

Summary
Vector competence (VC) refers to the efficiency of pathogen transmission by vectors. Each step in the infection of a mosquito vector constitutes a barrier to transmission that may impose bottlenecks on virus populations. West Nile virus (WNV) is maintained by multiple mosquito species with varying VC. However, the extent to which bottlenecks and VC are linked is poorly understood. Similarly, quantitative analyses of mosquito-imposed bottlenecks on virus populations are limited. We used molecularly barcoded WNV to quantify tissue-associated population bottlenecks in three variably competent WNV vectors. Our results confirm strong population bottlenecks during mosquito infection that are capable of dramatically reshaping virus population structure in a non-selective manner. In addition, we found that mosquitoes with differing VC uniquely shape WNV population structure: highly competent vectors are more likely to contribute to the maintenance of rare viral genotypes. These findings have important implications for arbovirus emergence and evolution.
Subject areas: Genetics, Virology
Graphical abstract
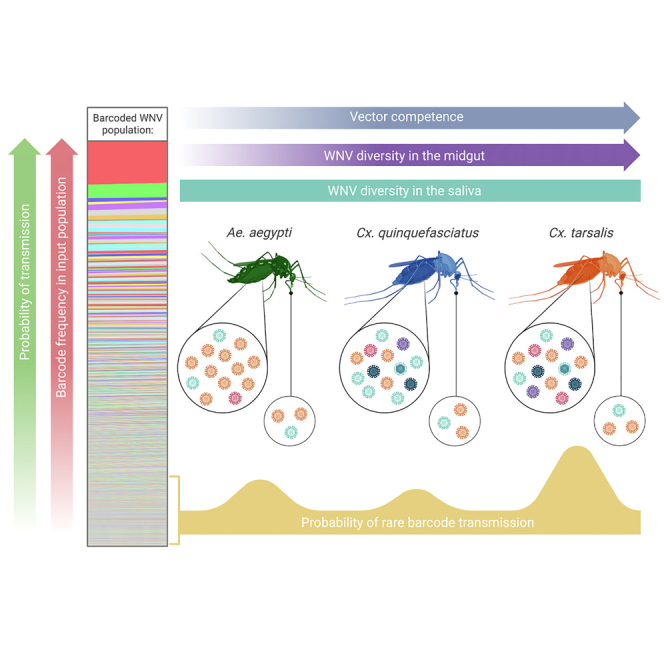
Highlights
-
•
WNV diversity in the midgut increases with increasing vector competence
-
•
Vector competence does not impact the diversity of transmissible WNV populations
-
•
Highly competent WNV vectors are more likely to transmit rare virus variants
Genetics; Virology
Introduction
West Nile virus (WNV, Flaviridae: flavivirus), an arthropod-borne virus (arbovirus), is the leading cause of arboviral disease in the contiguous United States.1 Since the introduction of WNV to the Western Hemisphere in 1999, it has spread rapidly and undergone dramatic population-level genetic changes as it adapted to local transmission cycles.2,3,4,5,6 Due to error-prone replication, RNA viruses such as WNV exist as genetically complex mutant swarms.7,8,9,10 This confers significant adaptive plasticity to these viruses as they encounter different selective environments within vertebrate and invertebrate hosts, enter and spread within new environments, and adapt to new environmental conditions.4,5,8,11,12,13 Previous work demonstrated that mosquito infection dramatically shapes WNV populations via selective pressures and single nucleotide polymorphism (SNP) accumulation in the viral genome, indicating that mosquito infection has a significant impact on the evolution of WNV.4,5,10,14,15 However, stochastic processes such as genetic drift, that are not generally selective, also have a major impact on RNA virus evolution within individual hosts.8,16,17
During infection of the mosquito vector, WNV sequentially infects the mosquito midgut and salivary glands, and ultimately is released into the saliva.8,13,18,19,20,21 Infection of and escape from the midgut and salivary glands constitute the main barriers to virus transmission within an individual mosquito.8,15,22 These barriers also impose stochastic reductions on the richness and complexity of the virus population (i.e., bottlenecks), which can negate changes that occur selectively by randomly selecting variants for onward transmission.8,15,19,20 Several studies have clearly demonstrated that tissue-level bottlenecks play an important role in shaping virus population structure in mosquitoes.18,19,20 Moreover, it has been established that mechanisms of evolution – such as bottlenecks – that alter the frequency of virus variants within the mosquito vector, are determinants of emergence.23
WNV is transmitted in nature by mosquito species of varying vector competence (VC) – the capacity to become infected with and transmit a virus. VC clearly influences the rate of virus evolution during mosquito infection.15,24,25,26 While it is known that bottlenecks play an important role in virus evolution, it remains unknown whether VC impacts these stochastic mechanisms of intrahost evolution.15,18,20,27 Additionally, quantitative characterization of bottlenecks and their impact on virus populations within the mosquito are scarce.8,16 Therefore, we determined the impact of VC on the severity of tissue-associated bottlenecks in three variably competent vectors of WNV.
We utilized a highly diverse, molecularly barcoded WNV (bcWNV) to mimic an RNA virus mutant swarm, allowing us to determine the extent to which bottlenecks and VC are related.28,29 Using bcWNV, we quantitatively measured population bottlenecks in three WNV vectors with varying VC; Culex tarsalis, Culex quinquefasciatus, and Aedes aegypti. Cx. tarsalis is the most competent WNV vector of the three species, followed by Cx. quinquefasciatus and Ae. aegypti.15,24 We examined alterations in virus population structure by measuring changes to barcode diversity in midguts, salivary glands and expectorated saliva from each of these species. We quantified how bottlenecks associated with infection, dissemination, and transmission influence the overall diversity of a WNV swarm, which gave insight into the impact of VC on the stringency of bottlenecks that occur during mosquito infection. In addition to providing a comprehensive and quantitative analysis of WNV population dynamics in three relevant mosquito species, we determined the probability of infection and transmission for barcodes of varying frequencies in the input population – demonstrating the effect initial proportion in the population has on successful virus transmission, and highlighting the stochastic nature of bottlenecks and their influence on virus transmission potential.
Results
Molecular characterization of barcode diversity
We utilized a virus barcoding method that has been employed previously with Zika virus.18,30 Barcoded-WNV contains degenerate nucleotides at the third coding position of 11 consecutive codons in the NS4b region of the genome.28 These changes are synonymous and do not alter the NS4b amino acid sequence (Figure 1A). We deep-sequenced the barcode region of our bcWNV stock and confirmed the barcodes contained varied proportions of A, T, C and G only at the third nucleotide positions, with no alterations to the barcode region amino acid sequence (Figure 1A). We then performed a bootstrap rarefaction to examine barcode richness as a function of read number (presented as subsample size) and found that sample richness increased as subsample size increased, with no notable flattening of the curve for any sample volume (50μl, 100μl, 200μl) (Figure 1B). This demonstrated that our sampling volume of 50μl and target coverage of 1.2 million paired-end reads per sample captured a subset of the most abundant barcodes, but not the full richness of the barcode population in each sample. We then evaluated barcode abundance and diversity of the bcWNV stock (Figures 1C and 1D). When visualizing individual barcodes, dominant barcodes (those comprising ∼10% of total barcodes) were consistently identified across all sample volumes, whereas low frequency barcodes (those detected only once) were variably captured (Figure 1C). The percent of barcodes occurring at certain abundances was similar across sample volumes, with barcodes occurring only once comprising ∼70% of the population, whereas those occurring 100+ times comprised only ∼0.2% (Figure 1D). To maximize the richness of our input population for downstream analyses, we merged the three stock samples by barcode (Figures 1C and 1D) and, for barcodes that overlapped between samples, retained the highest detected abundances. This merged input population was used as our input reference for all proceeding analyses.
Figure 1.

Confirming the diversity of the molecular barcode
(A) The barcode, located in the NS4B region of the WNV genome, contains degenerate nucleotides inserted at every third codon position for 11 consecutive codons. Degenerate nucleotides produce synonymous changes and do not alter the encoded amino acid. (created with weblogo.berkeley.edu) (B) Rarefaction analysis of sequenced bcWNV stock samples estimates barcode richness as a function of sequencing depth. RNA was extracted from 50, 100, and 200ul of virus, deep sequenced and (C) barcode frequencies were determined from abundance values and ranked from most to least frequent across three stock samples and a merged population containing unique barcodes from all three stock populations (D) and number and frequency of detected barcodes was determined.
