

Abstract
The understanding of the pathophysiology of bipolar disorder (BD) remains modest, despite recent advances in neurobiological research. The mitochondrial dysfunction hypothesis of bipolar disorder has been corroborated by several studies involving postmortem brain analysis, neuroimaging, and specific biomarkers in both rodent models and humans. Evidence suggests that BD might be related to abnormal mitochondrial morphology and dynamics, neuroimmune dysfunction, and atypical mitochondrial metabolism and oxidative stress pathways. Mitochondrial dysfunction in mood disorders is also associated with abnormal Ca2+ levels, glutamate excitotoxicity, an imbalance between pro- and antiapoptotic proteins towards apoptosis, abnormal gene expression of electron transport chain complexes, and decreased ATP synthesis. This paper aims to review and discuss the implications of mitochondrial dysfunction in BD etiology and to explore mitochondria as a potential target for novel therapeutic agents.
Keywords: Mitochondria, Bipolar disorder, Mania, Depression, Mitochondrial modulators
1. Introduction
Bipolar disorder (BD) is a chronic and debilitating psychiatric illness characterized by episodes of mania or hypomania, depression, and mixed states, often accompanied by psychotic features and functional impairment (Cotrena et al., 2016; Grande et al., 2016; Zarate et al., 2000). Despite the advances in neurobiological research, comprehension of the pathophysiology of BD remains modest. Several studies have suggested that the pathophysiology of BD is orchestrated by several mechanisms synergistically involving complex interactions between genetic predisposition and exposure to different environmental risk factors and psychosocial stressors (Manji et al., 2011). Numerous research findings have proposed diverse mechanisms in the pathophysiology of BD, including neurotrophic factors, epigenetic mechanisms, mitochondrial dysfunction, oxidative stress, inflammation, circadian rhythm abnormalities, and biological aging acceleration (Scaini et al., 2020).
In the past few years, BD has been suggested to be a progressive condition, characterized by cognitive impairment and functional decline (Knezevic and Nedic, 2013; Leboyer and Kupfer, 2010; Muneer, 2016). Neuroprogression in BD may be considered pathological rewiring of the brain that occurs in parallel with the functional and clinical deterioration over the course of the disorder (Berk, 2009; Berk et al., 2014, 2011b; Fries et al., 2012; Gama et al., 2013; Schneider et al., 2012). Evidence from the literature demonstrates that structural brain changes and cognitive deficits are not consistently observed at early stages of BD, appearing to become more evident with chronicity and number of episodes (El-Badri et al., 2001; Lyoo et al., 2006; Robinson and Ferrier, 2006; Strakowski et al., 2002). In this context, neuroprogression seems to be related to the cumulative effects of immune dysfunction, enhanced oxidative stress, neurotrophic support breakdown, mitochondrial dysfunction, and impairment of cellular resilience (Berk et al., 2011b; Fries et al., 2012; Kapczinski et al., 2008; Post et al., 2012). Staging models of BD have suggested that multiple episodes may lead to permanent alterations, including significant systemic toxicity, cognitive and functional impairment, and biological changes, which may be transduced to greater liability to relapse and poorer treatment response (Gama et al., 2013; Kapczinski et al., 2010; Post et al., 2012). However, such a progressive feature does not seem to be present across all patients, with some not experiencing as much cognitive or functional impairment as others (Passos et al., 2016), suggesting the existence of subgroups within BD.
Mitochondria are unique intracellular organelles that contain their own DNA (mtDNA) and are composed of inner and outer membranes with an intermembrane space and an intracellular matrix (Velot and Srere, 2000). Mitochondria are classically described as the powerhouse of the cell, as they are the source of adenosine triphosphate (ATP) through the process of oxidative phosphorylation via the electron transport chain (ETC) (Briere et al., 2004; Fang et al., 2016). The ETC is composed of five multimeric protein complexes (I-IV and ATP-synthase or complex V) that are responsible for ATP production through oxidative phosphorylation. Complex I (NADH: ubiquinone oxidoreductase) and complex II (succinate dehydrogenase) begin the process of oxidative phosphorylation by catalyzing the transfer of electrons from NADH and FADH2, respectively, to coenzyme Q (or ubiquinone). The transfer of electrons is serially conducted through complex III (ubiquinol: cytochrome c oxidoreductase), cytochrome c, and complex IV (cytochrome c oxidase), to the terminal acceptor, generating an electrochemical proton gradient that enhances ATP production in complex V via oxidative phosphorylation (Lenaz and Genova, 2010; Velot and Srere, 2000).
In addition to ATP generation, mitochondria play an important role in other cellular functions, including reactive oxygen species (ROS) production, cell death pathways (apoptosis), and calcium (Ca2+) homeostasis (Budd and Nicholls, 1998; Chakrabarty et al., 2018; Kadenbach et al., 2010). Mitochondria also modulate some of the effects of brain-derived neurotrophic factor (BDNF) in neurons and, therefore, exert an essential function regarding synaptic plasticity (Burkhalter et al., 2003). Multiple signaling pathways stimulate mitochondrial biogenesis, motility, shape, fusion and fission, and energy metabolism due to activation of neurotransmitters and growth factor receptors, which are ultimately related to memory and learning processes (Cheng et al., 2010; Ly and Verstreken, 2006). Moreover, mtDNA is associated with higher rates of damage compared to nuclear DNA due to exposure to ROS and insufficient repair mechanisms (Cuperfain et al., 2018).
Given the role of this organelle, it is not surprising that mitochondrial dysfunction has been implicated in a variety of neurological and psychiatric disorders, including BD (Cuperfain et al., 2018; Kim et al., 2019; Panchal and Tiwari, 2019; Pei and Wallace, 2018; Streck et al., 2014). The mitochondrial dysfunction hypothesis suggests that BD is triggered, in part, via mitochondria dysfunction, which is intimately linked to a wide range of processes associated with treatment outcomes and disease progression or severity, including inflammation, oxidative stress, stress response systems, and accelerated aging. Interestingly, studies in primary mitochondrial diseases have shown a high prevalence of self-reported affective syndromes, particularly BD (Colasanti et al., 2020; Koene et al., 2009; Morava et al., 2010). However, it remains unclear whether abnormal mitochondrial function is a cause or consequence (or even both) of BD. This paper aims to review and discuss, in more detail, the implications of mitochondrial dysfunction in the pathophysiology of BD and to explore the mitochondria as a potential target for novel therapeutic agents, providing an overview of recent findings on the mechanisms of mitochondrial dysfunction in BD (Fig. 1).
Fig. 1. Schematic representation of mitochondrial dysfunction in bipolar disorder.
