

Abstract
Background
Risperidone is the first new generation antipsychotic drug made available in a long‐acting injection formulation.
Objectives
To examine the effects of depot risperidone for treatment of schizophrenia or related psychoses in comparison with placebo, no treatment or other antipsychotic medication.
To critically appraise and summarise current evidence on the resource use, cost and cost‐effectiveness of risperidone (depot) for schizophrenia.
Search methods
We searched the Cochrane Schizophrenia Group's Register (December 2002, 2012, and October 28, 2015). We also checked the references of all included studies, and contacted industry and authors of included studies.
Selection criteria
Randomised clinical trials comparing depot risperidone with other treatments for people with schizophrenia and/or schizophrenia‐like psychoses.
Data collection and analysis
Two review authors independently selected trials, assessed trial quality and extracted data. For dichotomous data, we calculated the risk ratio (RR), with 95% confidence interval (CI). For continuous data, we calculated mean differences (MD). We assessed risk of bias for included studies and created 'Summary of findings' tables using GRADE.
Main results
Twelve studies, with a total of 5723 participants were randomised to the following comparison treatments:
Risperidone depot versus placebo
Outcomes of relapse and improvement in mental state were neither measured or reported. In terms of other primary outcomes, more people receiving placebo left the study early by 12 weeks (1 RCT, n=400, RR 0.74 95% CI 0.63 to 0.88, very low quality evidence), experienced severe adverse events in short term (1 RCT, n=400, RR 0.59 95% CI 0.38 to 0.93, very low quality evidence). There was however, no difference in levels of weight gain between groups (1 RCT, n=400, RR 2.11 95% CI 0.48 to 9.18, very low quality evidence).
Risperidone depot versus general oral antipsychotics
The outcome of improvement in mental state was not presented due to high levels of attrition, nor were levels of severe adverse events explicitly reported. Most primary outcomes of interest showed no difference between treatment groups. However, more people receiving depot risperidone experienced nervous system disorders (long‐term:1 RCT, n=369, RR 1.34 95% CI 1.13 to 1.58, very‐low quality evidence).
Risperidone depot versus oral risperidone
Data for relapse and severe adverse events were not reported. All outcomes of interest were rated as moderate quality evidence. Main results showed no differences between treatment groups with equivocal data for change in mental state, numbers leaving the study early, any extrapyramidal symptoms, weight increase and prolactin‐related adverse events.
Risperidone depot versus oral quetiapine
Relapse rates and improvement in mental state were not reported. Fewer people receiving risperidone depot left the study early (long‐term: 1 RCT, n=666, RR 0.84 95% CI 0.74 to 0.95, moderate quality evidence). Experience of serious adverse events was similar between groups (low quality evidence), but more people receiving depot risperidone experienced EPS (1 RCT, n=666, RR 1.83 95% CI 1.07 to 3.15, low quality evidence), had greater weight gain (1 RCT, n=666, RR 1.25 95% CI 0.25 to 2.25, low quality evidence) and more prolactin‐related adverse events (1 RCT, n=666, RR 3.07 95% CI 1.13 to 8.36, very low quality evidence).
Risperidone depot versus oral aripiprazole
Relapse rates, mental state using PANSS, leaving the study early, serious adverse events and weight increase were similar between groups. However more people receiving depot risperidone experienced prolactin‐related adverse events compared to those receiving oral aripiprazole (2 RCTs, n=729, RR 9.91 95% CI 2.78 to 35.29, very low quality of evidence).
Risperidone depot versus oral olanzapine
Relapse rates were not reported in any of the included studies for this comparison. Improvement in mental state using PANSS and instances of severe adverse events were similar between groups. More people receiving depot risperidone left the study early than those receiving oral olanzapine (1 RCT, n=618, RR 1.32 95% CI 1.10 to 1.58, low quality evidence) with those receiving risperidone depot also experiencing more extrapyramidal symptoms (1 RCT, n=547, RR 1.67 95% CI 1.19 to 2.36, low quality evidence). However, more people receiving oral olanzapine experienced weight increase (1 RCT, n=547, RR 0.56 95% CI 0.42 to 0.75, low quality evidence).
Risperidone depot versus atypical depot antipsychotics (specifically paliperidone palmitate)
Relapse rates were not reported and rates of response using PANSS, weight increase, prolactin‐related adverse events and glucose‐related adverse events were similar between groups. Fewer people left the study early due to lack of efficacy from the risperidone depot group (long term: 1 RCT, n=749, RR 0.60 95% CI 0.45 to 0.81, low quality evidence), but more people receiving depot risperidone required use of EPS‐medication (2 RCTs, n=1666, RR 1.46 95% CI 1.18 to 1.8, moderate quality evidence).
Risperidone depot versus typical depot antipsychotics
Outcomes of relapse, severe adverse events or movement disorders were not reported. Outcomes relating to improvement in mental state demonstrated no difference between groups (low quality evidence). However, more people receiving depot risperidone compared to other typical depots left the study early (long‐term:1 RCT, n=62, RR 3.05 95% CI 1.12 to 8.31, low quality evidence).
Authors' conclusions
Depot risperidone may be more acceptable than placebo injection but it is hard to know if it is any more effective in controlling the symptoms of schizophrenia. The active drug, especially higher doses, may be associated with more movement disorders than placebo. People already stabilised on oral risperidone may continue to maintain benefit if treated with depot risperidone and avoid the need to take tablets, at least in the short term. In people who are happy to take oral medication the depot risperidone is approximately equal to oral risperidone. It is possible that the depot formulation, however, can bring a second‐generation antipsychotic to people who do not reliably adhere to treatment. People with schizophrenia who have difficulty adhering to treatment, however, are unlikely to volunteer for a clinical trial. Such people may gain benefit from the depot risperidone with no increased risk of extrapyramidal side effects.
Plain language summary
Long‐acting preparation of risperidone for schizophrenia
Review question
Risperidone is a newer antipsychotic drug that was the first available as a long‐lasting injection (a depot injection). The review examines the clinical effects of depot risperidone for people with schizophrenia.
Background
People with schizophrenia often hear voices and see things (hallucinations) and have strange beliefs (delusions). People can also become withdrawn, socially isolated, tired and apathetic. The main treatment for these symptoms of schizophrenia is antipsychotic drugs. However, these drugs can have serious side effects, such as weight gain, uncontrollable shaking, tremors, spasms and tiredness. These side effects often mean that people stop taking their medication (non‐ compliance), which may lead to relapse.
Study characteristics
The review was updated in 2015 and includes 12 studies with 5723 people who received risperidone depot or a range of other treatments (placebo, general oral antipsychotics, oral risperidone, oral quetiapine, oral aripiprazole, oral olanzapine, atypical/newer depot antipsychotics, older depot antipsychotics).
Key results
It is difficult to know from the results of this review if depot risperidone is any more effective in treating the symptoms of schizophrenia than placebo or other treatments. For people who are happy to take oral medication, depot risperidone is about equal to oral risperidone. People on oral risperidone may continue to benefit if treated with depot risperidone, without the need to take tablets. However, in high doses, depot risperidone can have serious side effects, particularly movement disorders, uncontrollable shaking, spasms and tremors. Depot risperidone may bring this new antipsychotic to people who stop taking their tablets, so helping reduce relapse and with little increased risk of side effects.
Quality of the evidence
The quality of evidence presented is, in the main, low and at best moderate. There is the need for large, long‐term and well reported trials on depot risperidone for people with schizophrenia. Depot injections are often used on people who refuse treatment. Such people are difficult to include in studies.
Written by a consumer, Ben Gray, Senior Peer Researcher, McPin Foundation. http://mcpin.org/
Summary of findings
Summary of findings for the main comparison. RISPERIDONE DEPOT compared with PLACEBO for schizophrenia.
| RISPERIDONE DEPOT compared with PLACEBO for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: PLACEBO | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| PLACEBO | RISPERIDONE DEPOT | |||||
| Global state: Relapse ‐ long term ‐ not measured | See comment | See comment | Not estimable | ‐ | See comment | No study reported this outcome. |
| Mental state: clinically significant improvement in mental state ‐ long term1 ‐ not reported | See comment | See comment | Not estimable1 | ‐ | See comment | Study reported PANSS responder rate, but unusable due to high attrition. |
| Leaving the study early: Any reason ‐ all doses risperidone depot ‐ short term | 694 per 1000 | 513 per 1000 (437 to 611) | RR 0.74 (0.63 to 0.88) | 400 (1 study) | ⊕⊝⊝⊝ very low2,3 | |
| Adverse events: General: Severe adverse event ‐ any dose risperidone depot ‐ short term Spontaneous reporting by study participants | 235 per 10004 | 138 per 1000 (89 to 218) | RR 0.59 (0.38 to 0.93) | 400 (1 study) | ⊕⊝⊝⊝ very low2,5 | |
| Adverse events: Specific: Weight gain ‐ all doses of depot risperidone ‐ short term Spontaneous reporting by study participants | 20 per 1000 | 43 per 1000 (10 to 187) | RR 2.11 (0.48 to 9.18) | 400 (1 study) | ⊕⊝⊝⊝ very low2,6 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Not reported: only included study (Kane 2002*) reported PANSS responder rate, but these data were unusable due to high levels of attrition. 2 Risk of bias: 'very serious' ‐ high attrition in one included study (Kane 2002*) of greater than 50% overall. Research supported by Johnson and Johnson/ Janssen, producers of depot risperidone. 3 Imprecision: 'serious' ‐ only one small study reported data for this comparison. 4 Control risk: mean baseline presented for one individual study. 5 Imprecision: 'serious' ‐ adverse events were reported spontaneously by participants, rather than systematically assessed by the researchers. This could effect the precision of the results as there is only one study (Kane 2002*) addressing this comparison. 6 Imprecision: 'serious'‐ the method of measuring weight gain and threshold for reporting it were not described. This could effect the precision of the results as there is only one study (Kane 2002*) addressing this comparison.
Summary of findings 2. RISPERIDONE DEPOT compared with GENERAL ORAL ANTIPSYCHOTICS for schizophrenia.
| RISPERIDONE DEPOT compared with GENERAL ORAL ANTIPSYCHOTICS for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: GENERAL ORAL ANTIPSYCHOTICS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| GENERAL ORAL ANTIPSYCHOTICS | RISPERIDONE DEPOT | |||||
| Global state: Relapse (any reason) ‐ long term Number of participants relapsing in each treatment arm. | Moderate | RR 2.13 (0.84 to 5.43) | 63 (1 study) | ⊕⊝⊝⊝ very low2,3 | Criteria for relapse were derived from Csernansky 2002.<BR/> | |
| 161 per 10001 | 343 per 1000 (135 to 874) | |||||
| Mental state: clinically significant improvement in mental state ‐ long term | See comment | See comment | Not estimable | 0 (0) | See comment | Outcomes relating to mental state were unusable due to high study attrition. |
| Leaving the study early: Any reason ‐ long term | Study population | RR 1.24 (0.98 to 1.57) | 467 (2 studies) | ⊕⊕⊕⊝ moderate | ||
| 322 per 10004 | 399 per 1000 (315 to 505) | |||||
| Moderate | ||||||
| 387 per 10004 | 480 per 1000 (379 to 608) | |||||
| Adverse events: General: Severe adverse event ‐ any dose risperidone depot ‐ short term | See comment | See comment | Not estimable | 0 (0) | See comment | "Severe adverse events" were not explicitly reported. |
| Adverse events: Specific ‐ prolactin‐related ‐ long term It is unclear how adverse events were reported | Low | RR 10.27 (0.59 to 180.05) | 85 (1 study) | ⊕⊝⊝⊝ very low2,6 | ||
| 10 per 10005 | 103 per 1000 (6 to 1000) | |||||
| Moderate | ||||||
| 100 per 10005 | 1000 per 1000 (59 to 1000) | |||||
| High | ||||||
| 200 per 10005 | 1000 per 1000 (118 to 1000) | |||||
| Adverse events: Specific ‐ weight increase ‐ long term It is unclear how adverse events were reported | Study population | RR 1.33 (0.56 to 3.17) | 85 (1 study) | ⊕⊝⊝⊝ very low2,6 | ||
| 171 per 10004 | 227 per 1000 (96 to 541) | |||||
| Moderate | ||||||
| 171 per 10004 | 227 per 1000 (96 to 542) | |||||
| Adverse events: Nervous system disorders (inc. EPS) ‐ long term It is unclear how adverse events were reported | Study population | RR 1.34 (1.13 to 1.58) | 369 (1 study) | ⊕⊝⊝⊝ very low2,6 | ||
| 171 per 10004 | 227 per 1000 (96 to 541) | |||||
| Moderate | ||||||
| 171 per 10004 | 227 per 1000 (96 to 542) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: mean baseline presented for one individual study. 2 Risk of bias: 'very serious' ‐ a high level of attrition (> 50%), the open‐label nature of this study and the fact that it was supported by the manufacturers of depot risperidone result in a very serious risk of bias. 3 Imprecision: 'serious' ‐ the sample size for this outcome was small (n = 63). 4 Assumed risk: median control group risk from the studies. 5 Assumed risk: control risk relates to 'low' (0%). 6 Serious risk of imprecision due to the small sample size of this study.
Summary of findings 3. RISPERIDONE DEPOT compared with ORAL RISPERIDONE for schizophrenia.
| RISPERIDONE DEPOT compared with ORAL RISPERIDONE for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: ORAL RISPERIDONE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ORAL RISPERIDONE | RISPERIDONE DEPOT | |||||
| Global state: Relapse ‐ long term | See comment | See comment | Not estimable | 0 (0) | See comment | Outcomes relating to relapse were not available for this comparison. |
| Mental state: average PANSS total score at endpoint (non‐ITT data) PANSS total scores (30 to 210) Higher scores are worse. | The mean mental state: average PANSS total score at endpoint (non‐ITT data) in the intervention groups was 1.05 higher (0.77 lower to 2.88 higher) | 591 (2 studies) | ⊕⊕⊕⊝ moderate1 | |||
| Leaving the study early: Any reason ‐ short term | Study population | RR 1.28 (0.92 to 1.79) | 690 (2 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 145 per 10002 | 185 per 1000 (133 to 259) | |||||
| Moderate | ||||||
| 78 per 10002 | 100 per 1000 (72 to 140) | |||||
| Adverse events: General: Severe adverse event ‐ any dose risperidone depot ‐ short term | See comment | See comment | Not estimable | 0 (0) | See comment | "Severe adverse events" were not explicitly reported by these studies. |
| Adverse events: Movement disorder ‐ any extra pyramidal symptoms ‐ short term | Study population | RR 1.05 (0.59 to 1.88) | 640 (1 study) | ⊕⊕⊕⊝ moderate4 | ||
| 65 per 10003 | 69 per 1000 (39 to 123) | |||||
| Moderate | ||||||
| 65 per 10003 | 68 per 1000 (38 to 122) | |||||
| Adverse events: Specific: Mean (SD) weight increase in kg ‐ short term | The mean adverse events: specific: mean (SD) weight increase in kg ‐ short term in the control groups was 0.2 points | The mean adverse events: specific: mean (SD) weight increase in kg ‐ short term in the intervention groups was 0.2 higher (0.35 lower to 0.75 higher) | 640 (1 study) | ⊕⊕⊕⊝ moderate4 | ||
| Adverse events: Specific ‐ prolactin‐related | Moderate | RR 0.5 (0.15 to 1.65) | 640 (1 study) | ⊕⊕⊕⊝ moderate4 | ||
| 25 per 10003 | 12 per 1000 (4 to 41) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: 'serious' ‐ both studies received funding support from the manufacturers of risperidone depot 2 Assumed risk: median control group risk from the studies. 3 Assumed risk: mean baseline presented for one individual study. 4 Risk of bias: 'serious' ‐ this research was supported by the manufacturers of risperidone depot.
Summary of findings 4. RISPERIDONE DEPOT compared with ORAL QUETIAPINE for schizophrenia.
| RISPERIDONE DEPOT compared with ORAL QUETIAPINE for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: ORAL QUETIAPINE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ORAL QUETIAPINE | RISPERIDONE DEPOT | |||||
| Global state: Relapse ‐ long term | See comment | See comment | Not estimable | 0 (0) | See comment | Criteria for relapse were derived from Csernansky 2002. <BR/> Outcomes relating to relapse were reported, but were unusable due to study attrition. |
| Mental state: clinically significant improvement in mental state ‐ long term | See comment | See comment | Not estimable | 0 (0) | See comment | Outcomes relating to mental state were unusable due to high study attrition. |
| Leaving the study early: Any reason ‐ long term | Moderate | RR 0.84 (0.74 to 0.95) | 666 (1 study) | ⊕⊕⊕⊝ moderate2,3 | ||
| 644 per 10001 | 541 per 1000 (477 to 612) | |||||
| Adverse events: General ‐ serious Recorded at each follow‐up visit. | Moderate | RR 0.84 (0.62 to 1.13) | 666 (1 study) | ⊕⊕⊝⊝ low2,3,4 | ||
| 229 per 10001 | 192 per 1000 (142 to 259) | |||||
| Adverse events: Movement disorder ‐ any extra pyramidal symptom | Moderate | RR 1.83 (1.07 to 3.15) | 666 (1 study) | ⊕⊕⊝⊝ low2,3,4 | ||
| 56 per 10001 | 102 per 1000 (60 to 176) | |||||
| Adverse events: Specific: Mean (SD) weight increase in kg ‐ long term | The mean adverse events: specific: mean (SD) weight increase in kg ‐ long term in the intervention groups was 1.25 higher (0.25 to 2.25 higher) | 666 (1 study) | ⊕⊕⊝⊝ low2,3,4 | |||
| Adverse events: Specific ‐ prolactin‐related Reported by participants at follow‐up visits | Study population | RR 3.07 (1.13 to 8.36) | 666 (1 study) | ⊕⊕⊝⊝ low2,3,4 | ||
| 15 per 10001 | 46 per 1000 (17 to 124) | |||||
| Moderate | ||||||
| 15 per 10001 | 46 per 1000 (17 to 125) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: mean baseline risk used for one included study. 2 Risk of bias: 'serious' ‐ this study was supported by the manufacturers of risperidone depot. 3 Risk of bias: 'serious' ‐ this study was open‐label in nature. 4 Risk of bias: 'very serious' ‐ study attrition was high (> 50%).
Summary of findings 5. RISPERIDONE DEPOT compared with ORAL ARIPIPRAZOLE for schizophrenia.
| RISPERIDONE DEPOT compared with ORAL ARIPIPRAZOLE for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: ORAL ARIPIPRAZOLE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ORAL ARIPIPRAZOLE | RISPERIDONE DEPOT | |||||
| Global state: Relapse (any reason) ‐ long term Assessed by 5 blinded raters in accordance with study criteria (see comment). | Moderate | RR 1.05 (0.83 to 1.33) | 349 (1 study) | ⊕⊕⊝⊝ low2,3 | Criteria for relapse were derived from Csernansky 2002. | |
| 436 per 10001 | 458 per 1000 (362 to 580) | |||||
| Mental state: Average change scores‐ long term PANSS total score (30 to 210), higher scores are worse. | The mean mental state: average change scores‐ long term in the intervention groups was 0.1 lower (3.15 lower to 2.95 higher) | 349 (1 study) | ⊕⊕⊝⊝ low2,3 | |||
| Leaving the study early: Any reason ‐ long term | Study population | RR 0.83 (0.53 to 1.3) | 723 (2 studies) | ⊕⊝⊝⊝ very low5,6 | ||
| 387 per 10004 | 321 per 1000 (205 to 503) | |||||
| Moderate | ||||||
| 531 per 10004 | 441 per 1000 (281 to 690) | |||||
| Adverse events: General ‐ serious Unclear how these events were reported | Study population | RR 0.96 (0.66 to 1.39) | 729 (2 studies) | ⊕⊝⊝⊝ very low5,6 | ||
| 190 per 10004 | 182 per 1000 (125 to 264) | |||||
| Moderate | ||||||
| 177 per 10004 | 170 per 1000 (117 to 246) | |||||
| Adverse events: Movement disorder ‐ any extra pyramidal symptoms | Study population | RR 1.19 (0.91 to 1.55) | 729 (2 studies) | ⊕⊝⊝⊝ very low5,6 | ||
| 285 per 10004 | 339 per 1000 (259 to 442) | |||||
| Moderate | ||||||
| 196 per 10004 | 233 per 1000 (178 to 304) | |||||
| Adverse events: Specific ‐ weight increase | Moderate | RR 1.57 (0.38 to 6.45) | 374 (1 study) | ⊕⊕⊝⊝ low2,3 | ||
| 44 per 10001 | 69 per 1000 (17 to 284) | |||||
| Adverse events: Specific ‐ prolactin‐related | Study population | RR 9.91 (2.78 to 35.29) | 729 (2 studies) | ⊕⊝⊝⊝ very low5,6 | ||
| 9 per 10004 | 90 per 1000 (25 to 319) | |||||
| Moderate | ||||||
| 6 per 10004 | 59 per 1000 (17 to 212) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: mean baseline risk presented for one individual study. 2 Risk of bias: 'very serious' ‐ a number of the study authors were employed by the manufacturers of risperidone depot at the time of the study. 3 Risk of bias: 'serious' ‐ serious risk of bias due to the open nature label of the study. 4 Assumed risk: median control group risk from the studies. 5 Risk of bias: 'very serious' ‐ serious risk of bias as both studies were open‐label and supported by the manufacturers of risperidone depot. 6 Imprecision: 'serious' ‐ possibly serious risk of imprecision in Gaebel 2010* as the aripiprazole arm of this study was very small (n = 45) compared to the risperidone depot (n = 329) arm.
Summary of findings 6. RISPERIDONE DEPOT compared with ORAL OLANZAPINE for schizophrenia.
| RISPERIDONE DEPOT compared with ORAL OLANZAPINE for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: ORAL OLANZAPINE | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ORAL OLANZAPINE | RISPERIDONE DEPOT | |||||
| Global state: Relapse ‐ long term | See comment | See comment | Not estimable | 0 (0) | See comment | Outcomes relating to relapse were not reported for this comparison. |
| Mental state: Average change scores ‐ long term PANSS total score (30‐210), high scores are worse. | The mean mental state: average change scores ‐ long term in the intervention groups was 0.1 higher (3.96 lower to 4.16 higher) | 361 (1 study) | ⊕⊕⊝⊝ low1,2,3 | |||
| Leaving the study early: Any reason ‐ long term | Study population | RR 1.32 (1.1 to 1.58) | 618 (1 study) | ⊕⊕⊝⊝ low1,2,3 | ||
| 377 per 10004 | 497 per 1000 (414 to 595) | |||||
| Moderate | ||||||
| 377 per 10004 | 498 per 1000 (415 to 596) | |||||
| Adverse events: General ‐ serious | Moderate | RR 1.1 (0.8 to 1.51) | 547 (1 study) | ⊕⊕⊝⊝ low1,2,3 | ||
| 210 per 10004 | 231 per 1000 (168 to 317) | |||||
| Adverse events: Movement disorder ‐ any extra pyramidal symptoms | Moderate | RR 1.67 (1.19 to 2.36) | 547 (1 study) | ⊕⊕⊝⊝ low1,2,3 | ||
| 150 per 10004 | 250 per 1000 (179 to 354) | |||||
| Adverse events: Specific ‐ weight increase | Moderate | RR 0.56 (0.42 to 0.75) | 547 (1 study) | ⊕⊕⊝⊝ low1,2,3 | ||
| 360 per 10004 | 202 per 1000 (151 to 270) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: 'very serious' ‐ serious risk of bias due to study attrition in excess of 50%. 2 Risk of bias: 'serious' ‐ serious risk of bias as this study was supported by the manufacturers of risperidone depot, and some of the authors are employed by the same. 3 Risk of bias: 'serious' ‐ serious risk of bias due to the open‐label nature of the study. 4 Assumed risk: mean baseline risk from one included study.
Summary of findings 7. RISPERIDONE DEPOT compared with ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE) for schizophrenia.
| RISPERIDONE DEPOT compared with ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE) for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE) | RISPERIDONE DEPOT | |||||
| Global state: Relapse ‐ long term | See comment | See comment | Not estimable | 0 (0) | See comment | Outcomes relating to relapse were not reported for this comparison. |
| Mental state: PANSS responders (ITT data) ‐ medium term PANSS responders‐ participants achieving a >30% improvement in total score | Study population | RR 1.01 (0.83 to 1.23) | 1326 (2 studies) | ⊕⊕⊕⊝ moderate2,3 | ||
| 585 per 10001 | 591 per 1000 (486 to 720) | |||||
| Moderate | ||||||
| 619 per 10001 | 625 per 1000 (514 to 761) | |||||
| Leaving the study early: lack of efficacy ‐ long term | Study population | RR 0.60 (0.45 to 0.81) | 749 (1 study) | ⊕⊕⊝⊝ low2,3,4 | ||
| 361 per 10001 | 307 per 1000 (275 to 340) | |||||
| Moderate | ||||||
| 280 per 10001 | 238 per 1000 (213 to 263) | |||||
| Adverse events: Movement disorder requiring the use of anti‐EPS medication ‐ medium term | Study population | RR 1.46 (1.18 to 1.8) | 1666 (2 studies) | ⊕⊕⊕⊝ moderate2,3,5 | ||
| 122 per 10001 | 178 per 1000 (144 to 220) | |||||
| Moderate | ||||||
| 182 per 10001 | 266 per 1000 (215 to 328) | |||||
| Adverse events: Body weight (mean increase) ‐ medium/long term | The mean adverse events: body weight (mean increase) ‐ medium/long term in the intervention groups was 0.18 higher (0.36 lower to 0.72 higher) | 2350 (3 studies) | ⊕⊕⊝⊝ low2,3,4 | |||
| Adverse events: Any prolactin‐related ‐ medium term | Study population | RR 1.02 (0.61 to 1.71) | 1666 (2 studies) | ⊕⊕⊕⊝ moderate2,3 | ||
| 32 per 10001 | 33 per 1000 (20 to 55) | |||||
| Moderate | ||||||
| 48 per 10001 | 49 per 1000 (29 to 82) | |||||
| Adverse events: Any glucose‐related ‐ medium/long term | 10 per 10001 | 18 per 1000 (9 to 36) | RR 1.79 (0.89 to 3.61) | 2413 (3 studies) | ⊕⊕⊝⊝ low2,3,4 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Assumed risk: median control group risk from the studies. 2 Risk of bias: 'serious' ‐ Li 2011 was open‐label and supported by the manufacturer of risperidone depot. 3 Risk of bias: 'serious' ‐ Pandina 2011 was supported by the manufacturer of risperidone depot. 4 Risk of bias: 'serious' ‐ as the attrition rate of Fleischhacker 2011 was in excess of 50%, and the study was supported by the manufacturer of risperidone depot. 5 Possible imprecision: the rate of movement disorder requiring anti‐EPS medication may not be a reflection of the true rate of movement disorders.
Summary of findings 8. RISPERIDONE DEPOT compared with TYPICAL DEPOT ANTIPSYCHOTICS for schizophrenia.
| RISPERIDONE DEPOT compared with TYPICAL DEPOT ANTIPSYCHOTICS for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: Intervention: RISPERIDONE DEPOT Comparison: TYPICAL DEPOT ANTIPSYCHOTICS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| TYPICAL DEPOT ANTIPSYCHOTICS | RISPERIDONE DEPOT | |||||
| Global state: Relapse ‐ long term | See comment | See comment | Not estimable | 0 (0) | See comment | Outcomes relating to relapse were not reported for this comparison. |
| Mental state: Total average scores (PANSS, high score = worse) ‐ long term | The mean mental state: total average scores (PANSS, high score = worse) ‐ long term in the intervention groups was 1.8 higher (10.04 lower to 13.64 higher) | 43 (1 study) | ⊕⊕⊝⊝ low1,2 | |||
| Leaving the study early for any reason ‐ long term | Study population | RR 3.05 (1.12 to 8.31) | 62 (1 study) | ⊕⊕⊝⊝ low1,2 | ||
| 133 per 10003 | 407 per 1000 (149 to 1000) | |||||
| Moderate | ||||||
| 133 per 10003 | 406 per 1000 (149 to 1000) | |||||
| Adverse events: General: Severe adverse event | See comment | See comment | Not estimable | 0 (0) | See comment | "Severe adverse events" were not explicitly reported for this comparison. |
| Adverse events: related to movement disorder, weight gain, prolactin levels and glucose metabolism ‐ medium/long term ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Outcomes relating to specific adverse events were not reported in such as way as to be useable. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: 'serious' ‐ due to the open‐label nature of this study. 2 Imprecision: 'serious' ‐ due to the small size of the single study. 3 Assumed risk: median control group risk from the studies.
Background
Description of the condition
Schizophrenia is a major, often chronic, psychiatric disease that close to seven people in every 1000 will be affected by at some point during their lifetime (McGrath 2008). Antipsychotic drugs are effective for treating acute episodes and for preventing relapse (Davis 1977; Davis 1986). These drugs are usually given orally, but compliance is poor and ranges from 20% to 89% with an average of 50% (Fenton 1997; Young 1986). This means that, on average, half the patients treated with these drugs will not comply with prescribed medication. This is probably due to a combination of various factors such as the erosion of insight that accompanies psychotic illnesses, adverse effects and human nature.
Description of the intervention
Long‐acting depot antipsychotics, given by injection into the muscle, should be helpful in increasing compliance with medication. In studies comparing one depot with another, attrition rates are markedly lower than in studies comparing oral preparations, but in trials comparing an oral with a depot preparation, there are no differences in the attrition rates between groups (Adams 2001). This is likely to be due to a limitation in the design of the relevant studies, as people participating in randomised trials are more likely to be compliant. This is an area where 'real world' or 'pragmatic' randomised trials are indicated.
The newer generation of antipsychotics, often called atypical, seem to cause less of the movement disorders associated with older drugs. This group of compounds may be equally clinically effective (Small 1997), and be more acceptable to people with schizophrenia, than older drugs such as haloperidol (Leucht 1999; Marder 1994; Tollefson 1997) although this is disputed (Geddes 2000). Atypical drugs have gained popularity amongst clinicians but, along with their cost, a lack of a depot preparation has been cited as a significant obstacle to their frequent use (Sarfati 1999).
Risperidone is an atypical antipsychotic, first made available for the care of those with schizophrenia in 1986. Since then clinical trials have been conducted to evaluate its efficacy and safety and studies have indicated that it may be superior to older drugs (Marder 1994). When oral risperidone is compared with haloperidol, it appears to have marginal benefits in terms of clinical improvement and is less likely to cause movement disorders (Hunter 2003). Risperidone is the first newer drug to be available in a long‐acting injection formulation.
How the intervention might work
Risperidone is one of the new or second‐generation "atypical" antipsychotics, developed in the late 1980s. It is known to block dopamine D2 and 5HT2 (serotonin) receptors in the brain, with a high ratio of 5HT2 to D2 blockade. It also blocks alpha1 and alpha2 adrenoceptors, H1 receptors and has no effect on beta adrenoceptors, muscarinic cholinoceptors or peptidergic receptors (Janssen 1988).
The depot formulation of risperidone has unmodified risperidone encapsulated in biodegradable polymer microspheres, which are then suspended in an aqueous solution. Once the microspheres are injected into the muscle, the polymers begin to degrade and the drug is released at a set rate. This takes place over the course of several weeks, with the highest plasma concentration occurring approximately one month after injection (Ramstack 2003).
Why it is important to do this review
In terms of the costs of schizophrenia, this was estimated at about £6.7 billion in England in 2004/05, of which the direct costs were £2 million while the indirect costs accounted for the rest (Mangalore 2007). The cost of risperidone (depot) itself is expensive compared to other typical antipsychotics, at £142.76 for a 50 mg vial. The maximum monthly dose of risperidone (depot) is 100 mg per month, which costs £285.52 per month (BNF 2012). These newer, atypical antipsychotics in comparison are more expensive than typical antipsychotics, with olanzapine available at £13.11 for 28 x 5 mg tablets, and clozapine (Clozaril) at £21.56 for 28 x 100 mg tablets.
It is important to complement the clinical effectiveness of risperidone (depot) with its cost‐effectiveness. Davies et al. (Davies 2007) conducted a study on cost‐effectiveness of first‐generation antipsychotics (i.e. flupentixol, trifluoperazine, chlorpromazine) and the second‐generation antipsychotics (i.e. risperidone, olanzapine, amisulpiride). The study findings argue that there is no evidence to suggest that atypical (second‐generation) antipsychotics are more cost‐effective than typical (first‐generation) antipsychotics.
Objectives
To examine the effects of depot risperidone for treatment of schizophrenia or related psychoses in comparison with placebo, no treatment or other antipsychotic medication.
If possible, to critically appraise and summarise current evidence on the resource use, cost and cost‐effectiveness of risperidone (depot) for schizophrenia.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials (RCTs). Where a trial was described as 'double‐blind' but it was implied that the study was randomised, these trials were included in a sensitivity analysis. If there was no substantive difference within primary outcomes (see Types of outcome measures) when these 'implied randomisation' studies were added, then they were included in the final analysis. If there was a substantive difference, only clearly randomised trials were utilised and the results of the sensitivity analysis described in the text. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
People with schizophrenia and schizophrenia‐like disorders such as schizophreniform disorder, delusional disorder or schizoaffective disorder, diagnosed by any criteria. People with 'serious/chronic mental illness' or 'psychotic illness' were also included. Where possible, people with dementia, depression and problems primarily associated with substance misuse were excluded.
Types of interventions
1. Risperidone
Administered by long‐acting intramuscular injection, any dose.
2. Placebo or no treatment
3. Other antipsychotic drugs (depot)
Any dose, administered in depot form.
4. Other antipsychotic drugs (oral)
Any dose, administered in oral form. Oral drugs were divided into two subgroups; typical and atypical. For the purposes of this review atypicals were amisulpiride, aripiprazole, clozapine, clothiapine, loxapine, molindone, risperidone, olanzapine, quetiapine, sulpiride, zotepine, ziprasidone.
Types of outcome measures
All outcomes were reported for the short term (up to 12 weeks), medium term (13‐26 weeks), and long term (more than 26 weeks).
Primary outcomes
1. Global state
1.1 Relapse
2. Mental state
2.1 Clinically important change in general mental state
Secondary outcomes
1. Death ‐ suicide and natural causes
2. Global state
2.1 Time to relapse 2.2 Clinically important change in global state 2.3 Any change in global state 2.4 Average endpoint global state score 2.5 Average change in global state scores
3. Service outcomes
3.1 Hospitalisation 3.2 Time to hospitalisation 3.3 Duration of stay in hospital
4. Mental state
4.1 Change in general mental state 4.2 Average endpoint general mental state score 4.3 Average change in general mental state scores 4.4 Clinically important change in specific symptoms 4.5 Change in specific symptoms 4.6 Average endpoint specific symptom score 4.7 Average change in specific symptom scores
5. Leaving the study early
5.1 For specific reasons 5.2 For general reasons
6. General functioning
6.1 Clinically important change in general functioning 6.2 Any change in general functioning 6.3 Average endpoint general functioning score 6.4 Average change in general functioning scores 6.5 Clinically important change in specific aspects of functioning, such as social or life skills 6.6 Any change in specific aspects of functioning, such as social or life skills 6.7 Average endpoint specific aspects of functioning, such as social or life skills 6.8 Average change in specific aspects of functioning, such as social or life skills
7. Behaviour
7.1 Clinically important change in general behaviour 7.2 Any change in general behaviour 7.3 Average endpoint general behaviour score 7.4 Average change in general behaviour scores 7.5 Clinically important change in specific aspects of behaviour 7.6 Any change in specific aspects of behaviour 7.7 Average endpoint specific aspects of behaviour 7.8 Average change in specific aspects of behaviour
8. Adverse effects
8.1 Clinically important general adverse effects 8.2 Any general adverse effects 8.3 Average endpoint general adverse effect score 8.4 Average change in general adverse effect scores 8.5 Clinically important change in specific adverse effects 8.6 Any change in specific adverse effects 8.7 Average endpoint specific adverse effects 8.8 Average change in specific adverse effects
9. Engagement with services
9.1 Clinically important engagement 9.2 Any engagement 9.3 Average endpoint engagement score 9.4 Average change in engagement scores
10. Satisfaction with treatment
10.1 Recipient of care not satisfied with treatment 10.2 Recipient of care average satisfaction score 10.3 Recipient of care average change in satisfaction scores 10.4 Carer not satisfied with treatment 10.5 Carer average satisfaction score 10.6 Carer average change in satisfaction scores
11. Quality of life
11.1 Clinically important change in quality of life 11.2 Any change in quality of life 11.3 Average endpoint quality of life score 11.4 Average change in quality of life scores 11.5 Clinically important change in specific aspects of quality of life 11.6 Any change in specific aspects of quality of life 11.7 Average endpoint specific aspects of quality of life 11.8 Average change in specific aspects of quality of life
12. Economic outcomes
12.1 Average change in total cost of medical and mental health care 12.2 Total indirect and direct costs 12.3 Direct resource use: 12.3.1 Outpatients ‐ number of contacts (GP consultation, psychiatrist, psychologists, psychiatric nurse, counsellor, social worker) 12.3.2 Hospitalisation (taking battery of tests, patients’ physical, psychiatric and psychological profile and psychological assessment, number of days, relapse) 12.3.3 Medication (different types of antipsychotics to include dose and frequency, treatment of side effects) 12.3.4 Psychological therapies (different types of psychological therapies to include session numbers and frequency) 12.3.5 Other resources (day centres, night shelter) and transportation for medical care visits 12.4 Indirect resource use: 12.4.1 Family, relatives' and friends' resources 12.4.2 Police, criminal justice system 12.4.3 Benefits paid, social security payments 12.4.4 Employment agency workers, absence from work, loss of productivity 12.5 Cost‐effectiveness ratios represented by incremental cost‐effectiveness (ICER) 12.6 Cost‐utilities represented by incremental costs per quality‐adjusted life year (QALY) or disability‐adjusted life year (DALY) 12.7 Cost benefit represented by net Benefit Ratio, others.
13. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADE profiler (GRADEPRO) to import data from RevMan 5 (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
1. Relapse ‐ long term. 2. Clinically significant improvement in mental state ‐ long term. 3. Leaving the study early for any reason ‐ medium/long term. 4. Severe adverse effects ‐ medium/long term. 5. Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism ‐ medium/long term.
Search methods for identification of studies
Electronic searches
Cochrane Schizophrenia Group’s Trials Register
On October 28, 2015, the Trials Search Co‐ordinator (TSC) searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials using the following search strategy:
(*Risperidone* AND *Injection*) in Intervention Field of STUDY
In such a study‐based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, EMBASE, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
The review authors inspected references of all identified studies for more studies.
2. Personal contact
The review authors attempted to contact the first author of each study considered for inclusion in the review for more information regarding unpublished trials or any available data.
3. Drug companies
The review authors contacted Janssen‐Cilag Limited for further data.
Data collection and analysis
For details of previous data collection and analysis methods see Appendix 2.
Selection of studies
For this update, review author PH and TN (see Acknowledgements) independently inspected citations from the searches and identified relevant abstracts. A random 20% sample was independently re‐inspected by SS to ensure reliability. Where disputes arose, the full‐text report was acquired for more detailed scrutiny. If citations met inclusion criteria, we obtained full‐text reports for more detailed inspection. Again, a random 20% of reports were re‐inspected by SS in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification and added these studies to the list awaiting classification.
With regards to selecting studies for economic evaluations, review authors (SS and VF) categorised studies as per the following: Type A ‐ Full economic evaluation (within the framework of RCT): studies that focus on cost‐effectiveness analysis, cost‐utility analysis and cost‐benefit analysis. Type B ‐ Partial economic evaluation (within the framework of RCT): studies that focus on cost‐analysis and cost‐minimisation studies of Risperidone (depot). Type C ‐ Randomised trials that reported limited information, such as estimates of resources use or costs associated with Risperidone (depot).
Data extraction and management
1. Extraction
For this update PH, and TN extracted data from all included studies. In addition, to ensure reliability, SS independently extracted data from a random sample of these studies, comprising 10% of the total. Again, any disagreement was discussed, decisions documented and, if necessary, authors of studies contacted for clarification. With any remaining problems, we contacted editorial team (CEA) to help clarify issues and these final decisions were documented. Data presented only in graphs and figures were extracted whenever possible, but included only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary.
For the economic analysis had VF and SS found Type A and B studies (see Types of studies), they would have investigated whether appraisal had already been undertaken by NHS EED using their search tool derived for this purpose. If appraisal had not been undertaken, VF and SS would have applied the NHS EED tool to the data. In this current review, should there only be Type C studies available, we would extract outcome data directly from the already‐included effectiveness studies. We recognise that much information would be lacking to get results that are both valid and reliable.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument has not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, and we noted in Description of studies whether or not this was the case.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we used mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to relevant data before inclusion: a) standard deviations (SDs) and means are reported in the paper or obtainable from the authors; b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean, as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996); c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS) (Kay 1986), which can have values from 30 to 210), the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2 SD > (S‐S min), where S is the mean score and S min is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied. We entered skewed endpoint data from studies of less than 200 participants in additional tables rather than into an analysis. However, skewed data pose less of a problem when looking at mean if the sample size is large, we therefore, entered skewed endpoint data from studies with over 200 participants into syntheses.When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not and we entered skewed change data into analysis.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for risperidone depot. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we reported data where the left of the line indicates an unfavourable outcome. This was noted in the relevant graphs.
Assessment of risk of bias in included studies
For this update, PH and TN worked independently by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This new set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain additional information.