Barcoded WNV replication in cells and mosquitoes
To determine whether introduction of the barcode affects virus replication, we infected mammalian (Vero) and mosquito (CT, Cx. tarsalis derived) cells with bcWNV and the parental recombinant wild type WNV (rWT-WNV) at two multiplicities of infection (MOI) and measured extracellular viral RNA (vRNA) (Figure 2A). vRNA levels were not significantly different between the barcoded and WT viruses at both MOIs, in both cell types, on all days post-infection (Figure 2A). We next exposed Cx. tarsalis, Cx. quinquefasciatus, and Ae. aegypti to an infectious bloodmeal containing bcWNV, collected midguts, salivary glands, and saliva on multiple days post-infection and measured vRNA. All species became infected with bcWNV. However, midgut infection, dissemination (as measured by infection of salivary glands), and transmission (shedding into the saliva) rates were variable across species (Figure 2B; Table S1). At 8 days post-infection, Cx. tarsalis consistently had the highest infection rates of salivary glands, and saliva, followed by Ae. aegypti and Cx. quinquefasciatus (Figure 2B). By 12 days post-infection Cx. quinquefasciatus salivary gland infection rates overtook Ae. aegypti, however, Ae. aegypti maintained slightly higher transmission rates (Figure 2B). We examined vRNA levels in all samples and found that, on average, Cx. tarsalis attained the highest peak vRNA levels in the midgut and salivary glands (Figure 2C). While vRNA peak days and levels varied by species, lower competence vectors (Cx. quinquefasciatus and Ae. aegypti), despite having lower transmission rates (Figure 2B), did not have significantly lower levels of vRNA in the saliva compared to the higher competence species (Cx. tarsalis) (Figure 2C).
Figure 2.

Barcoded WNV efficiently infects, and replicates in cells and mosquitoes
(A) bcWNV (red) replication was evaluated relative to rWT-WNV (black) at MOIs of 0.1 (dashed line) and 1.0 (solid line) in mammalian (Vero) and insect cells (CT, Culex tarsalis-derived embryonic cell line) (n = 3, error bars denote mean ± SD). vRNA copies = WNV envelope copies per reaction. Cx. tarsalis (orange) and quinquefasciatus (blue), and Ae. aegypti (green) mosquitoes were infected with bcWNV. At the indicated days, saliva, salivary glands and midguts were collected from each species.
(B) Infection rates of midguts, salivary glands, and saliva were determined for all species at all timepoints.
(C) Levels of viral RNA were measured via qRT-PCR (n = 35–40), solid lines denote mean. vRNA copies = WNV envelope copies per sample. Within each time point, statistical significance between mosquito species was determined by two-way ANOVA with Tukey’s multiple comparisons test, ∗ = p < 0.05, ∗∗ = p < 0.005, ∗∗∗ = p < 0.0005, ∗∗∗∗ = p < 0.0001. Only significant relationships are shown.
Mosquito bottlenecks reduce richness and diversity of bcWNV populations
We next examined differences in barcode richness (number of unique barcodes) and complexity (Shannon’s index) within 10 individual mosquitoes per species at 4, 8, and 12 days post-infection using next-generation sequencing (Figures 3A–3C and S1). At 8 days post-infection, bcWNV population richness and complexity decreased significantly between the midgut and salivary glands (p < 0.005), and midgut and saliva (p < 0.001) in Cx. tarsalis (highest VC) (Figure 3A). A decrease in richness and complexity was seen between the midgut and salivary glands, (p = 0.002) and midgut and saliva (p < 0.05) in Cx. quinquefasciatus (Figure 3B). In Ae. aegypti the only significant decreases in richness and complexity were between the midgut and saliva (p = 0.02, p = 0.01) (Figure 3C). No significant reductions in richness nor complexity were observed between the salivary glands and saliva for any species (Figures 3A–3C). We observed similar trends at day 12 post-infection, and no significant reductions in population diversity in any species at day 4 post-infection (Figure S1).
Figure 3.

Quantifying the impact of intrahost bottlenecks on population richness and complexity
(A–C) 8-day post-infection bcWNV population richness and complexity in midgut, salivary gland, and saliva samples from Cx. tarsalis (A), Cx. quinquefasciatus (B), and Ae. aegypti (C) mosquitoes. Lines connecting points indicate that samples were collected from the same mosquito. Diversity indices were generated by applying the Shannon function from the QSutils package in R to barcode abundance vectors. Dashed line represents 0.
(D) Percent of preceding unique barcode population retained after each bottleneck in each species (all timepoints included). Solid lines denote mean. (A-D) Statistical significance determined using Kruskal-Wallis with Dunn’s multiple comparisons test, ∗ = p < 0.05, ∗∗ = p < 0.005, ∗∗∗ = p < 0.0005, ∗∗∗∗ = p < 0.0001. Only significant comparisons shown.
(E) Pearson’s correlation between vRNA and total barcodes, and (F) unique and total barcodes in all samples from each species. Pearson’s r values and significance values are displayed on plots.
We then examined what percent of population richness is retained after each bottleneck at all timepoints combined (Figure 3D). Culex species retained significantly more population richness (unique barcodes) between the input and midgut populations than Ae. aegypti (p < 0.0001), with Cx. tarsalis and Cx. quinquefasciatus retaining 10.0% and 4.4% respectively, compared to only 0.8% in Ae. aegypti (Figure 3D). Ae. aegypti retained significantly more population richness between the midgut and salivary gland populations than both Culex species (p < 0.005), and between the salivary gland and saliva populations than Cx. quinquefasciatus (p = 0.03) (Figure 3D). Cx. tarsalis sustained the greatest loss of richness between the midgut and salivary glands (97.5%), while Cx. quinquefasciatus and Ae. aegypti sustained the greatest loss of richness between the input and midgut populations (95.6% and 99.2% respectively). To further examine barcode dynamics and to ensure differences in richness/complexity were not an artifact of total barcodes detected, we compared vRNA, total barcodes and unique barcodes for all samples from each species (Figures 3E and 3F). We saw a significant, positive linear correlation between vRNA and total barcodes in all samples from all species, demonstrating that vRNA and total barcodes are analogous (Figure 3E). However, while we observed a significant, positive linear correlation between unique and total barcodes in all Cx. tarsalis and Cx. quinquefasciatus samples, (r > 0.7, p < 0.0001) there was no significant linear correlation between unique and total barcodes in Ae. aegypti samples (r = 0.13, p = 0.32) (Figure 3F).
Visual representation of barcode population dynamics
Visual inspection of barcode population dynamics in individual mosquitoes that expectorated detectable barcodes reinforced our quantitative findings in Figure 3. We saw decreases in barcode population complexity at days 4, 8, and 12 post-infection in individual mosquitoes for each species (Figure S2), with two representative plots for each species shown (Figure 4A). Midgut infection results in mosquito species-independent population bottlenecks - barcode populations in the midgut are always less complex than the input population (Figure 4A). Following this initial bottleneck, the barcode population is rapidly reduced to a small subset of midgut barcodes as the population infects the salivary glands, and sheds into the saliva (Figure 4A). Importantly, high barcode abundance in the input or midgut does not guarantee progression to the salivary glands and saliva (Figure 4A).
Figure 4.

Visualizing barcode dynamics across bottlenecks
(A) bcWNV barcode dynamics during mosquito infection in individual Cx. tarsalis, Cx. quinquefasciatus, and Ae. aegypti mosquitoes at days 8 and 12 post-infection. Barcodes were ranked from most to least frequent by each midgut sample. Color represents barcode rank and is not consistent across graphs. MG = midgut, SG = salivary glands, SL = saliva.
(B) bcWNV barcode maintenance in all midgut samples containing detectable barcodes, at each timepoint post-bloodfeed in Cx. tarsalis, Cx. quinquefasciatus, and Ae. aegypti mosquitoes. Barcodes were ranked from most to least frequent by the input population. Color represents both barcode rank and barcode sequence and is consistent within and across graphs.
Certain barcodes in our bcWNV stock were present at higher frequency than most, permitting examination of the relationship between starting population frequency and the likelihood of detection in various mosquito tissues and secretions. We visually compared all midguts to the input barcode population by ranking midgut barcodes by input abundance. In both Culex species, barcodes that are present at high frequencies in the input (i.e., the red barcode found at the top of the input column) are consistently maintained in midgut infection, and often become a highly abundant barcode in the midgut population (Figure 4B). The low complexity of barcode populations in Ae. aegypti midguts (consistent with Figure 4A) precludes inference from this visual comparison, as midgut populations in this species frequently comprise fewer than 10 unique barcodes (Figure 4B).