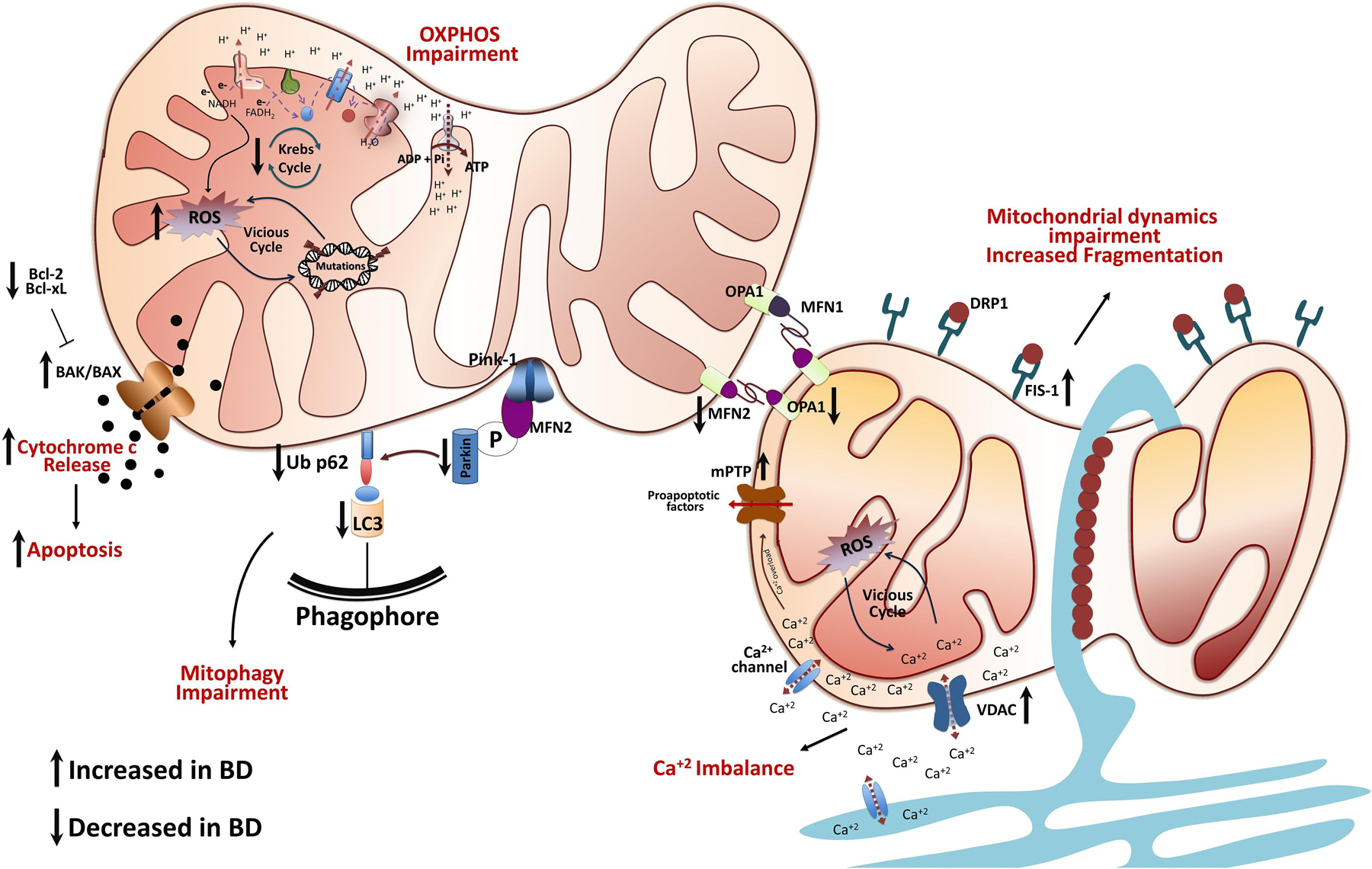
Several mitochondrial pathways, not all of which are shown here, interact simultaneously to cause cellular damage. Mitochondrial dysfunction in the pathophysiology of BD is based on oxidative phosphorylation impairment, a decrease in energy production, ROS production, mtDNA damage, Ca+2 imbalance, cytochrome c release, and mitochondrial dynamics and mitophagy impairment. Thus, mitochondrial dysfunction could initiate a vicious cycle where numerous systems and mechanisms intensify and accelerate cellular damage, that with time exacerbates the disease process, and could independently determine the course of the disease, progression, functional outcomes and premature aging.
2. Changes in mitochondrial bioenergetics and morphology in bipolar disorder
Mitochondria are the primary source of ATP synthesis and the major intracellular source of vital energy production in the brain. As a direct by-product of this ATP production, mitochondria also produce reactive oxygen species (ROS) that play an important role in cell signaling and regulation (Rhee, 1999; Thannickal and Fanburg, 2000). However, under pathological conditions, when cellular production of ROS overwhelms antioxidant capacity, oxidative stress occurs, causing damage to lipids, proteins, and DNA (Rhoads et al., 2006).
Over the past several decades, researchers have highlighted the role of this organelle in the pathophysiology of BD. A series of magnetic resonance spectroscopy (MRS) and phosphorus-31 MRS (31P-MRS) studies, Kato and colleagues have shown that patients with BD exhibited alterations in neurometabolites, including high-energy compounds, all of which are hallmarks of a decrease in mitochondrial energy production (Kato, 2005; Kato et al., 1998, 1992, 1994). In summary, patients with mood disorders present with lower levels of phosphocreatine (PCr), N-acetyl-aspartate (NAA), adenosine diphosphate (ADP), and ATP (Kato et al., 1992; Scaini et al., 2016). Moreover, studies have noted that negative correlations exist between NAA/Creatine + PCr or NAA levels and illness duration in BD (Chang et al., 2003; Deicken et al., 2003; Winsberg et al., 2000). Furthermore, some investigators have suggested that patients with BD exhibit an abnormality in enzymatic reaction rate in the creatine kinase (CK) system. These studies have shown a decrease in the forward rate constant of the CK enzyme without alterations in ATP or PCr levels, as well as downregulation of CK in postmortem brains of BD patients (Du et al., 2018; MacDonald et al., 2006). These findings are in line with a study indicating that individuals with BD maintain average brain concentrations of high-energy compounds at rest. However, when energy demand is increased, BD patients exhibited significant reductions in ATP but not in PCr, suggesting a failure to replenish ATP from PCr through CK enzyme catalysis during tissue activation (Yuksel et al., 2015).
Based on findings showing that BD patients present high lactate levels, a product of increased glycolysis, and reduced intracellular pH in the brain, studies have also suggested that in BD, during mitochondrial dysfunction, cells shift primarily to glycolytic metabolism to generate ATP (Dager et al., 2004; Dogan et al., 2018; Kato et al., 1998, 1992, 1994; Kuang et al., 2018). Supporting this hypothesis, several studies have reported mitochondrial-specific changes regarding the Krebs cycle and oxidative phosphorylation in BD. A metabolomics study showed that BD patients had higher serum levels of pyruvate, N-acetylglutamic acid, α-ketoglutarate, and arginine, whereas levels of β-alanine and serine were significantly lower (Yoshimi et al., 2016b). The same research group found evidence for abnormalities in the metabolism of isocitrate in cerebrospinal fluid (CSF) and postmortem brain tissue of BD patients. In line with these findings, Zverova et al. (2019) reported a significant decrease in citrate synthase (CS), complex II, and complex IV activities in platelets from BD patients during a depressive episode. In contrast, complex I activity and complex I/CS ratio were increased in these patients. Along the same lines, Valvassori et al. (2018) revealed that depressed BD patients present decreased complex II activity in peripheral blood mononuclear cells (PBMCs). On the other hand, studies on postmortem frontal cortex from patients with BD showed a decrease in complex I activity (Andreazza et al., 2010; Kim et al., 2015). Contrary to these reports, studies have reported that activity of complexes I, II-III and IV were unaltered in PBMCs and platelets from BD patients (Ben-Shachar et al., 1999; Gubert et al., 2013). Moreover, a study in drug naïve BD patients reported no significant alterations in citrate synthase, malate dehydrogenase, or succinate dehydrogenase (de Sousa et al., 2015).
