - GCaMP6s.
Calcium entry through voltage-gated channels triggers AMC
Ca2+ may enter the cell through voltage-gated calcium channels (VGCCs) in addition to the cut-ends of the injured axons.8 DRG neurons express five types of VGCCs: L, N, P/Q, R, and T types.18,19 We utilized pharmacological antagonists to inhibit these channels selectively. This approach unveiled that L- and N-type Ca2+ channels are involved at the earliest stages of the shrinkage following axotomy. Their respective blockers, nifedipine (NIF) or ω-conotoxin GVIA (CNX), effectively inhibited cell size reduction after axotomy (Figure 2A). NIF-mediated inhibition was most effective in the first 5 min while CNX exerted a more consistent inhibition. Blocking R-type channels with SNX-482 effectively prevented shrinking after 15–60 min post axotomy. Inhibition of P/Q-type channels with ω-agatoxin-IVA and T-type Ca2+ channels with mibefradil proved ineffective. The results of a prior work depolarizing rat DRG neurons with high K+ and examining the contribution of VGCCs to Ca2+ influx are consistent with the preponderance of L- and N-type channels in the early stages of shrinkage and the major contribution of R-type channels later on.18 VGCCs are classified as either high- or low-voltage-activated types. The level of their expression and expression ratio among subtypes of primary sensory neurons differ; these are subject to change in culture.19,20,21 Our findings demonstrate that Ca2+ influx through the high-voltage-activated L-, N-, and R-type channels induces shrinkage in cultured DRG neurons. Confirmation of these results in vivo remains to be investigated.
Reports indicate that axotomy can mobilize internal Ca2+ stores.22 To understand the contribution of endoplasmic reticulum-stored Ca2+ to cell shrinkage, we used thapsigargin (TG), a Ca2+ ATPase inhibitor, in a two-stage experiment described elsewhere.23 First, neurons were incubated with TG to empty the internal Ca2+ stores. Then, we performed axotomies and analyzed shrinkage to test the effect of extracellular Ca2+ alone. Eliminating internal Ca2+ release did not alter the shrinkage during the first 5min following axotomy (Figure 2A). However, cell shrinkage could not continue for more than 15 min without internal Ca2+ reserves.
In this study section, we demonstrated that extracellular Ca2+ coming through VGCCs initiates axotomy-induced shrinking, which is later maintained by Ca2+ levels from internal stores. The interplay of multiple mechanisms probably determines the net increase in cytosolic Ca2+. In this sense, the potential role of store-operated Ca2+ entry (SOCE), typically induced by depletion of internal stores, may also be considered.24 However, we obtained indirect evidence that SOCE is unlikely to share responsibility for axotomy-induced shrinkage. According to previous reports, SOCE is activated within 1 h of the TG application.25 In our experiments, the time from TG application to axotomy was 90 min, enough to induce SOCE. If SOCE played a significant role in the axotomy-induced increase in cytosolic Ca2+, it is expectable that shrinking would continue for 60 min after the axotomy. Therefore, we indirectly conclude that SOCE may not be essential for axotomy-induced shrinkage.
The following hypothesis was that the axotomy-induced elevation in cytosolic Ca2+ may lead to shrinkage through active contraction and the cytoskeletal reorganization necessary for size change. To provide evidence, we first conducted in vitro axotomies in Ca2+-free medium or in the presence of inhibitors of actin (cytochalasin E - CytE and swinholide A - SWH) or myosin (blebbistatin – BB and 2,3-butanedione monoxime - BDM). Lack of extracellular Ca2+ or addition of SWH, CytE, or BDM significantly inhibited neuronal shrinkage while BB was ineffective (Figure 2B). CytE prevents polymerization of actin while SWH severs F-actins.26,27 Expectedly, the effect of CytE was substantially less than SWH, indicating that the pre-existing F-actins may still mediate part of the contraction despite the elimination of new actin polymers. Failure of BB to counteract the shrinkage may be due to its high selectivity against myosin subtypes.28 However, BDM, a more general myosin inhibitor that blocks phosphorylation of myosin light chain (MLC) kinase,29 effectively blocked cell shrinkage. Since existing literature focuses on the contraction of axonal cytoskeleton following an injury,13 we also analyzed changes in the proximal part after axotomy. While elimination of Ca2+ in the culture medium significantly decreased the shrinkage in the proximal stump, which is normally quite substantial, myosin inhibition per se with BDM did not have this effect (Figure S3). We suggest that very high level of Ca2+ entered through the cut-end may activate some resealing mechanisms like vesicular plug formation with more severe cytoskeletal degradation30 that may hinder AMC.
RhoA signaling is not essential but contributes to the AMC
To understand how AMC is initiated, we next focused on the Rho GTPases and their downstream effectors, Rho-associated kinases (ROCKs), as potential signaling mechanisms. In non-muscle cells, such as neurons, RhoA and ROCK govern actomyosin contractility in a Ca2+-dependent manner and moreover they are activated by axotomy.31,32 We performed axotomy in the presence of RhoA inhibitor exoenzyme C3 and ROCK inhibitor Y-27632. RhoA inhibition significantly reduced shrinkage at the 15th and 60th min post axotomy, while ROCK inhibition decreased cell shrinkage only at 60 min (Figure 2C). Y-27632 displayed marginal effects, possibly because ROCK enhances AMC only through MLC by inactivating its inhibitor MLC phosphorylase.33 However, RhoA has an additional effector, diaphanous-related formin 1, which activates profilin, an actin-polymerizing and stabilizing factor.34 Our data indicate that RhoA activation contributes to AMC minutes after axotomy and is only partially responsible for the shrinkage. In a relevant study, an endothelial cell stretch model demonstrated a similar temporal relationship with Ca2+ increase, RhoA activation, and AMC.35 We suggest that axotomy-induced membrane depolarization is the first step in the likely mechanism of AMC. Next, Ca2+ influx directs the contraction while activating the RhoA system, which may aid in boosting actin polymerization and disinhibiting MLC.
Calpains digest the cytoskeleton to enable cell shrinkage
Beneath the plasma membrane lies a contractable actomyosin cortex that participates in various physiological processes, including alteration of cell shape and the control of intracellular hydrostatic pressure.36 To make changes in a cell’s size or shape, the tension created by AMC must overcome the opposing forces exerted by relatively static structural elements of the cytoskeleton. Therefore, we reasoned that axotomy might cause the activation of proteolytic enzymes, namely calpains, which could break down cytoskeletal proteins and reduce the opposing forces. To test this hypothesis, we axotomized cultured neurons in the presence of a general calpain inhibitor PD150606. We found that this approach almost completely prevented cell shrinkage (Figure 3A). Calpains are activated by increased intracellular Ca2+, targeting many functional and structural proteins, which make them accountable for various pathological changes in neurodegenerative processes.37,38 Although identifying calpain substrates is out of the scope of this study, we suspected that it might be spectrin because it forms a pre-tensed supporting mesh under the plasma membrane and tubulin, which constitutes the major solid network opposing the compression forces.37,39 On the other hand, reports suggest that actins and myosins are not the primary targets of calpains.40 Thus, we presumed that calpain inhibition did not preclude AMC, but neurons could not shrink due to the resistance of the intact cytoskeleton. Indeed, when calpains and AMC were blocked simultaneously, neurons’ response to axotomy was reversed to swelling instead of shrinking. In the earlier experiments, BDM alone prevented shrinkage but did not cause swelling (Figure 2B). How simultaneous inhibition of calpains led to the swelling is unclear. Yet, it may indicate that calpains’ actions after axotomy are possibly as diverse as their many substrates.40
Figure 3.