Differences in diversity of populations establishing infection in the midgut across species
We next compared richness and complexity of bcWNV populations across species to determine whether VC impacts bottleneck stringency. Midgut richness in Cx. tarsalis was significantly higher than Cx. quinquefasciatus at 8 days post-infection (p < 0.05), and significantly higher than Ae. aegypti at all timepoints (p < 0.002) (Figure 5A). Cx. quinquefasciatus midgut richness was significantly higher than Ae. aegypti midgut richness at days 4 and 8 post-infection (p < 0.05) (Figure 5A). Population complexity in Culex midguts was significantly higher than Ae. aegypti midguts at all time points (p < 0.05) (Figure 5B). The only significant difference in salivary gland population complexity was observed between Cx. tarsalis and Ae. aegypti at 12 days post-infection (p < 0.05) (Figure 5B). No significant differences in saliva population diversity were observed between species. To ensure that variability in sequencing coverage did not impact barcode abundance measurements, a subset of sample files were down-sampled to 750,000 reads. We replicated these analyses and findings on these down-sampled files (data not shown); confirming that coverage variability did not impact our results.
Figure 5.

Bottleneck severity, post-midgut infection, does not vary between vectors of differing competence
(A and B) Comparing bcWNV population richness (A) and complexity (B) in midgut, salivary gland, and saliva samples across Cx. tarsalis (orange), Cx. quinquefasciatus (blue), and Ae. aegypti (green) mosquitoes at days 4, 8, and 12 post-infection. Dashed line represents 0 and was added for easier visualization of low complexity values. Within each mosquito sample type, statistical significance between mosquito species was tested using Kruskal-Wallis with Dunn’s multiple comparisons test, ∗ = p < 0.05, ∗∗ = p < 0.005, ∗∗∗ = p < 0.0005, ∗∗∗∗ = p < 0.0001. Solid lines denote mean. Only significant comparisons shown.
Barcode dominance in the input does not guarantee transmission
Having determined that input frequency of a barcode influences barcode maintenance in the midgut of Culex species, we examined input frequency’s impact on barcode transmission (i.e., detection in the saliva). We plotted the input frequency of every barcode detected in all saliva samples, with the frequencies of the most (upper dashed line, 0.062) and least (lower dashed line, 0.000039) frequent barcodes in the input (Figure 6A). Transmissible barcodes had a large range of input frequencies, including one at the lowest input frequency, and we saw no significant differences in the starting frequency of transmissible barcodes between species (Figure 6A). Next, for all transmissible barcodes, we assessed the relationship between proportion in the input population and proportion of successful transmission events (number of saliva samples that contained a barcode/total positive saliva samples).19 There was a significant positive relationship between input proportion and proportion of successful transmission in both Cx. tarsalis and Cx. quinquefasciatus (r = 0.62, p < 0.0001 and r = 0.52, p = 0.0002 respectively) (Figure 6B), however, no significant relationship was observed in Ae. aegypti (r = 0.37, p > 0.05) (Figure 6B).
Figure 6.

The impact of barcode input frequency on infection and transmission probability
All points correspond to unique barcodes not mosquitoes.
(A) The input frequency of all barcodes detected in all saliva samples from each species were plotted and compared by Kruskal-Wallis with Dunn’s multiple comparisons test. Dashed lines represent the highest and lowest barcode frequencies in the input. Solid lines denote median.
(B) Relationship between barcode proportion in the input and proportion of successful transmission events (number of saliva samples that contained the barcode/total positive saliva samples) for all species was determined using Pearson’s correlation – Pearson’s r values and significance values are displayed on plots, with splines added for visualization.
(C) Midgut infection probabilities for Cx. tarsalis (orange), Cx. quinquefasciatus (blue), and Ae. aegypti (green) by abundance in the input population.
(D) Salivary gland infection probabilities for Cx. tarsalis (orange), Cx. quinquefasciatus (blue), and Ae. aegypti (green) by abundance in the input population.
(E) Transmission probabilities for Cx. tarsalis (orange), Cx. quinquefasciatus (blue), and Ae. aegypti (green) by abundance in the input population. All error bars denote ±SEM. Significance between mosquito species for infection/transmission probabilities was determined using Kruskal-Wallis with Dunn’s multiple comparisons test. ∗ = p < 0.05, ∗∗ = p < 0.005, ∗∗∗ = p < 0.0005, ∗∗∗∗ = p < 0.0001. Only significant comparisons shown.
Barcode infection and transmission probability is predominantly influenced by frequency in the input
We next wanted to quantify the likelihood of barcode infection and transmission based on frequency in the input. We calculated the probability a barcode would survive from the input to midgut, midgut to salivary glands, and salivary glands to saliva, based on that barcode’s abundance in the preceding population and the size of the proceeding population.
We found that barcodes of higher abundance in the input population consistently had greater probabilities of infecting the midgut and salivary glands, and eventually entering the saliva (transmission) (Figures 6C–6E; Table 1). Barcodes of all frequencies had a significantly higher probability of infecting the midgut in Cx. tarsalis (p < 0.0001), followed by Ae. aegypti, with the exception of the most abundant barcode quantile (p < 0.0001) (Figure 6C; Table 1). Cx. quinquefaciatus had the lowest midgut infection probabilities for all barcodes present at frequencies ≤0.0021 (≤99 barcodes in input). Barcodes present at frequencies >0.00063 (>30) in the input did not have significantly different probabilities of infecting the salivary glands of one Culex species over the other. However, barcodes present at frequencies ≤0.00018 (≤9) were significantly more likely to establish infection in Cx. tarsalis salivary glands (p < 0.0001) (Figure 6D). The probability of infecting Ae. aegypti salivary glands was significantly lower than both Culex species for all barcode frequencies, apart from barcodes with an abundance of 1 (p < 0.003) (Figure 6D). Transmission probabilities were comparable between Culex species for barcodes at frequencies ≥0.00021 (≥99), but barcodes at frequencies of 0.00018–0.000083 and 0.00002 (9-4, and 1) had a greater probability of transmission in Cx. tarsalis (p ≤ 0.0004) (Figure 6E; Table 1). Cx. tarsalis consistently had significantly higher transmission probabilities than Ae. aegypti for barcodes present at ≤0.0021 (≤99) (p < 0.009) (Figure 6E; Table 1). Barcodes at frequencies of ≤0.00018 (≤9) had the lowest transmission probability in Cx. quinquefaciatus (Figure 6E; Table 1). The highest observed probability of transmission for barcodes of the lowest frequency was 1.8x10−6 in Cx. tarsalis – significantly higher than Cx. quinquefaciatus and Ae. aegypti (p < 0.0001). (Figure 6E; Table 1). This is consistent with our finding that Cx. tarsalis was the only species to expectorate a barcode of the lowest frequency (Figure 6A).
Table 1.