Microarray and real-time quantitative polymerase chain reaction (qPCR) data revealed decreased expression of many messenger RNAs (mRNAs) encoding subunits of ETC complexes I to V in BD patients (Table 1). Studies have shown significant differences in mRNA levels for complex I subunits NDUFV1, NDUFV2, NDUFS1, NDUFS7 and NDUFS8 in patients with BD, yet a follow up study described no difference (Akarsu et al., 2015; Andreazza et al., 2010, 2013; Iwamoto et al., 2004; Konradi et al., 2004; Sun et al., 2006). In a re-analysis of microarray studies in postmortem brains, Scola et al. (2013) showed that subunits containing iron-sulfur clusters within the hydrophilic arm (NDUFV2, NDUFS8, and NDUFS7) were downregulated in BD patients, suggesting dysfunction of the electron transfer process. Subunits associated with complex III (UQCRC2), complex V (COX7A2, COX7B, COX5A, COX6C, and COX15), and complex IV (ATP5C1, ATP5G3, ATP5H, ATP5J, ATP5J2, and ATP5O) have also been reported as decreased in the prefrontal cortex and hippocampus, while in blood, increases in subunits of the ETC were observed (Beech et al., 2010; Iwamoto et al., 2004; Konradi et al., 2004; Sun et al., 2006). Moreover, Naydenov et al. (2007) showed that during glucose deprivation, genes associated with complex III and IV were downregulated in lymphocytes from patients with BD. However, a recent multivariate meta-analysis of mitochondrial complexes I and IV revealed that alterations in these complexes are not always consistent, suggesting brain region- and tissue-specific heterogeneity. In summary, they found moderate effects on complex I and small effects in complex IV (Holper et al., 2019). In addition to the alterations in genes of the ETC, Yoshimi et al. (2016a) showed downregulation of isocitrate dehydrogenase genes (IDH3A and IDH3B) in BD patients. Considering these findings, we hypothesized that besides downregulation in subunits of the ETC complexes, alterations in Krebs cycle enzymes cause a decrease in its velocity, limiting NADH production for complex I, which might cause maximal reduction of the rate and efficiency of electron transfer.
Table 1.
Summary of mitochondrial complex gene alterations in bipolar disorder.
|
| |||||
| Reference | Gene |
Description
|
Sample | Expression | |
| Iwamoto et al. (2004) | NDUFS1 |
Complex I |
NADH dehydrogenase (ubiquinone) Fe-S protein 1 |
Prefrontal cortex | ↓ |
| UQCRC2 | Complex III | Ubiquinol-cytochrome c reductase core protein II | |||
|
|
COX15 | Complex IV | Cytochrome c oxidase assembly homolog 15 |
|
|
| Konradi et al. (2004) | NDUFAB1 | Complex I | NADH dehydrogenase (ubiquinone) 1, α/β subcomplex 1 | Hippocampus | ↓ |
| COX7A2 | Complex IV | Cytochrome-c oxidase, subunit VIIa, polypeptide 2 | |||
| COX7B | Cytochrome-c oxidase, subunit VIIb | ||||
| ATP5G3 | Complex V | ATP synthase, mitochondrial Fo complex, subunit C3 | |||
| ATP5H | ATP synthase, mitochondrial Fo complex, subunit d | ||||
| ATP5J2 | ATP synthase, mitochondrial Fo complex, subunit F2 | ||||
| ATP5C1 | ATP synthase, mitochondrial F1 complex, ɣ polypeptide 1 | ||||
| ATP5O | ATP synthase, mitochondrial F1 complex, O subunit | ||||
| Sun et al. (2006) | NDUFS7 | Complex I | NADH dehydrogenase (ubiquinone) Fe-S protein 7 | Prefrontal cortex | ↓ |
| NDUFS8 | NADH dehydrogenase (ubiquinone) Fe-S protein 8 | ||||
| UQCRC2 | Complex III | ubiquinol-cytochrome c reductase core protein II | |||
| COX5A | Complex IV | cytochrome c oxidase subunit Va | |||
| COX6C | cytochrome c oxidase subunit Vic | ||||
| ATP5C1 | Complex V | ATP synthase, mitochondrial F1 complex, ɣ polypeptide 1 | |||
| ATP5J | ATP synthase, mitochondrial Fo complex, subunit F6 | ||||
| ATP5G3 | ATP synthase, mitochondrial Fo complex, subunit C3 | ||||
| Andreazza et al. (2010) | NDUFS7 | Complex I | NADH dehydrogenase (ubiquinone) Fe-S protein 7 | Prefrontal cortex | ↓ |
| Akarsu 2015 | NDUFV1NDUFV2NDUFS1 | Peripheral blood samples | ↑ | ||
|
| |||||
In addition to alterations in energy metabolism, recent attention has focused on the role of the mitochondrial quality control system in BD. This system is sustained through synthesis of new mitochondria, the processes of fusion and fission, and elimination of damaged mitochondria (Kornmann, 2014), which is responsible for the maintenance of a healthy mitochondrial population that is fundamental to cell health and viability (Boveris et al., 1972; Fischer et al., 2012). Previous research findings into mitochondrial morphology have reported that postmortem prefrontal cortical neurons from patients with BD and their peripheral cells display morphological abnormalities (more mitochondria of smaller size). Moreover, the same authors showed an abnormal pattern of clumping and marginalization with respect to the intracellular distribution of mitochondria in peripheral cells and atypically shaped mitochondria (ring- or cup-shaped mitochondrial profiles), suggesting subtle changes in the critical network architecture of mitochondria in cells (Cataldo et al., 2010). In an elegant study using a BD neuronal model based on iPSC technology, Mertens et al. (2015) corroborated previous findings showing that iPSC-derived fibroblasts from patients with BD exhibited smaller mitochondria than those from iPSC-derived fibroblasts of HCs.