Calpains digest the cytoskeleton to enable cell shrinkage and isotonic contraction pumps water out of the axotomized neuron
(A) Scatter dot plot shows size change 5 min after axotomy in the presence of calpain inhibitor PD150606 (PD) alone (n = 3/33), in combination with the myosin inhibitor butanedione monoxime (BDM) (dots, n = 4/41) and in control conditions (n = 6/65). Kruskal Wallis (A) and Mann-Whitney U-tests were used for comparisons.
(B) Representative three-dimensional confocal images show an axotomized neuron over time. Wavy arrows in the inlet point to depressions on the cell surface due to shrinkage (scale bar, 15 μm; arrowhead points to the site of axotomy) (See also Video S4).
(C) Mean volume of DRG neurons before and 5 min after axotomy (n = 3/22). Wilcoxon signed ranks test was used for comparison.
(D) Comparison of size change 5 min after axotomy with AQP inhibition (n = 3/20) to the control axotomy (n = 6/65).
(E) A Representative bright-field image shows the measurement of membrane tensions by pulling an attached polystyrene bead with an optical tweezer.
(F) Changes in the membrane tensions (T) due to axotomy (n = 4/48) and the effect of aquaporin (AQP) channel inhibition with HgCl2 (n = 3/20). Mann-Whitney U test was used for comparisons in D and F. Error bars: SEM; n = number of animals used/number of cells analyzed; for all tests: ∗p < 0.05, ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001.
Isotonic contraction pumps water out of the axotomized neuron
We next sought to understand whether the change in cell size after axotomy means a change in volume as the opposite is also possible since neurons may get elongated vertically while shrinking laterally. We cultured DRG neurons from EGFP mice and acquired three-dimensional images with a fast confocal microscope during axotomy. We determined that neuronal volumes were significantly reduced concordant with the change in size (Figures 3B and 3C, and Video S4). To explain the volume loss, we proposed that, by contraction, water may be extruded from the cells through aquaporin channels (AQP).41 Indeed, when we blocked AQPs with HgCl2, neuronal size did not change upon axotomy (Figure 3D). To further characterize the contraction, we measured the plasma membrane tensions of neurons before and after axotomy with a special microscope combining laser microdissection and an optical tweezer (Figure 3E). The slight increases in lateral and cortical tensions caused by axotomy were significantly augmented by inhibition of AQPs with HgCl2 (Figure 3F). This observation strongly suggests that axotomy causes an isotonic type of contraction characterized by a decreasing cell volume and only slightly elevated membrane tensions consistent with the law of Laplace.42 Increased membrane tensions show that cellular contraction becomes isometric in nature when volume change is inhibited (Figures 3D and 3F).
Shrinkage increases the survival chance of axotomized neurons in vitro and in vivo
Our results thus far provide evidence that neurons respond to axotomy by shrinking under the influence of AMC and calpain activity, which forces water out of the cell through AQP channels. Thus, we hypothesized that shrinkage could help neurons survive by preventing swelling. We ran several in vitro and in vivo experiments to test this hypothesis.
In culture, when the contraction was inhibited by BDM, more neurons died progressively over 24 h incubation period (Figure 4A and supplemental Text). The surviving axotomized neurons for the next 1 and 24 h were those that had shrunk, and those that died had not shrunk by the 5th min post axotomy (Figure 4B). The chance of survival after axotomy was correlated to the degree and persistence of shrinkage (Figure S4 and supplemental Text). Using in vivo models, we sought to confirm the biological relevance of these in vitro findings suggesting that shrinkage favors neuronal survival. Using the same technique utilized for in vivo axotomy,16 we constructed an imaging window over L2 DRG of Tau-EGFP mice whose neuronal soma was visibly labeled.43 The basic experimental paradigm was to cut the peripheral nerve emanating from the DRG, thus axotomizing all neurons. Propidium iodide was injected intravenously to monitor cell death. In some experiments, BDM was intrathecally administered before the nerve cut. With a two-photon microscope, we took images before, and 5 min, and 24 h after axotomy. The nerve cut caused an apparent reduction in size, which was even more significant after 24 h (Figure S5). BDM prevented neuronal shrinkage and significantly increased neuronal death compared to control animals, while the sham group with only BDM injection showed no neuron loss (Figures 4C, 4D, and S5). As demonstrated by in vitro experiments, surviving neurons were those that had shrunk. Expectedly, DRG neurons from Thy1-GFP animals pre-injected with BDM did not shrink upon single-cell axotomy in vivo (Figure S5).
Figure 4.

Shrinkage increases the survival chance of axotomized neurons in vitro and in vivo
(A) Cumulative death rates of axotomized and control neurons during 24 h incubation period with and without inhibition of AMC by butanedione monoxime (BDM) (n = 3/117 (BDM+ax), 3/100 (Control ax), 3/106 (BDM sham), 3/114 (Sham). Groups were compared using a repeated measures mixed model (See also Figure S4 and Video S5).
(B) Size changes of neurons that survived and died after in vitro axotomy (n = 16/303 (live 1 h), 16/113 (dead 1 h), 16/160 (live 24 h), 16/266 (dead 24 h)) and in vivo nerve cut (n = 6/11 (live), 6/10 (dead)). Note that in vitro data were pooled from various experimental groups, including those with pharmacological agents. Mann-Whitney U test was used for comparisons (See also Figure S4).
(C) Rate of cell death in DRGs 24 h after peripheral nerve cut or sham operation and effect of inhibition of AMC with BDM in vivo. A comparison of neuronal death rates was performed with Kruskal Wallis and Mann-Whitney U test, (n = 7 (nerve cut), 6 (nerve cut+BDM), 4 (sham + BDM)).
(D) Representative confocal images of DRGs from in vivo experiments in C. Arrows point to a few PI-stained dead neurons, while the thick arrow shows a large group of them (scale bar, 100 μm). (See also Figure S4). Error bars: SD; n = number of animals used/number of cells analyzed; for all tests: ∗∗∗p < 0.0001, ∗∗∗∗p < 0.00001.
An interesting observation we made but not quantitatively analyzed in this study was that contraction of soma could constrict and amputate the proximal stump when axotomy is very close, which represents a fast and efficient way of membrane resealing, potentially increasing the chance of survival when the risk of death is high (Video S5). In line with our observations, Kelley et al. demonstrated in an in vivo model that traumatic axonal injury close to neuronal soma in rat thalamus results in secondary axotomy within 15 min and is accompanied by an efficient resealing.44
Discussion
This study describes an axotomy-induced AMC coupled with proteolysis that shrinks injured neurons to pump out water and prevent deadly swelling. The postulated pathway begins with axotomy-induced membrane depolarization, opening high-voltage-activated Ca2+ channels. AMC and calpain enzymes are activated by elevated cytoplasmic Ca2+ (internal Ca2+ stores can contribute) and break down cytoskeletal proteins to lessen the soma’s ability to resist shrinking. The cell contraction is partially sustained by RhoA signaling and occurs isotonically, pumping water out of the cell through AQP channels.