Barcode infection and transmission probabilities by input frequency
| Barcode abundance in input: | 100+ | 99–31 | 30–10 | 9–4 | 3–2 | 1 | |
|---|---|---|---|---|---|---|---|
| Mean frequency: | 0.00558 | 0.00105 | 0.00032 | 0.00011 | 0.00005 | 0.00002 | |
| Frequency range: | 0.033–0.0021 | 0.0021–0.00065 | 0.00063–0.00021 | 0.00018–8.34E-05 | 6.26E-05–4.17E-05 | 2.09E-05 | |
| n total: | 3747 | 3527 | 3468 | 3379 | 3639 | 30188 | |
| n unique: | 14 | 70 | 224 | 613 | 1590 | 30188 | |
| Midgut Infection Probability | Cx. tarsalis | 0.9685 | 0.9187 | 0.7786 | 0.5273 | 0.2978 | 0.1498 |
| Cx. quinquefasciatus | 0.8719 | 0.7375 | 0.5212 | 0.2891 | 0.1441 | 0.0681 | |
| Ae. aegypti | 0.8627 | 0.8085 | 0.6199 | 0.3658 | 0.1900 | 0.0917 | |
| Salivary Gland Infection Probability | Cx. tarsalis | 0.4273 | 0.1979 | 0.0851 | 0.0228 | 0.0057 | 0.0015 |
| Cx. quinquefasciatus | 0.3967 | 0.2177 | 0.0895 | 0.0216 | 0.0037 | 0.0005 | |
| Ae. aegypti | 0.1018 | 0.0342 | 0.0146 | 0.0027 | 0.0008 | 0.0005 | |
| Transmission Probability | Cx. tarsalis | 0.0836 | 0.0218 | 0.0031 | 0.0006 | 2.59693E-05 | 1.83174E-06 |
| Cx. quinquefasciatus | 0.0823 | 0.0261 | 0.0037 | 2.56627E-05 | 2.45158E-06 | 6.62569E-09 | |
| Ae. aegypti | 0.0153 | 0.0040 | 0.0009 | 3.97347E-05 | 9.61687E-06 | 4.89198E-08 |
Discussion
In nature, both selective and stochastic processes control virus population structure.8,15,16,17,31 Arboviruses, including WNV, experience extreme population bottlenecks as they cycle between mosquito and avian hosts.14,16 The degree of WNV virus diversification, the strength of purifying and positive selection, and the ‘tightness’ of population bottlenecks is host dependent.8,15,32 However, precise quantification of the degree to which different vectors impose bottlenecks on virus populations remains elusive.16,20 Additionally, the degree to which population bottleneck size is dependent on mosquito VC has not been quantified using high resolution molecular tools. This is a significant shortcoming in the field considering the rapid globalization of arboviruses in recent decades, and the frequency with which they encounter new vectors of varying VC in different environments.12,32,33,34,35 Therefore, we sought to determine how tissue-associated bottlenecks, and their impact on virus population dynamics, varied between three WNV vectors of varying competence. The selected species represent high competence (Cx. tarsalis) moderate competence (Cx. quinquefasciatus) and low competence (Ae. aegypti) vectors of WNV.15,24,36,37 Cx. tarsalis and Cx. quinquefasciatus are known enzootic vectors of WNV that, in addition to being competent vectors, have high vectorial capacity for WNV due to their propensity to feed on birds, with mammal feeding occurring in a context-dependent manner.26,36,37,38 Ae. aegypti have low vectorial capacity for WNV due to their preference for feeding on humans and other mammals.39,40 However, despite being somewhat refractory to infection, Ae. aegypti are capable of transmitting WNV – making them an ideal model for bottlenecks in a low competence vector.24 This, in conjunction with WNV’s historical propensity to rapidly adapt to new ecological niches, makes Ae. aegypti:WNV a vector:virus pairing of great interest.3,6
We used a previously described barcoded West Nile virus (bcWNV) which mimics a genetically diverse mutant swarm, allowing us to quantify the impact of mosquito bottlenecks on viral populations and measure stochastic genetic restriction of the swarm.18,28,29 Sequencing our bcWNV stock confirmed a high degree of barcode genetic diversity and preservation of the native amino acid sequence. Importantly, we used unique molecular identifiers (UMIs) in our cDNA primers to uniquely tag RNA molecules and ensure that barcode abundance calculations were free of sequencing errors and amplification bias (Figure S3).41,42 Characterization of bcWNV revealed that bcWNV replicates comparably to unbarcoded WNV in vitro and was infectious to, and transmissible by, the mosquitoes in our study. Notably, results from VC studies conducted herein with bcWNV are extremely similar to those in a prior publication using unbarcoded WNV.15 These data indicate that bcWNV is highly genetically diverse and phenotypically comparable to wild-type WNV and therefore a suitable tool for our studies. Because the introduction of the barcode is coding-neutral and maintains similar fitness between each uniquely barcoded virus, the individual fitness of each barcode in our ‘input’ WNV population (i.e., that has been successfully produced and propagated) is likely approximately equal.19,20,28,29 However, RNA secondary structure and codon usage can impact the fitness of RNA viruses and result in alterations to the fitness of any given barcode.43,44,45 In nature, fitness would likely play a role in every step of mosquito infection and transmission.8,13 However, intrahost bottlenecks within mosquitoes make survival and maintenance of any variant challenging for even the most fit viruses in a population.13,16 Removing fitness as a variable by using a swarm of comparably fit barcoded viruses, allowed us to specifically quantify the impact of stochastic pressures on a virus population within mosquitoes.
We hypothesized based on prior literature that the diversity of barcode populations would decrease during stages of systemic mosquito infection (i.e., from input to midgut, midgut to salivary glands, etc.).7,8,15,18,19,20,21 We also reasoned that bottleneck size would decrease with decreasing VC (e.g., high diversity in highly competent Cx. tarsalis and lower diversity in less competent Ae. aegypti).24,26 We observed overall decreases in barcode diversity between the midgut and saliva populations in all species – confirming the occurrence of bottlenecks, consistent with previous studies of bottlenecks in mosquitoes, in all species of interest.18,19,20 However, Ae. aegypti was the only species in which we did not observe significant decreases in barcode diversity between the midgut and salivary glands due to the extremely low barcode diversity found in Ae. aegypti midguts. We found that the complexity of barcode populations infecting the mosquito midgut were significantly higher in Culex species than Ae. aegypti on all sampled days post infection. We also saw, on average, 13-fold and 5-fold higher barcode richness in Cx. tarsalis and Cx. quinquefasciatus midguts compared to Ae. aegypti. In contrast, we observed minimal differences in barcode diversity between salivary gland and saliva populations across different mosquito species with different VC. It seems likely that this is due to the severe initial reduction of genetic diversity imposed on the bcWNV population during midgut escape.20,22 This indicates that bottlenecks occurring downstream of midgut infection negate differences in diversity observed in the midguts of these mosquitoes, as all species end up expectorating comparably diverse virus populations. Moreover, these data support the observation that maintenance of WNV genetic diversity upon mosquito infection is associated with the degree of VC – with a larger proportion of standing viral genetic variation from the input population present in the midguts of our more competent vectors. This founding population is then subject to additional, non-species dependent, bottlenecks during systemic infection of the mosquito.
Reduction of input population richness upon midgut infection varied by species with Ae. aegypti sustaining the greatest reduction, followed by Cx. quinquefasciatus and Cx. tarsalis, further demonstrating the stringency of the midgut infection barrier in Ae. aegypti. Previous work with barcoded Zika virus (bcZIKV) – a virus for which Ae. aegypti is a highly competent vector – saw no significant decrease in bcZIKV population diversity between the input population and midgut.18 This further supports that Ae. aegypti’s low competence as a WNV vector impacts the diversity of the barcode population establishing infection in the midgut. Interestingly, we noted the most dramatic loss of unique barcodes between the midgut and salivary glands of Cx. tarsalis, our most highly competent vector. It has been previously shown that when the midgut is readily infected, as in Cx. tarsalis, a severe bottleneck occurs during midgut escape, which supports this finding.8,20,22 The most severe reduction in barcode richness between the salivary glands and saliva was seen in Cx. quinquefasciatus, which aligns with our findings that Cx. quinquefasciatus produced the fewest transmission positive saliva samples out of the three species (Figure 2B) and that low frequency barcodes have the lowest probability of transmission in Cx. quinquefasciatus (Figure 6E). Further, this finding aligns with a study of WNV VC that noted high midgut infection rates and low dissemination and transmission rates in Cx. quinquefasciatus.36
The cause of the significant differences in midgut population diversity between Culex and Ae. aegypti remain to be determined. Ae. aegypti was the only species in which no significant linear relationship existed between total and unique barcodes – indicating an overall dearth of barcode population diversity in that species. Interestingly, despite the low barcode diversity in Ae. aegypti, all Ae. aegypti samples contained more total barcodes than Cx. quinquefasciatus, and Ae. aegypti salivary glands contained more total barcodes than Cx. tarsalis. The lack of linear relationship between total and unique barcodes, in conjunction with the high number of total barcodes identified in Ae. aegypti tissues, demonstrates that low diversity in Ae. aegypti midguts is not due to poor virus replication. This is further supported by our finding that Ae. aegypti and Cx. quinquefaciatus achieved comparable peak vRNA levels in the midgut.
Successful midgut infection hinges upon a virus entering and replicating in midgut cells.22,26,46 Thus, the strong bottleneck/genetic restriction in Aedes midguts compared to Culex could be due to a smaller subset of midgut cells that are susceptible to infection. Further, genetic bases for infection and escape barriers have been identified and shown to have significant influence on the success of vector-virus pairings.21,22,26 It is possible that the Ae. aegypti – WNV pairing is impacted by unknown cellular, and genetic factors.26,47 Single-cell analyses of mosquito midguts at early timepoints post-infection could identify differences in cell population involvement in, and genetic response to, midgut infection. This would elucidate the establishment of population diversity between different species, and provide key insight into the physiology of infection within arbovirus vectors. Additionally, the mosquito microbiome is known to have a profound impact on both VC and vectorial capacity.26,48 Recent work has demonstrated that pre-activation of mosquito innate immune system by the microbiome can promote antiviral defense.49,50 This innate immune activity can, in turn, impede virus transmission.49 While our understanding of mosquito microbiome interactions with pathogens remains limited for most mosquito:virus pairings, the microbiomes of Culex species and Ae. aegypti are known to differ.48,51,52
An important remaining question concerns the likelihood of transmission of minority population variants given the bottlenecks that occur in mosquitoes. In other words: at what population frequencies does transmission of a WNV variant become either very likely or very unlikely? Previous work demonstrated that mosquitoes expectorate a unique virus population each time they feed, and that conditions that impact the frequency of virus variants within the mosquito vector are causal factors of emergence.23 This knowledge, combined with the stringent reductions in barcode population diversity that we observed upon escape into the saliva, suggests that bottlenecks play a role in the transmission of rare or unique variants regardless of their fitness.