Following these findings, Scaini et al. showed that BD patients present alterations in the levels of proteins governing mitochondrial fission and fusion, causing an imbalance in mitochondrial fission and fusion towards fission. The same group also showed impairment in the removal of damaged mitochondria via the mitophagy pathway, followed by apoptosis activation in BD patients (Scaini et al., 2019, 2017b). In summary, these studies revealed downregulation of the mitochondrial fusion-related proteins Mfn-2 and Opa-1 and upregulation of the fission protein Fis-1 in PBMCs from BD patients, suggesting that the imbalance in mitochondrial dynamics might explain the abnormal mitochondrial morphology observed in patients with BD. Moreover, mRNA expression and protein levels of Parkin, p62, and LC3B, which are mitophagy-related proteins, were decreased in PBMCs from BD patients, followed by higher levels of activated caspase-3. Taken together, these results suggest that in BD, the number of damaged mitochondria exceeds the capacity of mitophagy, causing apoptosis to become the dominant pathway to minimize tissue damage (Scaini et al., 2019, 2017b). The lack of mitophagy leads to a loss of mitochondrial quality control, resulting in a release of damage-associated molecular patterns (DAMPs) that induce sterile inflammation by interacting with Toll-like receptor 9 (TLR9) and promoting activation of NF-κB and the NLRP3 inflammasome (Nakahira et al., 2011; Pinti et al., 2014; Wu et al., 2019; Yuzefovych et al., 2019). Not surprisingly, inflammation has been associated with BD by many studies and research groups (Fries et al., 2019b)
As mentioned above, defects in the ETC result in increased ROS production, culminating in oxidative stress and changes in energy production that lead to a vicious cycle of cellular dysfunction and death (Ott et al., 2007; Sanz et al., 2006). In fact, oxidative stress has been implicated in BD, being explored as an agent of accelerated aging in BD (Fries et al., 2020). Several markers of oxidative stress were found to be altered in patients with BD, including glutathione peroxidase, 8-oxo-2′-deoxyguanosine (8-oxodG), 8-oxo-7,8-dihydroguanosine (8-oxoGuo), lipid peroxidation hydroperoxides, glutathione S-transferase, glutathione reductase, 3-nitrotyrosine, protein carbonyl, and thiobarbituric acid reactive substances (Andreazza et al., 2013; Ceylan et al., 2018; de Sousa et al., 2014; Gubert et al., 2013; Raffa et al., 2012; Rosa et al., 2014). A meta-analysis of studies that measured oxidative stress markers in BD patients compared to healthy controls showed that lipid peroxidation, DNA/RNA damage, and nitric oxide were significantly increased in BD patients (Brown et al., 2014). Another study demonstrated a significant increase in GSH concentrations in the anterior cingulate cortex (ACC) of BD patients (Das et al., 2019). Moreover, a recently published study focusing on cerebral spinal fluid revealed that 8-oxoGuo is statistically significantly higher at baseline and follow-up in BD, suggesting that CSF oxidative stress may represent state and trait markers in BD and may reflect neurobiological correlates of illness progression and sensitization (Knorr et al., 2019).
3. Bipolar disorder and changes in calcium and apoptosis
As mentioned above, mitochondria are the primary source of cellular energy. However, they are also responsible for other crucial processes for cellular function and survival, such as apoptosis and Ca2+ homeostasis (Zavodnik, 2016). Mitochondria play a part in intracellular Ca2+ signaling as modulators, buffers and sensors (Rizzuto et al., 2012). Ca2+ influx and efflux through the outer mitochondrial membrane occurs by the voltage-dependent anion channel (VDAC), the principal metabolite transport system across the outer mitochondrial membrane. While mitochondrial Ca2+ uniporter (MCU), mitochondrial ryanodine receptor (mRyR), and rapid mode of Ca2+ uptake (RaM) are responsible for Ca2+ influx through the inner mitochondrial membrane (IMM) (Bravo-Sagua et al., 2017). Ca2+ uptake in the mitochondrion has essential roles such as spatially remodeling intracellular Ca2+ signaling, controlling the rate of energy production, being conducive to cellular death, and neuronal excitability (Belosludtsev et al., 2019; Giorgi et al., 2018; Gorlach et al., 2015). On the other hand, Ca2+ overload causes endoplasmic reticulum stress and Ca2+ leakage, which affects mitochondrial uptake, causing an excessive Ca2+ taken up into mitochondria, resulting in a collapse of the mitochondrial membrane potential, termination of oxidative phosphorylation processes, osmotic changes, mitochondrial swelling, and inner membrane remodeling, mitochondrial permeability transition pore (mPTP) opening, cytochrome c release, activation of caspases and apoptosis, and subsequently cell death (Belosludtsev et al., 2019; Bravo-Sagua et al., 2017; Giorgio et al., 2018; Marchi et al., 2018).
Altered intracellular Ca2+ levels are a consistent finding in BD patients, and many studies point to the role of mitochondrial dysfunction, indicating the possibility that mitochondrial Ca2+ dysregulation is involved in the pathophysiology of BD (Kato, 2008). Several studies have investigated intracellular Ca2+ dynamics and alterations in the pathophysiology of BD. Most of these studies consistently found elevated basal (Ca2+B) and agonist-stimulated (Ca2+S) intracellular Ca2+ levels in the platelets, lymphocytes, B lymphoblast cell lines (BLCLs) and olfactory neurons of BD patients compared with healthy subjects (Corson et al., 2001; Dubovsky et al., 2014; Emamghoreishi et al., 1997; Hahn et al., 2005; Hough et al., 1999; Perova et al., 2008). Moreover, postmortem brain studies have also found altered mRNA expression of inositol monophosphate type II (IMPase) and a transient receptor potential channel (TRPM2), proteins that are involved in the Ca2+ homeostasis (Yoon et al., 2001a, 2001b). In fact, a recently systematic review and meta-analysis of studies of cellular calcium levels in BD found a robust, medium effect size elevation of basal and stimulated free intracellular Ca2+, suggesting altered Ca2+ functioning in the disorder (Harrison et al., 2019). In line with this, studies have proposed that chronic lithium at therapeutic doses blocks the increase in Ca2+ concentration, attenuating agonist-stimulated intracellular Ca2+ responses (Gould and Manji, 2005; Gurvich and Klein, 2002; Hashimoto et al., 2002; Li et al., 2002). Wasserman et al. (2004) showed that lithium exerts significant modulatory effects on intracellular Ca2+ dynamics in BLCLs from BD-I patients through the attenuation of receptor-G-protein coupled agonist (lysophosphatidic acid (LPA)) stimulation of Ca2+ mobilization (a G-protein-coupled receptor activated signaling pathway), and TG-evoked store-depletion-induced Ca2+ influx (a measure of SOCE).
More recently, Mertens et al. (2015) demonstrated that young hippocampal dentate gyrus-like neurons derived from iPSCs of patients with BD had smaller mitochondria than controls, as well as changes on the gene expression of pathways involving Ca2+ signaling, neuroactive ligand-receptor interaction, and protein kinase PKA/PKC signaling, in addition to changes in the action potential firing system. Moreover, using neuronal hyperactivity and lithium responsiveness as two indices, the same authors detected correlated changes in the PKA/PKC/AP and mitochondria genes in the BD neurons, indicating that these pathways might be related to neuronal hyperexcitability. Indeed, excess Ca2+ affects both neuronal excitability and signaling cascades regulating gene expression, leading to perturbation of multiple neuronal processes, such as dendrite development, synaptic plasticity, and excitatory/inhibitory balance (Greer and Greenberg, 2008). Therefore, these changes in Ca2+ dynamics described in patients with BD might be related to disturbances in homeostatic cellular physiology control.