We have noted the level of risk of bias in both the text of the review and in the Table 1.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI)*. It has been shown that RR is more intuitive (Boissel 1999) than odds ratios (ORs) and that ORs tend to be interpreted as RR by clinicians (Deeks 2000). We did not calculate the Number Needed to Treat/Harm (NNT/H). The NNT/H statistic with its CIs is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' tables, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes, we estimated mean difference (MD) between groups. We prefer not to calculate effect size measures (standardised mean difference SMD). However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, if we had included cross‐over trials, we planned only to use the data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involves more than two treatment arms, if relevant, the additional treatment arms were presented in comparisons. Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we did not reproduce these data or use them within analyses, except for the outcomes of leaving the study early and adverse events. If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we marked such data with (*) to indicate that such a result may well be prone to bias. This was the case for three studies (Gaebel 2010*; Kane 2002*Quinn 2012*).
2. Binary
In the case where attrition for a binary outcome was between 0% and 50%, and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). Those leaving the study early were all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes, the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ were used for those who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when data only from people who completed the study to that point were compared to the intention‐to‐treat analysis using the above assumptions.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported, we presented and used these data.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we first tried to obtain the missing values from the authors. If not available, where there are missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals available for group means, and either a P value or T value available for differences in mean, we can calculate them according to the rules described in the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011): When only the SE is reported, SDs are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, T or F values, confidence intervals, ranges or other statistics. If these formulae do not apply, we can calculate the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. However, we did not impute any data in this review.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data had been used in the trial, if less than 50% of the data had been assumed, we reproduced these data and indicated that they are the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, these were fully discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arise these were fully discussed.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
Heterogeneity between studies was investigated by considering the I2 method alongside the Chi2 'P' value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of an I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. 'P' value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 70% accompanied by a statistically significant Chi2 statistic, was interpreted as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for the heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We planned not to use funnel plots for outcomes where there were less than 10studies in each analysis, or where all studies were of similar sizes. If funnel plots had been possible, we planned to seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect these studies can either inflate or deflate the effect size. We chose the random‐effects model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses ‐ only primary outcomes
1.1 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of risperidone depot for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage and with similar problems.
2. Investigation of heterogeneity
If inconsistency was high, this was reported. First, we investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and removed outlying studies to see if heterogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, data were presented. If not, data were not pooled and issues discussed. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity were obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes, we included these studies and if there was no substantive difference when the implied randomised studies were added to those with a better description of randomisation, then all data were used from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we reported results and discussed them, but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings on primary outcomes when we used our assumption compared with complete data only. A sensitivity analysis was undertaken to test how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we reported results and discussed them, but continued to employ our assumption.
3. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included data from these trials in the analysis
4. Imputed values
Had we included cluster‐randomised trials, we planned to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in such trials.
If substantial differences were noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed‐effect and random‐effects
All data were synthesised using a random‐effects , however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether the greater weights assigned to larger trials with greater event rates, altered the significance of the results compared to the more evenly distributed weights in the fixed‐effect model.
Results
Description of studies
Salient features of the included and excluded studies are given in the tables (Characteristics of included studies; Characteristics of excluded studies).
Results of the search
1. Overall
The original published version of this review (Hosalli 2003) included two studies and excluded 11, with two studies awaiting classification. The search updates in 2010, 2012 and 2015 identified 181 references with no additional records identified through other sources. No duplicates were found. We screened 181 records. Twenty‐eight potentially relevant full‐text reports were obtained and scrutinised, and 17 of these reports did not meet the inclusion criteria (see Characteristics of excluded studies), and were added to the excluded studies. Ten studies were added to included studies and one study was added to awaiting classification (Figure 1).
1.
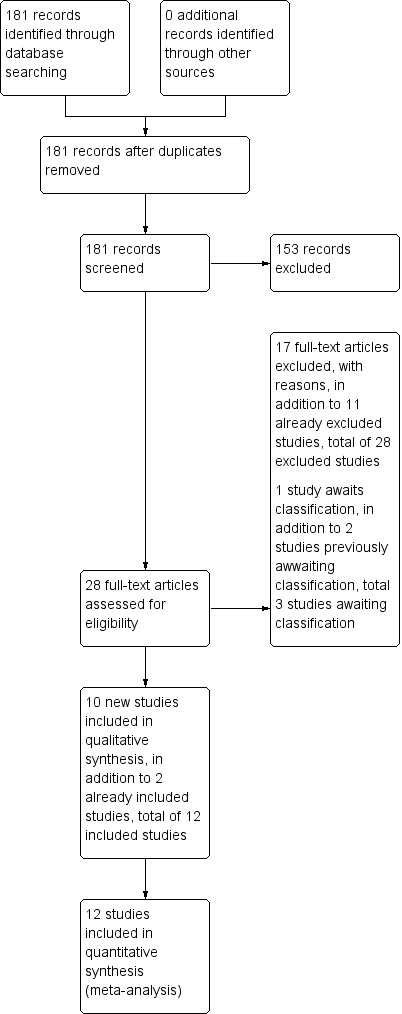
Study flow diagram: 2010 and 2012, 2015 updated search
Included studies
Twelve studies, reported as 15 conference presentations and 13 full‐text papers met the selection criteria and are included in this update.
1. Study design
All 12 included studies were randomised and eight featured some form of blinding ‐ ranging from blinding of raters in otherwise open‐label studies to blinding of all participants and investigators; four studies were expressly or implied as 'open‐label' but employed blinded raters (Bai 2006; Covell 2012; Li 2011; MacFadden 2010). Study duration also varied considerably; three studies were short term at 12 to 13 weeks (Bai 2006; Chue 2002; Pandina 2011); Covell 2012 was medium term, while seven followed up participants for two years (Fleischhacker 2011; Gaebel 2010*; Kane 2002*; Li 2011; MacFadden 2010; Quinn 2012*; Rosenheck 2011). One study (Keks 2007), combined short‐ and medium‐term outcomes, with analyses at 12 weeks and one‐year of follow‐up. The majority of studies took place in the community, and were well‐represented internationally, with studies conducted in Taiwan (Bai 2006) and the remaining as multi‐centre studies, conducted within the USA (Covell 2012; Kane 2002*; Rosenheck 2011), Canada (Quinn 2012*), China (Li 2011), Europe (Gaebel 2010*; Keks 2007); and international multi‐centre studies (Chue 2002; Fleischhacker 2011; MacFadden 2010; Pandina 2011).
2. Participants
A total of n = 5723 participants who received the intervention were included, with the majority of studies providing data as to male and female participants; a total of n = 3140 male and n = 2112 female participants were included, with a mean age of around 40 years. All studies used the Diagnostic and Statistical Mannual version 1V (DSM‐IV) (APA 2000) to define schizophrenia; so for at least six continuous months a participant must have shown some evidence of schizophrenia, and for at least one month must have shown at least two symptoms of frank psychosis. These symptoms would have included delusions, hallucinations, incoherent speech, disorganised or catatonic behaviour, or flat affect. To meet DSM IV criteria, the symptoms must be disabling in such a way that social and occupational functioning is impaired; these symptoms should not be the direct result of a physical disorder or of substance misuse. Most studies randomised people who had been experiencing schizophrenia for some years; often specifying that they need to have had schizophrenia for at least one year before diagnosis.
Further criteria for selecting participants varied from study to study, depending on exactly what was being investigated, but general exclusion criteria were reasonably consistent; people with a history of violence to themselves or others, or of recent suicide attempts were not permitted to take part. Nor were those with active DSM‐IV diagnosed substance dependence or general ill health, including serious psychiatric problems other than schizophrenia. Tolerance to risperidone was an important factor that all studies addressed; most potential participants underwent a screening period at the start of their trial to establish that they could tolerate risperidone; this was sometimes waived if they had already demonstrated this (i.e. they were currently treated with the drug).
3. Interventions
The prescribing of depot risperidone was consistent across the board; 25, 37.5 and 50 mg injections every two weeks were the most common dosages, with participants typically initiated on 25 mg/two weeks, which was then stepped up in 12.5 mg increments if their symptoms worsened. Three earlier studies (Chue 2002; Kane 2002*; Keks 2007) included depot risperidone up to 75 mg, but such high doses were not used by the most recent studies. Because it can take several weeks for therapeutic plasma levels of the drug to build‐up, oral supplementation, either with oral risperidone or the participant's previous oral antipsychotic, was typically used for the initial two to four weeks.
The trials involving oral antipsychotics either compared depot risperidone versus another specific single antipsychotic or to more than one antipsychotic. Only one study compared depot risperidone with placebo (Kane 2002*). Of the studies comparing depot risperidone with another single antipsychotic, two used oral risperidone at 2 mg to 6 mg per day (Bai 2006; Chue 2002); one study investigated5 mg to 20 mg/day oral olanzapine (Keks 2007), and one compared depot risperidone against 5 mg to 30 mg/day oral aripiprazole (MacFadden 2010). Covell 2012 compared either haloperidol decanoate or fluphenazine (no doses prescribed, but used at 'clinician's judgement'; Gaebel 2010* was mainly concerned with quetiapine at up to 750 mg/day, but also featured a smaller aripiprazole arm of 10 mg to 30 mg per day. The remaining three studies randomised patients to receive depot risperidone or to remain on their current oral antipsychotic; in the case of Quinn 2012*, only second‐generation "atypical" drugs were used, specifically risperidone, olanzapine and quetiapine; Rosenheck 2011 provides no details of which drugs were used.
Paliperidone palmitate (PP) is the active metabolite of risperidone, but features a different administration schedule and dosages to depot risperidone; PP doses can be given either in milligrams (mg), for the overall volume injected, or in milligram equivalents (mg eq), which refers to the fraction of the drug that is actually pharmacologically active. So 39 mg of PP is given as 25 mg eq, 78 mg is 50 mg eq, etc. The injection schedule of this drug only requires monthly intra‐muscular injections to maintain therapeutic levels and oral supplementation is not needed, so in the non open‐label PP studies (Fleischhacker 2011; Li 2011; Pandina 2011) oral placebo and bi‐weekly placebo injections were used.
4. Outcomes
4.1 Global state
The Clinical Global Impression (CGI) scale was utilised in some manner by the majority of the included studies, with data provided either as mean (SD or SE) endpoint or change scores, or as percentages of participants who were either not ill or mildly ill. Much data were unusable due to the high level of attrition in some studies and inconsistent reporting in others. Another rating scale used to assess global state was the Schedule for Deficit Syndrome (SDS) scale.
Other outcomes that give an impression of the general condition of somebody with schizophrenia, such as levels of relapse and remission, the proportion of participants who needed concomitant benzodiazepine and how long they were able to stay on the study drug are also included here.
Where relapse was included as an outcome, the investigators typically used the criteria for relapse in Csernansky 2002. Briefly, these consist of worsening of psychiatric condition (i.e. requiring hospitalisation or increased care), Positive and Negative Syndrome Scale (PANSS) or CGI scores that are markedly higher than the baseline, self‐harm or suicidal/homicidal ideation, discontinuing the study drug due to ineffectiveness, requiring additional antipsychotic medications or non protocol doses of the study drugs.
4.2 Mental state
PANSS was the key mental state assessment scale; each study reported changes in PANSS total scores, with some also including the subscales and factor scores. Unfortunately, as with CGI, high attrition prevented us from including PANSS scores from every study in the analysis.
4.3 General functioning
The Global Assessment of Functioning (GAF) scale was used by Bai 2006 and data for the Personal and Social Performance (PSP) scale were available from Li 2011 and Pandina 2011.
4.4 Service utilisation
The primary outcomes for Rosenheck 2011 centred around the use of medical and psychiatric health services; the data for these outcomes were obtained through the VA health services, which potentially side‐steps the issue of low follow‐up interview rates as these data would be available for participants who missed interviews but nonetheless remained a part of the study. Data on in‐patient and out‐patient care were available, and include rates of hospitalisation and mean (SD) numbers of visits to individual or group psychiatry sessions, as well as other psychiatric and general healthcare services.
4.5 Quality of life
It is disappointing that more studies did not include measures to assess quality of life (QoL) on the trial drugs. Several studies did make use of various QoL questionnaires, but high attrition means that the results could not be used. The one exception to this is in Bai 2006, which made use of the Medical Outcome Study Short‐Form Health Survey (SF‐36).
4.6 Economic outcomes
Very few studies involved any assessment of the economic outcomes associated with treatment with depot risperidone. The two exceptions are Rosenheck 2011 and Gaebel 2010* which presented these outcomes in papers published separately to the main study report. Lack of standard deviations and the format in which some of these outcomes were presented makes it difficult to use them in a quantitative analysis, but we have commented on the key findings in a qualitative manner.
4.7 Satisfaction with treatment
Only one trial addressed this in a manner that did not fall foul of the 50% attrition limit; Bai 2006 recorded patient satisfaction with their current treatment using a five‐point scale, very good to poor. However, no evidence to validate this method was presented, consequently we decided not to include the data.
4.8 Leaving the study early
All studies reported the number of participants leaving the study early from both groups, and provided a breakdown of the main reasons given for doing so.
4.9 Adverse effects
Adverse events were reported by all of the included studies. Chue 2002 reported overall rates of adverse events in both groups, and the numbers withdrawing from the study as a result of side effects. No details were given regarding the nature of these adverse events or how they were recorded. The abstracts available for this review state that body weight was measured and laboratory tests were undertaken. The reports state that there were no differences between oral and depot groups, but present no numbers. Chue 2002 also used the Extrapyramidal Symptom Rating Scale (ESRS), but again, no numerical data were reported. Kane 2002* reported rates of individual adverse events spontaneously reported by participants, and reported these for all people in the study, not just those who completed the trial. Median ESRS scores were also reported for each group at baseline and change at endpoint. Pain and swelling at injection sites rated by investigators and patients were also reported.
MacFadden 2010 gave rates of adverse events that occurred in >10% of participants in either group, as well as serious, prolactin‐ and glucose‐related events. The results of laboratory tests for levels of prolactin, glucose, cholesterol and triglycerides were also provided. Adverse events occurring in more than 5% of participants were reported in Rosenheck 2011; there was no discussion of how these events were identified or reported. They were also grouped under quite general subheadings, so "nervous system disorders" comprises all extrapyramidal symptoms (EPS), as well as the likes of headache, somnolence and dizziness.
5. Outcome measures used in this review
5.1 Global state
5.1.1 Global functioning. Clinical Global Impression ‐ CGI (Guy 1976) A rating instrument commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used with low scores indicating decreased severity and/or greater recovery.
5.1.2 Global Assessement of Functioning ‐ GAF (DSM‐IV‐TR, APA 2000)
The GAF rating scale is presented in DSM‐IV as a quick method for assessing psychological, social and occupational function to give an impression of an individual's overall level of functioning relating to their mental health. Scoring is from zero to 100. Very high scores indicate absent or minimal symptoms that have little effect on functioning, very low scores (below 20) indicate a serious danger that somebody will hurt themselves or others.
5.1.3 Schedule for Deficit Syndrome ‐ SDS (Kirkpatrick 1989)
This tool is used to evaluate the presence of negative or deficit‐related symptoms, SDS regards these as flattened affect, poverty of speech, diminished emotional range, curbing of interest and diminished sense of purpose. If a person with schizophrenia has had two or more of the listed symptoms for 12 months, and they were not caused by other factors such as substance dependence or depression, then they are diagnosed with deficit syndrome. Severity is rated from zero to four, higher scores are worse, for each criteria.
5.2 Mental state
5.2.1 Positive and Negative Symptom Scale ‐ PANSS (Kay 1987) This scale was developed to evaluate the positive, negative and general symptoms in schizophrenia. The PANSS has 30 items, and each item can be defined on a seven‐point scoring system varying from one (absent) to seven (extreme). This scale is divided into three subscales for measuring the severity of general psychopathology, positive symptoms (PANSS‐P) and negative symptoms (PANSS‐N). A low score indicates lesser severity.
5.3 General functioning
5.3.1 Personal and Social Performance Scale ‐ PSP (Nafees 2012)
The PSP scale measures an individual's functioning within society and is assessed through a 10 to 15 minute structured interview looking at four domains (socially useful activities, personal and social relationships, self‐care and disturbing and aggressive behaviours). An overall score between zero and 100 is derived from the individual factor scores; a higher score indicates better personal and social functioning.
5.4 Quality of life
5.4.1 Medical Outcome Study Short‐Form Health Survey ‐ SF‐36 (Ware 1992)
SF‐36 rates health‐related quality of life in eight main components (physical function, role limitations due to physical problems, bodily pain, general health, mental health, role limitations due to emotional problems, social function and vitality), each scored from zero to 100, higher scores are better.
5.5 Adverse effects
The majority of the following scales were used by the study investigators to obtain dichotomised results relating to specific adverse effects ‐ the data and analysis section of this review presents predominantly dichotomised data relating to adverse effects.
5.5.1 Extrapyramidal Symptom Rating Scale ‐ ESRS (Chouinard 1980) This scale consists of a questionnaire relating to parkinsonian symptoms (nine items), a physician's examination for parkinsonism and dyskinetic movements (eight items) and a clinical global impression of tardive dyskinesia. High scores indicate severe levels of movement disorder.
5.5.2 Barnes Akathisia Scale ‐ BARS (Barnes 1989)
This rating scale assesses drug‐induced akathisia (restlessness or the urge to move). Individuals are rated on their observable level of restlessness while sitting (objective), their awareness of the urge to move and the distress, if any, that this causes (subjective) and on a global assessment. Higher scores indicate more severe akathisia.
5.5.3 Abnormal Involuntary Movement Scale ‐ AIMS (Munetz 1988)
This 12‐item scale is used to assess the severity of dyskinesia in an individual. Items such as movement of the jaw or the extremities are rated on a scale from zero (none) to four (severe) and combined to give a total score.
5.5.4 The Udvalg for Kliniske Undersgelser side effects rating scale ‐ UKU (Lingjaerd 1987)
The UKU is a tool for recording side effects of neuroleptic treatment in a standardised manner, items are scored from zero, absent or normal, to three, severe.
5.5.5 Arizona Sexual Experiences Scale ‐ ASEX (McGahuey 2000)
The ASEX scale is a five‐item rating scale that quantifies sexual experiences (including drive/ arousal/ satisfaction from or ability to reach orgasm) with total scores ranging from five to 30, with higher scores indicating greater sexual dysfunction
Excluded studies
We have excluded a total of 28 studies (see Characteristics of excluded studies).
Studies awaiting assessment
Three studies await further clarification; Turner 2000 seemed to be an eligible study for the first edition of this review (Hosalli 2003), but while preparing the updated review we were unable to find any further details. Nasrallah 2002, both conference abstracts, reports QoL data with depot risperidone but it may be part of one of the included studies (Kane 2002*). Again, the authors have been contacted for more details. Segarra 2010 also appeared to be eligible, but we were unable to extract any usable data from the published conference poster and abstract. We have contacted Dr Segarra for more details of this study.
Ongoing studies
We contacted Janssen‐Cilag Limited for further information regarding ongoing studies and were told that such studies existed and the data would be forwarded to us. We are still awaiting these studies and data.
Risk of bias in included studies
2.

3.
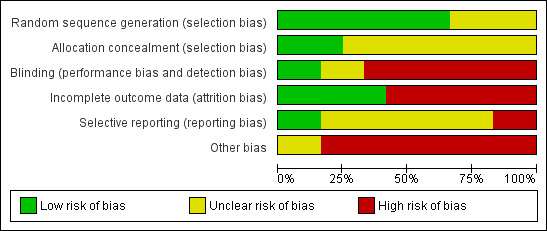
Allocation
All studies were randomised, but there was extreme inconsistency in how well the methods used were reported. Most studies also did not describe how allocation concealment was achieved. At one end of the spectrum, it is simply stated that participants were randomised (Bai 2006; MacFadden 2010; Quinn 2012*) with no further details, with other studies giving more details on the stratification factors used, e.g. site, PANSS score, previous antipsychotic medication (Chue 2002; Gaebel 2010*; Rosenheck 2011). While the implication is that all studies used computer‐generated schemes to randomise participants, this is only explicitly stated in a handful of cases. Descriptions of allocation concealment are neglected by most of the included studies, but methods were similar amongst studies that provide them. Interactive Voice Response Systems (IVRS) were used by Fleischhacker 2011, Keks 2007 and Pandina 2011; this is a system whereby the investigator phones a number, enters the patient details and receives a medication allocation based on the randomisation algorithm. Keks 2007 set the benchmark for minimising the risk of allocation bias, with a description of the randomisation algorithm and the use of an IVRS.
Blinding
The included studies utilised a number of types of blinding, each resulting in different amounts of risk for performance and detection bias.
The studies with the least risk were often described as "double blind"; they took measures to ensure that neither the participants nor the investigators monitoring them and rating outcomes would know which treatment a person was actually taking. The study of depot risperidone versus placebo (Kane 2002*) and one of those involving oral antipsychotics (Chue 2002) used placebo injections that were the same volume as those containing the study drug. Two of the paliperidone palmitate (PP) studies (Fleischhacker 2011; Pandina 2011) utilised placebos in the PP group so that both groups took the same number of tablets and had the same number of injections. Participants were also precluded from seeing the syringe when they received the injection, as the two drugs were different in appearance. The techniques used for blinding in these studies were of very high quality and present little risk of the blind being broken.
All other studies have a significantly higher risk of performance bias as the allocation of interventions was not blinded; this was somewhat mitigated against in Bai 2006, Li 2011, MacFadden 2010, and Rosenheck 2011, all of which used blinded raters to perform outcome assessments, often via video conference.
Incomplete outcome data
There is little risk of attrition bias emerging from incomplete outcome data in most studies. Drop‐out rates are uniformly reported and are normally accompanied by a breakdown of the main reasons for leaving the study early. Overall attrition is greater than 50% in some studies; where this is the case we have not used the data from affected outcomes. In Rosenheck 2011, the attrition rate was not simply explained; it was stated that 69.8% of the oral treatment group and 63.6% of the depot risperidone group completed the study, but what this actually means is never made explicitly clear. Indeed, the overall completion rate does not tally with the follow‐up interview rates, which are given as 60% at 12 months, 46% at 18 months and only 29% of participants completing the full two years of follow‐up interviews, with there being no difference between follow‐up rates across the two intervention groups. The implication is that patients could remain in the study and have their health service use available for analysis despite no longer taking part in the follow‐up interviews to assess secondary outcome measures. Generally, across studies, attrition was high, and therefore rated at a 'high' risk of bias ‐ three studies in particular reported losses of greater than 50% (Gaebel 2010*; Kane 2002*; Quinn 2012*).
Selective reporting
Gaebel 2010* stated in the methods section of the main study report that CGI‐S was used as an outcome measure, but this outcome is not reported in the results section. As CGI‐S is a very commonly used measure, we have contacted the study authors to try to obtain any unpublished data that may pertain to this, but at the time of writing have had no response.
Other potential sources of bias
Author conflicts of interest are reported for all studies, as are funding sources.
Most studies included in this review were either partially or fully funded and supported by Jassen‐Cilag/Johnson & Johnson Co. In addition to this, a significant number of authors and investigators are or were employees of Jassen. While it is understandable that a pharmaceutical company has an interest in research regarding their own products, it is perhaps alarming that the only studies that address the questions posed by this review are supported by a party with a vested interest in depot risperidone.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8
COMPARISON 1: RISPERIDONE DEPOT versus PLACEBO
For this comparison, we found only one relevant study (Kane 2002*, n = 400); apart from one outcome (leaving the study early), all data for this comparison from this 12‐week study, are 'short term'.
1.1 Mental state: 1. Change (exacerbation) in specific symptoms
Mental state data were presented as change in specific symptoms. For the outcomes of anxiety, hallucination and nervousness, no differences between groups were found; however, there was statistically significant difference in favour of depot risperidone for levels of agitation (risk ratio (RR) 0.60 95% confidence interval (CI) 0.39 to 0.92) and psychosis (RR 0.52 95% CI 0.33 to 0.83, Analysis 1.1).
1.1. Analysis.
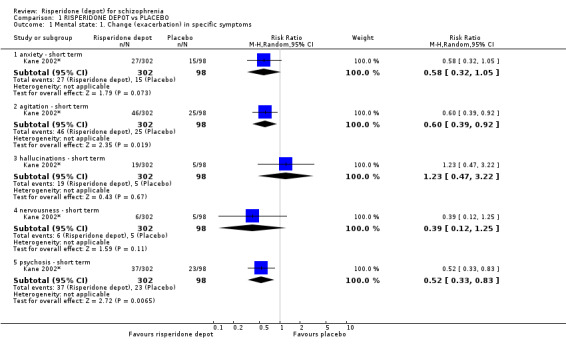
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 1 Mental state: 1. Change (exacerbation) in specific symptoms.
1.2 Leaving the study early: 1. Any reason (by time period)
The number of participants leaving the study early 'very early on' were similar between treatment groups, however data at 12 weeks showed more people left the placebo group early compared to those receiving risperidone depot (RR 0.74 95% CI 0.63 to 0.88, Analysis 1.2).
1.2. Analysis.
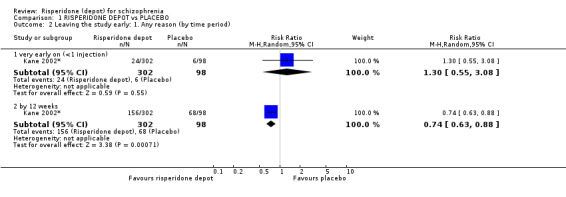
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 2 Leaving the study early: 1. Any reason (by time period).
1.3 Leaving the study early: 2. Any reason (by doses)
More participants left the study early for any reason from the placebo group compared to those receiving either 'all doses' risperidone depot (RR 0.74 95% CI 0.63 to 0.88), 25 mg risperidone depot (RR 0.74 95% CI 0.59 to 0.94), 50 mg risperidone depot (RR 0.74 95% CI 0.59 to 0.93) or 75 mg risperidone depot (RR 0.75 95% CI 0.60 to 0.94, Analysis 1.3).
1.3. Analysis.
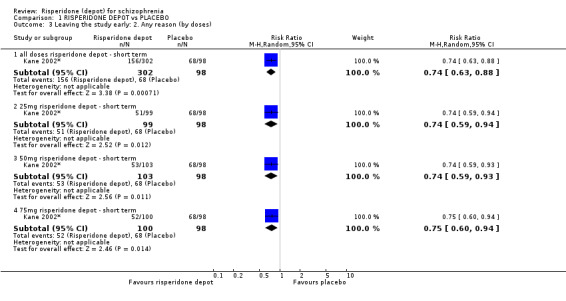
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 3 Leaving the study early: 2. Any reason (by doses).
1.4 Leaving the study early: 3. Due to insufficient response (by doses)
For leaving the study early due to insufficient response, there was no statistically significant difference between groups for those receiving 25 mg depot risperidone. However fewer participants left early from the risperidone depot groups for those receiving either all three doses combined (RR 0.53 CI 0.36 to 0.79), 50 mg (RR 0.48 CI 0.27 to 0.83) or 75 mg (RR 0.39 CI 0.21 to 0.72, Analysis 1.4).
1.4. Analysis.
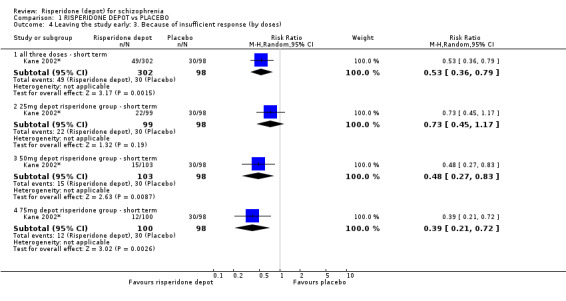
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 4 Leaving the study early: 3. Because of insufficient response (by doses).
1.5 Adverse events: 1. General: a. Death
No deaths were reported in the risperidone depot group, one death was reported in the placebo group. This difference was not statistically significant (RR 0.11 95% CI 0.00 to 2.65 Analysis 1.5).
1.5. Analysis.
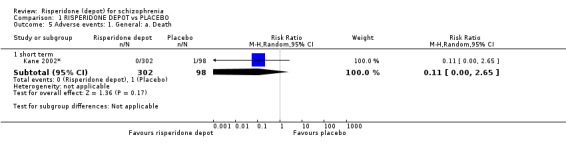
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 5 Adverse events: 1. General: a. Death.
1.6 Adverse events: 1. General: b. Severe adverse event (by doses)
There were no statistically significant differences between groups for severe adverse events by doses (25 mg, 50 mg or 75 mg) of depot risperidone (n = 400, Kane 2002*); however, 'any dose' of risperidone demonstrated a statistically significant difference, favouring risperidone depot, between groups (RR 0.59 CI 0.38 to 0.93, Analysis 1.6).
1.6. Analysis.
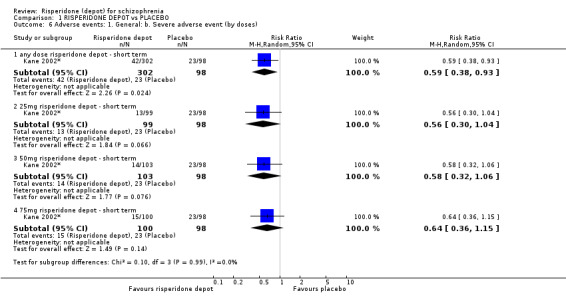
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 6 Adverse events: 1. General: b. Severe adverse event (by doses).
1.7 Adverse events: 1. General: c. Adverse event necessitating withdrawal from study (by doses)
There were no statistically significant difference between groups for adverse events necessitation withdrawal from the study by any dose, 25 mg, 50 mg or 75 mg (Analysis 1.7).
1.7. Analysis.
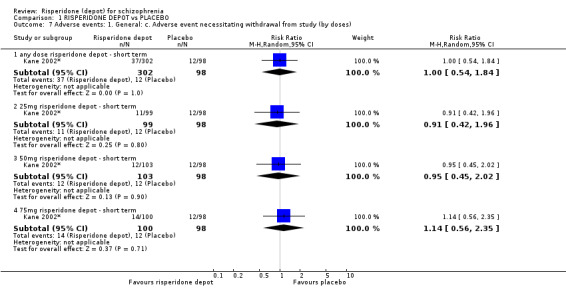
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 7 Adverse events: 1. General: c. Adverse event necessitating withdrawal from study (by doses).
1.8 Adverse events: 2. Specific: a. Cardiovascular
There was no statistically significant difference between groups for dizziness; however, there was a statistically significant difference between groups for tachycardia, with greater instances in the placebo group (RR 0.32 95% CI 0.11 to 0.98, Analysis 1.8).
1.8. Analysis.
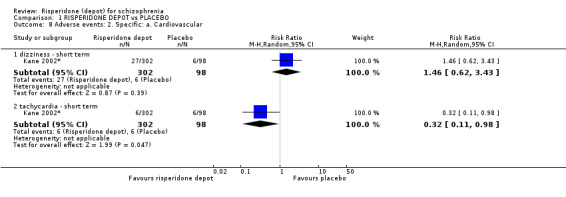
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 8 Adverse events: 2. Specific: a. Cardiovascular.
1.9 Adverse events: 2. Specific: b. Gastrointestinal
There were no statistically significant differences between groups for constipation, diarrhoea, nausea or vomiting (Analysis 1.9)
1.9. Analysis.
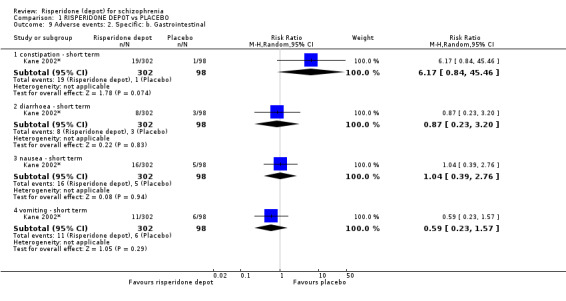
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 9 Adverse events: 2. Specific: b. Gastrointestinal.
1.10 Adverse events: 2. Specific: c. Movement disorders: a. Extrapyramidal disorder ‐ spontaneously reported (by doses)
There was no statistically significant difference between groups for extrapyramidal disorder when receiving 'all doses', 25 mg, 50 mg or 75 mg depot risperidone (Analysis 1.10).
1.10. Analysis.
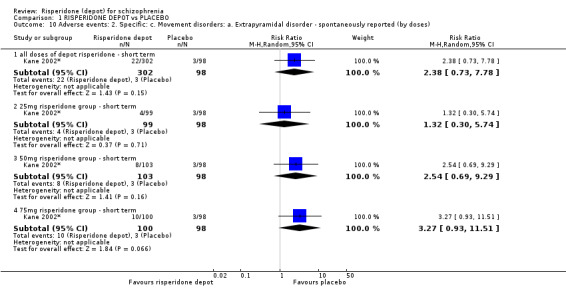
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 10 Adverse events: 2. Specific: c. Movement disorders: a. Extrapyramidal disorder ‐ spontaneously reported (by doses).
1.11 Adverse events: 2. Specific: d. Movement disorders: b. Hyperkinesia (by doses)
There was no statistically significant difference between groups for hyperkinesia when receiving 'all doses', 25 mg, 50 mg or 75 mg depot risperidone (Analysis 1.11).
1.11. Analysis.
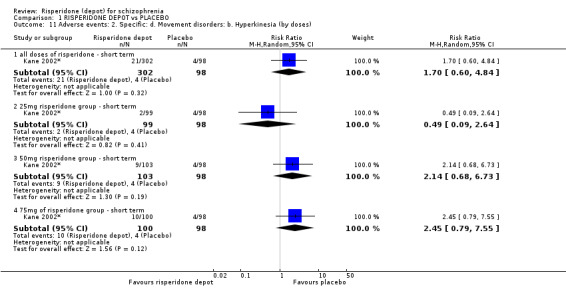
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 11 Adverse events: 2. Specific: d. Movement disorders: b. Hyperkinesia (by doses).
1.12 Adverse events: 2. Specific: e. Movement disorders: c. Hypertonia (by doses)
There was no statistically significant difference between groups for hypertonia when receiving 'all doses', 25 mg, 50 mg or 75 mg depot risperidone (Analysis 1.12).
1.12. Analysis.
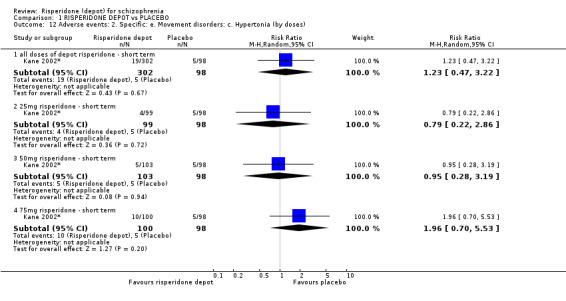
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 12 Adverse events: 2. Specific: e. Movement disorders: c. Hypertonia (by doses).
1.13 Adverse events: 2. Specific: f. Pain
There was no statistically significant difference between groups for headache or other 'unspecified' pain (Analysis 1.13).
1.13. Analysis.
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 13 Adverse events: 2. Specific: f. Pain.
1.14 Adverse events: 2. Specific: g. Salivation
There was no statistically significant difference between groups for either increased or decreased levels of salivation (Analysis 1.14).
1.14. Analysis.
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 14 Adverse events: 2. Specific: g. Salivation.
1.15 Adverse events: 2. Specific: h. Sleep disturbances
There was no statistically significant difference between groups for either insomnia or somnolence (Analysis 1.15).
1.15. Analysis.
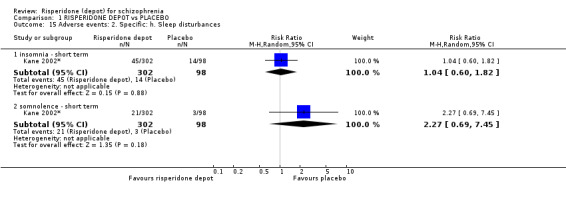
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 15 Adverse events: 2. Specific: h. Sleep disturbances.
1.16 Adverse events: 2. Specific: i. Weight gain
There was no statistically significant difference between groups for weight gain with any dose of depot risperidone (1 RCT, n = 400, RR 2.11, 95% CI 0.48 to 9.18) (Analysis 1.16).
1.16. Analysis.
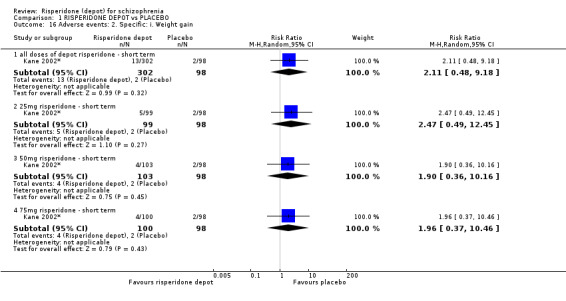
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 16 Adverse events: 2. Specific: i. Weight gain.
1.17 Adverse events: 2. Specific: j. Others
There was no statistically significant difference between groups for coughing, fatigue, injury or rhinitis (Analysis 1.17).
1.17. Analysis.
Comparison 1 RISPERIDONE DEPOT vs PLACEBO, Outcome 17 Adverse events: 2. Specific: j. Others.
COMPARISON 2: RISPERIDONE DEPOT versus GENERAL ORAL ANTIPSYCHOTICS
Two studies provided data for this comparison (Quinn 2012*; Rosenheck 2011, n = 467). Both studies were two years‐long, and are categorised as 'long term'.
2.1 Global state: 1. Relapse (any reason)
There was no statistically significant difference between groups for relapse (n = 63, Quinn 2012*, Analysis 2.1).
2.1. Analysis.
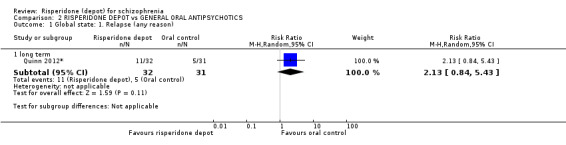
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 1 Global state: 1. Relapse (any reason).
2.2 Global state: 2. Needing use of benzodiazepine or sedative drugs
There was no statistically significant difference between groups for requiring use of benzodiazepines or sedative drugs (n = 369, Rosenheck 2011, Analysis 2.2).
2.2. Analysis.
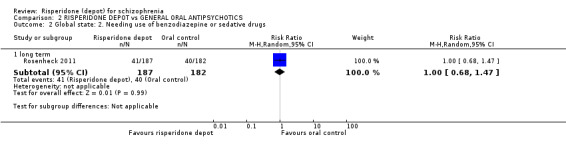
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 2 Global state: 2. Needing use of benzodiazepine or sedative drugs.
2.3 Service utilisation: 1. Hospitalisation
There was no statistically significant difference between groups for hospitalisation by long term (n = 369, Rosenheck 2011, Analysis 2.3).
2.3. Analysis.
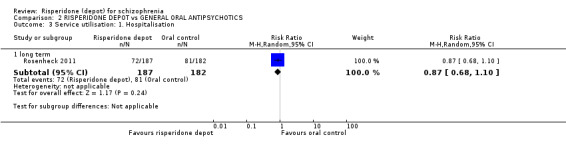
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 3 Service utilisation: 1. Hospitalisation.
2.4 Service utilisation: 2. Outpatient care ‐ number of outpatient visits (skewed data)
Data for this outcome are considerably skewed, and are best inspected by viewing (Analysis 2.4).
2.4. Analysis.
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 4 Service utilisation: 2. Outpatient care ‐ number of outpatient visits (skewed data).
| Service utilisation: 2. Outpatient care ‐ number of outpatient visits (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| long term | ||||
| Rosenheck 2011 | Risperidone depot | 122.4 | 130.9 | 187 |
| Rosenheck 2011 | Oral control | 136.5 | 137 | 182 |
2.5 Not receiving allocated study medication
There was no statistically significant difference between risperidone depot and general oral antipsychotics (n = 382, Rosenheck 2011, Analysis 2.5).
2.5. Analysis.
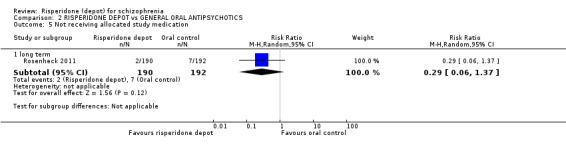
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 5 Not receiving allocated study medication.
2.6 Leaving the study early: 1. Any reason
There was no statistically significant difference between groups for leaving the study early (n = 467, two randomised controlled trials (RCTs), Analysis 2.6).
2.6. Analysis.
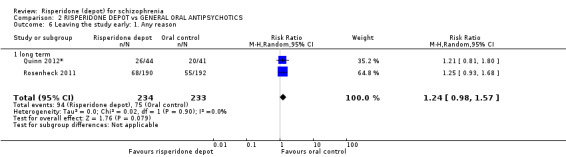
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 6 Leaving the study early: 1. Any reason.
2.7 Leaving the study early: 2. Specific
There was no statistically significant difference between groups for leaving the study early due to insufficient response or withdrawn consent (n = 382, Rosenheck 2011, Analysis 2.7).
2.7. Analysis.
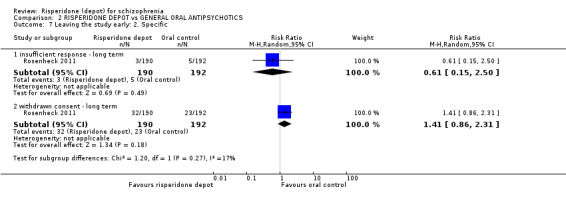
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 7 Leaving the study early: 2. Specific.
2.8 Adverse events: 1. General: a. Death
There was no statistically significant difference between groups for instances of death (n = 467, Rosenheck 2011, Analysis 2.8).
2.8. Analysis.
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 8 Adverse events: 1. General: a. Death.