The tell-tale sign of this protective response is a reduction in somatic size both in central and peripheral neurons. Various other studies have reported that neurons’ diameter decreases after axonal injury, which was often interpreted as atrophy.45,46,47,48,49,50,51,52 In in vivo experiments, DRG neurons shrank significantly within 5 min after the nerve cut but they were much smaller the next day (Figure S5B). This may suggest that once shrink, neurons may not recover to their original size for a long time and furthermore shrinkage may represent initial stage of atrophy. The overall death rate of axotomized neurons in vivo is a matter of debate due to the concerns about quantification methods. However, all studies are in agreement that surviving neurons are smaller compared to uninjured state.4 This consistent observation is quite in line with our findings.
In the existing literature, the contractile capacity of neurons has been mainly associated with neurites and growth cones.13,53 An exception relevant to our findings was a report by Wan et al. back in 1995, stating that “a plasma membrane-linked contractile machinery (presumably actomyosin) might contribute to the neurons’ mechano-osmotic robustness by restricting water influx” during hyposmotic challenge.54 We have demonstrated that water leaves the shrinking neurons via AQP channels. AQP1 is expressed in DRG neurons and upregulated after nerve injury,55 and AQP2 expression is induced after nerve constriction.56 HgCl2, which we used to block water efflux, is primarily an AQP1 inhibitor but not strictly selective.57 Therefore, we do not exactly know which subtypes of AQPs are responsible for water efflux.
Results of in vitro and in vivo experiments have proved that axotomy-induced shrinkage is a common phenomenon both in DRG and CNS neurons and in adult and immature ones. As the selected model, we investigated the mechanism of the shrinkage using DRG neurons. Although we did not conduct a differential analysis, heterogeneity among different subtypes of these neurons in shrinkage response to axotomy can be expected due to varying constitutional expression of molecular components of the event. On the other hand, our observation that the initial size of DRG neurons, which roughly defines different subpopulations,14 does not affect the unique consequence of the shrinkage—the increased chance of survival—suggests that phenotypic differences do not preclude the significance of the phenomenon. At this point, a standing question is whether shrinkage is a common protection mechanism against swelling in other cell types too. To test this, we attempted to cut extensions of cultured glial cells and fibroblasts but failed to measure size changes reliably for two major reasons. First, contrary to neurons, these cells are not stationary; they continuously move and change shape and laser transection makes them even more motile. Second, their soma is not clearly delineated due to flattened shape and extensive lamellipodia. On the other hand, a study has shown that depolarization of cancer cells causes swelling, contrary to our observations with neurons.42 This may suggest that a depolarization-mediated shrinking is not a universal response of all cell types to injury.
When released in excessive amounts, glutamate may lead to excitotoxicity through N-methyl-D-aspartate receptor (NMDAR), which is a common pathway leading to neuronal death in various nervous system diseases and injuries.58 A recent study has demonstrated that stretching plasma membrane can mechanically activate NMDAR independent of glutamate.59 By counteracting swelling, active shrinkage prevents membrane stretching; thus, it may block a potential mechanically induced excitotoxicity.
Mechanobiology of nerve cells is a rapidly growing field of research owing to detection of various mechanosensitive channels both in neurons and glial cells.60,61 Recent studies have shown that neurons respond to mechanical stimuli to their soma or neurites and even differentiate location and magnitude of the stimulus62 and that astrocytes transduce mechanical forces into chemical signals to regulate adult neurogenesis.63 In this sense, what mechanical effects a shrinking neuron may have on adjacent cells remains an important question.
In this study, we have revealed a critical phenomenon that protects neurons after axotomy. This mechanism may have far-reaching implications in various pathological conditions involving the nervous system, from direct traumas to neurodegenerative diseases associated with primary or secondary axonal injury.
Limitations of the study
We used a pharmacological approach to understand mechanism and significance of axonal shrinkage after axonal injury. Although we opted for selective antagonists where possible, the specificity of the results from such experiments is limited by proposed selectivity of the compounds. We are aware that genetical approaches would have provided more robust outputs; however, due to multiplicity of the investigated mechanisms, we did not find it feasible in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Ketamine HCl | Pfizer | Ketamine |
| Xylazine HCl | Bayer | Rompun |
| RPMI 1640 | Sigma | R8758 |
| Antibiotic antimycotic solution | Sigma | A5955 |
| Collagenase | Sigma | C7657 |
| Trypsin | Sigma | T4549 |
| DNase | Sigma | D5025 |
| Neurobasal A | Thermo Fisher | 10888022 |
| B27 | Thermo Fisher | 17504001 |
| GlutaMAX | Thermo Fisher | 35050061 |
| Poly-L-lysine | Sigma | P6282 |
| Percoll | Sigma | P4937 |
| Poly-D lysine | Sigma | P7280 |
| L-15 | Thermo Fisher | 11415064 |
| Hibernate A | Thermo Fisher | A1247501 |
| Gentamicin | Thermo Fisher | 15750060 |
| Papain | Sigma | P4762 |
| FBS | Thermo Fisher | 16000044 |
| Horse Serum | Thermo Fisher | 16050122 |
| Polystrene beads | Sigma | LB30 |
| Isoflurane | Adeka | ISOFLURANE USP |
| Kwik-sil | WPI | KWIK-SIL |
| 2,3-Butanedione monoxime (BDM) | Sigma | B0753 |
| Propidium Iodide | Thermo Fisher | P3566 |
| Clostridium Botulinum Exoenzyme C3 | Abcam | ab63835 |
| Cytochalasin E | Sigma | C2149 |
| Swinholide A | Sigma | 574776 |
| Blebbistatin | Sigma | B0560 |
| Nifedipine | Sigma | 481981 |
| ω-conotoxin GVIA | Sigma | 343781-M |
| ω -agatoxin-IVA | Sigma | A6719 |
| SNX-482 | Sigma | SML1150 |
| Mibefradil | Sigma | M5441 |
| Thapsigargin | Sigma | 239805 |
| PD150606 | Sigma | 513022 |
| HgCl2 | Sigma | M1136 |
| Y-27632 | Sigma | SCM075 |
| Tetrodotoxin | Sigma | T8024 |
| Experimental models: Organisms/strains | ||
| Mouse: B6 ACTb-EGFP | Jackson Laboratories | 003291, RRID: IMSR_JAX:003291 |
| Mouse: TauEGFP knock-in | Jackson Laboratories | 004779, RRID:IMSR_JAX:004779 |
| Mouse: Thy1-GFP line M | Jackson Laboratories | 007788, RRID:IMSR_JAX:007788 |
| Mouse: GCaMP6s - Ai96(RCL-GCaMP6s) | Jackson Laboratories | 024106, RRID:IMSR_JAX:024106 |
| Mouse: VGlut2-ires-cre | Jackson Laboratories | 016963, RRID:IMSR_JAX:016963 |
| Mouse: Balb/c | Jackson Laboratories | 000651, RRID:IMSR_JAX:000651 |
| Software and algorithms | ||
| Fiji | ImageJ | RRID:SCR_002285 |
| SPSS V24 | IBM | RRID:SCR_002865 |
| ZEN | Carl Zeiss | RRID:SCR_013672 |
| pClamp | Axon Instruments | RRID:SCR_011323 |
| SAS V9.4 | SAS Institute | RRID:SCR_008567 |
| GraphPad | Prism | RRID:SCR_002798 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Gürkan Öztürk (gozturk@medipol.edu.tr).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Animals
The use of animals in this study was approved by Istanbul Medipol University Animal Experimentation Ethical Committee. Maximum care was paid with total commitment to the 4R principles to use the minimum number of animals necessary for the research aims and to minimize suffering throughout the study. All procedures were conducted in compliance with European Council Directive 2010/63/EU. Breeding colonies of all mice strains were obtained from The Jackson Laboratory and bred at Istanbul Medipol University Experimental Animal Facility. These were general GFP - B6 ACTbEGFP (stock no: 003291, RRID: IMSR_JAX:003291),64 TauEGFP knock-in (stock no: 004779, RRID:IMSR_JAX:004779),43 Thy1-GFP line M (stock no: 007788, RRID:IMSR_JAX:007788),15 cross breeds of cre-dependent GCaMP6s - Ai96(RCL-GCaMP6s)65 (stock no: 024106, RRID:IMSR_JAX:024106) and VGlut2-ires-cre (stock no: 016963, RRID:IMSR_JAX:016963)66 and non-transgenic BALBc (stock no: 000651, RRID:IMSR_JAX:000651) mice. All animals are kept under 22–24oC temperature with 12 h light/12 h dark cycles. Food and water were provided ad libitum. Adult mice used in this study were 6–8 weeks old males and newborn mice were p1-3 days old. In in vivo studies, after single session experiments animals were immediately sacrificed by cervical dislocation while they are still under anesthesia. For evaluation of neuronal survival 24 h following peripheral nerve cut, skin was sutured to close the incision exposing only the vertebral window and animals were let recover from anesthesia on a heated pad after the first experimental session and sacrificed after final experiments.