Another fascinating aspect of Ca2 + signaling is its capacity to regulate cell death. One of the most well-studied Ca2+-induced cell death pathways is the cross-talk between the endoplasmic reticulum and mitochondria. As described above, mitochondrial Ca2 + overload induces several changes that culminate by mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c, which is an inducer of apoptosis and modulator of other proapoptotic factors, such as Smac/DIABLO, HtrA2/Omi, AIF and EndoG. Moreover, it is proposed that VDAC1, play crucial role in apoptosis activation mediated by cytochrome c release through transfers of apoptotic Ca2 + signals from the endoplasmic reticulum to mitochondria, as well as by the interaction with the Bcl-2 family of proteins (Shimizu et al., 2000b, 2001; Tsujimoto and Shimizu, 2000; Azoulay-Zohar et al., 2004).
The widely described presence of apoptosis in cells from BD patients occurs through several mechanisms, including Ca2+-mediated excitotoxicity, as mentioned above, increased oxidative stress parameters, and profound changes in mitochondrial structure and function (Giorgi et al., 2008). A postmortem brain study in BD patients revealed a significant decrease in levels of Bcl-2 and brain-derived neurotrophic factor (BDNF), whereas Bax, Bad and caspase-3 and −9 levels were significantly increased (Kim et al., 2010). The changes observed in postmortem brain studies parallel changes observed in peripheral cells of patients with BD. Bei et al. (2009) observed decreased cytosolic Bax, suggesting an increase in Bax activation and translocation to mitochondria to activate apoptosis, as well as decreased expression of HSP70, an antiapoptotic factor. In agreement with these findings, Moutsatsou et al. (2014) showed an increase in the BAX/Bcl2 ratio, cytochrome c release, and caspase-3 activity in manic and depressed patients compared to healthy controls. Likewise, higher levels of antiapoptotic proteins, Bcl-xL, survivin, and the Bcl-xL/Bak dimer, were significantly decreased in PBMCs from patients with BD, while active caspase-3 levels were significantly increased (Scaini et al., 2017a). The same authors showed that BD patients present higher levels of VDAC1 and TSPO (Scaini et al., 2019). Another study showed that in depressed patients, S100B levels correlated with cytochrome c release (Paraskevi et al., 2014). Furthermore, the percentage of cells in early apoptosis was higher in PMBCs from BD patients compared to controls (Fries et al., 2014). Taken together, could be hypothesized that in patients with BD, the selective placement of VDAC channels at ER/mitochondria contact sites facilitates mitochondrial Ca2 + accumulation leading to cell death. However, further experimental investigations are needed to estimate the functional relevance of the crosstalk between VDAC, mitochondrial Ca2 + dynamics, and ROS metabolism to the apoptosis activation in BD patients.
4. Mitochondria, redox system and circadian rhythmicity in bipolar Disorder. How do these Interrelate?
Several studies have demonstrate that, at the cellular level, mitochondrial oxidative phosphorylation and redox states are regulated in a circadian manner, and also signal back to the core clock, suggesting a regulation loop exists between clock machinery and metabolism (de Goede et al., 2018; Peek et al., 2013; Schmitt et al., 2018). Several studies have shown that suprachiasmatic nucleus neurons and astrocytes exhibit daily rhythms in cytochrome c oxidase activity, mitochondrial membrane potential and calcium release from mitochondria (Burkeen et al., 2011; Isobe et al., 2011; Marpegan et al., 2011). In fact, studies have described that metabolic byproducts (e.g.: NAD+, ATP) display 24 h oscillation and can in turn regulate the clock. The rhythms of these byproducts are believed to be dependent on the molecular clockwork because the activity of the rate-limiting enzyme nicotinamide phosphorribosyltransferase (NAMPT) is controlled by CLOCK–BMAL1 activator complex in conjunction with NAD + -dependent deacetylase sirtuin 3 (SIRT3) (Nakahata et al., 2009; Ramsey et al., 2009; Sahar et al., 2011). Moreover, it was shown that the transcriptional coactivator PGC-1α, which is the master regulator of mitochondrial biogenesis and energy metabolism, is an essential component of the circadian clock in the liver and muscle (Liu et al., 2007). In the same line, studies have also suggest that the activation of AMPK (adenosine monophosphate-dependent protein kinase), can directly modulate the core clock machinery by phosphorylation of the clock repressor proteins CRY and PER, targeting them for proteasomal (Jordan and Lamia, 2013). Of the note, evidence has also shown that alterations in mitochondrial morphology (i.e., fusion and fission) and mitochondrial abundance are under circadian clock control (Jacobi et al., 2015; Schmitt et al., 2018). These cycles of fission and fusion and, consequently, the rhythms of energy production are regulated by phosphorylation and activity of dynamin-related protein 1 (DRP1), which occurs in a circadian manner to drive mitochondrial network fragmentation (Schmitt et al., 2018).
So given the intimate connection between the circadian system and redox sensing, it seems reasonable to suspect that a disruption in metabolic/clock cross talk might lead to poor cellular health. At this point to be remember that mitochondrial dysfunction is a key element in the pathophysiology of BD. Moreover, the role of SIRT3 and PGC-1α in the context of BD phenotypes and treatments has been suggested but not studied in depth. A study using PGC-1α null mice showed that the lack of PGC-1α in GABAergic neurons causes an increase in the activity across tests that might be related to a mania-like phenotype. Geoffroy et al. (2016) performed a pharmacogenetic study and found an association between PGC-1α and lithium response in BD patients, but this association did not survive Bonferroni correction, requiring additional studies to provide more functional/biological data supporting these preliminary finding. Of the first consideration, BD are associated with major disruptions in circadian rhythms. Studies have demonstrate that genes such as Clock, Bmal1, and Per, which are intimately involved in the generation and regulation of circadian rhythms, have been linked to BD, as well as illness severity (Geoffroy, 2018; Maciukiewicz et al., 2014; McCarthy et al., 2012, 2013; Milhiet et al., 2014; Shi et al., 2008; Yang et al., 2009)
Taking together the evidence of the correlation between mitochondrial and circadian clock dysfunction, and the fact the cross-sectional research design of most studies precludes establishing a cause/effect relationship between mitochondrial dysfunction, circadian disruption, and BD, it remains undetermined what comes first. Of note, given that mitochondrial metabolism is connected, on the one hand, with circadian rhythms and, on the other hand with the pathogenesis of BD, it would be a thrilling challenge to highlight the impending interplay between mitochondrial network, the circadian rhythm in both physiological systems.