We found a significant relationship between barcode frequency and proportion of successful transmission events in Culex mosquitoes, the most significant WNV vectors included in this study. However, despite the relationship between starting frequency and successful transmission, higher barcode frequency (∼3%) in the input does not guarantee transmission, as we only identified our most frequent input barcode in 7 saliva samples. This is consistent with studies of bcZIKV in Ae. aegypti, that demonstrated that the highest frequency barcode in the input population was not consistently the highest frequency in the saliva.18 We found that infection and transmission probability decreased with barcode input frequency in all species – this aligns with previous work demonstrating that in a stochastic system, the main variable that influences viral dominance and/or maintenance throughout infection is frequency in the input population.17,19 However, we observed that, while rare, barcodes of the lowest frequency can escape into the saliva, and noted that rare barcodes (i.e., low frequency barcodes – present in the input population at frequencies from 2.1e−5 – 6.3e−5) had a non-zero chance of midgut and salivary gland infection and transmission in all species. This finding is supported by work characterizing WNV population dynamics in Cx. pipiens, which demonstrated that all variants in a starting population had a chance of surviving intrahost bottlenecks.19 Further, this finding highlights the stochasticity of mosquito transmission, as previous studies have demonstrated that stochastic transmission is marked by a non-zero chance of transmission for all viruses in a population.18,53 Interestingly, low frequency barcodes had a significantly higher chance of establishing infection in, and being transmitted by, Cx. tarsalis when compared to our lower competence vectors. This indicates that the most competent vector is the most likely to become infected with and promote the transmission of rare variants in a virus population. Given that the virus population that is being expectorated by a mosquito can ultimately establish infection in a new host, these findings suggest that mosquito infection, particularly in highly competent vectors, serves as an opportunity for rare virus variants to be transmitted and establish infection in a new host, which has important implications for WNV evolution and emergence.7,23 Moreover, bottlenecks and transmission of novel, low-frequency virus variants that go on to establish infection in new hosts, alter virus population structure in a non-selective manner.
The ongoing globalization of arboviruses has guaranteed that they will continue to encounter new vectors of varying VC.35 Additionally, it is likely that as viruses adapt to their local ecology and vectors, the interactions between virus and mosquito species (e.g., VC, bottleneck stringency, etc.) may be altered as well. The work presented here demonstrates that VC is linked to the stringency of intrahost population bottlenecks and that this in turn influences the maintenance of WNV population diversity upon midgut infection. Ultimately the virus that is relevant in nature is the one that gets transmitted; while VC does not appear to impact the diversity of transmissible virus populations, infection of a highly competent mosquito vector can promote the transmission of new or rare virus variants.
Limitations of the study
The severity and variability of the midgut infection and escape barriers across species makes it difficult to compare salivary gland associated bottlenecks across species. The salivary glands are critical sites of virus infection and replication as they are the compartment directly preceding virus transmission.21,22 Previous work has demonstrated the impact replication in the salivary glands has on WNV population diversification and the subsequent diversity of the transmissible population.15,16,23 Future work in this area could better examine salivary gland bottlenecks by infecting mosquitoes via intrathoracic injection and bypassing the midgut infection and escape barriers; an approach used previously in a study characterizing the impact of intrahost bottlenecks on Venezuelan equine encephalitis virus in its primary vector.20 This approach was not undertaken as part of this work because our focus was on biological transmission mechanisms, and as such we opted to mimic natural infection as closely as possible. Additionally, limitations associated with laboratory colony-derived mosquitoes may impact VC.54 Future studies with outbred or field-collected mosquitoes could be warranted to confirm the phenotype.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| barcoded West Nile virus (bcWNV), lineage I, strain 3356 | Colorado State University, Microbiology department | N/A |
| Deposited data | ||
| Sequencing files (.fastq) | Mendeley Data: https://doi.org/10.17632/88xx6596wz.1https://doi.org/10.17632/s7kthpysb7.1https://doi.org/10.17632/556zktntym.1. | N/A |
| Experimental models: Cell lines | ||
| Vero cells | ATCC | CVCL_0059 |
| CT (Culex tarsalis derived) cells | Aaron Brault, Centers for Disease Control and Prevention | CVCL_0F44 |
| Experimental models: Organisms/strains | ||
| Culex tarsalis mosquitoes | USA, CA | N/A |
| Culex quinquefasciatus mosquitoes | USA, FL | N/A |
| Aedes aegypti mosquitoes | Mexico, Poza Rica | N/A |
| Oligonucleotides | ||
| WNV_ENV_F70 - TCAGCGATCTCTCCACCAAAG | IDT | N/A |
| WNV_ENV_R70 - GGGTCAGCACGTTTGTCATTG | IDT | N/A |
| WNENV-prob - FAM/TGCCCGACC/ ZEN/ATGGGAGAAGCTC/3|ABkFQ/ |
IDT | N/A |
| ID_cDNAWNV_7374_Rev - GTCTCGTGGGCTCG GAGATGTGTATAAGAGACAGNNNNNNNNN cagtGCCATCCACTACAGCGTTCT |
IDT | N/A |
| 5'_ID_Primer_Rev - GTCTCGTGGGCTCGGA GATGTGTAT |
IDT | N/A |
| 5'_ID_WNV_7171_For - TCGTCGGCAGCGTCA GATGTGTATAAGAGACAGNNNNTTCCCCTT CGTCGATGTTGG |
IDT | N/A |
| 2ND round FP (N5XX) - AATGATACGGCGAC CACCGAGATCTACAC[i5]TCGTCGGCAGCGTC |
IDT | N/A |
| 2ND round RP (N7XXX) - CAAGCAGAAGACG GCATACGAGAT[i7]GTCTCGTGGGCTCGG |
IDT | N/A |
| Software and algorithms | ||
| Original code used for used for data processing, analysis, and visualization. | https://github.com/fitz-meyer/bcWNV_analysis.git | R - SCR_001905 GitHub - SCR_002630 |
| Statistical analyses | R version 4.2.2 Prism version 9.4.1 |
R - SCR_001905 Prism -SCR_002798 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Gregory Ebel (gregory.ebel@colostate.edu).
Materials availability
This study did not generate unique reagents.
Experimental model and study participant details
Cells
Vero (ATCC CCL-81) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (10,000 U/ml), and 50 μg/mL gentamicin at 37°C with 5% CO2 (18). CT, Cx. tarsalis-derived embryonic cell line cells were maintained in Schneider’s insect medium with 10% FBS, 1% penicillin-streptomycin (10,000 U/ml), and 50 μg/mL gentamicin at 28°C.18,55
Mosquito infection
Mosquito studies were conducted using three species of laboratory colony-derived mosquitoes; Culex quinquefasciatus (>50 passages), Culex tarsalis (>50 passages) and Ae. aegypti (>30 passages). West Nile virus infections in mosquitoes were performed under A-BSL3 conditions. Larvae were raised on a diet of powdered fish food. Mosquitoes were maintained at 26°C–27°C (Culex) or 28°C (Aedes) with a 16:8 (Culex) or 12:12 (Aedes) light:dark cycle.25 All species were maintained at 70–80% relative humidity, with water and sucrose provided ad libitum. Cx. tarsalis, Cx. quinquefasciatus, and Ae. aegypti mosquitoes were exposed to an infectious bloodmeal containing a 1:1 dilution of defibrinated calf’s blood and bcWNV stock at a final concentration of 3-6x107 pfu/ml. A final concentration of 1 mM ATP was added to Ae. aegypti bloodmeals to encourage feeding to repletion. All bloodmeals were provided in a hog’s gut membrane feeder, warmed by circulating 37°C water. Following 40–60 min of feeding, mosquitoes were anesthetized at −20°C, engorged females were separated into cartons corresponding to each timepoint and maintained on sucrose.