Adult sensory neuron cultures
Dorsal root ganglia (DRG) were removed from male adult (6–8 week old) mice, and primary sensory neurons were isolated as described before.67 Briefly, animals were anesthetized with an I.P. injection of ketamin (Pfizer, 100 mg/kg) and Xylazine (10 mg/kg) and sacrificed by cervical transection. The vertebral column was cut out and quickly transferred into a dissection plate with RPMI1640 medium (Sigma). DRGs were collected under a stereo microscope and digested by collagenase (Sigma, 100 U/mL, 50 min), trypsin (Sigma, 1 mg/mL, 15 min) and DNAse (Sigma, 50 μg/mL) treatments, after which they were triturated through pipet tips of narrowing bores for 15 min and finally through a 26-gauge injector needle. The cell suspension obtained was spun at 120 g for 3 min, and the pellet was resuspended in Neurobasal A supplemented with B27 (Thermo Fisher), glutamine (Glutamax-Thermo Fisher), and antibiotic solution (Sigma) (NBA-B27). To eliminate nonneuronal cells, a gradient centrifuge technique was used. The cell suspension was carefully pipetted on top of a three-layer percoll (Sigma) gradient (60%, 35%, and 10% from bottom to top) prepared in NBA-B27 and spun at 3000 g for 20 min in a centrifuge at 4°C. The neurons collected from 35% layer were washed with NBA-B27 and spun once more at 120 g for 3 min, after which the supernatant was discarded, and the pellet was resuspended in NBA-B27.The suspension was transferred on 35 mm diameter glass-bottomed Petri dishes (WPI) coated with poly-L-lysine and laminin and incubated at 37°C with 5% CO2.
Newborn hippocampal neuron cultures
Postnatal (p1-p3) mice of either sex were sacrificed with CO2, and brains were collected under aseptic conditions. Hippocampi were surgically removed in a dissection medium (L15, 1% antibiotic-antimycotic solution), cut into small pieces, and digested with papain (Sigma, 12.5 U/mL, 45 min) with agitation at 4°C. Afterward, DNAse (Sigma, 50 μg/mL) was added and tissues were triturated via serial pipetting, and papain was inhibited with 10% FBS containing dissection medium for 15 min at 4°C. Then, cells were pelleted by centrifugation at 180 g for 5 min at 4°C, the dissection medium was discarded, and cells were plated on poly-D-lysine coated glass bottom culture dishes and incubated in NBA-B27 at 37°C with 5% CO2.
Adult cortical and hippocampal neuron cultures
Cortex and hippocampi were removed from male adult (6–8 week old) mice and neurons were cultured using a protocol adapted from.68 Briefly, mice were anesthetized with an I.P. injection of ketamin (Pfizer) and sacrificed by cervical transection. Hippocampi and cortex were surgically removed in dissection medium (Hibernate A, 1% gentamicin), cut into small pieces and digested with papain (Sigma, 12.5 U/mL, 45 min) at 30°C. Papain was removed after incubation and and tissue pieces were triturated in 10% FBS and DNase (Sigma, 50 μg/mL) containing disection medium. Cells were pelleted by centrifugation at 180 g for 5 min at 4°C and resuspended in culture medium 10% horse serum containing NBA-B27. Cells were plated on poly-D-lysine coated glass bottom culture dishes and incubated in NBA-B27 at 37°C with 5% CO2 and 9% O2.
Method details
Microscopy and in vitro manipulations
For microscopic imaging, the following systems (all from Zeiss) were used: LSM780 NLO, LSM880, and LSM7 MP laser scanning confocal and two-photon microscopes, Cell Observer time-lapse imaging system, and Palm Combi system. Various modules of Zeiss Axiovision and Zen software were used for time-lapse imaging of multiple positions at desired intervals, three dimensional scanning, and Ca++ signal imaging. Axotomy was performed with the femtosecond infrared lasers of two-photon microscopes in vivo, and UV lasers fitted to LSM880 and Palm laser microdissection - optical tweezer combi system in vitro. All in vitro microscopy studies were performed under physiological conditions in stage – top chambers that provide an atmosphere at 37°C with 5% CO2.
In vitro axotomy
A brief pulse (0.5–1 s) of UV (337 nm or 355 nm) laser beam was focused on axons about 50–100 μm distal to the soma emitting approximately 1–30 pulses per second, each lasting 3 nsec and delivering approximately 300 μJ of energy. Laser beam was focused on the axons for about 1–2 s until the transection was visible. For sham operations, an empty spot 50 μm away from the cell body was irradiated similarly.
Membrane tension measurement
The optical tweezer controlled by Palm software with a force measurement module was used for membrane tension measurements. For this, poly-L-lysin-coated 3 μm-diameter polystyrene beads (Sigma) were carried and attached to the neuronal plasma membrane by the optical trap. Then they were pulled vertical or parallel to the plasma membrane with the optical trap to measure cortical and lateral membrane tensions, respectively. The force resisting the pull was measured in piconewtons and reported by the software.