2.9 Adverse events: 2. Specific
There was a statistically significant difference between groups for general disorders and administration site conditions (n = 369, Rosenheck 2011, RR 1.31 95% CI 1.02 to 1.69, Analysis 2.9) and headache (n = 85, Quinn 2012*, RR 2.80 95% CI 1.12 to 7.00 Analysis 2.9), both favouring oral antipsychotics, with no statistically significant difference between groups for other outcomes of anxiety; dizziness; fatigue/somnolence; insomnia; nausea/vomiting; prolactin‐related; weight increase (n = 85, Quinn 2012*; Analysis 2.9), as well as diabetes mellitus and gastrointestinal adverse events (n = 369, Rosenheck 2011, Analysis 2.9).
2.9. Analysis.
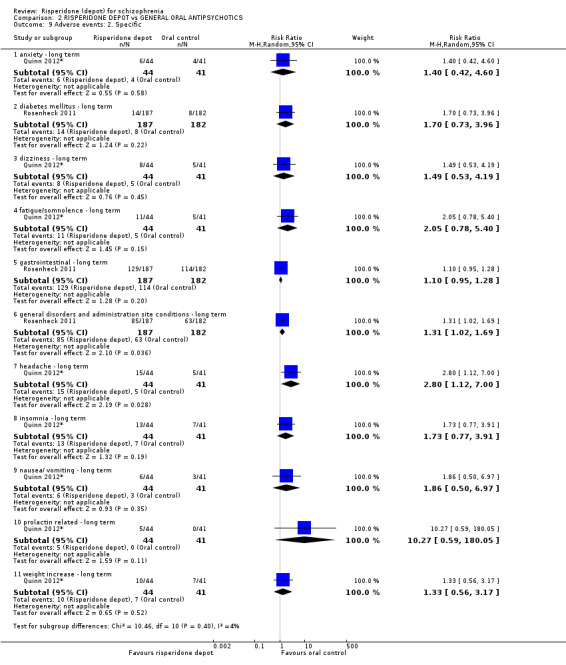
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 9 Adverse events: 2. Specific.
2.10 Adverse events: Nervous system disorders (including extrapyramidal symptoms (EPS))
There was a statistically significant difference between groups for nervous system disorders, favouring the unspecified oral antipsychotics (as prescribed by study's treating physician) (n = 369, Rosenheck 2011, RR 1.34 CI 1.13 to 1.58, Analysis 2.10).
2.10. Analysis.
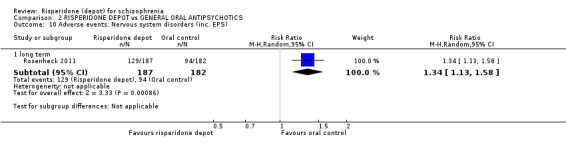
Comparison 2 RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS, Outcome 10 Adverse events: Nervous system disorders (inc. EPS).
COMPARISON 3: RISPERIDONE DEPOT versus ORAL RISPERIDONE
Two studies provided data for this comparison (Bai 2006; Chue 2002, n = 690); both studies were 12‐weeks long and categorised in the 'short term' (note: Chue 2002 had an eight‐week open‐label run‐in period in which participants were stabilised on oral risperidone).
3.1 Global state: 1. Moderate to severely ill at end of study period (Clinical Global Impression (CGI) rating)
There was no statistically significant difference between groups for being 'moderate to severely ill at the end of the study period' (n = 640, Chue 2002, Analysis 3.1).
3.1. Analysis.
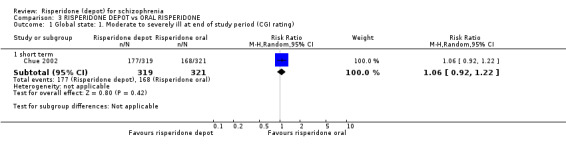
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 1 Global state: 1. Moderate to severely ill at end of study period (CGI rating).
3.2 Global state: 2. Mean change from baseline (CGI‐S, greater change = better outcome)
There was no statistically significant difference between groups in mean changes in CGI‐S score from baseline (n = 50, Bai 2006, Analysis 3.2).
3.2. Analysis.
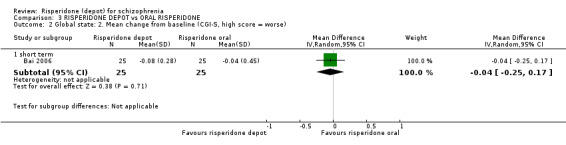
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 2 Global state: 2. Mean change from baseline (CGI‐S, high score = worse).
3.3 Global state: 3. Mean (SD) Global Assessment of Functioning (GAF) score change to endpoint
There was no statistically significant difference between risperidone depot and oral risperidone in mean endpoint scores using the GAF (n = 50, Bai 2006), Analysis 3.3).
3.3. Analysis.
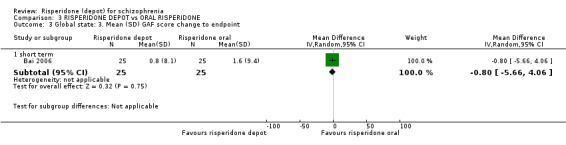
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 3 Global state: 3. Mean (SD) GAF score change to endpoint.
3.4 Global state: 4. Needing use of benzodiazepine or sedative drugs
There was no statistically significant difference between risperidone depot and oral risperidone for needing benzodiazepines or sedative drugs (n = 690, two RCTs, Analysis 3.4).
3.4. Analysis.
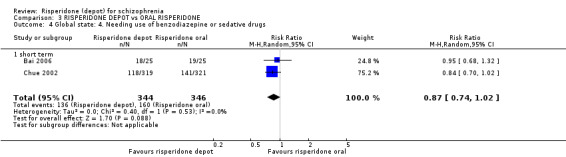
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 4 Global state: 4. Needing use of benzodiazepine or sedative drugs.
3.5 Mental state: 1. Average change/endpoint scores (Positive and Negative Syndrome Scale (PANSS))
There were no statistically significant differences between for average endpoint scores using the PANSS for mean total scores (2 RCTs, n = 591, MD 1.05, CI ‐0.77 to 2.88); average change in positive symptoms or negative symptoms; disorganised thoughts; hostility/excitement; or anxiety/depression (Analysis 3.5).
3.5. Analysis.
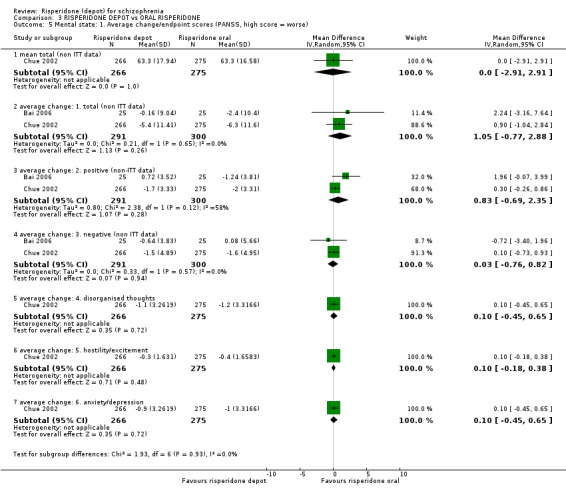
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 5 Mental state: 1. Average change/endpoint scores (PANSS, high score = worse).
3.6 Leaving the study early: 1. Any reason
There was no statistically significant difference between risperidone depot and oral risperidone for leaving the study early for any reason (n = 690, 2 RCTs, Analysis 3.6).
3.6. Analysis.
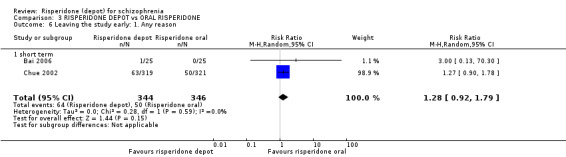
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 6 Leaving the study early: 1. Any reason.
3.7 Leaving the study early: 2. Specific
There were no statistically significant differences between groups for leaving the study early for adverse events; insufficient response; or withdrawn consent (n = 640, Chue 2002, Analysis 3.7).
3.7. Analysis.
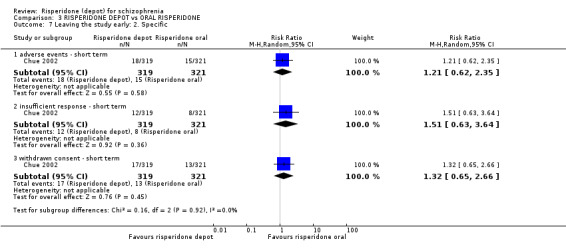
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 7 Leaving the study early: 2. Specific.
3.8 Quality of life: Mean (SD) SF‐36 score change/endpoint
In one small study, there was a statistically significant difference favouring depot risperidone for the social functioning component of the SF‐36 scale (n = 50, Bai 2006, mean difference (MD) 18.50 95% CI 3.98 to 33.02). There were no statistically significant differences between groups for remaining physical and mental components; vitality; general health; mental health; bodily pain; and physical function (n = 50, Bai 2006, Analysis 3.8).
3.8. Analysis.
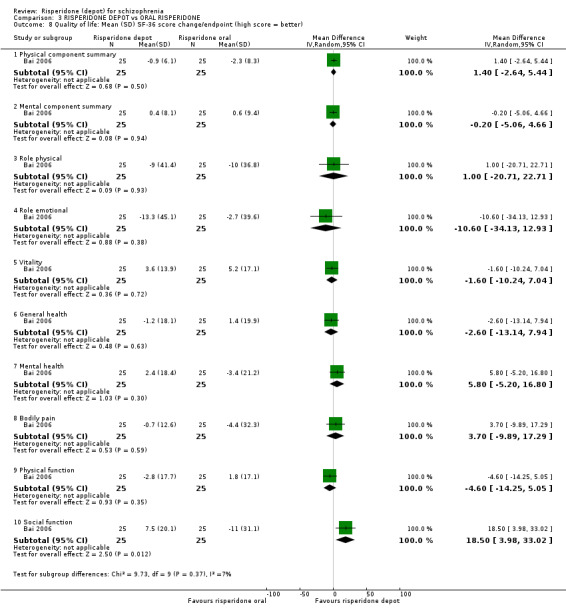
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 8 Quality of life: Mean (SD) SF‐36 score change/endpoint (high score = better).
3.9 Adverse events: 1. General
There were no statistically significant differences between groups for 'any' adverse events or death (n = 640, Chue 2002, Analysis 3.9).
3.9. Analysis.
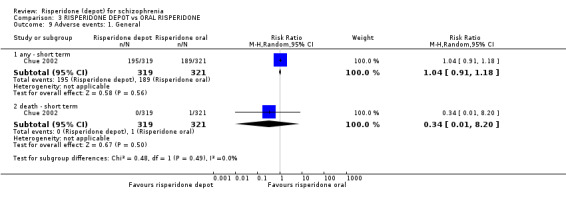
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 9 Adverse events: 1. General.
3.10 Adverse events: 1. General: Udvalg for Kliniske Undersgelser (UKU) (average change score)
There was a statistically significant difference between groups, favouring risperidone depot for adverse events using the UKU, short term (n = 50, Bai 2006, MD ‐1.99, 95% CI ‐3.59 to ‐0.39, Analysis 3.10).
3.10. Analysis.
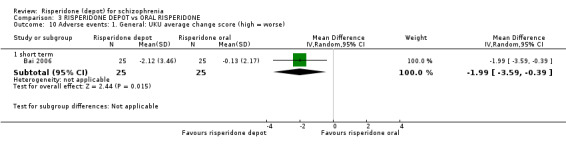
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 10 Adverse events: 1. General: UKU average change score (high = worse).
3.11 Adverse events: 2. Specific
There were no statistically significant differences between groups for specific adverse event outcomes of anxiety; psychosis; prolactin‐related; impotence/ejaculation failure; dysmenorrhoea; hyperprolactinaemia galactorrhoea; headache; insomnia or sexual dysfunction (n = 640, Chue 2002, Analysis 3.11).
3.11. Analysis.
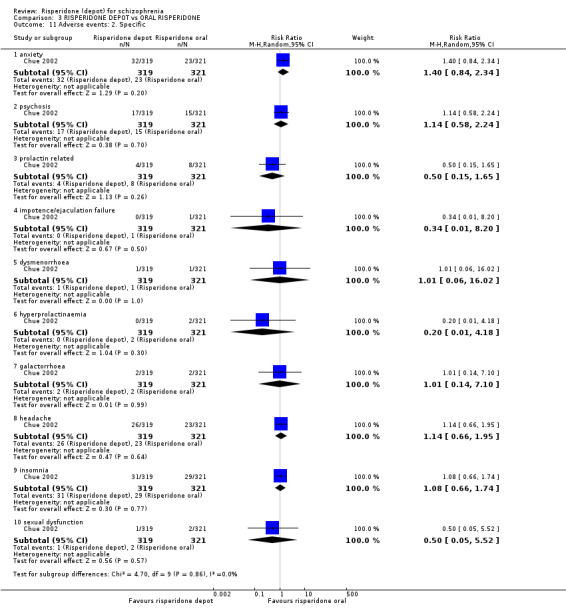
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 11 Adverse events: 2. Specific.
3.12 Adverse events: 2. Specific: Mean (SD) weight increase
There was no statistically significant difference between groups for mean weight increase (kg) (n = 640, Chue 2002, Analysis 3.12).
3.12. Analysis.
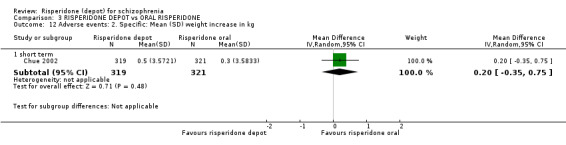
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 12 Adverse events: 2. Specific: Mean (SD) weight increase in kg.
3.13 Adverse events: 3. Movement disorder
There were no statistically significant differences between groups for EPS or tardive dyskinesia (n = 640, Chue 2002); nor was there any statistically significant difference between groups for participants requiring anti‐cholinergic drugs (n = 690, 2 RCTs, Analysis 3.13).
3.13. Analysis.
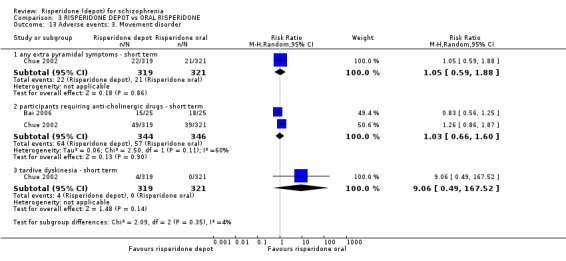
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 13 Adverse events: 3. Movement disorder.
3.14 Adverse events: Mean (SD) change in movement disorder rating scales
There were no statistically significant differences between groups for change in movement disorder rating scales using either Abnormal Involuntary Movement Scale (AIMS), Barnes Akathisia Rating Scale (BARS) or Simpson and Angus Rating scale (SAS) (n = 50, Bai 2006, Analysis 3.14).
3.14. Analysis.
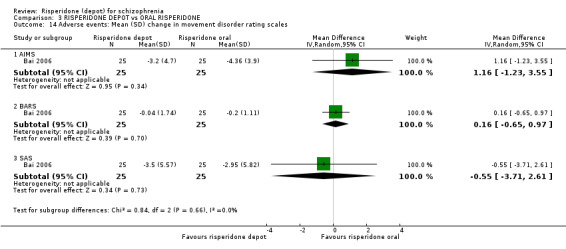
Comparison 3 RISPERIDONE DEPOT vs ORAL RISPERIDONE, Outcome 14 Adverse events: Mean (SD) change in movement disorder rating scales.
COMPARISON 4: RISPERIDONE DEPOT versus ORAL QUETIAPINE
One study provided data for this outcome (n = 666, Gaebel 2010*); as a two‐year study, results are labelled as 'long term'.
4.1 Leaving the study early: 1. Any reason
There was a statistically significant difference favouring risperidone depot for participants leaving the study early for any reason (n = 666, Gaebel 2010*, RR 0.84 95% CI 0.74 to 0.95, Analysis 4.1).
4.1. Analysis.
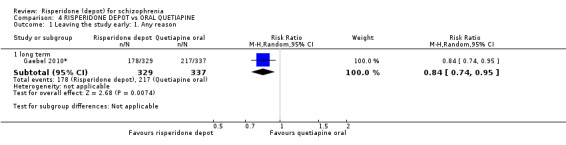
Comparison 4 RISPERIDONE DEPOT vs ORAL QUETIAPINE, Outcome 1 Leaving the study early: 1. Any reason.
4.2 Leaving the study early: 2. Specific
There was a statistically significant difference favouring risperidone depot for participants leaving the study early due to relapse (n = 666, Gaebel 2010*, RR 0.54, CI 0.40 to 0.73, Analysis 4.2).
4.2. Analysis.
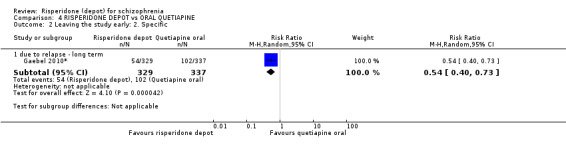
Comparison 4 RISPERIDONE DEPOT vs ORAL QUETIAPINE, Outcome 2 Leaving the study early: 2. Specific.
4.3 Adverse events: 1. General
There were no statistically significant differences between groups for any general adverse events; serious adverse events or death (n = 666, Gaebel 2010*, Analysis 4.3).
4.3. Analysis.
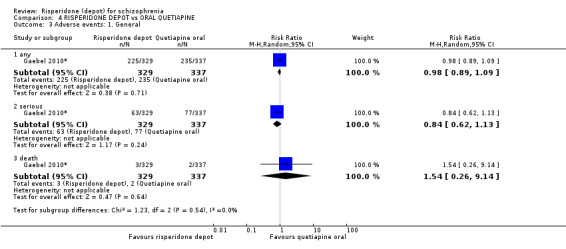
Comparison 4 RISPERIDONE DEPOT vs ORAL QUETIAPINE, Outcome 3 Adverse events: 1. General.
4.4 Adverse events: 2. Specifc
For specific adverse events, there were no statistically significant differences between groups for psychiatric symptoms; serious psychiatric symptoms; weight increase; or headache (n = 666, Gaebel 2010*). There was a statistically significant difference favouring risperidone depot for fatigue/somnolence (n = 666, Gaebel 2010*, RR 0.16 95% CI 0.07 to 0.38); however, there was a statistically significant difference favouring quetiapine oral for prolactin‐related adverse events (P = 0.03) (n = 666, Gaebel 2010*, RR 3.07 95% CI 1.13 to 8.36) and hyperprolactinaemia (n = 666, Gaebel 2010*, RR 8.81 95% CI 3.53 to 21.96, Analysis 4.4).
4.4. Analysis.

Comparison 4 RISPERIDONE DEPOT vs ORAL QUETIAPINE, Outcome 4 Adverse events: 2. Specifc.
4.5 Adverse events: 2. Specific: Mean (SD) weight increase in kg
There was a statistically significant difference between groups favouring oral quetiapine for weight increase (n = 666, Gaebel 2010*, MD 1.25 95% CI 0.25 to 2.25, Analysis 4.5).
4.5. Analysis.
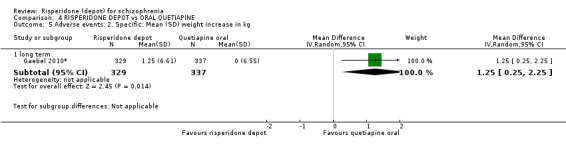
Comparison 4 RISPERIDONE DEPOT vs ORAL QUETIAPINE, Outcome 5 Adverse events: 2. Specific: Mean (SD) weight increase in kg.
4.6 Adverse events: 3. Movement disorder
There was a statistically significant difference between groups, favouring oral quetiapine for any EPS (n = 666, Gaebel 2010*), RR 1.83 95% CI 1.07 to 3.15); tremor (RR 5.12 95% CI 1.13 to 23.20), and parkinsonism (RR 2.56 95% CI 1.01 to 6.52). There was no statistically significant difference between groups for further outcomes of tardive dyskinesia; dystonia; and hyperkinesia (n = 666, Gaebel 2010*, Analysis 4.6).
4.6. Analysis.
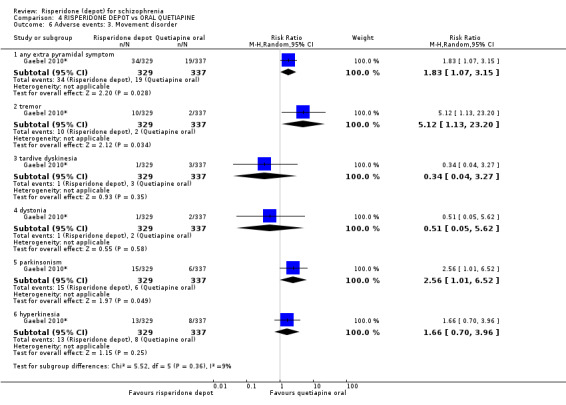
Comparison 4 RISPERIDONE DEPOT vs ORAL QUETIAPINE, Outcome 6 Adverse events: 3. Movement disorder.
COMPARISON 5: RISPERIDONE DEPOT versus ORAL ARIPIPRAZOLE
Two studies provided data for this comparison (n = 730, Gaebel 2010*; MacFadden 2010); both studies were two‐years long, and results are therefore listed as 'long term'.
5.1 Global state: 1. Relapse (any reason)
There was no statistically significant difference between risperidone depot and oral aripiprazole (n = 349, MacFadden 2010, RR 1.05 95% CI 0.83 to 1.33, Analysis 5.1).
5.1. Analysis.
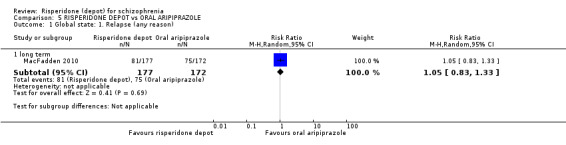
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 1 Global state: 1. Relapse (any reason).
5.2 Global state: 3. Mean time in remission (days)
There was no statistically significant difference between groups for mean time in remission (n = 348, MacFadden 2010, Analysis 5.2).
5.2. Analysis.
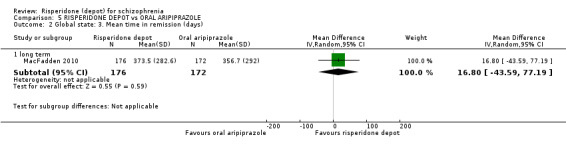
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 2 Global state: 3. Mean time in remission (days).
5.3 Mental state: 1. Average change scores (PANSS, negative change = good) 1. Total
There was no statistically significant difference between groups for change in mental state using PANSS (n = 349, MacFadden 2010, Analysis 5.3).
5.3. Analysis.
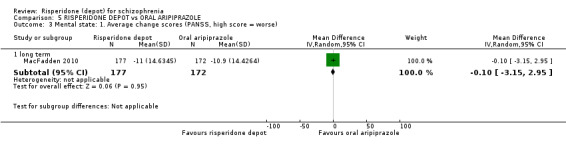
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 3 Mental state: 1. Average change scores (PANSS, high score = worse).
5.4 Leaving the study early: 1. Any reason
There was no significant difference between groups for leaving the study early for any reason (2 RCTs n = 723, RR 0.83 95% CI 0.53 to 1.30, Analysis 5.4). This outcome had important levels of heterogeneity (Chi2 = 5.67; df = 1; P = 0.02; I2 = 82%).
5.4. Analysis.
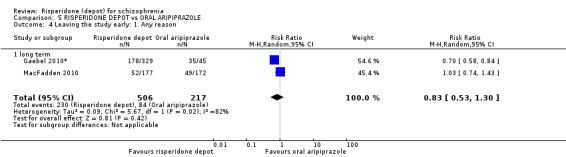
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 4 Leaving the study early: 1. Any reason.
5.5 Leaving the study early: 2. Specific
There were no statistically significant differences between groups for leaving the study early due to adverse events; withdrawn consent (n = 723, 2 RCTs); insufficient response; lost to follow‐up (n = 349, MacFadden 2010); or due to relapse (n = 374, Gaebel 2010*, Analysis 5.5).
5.5. Analysis.
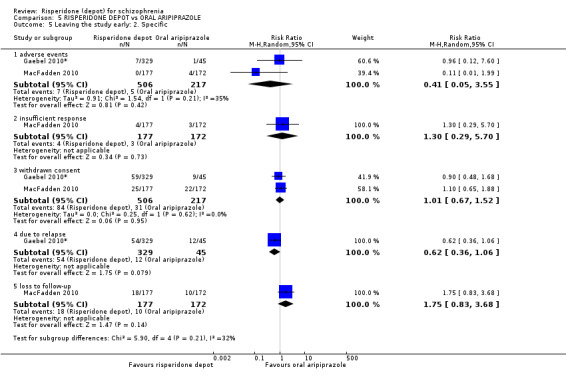
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 5 Leaving the study early: 2. Specific.
5.6 Adverse events: 1. General
There were no statistically significant differences between groups for 'any' adverse events; serious adverse events; or death (n = 729, 2 RCTs, Analysis 5.6)
5.6. Analysis.
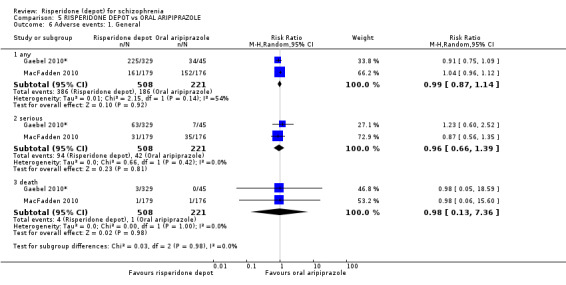
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 6 Adverse events: 1. General.
5.7 Adverse events: 2. Specific
There was a statistically significant difference favouring oral aripiprazole for prolactin‐related adverse events (n = 729, 2 RCTs, RR 9.91 CI 2.78 to 35.29); decreased appetite (1 RCT, n = 355, RR 1.78 CI 1.00 to 3.16), and dizziness (1 RCT, n = 355, RR 1.89 CI 1.00 to 3.58). Gaebel 2010* found a statistically significant difference favouring risperidone depot, with higher instances of gastrointestinal adverse effects (1 RCT, n = 374, RR 0.27 CI 0.14 to 0.55), and MacFadden 2010 had similar results for upper respiratory track infection (1 RCT, n = 355, RR 0.38 CI 0.16 to 0.89, Analysis 5.7).
5.7. Analysis.
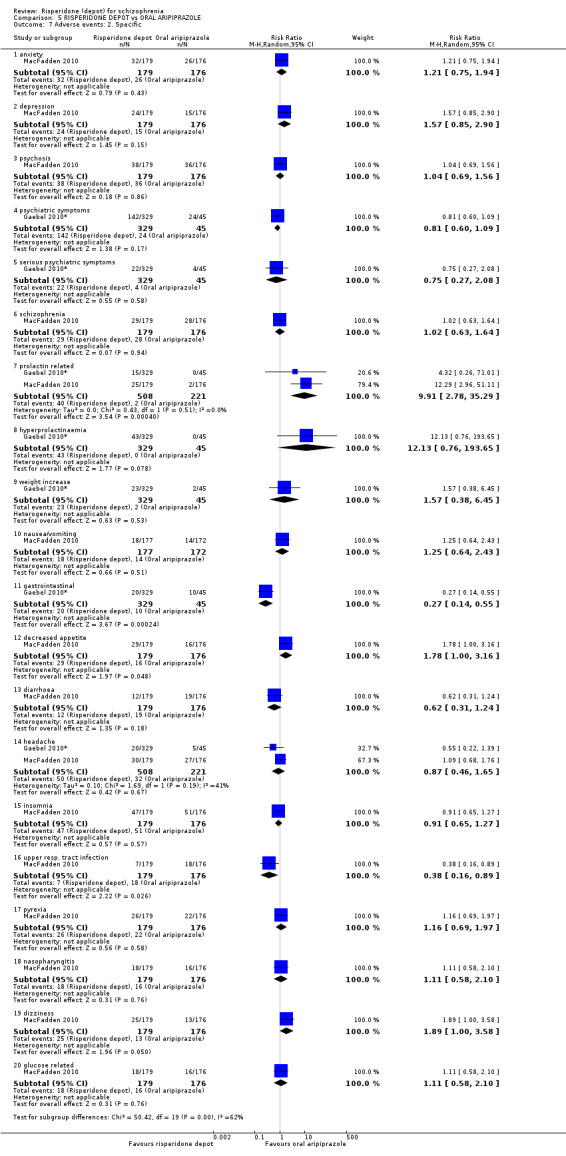
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 7 Adverse events: 2. Specific.
There were no statistically significant differences between groups for headache; anxiety; depression; psychosis; 'schizophrenia'; nausea/vomiting; diarrhoea; insomnia; pyrexia; nasopharyngitis; glucose‐related; psychiatric symptoms; 'serious' psychiatric symptoms; hyperprolactinaemia; or weight increase (Analysis 5.7).
5.8 Adverse events: 2. Specific 12. Mean (SD) weight increase in kg
MacFadden 2010 found no significant difference between risperidone depot and oral aripiprazole for long‐term mean weight increase (1 RCT, n = 355, MD 1.00 95% CI ‐0.42 to 2.42, Analysis 5.8).
5.8. Analysis.
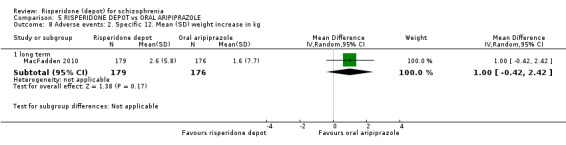
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 8 Adverse events: 2. Specific 12. Mean (SD) weight increase in kg.
5.9 Adverse events: 3. Movement disorder
There were no statistically significant differences between groups for any EPS, tremor or akathisia (Analysis 5.9).
5.9. Analysis.
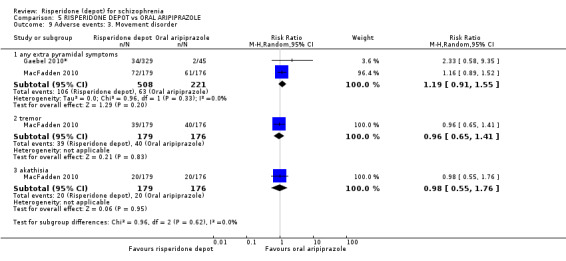
Comparison 5 RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE, Outcome 9 Adverse events: 3. Movement disorder.
COMPARISON 6: RISPERIDONE DEPOT versus ORAL OLANZAPINE
One study provided data for this comparison (n = 361, Keks 2007), which at 12 months is labelled as a 'long‐term' study.
6.1 Mental state: 1. Average change scores (PANSS, negative change = good)
There were no statistically significant differences between groups in average change scores using the PANSS total on short‐term or long‐term positive symptoms, negative symptoms, disorganised thoughts, hostility/excitement or anxiety/depression components (Analysis 6.1).
6.1. Analysis.
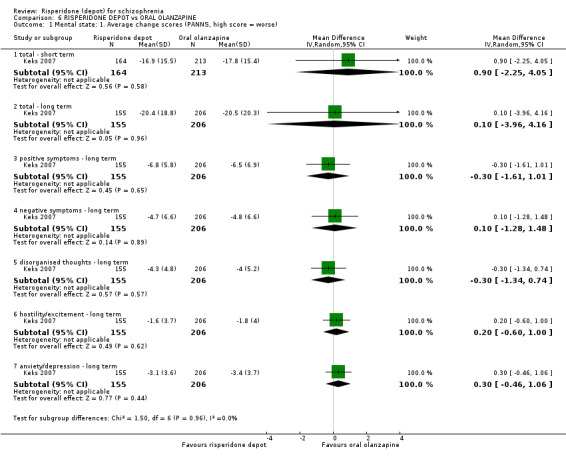
Comparison 6 RISPERIDONE DEPOT vs ORAL OLANZAPINE, Outcome 1 Mental state: 1. Average change scores (PANNS, high score = worse).
6.2 Leaving the study early: 1. Any reason
There were higher rates of leaving the study early in the risperidone (depot) group (1 RCT, n = 618, RR 1.32 95% CI 1.10 to 1.58, Analysis 6.2).
6.2. Analysis.
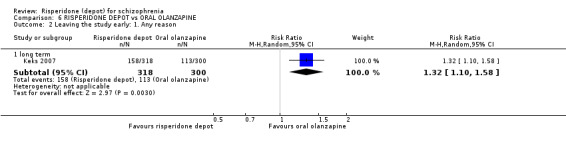
Comparison 6 RISPERIDONE DEPOT vs ORAL OLANZAPINE, Outcome 2 Leaving the study early: 1. Any reason.
6.3 Leaving the study early: 2. Specific
There was no statistically significant difference between groups when leaving the study early due to adverse events; insufficient response; or due to weight gain. There was a statistically significant difference between groups for leaving the study early due to withdrawn consent, with higher losses in the risperidone depot group (1 RCT, n = 547, RR 2.54 95% CI 1.56 to 4.16, Analysis 6.3).
6.3. Analysis.
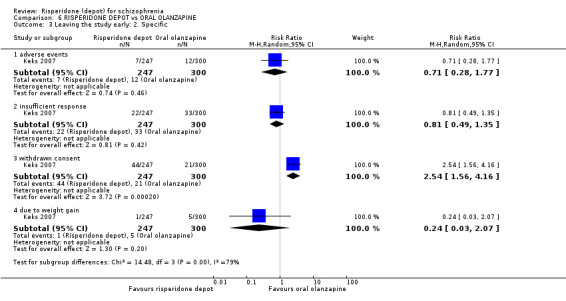
Comparison 6 RISPERIDONE DEPOT vs ORAL OLANZAPINE, Outcome 3 Leaving the study early: 2. Specific.
6.4 Adverse events: 1. General
There were no statistically significant differences between groups for serious adverse events or death (Analysis 6.4).
6.4. Analysis.
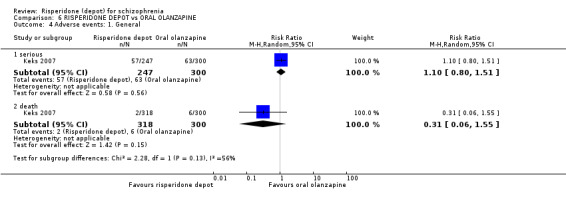
Comparison 6 RISPERIDONE DEPOT vs ORAL OLANZAPINE, Outcome 4 Adverse events: 1. General.
6.5 Adverse events: 2. Specific
There were no statistically significant differences between groups for specific adverse events including anxiety, depression, psychosis, headache, fatigue/somnolence, nasopharyngitis impotence/ejaculation failure, galactorrhoea, serious psychiatric symptoms, serious anxiety, suicide attempt, serious injury, diabetes mellitus, hypoglycaemia, and hyperglycaemia (Analysis 6.5).
6.5. Analysis.
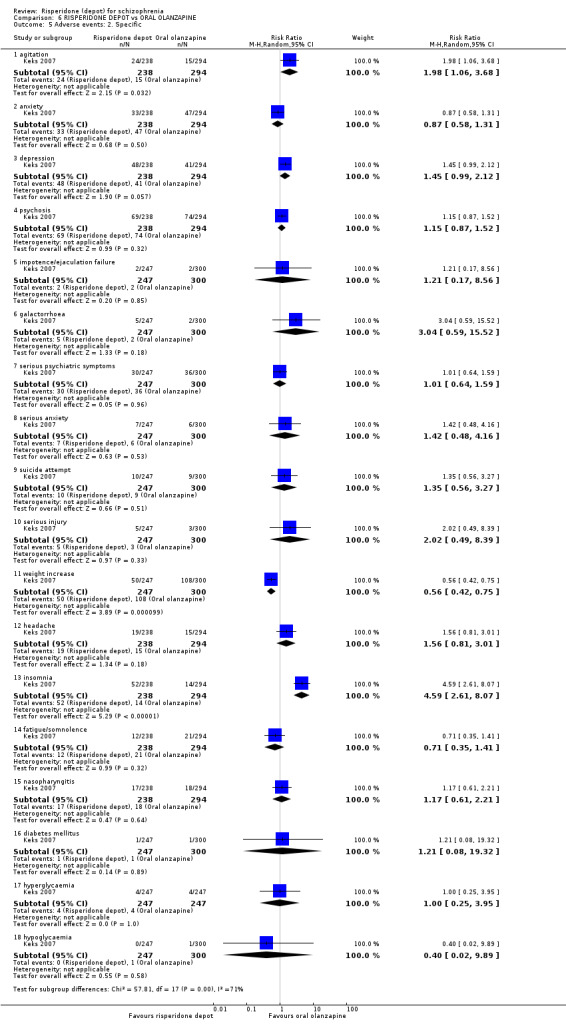
Comparison 6 RISPERIDONE DEPOT vs ORAL OLANZAPINE, Outcome 5 Adverse events: 2. Specific.
There was a statistically significant difference favouring risperidone depot for weight increase (1 RCT, n = 547, RR 0.56 95% CI 0.42 to 0.75), however with significant favour of oral olanzapine for agitation (1 RCT, n = 532, RR 1.98 95% CI 1.06 to 3.68), and levels of insomnia ((1 RCT, n = 532, RR 4.59 CI 2.61 to 8.07, Analysis 6.5).
6.6 Adverse events: 3. Movement disorder
There were no significant differences between groups for tardive dyskinesia; hypertonia; or dystonia (n = 547, Keks 2007). There was a statistically significant difference between groups, favouring oral olanzapine for EPS (1 RCT, n = 547, RR 1.67 CI 1.19 to 2.36), tremor (RR 2.29 95% CI 1.04 to 5.06); hyperkinesia (1 RCT, n = 547, RR 2.02 CI 1.01 to 4.06); and requiring antiparkinson drugs (1 RCT, n = 547, RR 1.26 CI 1.02 to 1.56, Analysis 6.6).
6.6. Analysis.
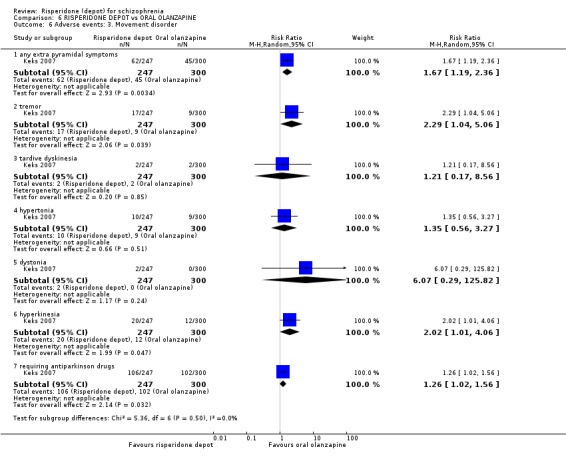
Comparison 6 RISPERIDONE DEPOT vs ORAL OLANZAPINE, Outcome 6 Adverse events: 3. Movement disorder.
COMPARISON 7: RISPERIDONE DEPOT versus ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES)
We pooled all studies comparing risperidone depot with all oral antipsychotics in order to more effectively present estimates of the effects for our primary outcomes in a more concise format (n = 2840, 7 RCTs).
7.1 Global state: 1. Relapse (any reason)
There was no significant difference between groups for relapse with either risperidone depot versus aripiprazole (n = 349, MacFadden 2010), or versus 'general oral antipsychotics' (n = 63, Quinn 2012*, Analysis 7.1).
7.1. Analysis.
Comparison 7 RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES), Outcome 1 Global state: 1. Relapse (any reason).
7.2 Mental state: 1. Average scores (PANSS, greater change = better outcome) 1. total
There was no statistically significant difference between groups in PANSS scores when risperidone depot was compared with oral risperidone (n = 591, 2 RCTs); oral olanzapine at short term (n = 377, Keks 2007) and long term (n = 361, Keks 2007); or oral aripiprazole (n = 349, MacFadden 2010, Analysis 7.2).
7.2. Analysis.
Comparison 7 RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES), Outcome 2 Mental state: 1. Average change scores (PANSS, high score = worse) 1. total.
7.3 Leaving the study early: 1. Any reason
There was no difference between groups in numbers leaving the study early when risperidone depot was compared with oral aripiprazole (n = 723, 2 RCTs); oral risperidone (n = 690, 2 RCTs); or 'any oral antipsychotic' (n = 382, Rosenheck 2011). Gaebel 2010* found a statistically significant difference between groups favouring risperidone depot when compared with oral quetiapine (1 RCT, n = 666, RR 0.84 95% CI 0.74 to 0.95), Quinn 2012* found similar results for 'any new generation antipsychotic' (1 RCT, n = 77, RR 0.72 95% CI 0.55 to 0.95). Keks 2007 found there was a statistically significant difference between groups favouring oral olanzapine (1 RCT n = 618 RR 1.32 95% CI 1.10 to 1.58, Analysis 7.3).
7.3. Analysis.

Comparison 7 RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES), Outcome 3 Leaving the study early: 1. Any reason.
7.4 Adverse events: 1. Death
There were no statistically significant differences between groups for the outcome of death with risperidone depot was compared with oral olanzapine (n = 618, Keks 2007); oral risperidone (n = 640, Chue 2002); 'any oral antipsychotic' (n = 382, Rosenheck 2011); oral aripiprazole (n = 729, 2 RCTs); or oral quetiapine (n = 666, Gaebel 2010*, Analysis 7.4).
7.4. Analysis.
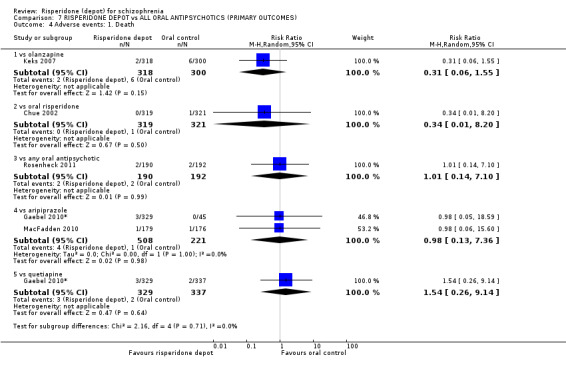
Comparison 7 RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES), Outcome 4 Adverse events: 1. Death.