Method details
Generation of rWT-WNV and bcWNV P2 stocks
The parental rWT-WNV (epidemic lineage I strain, 3356) and bcWNV P2 stocks were generated by infecting confluent flasks of Vero cells at an MOI of ∼0.5 with rWT-WNV or bcWNV P1 stock, as previously described.56,57 Supernatant was harvested from cells 3 days post-infection, aliquoted into single use stocks and stored at −80°C. Titers for each stock were determined by standard Vero cell plaque assay.30 RNA was extracted from the bcWNV P2 stock at volumes of 50, 100, and 200μl using a Direct-zol miniprep plus kit (Zymo) and sequenced as described below.
Growth curves
Vero and CT (Culex tarsalis derived) cells were infected with bcWNV and rWT-WNV at MOIs of 0.1 and 1.0, for 1 h, and washed three times with PBS before fresh growth media was added. Supernatant was sampled daily and RNA was extracted using the Omega MagBind Viral DNA/RNA 96 kit on the KingFisher Flex Magnetic Particle Processor (ThermoFisher). qRT-PCR was performed using One-Step SuperScript qRT-PCR kit (Invitrogen) and WNV envelope primer and probes (Table S2). RNA copies/ml was extrapolated using an RNA standard. The RNA standard was generated by amplifying ∼1 kb of sequence surrounding the envelope region of the WNV genome using a forward primer containing a T7 promoter sequence. The resulting amplicon was transcribed with T7 polymerase, and RNA was extracted and quantified.
Collection of mosquito tissues
At indicated time points, mosquitoes were cold-anesthetized and legs and wings were removed prior to salivating on capillaries containing immersion oil for 30 min. Capillaries were broken off into tubes containing 100ul of mosquito diluent and centrifuged at 15,000xg for 5 min at 4°C. Mosquitoes were then transferred to a dish containing 1X phosphate buffered saline (PBS) for salivary gland and midgut dissection. Salivary glands and midguts were rinsed in clean PBS before being transferred to individual tubes containing 200ul of mosquito diluent and a stainless-steel bead for homogenization. Tissue samples were homogenized using RetschMixerMill MM400 at a frequency of 29 cycles/second for 1 min. RNA was extracted using the Omega MagBind Viral DNA/RNA 96 kit on the KingFisher Flex Magnetic Particle Processor (ThermoFisher). Presence of viral RNA was assessed by qRT-PCR as described above. Midgut, salivary gland, and saliva samples from 10 mosquitoes per species per timepoint were selected for next-generation sequencing with preference given to those with detectable vRNA in their saliva.
Library preparation and sequencing
cDNA was generated using Superscript IV RT enzyme and kit (ThermoFisher), and a previously described custom cDNA primer containing a unique molecular identifier41,42 (Table S2; Figure S3). cDNA was purified using AMPure XP beads (Beckman) at a 1X concentration. All bead purifications were performed using the KingFisher Flex Magnetic Particle Processor. The barcoded region of the genome was amplified via PCR using custom primers and KAPA HiFi HotStart mastermix (Roche) (Table S2). Amplified samples were purified using AMPure XP beads at a 0.6X concentration. Samples were uniquely indexed with Illumina i5 and i7 indices via PCR using custom primers and KAPA HiFi HotStart mastermix (Table S2). Second-round amplification products were purified using AMPure XP beads at a 0.6X concentration. Samples underwent a final library amplification using a KAPA Library Amplification Kit (Roche) and were purified using AMPure XP beads at a 0.7X concentration prior to quantification and pooling. Sample concentrations were determined using Qubit dsDNA High Sensitivity kit (Thermo Fisher) and pooled by mass at 2 ng per sample. The final pool was concentrated using AMPure XP beads at a concentration of 1.4X. Size distribution (∼350 bp for this library) and concentration of the final pool were verified by TapeStation D1000 HS Screen Tape assay (Agilent) and KAPA library quantification (Roche) respectively. A pilot run was performed to determine target coverage and stock diversity. The pilot library was denatured and a 15% PhiX control was spiked in. The library was loaded at a concentration of 1.5p.m. and sequenced using a NextSeq 550 system, 300 cycle (2x150 paired-end) mid output kit (Illumina). A 40% PhiX control was spiked into all following libraries to offset the low diversity of the library and prevent low sequence quality. All libraries were loaded at a concentration of 1.5p.m. and sequenced using a NextSeq 550 system, 300 cycle high output kit (Illumina). Sequencing data files can be accessed through Mendeley Data: https://doi.org/10.17632/88xx6596wz.1, https://doi.org/10.17632/s7kthpysb7.1, https://doi.org/10.17632/556zktntym.1.
Quantification and statistical analyses
Analyses
The data manipulation and analyses of next generation sequencing data was performed as previously described.28,41 Briefly, sequence data was demultiplexed and trimmed of adaptor and index sequences. Paired-end reads were merged and aligned to the WNV genome, reads were grouped by UMI and consensus sequences for unique RNA molecules were generated (Figure S3). The barcode region was identified by upstream and downstream flanking regions, extracted from the consensus sequences, and abundance determined using custom Perl and Ruby scripts. Barcode population richness and complexity (Shannon’s index) for each sample were determined using custom R scripts (R version 4.2.2.); richness was determined by summing the number of unique barcodes detected in a sample, while complexity was determined by applying the Shannon function from the QSutils (quasispecies diversity) package to barcode abundance vectors.58
When calculating richness retention (Figure 3D) there were salivary gland and saliva samples in Ae. aegypti that appeared to retain greater than 100% of the unique barcodes identified in the preceding population – a finding consistent with previous studies employing a barcoded virus system.18 This system does not allow for the generation of novel barcodes after amplification and rescue of the initial barcoded virus stock, and any barcodes introduced by mutation are removed from the dataset prior to analysis. While some barcodes were found to be truly “parentless” (i.e., present in one compartment but not present in the upstream compartment) we found that many of these parentless barcodes were present in the raw reads from the sample file associated with the upstream compartment. These missing parent barcodes were present at low enough levels that they were removed from the dataset by previously described Primer ID cutoff rates.41,42 For every case where richness retention exceeded 100% due to low-level parent barcodes being filtered out of the dataset, we adjusted the percent retention value to 100%.
We merged all sample files by barcode and determined that the entire dataset contained >30,000 unique barcodes. As mentioned previously, this system does not allow for the generation of novel barcodes within the mosquito – leading us to determine that ∼20,000 unique barcodes not initially identified in our bcWNV stock population were present in the stock, but not detected. We merged the additional unique barcodes into our merged input population to create an estimated input population. Since we could infer presence but not abundance, we preserved the abundances of the merged input population and assigned the minimum abundance of 1 to all additional unique barcodes. This estimated input population was used for all probability calculations. All probability calculations are derived from barcode abundance, population size (number of total barcodes in the current population), and subsample size (number of total barcodes in the downstream population) and thus are based upon but not entirely representative of the data. Infection and transmission probabilities were calculated via custom R scripts, using the following equation:
These calculations were performed for all barcodes in all mosquitoes individually and then averaged to produce mean probabilities for each barcode in each species. We divided all barcodes present in the estimated input population at abundances >1 into quintiles containing ∼3500 total barcodes (±250 barcodes to prevent dividing abundance groups) (Table 1). We then averaged the mean probabilities for each species within each of the abundance quantiles (Table 1).
All scripts used for data processing, analysis, and visualization are available on GitHub: https://github.com/fitz-meyer/bcWNV_analysis.git. Statistical analyses were performed in GraphPad Prism version 9.4.1. Normality of datasets was measured by Shapiro-Wilk test (shapiro.test()) in R. Differences in sample richness and complexity by sample type and species were measured by Kruskal-Wallis with Dunn’s multiple comparisons test. Differences in infection and transmission probability were measured by Kruskal-Wallis with Dunn’s multiple comparisons test. Relationships between unique and total identified barcodes, and starting frequency and infection/transmission probability were measured by Pearson’s and Spearman’s correlation respectively. Additional statistical details can be found in the text and figure legends.
Acknowledgments
We thank Reyes Murrieta and Brooke Anderson for developing and assisting in the development of the barcode analysis pipeline, Matthew Aliota and Kasen Riemersma for sharing the scripts used to generate the barcode progression and maintenance plots, Nicole Sexton for assisting in parsing the library preparation methods we followed, and Mark Stenglein for his guidance in all things sequencing and assistance in generating the bootstrap rarefaction scripts.