Osmolarity experiments
To validate the cell volume and area changes, cells were exposed to hyper and hypo-osmolar conditions. DRG neurons were cultured on 30-mm round cover glasses (Glaswarenfabrik Karl Knecht Sondheim, Germany). Cells were incubated in iso-osmolar aCSF solution for 30 min before imaging and z stack images were taken using confocal microscope to get baseline volume and area measurements. Hyper-osmolar or hypo-osmolar aCSF solutions were administrated through a metal perfusion chamber (NGFI, Graz, Austria) and custom-made pump-driven perfusion system. The standard extracellular isotonic solution (270 mOsmol/L): 130 mM NaCl, 2 mM CaCl2, 10 mM HEPES and 13 mM D-glucose in dH2O. Hypotonic solution (210 mOsm/L): NaCl was reduced from 130 to 100 mM, HEPES was reduced from 10 mM to 5mM. Hypertonic solution (350 mOsm/L): Glucose was increased to 80 mM pH of all liquids was adjusted to 7.3 with NaOH. z stack images taken 30 min after hyper and hypo-osmolar aCSF applications to determine volume and area changes.
Electrophysiological recordings
Patch-clamp pipettes were made of borosilicate glass (Sutter Instrument Borosilicate Glass with flament. O.D.: 1.5 mm, I.D.: 1.17 mm 10 cm Length, Novato, CA, USA). The standard extracellular bath solution contains 130 mM NaCl, 2 mM CaCl2, 10 mM HEPES, and 13 mM D-glucose, pH adjusted with NaOH to 7.35. The osmolarity of the solution was 270 mOsmol/L. The pipette (internal) solution for patch-clamp recording contains 130 mM K-gluconate, 5 mM EGTA, and 10 mM HEPES (270 mOsmol/L; pH 7.2) (adjusted with NaOH). The pipette resistances were about 4–6 MΩ. Also, the neurons were held at a potential of −70 mV. The resistance was made to be giga-seal (2–10 GΩ), and then the whole cell configuration was started with negative pressure. The Whole-cell patch-clamp technique was used to perform current-clamp recording by using an amplifier (Axon CNS MultiClamp 700B). Low-pass-filtering at 5–10 kHz was used during data recording. Sampling rates for current and voltage records were 10–20 kHz. A Digidata 1550B interface (Axon Instruments, Foster City, CA, USA) was used for the digitization of data, and a computer was used for storage and further analysis. pClamp software (version 11; Axon Instruments) was used for the generation of stimulus, acquisition, and offline analysis of data. To record from neurons during axotomy, the patch clamp rig was mounted on a Palm laser microdissection microscope, and after a giga-seal was created, a selected axon of the patched neuron was cut with a laser beam as described earlier.
Calcium imaging
Cultured DRG neurons of GCaMP6s - Ai96 mice were imaged with a confocal microscope before, during, and immediately, then every 5 min after axotomy. Change in green fluorescence emission was analyzed using ZEN software (Carl Zeiss, Germany).
Use of blocking reagents in vitro
Neurons were incubated with the blockers for 1 h before microscopic manipulations except for exoenzyme C3, whose incubation period was 6 h. All inhibitors were purchased from Sigma, except exoenzyme C3, which was obtained from Abcam. Cytochalasin E (CytE, 200 ng/mL)27 and swinholide A (SWH, 500 ng/mL)69 were used to depolymerize actin. Myosin activity was blocked with blebbistatin (BB, 100 μM)70 and 2,3 butanedione monoxime (BDM, 50mM).29 Voltage-gated calcium channel subtypes L, N, P/Q, R, and T were blocked with nifedipine (NIF, 10μM),71 ω-conotoxin GVIA (CNX, 1μM),72 ω -agatoxin-IVA (AGX, 200 nM),73 SNX482(SNX, 200nM),73 mibefradil (MIB, 10μM)74 and Tetrodotoxin (TTX, 1 μM)75 was used for blocking sodium channels respectively. To test the contribution of internally stored Ca++ to the shrinkage, neurons were incubated with thapsigargin for 60 min (TG, 2μM)23,76 to empty internal Ca++ stores. Since store-operated Ca++ entry is induced by depletion of internal stores, these experiments were performed in Ca++ - free DMEM.23 Then the medium was replaced with NBA-B27, and 30 min later, axotomies were performed. PD150606 (50μM)77 was used as a calpain inhibitor. Aquaporin channels were blocked using HgCl2 (100μM).78 Ras homolog gene family member A (RhoA) and its downstream effector Rho-associated protein kinase (ROCK) were inhibited with clostridium botulinum exoenzyme C3 protein (C3, 500 nM)79 and Y-27632 (10 μM),80 respectively.
Vertebral window surgery
Construction of an imaging window over lumbar DRGs was performed according to the method described by Chen et al.16 Briefly, the mice were anesthetized with 2% isoflurane (30% O2), and a small incision was made in the dorsal skin at the level of L1-L3 of the spine, paraspinal muscles and ligaments connected to right transverse processes of L1 to L3 were carefully dissected using fine scissors to expose DRG. The mouse was then placed lying on its left side onto a custommade vertebral holder, which allows access to L2 DRG for in vivo microscopy. Carefully removing surrounding muscles and connective tissue around the L2 DRG using a fine tweezer, DRG was exposed. To achieve optical clearance and stabilization, DRG and surrounding bone structures were covered with silicon adhesive Kwik-Sil (World Precision Instruments), and sealed with a 3-mm diameter round cover glass. The cover glass was fixed to vertebrae using cyanoacrylate glue and dental cement.
Cranial window surgery
Cranial window surgery was performed similarly to the protocol described by Dombeck et al.17 Briefly, the mice were anesthesized with 2% isoflurane (30% O2), and ∼3 mm crainotomy was made at skull overlaying the dorsal hippocampus using a trephine drill (Meisinger, Germany). Afterward, the dura was removed carefully with a fine forceps. Kwik-Sil was applied and 3 mm diameter round cover glass sealed for two-photon imaging of cortical neurons.
For hippocampal imaging, cortex was aspirated slowly during continuous saline application. The extraction process was performed at a slow pace, gradually aspirating tissue in increments of approximately 50–100 microns until hippocampus is visible. A small amount of Kwik-Sil was gently applied to the surface and two-photon imaging was performed.
In vivo axotomy
Thy1-GFP line M mice were preferred for single cell axotomy as distinctly labeled neurons with clearly visible axons were easier to identify and analyze after the procedure. Single-cell axotomy was performed by irradiating axons with Ti:Sa laser about 100 μm distal to the cell body. For this purpose, the laser was adjusted for a single spot scan with 100 iterations at 820 nm, delivering ∼270mJ power at the focal plane.
Peripheral nerve cut
TauEGFP knock-in mice were used to perform axotomy in all neurons within the DRG by cutting the peripheral nerve of DRG. This strain was preferred for these experiments due to strong expression of EGFP in neuronal soma, which enabled deep imaging and analyzing size changes. Using the two-photon microscope (LSM 7MP), neurons in L2 DRG were imaged through the vertebral window with 10×/0.45NA air objective, and 820 nm excitation laser before and 15 min after its peripheral nerve was completely cut with surgical scissors. The same DRG of the same animal was imaged before and 5 min and 24hrs after the nerve cut.
Myosin inhibition during nerve cut
Intrathecal delivery of myosin inhibitor 2,3-Butanedione monoxime (BDM) was performed as previously described.81 Briefly, a Hamilton syringe (Hamilton 80314, Hamilton, Reno, NV) with a 32-gauge needle was inserted between the L2-L3 vertebrae to the subarachnoid space. Access to the intrathecal space was confirmed by reflux of cerebrospinal fluid. Five microliters of SF containing 50 mg/mL BDM was injected intrathecally 30 min prior to in vivo experiments. To determine the cell death in DRG neurons, 50 μL of 1/1000 propidium iodide (PI, Molecular Probes), which stains nuclei of dead cells, was injected intravenously via the tail vein 1 h before the microscopic imaging.