7.5 Adverse events: 1. General: a. any
There were no significant differences between groups for instances of 'any adverse events' when risperidone depot was compared with either oral aripiprazole (n = 729, 2 RCTs); oral risperidone (n = 640, Chue 2002); or oral quetiapine (n = 666, Gaebel 2010*, Analysis 7.5).
7.5. Analysis.
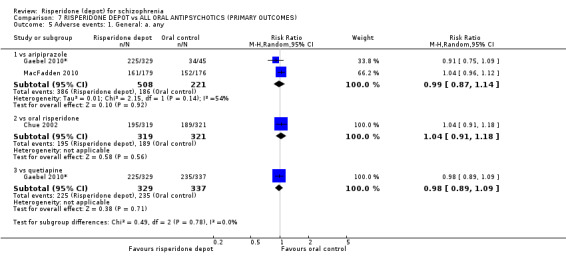
Comparison 7 RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES), Outcome 5 Adverse events: 1. General: a. any.
7.6 Adverse events: 1. General: b. serious
There were no significant differences between groups for instances of 'serious adverse events' when risperidone depot was compared with either oral quetiapine (n = 666, Gaebel 2010*); oral aripiprazole (n = 729, 2 RCTs); or oral olanzapine (n = 547, Keks 2007, Analysis 7.6).
7.6. Analysis.
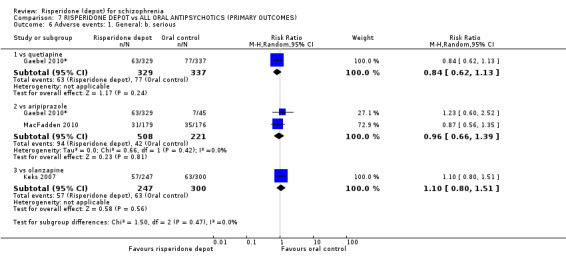
Comparison 7 RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES), Outcome 6 Adverse events: 1. General: b. serious.
7.7 Adverse events: 2. Movement disorder: a. any extrapyramidal symptoms (EPS)
There was no statistically significant difference between groups for instances of 'any EPS' when risperidone depot was compared with oral aripiprazole (n = 729, 2 RCTs) or oral risperidone (n = 640, Chue 2002). There were significantly more instances of EPS amongst participants receiving risperidone depot when compared with oral quetiapine Gaebel 2010* (1 RCT, n = 666, RR 1.83 95% CI 1.07 to 3.15), or oral olanzapine, Keks 2007 (1 RCT, n = 547, RR 1.67 95% CI 1.19 to 2.36, Analysis 7.7).
7.7. Analysis.
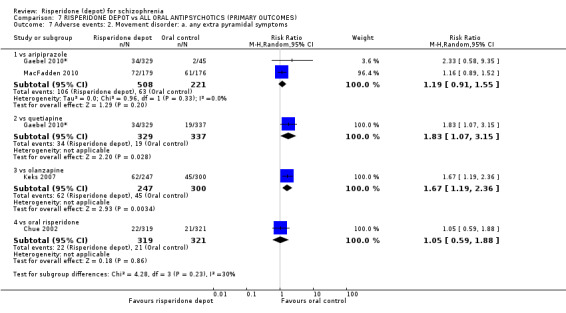
Comparison 7 RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES), Outcome 7 Adverse events: 2. Movement disorder: a. any extra pyramidal symptoms.
COMPARISON 8: RISPERIDONE DEPOT VERSUS ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE)
Three studies compared risperidone depot with paliperidone palmitate (Fleischhacker 2011; Li 2011; Pandina 2011), with a total of n = 2,421. Two of the included studies lasted for 12 months (Fleischhacker 2011; Li 2011), as a long‐term study; Pandina 2011 was categorised as a medium‐term study lasting for 13 weeks.
8.1 Global State: 1. CGI‐S mean change from baseline to endpoint (intention‐treat (ITT) data)
There was no statistically significant difference between groups at medium term (2 RCTs, n = 1326, MD ‐0.07 95% CI ‐ 0.26 to 0.11, Analysis 8.1). This outcome had important levels of heterogeneity (Chi2 = 2.23; df = 1; P = 0.135; I2 = 55%).
8.1. Analysis.
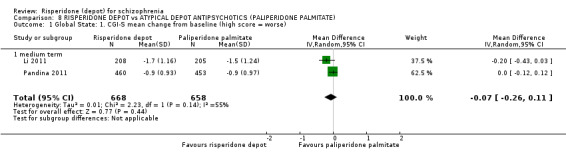
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 1 Global State: 1. CGI‐S mean change from baseline (high score = worse).
8.2 Global state: Schedule for Deficit Syndrome (SDS) scale (mean change from baseline to endpoint, ITT data)
There was no statistically significant difference between groups at medium term for mean change scores using the SDS scale, Pandina 2011, (1 RCT, n = 913, MD 0.1 CI ‐0.29 to 0.49, Analysis 8.2).
8.2. Analysis.
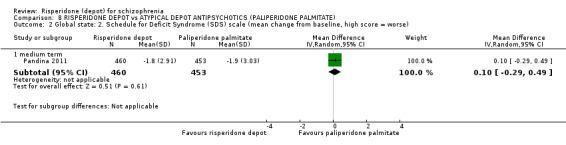
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 2 Global state: 2. Schedule for Deficit Syndrome (SDS) scale (mean change from baseline, high score = worse).
8.3 Mental state: 1. PANSS scores ‐ medium term
Medium term PANSS scores from two studies (n = 1326) demonstrate no statistically significant differences in mental state between groups using the PANSS. The majority of data relating to the subscale scores demonstrated moderate to substantial levels of heterogeneity (P = 0.05; I2 = 73% for anxiety/depression score for endpoint; P = 0.008; I2 = 86% for changes in positive symptom scores, Analysis 8.3).
8.3. Analysis.
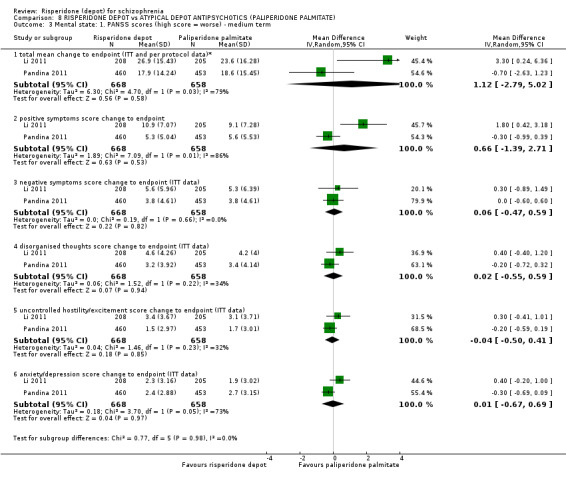
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 3 Mental state: 1. PANSS scores (high score = worse) ‐ medium term.
8.4 Mental state: Improved by 30% in total PANSS score (ITT data)
There was no significant difference between risperidone depot and atypical depot antipsychotics (paliperidone palmitate) at medium term (n = 1326, 2 RCTs), Analysis 8.4. This outcome had important levels of heterogeneity (Chi2 = 5.31; df = 1; P = 0.021; I2 = 81%).
8.4. Analysis.
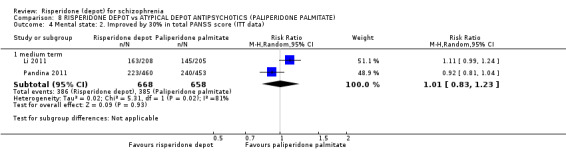
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 4 Mental state: 2. Improved by 30% in total PANSS score (ITT data).
8.5 General functioning: Personal and Social Performance (PSP) scale (mean change from baseline to endpoint)
There was no significant difference between groups in general functioning using the PSP scale at medium term (n = 1326, 2 RCTs, Analysis 8.5).
8.5. Analysis.
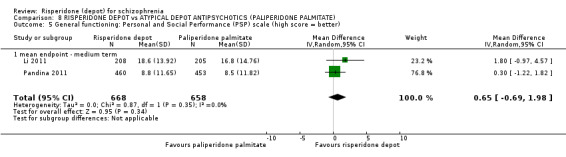
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 5 General functioning: Personal and Social Performance (PSP) scale (high score = better).
8.6 Leaving the study early: 1. Any reason
There were no significant differences between groups leaving the study early due to death; adverse events; patient choice/withdrawn consent; lost to follow‐up; pregnancy; 'other' or 'any reason' at medium term. Although there was no significant difference at medium term, significantly more people receiving paliperidone palmitate left the study early due to lack of efficacy in the long term (1 RCT n = 749, RR 0.60 95% CI 0.45 to 0.81, Analysis 8.6).
8.6. Analysis.
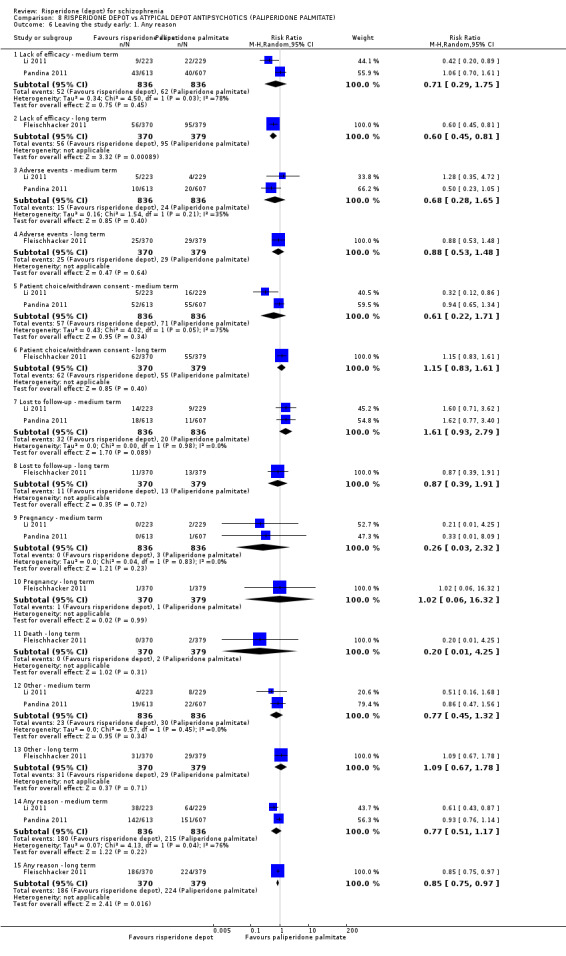
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 6 Leaving the study early: 1. Any reason.
8.7 Adverse events: 1. general
Fleischhacker 2011 found a statistically significant difference in the long term for overall rates of adverse events (1 RCT, n = 747, RR 0.74 95% CI 0.58, 0.95); however, there were no statistically significant differences between groups for general adverse events in the medium term for overall adverse events (n = 1666, 2 RCTs) or medium or long term for; worsening of schizophrenia (n = 1666, 2 RCTs), worsening of psychiatric disorders (n = 1214, Pandina 2011), or death (n = 2415, 3 RCTs), Analysis 8.7.
8.7. Analysis.
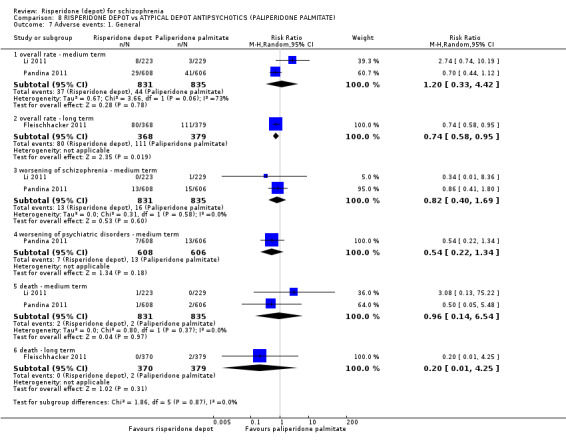
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 7 Adverse events: 1. General.
8.8 Adverse events: 2. specific
There were no significant differences between groups for overall rates of specific adverse effects in the medium or long term (n = 2413, 3 RCTs); insomnia in medium or long term (n = 1961, 2 RCTs), headache, psychotic disorder or 'worsening of schizophrenia' (n = 747, Fleischhacker 2011), somnolence, weight gain (n = 452, Li 2011) or tachycardia (n = 1199, 2 RCTs).
In the medium term, levels of anxiety were significantly greater in participants receiving paliperidone palmitate (1 RCT, n = 1214, RR 0.50 95% CI 0.26 to 0.96); however, at long term, levels of anxiety were significantly greater in participants receiving risperidone depot (1 RCT, n = 747, RR 1.49 95% CI 1.01 to 2.20). People receiving risperidone depot also reported significantly higher instances of constipation at medium term (1 RCT, n = 1214, RR 3.79 CI 1.42 to 10.08). Results were statistically significant for greater pain in injection site at medium term for people receiving paliperidone palmitate (n = 1666, 2 RCTs, RR 0.16 CI 0.07 to 0.38, Analysis 8.8).
8.8. Analysis.
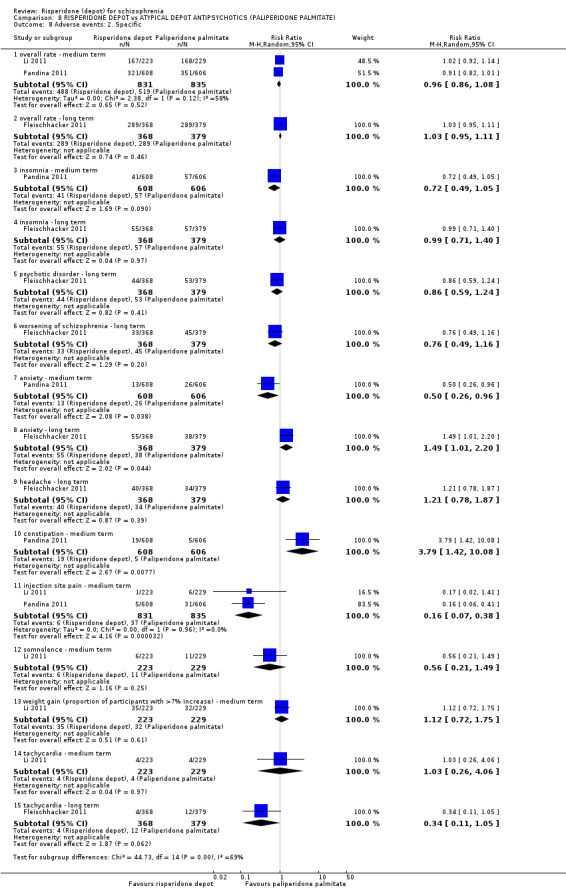
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 8 Adverse events: 2. Specific.
8.9 Adverse events: 3. Prolactin‐related
There were no statistically significant differences between groups in prolactin‐related adverse events, including amenorrhoea (n = 784, 2 RCTs); galactorrhoea or amenorrhoea‐galactorrhoea syndrome (n = 271, Li 2011); hyperprolactinaemia or increase in serum prolactin (n = 452, Li 2011); erectile dysfunction (n = 701, Li 2011); or 'any' prolactin‐related (n = 1666, 2 RCTs). Fleischhacker 2011 found that more men receiving risperidone depot experienced a statistically significant abnormally high level of prolactin (1 RCT n = 424, RR 1.68 95% CI 1.32 to 2.14),,Analysis 8.9, with no difference between groups for women.
8.9. Analysis.
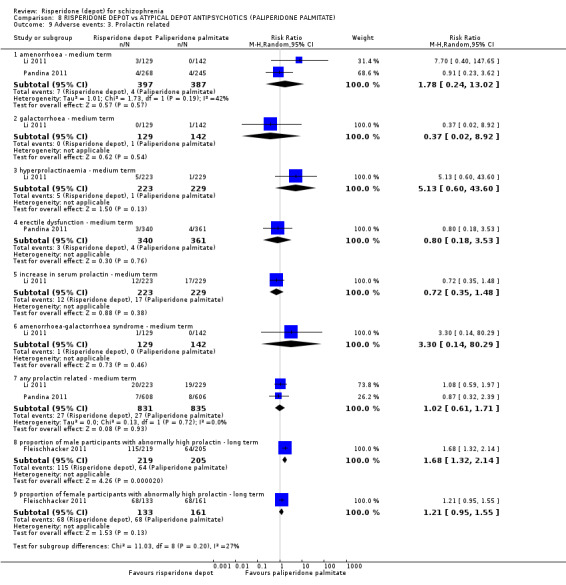
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 9 Adverse events: 3. Prolactin related.
8.10 Adverse events: 4. Movement disorder
In the medium term, there were no significant differences between groups with instances of tardive dyskinesia (n = 1214, Pandina 2011). There were also no significant differences between groups for instances of akathisia and neuroleptic malignant syndrome at medium term (n = 452, Li 2011). Remaining movement disorders were more prevalent in people receiving risperidone depot compared to paliperidone palmitate, with statistically significant differences between groups for tremor (1 RCT, n = 452, RR 1.71 95% CI 1.07 to 2.74) and hyperkinesia (1 RCT, n = 747, RR 1.66 95% CI 1.0 to 2.73). There was a statistically significant difference (P = 0.0004) favouring paliperidone palmitate for requiring use of anti‐EPS medication at medium term (n = 1666, 2 RCTs, RR 1.46 CI 1.18 to 1.80, Analysis 8.10).
8.10. Analysis.
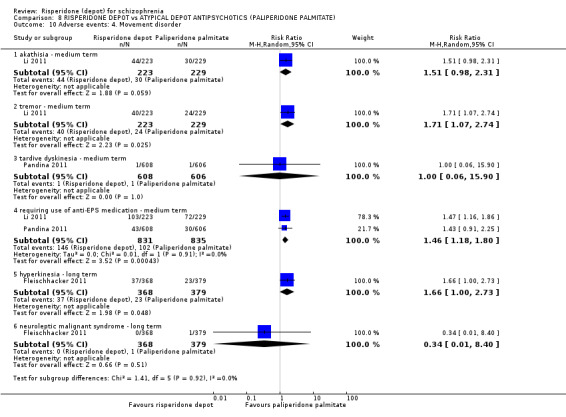
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 10 Adverse events: 4. Movement disorder.
8.11 Adverse events: 5. Body weight (mean increase)
For this outcome we found three relevant trials (n = 2350). There was no statistically significant difference between groups at medium term (n = 1666, 2 RCTs), however, Fleischhacker 2011 found a statistically significant difference in favour of paliperidone palmitate at long term (1 RCT, n = 684, MD 1.00 95% CI 0.13 to 1.87, Analysis 8.11).
8.11. Analysis.
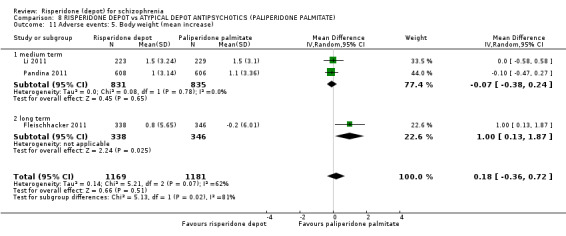
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 11 Adverse events: 5. Body weight (mean increase).
8.12 Adverse events: 6. Mean prolactin level increase (ng/mL)
In this subgroup we found three relevant trials; there were no significant differences between male (n = 1125) and female (n = 807) subgroups in mean prolactin level increase (Analysis 8.12).
8.12. Analysis.
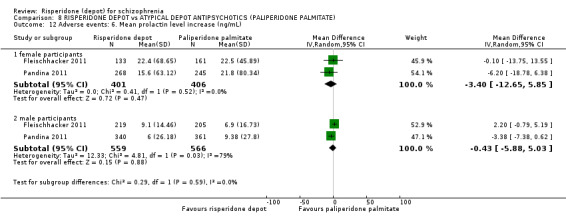
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 12 Adverse events: 6. Mean prolactin level increase (ng/mL).
8.13 Adverse events: 7. Glucose‐related
There were no statistically significant differences between groups for glucose‐related adverse events, including increased blood glucose; hyperglycaemia; diabetes mellitus; glycosuria; ketonuria; urine ketone; hypoglycaemia (n = 747, Fleischhacker 2011); and 'any' glucose‐related adverse events at medium term (n = 1666, 2 RCTs) and long term (n = 747, 1 RCT, Analysis 8.13).
8.13. Analysis.
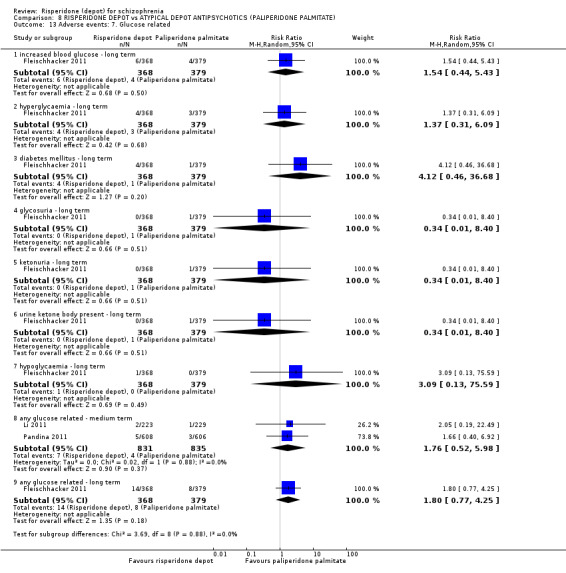
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 13 Adverse events: 7. Glucose related.
8.14 Adverse events: 8. Injection site pain (mean (SD) Visual Analogue Scale score (0‐100 mm))
In one study (Fleischhacker 2011), there were no significant differences in mean scores for injection site pain between groups at baseline and endpoint (n = 474, Analysis 8.14).
8.14. Analysis.
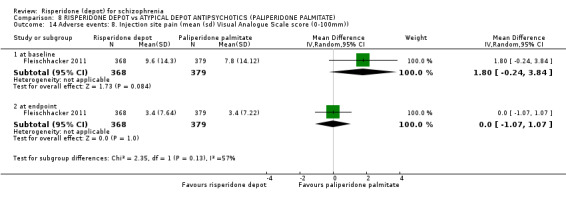
Comparison 8 RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE), Outcome 14 Adverse events: 8. Injection site pain (mean (sd) Visual Analogue Scale score (0‐100mm)).
COMPARISON 9: RISPERIDONE DEPOT versus TYPICAL DEPOT ANTIPSYCHOTICS
One study provided data for this outcome (Covell 2012, n = 62), which, as a six‐month study, is categorised as medium term.
9.1 Mental state: 1. Average scores (PANNS, high score = worse) 1. total
There were no statistically significant differences between groups in total average change score using PANSS at either short (n = 49), medium (n = 46), or long term (n = 43, Analysis 9.1).
9.1. Analysis.
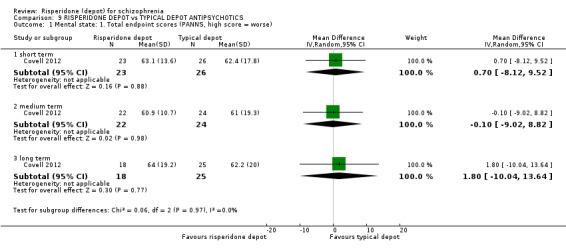
Comparison 9 RISPERIDONE DEPOT vs TYPICAL DEPOT ANTIPSYCHOTICS, Outcome 1 Mental state: 1. Total endpoint scores (PANNS, high score = worse).
9.2 Leaving the study early
There were no significant differences between groups for leaving the study early due to increased psychiatric symptoms; due to EPS effects; due to weight gain and hypertension; or due to participant preference (n = 62, Covell 2012). There was a statistically significant difference between groups for leaving the study early before beginning assigned treatment (RR 7.50 CI 1.00 to 56.44) and by six months (RR 3.05 CI 1.12 to 8.31, Analysis 9.2).
9.2. Analysis.
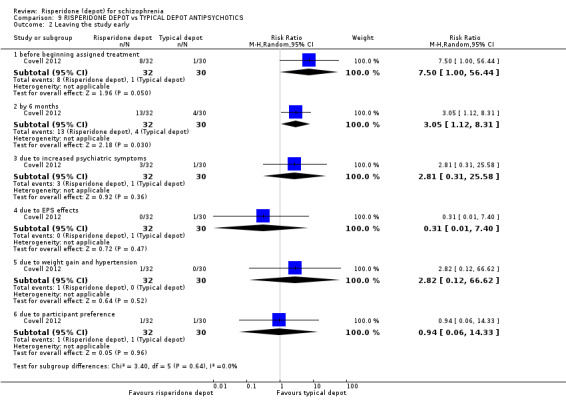
Comparison 9 RISPERIDONE DEPOT vs TYPICAL DEPOT ANTIPSYCHOTICS, Outcome 2 Leaving the study early.
9.3 Hospitalisation by six months
There was no significant difference between groups for hospitalisation by medium term (n = 62, Covell 2012, Analysis 9.3).
9.3. Analysis.
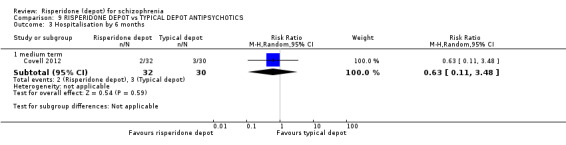
Comparison 9 RISPERIDONE DEPOT vs TYPICAL DEPOT ANTIPSYCHOTICS, Outcome 3 Hospitalisation by 6 months.
9.4 Adverse events: 1. Continuous outcomes (skewed)
All continuous outcome data for change in body mass index (BMI) and prolactin endpoint levels are skewed and are best inspected by viewing (Analysis 9.4).
9.4. Analysis.
Comparison 9 RISPERIDONE DEPOT vs TYPICAL DEPOT ANTIPSYCHOTICS, Outcome 4 Adverse events: 1. Continuous outcomes (skew).
| Adverse events: 1. Continuous outcomes (skew) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Change in BMI ‐ short term (skew) | ||||
| Covell 2012 | Risperidone depot | 1.29 | 1.9 | 23 |
| Covell 2012 | Typical depot antipsychotics | 0.48 | 1.4 | 26 |
| Change in BMI ‐ medium term (skew) | ||||
| Covell 2012 | Risperidone depot | 1.53 | 2.2 | 22 |
| Covell 2012 | Typical depot antipsychotics | 0.53 | 1.3 | 24 |
| Change in BMI ‐ long term (skew) | ||||
| Covell 2012 | Risperidone depot | 1.04 | 2.0 | 17 |
| Covell 2012 | Typical depot antipsychotics | ‐0.28 | 1.7 | 24 |
| Prolactin endpoint levels (ng/mL) ‐ short term (skew) | ||||
| Covell 2012 | Risperidone depot | 22.5 | 19.1 | 19 |
| Covell 2012 | Typical depot antipsychotics | 15.1 | 7.6 | 22 |
| Prolactin endpoint levels (ng/mL) ‐ medium term (skew) | ||||
| Covell 2012 | Risperidone depot | 23.4 | 13.8 | 18 |
| Covell 2012 | Typical depot antipsychotics | 16 | 7.5 | 21 |
| Prolactin endpoint levels (ng/mL) ‐ long term (skew) | ||||
| Covell 2012 | Risperidone depot | 19 | 10.6 | 14 |
| Covell 2012 | Typical depot antipsychotics | 15.2 | 5.1 | 18 |
9.5 Adverse events: 2. Sexual experiences (Arizona Sexual Experiences Scale( ASEX), high score = worse)
There were no statistically significant differences between groups in sexual experiences scores using ASEX at either short (n = 44), medium (n = 41), or long term (n = 40, Analysis 9.5).
9.5. Analysis.
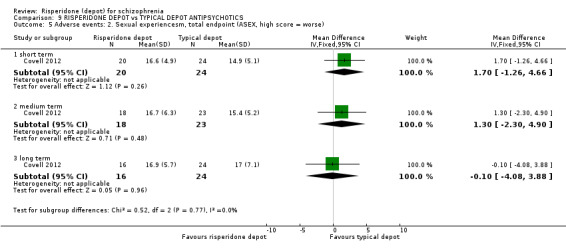
Comparison 9 RISPERIDONE DEPOT vs TYPICAL DEPOT ANTIPSYCHOTICS, Outcome 5 Adverse events: 2. Sexual experiencesm, total endpoint (ASEX, high score = worse).
10. SENSITIVITY ANALYSIS
10.1 Implication of randomisation
10.1.1 Global state: relapse
Both included studies for this outcome were rated as an 'unclear' risk of bias for randomisation and made mention that participants were randomised; however methods of randomisation were not fully described in either study (MacFadden 2010; Quinn 2012*).
10.1.2 Mental state: average scores (PANSS)
Two studies that implied randomisation were removed from data and analysis (Bai 2006; MacFadden 2010); there was no significant difference in the estimate of effect after the removal of these studies.
10.2 Assumptions for lost binary data
10.2.1 Global state: relapse
For our binary outcome of relapse, there was no significant difference in results when completer‐only data were compared with ITT data.
10.3 Risk of bias
All included studies were rated as a 'high' risk of bias across at least one domain; therefore there were no data left to compare for either outcome of relapse, or no clinically important change in mental state.
10.4 Imputed values
We did not include any cluster‐randomised trials, and therefore no values were imputed for intra‐class correlation coefficients (ICC) for either relapse, or no clinically important change in mental state.
10.5 Fixed‐effect and random‐effects
There was no difference in data when synthesised using a fixed‐effect model for both outcomes of relapse and no clinically important change in mental state.
Discussion
Summary of main results
1. Risperidone depot versus placebo
1.1 Relapse
No direct evidence exists regarding the risk of relapse for risperidone depot versus placebo. The closest available data relate to change in mental state, which is discussed below.
1.2 Clinically significant improvement in mental state
Clinically significant improvement in the context of Kane 2002* was defined as being a 20% improvement in total Positive and Negative Syndrome Scale (PANSS) score, but evidence pertaining to this cannot be used in this analysis as more than half of the data were assumed by the authors.To indirectly assess clinically significant changes in mental state, it may possible to look at the incidence of related adverse events; depot risperidone did result in significantly decreased reports of agitation and psychosis, the latter especially could be indicative of a clinical differences. Using adverse events from this study in this way is hampered by the fact that they were only recorded when spontaneously reported by a participant.
1.3 Leaving the study early for any reason
Four hundred people were randomised and 370 participants had at least one injection and one post‐baseline assessment. There was no difference between depot risperidone and placebo group for the outcome of leaving the study before one injection (n = 400, RR 1.30 CI 0.55 to 3.08). Fifty‐six per cent of people did not complete the three‐month study (68% in the placebo group, 52% in the depot groups). The attrition rate was higher for those allocated placebo compared with people allocated risperidone depot. There did not seem to be any dose‐related effects, although when the reason for leaving was lack of efficacy, there seemed to be some differences. Higher doses of risperidone depot resulted in slightly less attrition. People randomised to the placebo were more likely to leave the study early than those in the depot risperidone group, and the level of attrition for any reason did not differ between the three doses of risperidone depot. The numbers of people leaving early due to lack of efficacy may be dose‐related; half as many people gave it as their reason in the 75 mg group compared to the 25 mg group. This suggests that there were people for whom higher doses of depot risperidone are required to produce a therapeutic effect.
1.4 Severe adverse effects
Severe adverse events were common in Kane 2002*, occurring in 13% to 23% of participants and significantly more often in the placebo group. The authors define a severe adverse event as anything that resulted in death or was life‐threatening, required hospitalisation or prolongation of hospitalisation, resulted in persistent or significant disability or incapacity, or resulted in congenital anomaly or birth defect. No details are given about specific severe adverse events that were reported in each group; this raises the serious possibility that very different events have been bundled together. This in turn could conceal evidence regarding important effects of using depot risperidone, but there is no way to tell with the data reported as they are.
1.5 Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism
Rates of movement disorder did not differ between depot risperidone and the placebo; the data did show a trend towards more extrapyramidal symptoms (EPS) with increasing doses of depot risperidone, but even at the 75 mg dose of risperidone, this fell short of statistical significance (Analysis 1.10). More studies, with larger sample sizes, would probably produce a significant result, but it is unlikely that such studies will ever be conducted.
The number of participants experiencing weight gain did not differ between the interventions.
2. Risperidone depot versus general oral antipsychotics
2.1 Relapse
Quinn 2012* found no significant difference in relapse rates between risperidone depot and general oral antipsychotics.
2.2 Clinically significant improvement in mental state
Data relating directly to mental state (such as PANSS scores) are not useable due to the high levels of attrition in both Quinn 2012* and Rosenheck 2011. Alternative outcomes that may give some indirect indication of the mental state of participants include the need for benzodiazapine or sedative drugs and the rate of hospitalisation. Neither of these outcomes differed significantly between the treatment groups.
2.3 Leaving the study early for any reason
There was no significant difference in the overall rates of study attrition when comparing risperidone depot with general oral antipsychotics, however the general trend was towards risperidone depot group participants being more likely to leave, with the lower limit of the 95% confidence interval only just falling to the left of the line of no effect (Analysis 2.6).
2.4 Severe adverse effects
Outcomes relating to severe adverse events were not available.
2.5 Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism
There was no difference between risperidone depot and general oral antipsychotics for adverse events related to weight gain, prolactin or glucose metabolism. "Nervous system disorders" were more likely to occur in those taking risperidone depot than those on oral treatment; it is unclear precisely what proportion of these events involved movement disorder.
3. Risperidone depot versus oral risperidone
3.1 Relapse
Relapse was not an outcome addressed by any usable outcomes from these studies.
3.2 Clinically significant improvement in mental state
3.3 Leaving the study early for any reason
Leaving the study early did not occur any more in one treatment arm over the other. The attrition rate in Bai 2006 was very low, with only one participant from the risperidone depot group and none from the oral group leaving early; this is probably due to its setting being an inpatient psychiatric unit, where the constant presence of various healthcare professionals would ensure that patients adhered to their prescribed medication. Attrition in Chue 2002, a study taking place in the community, was predictably higher.
3.4 Severe adverse effects
No distinction was made between serious and general adverse events in either study.
3.5 Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism
There was no difference between depot and oral risperidone regarding the number experiencing any EPS, which occurred in 6.5% to 6.8% of all participants in Chue 2002. Tardive dyskinesia was reported four times in the depot group of Chue 2002 and not at all in the oral group, but this was not quite enough to establish a statistical difference between the two.
Prolactin‐related adverse events were rare, occurring in 1.2% to 2.5% of participants; with between zero and two people in each group experiencing each of: sexual dysfunction and impotence, galactorrhoea and dysmenorrhoea. The number of participants who experienced a significant amount of weight gain is not known, but there was a small mean increase of 0.5 kg in the depot group and 0.3 kg in the oral group.
4. Risperidone depot versus oral quetiapine
4.1 Relapse
Time to relapse was measured as a primary outcome by Gaebel 2010*, but the high level of attrition in the study made most of the data unsuitable for inclusion in our analysis. One outcome that can be included is the number of participants leaving the study early for the specific reason of relapse; individuals in the oral quetiapine group were 46% more likely to leave the study due to relapse than the risperidone depot group.
4.2 Clinically significant improvement in mental state
Outcomes relating to mental state suffer from the same high attrition rates that affect so many of the included studies.
4.3 Leaving the study early for any reason
Participants in the oral quetiapine group were significantly more likely to leave the study early for any reason,
4.4 Severe adverse effects
There was no difference between the two interventions for the rate at which severe adverse events were reported, and the overall rate of severe adverse events in Gaebel 2010* appears broadly congruent with the other included studies.
4.5 Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism
Risperidone depot caused more EPS than oral quetiapine among the study population, with ˜10% of the risperidone group reporting any EPS symptom compared to 5.6% of the quetiapine group. The specific symptoms of tremor and "parkinsonism" were more common with risperidone depot.
Similar numbers of people in both groups experienced weight gain through the study period, but the mean (SD) increase in weight appears to be greater for risperidone depot.
Significantly more prolactin‐related problems occurred in those taking depot risperidone. The rate of hyperprolactinaemia was almost nine times greater than the quetiapine group, and risperidone group members were three times more likely to experience side effects related to prolactin.
5. Risperidone depot versus oral aripiprazole
5.1 Relapse
MacFadden 2010 showed no difference between the two interventions, with just under half of each group relapsing during the study period. These data cannot be compared directly to the relapse data from Gaebel 2010* due to the attrition level of that study, but the number of individuals leaving the study early due to relapse in Gaebel 2010* did trend towards favouring risperidone depot for relapse prevention, the very small size of the aripiprazole arm in this study prevents this outcome from reaching statistical significance.
5.2 Clinically significant improvement in mental state
Data relating to PANSS responder rates were not available or usable for either study, but MacFadden 2010 reported that the overall change in PANSS total score through the study period was virtually the same for both groups, with a mean reduction of around 11 points.
5.3 Leaving the study early for any reason
There was substantial heterogeneity relating to this outcome. Meta‐analysis shows no difference between risperidone depot and oral aripiprazole when leaving the study early for any reason, but the participants in each study differed markedly. Drop‐out rates in MacFadden 2010 were very similar (n = 349 RR 1.03 [0.74, 1.43]), with around 30% attrition overall, whereas in Gaebel 2010*, those in the aripiprazole group were significantly more likely to leave (n = 384 RR 0.70 [0.58, 0.84]), with 78% dropping out of that group. It is unclear what has contributed to this disparity.
5.4 Severe adverse effects
Rates of serious adverse events were the same for both interventions.
5.5 Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism
Rates of movement disorder did not differ significantly overall between the risperidone depot and oral aripiprazole groups, but this probably does not tell the whole story. Risperidone depot was associated with movement disorder in a greater proportion of participants than aripiprazole in Gaebel 2010*, but the substantial disparity in the size of the two analysis sets prevents those results from achieving statistical significance (n = 384 RR 2.33 [0.58, 9.35]). There were also differences between the studies, with substantially more participants in both groups reporting movement disorder in MacFadden 2010. It is not clear why this is so; whether it is due to methodological differences in the way such events were defined and the data collected, differences in the make up of the study population and their susceptibility to EPS effects or some other factors.
Glucose‐related events were reported by 9% to 10% of those in MacFadden 2010, with no difference between the groups. Weight gain was also broadly similar for both interventions, but prolactin‐related adverse events were much more common with risperidone depot and there were no incidences of hyperprolactinaemia with aripiprazole
6. Risperidone depot versus oral olanzapine
6.1 Relapse
The only study comparing risperidone depot to oral olanzapine (Keks 2007) did not include relapse as an outcome.
6.2 Clinically significant improvement in mental state
There is some discrepancy between the stated levels of study attrition (i.e. the completion rate) and the number of participants for whom outcome data are available up to the end of the study period. For depot risperidone, average endpoint PANSS scores are provided for 155 participants, with last observation carried forwards (LOCF) analysis used. However, the number of participants for whom the mean PANSS score change at 12 months (the end of the study) is given was only 116. We are not certain how the difference between the number of participants who completed the study from this group (n = 160) and the number for whom data are available at 12 months (n = 116) can be so great.
6.3 Leaving the study early for any reason
Participants taking risperidone depot had a significantly increased risk of leaving the study early; about half of the risperidone depot group compared to a third of the olanzapine arm. Much of this difference can be explained by the decision of the investigators to alter the protocol and exclude the 75 mg risperidone depot group after randomisation had taken place, resulting in 66 participants being excluded from the main analysis. If this is taken into account, the rates of participants leaving the study for other reasons, such as insufficient response, are similar for each intervention.
6.4 Severe adverse effects
Severe adverse events were reported at similar rates for both interventions, but what made a particular adverse event "severe" is not explained. Adverse events such as anxiety are reported as both general and severe adverse events Presumably the investigators in Keks 2007 had some criteria to differentiate severe adverse events, but these are not explained.
6.5 Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism
Movement disorder was a common adverse event in both groups, but was significantly more common in those receiving risperidone depot, with around 25% experiencing it to some degree, compared to 15% of the olanzapine group. Perhaps unsurprisingly, the usage of anti‐parkinsonian medications was greater in the depot risperidone group as well. Reporting of EPS was not clear in this study; the overall percentage of participants experiencing EPS was given, but it is difficult to see how these data correspond to those in a table which breaks down EPS into separate symptoms, e.g. hyperkinesia, tardive dyskinesia, tremor, etc. In particular, "Extrapyramidal disorder" is listed as a specific outcome in this table, but was apparently only reported by 8% of the risperidone depot group and 4% of the olanzapine group. There is no way to discern precisely what this item encompasses, nor what differentiates it from the overall rate of EPS. We have contacted the study authors for clarification on this point, but have yet to receive a response.
Olanzapine caused significantly more weight gain than risperidone depot in the study population; more than 1/3 of participants taking olanzapine are reported to have experienced weight gain greater than 7% of their bodyweight (mean weight gain overall of 4.0 kg), compared to a 1/5 of the risperidone group (mean increase of 1.7 kg overall).
Prolactin‐related adverse events were rare and did not differ between the interventions; there were two instances of impotence or ejaculation failure in each arm, and similarly low levels of galactorrhoea. One new diagnosis of diabetes mellitus was made in each group during the study period.
7. Risperidone depot versus all oral antipsychotics (primary outcomes)
7.1 Relapse
Data from the three trials (Gaebel 2010*, MacFadden 2010, Quinn 2012*) that measured relapse directly suggest that it occurs at the same rate when depot risperidone treatment is compared to aripiprazole or a variety of second‐generation antipsychotics. When compared to quetiapine, however, depot risperidone treatment leads to only half as many cases of relapse. The precise methods used to define this outcome vary between Gaebel 2010* and MacFadden 2010, but they are similar enough in appearance (factors such as hospitalisation, PANSS and CGI change being common between the two) that we can be confident they are measuring the same thing. Quinn 2012* is more problematic, as no definition of relapse is given; such methodological details are probably too much to be asking of a single poster and an abstract, but if a full report becomes available it will be important to confirm this.