The research reported in this publication was supported by National Institutes of Health grant R01- AI067380 (G.D.E), National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number T32-AI162691, and by Colorado State University’s Office of the Vice President for Research’s “Accelerating Innovations in Pandemic Disease” initiative, made possible through support from The Anschutz Foundation. The content is solely the responsibility of the authors.
Author contributions
E.A.F. – conceived and designed the analysis, collected the data, contributed data or analysis tools, performed the analysis, wrote the paper (original draft preparation).
E.N.G. – collected the data, contributed data or analysis tools, wrote the paper (review and editing).
J.W-L. – conceived and designed the analysis, contributed data or analysis tools.
M.L.K. – contributed data or analysis tools.
Z.A. - contributed data or analysis tools.
K.P. – collected the data.
M.Y. - contributed data or analysis tools.
G.D.E. - conceived and designed the analysis, wrote the paper (review and editing).
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107711.
Supplemental information
Data and code availability
-
•
Sequencing data files have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
All original code used for data processing, analysis, and visualization has been deposited on GitHub. Link to repository is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.McDonald E., Martin S.W., Landry K., Gould C.V., Lehman J., Fischer M., Lindsey N.P. West Nile Virus and Other Domestic Nationally Notifiable Arboviral Diseases — United States, 2018. MMWR Morb. Mortal. Wkly. Rep. 2019;68:673–678. doi: 10.15585/mmwr.mm6831a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray K.O., Mertens E., Desprès P. West Nile virus and its emergence in the United States of America. Vet. Res. 2010;41:67. doi: 10.1051/vetres/2010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moudy R.M., Meola M.A., Morin L.-L.L., Ebel G.D., Kramer L.D. A Newly Emergent Genotype of West Nile Virus Is Transmitted Earlier and More Efficiently by Culex Mosquitoes. Am. J. Trop. Med. Hyg. 2007;77:365–370. [PubMed] [Google Scholar]
- 4.Ciota A.T., Lovelace A.O., Jia Y., Davis L.J., Young D.S., Kramer L.D. Characterization of mosquito-adapted West Nile virus. J. Gen. Virol. 2008;89:1633–1642. doi: 10.1099/vir.0.2008/000893-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duggal N.K., Langwig K.E., Ebel G.D., Brault A.C. On the Fly: Interactions between Birds, Mosquitoes, and Environment That Have Molded West Nile Virus Genomic Structure over Two Decades. J. Med. Entomol. 2019;56:1467–1474. doi: 10.1093/jme/tjz112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brault A.C. Changing patterns of West Nile virus transmission: Altered vector competence and host susceptibility. Vet. Res. 2009;40:43. doi: 10.1051/vetres/2009026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patterson E.I., Khanipov K., Rojas M.M., Kautz T.F., Rockx-Brouwer D., Golovko G., Albayrak L., Fofanov Y., Forrester N.L. Mosquito bottlenecks alter viral mutant swarm in a tissue and time-dependent manner with contraction and expansion of variant positions and diversity. Virus Evol. 2018;4:vey001. doi: 10.1093/ve/vey001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weaver S.C., Forrester N.L., Liu J., Vasilakis N. Population bottlenecks and founder effects: implications for mosquito-borne arboviral emergence. Nat. Rev. Microbiol. 2021;19:184–195. doi: 10.1038/s41579-020-00482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Domingo E., Holland J.J. RNA VIRUS MUTATIONS AND FITNESS FOR SURVIVAL. Annu Rev Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 10.Brackney D.E., Beane J.E., Ebel G.D. RNAi targeting of West Nile virus in mosquito midguts promotes virus diversification. PLoS Pathog. 2009;5:e1000502. doi: 10.1371/journal.ppat.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitzpatrick K.A., Deardorff E.R., Pesko K., Brackney D.E., Zhang B., Bedrick E., Shi P.-Y., Ebel G.D. Population variation of West Nile virus confers a host-specific fitness benefit in mosquitoes. Virology. 2010;404:89–95. doi: 10.1016/j.virol.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsetsarkin K.A., Vanlandingham D.L., McGee C.E., Higgs S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 2007;3 doi: 10.1371/journal.ppat.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forrester N.L., Coffey L.L., Weaver S.C. Arboviral bottlenecks and challenges to maintaining diversity and fitness during mosquito transmission. Viruses. 2014;6:3991–4004. doi: 10.3390/v6103991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jerzak G., Bernard K.A., Kramer L.D., Ebel G.D. Genetic variation in West Nile virus from naturally infected mosquitoes and birds suggests quasispecies structure and strong purifying selection. J. Gen. Virol. 2005;86:2175–2183. doi: 10.1099/vir.0.81015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grubaugh N.D., Weger-Lucarelli J., Murrieta R.A., Fauver J.R., Garcia-Luna S.M., Prasad A.N., Black W.C., Ebel G.D. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host. Cell Host Microbe. 2016;19:481–492. doi: 10.1016/j.chom.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutiérrez S., Michalakis Y., Blanc S. Virus population bottlenecks during within-host progression and host-to-host transmission. Curr. Opin. Virol. 2012;2:546–555. doi: 10.1016/j.coviro.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 17.McCrone J.T., Woods R.J., Martin E.T., Malosh R.E., Monto A.S., Lauring A.S. Stochastic processes constrain the within and between host evolution of influenza virus. Elife. 2018;7 doi: 10.7554/eLife.35962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weger-Lucarelli J., Garcia S.M., Rückert C., Byas A., O’Connor S.L., Aliota M.T., Friedrich T.C., O’Connor D.H., Ebel G.D. Using barcoded Zika virus to assess virus population structure in vitro and in Aedes aegypti mosquitoes. Virology. 2018;521:138–148. doi: 10.1016/j.virol.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciota A.T., Ehrbar D.J., Van Slyke G.A., Payne A.F., Willsey G.G., Viscio R.E., Kramer L.D. Quantification of intrahost bottlenecks of West Nile virus in Culex pipiens mosquitoes using an artificial mutant swarm. Infect. Genet. Evol. 2012;12:557–564. doi: 10.1016/j.meegid.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forrester N.L., Guerbois M., Seymour R.L., Spratt H., Weaver S.C. Vector-Borne Transmission Imposes a Severe Bottleneck on an RNA Virus Population. PLoS Pathog. 2012;8:e1002897. doi: 10.1371/journal.ppat.1002897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez-Vargas I., Olson K.E., Black W.C. The genetic basis for salivary gland barriers to arboviral transmission. Insects. 2021;12:73. doi: 10.3390/insects12010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpenter A., Clem R.J. Factors Affecting Arbovirus Midgut Escape in Mosquitoes. Pathogens. 2023;12 doi: 10.3390/pathogens12020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grubaugh N.D., Fauver J.R., Rückert C., Weger-Lucarelli J., Garcia-Luna S., Murrieta R.A., Gendernalik A., Smith D.R., Brackney D.E., Ebel G.D. Mosquitoes Transmit Unique West Nile Virus Populations during Each Feeding Episode. Cell Rep. 2017;19:709–718. doi: 10.1016/j.celrep.2017.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turell M.J., O’guinn M.L., Dohm D.J., Jones J.W. Vector Competence of North American Mosquitoes (Diptera: Culicidae) for West Nile Virus. J. Med. Entomol. 2001;38:130–134. doi: 10.1603/0022-2585-38.2.130. [DOI] [PubMed] [Google Scholar]
- 25.Weger-Lucarelli J., Rückert C., Chotiwan N., Nguyen C., Garcia Luna S.M., Fauver J.R., Foy B.D., Perera R., Black W.C., Kading R.C., Ebel G.D. Vector Competence of American Mosquitoes for Three Strains of Zika Virus. PLoS Neglected Trop. Dis. 2016;10 doi: 10.1371/journal.pntd.0005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramer L.D., Ciota A.T. Dissecting vectorial capacity for mosquito-borne viruses. Curr. Opin. Virol. 2015;15:112–118. doi: 10.1016/j.coviro.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brackney D.E., Pesko K.N., Brown I.K., Deardorff E.R., Kawatachi J., Ebel G.D. West Nile Virus Genetic Diversity is Maintained during Transmission by Culex pipiens quinquefasciatus Mosquitoes. PLoS One. 2011;6 doi: 10.1371/journal.pone.0024466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frank, D.T., Byas, A.D., Murrieta, R., Weger-Lucarelli, J., Rückert, C., Gallichotte, E., Yoshimoto, J.A., Allen, C., Bosco-Lauth, A.M., Graham, B., et al. Intracellular Diversity of WNV within Circulating Avian Peripheral Blood Mononuclear Cells Reveals Host-dependent Patterns of Polyinfection. Preprint at bioRxiv. 10.1101/2023.01.27.525959 [DOI] [PMC free article] [PubMed]