5. Genetic evidence for mitochondrial dysfunction in bipolar disorder
As mentioned above, mitochondria are organelles that contain their own DNA, which is responsible for encoding genes that include proteins involved in oxidative phosphorylation, as well as some unique transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) (Calvo et al., 2016; Taanman, 1999). Although mitochondria have their own DNA, cellular metabolism and mitochondrial function require a combination of proteins that are also encoded in the nucleus (Cuperfain et al., 2018). Thus, both nuclear DNA (nDNA) and mtDNA must be analyzed in a genetic investigation of mitochondrial dysfunction.
Several studies suggest that BD possesses an important genetic component, with twin studies showing heritability rates as high as 70–80% (Edvardsen et al., 2008; McGuffin et al., 2003). The high genetic component is also shown by the increased risk of BD in first-degree relatives of patients (Gottesman et al., 2010). Accordingly, children of parents diagnosed with BD have been shown to have a significantly increased risk to develop the disorder compared to offspring of control parents (Duffy et al., 2019; Rasic et al., 2014) and often present signs and symptoms that may accompany familial risk in the absence of a BD diagnosis (Axelson et al., 2015; Diler et al., 2017; Dong et al., 2019; Levenson et al., 2015; Manelis et al., 2016).
Recent studies have investigated specific mutations related to mitochondrial function in mtDNA and nDNA, such as single nucleotide polymorphisms (SNPs), deletions, copy number variations, as well as epigenetics and haplogroups, which may be correlated with the pathophysiology of psychiatric disorders, including BD (Cuperfain et al., 2018; Kasahara and Kato, 2018; Schulmann et al., 2019). Munakata et al. (2004) suggested that the 3644C mutation is associated with increased risk for BD, by decreasing mitochondrial membrane potential and complex I activity, due to a 3644 T → C that causes a V113A amino acid substitution in NADH-ubiquinone dehydrogenase subunit I (ND1). It was also identified that in postmortem brains of BD patients, there was increased gene expression of mitochondrial leucyl-tRNA synthetase, which is related to the accumulation of 3243A → G in mtDNA (Munakata et al., 2005). Moreover, different amino acid substitutions in specific candidate genes and haplogroups in BD patients have been reported (Kato and Kato, 2000; Kato et al., 2001; Kazuno et al., 2009).
Genome-wide association studies (GWAS) have showed that genes encoding ankyrin 3 (ANK3) and the α-calcium channel subunit (CAC-NA1C) are considerable genetic risk factors for the development of BD (Fiorentino et al., 2014). In line with this, Michels et al., 2018 showed that downregulation of CACNA1C expression protected cells from oxidative stress by inhibiting excessive mitochondrial ROS generation and Ca2+ influx in mouse hippocampal cells. Recently, a trio-based WES study for BD published by Kataoka et al. (2016) identified potential roles of de novo protein-altering mutations and calcium-related genes in the disease etiology most of which create non-functioning proteins, predominantly in genes for calcium-binding proteins. The same authors described that earlier age at onset in carriers of protein-altering de novo mutations, and the rate of de novo mutations was higher in BD-I, suggesting a role for these mutations in BD (Kataoka et al., 2016). Schulmann et al. (2019) demonstrated that the odds ratios for BD risk decreased from 1.1 to 0.38 by adding a mtSNP minor allele compared to the mtSNP major allele for the nuclear gene AK5, a brain-specific gene involved in the synthesis of ADP and the cytochrome B oxidase gene (CYTB), suggesting. These findings suggest that both genomes jointly determine the cellular energy production requirements, impairment of which is associated with risk for BD (Schulmann et al., 2019).
Recently Fries et al. (2017) showed that BD patients may display accelerated epigenetic aging, most likely due to high mtDNA copy number (mtDNAcn), although no association with telomere shortening was observed (Fries et al., 2017). The same group also demonstrated a reduction in mtDNA copy number and an epigenetic aging acceleration in postmortem hippocampus from BD patients compared to controls among older subjects (Fries et al., 2019a). However, a meta-analysis identified no significant differences between mtDNA copy numbers in BD patients. Another meta-analysis, including a low level of heterogeneity, revealed significantly lower mtDNAcn in patients (Yamaki et al., 2018). Recent studies have also described a downregulation of DNA polymerase gamma (POLG) and 8-oxoguanine-DNA glycosylase 1 (OGG1), a DNA repair enzyme, in patients with BD, suggesting that this may be one of the mechanisms responsible for reduced mtDNA copy number (Ceylan et al., 2018; Munkholm et al., 2015).
6. Effects of mood stabilizers on mitochondria
Multiple drugs, such as lithium, some anticonvulsants and antipsychotics, have been shown to be effective in treating or preventing manic and depressive episodes; however, full comprehension of their precise mechanisms of action in BD remains unknown with little progress being made in developing novel medications. It has been postulated that mood stabilizers likely enhance energy metabolism by changing intracellular signaling pathways (Hroudova and Fisar, 2011), and there are several explanations for their neuroprotective properties. Among mood stabilizers, lithium seems to be the most investigated compound.
In vitro studies showed that lithium might stabilize mitochondrial membrane potential and reduce DNA damaging effects. Lithium may also delay Ca + 2-induced apoptosis by antagonizing the mPTP, as demonstrated in isolated rat mitochondria (Shalbuyeva et al., 2007). Findings of increased grey matter in BD subjects following lithium treatment is congruent with its antiapoptotic properties (Moore et al., 2000). Lithium also decreased levels of DNA methylation in BD patients (Huzayyin et al., 2014), increased levels of glutathione transferase (Clay et al., 2011; Cui et al., 2007; Shao et al., 2005), reduced apoptosis and enhanced catalase activity (Machado-Vieira et al., 2007).
Impairment of complex I function is related to increased generation of ROS (Sharma et al., 2011), and lithium stimulates the activity of this complex in the prefrontal cortex (Valvassori et al., 2010). Another study by Scola et al. (2014) demonstrated that lithium attenuated complex I dysfunction on DNA methylation and hydroxymethylation induced by rotenone in rat primary cortical neurons. In addition to complex I, lithium has also been observed to increase the activity of mitochondrial chain enzyme complexes II and III in human frontal cortex (Maurer et al., 2009). In addition to the numerous benefits related to lithium use, this medication is also associated with several side effects, and some studies have reported negative outcomes involving lithium and mitochondrial function. For example, one study reported reduced synthesis of antioxidants and decreased mitochondrial membrane potential in rat hepatocytes (Eskandari et al., 2012). The cardiotoxic adverse effects of lithium may be partially explained by inhibition of complex II activity and ATP formation, enhanced ROS generation and increased activity of proapoptotic caspase-3 (Salimi et al., 2017).
Valproate and lithium seem to increase the expression of the antiapoptotic Bcl-2, which leads to the inhibition of proapoptotic enzymes, such as caspase 3 (Chen et al., 1999). In addition, both drugs are related to inhibition of glycogen synthase kinase-3 (GSK-3) enzyme activity, which is responsible for modulating gene expression of proteins involved in apoptosis, synaptic plasticity and cellular resilience (Bachmann et al., 2009; Kato, 2011; Kazuno et al., 2008; Zarate et al., 2006). Lithium and valproate were found to increase production of BDNF, decrease glutamate-induced excitotoxicity and inhibit NMDA receptor-mediated Ca2 + influx (Hashimoto et al., 2002; Nonaka et al., 1998). Valproate also seems to influence mitochondrial epigenetics due to its potent inhibition of histone deacetylase (Chen et al., 2012).