The quality of some of this evidence is also questionable; Gaebel 2010* and Quinn 2012* in particular had a high risk of bias due to their nature as open‐label studies with no evidence of rater blinding. One perhaps very serious methodological weakness of Gaebel 2010* that may have had an impact on the effect size reported was that follow‐up methods differed between intervention groups. Depot risperidone participants attended physical appointments to receive their medication and undergo assessment of certain outcomes every two weeks, whereas those in the oral quetiapine and aripiprazole groups received phone calls for the same purpose. The depot group participants therefore spent more time with healthcare professionals during the course of the study; even if the exact same questions were asked at in‐person and telephone check‐ups, seeing a patient in the flesh may have made it easier to detect clinical deterioration and adjust doses of concomitant medications and the study drug before a slight deterioration has time to snowball into a full relapse.
The regular attention that comes with being prescribed depot rather than oral antipsychotics may be an important aspect of the intervention, but in a randomised clinical trial (RCT) of two drugs there is a case for limiting potential confounders so that the only effect measured is that of the drugs, not that of the drugs plus the intensity of care provided. More studies are needed that address the issue of relapse directly; a RCT comparing relapse and hospitalisation rates between depot and oral risperidone is of particular importance. Using only the currently available evidence comparing depot risperidone with oral quetiapine, it is not possible to tell whether the significant difference in relapse rates is due mainly to the difference in administration methods or due to the different pharmacological properties of risperidone and quetiapine. If oral risperidone is as effective as the depot preparation at preventing relapse, then economic considerations may take on greater importance in prescribing decisions.
7.2 Clinically significant improvement in mental state
No comparisons with usable data from these studies give any direct indication as to how many participants achieved clinically significant improvements in mental state, but data regarding average mental state scores are available. Risperidone depot is not significantly different from oral risperidone, olanzapine or aripiprazole in its effect on mental state, as measured through all domains of PANSS, with the average scores for participants in all study arms either improving or remaining largely the same (Analysis 7.2).
7.3 Leaving the study early for any reason
People randomised to risperidone depot were as likely to leave the study early as those assigned to aripiprazole (n = 723, RR 0.83, 95% CI 0.53 to 1.30), oral risperidone (n = 690, RR 1.28, 95% CI 0.92 to 1.79), or any oral antipsychotic (n = 382, RR 1.25, 95% CI 0.93 to 1.68). Compared to oral quetiapine and any new‐generation oral antipsychotic, participants in the risperidone depot group were significantly less likely to leave the study early (n = 666, RR 0.84, 95% CI 0.74 to 0.95 and n = 77, RR 0.72, 95% CI 0.55 to 0.95, respectively), and significantly more likely to drop out than participants randomised to olanzapine (n = 618, RR 1.32, 95% CI 1.10 to 1.58).
7.4 Severe adverse effects
There is great inconsistency in the reporting of serious adverse effects amongst these studies, which makes any comparisons problematic. When severe effects were reported there were no differences in the rates at which they occurred when comparing depot risperidone to quetiapine, aripiprazole or olanzapine. It is not clear, though, whether the outcome of severe adverse effects if comparable between these studies; of the three trials, none provide a definition of what they consider to be a serious adverse event and the reporting of the specific events is often very vague. Keks 2007 is the only study to explicitly list the severe adverse events that were reported by more than 2% of either group; of the other two studies, Gaebel 2010* only gives an overall rate and the rate of the most common serious adverse events (psychiatric symptoms), while MacFadden 2010 states that psychotic disorder and schizophrenia were the most common serious adverse events but gives no details on their precise incidence.
With severe adverse events reported in such a scattergun manner throughout these trials, it is impossible to perceive any trends or patterns of severe adverse events emerging for any of the interventions. A rare, but serious, side effect (or side effects) may occur consistently at a rate below the reporting threshold yet remain unreported in the literature.
7.5 Economic costs of treatment
An analysis of Medical Resource Utilisation (MRU) using a small subset of relapsed participants from Gaebel 2010* determined that each individual cost their country's health service an average of €7592 in the three months following their relapse and that this figure was €1525 higher in people who required psychiatric hospitalisation during this time.
Drawing too many conclusions from these data are dangerous; this analysis is available only as a single abstract. As such, details on the methods used to produce these figures are sparse; of particular concern is that fact that Gaebel 2010* was spread out over 124 centres in 25 European countries. It is quite conceivable that there is significant heterogeneity in the health services of these countries that may affect how and where money is spent. The full version of the paper may be able to address this, but until then it is unclear exactly how applicable this evidence is.
Despite this, it is still clear that relapse, especially if it involves hospitalisation, is expensive. Using administrative data from the Veterans Administration (VA), the researchers in Rosenheck 2011 were able to determine the mean (SD) cost of in‐ and out‐patient care, as well as medication costs, in each treatment group per quarter (three months) of follow‐up). They found that the overall costs relating to service use for each group were similar, a result that seems congruent with the finding that there was no difference between groups in levels of post‐randomisation hospitalisation or out‐patient care. Medication costs, however, were substantially higher for depot risperidone. The study drug itself cost more than the control group's medication, but further to this, depot group participants also received nearly $500 of extra oral antipsychotics. They also required $357 more in concomitant medications, leading to approximately $1000 more per quarter being spent on each patient with no quantifiable difference in outcomes.
Based solely on these data, it is not possible to claim that depot risperidone is a cost‐effective treatment option for schizophrenia. Generalising these results specifically in terms of absolute amounts spent may be an issue; the study they are based on was of a 90% male population of military veterans with unstable schizophrenia, whose needs could differ substantially from patients in other situations. There is also a reasonable risk that the unblinded nature of follow‐up could have lead to clinical decisions (e.g. whether to hospitalise) being made differently depending on the intervention a person was using, leading to performance bias. However, the data regarding the price of depot risperidone compared to oral treatment are important.
In the future, we plan to undertake full economic analysis to complement the above findings.
8. Risperidone depot versus atypical depot antipsychotics (paliperidone palmitate (PP))
8.1 Relapse
As with placebo, there is no direct evidence regarding relapse for this comparison.
8.2 Clinically significant improvement in mental state
A clinically significant difference in mental state was considered to be a 30% or greater reduction in total PANSS score by the three studies. For Fleischhacker 2011, however, the high level of overall study attrition (55% did not complete) combined with the use of LOCF data means that more than half of the data used to determine this outcome was based on assumptions. Synthesis of the data from Li 2011 and Pandina 2011 shows there is no difference between depot risperidone and paliperidone palmitate for the number of PANSS responders, though Li 2011 did report a greater improvement in total PANSS (RR 3.30 CI 0.24 to 6.36) and positive symptom (P = 0.01, RR 1.80 CI 0.42 to 3.18, Analysis 8.4) scores for those receiving PP.
The PANSS responder rates reported by the two studies demonstrate a significant level of heterogeneity, with around 50% of participants in Pandina 2011 compared to 70% to 78% in Li 2011, and it is not immediately apparent what has caused this difference. Their respective designs are very similar, with common treatment period duration, the same range of doses used for each intervention and the average final dose for each treatment was also similar (the mean (SD) final PP dose was 104.5 (30.51) mg eq in Pandina 2011 and 115.8 (9.07) mg eq in Li 2011, but this difference can probably be accounted for by the analysis set used to determine these figures. Li 2011 gave the mean for the per protocol analysis set, while Pandina 2011 gave it for the safety analysis set, which includes participants who left the study after only one dose who therefore would not have been titrated onto the higher doses yet). The two important differences are probably blinding and sample size; Li 2011 randomised 452 people and was open‐label, whereas Pandina 2011 had more than double the study population (n = 1220) and utilised a double‐blind approach.
8.3 Leaving the study early for any reason
Participants assigned to depot risperidone were overall less likely to leave the study early for any reason, but it is difficult to reason exactly why that is. It could be related to efficacy; data from mental and global state outcomes show that depot risperidone and PP are essentially equivalent in their effects, yet there was a (not quite statistically significant) trend towards PP group participants being more likely to cite lack of efficacy when leaving the study. As PP is a newer drug (albeit a derivative of risperidone), it is conceivable that the dosing guidelines are less well supported, or that people taking PP were less likely to be dosed appropriately than those allocated to depot risperidone.
8.4 Severe adverse effects
Serious adverse events occurred at the same rate for depot risperidone and paliperidone palmitate, being reported by 10% to 13% of participants in all three studies. If the two‐year long trial (Fleischhacker 2011) is excluded from this analysis to leave only the shorter follow‐up studies, the rate of serious adverse events goes down to below 5%, which suggests that serious adverse events may be more likely to occur after being on either of the study drugs for a prolonged period. Fleischhacker 2011 also featured significantly more severe adverse events in the paliperidone palmitate group.
Differences in study design other than the length of follow‐up are unlikely to account for the excess severe adverse events as all three studies shall very similar methodology, with the exception that Fleischhacker 2011 & Pandina 2011 are double‐blind studies, while Li 2011 is open‐label. This finding is important because most people who are prescribed antipsychotics for schizophrenia are presumably expecting to take them for more than 13 weeks, so the short‐term studies might not present an accurate representation of the risk.
Fleischhacker 2011 also raises the possibility that while the two drugs may be similar in the short term, over time, the risk of serious adverse effects may be greater for PP; more long‐term studies of depot risperidone and paliperidone palmitate are needed to assess this.
8.4.1 Adverse events related to movement disorder, weight gain, prolactin levels and glucose metabolism
Keks 2007 has very strange analysis sets
8.5 Economic costs of treatment
Cost or cost‐effectiveness outcomes are not included in any of the three studies comparing depot risperidone versus paliperidone palmitate.
9. Risperidone depot versus typical depot antipsychotics
9.1 Relapse
Covell 2012 did not directly assess rates of relapse for this comparison, but if we use rates of hospitalisation as a proxy for this outcome there was no difference between risperidone depot and typical depots (n = 62, Covell 2012, Analysis 9.3).
9.2 Clinically significant improvement in mental state
The single study making this comparison (Covell 2012) did not provide details of the number of participants making a clinically significant improvement while on the study drugs, instead providing average PANNS total scores for each treatment arm at short‐, medium‐ and long‐term (for this study) follow‐up. These data showed no difference between the two groups at any point, but it is not possible to say anything about how many participants experienced a significant change in their mental state. As the study population were all initially being treated with typical depot antipsychotics, it is unlikely that much by way of clinically significant change took place.
9.3 Leaving the study early for any reason
Participants randomised to receive depot risperidone were more likely to leave the study, both before commencement of their assigned treatment (RR 7.50 95 CI 1.00 to 56.44) and by six months (RR 3.05 95% CI 1.12 to 8.31, Analysis 9.2). As the researchers themselves noted, this can probably be explained by the study design; being randomised to risperidone depot came with a requirement to switch their medication, whereas those in the typical depot group were allowed to stay on the antipsychotics they had already been taking. It is understandable that some people would not feel comfortable switching and would therefore leave the study early, but it is not possible to differentiate these individuals from those who left for other reasons.
9.4 Serious adverse effects
Data relating to serious adverse effects are not available for Covell 2012. The adverse effects data that were collected are skewed, and in accordance with our methodology, cannot be included in the analysis.
9.5 Economic costs of treatment
There are no studies addressing the economic costs associated with risperidone depot compared to typical depot antipsychotics.
Overall completeness and applicability of evidence
Naturalistic protocols more closely resemble real‐life clinical practice, but very high attrition in some of these makes the results less useful.
The applicability of the evidence in this review to real‐life practice may be somewhat hindered by the nature of the participants recruited to the included studies, more specifically the question of whether or not they really reflect the types of patients in the real world who would most benefit from taking depot antipsychotics over oral preparations.
Depot antipsychotics are intended for people that do not want, or are unable, to take daily oral drugs. If an individual has enough stability and insight in their day to day life to adhere to an oral regimen, then they probably would not gain any greater freedom from their symptoms through a depot. In the real world, depots are prescribed for patients for whom the severity of their illness prevents this level of stability and control, and who may benefit from having their medication delivered consistently in a manner that is "out of their hands", so to speak. The people recruited to most, if not all, of these studies did not appear to be that ill, with baseline PANSS total scores consistently in the 60 to 80 range and recruitment criteria that excluded those with substance abuse problems, a history of violent or suicidal behaviour or comorbid psychiatric problems. The results can only really speak for the effects of risperidone depot on patients for whom receiving their medication as bi‐weekly injections or daily oral preparations may make very little difference to the amount of drug delivered to their system.
Quality of the evidence
Kane 2002* features a very high level of attrition from the study, almost 70% in the placebo group and just over 50% in the active treatment group. The consequence of this is that the authors conclusion, that depot risperidone is significantly more efficacious than placebo for improving the symptoms of schizophrenia, is based mainly on assumptions. Using the evidence from this study alone, there is nothing to support the idea that depot risperidone is any better than placebo. Yet despite the problems with this study, which was included in the previously published version of this review (Hosalli 2003), a decade later it is still the only randomised controlled trial that compares depot risperidone versus placebo. While the other results in this review demonstrate that depot risperidone is very likely as effective as other antipsychotics, it is worrying that these trials all took place in an environment where the superiority of depot risperidone was taken as read.
Adverse event reporting was inconsistent amongst the included studies; specifically there was no evidence that the criteria used to define "serious adverse events/effects" were the same from study to study. This makes it difficult to compare these outcomes between studies to produce generalisable results.
Open‐label study designs are associated with a high risk of performance bias; whereby the participants' knowledge of which intervention they are receiving affects their response to it. Most of the studies in this review utilised this design, resulting in widespread risk of bias that may downgrade the quality of the evidence.
Potential biases in the review process
The review protocol and process of study selection were strictly adhered to throughout the entire review process, and the process for searching for studies was thorough and data were extracted independently. We contacted authors of included studies to obtain details of ongoing or unpublished studies, but there remains a possibility that other unpublished trials of the intervention exist for which the review authors do not currently have access.
Agreements and disagreements with other studies or reviews
To the authors' knowledge, this is the first systematic review to comprehensively meta‐analyse RCTs relating to risperidone depot for schizophrenia.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
Based on the evidence from the one study (Kane 2002*), depot risperidone may be more acceptable than placebo injection but it is hard to know if it is any more effective in controlling the symptoms of schizophrenia. The active drug, especially at higher doses, may be associated with more movement disorders than placebo. People already stabilised on oral risperidone may continue to maintain benefit if treated with depot risperidone and avoid the need to take tablets, at least in the short term. In people who are happy to take oral medication the depot, risperidone is approximately equal to oral risperidone as seen within the considerable limitations of the two relevant studies. It is possible that the depot formulation, however, can bring a second‐generation antipsychotic to people who do not adhere to treatment. People with schizophrenia who have difficulty adhering to treatment, however, are unlikely to volunteer for a clinical trial. Such people may gain benefit from the depot risperidone with no increased risk of extrapyramidal side effects (EPS).
2. For clinicians
For reasonably well and stable people, it may mean they can avoid taking regular oral doses but adverse affects are not well reported. When given to more severely ill people, few benefits were demonstrated in the short term, although it may increase compliance with injections compared with placebo. Use of depot risperidone, especially at the higher doses, is weakly associated with movement disorders.
Review of the evidence on the efficacy of depot formulations of first‐generation antipsychotics in comparison with oral formulations indicates that there is only a very modest advantage of depot. There is very little difference between depot and oral formulations in most studies. Patients who volunteer for research are often co‐operative patients who will take their medication, particularly if they are seen every few weeks for ratings, reminding them of the importance of adherence to the medication schedule. It is possible that depot risperidone may have a unique benefit in non‐compliant patients, but the included studies do not address this issue.
3. For managers
Currently, no data are available on satisfaction with care or long‐term benefits and we know of no cost‐benefit analysis of depot risperidone. In view of this, it is unclear whether the increased costs, which would be incurred purchasing the drug and the additional arrangements needed to administer it, would be justified.
Implications for research.
1. General
Researchers should provide more details when reporting trials so that it is clear how many people really made significant progress. There should be more effort to assess people at the end of designated study period even if they have failed to complete all follow‐up ratings. See Table 9 for a suggested design of future study.
1. Suggested design of study.
| Methods | Allocation: randomised, fully explicit description of methods of randomisation and allocation concealment. Blinding: single, tested. Setting: community rather than hospital. Duration: 12 weeks treatment, and then follow‐up to at least 52 weeks. |
| Participants | Diagnosis: schizophrenia (ICD/DSM/CCMD). N = 300.* Age: adults. Sex: both. |
| Interventions | 1. Depot risperidone. N = 150. 2. Standard care. N = 150. |
| Outcomes | General: time to all‐cause treatment failure marked by its discontinuation, relapse, general impression of clinician (CGI), career/other, compliance with treatment., healthy days, Mental state: BPRS and PANSS. Global state: CGI (Clinical Global Impression). Quality of life. QOL (Quality of Life Questionnaire). Family burden: FBQ (Family Burden Questionnaire). Social functioning: return to everyday living for 80% of time.* Adverse events: any adverse event recorded. Economic outcomes. |
| Notes | * Powered to be able to identify a difference of ˜ 20% between groups for primary outcome with adequate degree of certainty. |
BPRS: Brief Psychiatric Rating Scale CGI: Clinical Global Impression PANSS: Positive and Negative Syndrome Scale
2. Specific
2.1 Reviews
Excluded studies with data relevant to other reviews are highlighted in Table 10. Existing reviews are always in the process of update and the older broad multi‐comparison titles may be broken down into smaller single comparison reviews. Several of the good studies we had to exclude would find a home in these reviews.
2. Excluded studies and suggestions for relevant reviews.
| Excluded study | Comparison | Existing review | Suggested future review titles |
| Bouchard 2000; Gallhofer 1995; Kogeorgos 1995; | Oral risperidone vs conventional antipsychotic drugs (haloperidol, fluphenazine, chlorpromazine, trifluoperazine), not depot risperidone. | Risperidone vs typical antipsychotic medication for schizophrenia (Hunter 2003). | Risperidone vs haloperidol, risperidone vs fluphenazine, risperidone vs chlorpromazine, risperidone vs trifluoperazine. |
| Littrell 1999; Ritchie 1999; Robinson 2000 | Oral risperidone vs atypical antipsychotics (olanzapine), not depot risperidone. | Risperidone vs olanzapine for schizophrenia (Jayaram 2006). | |
| Macfadden 2008; Simpson 2006 | Dose comparison (25 mg vs 50 mg of risperidone depot). | Risperidone dose for schizophrenia (Li 2009). | This could also generate further comparisons for this current review. |
2.2 Trials
This review highlights the need for good quality controlled clinical trials to assess the effect and clinical outcomes of using depot risperidone for people with schizophrenia. Such studies are difficult and need different designs if they are to be informative. Depots are used for people who do not want to take oral medication. Such people are difficult to randomise. If they are avoided, in the case of Chue 2002, the reasonably complete results are difficult to apply. If this difficult group is not avoided, as in the case of Kane 2002*, the explanatory design results in such a degree of attrition as to render results almost entirely without meaning. Pragmatic design could help deal with these problems. People appropriate for depot risperidone, if in agreement, could be randomised to an oral antipsychotic or depot risperidone. Outcomes could then be gathered for events that would be recorded in routine data (discontinuation of medication, specific adverse events, hospitalisation, global improvement whether or not the person stayed on the allocated treatment).
According to the manufacturers of risperidone, Janssen‐Cilag Limited, the reason why there are so few studies on depot risperidone is that the compound is unmodified, and is merely delivered differently. More real world trials are required, to establish the efficacy and safety of depot risperidone.
Feedback
Response from Janssen‐Cilag Ltd
Summary
Dear Editor
The purpose of this letter is to provide comment on the recent review of Risperdal Consta long‐acting injectable in Schizophrenia. We appreciate the opportunity to discuss issues with the review's conclusions and provide insight into possible solutions to any issues raised.
The content of this discussion will be based on the following key assertions:
‐ That the review's conclusions are overly critical of available data; ‐ That the review's conclusions regarding reliable evidence are potentially misleading and are based on incomplete information;
Comment on these assertions will be focused on the following specific issues:
1. Application of strict methodological rules to placebo controlled trials in schizophrenia leading to exclusion of any data where patient drop outs are >50%. This led to exclusion of key data from Kane et al 2002, where overall drop rates were only 56%.
Additional analysis will be provided for consideration, including:
‐ Mean change in total PANSS ‐ Results (from total patient cohort and risperidone long‐acting injectable monotherapy cohort) up to the Cochrane methodology‐defined point where 50% of patients had dropped out (between 6 and 10 weeks and at end‐point), show statistically significant differences between risperidone long‐acting injectable and placebo. These analyses report on the total 'starting' patient cohort, and separately for the risperidone long‐acting injectable 'monotherapy' subset.
‐ Population analysis for clinical improvement associated with risperidone long‐acting injectable showing a conclusive treatment effect unrelated to demographic expectations and low NNT values for significant reduction in Total PANSS.
‐ Additional testing of an "assumption" of stability after leaving the study by using last observation carried forward (potential impact of drop outs) showed no impact on the conclusions of the study related to the relative comparison of risperidone long‐acting injectable to placebo.
2. Identification of specific outcome measures in the review appear to provide potentially misleading or erroneous conclusions.
3. Conclusions based on critique and dismissal of non‐inferiority data cause isolation of the effectiveness of risperidone long‐acting injectable from established benefits of risperidone compound.
4. Discussion of the risks associated with categorization of risperidone long‐acting injectable as a "depot".
Detailed comments 1. Exclusion of Kane et al 2002 due to drop out rates
We acknowledge the transparent nature of Cochrane methodology and the corresponding universal application of the 50% drop out rate criterion.
Previous Cochrane reviews (1, 2, 3) of atypicals have included studies of shorter duration (e.g. 6‐8 weeks). However, concluding that the Kane study provides a lack of reliable data based on an overall drop out rate of 56% over the longer period of 12 weeks, is overly critical. Post‐hoc analysis shows that, had this study been ended at 6 weeks the criterion would have been satisfied and, the data would therefore have been included in this review.
To this end, we include in Appendix A the Weighted‐mean Difference analyses of mean change in total PANSS from baseline (using Forrest plots) of the Kane 2002* (4) data at 6 weeks, where drop outs were less than 50% overall, as well as the end‐point last observation carried forward (LOCF) data. Significant effect was seen with both doses at end‐point (‐8.60 for 25mg/fortnight; ‐11.20 for 50 mg/fortnight) and for 50mg/fortnight at 6 weeks (Where n>50%), of ‐8.30.
We also include these analyses for the sub‐population known as 'monotherapy'. This included only those patients who underwent a PANSS evaluation after 18 days of oral risperidone supplementation and then continued in the study, thus reflecting the effects ‐ from treatment baseline ‐ of risperidone long‐acting injectable alone. Again, both doses were associated with significant effect at end‐point (‐11.90 for 25 mg/fortnight and ‐9.60 for 50mg/fortnight) and significantly for 25mg at 10 weeks (n>50%), being ‐9.10.
Empirical analysis (Chi‐squared test) of percentages of 'responders (showing a 20% or greater improvement in Total PANSS)' showed a significant advantage in the risperidone long‐acting injectable treatment group at 6 weeks and at end‐point for all doses taken together (Appendix B). Furthermore, for this sample, NNT's for a 20% or greater drop in Total PANSS versus placebo were 5.6 (+/‐ 0.17 for 95% confidence) at 6 weeks and 3.3 (+/‐ 0.10) at End Point. These results clearly indicate a significant difference from the placebo group. Additional analyses were conducted to assess the impact from the level of dropouts in the study on conclusions of Kane et al, 2002. This was an attempt to assess whether the assumption inherent in the regulatory‐driven primary LOCF analysis were valid for this trial.
Longitudinal analysis of observed cases were entirely consistent with primary analysis, supporting the overall conclusions. In addition, analysis further to that in the Study Report for Kane et al (2002), tested assumptions more conservative than for LOCF. By adding an assumed 10 points in PANSS to the endpoint change from baseline for all drop outs (i.e. endpoint levels for drop outs were assumed to be 10 points worse at 12 weeks than reported in primary analysis at endpoint), the mean change from baseline for each group was different as would be expected, but the between group comparisons with placebo were still significant.
Also, conclusions did not change when testing the outcome of achieving at least a 20% improvement in total PANSS, where each drop out was set to "No".
As there were differential drop out rates in each group (itself an outcome measure of benefit included in all Cochrane reviews), based on the above, the only way it seems that the conclusions would be altered is if we assume the placebo drop outs would have improved at 12 weeks had they stayed in the trial, and the risperidone long‐acting injectable patients would have become worse. Concluding a lack of reliable evidence of clinical benefit of risperidone long‐acting injectable based on this assumption, is misleading and overly critical.
Recommendations We would request that the total PANSS mean change data from Kane et al 2002 (4) be included in the graphs section, also the population analyses for 'responders' and reviewed in the results text, highlighting the statistically significant differences in favour of risperidone long‐acting injectable over placebo. Conclusions should also be altered to reflect that there is reliable RCT data supporting significant benefits of risperidone long‐acting injectable for schizophrenia patients.
2. Specific outcome measures
Adverse Events: The review rightly highlights the need for consistency and greater clarity in the reporting of adverse events (Discussion 3.2 'Problematic outcomes', p20) and yet draws some analytical conclusions from such spontaneously reported data (Sections 2.4.2, p18 and Section 4.4, p21). The validity of and ‐ even more thorough ‐ the reporting of adverse events, and the interpretation of these reports as clinical outcomes has to be questioned in the absence of standardised techniques for assessing prevalence and severity of these events.
Trial discontinuation: 'Poor compliance' implies cessation of taking the medication, which is very different from protocol violation. In Analysis: 02.04 ('Poor compliance', p47), the three risk‐ratio analyses from the data of Chue et al's 2002 study, show groups of patients who for various reasons were non‐compliant with the trial and left the study: this does not necessarily mean that they were non‐compliant with the medication and this should be clarified.
3. Dismissal of non‐inferiority data and exclusion of benefits already clearly established with risperidone
The results reported for the Chue et al 2002 study (5), critically imply no clear differences between risperidone long‐acting injectable and risperidone oral when taken over 12 weeks, without acknowledging that this trial was designed apriori to investigate the hypothesis of non‐inferiority.
Further, there is no acknowledgement of the inference of benefits to risperidone long‐acting injectable supported by the established safety and effectiveness of risperidone oral in treatment of schizophrenia (which, in turn, is supported by the most recently updated Cochrane review of risperidone versus typical antipsychotics). We find this inconsistent with the review's acknowledgement that the data from Chue et al's study (2002) established non‐inferiority of risperidone long‐acting injectable compared to oral risperidone ('Main Results', p5) and with the statement ‐ quite correct ‐ that risperidone long‐acting injectable "encapsulates unmodified risperidone" and provides "therapeutic blood levels of the drug.." ('background', p7).
Again, the conclusion that there is no reliable evidence to support the claim that risperidone long‐acting injectable is beneficial for people with schizophrenia is misleading and potentially harmful.
Recommendations That the review should be modified to recognize that, based on a study designed to show non‐inferiority, there is clinical equivalence between risperidone long‐acting injectable and daily‐administered oral risperidone over 12 weeks of therapy. In addition, is would be useful to recognize that advice and requirements of regulatory authorities were strictly adhered to in designing the study, and took into account the delicate ethico‐clinical balances involved when designing placebo‐controlled trials with this group of severely unwell patients (a point also self‐evident from the placebo drop‐out rates).
4. Classification of risperidone long‐acting injectable as a depot
Risperidone long‐acting injectable is the first atypical long‐acting compound available for schizophrenia patients. It contains a microspheres technology with a water‐based delivery system, clearly differentiating it from traditional oil‐based antipsychotic depot medications.
Conventional depot medications, due to their long‐standing availability, have established dosage and administration techniques. Labelling risperidone long‐acting injectable as "Depot risperidone" is misleading and potentially harmful, as physicians may assume that mode of administration, plasma profile and drug metabolism detriments associated with depot injections apply to risperidone long‐acting injectable. These detriments include 'scarring' and subcutaneous nodules due to oil‐based residual impurities, pain at site of injection and during the injection, the need to use the painful z‐track technique and post‐injection seepage of the oily vector. Due to its advanced microspheres technology and aqueous formulation, these major shortcomings are not seen with Risperdal long‐acting injectable.
It is clear, therefore, that it is desirable at all levels to separate a description of risperidone long‐acting injection from that for the older and problematic depots. Specific concerns are that the confusion caused by this mislabelling could, for example, discourage people prescribing from using risperidone long‐acting injection because of their or their patients' previous bad experiences with typical depots. Market research has shown that the term 'depot' has a negative connotation for many patients for the reasons outlined above, and because, over time, depots have been reserved clinically for 'the worst of the worst' ‐type of patient. The word 'depot' then, is widely seen as a pejorative term. Lastly, the mental image generated by the word 'depot' focuses more with the physical aspects of the older and less sophisticated oily injection that the intramuscular site from which the drug is released.
Together, these issues lead to an unnecessary and inaccurate stigmatization and potential limitation in use of risperidone long‐acting injection due to the imposition of a convenient but misleading label.
Recommendations The use of "depot risperidone" should be replaced everywhere in the review to risperidone long‐acting injectable.
Overall conclusions and recommendations
‐ Our further analyses of data from the placebo‐controlled trial (Kane et al, 2002) demonstrate convincing evidence that risperidone long‐acting injectable is beneficial for people with schizophrenia when compared with those taking injectable placebo, both in the short and long‐term. The review should acknowledge this and the conclusion (p6) that "there is no reliable evidence that risperidone long‐acting injectable is beneficial for people with schizophrenia" should be withdrawn.
‐ Risperidone long‐acting injectable represents a novel and unique delivery of the same active antipsychotic agent as exits in oral formulations of risperidone. Since this fact, as well as the clinical equivalence of risperidone long‐acting injectable and oral risperidone are acknowledged in the review, by inference it cannot be safely stated that "there is no reliable evidence that risperidone long‐acting injectable is beneficial for people with schizophrenia". A further reason that this conclusion should be withdrawn.
‐ The use of "depot risperidone" should be replaced everywhere in the review with "risperidone long‐acting injectable" since it does not accurately describe risperidone long‐acting injectable and may lead to dangerous clinical confusion.
‐ Finally, conclusions based on spontaneously reported non‐clinical events interpreted as outcomes should be treated with caution, as should the assumption that trial protocol violation equated to poor compliance with medication.
(1). Joy CB, Adams CE, Lawrie SM. Haloperidol versus placebo for schizophrenia (Cochrane Review). In: The Cochrane Library, Issue 1, 2004. Chichester, UK: John Wiley & Sons, Ltd.
(2). Duggan L, Fenton M, Dardennes RM, El‐Dosoky A, Indran S. Olanzapine for schizophrenia (Cochrane Review). In: The Cochrane Library, Issue 1, 2004. Chichester, UK: John Wiley & Sons, Ltd.
(3). Srisurapanont M, Disayavanish C, Taimkaew K. Quetiapine for schizophrenia (Cochrane Review). In: The Cochrane Library, Issue 1, 2004. Chichester, UK: John Wiley & Sons, Ltd.
(4). Cited as: Kane, J. et al, Am J Psychiatry 2003, 160 (6) 1125‐32.
(5).Cited as: Chue, P. et al, Schizophrenia Research 2002, 3 (Suppl 1) 174.
Reply
We would like to thank the commentators for their comments and for acknowledging the transparent nature of Cochrane methodology. We would like to discuss the issues raised by them.
1. Exclusion of data from Kane 2002*
We too are concerned with excluding data. We do not, however, agree that loss of 56% of people by 12 weeks should be described as "only 56%" and feel this degree of complacency is lamentable. The Agency for Health Care Policy and Research (AHCPR) proposed criteria for assessing a randomised controlled trial's quality that included dropout rates suggests that any study of less than three months' duration with a dropout rate exceeding 10% (15% for study of more than three months) should be considered as flawed to a major degree (Hadorn 1996).
The commentators correctly point out that other Cochrane reviews have included studies of shorter duration. The designers of Kane 2002* felt that outcomes were meaningful at 12 weeks. We agree. Recalculating now to make the study fit into the less than 50% attrition category contradicts the protocol of the trial. It is a data‐driven exercise.
The level of attrition acceptable to reviewers does differ. Partly this is a function of the participants involved, the interventions and the outcome. So for the acute treatment of aggression secondary to psychosis outcomes may be measured in hours and almost any loss to follow‐up unacceptable. For longer studies some attrition is deemed acceptable. We thought that the cut off of 50% to be generous. We are sorry that the commentators do not.
We would like to thank the commentators for their sensitivity analysis around their assumptions. First, it seems problematic to us that these analysis cause conclusions to be drawn when over half the data are assumed. Second, we are unsure if a 20% improvement is clinically meaningful and would be more interested in levels of 40% and above. It has been noted in other Cochrane Reviews relevant to risperidone that the 20% cut off may have little clinical meaning (Hunter 2004).
2. Specific outcome measures
Having been criticized for NOT reporting data, because of large loss to follow‐up, we are now criticized for reporting data that are there and usable. We are glad that commentators agree that greater clarity is needed in reporting of adverse events and that it is not the best way to interpret clinical outcomes from spontaneously reported data. We hope that the commentators are in a better position to influence trial design to ensure better reporting in their own trials in the future.
With regard to analysis 02.04, we note the comment that non‐compliance does not necessarily mean poor compliance with medication and have amended this in the text.
Current text reads "Compliance was measured in several ways. Most people received at least four injections (83.4% in the depot group and 85.6% in the oral risperidone group, n=640, RR <4 injections or "major protocol violation" 1.16 CI 0.81 to 1.67). There was no difference between groups in the rate of discontinuation before the end of the 12‐week study (n=640, RR 1.27 CI 0.90 to 1.78)."
Amended text reads "Compliance was measured in several ways. Most people received at least four injections (83.4% in the depot group and 85.6% in the oral risperidone group, n=640, RR <4 injections or "major protocol violation" 1.16 CI 0.81 to 1.67). There was no difference between groups in the rate of discontinuation before the end of the 12‐week study (n=640, RR 1.27 CI 0.90 to 1.78). Please note that 'compliance' in this context could apply to protocol violation for many reasons only one of which would be non‐compliance with the study drugs."
3. Dismissal of non‐inferiority data and exclusion of benefits already clearly established with risperidone
The commentators are correct to point out that we have not worded the conclusions well. We intended to state that there are no clear differences between the depot preparation of risperidone and oral risperidone in people whose symptoms are already controlled using oral risperidone. This does not imply that actively symptomatic patients would benefit from the depot preparation. We have stated in the conclusion that "People already stabilised on oral risperidone may continue to maintain benefit if treated with depot risperidone and avoid the need to take tablets, at least in the short term." As we have not been clear enough in the conclusions we have reworded them. We have also emphasised the possible benefits for people who are non compliant with medications in the real world who are unlikely to voulnteer for a clinical trial.
Current version of conclusions in the ABSTRACT reads "There is no reliable data to support the claim that depot risperidone is beneficial for people with schizophrenia. For reasonably well, stable people it may mean that the need for regular oral doses can be avoided, but adverse affects are not well reported. For more severely ill people, few benefits are evident although it may increase compliance with injections in comparison with placebo. Use of depot risperidone, especially at the higher doses, is weakly associated with movement disorders."
Updated version in response to this comment reads "For reasonably well, stable people use of the depot formulation may mean that the need for regular doses of oral risperidone can be avoided, but adverse affects of the depot formulation are not well reported. For such people, depot risperidone may be as effective as the oral preparation, although data are few. For more severely ill people, few benefits of depot risperidone are evident although it may increase compliance with injections in comparison with placebo. Use of depot risperidone, especially at the higher doses, is weakly associated with movement disorders."
Current version of conclusions in the IMPLICATIONS FOR PRACTICE reads Implications for practice For people with schizophrenia There are only two studies on which to base an informed choice about depot risperidone. Depot risperidone may be more acceptable than placebo injection but it is hard to know if it is any more effective in controlling the symptoms of schizophrenia. The active drug, especially higher doses, may be associated with more movement disorders than placebo. People already stabilised on oral risperidone may continue to maintain benefit if treated with depot risperidone and avoid the need to take tablets, at least in the short term. For clinicians There is no reliable data to support the claim that depot risperidone is beneficial to people with schizophrenia. For reasonably well and stable people it may mean they can avoid taking regular oral doses but adverse affects are not well reported. When given to more severely ill people, few benefits were demonstrated in the short term, although it may increase compliance with injections compared with placebo. Use of depot risperidone, especially at the higher doses, is weakly associated with movement disorders.
Updated version in response to this comment reads For people with schizophrenia There are only two studies on which to base an informed choice about depot risperidone. Depot risperidone may be more acceptable than placebo injection but it is hard to know if it is any more effective in controlling the symptoms of schizophrenia. The active drug, especially higher doses, may be associated with more movement disorders than placebo. People already stabilised on oral risperidone may continue to maintain benefit if treated with depot risperidone and avoid the need to take tablets, at least in the short term. In people who are happy to take oral medication the depot risperidone is approximately equal to oral risperidone as seen within the considerable limitations of the relevant study. It is possible that the depot formulation, however, can bring a second‐generation antipsychotic to people who do not reliable adhere to treatment. People with schizophrenia who have difficulty adhering to treatment, however, are unlikely to volunteer for a clinical trial. Such people may gain benifit from the depot risperidone with no increased risk of extrapyramidal side effects.
For clinicians For reasonably well and stable people it may mean they can avoid taking regular oral doses but adverse affects are not well reported. When given to more severely ill people, few benefits were demonstrated in the short term, although it may increase compliance with injections compared with placebo. Use of depot risperidone, especially at the higher doses, is weakly associated with movement disorders. Review of the evidence on the efficacy of depot formulations of first‐generation antipsychotics in comparison with oral indicates that there is only a very modest advantage of depot. There is very little difference between depot and oral in most studies. Patients who volunteer for research are often cooperative patients who will take their medication particularly if they are seen every few weeks for ratings, reminding them of the importance of adherence to the medication schedule. The importance of the two risperidone depot studies is to indicate that this preparation may be effective within the limitations of the methodology. It is possible that depot risperidone may have a unique benefit in non‐compliant patients but the included studies do not address this issue.
4. We do not agree that the labelling of risperidone long acting injection as depot risperidone is misleading. The difference in the meaning of depot injection and long acting injection is none for all practicable purpose. The debate is more of semantics. The Oxford English Dictionary defines the physiological meaning of 'depot' as "the site of an accumulation or deposit of a substance (esp. fat) in an animal body….applied to any substance stored for eventual absorption by the organism, or to an action or process concerned with the deposition of such a substance." (OED 1989) We acknowledge that the risperidone preparation that is the focus of this review is not deposited in fat. It is, however, clearly comes with the meaning of "deposited in an animal body and a substance stored for eventual absorption by the organism."
The word depot may have negative connotation within psychiatry but it does not take away from the fact depot means a long acting injectable preparation. For marketing purposes the manufactures may want to use different terms/names to differentiate it from other drugs, but from a clinical point of view it is a depot preparation which means a long acting injection, whatever may be its technical differences from the other drugs.
Nowhere in the reports on risperidone injectable preparations it has been claimed that it is not a depot. The term long acting injection has been used as a synonym for the depot in these reports. For example Chue 2002 states… "Currently, however, only typical psychotropics have been available as long acting formulations. Risperidone is the first atypical psychotropic medication available in a long acting formulation". Therefore there is no justification to alter the word "depot" to "long acting injection" throughout the review.
Hardon 1996 Hardon DC, Baker D, Hodges JS, Hicks N. Rating the quality of evidence for clinical practice guidelines. Journal of Clinical Epidemiology 1996;49:749‐54.
Hunter 2004 Hunter RH, Joy CB, Kennedy E, Gilbody SM, Song F. Risperidone versus typical antipsychotic medication for schizophrenia (Cochrane Review). In: The Cochrane Library, Issue 2, 2004. Chichester, UK: John Wiley & Sons, Ltd.
OED Oxford English Dictionary, 2nd edition, 1989. http://dictionary.oed.com
Contributors
Mike Ingham, Senior Health economics Director Janssen‐Cilag Ltd, Beerse Belgium
Clive M. Rogers, Clinical Evidence Liaison Service Manager Janssen‐Cilag Ltd, Saunderton UK
What's new
| Date | Event | Description |
|---|---|---|
| 25 January 2016 | New citation required and conclusions have changed | Results of update searches provide more data and evidence. |
| 28 October 2015 | New search has been performed | Search re‐run (October 2015), seven studies added to Characteristics of excluded studies. |
History
Protocol first published: Issue 2, 2003 Review first published: Issue 4, 2003
| Date | Event | Description |
|---|---|---|
| 17 February 2014 | Amended | Results of updated search (December 2010, May 2012) added to review, 10 new studies added to Characteristics of included studies and one study to the Characteristics of studies awaiting classification pending update of review. |
| 30 October 2008 | Amended | Converted to new review format. |
Acknowledgements
The Cochrane Schiozphrenia Group provide a template for the Methods section, we have used this template and adapted for our criteria.
We would like to thank Thomas Neely who contributed to writing, trial selection and data extraction for the 2015 version. We would also like to thank Nancy Covell for her assistance in obtaining further information to an included study, and Nicholas Keks for providing further information relating to statistical analysis in his study.
We would also like to thank Ben Gray for writing our Plain language summary, and copy editors for making the review read better and look consistent.
“This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Schizophrenia Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.”