Quantification and statistical analysis
Image analysis
Cross sectional areas (referred to as “size”) from two dimensional images and volumes of neurons from serial optical sections and fluorescence intensities were measured using Fiji82 and ZEN (Carl Zeiss, Germany) software. Image analyses were performed blind to the experimental condition.
Statistical analysis
IBM SPSS V24, SAS V9.4 and GraphPad Prism were used. The distribution of the data was evaluated with Kolmogorov-Smirnov test. Paired t-test for ratios or Wilcoxon test was used to compare neuronal size before and after axotomy. Though some datasets had a normal distribution, most of them did not, therefore, non-parametric tests were used for group comparisons. The distribution of a continuous marker was compared among two or more levels of a factor of interest by using Mann-Whitney test. Correlation analysis was used to analyze relationship between two variables. Change of marker overtime was modeled through random-coefficient models using the MIXED procedure in SAS, where spatial-power of time-lag covariance structure was used to account for the correlation structure among measurements. Similarly, repeated-measures models with variance-components covariance structure were also constructed using the MIXED procedure. To estimate the impact of a given predictor on the likelihood of survival, univariable Logistic regression was used and the significance of the predictor was assessed through both p values and Area Under the curve (AUC). A p < 0.05 is considered significant. Details of statistical methods used for individual experiments can be found in figure legends.
Acknowledgments
This research was funded by Yüzüncü Yıl University Directorate of Scientific Research Projects, grant TF073 (G.Ö.) and the Scientific and Technological Research Council of Turkey (TÜBİTAK), grant 107S358 (G.Ö.).
Author contributions
Conceptualization: G.Ö., M.Ş.A.; Methodology: G.Ö., A.H., M.K., E. Erdoğan; Investigation: G.Ö., M.Ş.A., S.B., E.N.Y., C.Ö., E.Y.K., E.K.O., N.A., N.C., E. Erdoğan, A.H.; Visualization: G.Ö., M.Ş.A., E.N.Y., E. Erdoğan, E. Eroglu; Funding acquisition: G.Ö.; Project administration and supervision: G.Ö.; Writing – original draft: G.Ö., Writing – review & editing: G.Ö., M.K., E. Eroglu.
Declaration of interests
Authors declare that they have no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107715.
Contributor Information
Emrah Eroglu, Email: emrah.eroglu@medipol.edu.tr.
Gürkan Öztürk, Email: gozturk@medipol.edu.tr.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Hill C.S., Coleman M.P., Menon D.K. Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends Neurosci. 2016;39:311–324. doi: 10.1016/j.tins.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greer J.E., Hånell A., McGinn M.J., Povlishock J.T. Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment. Acta Neuropathol. 2013;126:59–74. doi: 10.1007/s00401-013-1119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medana I.M., Esiri M.M. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–530. doi: 10.1093/brain/awg061. [DOI] [PubMed] [Google Scholar]
- 4.Rodemer W., Selzer M.E. Role of axon resealing in retrograde neuronal death and regeneration after spinal cord injury. Neural Regen. Res. 2019;14:399–404. doi: 10.4103/1673-5374.245330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cengiz N., Oztürk G., Erdoğan E., Him A., Oğuz E.K. Consequences of neurite transection in vitro. J. Neurotrauma. 2012;29:2465–2474. doi: 10.1089/neu.2009.0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandolesi G., Madeddu F., Bozzi Y., Maffei L., Ratto G.M. Acute physiological response of mammalian central neurons to axotomy: ionic regulation and electrical activity. FASEB J. 2004;18:1934–1936. doi: 10.1096/fj.04-1805fje. [DOI] [PubMed] [Google Scholar]
- 7.Rungta R.L., Choi H.B., Tyson J.R., Malik A., Dissing-Olesen L., Lin P.J.C., Cain S.M., Cullis P.R., Snutch T.P., MacVicar B.A. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. 2015;161:610–621. doi: 10.1016/j.cell.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 8.LoPachin R.M., Lehning E.J. Mechanism of calcium entry during axon injury and degeneration. Toxicol. Appl. Pharmacol. 1997;143:233–244. doi: 10.1006/taap.1997.8106. [DOI] [PubMed] [Google Scholar]
- 9.Yu S.P., Choi D.W. Na(+)-Ca2+ exchange currents in cortical neurons: concomitant forward and reverse operation and effect of glutamate. Eur. J. Neurosci. 1997;9:1273–1281. doi: 10.1111/j.1460-9568.1997.tb01482.x. [DOI] [PubMed] [Google Scholar]
- 10.Plotegher N., Filadi R., Pizzo P., Duchen M.R. Excitotoxicity Revisited: Mitochondria on the Verge of a Nervous Breakdown. Trends Neurosci. 2021;44:342–351. doi: 10.1016/j.tins.2021.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Blaesse P., Airaksinen M.S., Rivera C., Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Hasbani M.J., Hyrc K.L., Faddis B.T., Romano C., Goldberg M.P. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Exp. Neurol. 1998;154:241–258. doi: 10.1006/exnr.1998.6929. [DOI] [PubMed] [Google Scholar]
- 13.Mutalik S.P., Joseph J., Pullarkat P.A., Ghose A. Cytoskeletal Mechanisms of Axonal Contractility. Biophys. J. 2018;115:713–724. doi: 10.1016/j.bpj.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Moraes E.R., Kushmerick C., Naves L.A. Morphological and functional diversity of first-order somatosensory neurons. Biophys. Rev. 2017;9:847–856. doi: 10.1007/s12551-017-0321-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., Sanes J.R. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 16.Chen C., Zhang J., Sun L., Zhang Y., Gan W.B., Tang P., Yang G. Long-term imaging of dorsal root ganglia in awake behaving mice. Nat. Commun. 2019;10:3087. doi: 10.1038/s41467-019-11158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dombeck D.A., Harvey C.D., Tian L., Looger L.L., Tank D.W. Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat. Neurosci. 2010;13:1433–1440. doi: 10.1038/nn.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs A., Rigaud M., Sarantopoulos C.D., Filip P., Hogan Q.H. Contribution of calcium channel subtypes to the intracellular calcium signal in sensory neurons: the effect of injury. Anesthesiology. 2007;107:117–127. doi: 10.1097/01.anes.0000267511.21864.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scroggs R.S., Fox A.P. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J. Physiol. 1992;445:639–658. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carbone E., Lux H.D. A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature. 1984;310:501–502. doi: 10.1038/310501a0. [DOI] [PubMed] [Google Scholar]