- 29.Fitzmeyer E.A., Gallichotte E.N., Ebel G.D. Scanning barcodes: A way to explore viral populations. PLoS Pathog. 2023;19 doi: 10.1371/journal.ppat.1011291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sexton N.R., Bellis E.D., Murrieta R.A., Spangler M.C., Cline P.J., Weger-Lucarelli J., Ebel G.D. Genome Number and Size Polymorphism in Zika Virus Infectious Units. J. Virol. 2021;95 doi: 10.1128/JVI.00787-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novella I.S., Presloid J.B., Smith S.D., Wilke C.O. Specific and Nonspecific Host Adaptation during Arboviral Experimental Evolution. Microb. Physiol. 2011;21:71–81. doi: 10.1159/000332752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coffey L.L., Vasilakis N., Brault A.C., Powers A.M., dé ric Tripet F., Weaver S.C. Arbovirus Evolution in Vivo Is Constrained by Host Alternation. Proc. Natl. Acad. Sci. USA. 2008;105:6970–6975. doi: 10.1073/pnas.0712130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pybus O.G., Tatem A.J., Lemey P. Virus evolution and transmission in an ever more connected world. Proc. Biol. Sci. 2015;282 doi: 10.1098/rspb.2014.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franklinos L.H.V., Jones K.E., Redding D.W., Abubakar I. The effect of global change on mosquito-borne disease. Lancet Infect. Dis. 2019;19:e302–e312. doi: 10.1016/S1473-3099(19)30161-6. [DOI] [PubMed] [Google Scholar]
- 35.Coffey L.L., Forrester N., Tsetsarkin K., Vasilakis N., Weaver S.C. Factors shaping the adaptive landscape for arboviruses: Implications for the emergence of disease. Future Microbiol. 2013;8:155–176. doi: 10.2217/fmb.12.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sardelis M.R., Turell M.J., Dohm D.J., O’guinn M.L. Vector Competence of Selected North American Culex and Coquillettidia Mosquitoes for West Nile Virus. Emerg. Infect. Dis. 2001;7:1018–1022. doi: 10.3201/eid0706.010617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kent R., Juliusson L., Weissmann M., Evans S., Komar N. Seasonal Blood-Feeding Behavior of Culex tarsalis (Diptera: Culicidae) in Weld County, Colorado, 2007. J. Med. Entomol. 2009;46:380–390. doi: 10.1603/033.046.0226. [DOI] [PubMed] [Google Scholar]
- 38.Apperson C.S., Hassan H.K., Harrison B.A., Savage H.M., Aspen S.E., Farajollahi A., Crans W., Daniels T.J., Falco R.C., Benedict M., et al. Host feeding patterns of established and potential mosquito vectors of West Nile virus in the Eastern United States. Vector Borne Zoonotic Dis. 2004;4:71–82. doi: 10.1089/153036604773083013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sene N.M., Diouf B., Gaye A., Ndiaye E.H., Ngom E.M., Gueye A., Seck F., Diagne C.T., Dia I., Diallo D., Diallo M. Blood Feeding Patterns of Aedes aegypti Populations in Senegal. Am. J. Trop. Med. Hyg. 2022;106:1402–1405. doi: 10.4269/ajtmh.21-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ponlawat A., Harrington L.C. Blood Feeding Patterns of Aedes aegypti and Aedes albopictus in Thailand. J. Med. Entomol. 2005;42:844–849. doi: 10.1093/jmedent/42.5.844. [DOI] [PubMed] [Google Scholar]
- 41.Zhou S., Jones C., Mieczkowski P., Swanstrom R. Primer ID Validates Template Sampling Depth and Greatly Reduces the Error Rate of Next-Generation Sequencing of HIV-1 Genomic RNA Populations. J. Virol. 2015;89:8540–8555. doi: 10.1128/jvi.00522-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jabara C.B., Jones C.D., Roach J., Anderson J.A., Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. USA. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sabi R., Tuller T. Modelling the Efficiency of Codon–tRNA Interactions Based on Codon Usage Bias. DNA Res. 2014;21:511–526. doi: 10.1093/dnares/dsu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernández-Sanlés A., Ríos-Marco P., Romero-López C., Berzal-Herranz A. Functional Information Stored in the Conserved Structural RNA Domains of Flavivirus Genomes. Front. Microbiol. 2017;08 doi: 10.3389/fmicb.2017.00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scroggs S.L.P., Grubaugh N.D., Sena J.A., Sundararajan A., Schilkey F.D., Smith D.R., Ebel G.D., Hanley K.A., Lakdawala S. Endless Forms: Within-Host Variation in the Structure of the West Nile Virus RNA Genome during Serial Passage in Bird Hosts. mSphere. 2019;4 doi: 10.1128/mSphere. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith D.R., Adams A.P., Kenney J.L., Wang E., Weaver S.C. Venezuelan equine encephalitis virus in the mosquito vector Aedes taeniorhynchus: Infection initiated by a small number of susceptible epithelial cells and a population bottleneck. Virology. 2008;372:176–186. doi: 10.1016/j.virol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franz A.W.E., Kantor A.M., Passarelli A.L., Clem R.J. Tissue barriers to arbovirus infection in mosquitoes. Viruses. 2015;7:3741–3767. doi: 10.3390/v7072795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cansado-Utrilla C., Zhao S.Y., McCall P.J., Coon K.L., Hughes G.L. The microbiome and mosquito vectorial capacity: rich potential for discovery and translation. Microbiome. 2021;9:111. doi: 10.1186/s40168-021-01073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cirimotich C.M., Ramirez J.L., Dimopoulos G. Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe. 2011;10:307–310. doi: 10.1016/j.chom.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramirez J.L., Souza-Neto J., Torres Cosme R., Rovira J., Ortiz A., Pascale J.M., Dimopoulos G. Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS Neglected Trop. Dis. 2012;6:e1561. doi: 10.1371/journal.pntd.0001561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hegde S., Khanipov K., Albayrak L., Golovko G., Pimenova M., Saldaña M.A., Rojas M.M., Hornett E.A., Motl G.C., Fredregill C.L., et al. Microbiome interaction networks and community structure from laboratory-reared and field-collected Aedes aegypti, Aedes albopictus, and Culex quinquefasciatus mosquito vectors. Front. Microbiol. 2018;9:2160. doi: 10.3389/fmicb.2018.02160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duguma D., Hall M.W., Rugman-Jones P., Stouthamer R., Terenius O., Neufeld J.D., Walton W.E. Developmental succession of the microbiome of Culex mosquitoes Ecological and evolutionary microbiology. BMC Microbiol. 2015;15:140. doi: 10.1186/s12866-015-0475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varble A., Albrecht R.A., Backes S., Crumiller M., Bouvier N.M., Sachs D., García-Sastre A., Tenoever B.R. Influenza a virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe. 2014;16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ross P.A., Endersby-Harshman N.M., Hoffmann A.A. A comprehensive assessment of inbreeding and laboratory adaptation in Aedes aegypti mosquitoes. Evol. Appl. 2019;12:572–586. doi: 10.1111/eva.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schirtzinger E.E., Jasperson D.C., Swanson D.A., Mitzel D., Drolet B.S., Richt J.A., Wilson W.C. Establishment of a Culex tarsalis (Diptera: Culicidae) Cell Line and its Permissiveness to Arbovirus Infection. J. Med. Entomol. 2023;60:239–244. doi: 10.1093/jme/tjac155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi P.-Y., Tilgner M., Lo M.K., Kent K.A., Bernard K.A. Infectious cDNA Clone of the Epidemic West Nile Virus from New York City. J. Virol. 2002;76:5847–5856. doi: 10.1128/jvi.76.12.5847-5856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weger-Lucarelli J., Duggal N.K., Bullard-Feibelman K., Veselinovic M., Romo H., Nguyen C., Rückert C., Brault A.C., Bowen R.A., Stenglein M., et al. Development and Characterization of Recombinant Virus Generated from a New World Zika Virus Infectious Clone. J. Virol. 2017;91 doi: 10.1128/JVI.01765-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gregori J., Perales C., Rodriguez-Frias F., Esteban J.I., Quer J., Domingo E. Viral quasispecies complexity measures. Virology. 2016;493:227–237. doi: 10.1016/j.virol.2016.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sequencing data files have been deposited at Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
All original code used for data processing, analysis, and visualization has been deposited on GitHub. Link to repository is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.