Appendices
Appendix 1. Previous searches
1.1 Search in 2002
1.1.1 Cochrane Schizophrenia Group's Register
We searched the Cochrane Schizophrenia Group's Register (December 2002) using the phrase:
[(risp* or * risp * or 9‐OH‐risperidone*) and (* depot* or * microsph* or * micro‐sp* or * long‐acting* or * long act*) in title, abstract, index terms of REFERENCE] or [(depot and risp*) in interventions of STUDY]
The Schizophrenia Group's Trials Register is based on regular searches of BIOSIS Inside, CENTRAL CINAHL, EMBASE, MEDLINE, PsycINFO, hand searching of relevant journals and conference proceedings, and searches of several key grey literature sources. A full description is given in the group's module.
1.2 Search in 2010
1.2.1 Cochrane Schizophrenia Group's Register
We searched the register using the phrase:
[((risp* or * risp * or 9‐OH‐risperidone*) and (* depot* or * microsph* or * micro‐sp* or * long‐acting* or * long act*) in title, abstract, index terms of REFERENCE) or ((depot and risp*) in interventions of STUDY)]
This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group's Module).
1.3 Search in 2012
1.3.1 Electronic searches
1.3.1.1 Cochrane Schizophrenia Group Trials Register (October 2012)
Using the phrase:
[(risp* or * risp * or *9‐OH‐risperidone*) and (* depot* or * microsph* or * micro‐sp* or * long‐acting* or * long act*) in title, abstract, index terms of REFERENCE] or [((*depot* or *long* or *LAI*) and *risp*) in interventions of STUDY]
1.3.1.2 Economic study search of Cochrane Schizophrenia Group Health Economic Database (2013)
For the economic search, we replicated the above strategy in the Cochrane Schizophrenia Group Health Economic Database (CSzGHED) on 31 January 2014. The database of studies relates to cost‐effectiveness of schizophrenia treatments. This database was constructed from systematic searches of four databases: Health Economic Evaluation Database (HEED), National Health Services Health Economic Database (NHS EED), Cost‐Effectiveness Analysis Registry (CEA) and EconLit as well as Cochrane Registry.
1.3.2 Searching other resources
1.3.2.1 Reference searching
The reviewers inspected references of all identified studies for more studies.
1.3.2.2 Personal contact
The reviewers attempted to contacted the first author of each study considered for inclusion in the review for more information regarding unpublished trials or any data available.
1.3.2.3 Drug companies
The reviewers contacted the Janssen‐Cilag Limited for further data.
Appendix 2. Previous data collection and analysis
1. Selection of trials Two reviewers (PH, JD) independently inspected the citations identified from the search. Potentially relevant abstracts were identified and full papers ordered and reassessed for inclusion and methodological quality. Any disagreement was discussed and reported.
2. Quality assessment Trials were allocated to three quality categories, as described in the Cochrane Collaboration Handbook (Clarke 2002) by each reviewer, again, working independently. When disputes arose as to which category a trial was allocated, resolution was attempted by discussion. When this was not possible, and further information was necessary, data were not entered into the analyses and the study was allocated to the list of those awaiting assessment. Only trials in Category A or B were included in the review.
3. Data management
3.1 Data extraction Two reviewers (PH, JD) independently extracted data and, where further clarification was needed, contacted authors of trials to provide missing data.
3.2 Intention to treat analysis Data were excluded from studies where more than 50% of participants in any group were lost to follow‐up (this did not include the outcome of 'leaving the study early'). In studies with less than 50% drop‐out rate, people leaving early were considered to have had the negative outcome, except for the event of death. The impact of including studies with high attrition rates (25‐50%) was analysed in a sensitivity analysis for primary outcomes. If inclusion of data from this latter group did result in a substantive change in the estimate of effect, the data were not added to trials with less attrition, but presented separately.
4. Data analysis
4.1 Binary data For binary outcomes a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI) was calculated. The number needed to treat statistic (NNT) was also calculated. If heterogeneity was found (see section 5) a random effects model was used.
4.2 Continuous data 4.2.1 Intention‐to‐treat analyses versus analyses that only take into account those who completed the study: in the case of continuous data, it was supposed that in many cases an intention‐to‐treat analysis would not be available, so an analysis was presented on those who completed the study.
4.2.2 Rating scales: a wide range of instruments is available to measure mental health outcomes. These instruments vary in quality and many are not valid, or even ad hoc. For outcome instruments some minimum standards have to be set. Continuous data from rating scales were included only if the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000), the instrument was either a self report or completed by an independent rater or relative (not the therapist), and the instrument could be considered a global assessment of an area of functioning. However, as it was expected that therapists would frequently also be the rater, such data was tagged as 'prone to bias'.
4.2.3 Normal distribution of data: mental health continuous data are often not normally distributed. Most statistics assume a normal distribution. To avoid including non‐normally distributed data in the statistical analysis, the following criteria are applied to all data before inclusion:
a. Standard deviations and means were reported or derivable from data in the paper, or were obtainable from the authors. b. When a scale started from zero, the standard deviation, when multiplied by two, was less than the mean (as otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution (Altman 1996). Endpoint scores on scales often have a finite start and end point and this rule can be applied to them. c. When continuous data are presented on a scale which includes a possibility of negative values (such as change on a scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not. It is thus preferable to use scale endpoint data, which typically cannot have negative values. If endpoint data were not available, reviewers chose to use change data, because the statistics used in Metaview are rather robust towards skew. d. If a scale starts from a positive value (such as PANSS, which can have values from 30‐210) the calculation described above in (b) should be modified to take the scale starting point into account. In these cases skew is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score.
4.2.4 Endpoint versus change data: endpoint scale‐derived data are finite, ranging from one score to another. Change data are more problematic and for it the rule described above does not hold. Although most change scores are likely to be skewed, this cannot be proven so they were presented in MetaView. Where both endpoint and change were available for the same outcome, we presented the former in preference.
4.2.5 Summary statistic: for continuous outcomes, a weighted mean difference (WMD) between groups was estimated. Again, a random effects model was used.
4.3 Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) ‐ whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated ‐ causing type I errors (Bland 1997, Gulliford 1999). Secondly, RevMan does not currently support meta‐analytic pooling of clustered dichotomous data, even when these are correctly analysed by the authors of primary studies, since the 'design effect' (a statistical correction for clustering) cannot be incorporated.
Where clustering was not accounted for in primary studies, we presented data in a table, with an asterisk (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies, to seek intra‐class correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering had been incorporated into the analysis of primary studies, then we presented these data in a table. No further secondary analysis (including meta‐analytic pooling) will be attempted until there is consensus on the best methods of doing so, and until RevMan, or any other software, allows this. A Cochrane Statistical Methods Workgroup is currently addressing this issue. In the interim, individual studies were very crudely classified as positive or negative, according to whether a statistically significant result (p<0. 05) was obtained for the outcome in question, using an analytic method that allows for clustering.
5. Test for heterogeneity A Chi‐square test was used, as well as visual inspection of graphs, to investigate the possibility of heterogeneity. A significance level less than 0.10 was interpreted as evidence of heterogeneity. If heterogeneity was found, the data were re‐analysed using a random effects model to see if this made a substantial difference. If it did, the studies responsible for heterogeneity were not added to the main body of homogeneous trials, but summated and presented separately and reasons for heterogeneity investigated.
6. Addressing publication bias Data from all included studies were entered into a funnel graph (trial effect against trial size) in an attempt to investigate the likelihood of overt publication bias (Egger 1997).
7. Sensitivity analyses The effect of including studies with high attrition rates was analysed in a sensitivity analysis.
8. General Where possible, reviewers entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for depot risperidone.
Appendix 3. Previous description of studies
4. Included studies Two studies, reported as nine conference presentations and one full paper met the selection criteria and are included.
4.1 Study design All included studies were randomised and X featured some form of blinding, though the extent of this varied widely.
Chue 2002 involved an eight‐week run in period preceding randomisation. In the first two weeks, antipsychotic drugs other than risperidone were discontinued and oral risperidone introduced. In the next two weeks the risperidone dose was optimised and then people continued on this dose of oral risperidone for another four weeks before randomisation. Kane 2002* had a two‐week run in period preceding randomisation. The first week was a screening week followed by seven days during which people were started on oral risperidone and the dose was titrated to 4mg/day.
4.2 Participants People entering both studies met the criteria for DSM IV schizophrenia, so for at least six continuous months a participant must have shown some evidence of schizophrenia, and for at least one month must have shown at least two symptoms of frank psychosis. These symptoms would include delusions, hallucinations, incoherent speech, disorganised or catatonic behaviour, or flat affect. To meet DSM IV criteria, the symptoms must be disabling in such a way that social and occupational functioning is impaired; these symptoms should not be the direct result of a physical disorder or of substance misuse.
For Kane 2002* people who had substance dependence, tardive dyskinesia or a history of neuroleptic malignant syndrome, ECG abnormalities, suicidal ideas or risk of violent behaviour were excluded. Patients who had a history of unresponsiveness to risperidone were also excluded. Chue 2002 also stipulated that participants should have a total PANSS score of at least 50. This, in addition to the fulfilment the DSM IV criteria, means that people with at least some active symptoms of illness were included. In effect, despite the rigorous entry criteria, nearly 47% of people entering this study were rated by the authors as ''not ill'' or only ''mildly ill'' at baseline assessment on the CGI scale before randomisation. Chue 2002 randomised 640 people. Kane 2002*, however, randomised 400 people who appeared to be more severely ill. They had a baseline PANSS score in the range of 60‐120, with an average of about 80.
In both studies, participants were mainly men (about 70%) with an average age of about 40 years.
4.3 Interventions Chue 2002 randomised people to an active injection every two weeks and placebo tablets daily, or a placebo injection every two weeks and active tablets daily. Depending on the optimal stabilisation dose the person was randomised to continue that oral regimen or start the 'equivalent' dose of depot. For example, 2mg of oral risperidone per day was taken as being equivalent to 25mg of depot risperidone every two weeks. It is not clear, however, how the conversion dose was arrived at.
Kane 2002* randomised people to either a placebo injection or 25mg or 50mg or 75mg of depot risperidone every two weeks. People also received either placebo tablets or 2mg or 4mg or 6mg of oral risperidone respectively for the first three weeks after randomisation.
4.4 Outcomes 4.4.1 Global improvement Chue 2002 reported global improvement in the form of the percentage of people who were not ill or mildly ill on the Clinical Global Impression (CGI) scale at the end of the study period. Throughout this study results are reported for oral and depot groups as a whole, and not for specific dosage groups of depot. Chue 2002 did not report mean or change scores in the abstracts available for this review. Kane 2002* also used the CGI but reported average change from baseline to endpoint and data were unusable due to the substantial attrition.
4.4.2 Mental state Kane 2002* interpreted an improvement of more than 20% in PANSS total score as clinically important. This study also reported average change at endpoint from baseline in PANSS total, PANSS positive and PANSS negative but again so much data were lost because people left the study early that the results of the PANSS were unusable. Chue 2002 reported average change scores on PANSS total at endpoint in both the composite oral and depot groups. Chue 2002 did not seem to stipulate cut off points as 'clinically important improvement'.
4.4.3 Leaving the study early Both studies reported numbers discontinuing the study and specific reasons for this, such as adverse events, compliance problems and insufficient responses.
4.4.4 Adverse effects Chue 2002 reported overall rates of adverse events in both groups, and the numbers withdrawing from the study as a result of side effects. No details were given regarding the nature of these adverse events or how they were recorded. The abstracts available for this review state that body weight was measured and laboratory tests were undertaken. The reports state that there were no differences between oral and depot groups, but present no numbers. Chue 2002 also used the Extrapyramidal Symptom Rating Scale (ESRS) but, again, no numerical data were reported. Kane 2002* reported rates of individual adverse events spontaneously reported by participants, and reported these for all people in the study, not just those who completed the trial. Median ESRS scores were also reported for each group at baseline and change at endpoint. Pain and swelling at injection sites rated by investigators and patients were also reported.
4.4.5 Outcome measures used in this review
Global functioning. Clinical Global Impression ‐ CGI (Guy 1976) A rating instrument commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used with low scores indicating decreased severity and/or greater recovery.
Positive and Negative Symptom Scale ‐ PANSS (Kay 1987) This scale was developed to evaluate the positive, negative and general symptoms in schizophrenia. The PANSS has 30 items, and each item can be defined on a seven‐point scoring system varying from one (absent) to seven (extreme). This scale is divided into three subscales for measuring the severity of general psychopathology, positive symptoms (PANSS‐P) and negative symptoms (PANSS‐N). A low score indicates lesser severity.
Extrapyramidal Symptom Rating Scale ‐ ESRS (Chouinard 1980) This consists of a questionnaire relating to parkinsonian symptoms (nine items), a physician's examination for parkinsonism and dyskinetic movements (eight items) and a clinical global impression of tardive dyskinesia. High scores indicate severe levels of movement disorder.
4.4.6 Missing outcomes There are no data for outcomes beyond three months. Neither are data available on general functioning and change in behaviour. Nor were there any details on service outcomes, engagement with services, satisfaction with services, quality of life or economic outcomes.
Appendix 4. Previous Chue write up
3. COMPARISON: 2. DEPOT RISPERIDONE vs ORAL RISPERIDONE
Chue 2002 compared depot risperidone against oral risperidone
3.1 Global improvement The study did not report mean scores on the CGI scale. The trialists did report percentage of people mildly ill or not ill in both the depot risperidone and oral risperidone groups at the end of the study period, as rated using the CGI (about 57% as read from the graph, compared to 47% at baseline). Hence 43% must have been moderately ill or severely ill at the end of the study period. There was no difference between the depot group and the oral group (n=640, RR 1.06 CI 0.92 to 1.22).
3.1.1 Mental state Chue 2002 reported both average end score and change across time. For endpoint score there was no difference between groups (MD 0.00 CI ‐2.91 to 2.91), nor was there any difference between depot and oral risperidone for average change in the total PANSS score (n=541, WMD ‐0.90 CI ‐2.84 to 1.04), PANSS positive (WMD ‐0.30 CI ‐0.86 to 0.26) and PANSS negative scores (WMD ‐0.10 CI ‐0.93 to 0.73).
3.2 Poor compliance Compliance was measured in several ways. Most people received at least four injections (83.4% in the depot group and 85.6% in the oral risperidone group, n=640, RR <4 injections or "major protocol violation" 1.16 CI 0.81 to 1.67). There was no difference between groups in the rate of discontinuation before the end of the 12‐week study (n=640, RR 1.27 CI 0.90 to 1.78). Please note that 'compliance' in this context could apply to protocol violation for many reasons only one of which would be non‐compliance with the study drugs."
3.3 Adverse effects Adverse events are reported in order of severity. One death was reported in the oral risperidone group (n=640, RR death 1.04 CI 0.91 to 1.18). Low proportions of people had to withdraw from the study due to adverse events and there were no differences between the oral and depot preparation (n=640, RR 1.21 CI 0.62 to 2.35). Over half of both groups reported some adverse effects (n=640, RR 1.04 CI 0.91 to 1.18)
Appendix 5. Previous discussion
4. COMPARISON: 1. DEPOT RISPERIDONE vs PLACEBO
4.1 Global improvement No meaningful conclusions can be drawn as more than 50% of patients did not complete the trial. Hence the authors' conclusion that depot risperidone is superior to placebo is based on very limited data. Currently clinicians, recipients of care and researchers do not know if risperidone depot is any better than placebo in terms of global improvement in the short term.
4.2 Mental state The main mental state outcome (20% improvement in the PANSS total score) conveys no useful information as half the data are based on an assumption. From data presented on adverse effects, it is possible to get some data on mental state. Risperidone depot does not seem to affect symptoms of anxiety or nervousness but it may decrease agitation. There is no evidence that depot risperidone effects hallucination but the frequency of 'psychosis' was reduced. We are unsure what this means when both are reported as adverse effects. Overall the information regarding the effects on mental state of long acting risperidone compared with placebo is poor.
4.3 Leaving the study early. The majority of participants did not complete the 12‐week study period which makes it difficult to believe that depot risperidone might improve compliance. The drop out rate was higher in the placebo group but the NNT was six. Six people have to be treated with risperidone depot to avoid one person being leaving care when compared to the attrition from placebo injection treatment.
4.4 Adverse effects Only spontaneously reported adverse events in more than 5% of the participants were reported. Adverse effects which were not reported by the patient, or that were infrequent might have gone unnoticed. Serious adverse effects (those that resulted in death or were life threatening, required hospitalisation or prolongation of hospitalisation, resulted in persistent or significant disability or incapacity, or resulted in congenital anomaly or birth defect) were reported in such a way that the reviewers were left in some doubt about safety. Firstly, overall, these were common (13‐23%). Although there was no collective difference between the experimental and control groups it is feasible that those allocated to placebo needed 'prolongation of hospitalisation' and those given the depot drug encountered 'life threatening' effects. The lack of statistical difference in the 'lumped' data could mask real and disturbing effects. This review does not reassure users of long acting risperidone as regards safety.
The adverse effects that were reported clearly tended to suggest that the depot compound did cause some unwanted effects and that there may be a dose effect. The movement disorder effects were convincing of this. This 'atypical' drug seems to cause extrapyramidal effects, hyperkinesis and hypertonia, especially at the higher doses.
This depot may also cause more sleepiness and weight gain than placebo, but, as for all these adverse effects, more data are needed to confirm this.
5. COMPARISON: 2. DEPOT RISPERIDONE vs ORAL RISPERIDONE
Chue 2002 compared depot risperidone with oral risperidone. The main problem with this study is that it involved well people who are unlikely to be those for whom depot is very relevant.
5.1 Global improvement Data were difficult to extract from the conference proceedings and may have to be revised once the full paper is published, but there seems to be very little difference between the depot and oral forms of risperidone in terms of global improvement. This is encouraging, suggesting that the depot form is as effective as the oral. People already doing well on oral risperidone will continue to do so with depot risperidone.
5.2 Mental state Depot risperidone is similar to oral risperidone in terms of the changes in PANSS scores, thus confirming the impression that there is little difference between the oral and depot preparations for people who are compliant.
5.3 Poor compliance One major reason for giving a depot is to aid poor compliance. For this client group, there was no difference between the oral and depot groups in terms of several ways of measuring compliance. This probably reflects the design of the study where only compliant people were asked to participate. This greatly reduces the value of the study for generalising to real world circumstances.
5.4 Adverse effects. Again there is no clear difference between the oral and depot forms of risperidone, although more data may be available in the fully published paper. However, it should be noted that over half of both groups reported some adverse effects.
COMPARISON: 3. DEPOT RISPERIDONE VS PALIPERIDONE PALMITATE
Summary of main results
Overall completeness and applicability of evidence
3. Limited data
3.1 Loss of data Schizophrenia is often a chronic illness, which may require medication on a long‐term basis. In Kane 2002*, 56% of patients left the study in the first 12 weeks. We will discuss the differential loss to follow‐up below, but it would be difficult to encourage long‐term use of depot risperidone based on the findings of this study. It is likely that this huge loss of patients, greater than would be expected in clinical practice, may result from the limitations of study design where a rigid protocol is imposed on people who are unwell. When a similar protocol is implemented on reasonably well people attrition is less (17%, Chue 2002). Clinicians prefer depot for people who are already having difficulty in complying with treatment. In such a situation clinical common sense indicates that depot preparation may be more helpful, but pragmatic trials are required to confirm this.
3.2 Missing outcomes Risperidone depot is one of the options for the long‐term treatment of people with schizophrenia. However, there are no outcomes rated beyond 12 weeks in the current studies, and much of the three‐month data are 'carried forward' from the true time the person decided to leave the study. There are hardly any data on general functioning and change in behaviour, and none on service outcomes, engagement with services, satisfaction with services, quality of life or costs. It would seem important to address these deficiencies.
3.3 Problematic outcomes More clarity is needed in the reporting of adverse effects. In Kane 2002*, only spontaneously reported adverse events occurring in more than 5% of patients are recorded. This raises the possibility that some rare but clinically important adverse events may have been under‐reported. In the conference proceedings we have for Chue 2002, no specific adverse effects, except death, are reported. We recognise that it is a huge task to report every adverse event but unless careful attention is paid to rare adverse events they might go unnoticed.
The Kane 2002* full paper also groups severe adverse effects in an unusual way. It is possible that the lumping together of several 'severe' effects, some of which may be not as severe as others, could mask real effects of the interventions.
2. Applicability 2.1 Diagnosis Both the included studies used DSM IV operational criteria to help select participants. The use of these criteria means that participants are homogenous and that the study subsequently has greater internal validity, but external validity, i.e. applicability to the every day world of psychiatric care, is likely to be limited.
2.2 Severity of illness Chue 2002 included only people who were already stabilised on oral risperidone. Even though the inclusion criteria stipulated a PANSS score of >50, nearly half the participants were described as mildly ill or not ill at all on the CGI scale at baseline. From this study one can only infer the effects of depot risperidone on stable, reasonably well people. This study does not answer questions as to whether depot risperidone is helpful for people who are very ill. Kane 2002*, however, includes patients who were experiencing more symptoms, as observed by the high baseline PANSS total score.
Quality of the evidence
1. Quality of studies It is disappointing that the reporting of studies was not better. Perhaps to hope that CONSORT requirements (Moher 2001) should be met in conference proceedings is ambitious, but at least they should be considered when they are published in full. Both studies appear to be vulnerable to inclusion bias in favour of risperidone depot.
Data and analyses
Comparison 1. RISPERIDONE DEPOT vs PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. Change (exacerbation) in specific symptoms | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 anxiety ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.32, 1.05] |
| 1.2 agitation ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.39, 0.92] |
| 1.3 hallucinations ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.47, 3.22] |
| 1.4 nervousness ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.12, 1.25] |
| 1.5 psychosis ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.33, 0.83] |
| 2 Leaving the study early: 1. Any reason (by time period) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 very early on (<1 injection) | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.55, 3.08] |
| 2.2 by 12 weeks | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.63, 0.88] |
| 3 Leaving the study early: 2. Any reason (by doses) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 all doses risperidone depot ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.63, 0.88] |
| 3.2 25mg risperidone depot ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.59, 0.94] |
| 3.3 50mg risperidone depot ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.59, 0.93] |
| 3.4 75mg risperidone depot ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.60, 0.94] |
| 4 Leaving the study early: 3. Because of insufficient response (by doses) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 all three doses ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.36, 0.79] |
| 4.2 25mg depot risperidone group ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.45, 1.17] |
| 4.3 50mg depot risperidone group ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.27, 0.83] |
| 4.4 75mg depot risperidone group ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.21, 0.72] |
| 5 Adverse events: 1. General: a. Death | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.00, 2.65] |
| 6 Adverse events: 1. General: b. Severe adverse event (by doses) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 any dose risperidone depot ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.38, 0.93] |
| 6.2 25mg risperidone depot ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.30, 1.04] |
| 6.3 50mg risperidone depot ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.32, 1.06] |
| 6.4 75mg risperidone depot ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.36, 1.15] |
| 7 Adverse events: 1. General: c. Adverse event necessitating withdrawal from study (by doses) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 any dose risperidone depot ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.54, 1.84] |
| 7.2 25mg risperidone depot ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.42, 1.96] |
| 7.3 50mg risperidone depot ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.45, 2.02] |
| 7.4 75mg risperidone depot ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.56, 2.35] |
| 8 Adverse events: 2. Specific: a. Cardiovascular | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 dizziness ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.62, 3.43] |
| 8.2 tachycardia ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.11, 0.98] |
| 9 Adverse events: 2. Specific: b. Gastrointestinal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 constipation ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 6.17 [0.84, 45.46] |
| 9.2 diarrhoea ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.23, 3.20] |
| 9.3 nausea ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.39, 2.76] |
| 9.4 vomiting ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.23, 1.57] |
| 10 Adverse events: 2. Specific: c. Movement disorders: a. Extrapyramidal disorder ‐ spontaneously reported (by doses) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 all doses of depot risperidone ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 2.38 [0.73, 7.78] |
| 10.2 25mg risperidone group ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.30, 5.74] |
| 10.3 50mg risperidone group ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 2.54 [0.69, 9.29] |
| 10.4 75mg risperidone group ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 3.27 [0.93, 11.51] |
| 11 Adverse events: 2. Specific: d. Movement disorders: b. Hyperkinesia (by doses) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 all doses of risperidone ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.70 [0.60, 4.84] |
| 11.2 25mg risperidone group ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.09, 2.64] |
| 11.3 50mg risperidone group ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 2.14 [0.68, 6.73] |
| 11.4 75mg of risperidone group ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 2.45 [0.79, 7.55] |
| 12 Adverse events: 2. Specific: e. Movement disorders: c. Hypertonia (by doses) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 all doses of depot risperidone ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.47, 3.22] |
| 12.2 25mg risperidone ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.22, 2.86] |
| 12.3 50mg risperidone ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.28, 3.19] |
| 12.4 75mg risperidone ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.96 [0.70, 5.53] |
| 13 Adverse events: 2. Specific: f. Pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 headache ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [0.88, 2.80] |
| 13.2 pain ‐ unspecified ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.38 [0.48, 4.00] |
| 14 Adverse events: 2. Specific: g. Salivation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 decreased ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 2.92 [0.37, 22.76] |
| 14.2 increased ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 2.92 [0.37, 22.76] |
| 15 Adverse events: 2. Specific: h. Sleep disturbances | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 insomnia ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.60, 1.82] |
| 15.2 somnolence ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 2.27 [0.69, 7.45] |
| 16 Adverse events: 2. Specific: i. Weight gain | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 all doses of depot risperidone ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 2.11 [0.48, 9.18] |
| 16.2 25mg risperidone ‐ short term | 1 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 2.47 [0.49, 12.45] |
| 16.3 50mg risperidone ‐ short term | 1 | 201 | Risk Ratio (M‐H, Random, 95% CI) | 1.90 [0.36, 10.16] |
| 16.4 75mg risperidone ‐ short term | 1 | 198 | Risk Ratio (M‐H, Random, 95% CI) | 1.96 [0.37, 10.46] |
| 17 Adverse events: 2. Specific: j. Others | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 coughing ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.32, 2.95] |
| 17.2 fatigue ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 8.82 [0.53, 147.05] |
| 17.3 injury ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.13, 1.10] |
| 17.4 rhinitis ‐ short term | 1 | 400 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.47, 2.17] |
Comparison 2. RISPERIDONE DEPOT vs GENERAL ORAL ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Relapse (any reason) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 long term | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 2.13 [0.84, 5.43] |
| 2 Global state: 2. Needing use of benzodiazepine or sedative drugs | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 long term | 1 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.68, 1.47] |
| 3 Service utilisation: 1. Hospitalisation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 long term | 1 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.68, 1.10] |
| 4 Service utilisation: 2. Outpatient care ‐ number of outpatient visits (skewed data) | Other data | No numeric data | ||
| 4.1 long term | Other data | No numeric data | ||
| 5 Not receiving allocated study medication | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 long term | 1 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.06, 1.37] |
| 6 Leaving the study early: 1. Any reason | 2 | 467 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.98, 1.57] |
| 6.1 long term | 2 | 467 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.98, 1.57] |
| 7 Leaving the study early: 2. Specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 insufficient response ‐ long term | 1 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.15, 2.50] |
| 7.2 withdrawn consent ‐ long term | 1 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [0.86, 2.31] |
| 8 Adverse events: 1. General: a. Death | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 long term | 1 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.14, 7.10] |
| 9 Adverse events: 2. Specific | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 anxiety ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.42, 4.60] |
| 9.2 diabetes mellitus ‐ long term | 1 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 1.70 [0.73, 3.96] |
| 9.3 dizziness ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [0.53, 4.19] |
| 9.4 fatigue/somnolence ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 2.05 [0.78, 5.40] |
| 9.5 gastrointestinal ‐ long term | 1 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.95, 1.28] |
| 9.6 general disorders and administration site conditions ‐ long term | 1 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.02, 1.69] |
| 9.7 headache ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 2.80 [1.12, 7.00] |
| 9.8 insomnia ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [0.77, 3.91] |
| 9.9 nausea/ vomiting ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 1.86 [0.50, 6.97] |
| 9.10 prolactin related ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 10.27 [0.59, 180.05] |
| 9.11 weight increase ‐ long term | 1 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.56, 3.17] |
| 10 Adverse events: Nervous system disorders (inc. EPS) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 long term | 1 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [1.13, 1.58] |
Comparison 3. RISPERIDONE DEPOT vs ORAL RISPERIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Moderate to severely ill at end of study period (CGI rating) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.92, 1.22] |
| 2 Global state: 2. Mean change from baseline (CGI‐S, high score = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 short term | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.25, 0.17] |
| 3 Global state: 3. Mean (SD) GAF score change to endpoint | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐0.8 [‐5.66, 4.06] |
| 4 Global state: 4. Needing use of benzodiazepine or sedative drugs | 2 | 690 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.74, 1.02] |
| 4.1 short term | 2 | 690 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.74, 1.02] |
| 5 Mental state: 1. Average change/endpoint scores (PANSS, high score = worse) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 mean total (non ITT data) | 1 | 541 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐2.91, 2.91] |
| 5.2 average change: 1. total (non ITT data) | 2 | 591 | Mean Difference (IV, Random, 95% CI) | 1.05 [‐0.77, 2.88] |
| 5.3 average change: 2. positive (non‐ITT data) | 2 | 591 | Mean Difference (IV, Random, 95% CI) | 0.83 [‐0.69, 2.35] |
| 5.4 average change: 3. negative (non ITT data) | 2 | 591 | Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.76, 0.82] |
| 5.5 average change: 4. disorganised thoughts | 1 | 541 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.45, 0.65] |
| 5.6 average change: 5. hostility/excitement | 1 | 541 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.18, 0.38] |
| 5.7 average change: 6. anxiety/depression | 1 | 541 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.45, 0.65] |
| 6 Leaving the study early: 1. Any reason | 2 | 690 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.92, 1.79] |
| 6.1 short term | 2 | 690 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.92, 1.79] |
| 7 Leaving the study early: 2. Specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 adverse events ‐ short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.62, 2.35] |
| 7.2 insufficient response ‐ short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.63, 3.64] |
| 7.3 withdrawn consent ‐ short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.65, 2.66] |
| 8 Quality of life: Mean (SD) SF‐36 score change/endpoint (high score = better) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Physical component summary | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 1.4 [‐2.64, 5.44] |
| 8.2 Mental component summary | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐5.06, 4.66] |
| 8.3 Role physical | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 1.0 [‐20.71, 22.71] |
| 8.4 Role emotional | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐10.60 [‐34.13, 12.93] |
| 8.5 Vitality | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐1.6 [‐10.24, 7.04] |
| 8.6 General health | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐2.60 [‐13.14, 7.94] |
| 8.7 Mental health | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 5.8 [‐5.20, 16.80] |
| 8.8 Bodily pain | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 3.70 [‐9.89, 17.29] |
| 8.9 Physical function | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐4.6 [‐14.25, 5.05] |
| 8.10 Social function | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 18.5 [3.98, 33.02] |
| 9 Adverse events: 1. General | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 any ‐ short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.91, 1.18] |
| 9.2 death ‐ short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.20] |
| 10 Adverse events: 1. General: UKU average change score (high = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.1 short term | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐1.99 [‐3.59, ‐0.39] |
| 11 Adverse events: 2. Specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 anxiety | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.84, 2.34] |
| 11.2 psychosis | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.58, 2.24] |
| 11.3 prolactin related | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.15, 1.65] |
| 11.4 impotence/ejaculation failure | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.20] |
| 11.5 dysmenorrhoea | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.06, 16.02] |
| 11.6 hyperprolactinaemia | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.18] |
| 11.7 galactorrhoea | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.14, 7.10] |
| 11.8 headache | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.66, 1.95] |
| 11.9 insomnia | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.66, 1.74] |
| 11.10 sexual dysfunction | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.05, 5.52] |
| 12 Adverse events: 2. Specific: Mean (SD) weight increase in kg | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12.1 short term | 1 | 640 | Mean Difference (IV, Random, 95% CI) | 0.2 [‐0.35, 0.75] |
| 13 Adverse events: 3. Movement disorder | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 any extra pyramidal symptoms ‐ short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.59, 1.88] |
| 13.2 participants requiring anti‐cholinergic drugs ‐ short term | 2 | 690 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.66, 1.60] |
| 13.3 tardive dyskinesia ‐ short term | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 9.06 [0.49, 167.52] |
| 14 Adverse events: Mean (SD) change in movement disorder rating scales | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 14.1 AIMS | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 1.16 [‐1.23, 3.55] |
| 14.2 BARS | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.65, 0.97] |
| 14.3 SAS | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐0.55 [‐3.71, 2.61] |
Comparison 4. RISPERIDONE DEPOT vs ORAL QUETIAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 long term | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.74, 0.95] |
| 2 Leaving the study early: 2. Specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 due to relapse ‐ long term | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.40, 0.73] |
| 3 Adverse events: 1. General | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 any | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.89, 1.09] |
| 3.2 serious | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.62, 1.13] |
| 3.3 death | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.26, 9.14] |
| 4 Adverse events: 2. Specifc | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 psychiatric symptoms | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.84, 1.19] |
| 4.2 prolactin related | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 3.07 [1.13, 8.36] |
| 4.3 hyperprolactinaemia | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 8.81 [3.53, 21.96] |
| 4.4 serious psychiatric symptoms | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.58, 1.16] |
| 4.5 weight increase | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.63, 1.99] |
| 4.6 headache | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.64, 2.26] |
| 4.7 fatigue/somnolence | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.07, 0.38] |
| 5 Adverse events: 2. Specific: Mean (SD) weight increase in kg | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 long term | 1 | 666 | Mean Difference (IV, Random, 95% CI) | 1.25 [0.25, 2.25] |
| 6 Adverse events: 3. Movement disorder | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 any extra pyramidal symptom | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.07, 3.15] |
| 6.2 tremor | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 5.12 [1.13, 23.20] |
| 6.3 tardive dyskinesia | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.04, 3.27] |
| 6.4 dystonia | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.05, 5.62] |
| 6.5 parkinsonism | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 2.56 [1.01, 6.52] |
| 6.6 hyperkinesia | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.66 [0.70, 3.96] |
Comparison 5. RISPERIDONE DEPOT vs ORAL ARIPIPRAZOLE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Relapse (any reason) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 long term | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.83, 1.33] |
| 2 Global state: 3. Mean time in remission (days) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 long term | 1 | 348 | Mean Difference (IV, Random, 95% CI) | 16.80 [‐43.59, 77.19] |
| 3 Mental state: 1. Average change scores (PANSS, high score = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 long term | 1 | 349 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐3.15, 2.95] |
| 4 Leaving the study early: 1. Any reason | 2 | 723 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.53, 1.30] |
| 4.1 long term | 2 | 723 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.53, 1.30] |
| 5 Leaving the study early: 2. Specific | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 adverse events | 2 | 723 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.05, 3.55] |
| 5.2 insufficient response | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.29, 5.70] |
| 5.3 withdrawn consent | 2 | 723 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.67, 1.52] |
| 5.4 due to relapse | 1 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.36, 1.06] |
| 5.5 loss to follow‐up | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.83, 3.68] |
| 6 Adverse events: 1. General | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 any | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.87, 1.14] |
| 6.2 serious | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.66, 1.39] |
| 6.3 death | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.13, 7.36] |
| 7 Adverse events: 2. Specific | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 anxiety | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.75, 1.94] |
| 7.2 depression | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [0.85, 2.90] |
| 7.3 psychosis | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.69, 1.56] |
| 7.4 psychiatric symptoms | 1 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.60, 1.09] |
| 7.5 serious psychiatric symptoms | 1 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.27, 2.08] |
| 7.6 schizophrenia | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.63, 1.64] |
| 7.7 prolactin related | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 9.91 [2.78, 35.29] |
| 7.8 hyperprolactinaemia | 1 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 12.13 [0.76, 193.65] |
| 7.9 weight increase | 1 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [0.38, 6.45] |
| 7.10 nausea/vomiting | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.64, 2.43] |
| 7.11 gastrointestinal | 1 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.14, 0.55] |
| 7.12 decreased appetite | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.78 [1.00, 3.16] |
| 7.13 diarrhoea | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.31, 1.24] |
| 7.14 headache | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.46, 1.65] |
| 7.15 insomnia | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.65, 1.27] |
| 7.16 upper resp. tract infection | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.16, 0.89] |
| 7.17 pyrexia | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.69, 1.97] |
| 7.18 nasopharyngitis | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.58, 2.10] |
| 7.19 dizziness | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.89 [1.00, 3.58] |
| 7.20 glucose related | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.58, 2.10] |
| 8 Adverse events: 2. Specific 12. Mean (SD) weight increase in kg | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 long term | 1 | 355 | Mean Difference (IV, Random, 95% CI) | 1.0 [‐0.42, 2.42] |
| 9 Adverse events: 3. Movement disorder | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 any extra pyramidal symptoms | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.91, 1.55] |
| 9.2 tremor | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.65, 1.41] |
| 9.3 akathisia | 1 | 355 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.55, 1.76] |
Comparison 6. RISPERIDONE DEPOT vs ORAL OLANZAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. Average change scores (PANNS, high score = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 total ‐ short term | 1 | 377 | Mean Difference (IV, Random, 95% CI) | 0.90 [‐2.25, 4.05] |
| 1.2 total ‐ long term | 1 | 361 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐3.96, 4.16] |
| 1.3 positive symptoms ‐ long term | 1 | 361 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.61, 1.01] |
| 1.4 negative symptoms ‐ long term | 1 | 361 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐1.28, 1.48] |
| 1.5 disorganised thoughts ‐ long term | 1 | 361 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.34, 0.74] |
| 1.6 hostility/excitement ‐ long term | 1 | 361 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.60, 1.00] |
| 1.7 anxiety/depression ‐ long term | 1 | 361 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.46, 1.06] |
| 2 Leaving the study early: 1. Any reason | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 long term | 1 | 618 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [1.10, 1.58] |
| 3 Leaving the study early: 2. Specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 adverse events | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.28, 1.77] |
| 3.2 insufficient response | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.49, 1.35] |
| 3.3 withdrawn consent | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 2.54 [1.56, 4.16] |
| 3.4 due to weight gain | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 0.24 [0.03, 2.07] |
| 4 Adverse events: 1. General | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 serious | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.80, 1.51] |
| 4.2 death | 1 | 618 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.06, 1.55] |
| 5 Adverse events: 2. Specific | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 agitation | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [1.06, 3.68] |
| 5.2 anxiety | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.58, 1.31] |
| 5.3 depression | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [0.99, 2.12] |
| 5.4 psychosis | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.87, 1.52] |
| 5.5 impotence/ejaculation failure | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.17, 8.56] |
| 5.6 galactorrhoea | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 3.04 [0.59, 15.52] |
| 5.7 serious psychiatric symptoms | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.64, 1.59] |
| 5.8 serious anxiety | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.48, 4.16] |
| 5.9 suicide attempt | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.56, 3.27] |
| 5.10 serious injury | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [0.49, 8.39] |
| 5.11 weight increase | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.42, 0.75] |
| 5.12 headache | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.56 [0.81, 3.01] |
| 5.13 insomnia | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 4.59 [2.61, 8.07] |
| 5.14 fatigue/somnolence | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.35, 1.41] |
| 5.15 nasopharyngitis | 1 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.61, 2.21] |
| 5.16 diabetes mellitus | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.08, 19.32] |
| 5.17 hyperglycaemia | 1 | 494 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.25, 3.95] |
| 5.18 hypoglycaemia | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.02, 9.89] |
| 6 Adverse events: 3. Movement disorder | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 any extra pyramidal symptoms | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [1.19, 2.36] |
| 6.2 tremor | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 2.29 [1.04, 5.06] |
| 6.3 tardive dyskinesia | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.17, 8.56] |
| 6.4 hypertonia | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.56, 3.27] |
| 6.5 dystonia | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 6.07 [0.29, 125.82] |
| 6.6 hyperkinesia | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [1.01, 4.06] |
| 6.7 requiring antiparkinson drugs | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [1.02, 1.56] |
Comparison 7. RISPERIDONE DEPOT vs ALL ORAL ANTIPSYCHOTICS (PRIMARY OUTCOMES).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Relapse (any reason) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 vs aripiprazole ‐ long term | 1 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.83, 1.33] |
| 1.2 vs general oral antipsychotics ‐ long term | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 2.13 [0.84, 5.43] |
| 2 Mental state: 1. Average change scores (PANSS, high score = worse) 1. total | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 vs oral risperidone (non ITT data) ‐ short term | 2 | 591 | Mean Difference (IV, Random, 95% CI) | 1.05 [‐0.77, 2.88] |
| 2.2 vs olanzapine ‐ short term | 1 | 377 | Mean Difference (IV, Random, 95% CI) | 0.90 [‐2.25, 4.05] |
| 2.3 vs olanzapine ‐ long term | 1 | 361 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐3.96, 4.16] |
| 2.4 vs aripiprazole ‐ long term | 1 | 349 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐3.15, 2.95] |
| 3 Leaving the study early: 1. Any reason | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 vs aripiprazole | 2 | 723 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.53, 1.30] |
| 3.2 vs quetiapine | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.74, 0.95] |
| 3.3 vs oral risperidone | 2 | 690 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.92, 1.79] |
| 3.4 vs any new generation antipsychotic | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.55, 0.95] |
| 3.5 vs olanzapine | 1 | 618 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [1.10, 1.58] |
| 3.6 vs any oral antipsychotic | 1 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.93, 1.68] |
| 4 Adverse events: 1. Death | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 vs olanzapine | 1 | 618 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.06, 1.55] |
| 4.2 vs oral risperidone | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.20] |
| 4.3 vs any oral antipsychotic | 1 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.14, 7.10] |
| 4.4 vs aripiprazole | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.13, 7.36] |
| 4.5 vs quetiapine | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.26, 9.14] |
| 5 Adverse events: 1. General: a. any | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 vs aripiprazole | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.87, 1.14] |
| 5.2 vs oral risperidone | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.91, 1.18] |
| 5.3 vs quetiapine | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.89, 1.09] |
| 6 Adverse events: 1. General: b. serious | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 vs quetiapine | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.62, 1.13] |
| 6.2 vs aripiprazole | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.66, 1.39] |
| 6.3 vs olanzapine | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.80, 1.51] |
| 7 Adverse events: 2. Movement disorder: a. any extra pyramidal symptoms | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 vs aripiprazole | 2 | 729 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.91, 1.55] |
| 7.2 vs quetiapine | 1 | 666 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.07, 3.15] |
| 7.3 vs olanzapine | 1 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [1.19, 2.36] |
| 7.4 vs oral risperidone | 1 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.59, 1.88] |
Comparison 8. RISPERIDONE DEPOT vs ATYPICAL DEPOT ANTIPSYCHOTICS (PALIPERIDONE PALMITATE).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global State: 1. CGI‐S mean change from baseline (high score = worse) | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.26, 0.11] |
| 1.1 medium term | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.26, 0.11] |
| 2 Global state: 2. Schedule for Deficit Syndrome (SDS) scale (mean change from baseline, high score = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 medium term | 1 | 913 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.29, 0.49] |
| 3 Mental state: 1. PANSS scores (high score = worse) ‐ medium term | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 total mean change to endpoint (ITT and per protocol data)* | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | 1.12 [‐2.79, 5.02] |
| 3.2 positive symptoms score change to endpoint | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | 0.66 [‐1.39, 2.71] |
| 3.3 negative symptoms score change to endpoint (ITT data) | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.47, 0.59] |
| 3.4 disorganised thoughts score change to endpoint (ITT data) | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.55, 0.59] |
| 3.5 uncontrolled hostility/excitement score change to endpoint (ITT data) | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.50, 0.41] |
| 3.6 anxiety/depression score change to endpoint (ITT data) | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.67, 0.69] |
| 4 Mental state: 2. Improved by 30% in total PANSS score (ITT data) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 medium term | 2 | 1326 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.83, 1.23] |
| 5 General functioning: Personal and Social Performance (PSP) scale (high score = better) | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | 0.65 [‐0.69, 1.98] |
| 5.1 mean endpoint ‐ medium term | 2 | 1326 | Mean Difference (IV, Random, 95% CI) | 0.65 [‐0.69, 1.98] |
| 6 Leaving the study early: 1. Any reason | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Lack of efficacy ‐ medium term | 2 | 1672 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.29, 1.75] |
| 6.2 Lack of efficacy ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.45, 0.81] |
| 6.3 Adverse events ‐ medium term | 2 | 1672 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.28, 1.65] |
| 6.4 Adverse events ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.53, 1.48] |
| 6.5 Patient choice/withdrawn consent ‐ medium term | 2 | 1672 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.22, 1.71] |
| 6.6 Patient choice/withdrawn consent ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.83, 1.61] |
| 6.7 Lost to follow‐up ‐ medium term | 2 | 1672 | Risk Ratio (M‐H, Random, 95% CI) | 1.61 [0.93, 2.79] |
| 6.8 Lost to follow‐up ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.39, 1.91] |
| 6.9 Pregnancy ‐ medium term | 2 | 1672 | Risk Ratio (M‐H, Random, 95% CI) | 0.26 [0.03, 2.32] |
| 6.10 Pregnancy ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.06, 16.32] |
| 6.11 Death ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.25] |
| 6.12 Other ‐ medium term | 2 | 1672 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.45, 1.32] |
| 6.13 Other ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.67, 1.78] |
| 6.14 Any reason ‐ medium term | 2 | 1672 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.51, 1.17] |
| 6.15 Any reason ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.75, 0.97] |
| 7 Adverse events: 1. General | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 overall rate ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.33, 4.42] |
| 7.2 overall rate ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.58, 0.95] |
| 7.3 worsening of schizophrenia ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.40, 1.69] |
| 7.4 worsening of psychiatric disorders ‐ medium term | 1 | 1214 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.22, 1.34] |
| 7.5 death ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.14, 6.54] |
| 7.6 death ‐ long term | 1 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.25] |
| 8 Adverse events: 2. Specific | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 overall rate ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.86, 1.08] |
| 8.2 overall rate ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.95, 1.11] |
| 8.3 insomnia ‐ medium term | 1 | 1214 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.49, 1.05] |
| 8.4 insomnia ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.71, 1.40] |
| 8.5 psychotic disorder ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.59, 1.24] |
| 8.6 worsening of schizophrenia ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.49, 1.16] |
| 8.7 anxiety ‐ medium term | 1 | 1214 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.26, 0.96] |
| 8.8 anxiety ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.01, 2.20] |
| 8.9 headache ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.78, 1.87] |
| 8.10 constipation ‐ medium term | 1 | 1214 | Risk Ratio (M‐H, Random, 95% CI) | 3.79 [1.42, 10.08] |
| 8.11 injection site pain ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.07, 0.38] |
| 8.12 somnolence ‐ medium term | 1 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.21, 1.49] |
| 8.13 weight gain (proportion of participants with >7% increase) ‐ medium term | 1 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.72, 1.75] |
| 8.14 tachycardia ‐ medium term | 1 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.26, 4.06] |
| 8.15 tachycardia ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.11, 1.05] |
| 9 Adverse events: 3. Prolactin related | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 amenorrhoea ‐ medium term | 2 | 784 | Risk Ratio (M‐H, Random, 95% CI) | 1.78 [0.24, 13.02] |
| 9.2 galactorrhoea ‐ medium term | 1 | 271 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.02, 8.92] |
| 9.3 hyperprolactinaemia ‐ medium term | 1 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 5.13 [0.60, 43.60] |
| 9.4 erectile dysfunction ‐ medium term | 1 | 701 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.18, 3.53] |
| 9.5 increase in serum prolactin ‐ medium term | 1 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.35, 1.48] |
| 9.6 amenorrhoea‐galactorrhoea syndrome ‐ medium term | 1 | 271 | Risk Ratio (M‐H, Random, 95% CI) | 3.3 [0.14, 80.29] |
| 9.7 any prolactin related ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.61, 1.71] |
| 9.8 proportion of male participants with abnormally high prolactin ‐ long term | 1 | 424 | Risk Ratio (M‐H, Random, 95% CI) | 1.68 [1.32, 2.14] |
| 9.9 proportion of female participants with abnormally high prolactin ‐ long term | 1 | 294 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.95, 1.55] |
| 10 Adverse events: 4. Movement disorder | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 akathisia ‐ medium term | 1 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.98, 2.31] |
| 10.2 tremor ‐ medium term | 1 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [1.07, 2.74] |
| 10.3 tardive dyskinesia ‐ medium term | 1 | 1214 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.06, 15.90] |
| 10.4 requiring use of anti‐EPS medication ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [1.18, 1.80] |
| 10.5 hyperkinesia ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 1.66 [1.00, 2.73] |
| 10.6 neuroleptic malignant syndrome ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.40] |
| 11 Adverse events: 5. Body weight (mean increase) | 3 | 2350 | Mean Difference (IV, Random, 95% CI) | 0.18 [‐0.36, 0.72] |
| 11.1 medium term | 2 | 1666 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.38, 0.24] |
| 11.2 long term | 1 | 684 | Mean Difference (IV, Random, 95% CI) | 1.0 [0.13, 1.87] |
| 12 Adverse events: 6. Mean prolactin level increase (ng/mL) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12.1 female participants | 2 | 807 | Mean Difference (IV, Random, 95% CI) | ‐3.40 [‐12.65, 5.85] |
| 12.2 male participants | 2 | 1125 | Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐5.88, 5.03] |
| 13 Adverse events: 7. Glucose related | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 increased blood glucose ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.44, 5.43] |
| 13.2 hyperglycaemia ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.31, 6.09] |
| 13.3 diabetes mellitus ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 4.12 [0.46, 36.68] |
| 13.4 glycosuria ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.40] |
| 13.5 ketonuria ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.40] |
| 13.6 urine ketone body present ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.40] |
| 13.7 hypoglycaemia ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 3.09 [0.13, 75.59] |
| 13.8 any glucose related ‐ medium term | 2 | 1666 | Risk Ratio (M‐H, Random, 95% CI) | 1.76 [0.52, 5.98] |
| 13.9 any glucose related ‐ long term | 1 | 747 | Risk Ratio (M‐H, Random, 95% CI) | 1.80 [0.77, 4.25] |
| 14 Adverse events: 8. Injection site pain (mean (sd) Visual Analogue Scale score (0‐100mm)) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 14.1 at baseline | 1 | 747 | Mean Difference (IV, Random, 95% CI) | 1.80 [‐0.24, 3.84] |
| 14.2 at endpoint | 1 | 747 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐1.07, 1.07] |
Comparison 9. RISPERIDONE DEPOT vs TYPICAL DEPOT ANTIPSYCHOTICS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. Total endpoint scores (PANNS, high score = worse) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 1 | 49 | Mean Difference (IV, Random, 95% CI) | 0.70 [‐8.12, 9.52] |
| 1.2 medium term | 1 | 46 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐9.02, 8.82] |
| 1.3 long term | 1 | 43 | Mean Difference (IV, Random, 95% CI) | 1.80 [‐10.04, 13.64] |
| 2 Leaving the study early | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 before beginning assigned treatment | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 7.50 [1.00, 56.44] |
| 2.2 by 6 months | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 3.05 [1.12, 8.31] |
| 2.3 due to increased psychiatric symptoms | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 2.81 [0.31, 25.58] |
| 2.4 due to EPS effects | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.40] |
| 2.5 due to weight gain and hypertension | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 2.82 [0.12, 66.62] |
| 2.6 due to participant preference | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.06, 14.33] |
| 3 Hospitalisation by 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 medium term | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.11, 3.48] |
| 4 Adverse events: 1. Continuous outcomes (skew) | Other data | No numeric data | ||
| 4.1 Change in BMI ‐ short term (skew) | Other data | No numeric data | ||
| 4.2 Change in BMI ‐ medium term (skew) | Other data | No numeric data | ||
| 4.3 Change in BMI ‐ long term (skew) | Other data | No numeric data | ||
| 4.4 Prolactin endpoint levels (ng/mL) ‐ short term (skew) | Other data | No numeric data | ||
| 4.5 Prolactin endpoint levels (ng/mL) ‐ medium term (skew) | Other data | No numeric data | ||
| 4.6 Prolactin endpoint levels (ng/mL) ‐ long term (skew) | Other data | No numeric data | ||
| 5 Adverse events: 2. Sexual experiencesm, total endpoint (ASEX, high score = worse) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 44 | Mean Difference (IV, Fixed, 95% CI) | 1.70 [‐1.26, 4.66] |
| 5.2 medium term | 1 | 41 | Mean Difference (IV, Fixed, 95% CI) | 1.30 [‐2.30, 4.90] |
| 5.3 long term | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐4.08, 3.88] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bai 2006.