Platelets and lymphocytes from BD subjects have been associated with increased levels of intracellular Ca2 +. In this context, valproate and carbamazepine seem to contribute to the regulation of intracellular Ca2 + levels by enhancing expression of ER stress proteins, which potentiates neuronal resistance to lethal fluctuations in intracellular Ca2 + and cytotoxic insults (Cikankova et al., 2019, 2017). Other studies have demonstrated that pretreatment with valproate prevented amphetamine-induced citrate synthase and succinate dehydrogenase inhibition in the brain of rats (Correa et al., 2007; Feier et al., 2013). Another study reported that valproate reversed metabolic alterations induced by ouabain administration in a model of mania (Lopes-Borges et al., 2015).
Some researchers suggest that mitochondria may also play a role in the side effects related to the use of mood stabilizers. The toxicity of valproate can be partially explained by several different mechanisms. An investigation of the effects of valproate on isolated rat mitochondria demonstrated that it might induce oxidative stress as a result of decreased mitochondrial respiratory chain complex II. Additionally, valproate induces the opening of mitochondrial ion channels and membrane pores, with consequent release of cytochrome c and induction of apoptosis (Jafarian et al., 2013). A recent study involving isolated pig brain mitochondria showed that complex I activity was significantly inhibited in response to lithium and carbamazepine, while activity of complex IV was decreased after exposure to carbamazepine. The activities of complex II and III were unaffected by any tested drug (Cikankova et al., 2019). Furthermore, carbamazepine may reduce mitochondrial ATP production and inhibit Ca2 + -induced cellular swelling in rat liver cells (Finsterer and Scorza, 2017). Moreover, lamotrigine seems to prevent opening of the mPTP and increase levels of glutathione (Kim et al., 2007).
7. Mitochondria as a therapeutic target in BD
In addition to the impact of mood stabilizers on cellular metabolism, the mitochondrial dysfunction hypothesis of BD has been contributing to recent clinical trials involving mitochondrial modulators. However, there are some limitations for the use of mitochondrial modulators in the clinical practice. Most of the clinical trials, as described in Table 2, recruited an insufficient number of participants. Therefore, the questionable power of these studies could partially explain some of the intriguing results about the effectiveness of mitochondrial agents. Another hypothesis for the limited use of mitochondrial modulators is that mitochondrial dysfunction may be only present in a fraction of BD patients, which corresponds to a mitochondrial endophenotype subgroup of the illness. This may explain the insufficient effectiveness of therapeutic agents when used in a broader patient population. In addition, it remains unclear if mitochondrial dysfunction is a consequence of bipolar disorder, instead of its cause, which could theoretically explain the fair effect of mitochondrial agents described in clinical trials. Ultimately, it is possible that mitochondrial modulators interact with different psychotropic drugs, such as mood stabilizers and antipsychotics, with a consequent insufficient bioavailability of mitochondrial agents in the central nervous system.
Table 2.
Clinical and preclinical trials of mitochondrial modulators in Bipolar Disorder.
| Author(Year) | N | Sample | Design | Duration | Assessments | Main findings |
|---|---|---|---|---|---|---|
| Berk et al. (2008) | 75 | Patients with bipolar disorder | Double-blind randomized placebo controlled | 6 months | MADRS, BDRS, YMRS, CGI-BP, GAF, SOFAS, SLICE/LIFE, LIFE-RIFT, Q-LES-Q | Positive results showed improvements on most rating scales in NAC group compared with placebo group |
| Berk et al. (2011) | 149 | Patients with moderate depression | Open-label study | 2 months | MADRS, BDRS, YMRS, CGI, CGI-BP, GAF, SOFAS, SLICE/LIFE, LIFE-RIFT, Q-LES-Q | Significant reduction in depressive symptoms in patients with bipolar depression |
| Berk et al. (2012) | 149 | Patients with bipolar disorder | Double-blind randomized placebo-controlled trial | 8 weeks | MADRS, BDRS, YMRS, CGI, CGI-BD, PGI, GAF, SOFAS, SLICE/LIFE, LIFE-RIFT, Q-LES-Q | No significant differences in recurrence, symptomatic outcomes, quality of life measures during the maintenance phase of the trial between the groups receiving adjunctive NAC or placebo in addition to treatment as usual. |
| Magalhaes et al. (2011) | 14 | Patients with bipolar disorder type II | Double-blind randomized placebo-controlled | 24 weeks | BDRS, MADRS, YMRS, CGI, GAF, SOFAS, SLICE-LIFE, LIFE-RIFT, Q-LES-Q | NAC group showed significant improvement on measures of symptom severity, functioning and quality of life and depressive symptoms |
| Magalhaes et al. (2013) | 15 | Patients with bipolar disorder in (mania or hypomania) | Double-blind randomized placebo-controlled | 24 weeks | BDRS, MADRS, YMRS, CGI, GAF, SOFAS, SLICE-LIFE, LIFE-RIFT, Q-LES-Q | NAC tended to have less manic symptoms at endpoint |
| Pettegrew et al. (2002) | 2 | Mildly depressed elderly subjects | Open-label study | 12 weeks | HDRS, MMSE, CIRS, CIRS and UKU | After 12 weeks of ALCAR treatment, subjects show remission, normalization of PME and elevation of PCr levels |
| Zanardi and Smeraldi (2006) | 193 | Patients with dysthymic disorder | Double-blind randomized parallel-group | 12 weeks | HAM-D, CDRS, MADRS, CGI, CGI-S, CGI-I | Treatment with ALCAR for 12 weeks was associated with improvements of depressive symptoms |
| Brennan et al. (2013) | 40 | Patients with bipolar depression | Double-blind randomized placebo-controlled | 12 weeks | MADRS, HAM-D, CGI-S, YMRS, 31P-MRS scan | ALCAR and ALA does not have antidepressant effects in depressed bipolar patients |
| Forester et al. (2012) | 10 | Older Patients (>55 years old) with bipolar depression | Open-label study | 2 weeks | MADRS, YMRS, CGI, GDS, Rate constant of creatine kinase | CoQ10 improvements depression symptom severity in BD patients, but not demonstrate significant differences in the rate constant of creatine kinase |
| Stoll et al. (1999) | 30 | Patients with bipolar disorder | Double-blind randomized placebo-controlled | 4 months | Kaplan-Meier survival, YMRS, HAM-D, CGI, GAS | Omega-3 group showed longer remission and improvements of depressive symptoms, bipolar symptoms and global functioning |
| Frangou et al. (2006) | 25 | Patients with bipolar depression | Double-blindplacebo-controlled trial | 12 weeks | HAM-D, YMRS, CGI | Treatment with ethyl-EPA over a 12 week period was associated with increased NAA levels |
| Murphy et al. (2012) | 45 | Outpatients with bipolar disorder type I | Double-blind, randomized add-on Clinical Trial | 4 months | MADRS, YMRS, GAF | Supplementation with omega-3 did not produce a significant improvement in mood symptoms over an extended period of treatment |
| Roitman et al. (2007) | 10 | Patients with resistant depression | Open-label | 4 weeks | HAM-D, HAS, CGI | Creatine supplementation causes a significant reduction in depressive symptoms in unipolar depression, but precipitated manic episodes bipolar patients Patients with bipolar disorder type II |
ALCAR: Acetyl-L-carnitine; ALA: Alpha-lipoic acid; BDRS: Bipolar Depression Rating Scale; CGI: Clinical Global Impression; CGI-BP: CGI-Improvement scale for bipolar disorder; CoQ10: Coenzyme Q10; GAF: Global Assessment of Functioning Scale; GAS: Global Assessment Scale; Ethyl-EPA: Ethyl-eicosapentanoic acid; HAM-D: Hamilton Depression Rating Scale; HAS: Hamilton Anxiety Scale; LIFE-RIFT: Longitudinal Interval Follow-up Evaluation-Range of Impaired Functioning Tool; MADRS: Montgomery Åsberg Depression Rating Scale; NAC: N-acetyl-cysteine; NAA: N-acetyl-aspartate; PGI: Patient Global Impression; Q-LES-Q: Quality of Life Enjoyment and Satisfaction Questionnaire; SLICE/LIFE: Streamlined Longitudinal Interview Clinical Evaluation from the Longitudinal Interval Follow-up Evaluation; SOFAS: Social and Occupational Functioning Assessment Scale; YMRS: Young Mania Rating Scale.