- 21.Scroggs R.S., Fox A.P. Multiple Ca2+ currents elicited by action potential waveforms in acutely isolated adult rat dorsal root ganglion neurons. J. Neurosci. 1992;12:1789–1801. doi: 10.1523/JNEUROSCI.12-05-01789.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rigaud M., Gemes G., Weyker P.D., Cruikshank J.M., Kawano T., Wu H.E., Hogan Q.H. Axotomy depletes intracellular calcium stores in primary sensory neurons. Anesthesiology. 2009;111:381–392. doi: 10.1097/ALN.0b013e3181ae6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bird G.S., DeHaven W.I., Smyth J.T., Putney J.W., Jr. Methods for studying store-operated calcium entry. Methods. 2008;46:204–212. doi: 10.1016/j.ymeth.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gemes G., Bangaru M.L.Y., Wu H.E., Tang Q., Weihrauch D., Koopmeiners A.S., Cruikshank J.M., Kwok W.M., Hogan Q.H. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J. Neurosci. 2011;31:3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mercer J.C., Dehaven W.I., Smyth J.T., Wedel B., Boyles R.R., Bird G.S., Putney J.W., Jr. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor. J. Biol. Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bubb M.R., Spector I., Bershadsky A.D., Korn E.D. Swinholide A is a microfilament disrupting marine toxin that stabilizes actin dimers and severs actin filaments. J. Biol. Chem. 1995;270:3463–3466. doi: 10.1074/jbc.270.8.3463. [DOI] [PubMed] [Google Scholar]
- 27.Ruthel G., Hollenbeck P.J. Growth cones are not required for initial establishment of polarity or differential axon branch growth in cultured hippocampal neurons. J. Neurosci. 2000;20:2266–2274. doi: 10.1523/JNEUROSCI.20-06-02266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Limouze J., Straight A.F., Mitchison T., Sellers J.R. Specificity of blebbistatin, an inhibitor of myosin II. J. Muscle Res. Cell Motil. 2004;25:337–341. doi: 10.1007/s10974-004-6060-7. [DOI] [PubMed] [Google Scholar]
- 29.Siegman M.J., Mooers S.U., Warren T.B., Warshaw D.M., Ikebe M., Butler T.M. Comparison of the effects of 2,3-butanedione monoxime on force production, myosin light chain phosphorylation and chemical energy usage in intact and permeabilized smooth and skeletal muscles. J. Muscle Res. Cell Motil. 1994;15:457–472. doi: 10.1007/BF00122119. [DOI] [PubMed] [Google Scholar]
- 30.Eddleman C.S., Ballinger M.L., Smyers M.E., Godell C.M., Fishman H.M., Bittner G.D. Repair of plasmalemmal lesions by vesicles. Proc. Natl. Acad. Sci. USA. 1997;94:4745–4750. doi: 10.1073/pnas.94.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 32.Zhang G., Hu J., Rodemer W., Li S., Selzer M.E. RhoA activation in axotomy-induced neuronal death. Exp. Neurol. 2018;306:76–91. doi: 10.1016/j.expneurol.2018.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 34.Lu Q., Lu L., Chen W., Chen H., Xu X., Zheng Z. RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2015;253:669–680. doi: 10.1007/s00417-015-2985-3. [DOI] [PubMed] [Google Scholar]
- 35.Miroshnikova Y.A., Manet S., Li X., Wickström S.A., Faurobert E., Albiges-Rizo C. Calcium signaling mediates a biphasic mechanoadaptive response of endothelial cells to cyclic mechanical stretch. Mol. Biol. Cell. 2021;32:1724–1736. doi: 10.1091/mbc.E21-03-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart M.P., Helenius J., Toyoda Y., Ramanathan S.P., Muller D.J., Hyman A.A. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature. 2011;469:226–230. doi: 10.1038/nature09642. [DOI] [PubMed] [Google Scholar]
- 37.Bevers M.B., Neumar R.W. Mechanistic role of calpains in postischemic neurodegeneration. J. Cereb. Blood Flow Metab. 2008;28:655–673. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- 38.Vosler P.S., Brennan C.S., Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krieg M., Dunn A.R., Goodman M.B. Mechanical control of the sense of touch by beta-spectrin. Nat. Cell Biol. 2014;16:224–233. doi: 10.1038/ncb2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan S.L., Mattson M.P. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J. Neurosci. Res. 1999;58:167–190. [PubMed] [Google Scholar]
- 41.Papadopoulos M.C., Verkman A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013;14:265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yellin F., Li Y., Sreenivasan V.K.A., Farrell B., Johny M.B., Yue D., Sun S.X. Electromechanics and Volume Dynamics in Nonexcitable Tissue Cells. Biophys. J. 2018;114:2231–2242. doi: 10.1016/j.bpj.2018.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tucker K.L., Meyer M., Barde Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 44.Kelley B.J., Farkas O., Lifshitz J., Povlishock J.T. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp. Neurol. 2006;198:350–360. doi: 10.1016/j.expneurol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Agarwala S., Kalil R.E. Axotomy-induced neuronal death and reactive astrogliosis in the lateral geniculate nucleus following a lesion of the visual cortex in the rat. J. Comp. Neurol. 1998;392:252–263. doi: 10.1002/(sici)1096-9861(19980309)392:2<252::aid-cne7>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 46.Armstrong D.M., Terry R.D., Deteresa R.M., Bruce G., Hersh L.B., Gage F.H. Response of septal cholinergic neurons to axotomy. J. Comp. Neurol. 1987;264:421–436. doi: 10.1002/cne.902640309. [DOI] [PubMed] [Google Scholar]
- 47.Giehl K.M., Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy-induced death of rat corticospinal neurons in vivo. Eur. J. Neurosci. 1996;8:1167–1175. doi: 10.1111/j.1460-9568.1996.tb01284.x. [DOI] [PubMed] [Google Scholar]
- 48.Li L., Houenou L.J., Wu W., Lei M., Prevette D.M., Oppenheim R.W. Characterization of spinal motoneuron degeneration following different types of peripheral nerve injury in neonatal and adult mice. J. Comp. Neurol. 1998;396:158–168. [PubMed] [Google Scholar]
- 49.Ota T., Hara H., Miyawaki N. Brain-derived neurotrophic factor inhibits changes in soma-size of retinal ganglion cells following optic nerve axotomy in rats. J. Ocul. Pharmacol. Ther. 2002;18:241–249. doi: 10.1089/108076802760116160. [DOI] [PubMed] [Google Scholar]
- 50.Rich K.M., Disch S.P., Eichler M.E. The influence of regeneration and nerve growth factor on the neuronal cell body reaction to injury. J. Neurocytol. 1989;18:569–576. doi: 10.1007/BF01187077. [DOI] [PubMed] [Google Scholar]
- 51.McBride R.L., Feringa E.R., Smith B.E. The fate of prelabeled Clarke's column neurons after axotomy. Exp. Neurol. 1988;102:236–243. doi: 10.1016/0014-4886(88)90099-4. [DOI] [PubMed] [Google Scholar]
- 52.McBride R.L., Feringa E.R., Garver M.K., Williams J.K., Jr. Prelabeled red nucleus and sensorimotor cortex neurons of the rat survive 10 and 20 weeks after spinal cord transection. J. Neuropathol. Exp. Neurol. 1989;48:568–576. doi: 10.1097/00005072-198909000-00007. [DOI] [PubMed] [Google Scholar]
- 53.Brown J., Bridgman P.C. Role of myosin II in axon outgrowth. J. Histochem. Cytochem. 2003;51:421–428. doi: 10.1177/002215540305100403. [DOI] [PubMed] [Google Scholar]