| Methods | Allocation: random. Blinding: single blind, rater blind. Duration: 12 weeks. Design: parallel. Setting: inpatient, large psychiatric teaching hospital, Taiwan. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). Age: 18‐65 years. N = 50. Sex: 25 M and 25 F. History: symptomatically stable defined as PANSS total < 80, CGI‐no change in score between screening and baseline, previous treatment with oral risperidone for > 3 months. Included: 'good health' based on physical examination, medical history and blood biochemistry and haematology. Exclusion: History of NMS or organic CNS disorder; current seizure disorder; current risk of violent behaviour against others; current suicidal ideas or suicidal ideas in the last 6 months. Consent and ethics: study performed in accordance with the Declaration of Helsinki and approved by Ethics Review Committee. All participants provided written informed consent before starting the study. |
|
| Interventions | 1. Risperidone depot: 25 mg, 37.5 mg or 50 mg once every 2 weeks, n = 25. 2. Risperidone oral: mean 3.8 +/‐ 1.6 mg/day, n = 25. |
|
| Outcomes | Quality of life (SF‐36). Adverse events (recorded spontaneously); AIMS; BARS; SAS; UKU; movement disorder. Mental state: PANSS. Global state: CGI‐S, GAF. Leaving study early. Unable to use ‐ Satisfaction with treatment ‐ non‐peer reviewed scale. Pain at injection site (visual analogue scale) ‐ no SD reported. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "this trial was a randomized, parallel‐group, rater‐blind study of 52 weeks duration." No information on how randomisation was achieved. |
| Allocation concealment (selection bias) | Unclear risk | No information. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Implied to be an open‐label study. Not explicit, but implied open‐label with regard to participants and study drug administrators. Detection: "rater blind", but no information on how the blinding was maintained. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | N = 49 participants completed the study (98%). |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in methods appear to be reported. |
| Other bias | High risk | Funding: supported by Jassen‐Cilag Taiwan, Johnson & Johnson co. |
Chue 2002.
| Methods | Allocation: random.
Blinding: double.*
Duration: 12 weeks (preceded by 8‐week open‐label run‐in).
Design: parallel, international multi‐centre. Setting: inpatient, 95 sites, UK, mainland Europe, North America, Africa. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). Age: mean 40 ± 15 years, range 18‐65. N = 640.** Sex: 415 M, 225 F. History: inpatient or outpatient; PANSS score ≥ 50, but ˜47% "not ill or only mildly ill" on CGI, stabilised 8/52 on oral risperidone. Included: stable CGI scores for the previous 4 weeks of the oral risperidone run‐in period. Excluded: moderate or severe symptoms of tardive dyskinesia at study entry; history of neuroleptic malignant syndrome; known to be unresponsive to risperidone; required mood stabilisers; treated with clozapine in past 2 months before screening; treated with a depot antipsychotic within one treatment cycle of screening, or with antidepressants within 30 days before run‐in period. Consent and ethics: study performed in accordance with the Declaration of Helsinki; consent obtained from participant, relative, guardian or legal representative at study entry. |
|
| Interventions | 1. Risperidone depot: 25, 50 or 75 mg, every two weeks + daily placebo tablets, n = 319. 2. Risperidone oral: 2, 4 or 6 mg/day + placebo injections every two weeks, n = 321. | |
| Outcomes | Global state: needing use of benzodiazepines or sedative drugs; CGI (dichotomised). Mental state: PANSS. Leaving the study early. Adverse events: others as reported by participants; death. Unable to use ‐ Global state: CGI (data on subgroups only). Adverse events: ESRS (no usable data). Body weight: change (no usable data). Pain at injection site (no usable data). Physiological tests: including ECG (no usable data). |
|
| Notes | * Blindness was maintained with different doses by using the same volume of diluent. ** Numbers randomised not consistent in presentations (426 vs 640). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was stratified according to site, PANSS score, ESRS total score, use of depot antipsychotics in the previous 6 months and daily dose of oral risperidone at randomization" (p112). |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Placebo tablets and injections used to blind participants. "Double blind" stated but it is not clearly expressed who exactly was blinded, cannot be sure if rater blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | N = 541 participants completed the study (85%). Completer‐only data for PANSS. |
| Selective reporting (reporting bias) | Unclear risk | All outcomes reported, however not all presented as usable data, particularly continuous data, with no means or SD. |
| Other bias | High risk | Funding: supported by Janssen Research Foundation, Beerse, Belgium. |
Covell 2012.
| Methods | Allocation: random. Blinding: open‐label, blinded clinical raters. Duration: 6 months (+ 6 months naturalistic follow‐up). Design: parallel. Setting: National Institute of Mental Health Schizophrenia Trials Network and five sites in Conneticut's public mental health system, USA. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). Age: ≥18 years, mean age 48 yrs (risperidone depot: 48.5 ± 12.2; haloperidol/fluphenazine depot: 47.3 ± 9.1). N = 62. Sex: 44 M (22 in each group), 18 F. History: currently taking fluphenazine decanoate or haloperidol decanoate, "may benefit from changing medication" and "willing to change", able to afford own medication, at least one 3‐monthly clinic visit in past 6 months. Included: eligible patients were those who might benefit from switching to risperidone microspheres (with sub‐optimal response to treatment because of persistent psychopathology or significant side effects); people where change in medical opinion was a reasonable clinical opinion, but not required; willingness to change antipsychotic medication; access to medication without financial burden; at least 1 clinic visit every 3 months for past 6 months. Excluded: symptom severity indicating immediate change; exacerbation in previous 3/12; pregnancy; pending criminal charges; non‐independent living; antipsychotic polypharmacy. Consent and ethics: written informed consent after thorough description of the study to participants and assessment of understanding consent materials. |
|
| Interventions | 1. Risperidone depot: 25, 37.5 or 50 mg/ 2 weeks, n = 32. 2. Haloperidol decanoate OR fluphenazine*, n = 30. *No data on dosages actually prescribed in this arm ‐ "clinician's judgement". |
|
| Outcomes | Primary ‐ time to all‐cause treatment discontinuation. Mental state: PANSS (completer‐only). Hospitalisation by 6 months. Adverse events: Arizona Sexual Experiences Scale; weight; prolactin (completer‐only, skew data). Unable to use ‐ AIMS (adapted scale used). Subjective Side‐effect Rating Scale (no data reported). EPS (SAS); tardive dyskinesia (incomplete data for all participants ‐ only 44% accounted for). Tardive dyskinesia (more than 50% participants did not complete assessment). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised, quote, "stratified by gender and baseline decanoate. No exceptions were made to the predetermined randomisation streams" (p670). |
| Allocation concealment (selection bias) | Unclear risk | No details given of how this was achieved. Quote, "eligible patients were those who might benefit from a switch to risperidone microspheres" (p670). |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label study with assessment by blinded clinical raters. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data relating to loss to follow‐up are given and overall attrition is < 50%. Lost to follow‐up n = 8: reasons for discontinuation included increase in psychiatric symptoms (n = 4), EPS concerns (n = 1), participant preference (n = 2), hypertension and weight gain (n = 1). However, not all participants completed continuous outcome measures; LOCF carried forward used in primary study citation, completer‐only data provided with means and SD for additional requested information (unpublished). |
| Selective reporting (reporting bias) | High risk | Not all data reported, including means and SDs for most continuous outcomes. |
| Other bias | Unclear risk | Funding: quote, "research presented in this article was funded by the National Institute of Mental Health grant number MH71663 and MH59312." One author (Schooler) "has previously received grant/research support from... Janssen‐Cilag, and Johnson & Johnson." |
Fleischhacker 2011.
| Methods | Allocation: random. Blinding: double. Duration: 53 weeks. Design: parallel. Setting: international, multi‐centre, 19 countries: North America, Australia, New Zealand, Western and Eastern Europe. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). Age: ≥ 18 years of age. N = 749. Sex: 444 M, 305 F. History: DSM‐IV diagnosis of schizophrenia for at least one year before screening. Included: PANSS score between 60‐120; acutely symptomatic at screening and baseline; BMI ≥ 15 kg. Excluded: DSM‐IV Axis I diagnosis other than schizophrenia; decrease of ≥ 25% in the PANSS total score between screening and baseline; substance dependence during the three months preceding screening; history of treatment resistance; history of neuroleptic malignant syndrome or any significant or unstable systematic disease; suicidal or violent behaviour; pregnant or nursing or women planning pregnancy. Consent and ethics: Independent Ethics Committee or Institutional Review Board at each study site approved the protocol. Study conducted in accordance with Declaration of Helsinki and consistent with Good Clinical Practice (GCP) and applicable regulatory requirements. All participants provided written informed consent before entry. |
|
| Interventions | 1. Risperidone depot*: IM, 25, 37.5 mg or 50 mg (placebo injections matched to risperidone depot on day 1, with first active injection delayed until day 8), n = 370. 2. Paliperidone palmitate (PP): IM, 25, 50, 75 or 100 mg eq (placebo injections matched to PP on day 22 then monthly thereafter), n = 379. *1‐6 mg oral risperidone/ placebo supplementation was given for the first 4 weeks of the double‐blind treatment period. Oral risperidone (1‐4 mg) supplementation was also given at any dose increase from day 36 onwards, continuing up to week 3. |
|
| Outcomes | Adverse effects: various events, AIMS, BARS, SAS scores, laboratory results: various mean change in serum levels, EKGs, evaluations of injection site (all adverse data relate to participants who received at least one dose of the study drug). Leaving the study early. Death. Unable to use ‐ Primary outcome: Non inferiority of PP with risperidone (high attrition and unable to obtain data). Secondary outcomes: average change in PANSS total score (high attrition and unable to obtain data). Global state: relapse, change in CGI‐S (high attrition and unable to obtain data). Social functioning: Change in PSP (high attrition and unable to obtain data). Mental state: average change in PANSS (high attrition and unable to obtain data). |
|
| Notes | Antiparkinson medication (at permitted maximum daily doses) as rescue treatment for EPS; oral lorazepam (2‐6 mg) or other short‐acting benzodiazepines for agitation, anxiety or sleep difficulties; oral propranolol for akithisia were permitted. Antidepressents permitted if used at a stable dose for at least 30 days before screening. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: "computer generated randomisation schedule (prepared by the sponsor), balanced by using permuted blocks of treatments, stratified by centre and implemented using an interactive voice response system (IVRS)". |
| Allocation concealment (selection bias) | Low risk | Use of interactive voice response system. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Participants were not allowed to view the preparation or administration of the injection. Blinded raters used to measure outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High study attrition (55%): PP group drop‐out rate at n = 224 (n = 95 lack of efficacy; n = 29 adverse events; n = 55 patient choice; n = 13 lost to follow‐up; n = 1 pregnancy; n = 2 death; n = 29 other); risperidone depot group drop‐out rate at n = 186 (n = 56 lack of efficacy; n = 25 adverse events; n = 62 patient choice; n = 11 lost to follow‐up; n = 1 pregnancy; n = 0 death; n‐31 other). |
| Selective reporting (reporting bias) | Unclear risk | Missing outcomes in a supplementary table (not obtainable ‐ contact made with study author, but no reply). |
| Other bias | High risk | Funding: sponsored by Johnson & Johnson Pharmaceutical Research and Development, L.L.C. The sponsor provided a formal review of the manuscript. Two study authors (Mr Remmerie and Dr Eerdekens) were employees of Johnson & Johnson Pharmaceutical Research and Developement, Division of Janssen Pharmaceutica. |
Gaebel 2010*.
| Methods | Allocation: random. Blinding: open‐label. Duration: 2 year. Design: parallel. Setting: international, multi‐centre, 25 countries (Europe). |
|
| Participants | Diagnosis: schizophrenia or schizoaffective (DSM‐IV). N = 710. Age: ≥ 18 years, mean 40.6 +/‐ 12.5 in depot risperidone group; 42.6 +/‐ 13.1 quetiapine group; 40.9 +/‐ 12.94 in aripiprazole group. Sex: 442 M, 270 F. History: symptomatically stable. Included: switching therapy because of insufficient symptom control with current treatment, side effects or patient request; symptomatically stable‐ using stable dose of antipsychotic for ≥ 4 weeks and living in same residence for ≥ 30 days. Excluded: previous non‐response to risperidone, quetiapine or ≥ 2 antipsychotics despite adequate drug plasma levels; DSM IV axis I diagnosis other than schizophrenia or schizoaffective disorder; phenylketonuria or hypersensitivity to risperidone or quetiapine; drug or alcohol dependence during preceding 1 month; acute risk of suicide or history of suicide attempt. Consent and ethics: study conducted in accordance with guidelines of the International Conference on Harmonisation for Good Clinical Practice. Study protocol and consent were approved by ethics committees/ institutional review boards; informed consent obtained from all participants before enrolment. |
|
| Interventions | 1. Risperidone depot: 25‐50 mg IM every 2 weeks, n = 329. 2. Quetiapine oral: 25 mg twice a day: day 1, 300‐ 400 mg by day 4, max 750 mg a day, n = 337. 3. Aripiprazole oral: 10‐30 mg per day, n = 46. |
|
| Outcomes | Adverse events. Leaving the study early. Death. Unable to use ‐ (all due to high attrition) Time to relapse. Global state: CGI. Mental state: PANSS. Functioning assessment: SOFAS. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified randomisation according to previous treatment. |
| Allocation concealment (selection bias) | Unclear risk | No details given. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label study. No evidence for any rater blinding‐ follow‐up methods differed between treatment groups (phone calls for quetiapine group and in person with depot risperidone). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High attrition: treatment completed by n = 151/329 in risperidone depot; n = 120/337 oral quetiapine; n = 9 oral aripiprazole. Total follow‐up of n = 280/710 (39%) ‐ only leaving study early and adverse event data used. |
| Selective reporting (reporting bias) | Unclear risk | Some outcomes comprising relapse are not reported on their own. |
| Other bias | High risk | Funding: study sponsored by Janssen‐Cilag Medical Affairs EMEA. |
Kane 2002*.
| Methods | Allocation: random.
Blinding: double.
Duration: 12 weeks (preceded by up to 4 mg risperidone/day for 1 week).
Design: parallel. Setting: inpatient and outpatient, multi‐centre (41 centres in the USA). |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV).
N = 400.
Age: 18 to 55 years of age; mean˜37 ± 20 years.
Sex: 301 M, 99 F.
History: diagnosis of schizophrenia. Included: baselines PANSS of 60 to 120; good general health; standard laboratory test results 'within reference ranges or not clinically significant'. Excluded: received depot in past 120 days before start of trial; substance dependant diagnosis; tardive dyskinesia; history of neuroleptic malignant syndrome; clinically significant ECG abnormality; pregnant (or likely to become pregnant) or lactating; at risk of violent behaviour; current suicide ideation; history of severe drug sensitivity/ allergy (sensitivity to risperidone); people who were unresponsive to risperidone. Consent: trial conducted in accordance with 'current ICH‐Good Clinical Practice guidelines and the Declaration of Helsinki and its subsequent revisions'; written informed consent obtained from each participant or guardian/ legal representative. |
|
| Interventions | 1. Risperidone depot: 25 mg 2 weekly + 2 mg/day oral risperidone for 3/52, n = 99. 2. Risperidone depot: 50 mg 2 weekly + 4 mg/day oral risperidone for 3/52, n = 100. 3. Risperidone depot: 75 mg 2 weekly + 6 mg/day oral risperidone for 3/52, n = 100. 4. Placebo injections: 2 weekly + placebo tablets for first 3/52, n = 100. | |
| Outcomes | Adverse events: ESRS and others as reported by participants. Leaving the study early. Unable to use ‐ Global state: CGI. Mental state: 20% reduction PANSS. Body weight: change. Pain at injection site. Physiological tests: including ECG. Mental state: change PANSS (no SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: quote, "a dynamic method (22) was used to randomly assign patients to treatment groups. Stratification factors included investigator, inpatient/outpatient status, and Positive and Negative Syndrome Scale (23) total score at randomization." |
| Allocation concealment (selection bias) | Unclear risk | No details of allocation concealment. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Double blind study" but not clear who exactly was blinded, no indication given of whether the raters were blinded. Study controlled with "placebo injections that were identical in appearance [to the study drug injections]". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Greater than 50% attrition by study end. |
| Selective reporting (reporting bias) | Unclear risk | Unclear risk |
| Other bias | High risk | Funding: "supported by Johnson & Johnson Pharmaceutical Research and development, Titusville, N.J." |
Keks 2007.
| Methods | Allocation: random. Blinding: open‐label. Duration: 12 months. Design: parallel. Setting: international, multi‐centre (48 centres in Australia, Belguim, France, Germany, Greece, Luxumbourg, Poland, Russia, Spain, The Netherlands and UK). |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM‐IV). N = 629* (n = 11 not treated). Age: mean˜35 years, minimum 18 years. Sex: 312 M, 235 F. History: acute exacerbation of psychosis in previous 2 months and another episode during previous 2 years. Excluded: prior treatment with clozapine or depot antipsychotic. Included: PANSS total score > 50; at least 18 years of age; BMI not exceeding 40 mg/kg2. resistance or sensitivity to risperidone or olanzapine pregnant or breast feeding women, child bearing age women if not using contraception Consent: study protocol and amendments reviewed by independent ethics committees/ institutional review boards; conducted in accordance with Declaration of Helsinki and guidelines of International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Written informed consent required. |
|
| Interventions | 1. Risperidone depot: 25, 50 or 75 mg*, n = 318. 2. Oral olanzapine: 5‐20 mg/day, n = 300. |
|
| Outcomes | Mental state: PANSS. Specific adverse events; movement disorder; death and serious adverse events. Leaving the study early. |
|
| Notes | *After study initiation protocol was amended to restrict the doses of depot risperidone to 25 or 50 mg; 64 patients who had already received 75 mg of depot risperidone were withdrawn from the study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised, "using a central dynamic randomisation procedure. Randomisation was based on a minimisation algorithm that used a probability of assignment other than 0.5 to maintain balance of treatment groups within levels of each stratification factor" (p132). stratification factors of PANSS total score, number of previous psychiatric hospitalisations, BMI, inpatient or outpatient status, using a central dynamic randomisation procedure. |
| Allocation concealment (selection bias) | Low risk | Interactive voice response system (IVRS) used to obtain randomisation number. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label study. No evidence of rater blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | N = 618 originally randomised and treated; n = 264 (42%) completed at 12 months. LOCF used for endpoint data including n = 361 participants (58%). |
| Selective reporting (reporting bias) | High risk | Protocol amended to restrict risperidone doses to 20 or 50 mg after investigators found that, quote, "75 mg doses provide no greater benefit than lower doses" (p132). The n = 64 participants receiving 75 mg doses completed the study, their data were withdrawn and they were invited to enrol in an open‐label extension study. |
| Other bias | High risk | Funding: "M.I., A.K. and K.K. are employees of Johnson & Johnson... study was supported by Johnson & Johnson Pharmaceutical Research and Development" (p138). |
Li 2011.
| Methods | Allocation: random. Blinding: open‐label. Duration: 12 weeks (with 7 week washout pre‐randomisation). Design: parallel. Setting: multi‐centre, China. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 452. Age: ≥18 years of age. Sex: 181 M, 271 F. History: DSM‐IV diagnosis of schizophrenia for at least one year before screening. Included: PANSS total score between 60 to 120; BMI of 17.0 kg/m2 or greater. Excluded: DSM IV axis I diagnosis other than schizophrenia; 25% decrease in total PANSS score between screening and baseline; active substance dependence within 3 months; significant risk of suicidal or violent behaviour; presence or history of any significant or unstable systemic disease; history of treatment resistance towards two different antipsychotic treatments; pregnancy or planning; clozapine within 3 months before baseline; RIS‐LAI within 6 weeks before screening; PP within 10 months before baseline; ECT within 60 days before screening. Consent: Independent Ethics Committee or Institutional Review Board at each study site approved the protocol; study conducted in accordance with ethical principles of Declaration of Helsinki, consistent Good Clinical Practices and applicable regulatory requirements. Written informed consent required. |
|
| Interventions | 1. Risperidone depot: once every two weeks, mean dose 29.8 ± 4.67 mg, n = 223. 2. Paliperidone palmitate: once monthly injections, mean dose 115.8 ± 9.07 mg, n = 229. (Plus supplementary oral risperidone for risperidone depot participants: mean daily dose 2.5 ± 0.98 mg from days 1 to 28; 1.8 ± 0.52 mg from day 36 to 57; 1.7 ± 0.47 mg from day 64 to 85).* |
|
| Outcomes | Global state: CGI‐S scale score change from baseline. Mental state: change in PANSS total score; number of patients with a 30% or more reduction in PANSS total score. Adverse events: treatment‐emergent adverse events; EPSE; prolactin‐related. General functioning; Personal and Social Performance (PSP). |
|
| Notes | *Other medications permitted, including: antiparkinson medication; benzodiazepines; beta‐blockers; treatment for insomnia; topical anaesthetic cream; antidepressants; individual psychotherapy. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: (1:1) "based on a computer‐generated randomization schedule balanced by using permuted blocks of treatments." |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label. Rater blinding: "all efficacy assessments were administered and evaluated by independent, blinded and trained raters at each site." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | N = 350 (77%) completed the study; n = 64 withdrawn from the PP groups for adverse events (n = 4), pregnancy (n = 2), protocol deviation (n = 3), lack of efficacy (n = 22), lost to follow‐up (n = 9), withdrew consent (n = 16), other reasons (n = 8). From the risperidone depot group, n = 38 were withdrawn for adverse events (n = 5), protocol deviation (n = 1), lack of efficacy (n = 9), lost to follow‐up (n = 14), withdrew consent (n = 5), other reasons (n = 4). ITT used. |
| Selective reporting (reporting bias) | Unclear risk | Not known. |
| Other bias | High risk | Funding: "funded by Xian‐Janssen Pharmaceutical Limited, Beijing, PR China. The sponsor provided a formal review of the manuscript." A number of the authors were employed by Xian‐Janssen or Johnson & Johnson at time of publication. |
MacFadden 2010.
| Methods | Allocation: random. Blinding: open‐label, rater blind. Duration: 2 years. Design: parallel. Setting: international, multi‐centre (USA, South America, India). |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 355. Age: ≥18 years of age. Sex: 210 M, 139 F. History: not described. Included: DSM‐IV schizophrenia not controlled by current medication (judged by clinician); 2+ relapses (defined as "psychiatric hospitalisation caused by worsening of psychiatric symptoms; a change in antipsychotic treatment or significant increase in antipsychotic dose because of inadequate efficacy; a newly emergent, clinically important symptom such as 'suicidality'; or a clinically notable increase in the frequency or intensity of subject contact") in the past 2 years. Stable for 2 months before randomisation. Excluded: PANSS ≥100, current hospitalisation, major medication changes, or worsening of psychiatric symptoms within two months before study entry. Current treatment with clozapine, carbamazepine or depot antipsychotics. Evidence of alcohol or drug dependence (DSM‐IV Axis I criteria) within six months before entry. Consent: study conducted in accordance with Declaration of Helsinki and Good Clinical Practice; approved by Institutional Review Board or independent ethics committee at each centre. Written informed consent required. |
|
| Interventions | 1. Risperidone depot: 25‐50 mg/2 weeks, n = 177. 2. Oral aripiprazole: 5‐30 mg/daily, n = 172.* |
|
| Outcomes | Global state: mean time to relapse/time in remission. Mental state: PANSS. Specific adverse events; weight; movement disorders; death; serious adverse events. Laboratory tests. |
|
| Notes | *Other medications permitted, including antidepressants, anxiolytics, mood stabilisers. At clinician's judgement, if psychotics symptoms worsened, another antipsychotic was added (excluding clozapine) for up to seven days; this treatment continued if considered appropriate by investigators. If this proved ineffective, the investigator had the option to use another different secondary antipsychotic. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised: "subjects were randomly assigned in a 1:1 ratio" but no details on how this was achieved. |
| Allocation concealment (selection bias) | Unclear risk | Concealment not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label, participants and study drug administrators were not blinded, but with blinded raters. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "The proportions of injectable RLAT and aripiprazole subjects who discontinued the study before completing two years were 29.6% and 28.4%, respectively" and reasons for discontinuation are given. Of the original n = 355 randomised, n = 346 were included in ITT analysis. |
| Selective reporting (reporting bias) | Unclear risk | None detected. |
| Other bias | High risk | Study authors employed by Janssen: "Dr. Macfadden was with Ortho‐McNeil Janssen Scientific Affairs, LLC, Titusville, New Jersey, at the time of this analysis; Drs. Ma and Haskins are with Johnson & Johnson Pharmaceutical Research and Development, LLC, Titusville, New Jersey; and Drs. Bossie and Alphs are with Ortho‐McNeil Janssen Scientific Affairs, LLC, Titusville, New Jersey." |
Pandina 2011.
| Methods | Allocation: random. Blinding: double. Duration: 13 weeks. Design: parallel, double dummy, non‐inferiority comparative study. Setting: international multi‐centre, 89 centres from 14 countries (Bulgaria; Czech Republic; Estonia; Hungary; Lithuania; Poland; Russia; Ukraine; USA; Austria; France; Germany; Spain; India). |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 1220. Age: ≥18 years of age. Sex: 701 M, 513 F. History: 65% PP and 69% RIS‐LAI participants were receiving atypical antipsychotics prior to study, with oral risperidone used by similar percentage of participants in each group; Included: PANSS total score between 60 and 120, BMI ≥17 kg/m2 and <40 kg/m2; schizophrenia DSM‐IV criteria for >1 year. Excluded: DSM‐IV Axis I diagnoses other than schizophrenia; decrease in at least 25% in PANSS total from screening to baseline; substance dependence within 3 months before screening; history of treatment resistance; significant unstable systemic disease; suicidal or violent behaviour; previously received injections of PP and treatment with any other 'disallowed' medications (including mood stabilisers, lithium and anticonvulsants); exposure to an experimental drug, biologic or medical device within past 6 months pre‐screening; pregnancy/ planning or currently nursing. Consent: Independent Ethics Committee or Institutional Review Board at each study site approved protocol and amended protocol. Study conducted in accordance with Declaration of Helsinki and Good Clinical Practice guidelines. Written informed consent required. |
|
| Interventions | 1. Risperidone depot: bi‐weekly, oral supplementation (1 mg; mean final dose 3.3 ± 1.59 mg) and placebo injections (matched to PP); 25, 37.5 and 50 mg; mean final dose 31.7 ± 9.28 mg, n = 613. 2. Paliperidone palmitate (PP): initiation regimen*, monthly PP injections, placebo injections (matched to RIS) and placebo oral supplementation; 50, 100 and 150 mg equivalents; mean final dose 104.5 ±3 0.51 mg, n = 607. |
|
| Outcomes | Global state: CGI‐ S score change; Shedule for Deficit Syndrome. Mental state: PANSS total score change; responder rate with more than 30% reduction in PANSS. Adverse events: treatment‐emergent adverse events; EPS rating scales; Simpson and Angus Rating scale; BARS; AIMS. General functioning: Personal and Social Performance (PSP). |
|
| Notes | *PP deltoid injections day 1, 150 mg eq, day 8, 100 mg eq and subsequent flexible dosing (50, 100 or 150 mg eq) once a month | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised: 1:1, computer‐generated randomisation scheme, stratified by centre, implemented by an interactive voice response system. |
| Allocation concealment (selection bias) | Low risk | Interactive voice response system used. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Study drug administrator was the only person to contact IVRS to receive medication number and was not allowed to communicate patient‐related information to study site personal or to perform any efficacy and safety assessment. Patient and staff performing study‐related procedures were to be precluded from seeing the contents of syringe or observing the injection. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | N = 927 (76%) completed the study (n = 456 in PP; n = 471 in RIS‐LAI); n = 151 withdrawn from PP group (n = 55 withdrawn consent, n = 40 lack of efficacy, n = 20 adverse events, n = 11 lost to follow‐up, n = 2 death, n = 1 pregnancy, n = 22 'other'); n = 142 withdrawn from RIS‐LAI group (n = 52 withdrawn consent, n = 43 lack of efficacy, n = 10 adverse event, n = 18, lost to follow‐up, n = 19 'other'). 'Safety analysis' conducted (n = 1214), which included all participants that had received at least one dose of the study drug. |
| Selective reporting (reporting bias) | Unclear risk | Unclear. |
| Other bias | High risk | Funding: "Johnson & Johnson Pharmaceutical Research and Development, L.L.C. funded this study and was responsible for study design and data collection, analysis and its interpretation..." (p225). Many of the authors are employees of Johnson & Johnson Pharmaceutical Research and Development, L.L.C. |
Quinn 2012*.
| Methods | Allocation: random. Blinding: open‐label. Duration: 24 months. Design: parallel. Setting: multi‐centre, Canada. |
|
| Participants | Diagnosis: schizophrenia, schizophreniform disorder or schizoaffective disorder (DSM‐IV). N = 85. Age: risperidone depot mean 22.5 +/‐ 3.12 years of age; oral SGA mean 23.0 +/‐ 2.93 years of age. Sex: 65 M, 12 F. History: early onset (within the past 3 years) of psychosis. Included: no inclusion criteria stated. Excluded: no exclusion criteria stated. Consent: no details. |
|
| Interventions | 1. Risperidone depot: every 2 weeks, median dose at 18 weeks 25 mg; at 9, 12 and final visit 37.5 mg, n = 45. 2. Oral second generation antipsychotics (risperidone, olanzapine or quetiapine) (dosage not specified), n = 40. |
|
| Outcomes | Leaving the study early (discontinuation). Global state (relapse). Specific adverse events. Unable to use ‐ Mental state: PANSS; Global state: CGI‐S; Anxiety: Hamilton Anxiety scale; SAFS (unclear as to participant numbers within groups). Time to stabilisation (no SD). |
|
| Notes | Note: extractable data limited due to only available results from this study derived from conference poster. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote, "patients were randomized" ‐ no further details. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label described ‐ no further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | N = 46 (54%) discontinued the study; |
| Selective reporting (reporting bias) | Unclear risk | Conference poster, therefore results for all outcomes were not provided. |
| Other bias | High risk | Quinn AM and Mitchell D are both employees of Janssen Inc and Johnson and Johnson Stockholders; Camacho F is consultant to Janssen Inc; Chue P has received research and travel grants from Janssen, Pfizer, Eli Lilly, AstraZeneca, Sunovion, Lundbeck, GlaxoSmithKline, Bristol Mayers Squibb, Mylan, Novartis and Hoffman La Roche, Mella. |
Rosenheck 2011.
| Methods | Allocation: random. Blinding: single (rater). Duration: 2 years. Design: parallel. Setting: multi‐centre, 14 Veteran Affairs (VA) medical centres, inpatient, USA. |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM‐IV). N = 382. Age: ≥ 18 years of age. Sex: not stated. History: 64% participants reported problems with medication adherence in past (43% patient‐reported, and 60% physician‐reported); 37% participants reported active problems with alcohol or drug use (25% patient‐reported, and 36% physician‐reported). Included: at risk of hospitalisation as evidenced by current hospitalisation; hospitalisation in the previous 2 years, or increased use of services to prevent relapse. Excluded: detoxification in the previous month; past intolerance to risperidone or IM injections; current treatment with long‐acting injectable antipsychotics; oral clozapine, warfarin or a combination of those agents; serious medical conditions; unstable living arrangements; and a history of assault or suicidal behavior requiring urgent intervention. Consent: guardian or participant consent permitted; participants' decisional capacity assessed with MacArthur Competence Assessment Tool. |
|
| Interventions | 1. Risperidone depot: 25 mg to 50 mg every 2 weeks; dosage increments of 12.5 mg permitted every 4 weeks at discretion of treating physician, n = 190. 2. Oral antipsychotics: as prescribed by treating physician, n = 192*. |
|
| Outcomes | Service utilisation: hospitalisation; outpatient care. Global state: use of benzodiazepines or sedative drugs. Adverse events: death; other specific events. Not receiving allocation study medication. Leaving the study early: any reason. Unable to use ‐ Global state: CGI (follow‐up rates less than 50%). Metal State: Total PANSS score and Positive, negative and general subscale (follow‐up rates less than 50%). Quality of life: Heinrichs‐Carpenter Quality of Life Scale, Personal and Social Performance scale (PSP), Quality of well being scale (follow‐up rates less than 50%). Adverse events: BARS; Abnormal involuntary movements rating scale; Simpson and Angus rating scale for extrapyramidal side effects (follow‐up rates less than 50%). |
|
| Notes | *Concomitant psychotropic medication (anti‐anxiety, anti‐depressants, oral antipsychotics and mood stabilisers, as well as anticholinergic medications were permitted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly permuted blocks of various size, centrally conducted and stratified according to site. |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Single blind (implied). Blinded video conference assessment for some measures, but others assessed in unblinded meetings. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Of n = 382 randomised, n = 237 completed the study; including n = 75 oral antipsychotic treatment (n = 7 declined participation and did not receive intervention; n = 65 'lost to follow‐up or discontinued'; n = 3 excluded because participant did not have a Social Security number or baseline data), and n = 74 in risperidone depot group (n = 2 declined participation and did not receive intervention; n = 71 'lost to follow‐up or discontinued'; n = 1 excluded because participant did not have a Social Security number or baseline data). ITT analysis used ‐ follow‐up rates in this analysis group included n = 223 (60%) at year 1; n = 170 (46%) at 18 months; n = 107 (29%) at 24 months. Of the deaths, in the risperidone group, n = 1 died in his sleep and n = 1 took his own life; in the oral antipsychotic group, n = 1 died from chronic obstructive pulmonary disease and n = 1 from accidental drowning. |
| Selective reporting (reporting bias) | Low risk | Supplemental pages cover all outcomes. |
| Other bias | Unclear risk | Industry funded study but stated that Janssen had no involvement beyond financial and intervention drug provision. |
AIMS: Abnormal Involuntary Movement Scale BARS: Barnes Akathisia Rating Scale BMI: body mass index CGI: Clinical Global Impression CNS: central nervous system DSM‐IV: Diagnostic and Statistical Mannual version 1V EPS: extrapyramidal symptoms ESRS: Extrapyramidal Symptom Rating Scale GAF: Global Assessment of Functioning IM: intramuscular ITT: intention to‐treat LOCF: last observation carried forward N =: number of participants NMS: neuroleptic malignant syndrome PANSS: Positive And Negative Symptom Scale PP: paliperidone palmitate PSP: Personal and Social Performance Scale RIS‐LAI: risperidone long‐acting injectable SAFS: Social and Functioning Assessment Scale SAS: Simpson and Angus Rating scale SD: Standard Deviation SF36: short form 36 SGA: Second‐generation antipsychotic SOFAS: Social and Occupational Functioning Assessment Scale UKU:Udvalg for Kliniske Undersgelser side effects rating scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Agid 2010 | Allocation: not a randomised controlled trial. |
| Bouchard 2000 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral risperidone versus conventional antipsychotic drugs (not depot risperidone). |
| Canas 2010 | Allocation: not randomised; review article. |
| DeMartinis 2012a | Allocation: randomised. Participants: people with schizophrenia. Intervention: not depot risperidone. 1. PF‐02545920: 5 mg and 15 mg (titrated fixed doses. 3 mg (titrated). 2. Risperidone (oral) twice a day: 3 mg titrated. 3. Placebo. |
| Eerdekens 2002 a | Allocation: not randomised; open‐label. |
| Eerdekens 2002 b | Allocation: not randomised; review. |
| Gallhofer 1995 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone versus haloperidol or fluphenazine, not depot risperidone. |
| Geffen 2012 | Allocation: randomised. Participants: people with chronic schizophrenia. Intervention: BL‐1020: 10 mg/day, BL‐1020: 20 ‐ 30 mg/day, risperidone (oral): 2 ‐ 8 mg/day, placebo, not depot risperidone |
| Gefvert 2001 | Allocation: not a randomised controlled trial. |
| Kogeorgos 1995 | Allocation: randomised. Participants: people with schizophrenia. Interventions: sulpiride or risperidone versus chlorpromazine, trifluoperazine or haloperidol, not depot risperidone. |
| Koola 2009 | Allocation: randomised. Participants: people with schizophrenia. Intervention: long‐acting injectable risperidone or oral atypical antipsychotics. Outcomes: no useable data, only levels of insight and relapse at baseline. |
| Lindenmayer 1995 | Allocation: non‐randomised comparison of two samples taken from randomised trials. |
| Litman 2014 | Allocation: randomised.
Participants: people with schizophrenia. Intervention: AZD8529 40 mg, risperidone (oral) 4 mg, or placebo, not depot risperidone. |
| Littrell 1999 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone versus olanzapine, not depot risperidone. |
| Liu 2014f | Allocation: randomised. Participants: people with early stage schizophrenia. Intervention: minocycline or placebo, not depot risperidone. |
| Lloyd 2010 | Allocation: not randomised. |
| Macfadden 2008 | Allocation: randomised. Participants: people with schizophrenia. Interventions: 25 mg or 50 mg of risperidone depot no other comparison group (post‐hoc analysis from another study). |
| McClure 2009a | Allocation: randomised. Participnats: females with schizotypal personality disorder, not schizophrenia. |
| Pikalov 2012a | Allocation: not randomised, a review of studies. |
| Procyshyn 2010 | Allocation: non‐random, pilot study. |
| Ritchie 1999 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone versus olanzapine, not depot risperidone. |
| Robinson 2000 | Allocation: randomised. Participants: people with schizophrenia. Interventions: risperidone versus olanzapine, not depot risperidone. |
| Schmechtig 2010 | Allocation: randomised. Participants: people with subclinical levels of schizophrenia‐like symptoms (high schizotypy). Intervention: nicotine, risperidone, amisulpride or placebo, not depot risperidone. |
| Simpson 2006 | Allocation: randomised. Participants: people with schizophrenia. Interventions: two doses of depot risperidone, no control. |
| Vaughan 2000 | Allocation: randomised. Participants: people with schizophrenia. Interventions: not a drug trial, study of effect of community treatment orders. |
| Verma 2010 | Interventions: participants switched to depot risperidone with no other comparison group. |
| Weiden 2007 | Allocation: randomised to recommendation of treatment. |
| Wiffen 2010 | Allocation: non‐randomised; depot risperidone was the only intervention. |
Characteristics of studies awaiting assessment [ordered by study ID]
Nasrallah 2002.
| Methods | Randomised. |
| Participants | People with schizophrenia. |
| Interventions | Long‐acting risperidone, placebo. |
| Outcomes | Unsure |
| Notes | Both conference abstracts, reports quality of life data with depot risperidone but it may be part of one of the included studies (Kane 2002*). Authors have been contacted for more details |
Segarra 2010.
| Methods | Allocation: randomised. Blinding: unclear. Setting: not stated. Duration: not stated. |
| Participants | Diagnosis: schizophrenia, first episode N = 87 Age: not stated Sex: not stated History: not stated Included: not stated Excluded: not stated Consent: not stated |
| Interventions | 1. Risperidone depot, n = 18 2. Oral antipsychotic treatment, n = 21 |
| Outcomes | PAS Scale, neuropsychological battery, diagnostic assessment (SCID‐I) and stability at one year follow‐up, clinical assessment (PANSS; CGI; SUMD; HDRS and YMRS), functional assessment (GAF), quality of life (WHO/DAS), hospitalisations, urgency episodes and treatment compliance |
| Notes | Unable to extract any usable data from the published conference poster and abstract. Number of total included participants does not match numbers randomised. |
Turner 2000.
| Methods | Allocation: unsure |
| Participants | Diagnosis: schizophrenia |
| Interventions | 1. Risperidone depot 2. Risperidone tablets |
| Outcomes | Unsure. |
| Notes | Unable to find any details: authors have been contacted for more details |
CGI: Clinical Global Impression GAF: Global Assessment of Functioning HDRS: Hamilton Rating Scale for Depression PANSS: Positive and Negative Syndrome Scale PAS: Psychogeriatric Assessment Scale SCID‐1: Structured Clinical Interview for DSM‐IV Axis I Disorders SUMD: Scale to Assess Unawareness of Mental Disorder YMRS: Young Mania Rating Scale
Contributions of authors
Prakash Hosalli ‐ initiated the review, selected studies, extracted data, wrote the protocol and the review.
Steph Sampson ‐ checked data extraction, contributed towards review update development, economic evaluation of included studies.
Vivek Furtado ‐ economic evaluation of included studies.
John Davis ‐ helped write the protocol, independently selected and extracted data and helped write the review.
Sources of support
Internal sources
Leeds Community Mental Health NHS trust, UK.
University of Leeds, UK.
University of Illinois at Chicago, USA.
External sources
-
National Institute for Health Research (NIHR), UK.
Cochrane Collaboration Programme Grant 2011; Reference number: 10/4001/15
Declarations of interest
Prakash Hosalli ‐ none known.
Steph Sampson ‐ none known.
Vivek Furtado ‐ none known.
John Davis ‐ none known.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Bai 2006 {published data only}
- Bai YM, Chen TT, Wu B, Hung CH, Lin WK, Hu TM, et al. A comparative efficacy and safety study of long‐acting risperidone injection and risperidone oral tablets among hospitalized patients: 12‐week randomized, single‐blind study. Pharmacopsychiatry 2006;39(4):135‐41. [DOI] [PubMed] [Google Scholar]
Chue 2002 {published and unpublished data}
- Chue P, Eerdekens M, Augustyns I, Lachaux B, Molcan P, Eriksson L, et al. Efficacy and safety of long‐acting risperidone microspheres and risperidone oral tablets. Schizophrenia Research (Abstracts of the 11th Biennial Winter Workshop on Schizophrenia) 2002;3(Suppl 1):174. [Google Scholar]
- Chue P, et al. Maintenance of efficacy when switching from oral risperdal to risperdal consta RIS‐INT‐61. Promotional slides on file from Janssen‐Cliag UK Ltd 2002.
Covell 2012 {published data only}
- Covell NH, McEvoy JP, Schooler NR, Stroup TS, Jackson CT, Rojas IA, et al. Effectiveness of switching from long‐acting injectable fluphenazine or haloperidol decanoate to long‐acting injectable risperidone microspheres: an open‐label, randomized control trial. Journal of Clinical Psychiatry 2012;73(5):669‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fleischhacker 2011 {published data only}
- Fleischhacker WW, Gopal S, Lane R, Gassmann‐Mayer, Lim P, Hough D, et al. A randomized trial of paliperidone palmitate and risperidone long acting injectable in schizophrenia. International Journal of Neuropsychopharmacology 2012;15:107‐18. [DOI] [PubMed] [Google Scholar]
Gaebel 2010* {published data only (unpublished sought but not used)}
- Gaebel W, Bergmans P, Arce R, Rouillon F, Cordes J, Eriksson L, et al. Relapse prevention in schizophrenia and schizoaffective disorder with risperidone long‐acting injectable versus quetiapine:Randomized, long‐term, open‐label, clinical trial results (constatre). Proceedings of the 17th European Psychiatric Association, EPA Congress; 2009 Jan 24‐28; Lisbon Portugal 2009;24:S1020. [Google Scholar]
- Gaebel W, Schreiner A, Bergmans P, Arce R, Rouillon F, Cordes J, et al. Relapse prevention in schizophrenia and schizoaffective disorder with risperidone long‐acting injectable vs quetiapine: Results of a long‐term, open‐label, randomized clinical trial. Neuropsychopharmacology 2010;35(12):2367‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medori R, Wapenaar R, Arce R, Rouillon F, Gaebel W, Cordes J, et al. Relapse prevention and effectiveness in schizophrenia of risperidone long‐acting injectable (rlai) versus quetiapine or aripiprazole. Proceedings of the 161st Annual Meeting of the American Psychiatric Association; 2008 May 3‐8; Washington DC, USA. 2008.
- Rouillon F, Eriksson L, Burba B, Raboch J, Kaprinis G, Schreiner A. Functional recovery in schizophrenia and schizoaffective disorder: Results from the risperidone long‐acting injectable versus quetiapine relapse prevention trial (constatre). Proceedings of the 17th European Psychiatric Association, EPA Congress; 2009 Jan 24‐28; Lisbon Portugal 2009;24:S1023. [Google Scholar]
- Schreiner A, Rouillon F, Eriksson L, Burba B, Raboch J, Kaprinis G, et al. Functional improvement in schizophrenia and schizoaffective disorder: results from the risperidone long‐acting injectable versus quetiapine relapse prevention trial (ConstaTRE). Biological Psychiatry 2009:382.
- Schreiner A, Arce R, Eding E, Marques‐Teixeira J, Milanova V, Rancans E, et al. Descriptive analyses of the aripiprazole arm in the risperidone long‐acting injectable versus quetiapine relapse prevention trial (ConstaTRE). Biological Psychiatry 2009:381. [DOI] [PubMed]
- Smeraldi E, Cavallaro R, Folnegovic Smalc V, Bidzan L, Ceylan E, Schreiner A, et al. Long‐term remission in schizophrenia and schizoaffective disorder: Results from the risperidone long‐acting injectable versus quetiapine relapse prevention trial (constatre). Proceedings of the 17th European Psychiatric Association, EPA Congress; 2009 Jan 24‐28; Lisbon Portugal 2009;24:S1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeraldi E, Cavallaro R, Smalc VF, Bidzan L, Ceylan ME, Schreiner A, et al. Long‐term remission in schizophrenia and schizoaffective disorder: results from the risperidone long‐acting injectable versus quetiapine relapse prevention trial (ConstaTRE). Biological Psychiatry 2009:389. [DOI] [PMC free article] [PubMed]
- Arce Cordón R, Eding E, Marques‐Teixeira J, Milanova V, Rancans E, Schreiner A. Descriptive analyses of the aripiprazole arm in the risperidone long‐acting injectable versus quetiapine relapse prevention trial (ConstaTRE). European Archives of Psychiatry and Clinical Neuroscience March 2010;262:139‐49. [DOI] [PubMed] [Google Scholar]
Kane 2002* {published data only}
- Anonymous. Injectible, long‐acting risperidone effective. Journal of Pharmacy Technology 2001;4:157. [Google Scholar]
- Kane J, Eerdekens M, Keith S, Lesem M, Karcher K, Lindenmayer J‐P. Efficacy and safety of a novel long‐acting risperidone formulation. European Psychiatry (11th Association of European Psychiatrists Congress, 2002 4‐8th May, Stockholm, Sweden) 2002;17(Suppl 1):S193. [Google Scholar]
- Kane J, Eerdekens M, Keith S, Lesem M, Karcher K, Lindenmayer JP. Long‐acting risperidone microspheres for treatment of patients with schizophrenia. European Neuropsychopharmacology (Abstracts of the 14th Congress of the European College of Neuropsychopharmacology; 2001 Oct 13‐17; Istanbul, Turkey) 2001;11(3):291. [Google Scholar]
- Kane J, Eerdekens M, Keith SJ, Lesem M, Karcher K, Lindenmayer JP. Efficacy and safety of risperdal consta, the long‐acting injection risperidone formulation. International Journal of Neuropsychopharmacology (Abstracts of the 23rd Congress of the Collegium Internationale Neuro‐Psychopharmacologicum; 2002 Jun 23‐27; Montreal, Canada) 2002;5(Suppl 1):S188. [Google Scholar]
- Kane JM, Eerdekens M, Keith SJ, Lesem M, Karcher K, Lindenmayer J‐P. Efficacy and safety of a novel long‐acting risperidone microspheres formulation. Schizophrenia Research (Abstracts of the 11th Biennial Winter Workshop on Schizophrenia) 2002;53(3 Suppl.1):174. [Google Scholar]
- Kane JM, Eerdekens M, Keith SJ, Lesem M, Karcher K, Lindenmayer JP. Efficacy and safety of risperdal consta (TM), a long‐acting injection risperidone formulation RIS‐USA‐121. Promotional slides on file from Janssen‐Cliag UK Ltd 2002.
- Kane JM, Eerdekens M, Keith SJ, Lesem M, Karcher K, Lindenmayer JP. Long‐acting injectable risperidone: Efficacy and safety. European Neuropsychopharmacology: Journal of the European College of Neuropsychopharmacology (15th ECNP Congress; October 5‐9, 2002, Barcelona‐Spain 2002;12(Supplement 3):S325. [Google Scholar]
- Kane JM, Eerdekens M, Lindenmayer JP, Keith SJ, Lesem M, Karcher K. Long‐acting injectable risperidone: efficacy and safety of the first long‐acting atypical antipsychotic. American Journal of Psychiatry 2003;160(6):1125‐32. [DOI] [PubMed] [Google Scholar]
Keks 2007 {published data only (unpublished sought but not used)}
- Keks N, Ingham M, Khan A, Karcher K. Long‐acting injectable risperidone v. olanzapine tablets for schizophrenia or schizoaffective disorder: Randomised, controlled, open‐label study. British Journal of Psychiatry 2007;191:131‐9. [DOI] [PubMed] [Google Scholar]
- Keks N, Khan A, Augustyns I, Rabinowitz J, Ingham M. Non‐inferiority efficacy trial of risperidone long‐acting injection (RLAI) vs. olanzapine tablets (OLA). Proceedings of the 13th Biennial Winter Workshop on Schizophrenia Research; 2006 Feb 4‐10; Davos, Switzerland. 2006.
Li 2011 {published data only}
- Li H, Rui Q, Ning X, Xu H, Gu N. A comparative study of paliperidone palmitate and risperidone long‐acting injectable therapy in schizophrenia. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2011;35:1002‐8. [DOI] [PubMed] [Google Scholar]
MacFadden 2010 {published data only}
- MacFadden W, Yi‐Wen M, Haskins JT, Bossie CA, Alphs L. A prospective study comparing the long‐term effectiveness of injectable risperidone long‐acting therapy and oral aripiprazole in patients with schizophrenia. Psychiatry 2010;7(11):23‐31. [PMC free article] [PubMed] [Google Scholar]
Pandina 2011 {published data only}
- Pandina G, Lane R, Gassmann‐Mayer C, Hough D, Remmerie B, Simpson G. A randomised double blind study of flexible doses of paliperidone palmitate and risperidone long acting therapy in patients with schizophrenia. Proceedings of the 27th International College of Neuropsychopharmacology Congress; 2010 June 6‐10; Hong Kong. 2010.
- Pandina G, Lane R, Gopal S, Gassman‐Mayer C, Hough D, Remmerie B, et al. A randomized double blind study of flexible doses of paliperidone palmitate and risperidone long acting therapy in patients with schizophrenia. Biological Psychiatry 2010;67(9):77‐8. [Google Scholar]
- Pandina G, Lane R, Gopal S, Gassmann‐Mayer C, Hough D, Remmerie B, et al. A double‐blind study of paliperidone palmitate and risperidone long‐acting injectable in adults with schizophrenia. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2011;35:218‐26. [DOI] [PubMed] [Google Scholar]
Quinn 2012* {published data only}
- Quinn A, Camacho F, Mitchell D, Chue P, Malla A. Open‐label randomized exploratory investigation of Risperdal* Consta* and oral antipsychotics treatment of early psychosis. Autumn Conference of the International Society for CNS Clinical Trials and Methodology, October 1‐3. Marina Del Rey, California, 2012.
Rosenheck 2011 {published data only}
- Barnett PG, Scott JY, Krystal JH, Rosenheck RA. Cost and cost‐effectiveness in a randomized trial of long‐acting risperidone for schizophrenia. Journal of Clinical Psychiatry 2012;73:696‐702. [DOI] [PubMed] [Google Scholar]
- Rosenheck RA, Krystal JH, Lew R, Barnett PG, Fiore L, Valley D, et al. Long‐ acting risperidone and oral antipsychotics in unstable schizophrenia. New England Journal of Medicine March 3, 2011;364(9):842‐51. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Agid 2010 {published data only}
- Agid O, Foussias G, Remington G. Long acting injectable antipsychotics in the treatment of schizophrenia: Their role in relapse prevention. Expert Opinion on Pharmacotherapy 2010;11(14):2301‐17. [DOI] [PubMed] [Google Scholar]
Bouchard 2000 {published data only}
- Bouchard RH, Merette C, Pourcher E, Demers MF, Villeneuve J, Roy Gagnon MH, et al. The Quebec Schizophrenia Study Group. Longitudinal comparative study of risperidone and conventional neuroleptics for treating patients with schizophrenia. Journal of Clinical Psychopharmacology 2000;20(3):295‐304. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Canas 2010 {published data only}
- Canas F, Moller HJ. Long acting atypical injectable antipsychotics in the treatment of schizophrenia: Saftey and tolerability review. Expert Opinion on Drug Saftey 2010;9(5):683‐97. [DOI] [PubMed] [Google Scholar]
DeMartinis 2012a {published data only}
- DeMartinis N, Banerjee A, Kumar V, Boyer S, Schmidt C, Arroyo S. Results of a phase 2a proof‐of‐concept trial with a PDE10A inhibitor in the treatment of acute exacerbation of schizophrenia. Schizophrenia Research 2012;136:S262. [Google Scholar]
Eerdekens 2002 a {published data only}
- Chue P, Devos E, Duchesne I, Leal A, Mehnert A. Hospitalization rates in patients during long‐term treatment with long‐acting risperidone injection. Poster presented at the XXIII CINP Congress, Montreal, Canada. June 23‐ 27, 2002.
- Eerdekens M, Fleischhacker WW, Xie Y, Gefvert O. Long‐term safety of long‐acting risperidone microspheres. Schizophrenia Research (Abstracts of the 11th Biennial Winter Workshop on Schizophrenia) 2002;3(Suppl 1):174. [Google Scholar]
- Fleischhacker WW, Eerdekens M, Xie Y, Beauclair L, Sauret H, Chrzanowski W, et al. Long‐term saftey and efficacy of risperdal consta(TM) a long‐acting injection formulation of risperidone. ACNP, Hawaii, USA, December 2001 (Promotional slides on file from Janssen‐Cliag UK Ltd) 2002.
Eerdekens 2002 b {published data only}
- Eerdekens M. Treatment delivery: a hope for the future. Nordic Journal of Psychiatry (Abstracts of The Scandinavian College of Neuro‐Psychopharmacology Annual Conference; 2002 10‐13 Apr; Juan‐Les‐Pins, France) 2002;56(2):1. [Google Scholar]
Gallhofer 1995 {published data only}
Geffen 2012 {published data only}
- Geffen Y, Keefe R, Rabinowitz J, Anand R, Davidson M. Bl‐1020, a new ‐aminobutyric acid‐enhanced antipsychotic: Results of 6‐week, randomized, double‐blind, controlled, efficacy and safety study. Journal of Clinical Psychiatry 2012;73(9):e1168‐e74. [DOI] [PubMed] [Google Scholar]
Gefvert 2001 {published data only}
- Gefvert O, Nyberg S, Persson P, Helklin L, Bjorner A. Pharmacokinetics, D2 receptor occupancy, and clinical effects of a long‐acting injectable formulation of risperidone in patients with schizophrenia. Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; LA, USA. Marathon Multimedia, 2001.
Kogeorgos 1995 {published data only}
- Kogeorgos J, Kanellos P, Michalakeas A, Ioannidis J. Sulpiride and risperidone vs. "classical neuroleptics" in schizophrenia: a follow‐up study. 8th Congress of the European College of Neuropsychopharmacology; 1995 Sep 30 ‐ Oct 4; Venice, Italy. 1995.
Koola 2009 {published data only}
- Koola MM, Bustillo J, Lauriello J. Insight and its to baseline characteristics of schizophrenia patients randomized to long acting injectable risperidone or oral atypical antipsychotics: results from the proactive study. Proceedings of the 12th International Congress on Schizophrenia Research; 2009 Mar 28‐ Apr 1; San Diego, CA. Oxford Univ Press, 2009.
Lindenmayer 1995 {published data only}
- Lindenmayer J, Grochowski S, Hyman RB. Five factor model of schizophrenia ‐ replication across samples. Schizophrenia Research 1995;14(3):229‐34. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Litman 2014 {published data only}
- Litman R, Smith M, Doherty J, Cross A, Raines S, Zukin S. AZD8529, a positive allosteric modulator at the MGGLUR2 receptor, does not improve symptoms in schizophrenia: A proof of principle study. European Neuropsychopharmacology 2014;24:S508‐9. [DOI] [PubMed] [Google Scholar]
- Litman RE, Smith MA, Doherty J, Cross A, Raines S, Zukin S. AZD8529, a positive allosteric modulator at the mGLuR2 receptor, does not improve symptoms in schizophrenia: A proof of principle study. Schizophrenia Research 2014;153:S176. [DOI] [PubMed] [Google Scholar]
Littrell 1999 {published data only}
- Littrell KH. Patients switched from depot antipsychotics to oral risperidone or olanzapine: an open‐label randomized trial. 152nd Annual Meeting of the American Psychiatric Association; 1999 May 15‐20; Washington, USA. 1999. [MEDLINE: ]
Liu 2014f {published data only}
- 刘芳, 张炳奎, 谢丽琴, 郑英君, 欧建军, 吴仁容, et al. 米诺环素辅助治疗早期精神分裂症阴性症状的双盲、随机、对照研究. 长治医学院学报 2014; Vol. 27, issue 1:30‐2.
Lloyd 2010 {published data only}
- Lloyd K, Latif MA, Simpson S, Shrestha KL. Switching stable patients with schizophrenia from depot and oral antipsychotics to long acting injectable risperidone: Efficacy, quality of life and functional outcome. Human Psychopharmacology 2010;25(3):243‐52. [DOI] [PubMed] [Google Scholar]
Macfadden 2008 {published data only}
- MacFadden W, Bossie CA, Turkoz I, Haskins JT. Risperidone long acting therapy in stable patients with recently diagnosed schizophrenia. International Clinical Psychopharmacology 2010;25(2):75‐82. [DOI] [PubMed] [Google Scholar]
- Macfadden W, Bossie C, Turkoz I, Diekamp B, Ibach B, Haskins JT. Effect of long acting injectable risperidone on clinical outcomes in recently diagnosed stable schizophrenia patients. Proceedings of the XXVI Collegium Internationale Neuro Psychopharmacologicum congress; 2008 Jul 13‐17; Munich, Germany. 2008.
- Macfadden W, Bossie C, Turkoz I, Dorson P, Haskins T. Effect of long acting injectable risperidone on clinical outcomes in stable schizophrenia patients with early illness. Proceedings of the 161st Annual Meeting of the American Psychiatric Association; 2008 May 3‐8; Washington DC, USA 2008.
McClure 2009a {published data only}
- McClure MM, Koenigsberg HW, Reynolds D, Goodman M, New A, Trestman R, et al. The effects of risperidone on the cognitive performance of individuals with schizotypal personality disorder. Journal of Clinical Psychopharmacology 2009;29(4):396‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pikalov 2012a {published data only}
- Pikalov A, Cucchiaro J, Ogasa M, Silva R, Hsu J, Xu J, et al. Effect of lurasidone on weight and metabolic parameters: A comprehensive database analysis. Schizophrenia Research 2012;136:S278. [Google Scholar]
Procyshyn 2010 {published data only}
- Procyshyn RM, Barr AM, Flynn S, Schenk C, Ganesan S, Honer WG. Long acting injectable risperidone in treatment refractory patients: A 14 week open label pilot study. Schizophrenia Research 2010;123(2‐3):273‐5. [DOI] [PubMed] [Google Scholar]
Ritchie 1999 {published data only}
- Ritchie C, Ames D, Chiu E, O'Connor D, Hall K, Hassett A. Schizophrenia cohort study of olanzapine v risperidone in the elderly (score): analysis of conversion period. International Psychogeriatrics (Abstracts of the 9th Congress of the International Psychogeriatric Association, "Challenges for the New Millennium: Professional, Cultural and Regional Diversity"; 1999 Aug 15‐20; Vancouver, Canada) 1999;11(Suppl 1):157‐8. [Google Scholar]
Robinson 2000 {published data only}
- Robinson G, Wheeler A, Byrd J, Visser S. Longer‐term effects of switching from typical to atypical antipsychotics in patients with stable schizophrenia. Journal of the European College of Neuropsychopharmacology 2000;10(Suppl 3):S291. [Google Scholar]
Schmechtig 2010 {published data only}
- Schmechtig A, Dourish C, Craig K, Dawson GR, Williams S, Deakin W, et al. Effects of risperidone, amisulpride and nicotine on eye movement control and their modulation by high schizotypy. Pharmacopsychiatry 2011;21:A99. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Schmechtig A, Lees J, Dawson G, Dourish C, Craig K, Deakin B, et al. Effects of high schizotypy on control of eye movements: Modulation by antipsychotic drugs and nicotine. Proceedings of the 49th Annual Meeting of the Americam College of Neuropsychopharmacology; 2010 Dec 5‐9; Miami, Florida. 2010.
Simpson 2006 {published data only}
- Simpson GM, Mahmoud RA, Lasser RA, Kujawa M, Bossie CA, Turkoz I, et al. A 1‐year double‐blind study of 2 doses of long‐acting risperidone in stable patients with schizophrenia or schizoaffective disorder. Journal of Clinical Psychiatry 2006;67:1194‐203. [DOI] [PubMed] [Google Scholar]
Vaughan 2000 {published data only}
- Vaughan K, McConaghy N, Wolf C, Myhr C, Black T. Community Treatment Orders: Relationship to clinical care, medication compliance, behavioural disturbance and readmission. Australian and New Zealand Journal of Psychiatry 2000;5:801‐8. [DOI] [PubMed] [Google Scholar]
Verma 2010 {published data only}
- Verma S, Subramaniam M, Abdin E, Sim K, Su A, Lee N, et al. Saftey and efficacy of long acting injectable risperidone in patients with schizophrenia spectrum disorders: A 6 month open label trial in Asian patients. Human Psychopharmacology 2010;25(3):230‐5. [DOI] [PubMed] [Google Scholar]
Weiden 2007 {published data only}
- Weiden PJ, Goldfinger SM, Hindi A, Sunakawa A, Schooler NR. Acceptance of a recommendation of a long acting antipsychotic route in first episode schizophrenia: initial findings of a prospective randomized study. Proccedings of the 160th Annual Meeting of the American Psychiatric Association; 2007 May 19‐24; San Diego, CA. 2007.
- Weiden PJ, Schooler NR, Weedon JC, Elmouchtari A, Sunakawa, Goldfinger SM. A randomized controlled trial of long acting injectable risperidone vs continuation on oral atypical antipsychotics for first episode schizophrenia patients: initial adherence outcome. Journal of Clinical Psychiatry 2009;70(10):1397‐406. [DOI] [PubMed] [Google Scholar]
- Weiden PJ, Schooler NR, Weedon JC, Elmouchtari A, Sunakawa‐McMillan A. Maintenance treatment with long‐acting injectable risperidone in first‐episode schizophrenia: A randomized effectiveness study. Journal of Clinical Psychiatry 2012;73(9):1224‐33. [DOI] [PubMed] [Google Scholar]
Wiffen 2010 {published data only}
- Wiffen BDR, Rabinowitz J, Fleischhacker WW, David AS. Insight : Demographic differences and associations with one‐year outcome in schizophrenia and schizoaffective disorder. Clinical Schizophrenia and Related Psychoses 2010;4(3):169‐75. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Nasrallah 2002 {published data only}
- Nasrallah H, Duchesne I, Mehnert A, Janagap C. Long‐acting risperidone improves quality of life. Proceedings of the 12th World Congress of Psychiatry; 2002 Aug 24‐29; Yokohama, Japan. 2002.
- Nasrallah H, Duchesne I, Mehnert A, Janagap C. Long‐acting risperidone injection improves quality of life. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):S189. [Google Scholar]
- Nasrallah H, Duchesne I, Mehnert A, Janagap C. Long‐acting, injectable risperidone ‐ the first long‐acting, atypical antipsychotic ‐ improves quality of life. European Neuropsychopharmacology 2002;12(Suppl 3):S282. [Google Scholar]
- Nasrallah HA, Duchesne I, Mehnert A, Janagap C, Eerdekens M. Correction. Journal of Clinical Psychological Medicine [临床精神医学杂志] 2004;65(8):1150. [Google Scholar]
- Nasrallah HA, Duchesne I, Mehnert A, Janagap C, Eerdekens M. Health‐related quality of life in patients with schizophrenia during treatment with long‐acting, injectable risperidone. Journal of Clinical Psychiatry 2004;65(4):531‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Segarra 2010 {published data only}
- Segarra R, Ojeda N, Garcia J, Pena J, Bravo E, Eguiluz JI. Risperidone injectable long acting treatment vs other oral antipsychotics in first episode psychosis: One year longitudinal study. Schizophrenia Research. 2010; Vol. 117, issue 2‐3:495.
Turner 2000 {published data only}
- Turner T. Risperidone depot v risperidone tablets: a non‐inferiority efficacy trial in subjects with schizophrenia. National Research Register (http://www.update‐software.com/National/) 2000.
Additional references
Adams 2001
- Adams CE, Fenton MK, Quraishi S, David AS. Systematic meta‐review of depot antipsychotic drugs for people with schizophrenia. British Journal of Psychiatry 2001;179:290‐9. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and sStatistical Manual of Mental Disorders : DSM‐IV‐TR. Washington, DC: American Psychiatric Association, 2000. [Google Scholar]
Barnes 1989
- Barnes TR. A rating scale for drug‐induced akathisia. British Journal of Psychiatry 1989;154:672‐6. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315(7108):600. [DOI] [PMC free article] [PubMed] [Google Scholar]
BNF 2012
- BMJ Group and Pharmaceautical Press. British National Formulary No. 63. Antipsychotic drugs. BNF 2012.
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
CEA
- Cost‐Effectiveness Analysis Registry (CEA). https://research.tufts‐nemc.org/cear4/ accessed 11/09/13.
Chouinard 1980
- Chouinard G, Ross‐Chouinard A, Annable L. Extrapyramidal symptom rating scale. Canadian Journal of Neurological Science 1980;7:233. [Google Scholar]
Clarke 2002
- Clarke M, Oxman AD. Cochrane Collaboration Handbook. Cochrane Database of Systematic Reviews. Oxford: Update Software, 2002, issue 1.
Csernansky 2002
- Csernansky JG, Mahmood R, Brenner R, Risperidone‐USA‐79 Study Group. A comparison of risperidone and haloperidol for the prevention of relapse in patients with schizophrenia. New England Journal of Medicine 2002;346:16‐22. [DOI] [PubMed] [Google Scholar]
Davies 2007
- Davies LM, Lewis S, Jones PB, Barnes TR, Gaughran F, Hayhurst K, et al. Cost‐effectiveness of first‐ v. second‐generation antipsychotic drugs: results from a randomised controlled trial in schizophrenia responding poorly to previous therapy. British Journal of Psychiatry 2007;191:14‐22. [DOI] [PubMed] [Google Scholar]
Davis 1977
- Davis JM, Casper R. Antipsychotic drugs: clinical pharmacology and therapeutic use. Drugs 1977;14(4):260‐82. [DOI] [PubMed] [Google Scholar]
Davis 1986
- Davis JM, Andriukaitis S. The natural course of schizophrenia and effective maintenance drug treatment. Journal of Clinical Psychopharmacology 1986;6(1 Suppl):2S‐10S. [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Fenton 1997
- Fenton WS, Blyler CR, Heinssen RK. Determinants of medication compliance in schizophrenia: empirical and clinical findings. Schizophrenia Bulletin 1997;23(4):637‐51. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Geddes 2000
- Geddes J, Freemantle N, Harrison P, Bebbington P. Atypical antipsychotics in the treatment of schizophrenia: systematic overview and meta‐regression analysis. BMJ 2000;321(7273):1371‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149(9):876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. Early clinical drug evaluation (ECDEU) assessment manual for psychopharmacology. ECDEU Assessment Manual for Psychopharmacology. Washington, DC: National Institute of Mental Health, 1976:217‐22.
HEED
- Health Economic Evaluation Database (HEED). Online ISBN: 9780470510933. [DOI: 10.1002/9780470510933] [DOI]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Hunter 2003
- Hunter R, Kennedy E, Song F, Gadon L, Irving CB. Risperidone versus typical antipsychotic medication for schizophrenia. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000440] [DOI] [PMC free article] [PubMed] [Google Scholar]
Janssen 1988
- Janssen PA, Niemegeers CJ, Awouters F, Schellekens KH, Megens AA, Meert TF. Pharmacology of risperidone (R 64 766), a new antipsychotic with serotonin‐S2 and dopamine‐D2 antagonistic properties. Journal of Pharmacology and Experimental Therapeutics 1988;244(2):685‐93. [PubMed] [Google Scholar]
Jayaram 2006
- Jayaram MB, Hosalli P, Stroup TS. Risperidone versus olanzapine for schizophrenia. Cochrane Database of Systematic Reviews 2006, Issue 2. [DOI: 10.1002/14651858.CD005237.pub2] [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13:261‐76. [DOI] [PubMed] [Google Scholar]
Kirkpatrick 1989
- Kirkpatrick B, Buchanan RW, McKenny PD, Alphs LD, Carpenter WT. The schedule for the deficit syndrome: An instrument for research in schizophrenia. Psychiatry Research 1989;30(2):119‐23. [DOI] [PubMed] [Google Scholar]
Leucht 1999
- Leucht S, Pitschel‐Walz G, Abraham D, Kissling W. Efficacy and extrapyramidal side‐effects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta‐analysis of randomized controlled trials. Schizophrenia Research 1999;35(1):51‐68. [DOI] [PubMed] [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2009
- Li C, Xia J, Wang J. Risperidone dose for schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD007474.pub2] [DOI] [PubMed] [Google Scholar]
Lingjaerd 1987
- Lingjaerd O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale: A new comprehensive rating scale for psychotropic drugs and a cross‐sectional study of side effects in neuroleptic‐treated patients. Acta Psychiatrica Scandinavica 1987;76(s334):1‐100. [DOI] [PubMed] [Google Scholar]
Mangalore 2007
- Mangalore R, Knapp M. Cost of schizophrenia in England. Journal of Mental Health Policy and Economics 2007;10(1):23‐41. [PubMed] [Google Scholar]
Marder 1994
- Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. American Journal of Psychiatry 1994;151(6):825‐35. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
McGahuey 2000
- McGahuey CA, Gelenberg AJ, Laukes CA, Moreno FA, Delgado PL, McKnight KM, et al. The Arizona Sexual Experience Scale (ASEX): reliability and validity. Journal of Sex and Marital Therapy 2000;26(1):25‐40. [DOI] [PubMed] [Google Scholar]
McGrath 2008
- McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiologic Reviews 2008;30(1):67‐76. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D, CONSORT Group. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. 2001 JAMA;285(15):1987‐91. [DOI] [PubMed] [Google Scholar]
Munetz 1988
- Munetz MR, Benjamin S. How to examine patients using the Abnormal Involuntary Movement Scale. Hospital Community Psychiatry 1988;39(11):1172‐7. [DOI] [PubMed] [Google Scholar]
Nafees 2012
- Nafees B, Hanswijck de Jonge P, Stull D, Pascoe K, Price M, Clarke A, et al. Reliability and validity of the Personal and Social Performance scale in patients with schizophrenia. Schizophrenia Research 2012;140(1‐3):71‐6. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Ramstack 2003
- Ramstack JM, Grandolfi GP, Mannaert E, D'Hoore P, Lasser RA. Risperdal CONSTA™: Prolonged‐release injectable delivery of risperidone using Medisorb® Microsphere Technology. Schizophrenia 2003;60:314. [Google Scholar]
Sarfati 1999
- Sarfati Y, Olivier V, Bouhassira M. New antipsychotics in the treatment of schizophrenia. A European survey. Encephale 1999;25(6):658‐66. [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Small 1997
- Small JG, Hirsch SR, Arvanitis LA, Miller BG, Link CG, Seroquel Study Group. Quetiapine in patients with schizophrenia. A high‐ and low‐dose double‐ blind comparison with placebo. Archives of General Psychiatry 1997;54(6):549‐57. [DOI] [PubMed] [Google Scholar]
Tollefson 1997
- Tollefson GD, Beasley CM Jr, Tamura RN, Tran PV, Potvin JH. Blind, controlled, long‐term study of the comparative incidence of treatment‐emergent tardive dyskinesia with olanzapine or haloperidol. American Journal of Psychiatry 1997;154(9):1248‐54. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). I. Conceptual framework and item selection.. Medical Care 1992;30(6):473‐83. [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
Young 1986
- Young JL, Zonana HV, Shepler L. Medication noncompliance in schizophrenia: codification and update. Bulletin of the American Academy of Psychiatry and the Law 1986;14(2):105‐22. [PubMed] [Google Scholar]
References to other published versions of this review
Hosalli 2003
- Hosalli P, Davis JM. Depot risperidone for schizophrenia. Cochrane Database of Systematic Reviews 2003, Issue 4. [DOI: 10.1002/14651858.CD004161; PUBMED: 14584007] [DOI] [PubMed] [Google Scholar]