- 54.Wan X., Harris J.A., Morris C.E. Responses of neurons to extreme osmomechanical stress. J. Membr. Biol. 1995;145:21–31. doi: 10.1007/BF00233304. [DOI] [PubMed] [Google Scholar]
- 55.Zhang H., Verkman A.S. Aquaporin-1 water permeability as a novel determinant of axonal regeneration in dorsal root ganglion neurons. Exp. Neurol. 2015;265:152–159. doi: 10.1016/j.expneurol.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buffoli B., Borsani E., Rezzani R., Rodella L.F. Chronic constriction injury induces aquaporin-2 expression in the dorsal root ganglia of rats. J. Anat. 2009;215:498–505. doi: 10.1111/j.1469-7580.2009.01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verkman A.S., Anderson M.O., Papadopoulos M.C. Aquaporins: important but elusive drug targets. Nat. Rev. Drug Discov. 2014;13:259–277. doi: 10.1038/nrd4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pál B. Involvement of extrasynaptic glutamate in physiological and pathophysiological changes of neuronal excitability. Cell. Mol. Life Sci. 2018;75:2917–2949. doi: 10.1007/s00018-018-2837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belin S., Maki B.A., Catlin J., Rein B.A., Popescu G.K. Membrane Stretch Gates NMDA Receptors. J. Neurosci. 2022;42:5672–5680. doi: 10.1523/JNEUROSCI.0350-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delmas P., Parpaite T., Coste B. PIEZO channels and newcomers in the mammalian mechanosensitive ion channel family. Neuron. 2022;110:2713–2727. doi: 10.1016/j.neuron.2022.07.001. [DOI] [PubMed] [Google Scholar]
- 61.Momin A., Bahrampour S., Min H.K., Chen X., Wang X., Sun Y., Huang X. Channeling Force in the Brain: Mechanosensitive Ion Channels Choreograph Mechanics and Malignancies. Trends Pharmacol. Sci. 2021;42:367–384. doi: 10.1016/j.tips.2021.02.006. [DOI] [PubMed] [Google Scholar]
- 62.Gaub B.M., Kasuba K.C., Mace E., Strittmatter T., Laskowski P.R., Geissler S.A., Hierlemann A., Fussenegger M., Roska B., Müller D.J. Neurons differentiate magnitude and location of mechanical stimuli. Proc. Natl. Acad. Sci. USA. 2020;117:848–856. doi: 10.1073/pnas.1909933117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chi S., Cui Y., Wang H., Jiang J., Zhang T., Sun S., Zhou Z., Zhong Y., Xiao B. Astrocytic Piezo1-mediated mechanotransduction determines adult neurogenesis and cognitive functions. Neuron. 2022;110:2984–2999.e8. doi: 10.1016/j.neuron.2022.07.010. [DOI] [PubMed] [Google Scholar]
- 64.Okabe M., Ikawa M., Kominami K., Nakanishi T., Nishimune Y. Green mice' as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 65.Madisen L., Garner A.R., Shimaoka D., Chuong A.S., Klapoetke N.C., Li L., van der Bourg A., Niino Y., Egolf L., Monetti C., et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron. 2015;85:942–958. doi: 10.1016/j.neuron.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vong L., Ye C., Yang Z., Choi B., Chua S., Jr., Lowell B.B. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Öztürk G., Cengiz N., Erdoğan E., Him A., Oğuz E.K., Yenidünya E., Ayşit N. Two distinct types of dying back axonal degeneration in vitro. Neuropathol. Appl. Neurobiol. 2013;39:362–376. doi: 10.1111/j.1365-2990.2012.01295.x. [DOI] [PubMed] [Google Scholar]
- 68.Brewer G.J., Torricelli J.R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- 69.Bourne J., Morgan J.R., Pieribone V.A. Actin polymerization regulates clathrin coat maturation during early stages of synaptic vesicle recycling at lamprey synapses. J. Comp. Neurol. 2006;497:600–609. doi: 10.1002/cne.21006. [DOI] [PubMed] [Google Scholar]
- 70.Gallo G. Myosin II activity is required for severing-induced axon retraction in vitro. Exp. Neurol. 2004;189:112–121. doi: 10.1016/j.expneurol.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 71.Bellono N.W., Bayrer J.R., Leitch D.B., Castro J., Zhang C., O'Donnell T.A., Brierley S.M., Ingraham H.A., Julius D. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell. 2017;170:185–198.e16. doi: 10.1016/j.cell.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abdulla F.A., Smith P.A. Axotomy- and autotomy-induced changes in Ca2+ and K+ channel currents of rat dorsal root ganglion neurons. J. Neurophysiol. 2001;85:644–658. doi: 10.1152/jn.2001.85.2.644. [DOI] [PubMed] [Google Scholar]
- 73.Murali S.S., Napier I.A., Mohammadi S.A., Alewood P.F., Lewis R.J., Christie M.J. High-voltage-activated calcium current subtypes in mouse DRG neurons adapt in a subpopulation-specific manner after nerve injury. J. Neurophysiol. 2015;113:1511–1519. doi: 10.1152/jn.00608.2014. [DOI] [PubMed] [Google Scholar]
- 74.Pan Z.H., Hu H.J., Perring P., Andrade R. T-type Ca(2+) channels mediate neurotransmitter release in retinal bipolar cells. Neuron. 2001;32:89–98. doi: 10.1016/s0896-6273(01)00454-8. [DOI] [PubMed] [Google Scholar]
- 75.Wrosch J.K., Einem V.v., Breininger K., Dahlmanns M., Maier A., Kornhuber J., Groemer T.W. Rewiring of neuronal networks during synaptic silencing. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-11729-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adasme T., Paula-Lima A., Hidalgo C. Inhibitory ryanodine prevents ryanodine receptor-mediated Ca(2)(+) release without affecting endoplasmic reticulum Ca(2)(+) content in primary hippocampal neurons. Biochem. Biophys. Res. Commun. 2015;458:57–62. doi: 10.1016/j.bbrc.2015.01.065. [DOI] [PubMed] [Google Scholar]
- 77.Van den Bosch L., Van Damme P., Vleminckx V., Van Houtte E., Lemmens G., Missiaen L., Callewaert G., Robberecht W. An alpha-mercaptoacrylic acid derivative (PD150606) inhibits selective motor neuron death via inhibition of kainate-induced Ca2+ influx and not via calpain inhibition. Neuropharmacology. 2002;42:706–713. doi: 10.1016/s0028-3908(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 78.Zhang H., Verkman A.S. Aquaporin-1 tunes pain perception by interaction with Na(v)1.8 Na+ channels in dorsal root ganglion neurons. J. Biol. Chem. 2010;285:5896–5906. doi: 10.1074/jbc.M109.090233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rohrbeck A., von Elsner L., Hagemann S., Just I. Uptake of clostridium botulinum C3 exoenzyme into intact HT22 and J774A.1 cells. Toxins. 2015;7:380–395. doi: 10.3390/toxins7020380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maddox A.S., Burridge K. RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J. Cell Biol. 2003;160:255–265. doi: 10.1083/jcb.200207130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ogawa N., Terashima T., Oka K., Chan L., Kojima H. Gene therapy for neuropathic pain using dorsal root ganglion-targeted helper-dependent adenoviral vectors with GAD67 expression. Pain Rep. 2018;3:e695. doi: 10.1097/PR9.0000000000000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The neuron expresses Ca++ sensor - GCaMP6s.
The neuron expresses Ca++ sensor - GCaMP6s.
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.