As described below, several metabolic antioxidants have been tested as potential add-on treatments in BD, including N-acetylcysteine (NAC), acetyl-L-carnitine (ALCAR), alpha-lipoic acid (ALA), creatine monohydrate, omega-3 fatty acids, and ketogenic diet.
7.1. N-Acetylcysteine
NAC is known for its beneficial properties, such as participation in glutathione production, anti-inflammatory effects, neurogenesis enhancement, as well as modulation of apoptosis and glutamate pathways (Samuni et al., 2013). According to Fernandes et al. (2016), adjuvant NAC treatment seems beneficial for bipolar depression. It was also found that a 24-week administration of NAC, as an adjuvant treatment, decreased depressive symptoms in BD patients, with more pronounced effects after 20 weeks of administration, suggesting long-term NAC treatment (Berk et al., 2008). An open label study investigating the efficacy of maintenance treatment with NAC in BD patients with depressive episodes reported a statistically significant reduction in the severity of depressive symptoms at the end of the 8-week treatment (Berk et al., 2011a). The same authors conducted a double-blind randomized placebo trial investigating the efficacy of adjuvant NAC in the maintenance treatment of bipolar depression and found no significant differences in recurrence or symptomatic outcomes during the maintenance phase of the trial (Berk et al., 2012). In contrast, Magalhaes et al. (2011) reported that NAC resulted in significant improvement in symptom severity, function and quality of life, and depressive symptoms after 24 weeks of treatment in patients with type-II BD in a randomized placebo-controlled trial. Another placebo-controlled, randomized clinical trial concluded that administration of NAC for 24 weeks resulted in attenuated manic symptoms in the NAC group and worsening depressive symptoms in the placebo group at endpoint (Magalhaes et al., 2013). On the other hand, there are recent negative trials showing no significant differences between groups with respect to primary outcomes (Berk et al., 2012, 2019; Ellegaard et al., 2019).
7.2. Acetyl-L-Carnitine and Alpha-Lipoic acid
ALCAR and ALA are compounds that play critical roles in modulating the cellular stress response by improving mitochondrial function and reducing mitochondrial free-radical production and inflammation, exhibiting antioxidant and anti-inflammatory properties (Liu, 2008; Liu et al., 1993; Scafidi et al., 2010a, 2010b). Overall, ALCAR and ALA are promising agents for the treatment and/or prevention of neurodegenerative disorders (Dos Santos et al., 2019; Hager et al., 2007; Molz and Schroder, 2017; Palacios et al., 2011; Soczynska et al., 2008).
In BD, several early clinical trials suggest the ALCAR has significantly greater efficacy than placebo as an augmentation treatment for depressive disorders (Pettegrew et al., 2002; Zanardi and Smeraldi, 2006), while a randomized placebo-controlled trial using ALCAR and ALA concluded that there was no statistically significant difference between ALCAR/ALA compared to placebo groups with respect to changes in the Montgomery-Asberg Depression Rating Scale (MADRS) scores (Brennan et al., 2013).
7.3. Creatine
It has been proposed that oral intake of creatine may provide substrate to produce ATP by increasing brain concentrations of creatine and PCr (Lyoo et al., 2003). Creatine also attenuates decreases in N-acetylaspartate (NAA), a marker of impaired mitochondrial function, and inhibits activation of the mitochondrial permeability transition, suggesting neuroprotective effects (Ferrante et al., 2000; Hemmer and Wallimann, 1993; O’Gorman et al., 1996). Despite limited evidence in mood disorders, Roitman et al. (2007) showed that creatine supplementation was associated with a significant reduction in depressive symptoms in unipolar depression, but in two bipolar depression patients, its supplementation precipitated manic episodes. Similarly, a randomized, double-blind, placebo-controlled trial demonstrated significant superiority of creatine add-on vs. placebo on the rates of remission in the completers and of partial response and remission, but two patients transitioned to hypomania/mania early during the trial. Thus, the authors suggested that creatine supplementation may have a role in treatment of the depressive phase of the illness (Toniolo et al., 2018). However, further investigation with larger sample sizes should be conducted to verify the efficacy of creatine add-on to standard pharmacotherapy and to determine in which mood state and dosage it might ideally exert its neuroprotective effects.
7.4. Omega-3 fatty acids
Evidence suggests that erythrocytes from BD patients exhibit decreased levels of omega-3 fatty acids, such as docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA) and α-linolenic acid compared to healthy controls (Balanza-Martinez et al., 2011). Moreover, Sobczak et al. (2004) showed a trend towards lower levels of omega-3 fatty acids in first-degree relatives of BD patients. A double-blind randomized trial using supplementation with omega-3 fatty acids also reported longer remission and improvements of depressive symptoms, bipolar symptoms and global functioning compared to the placebo group (Stoll et al., 1999). Furthermore, a 12-week, double-blind study reported that ethyl-EPA is an effective adjuvant treatment in bipolar depression (Frangou et al., 2006). More recently, however, Murphy et al. (2012) reported that omega-3 fatty acids did not mediate a significant improvement in mood symptoms over an extended period of treatment.
A systematic review on clinical trials assessing nutraceuticals confirmed that omega-3 fatty acids may be useful in the treatment of depressive symptoms of BD (Fusar-Poli et al., 2019). However, sample sizes were small, reducing the chance of positive results (Sarris et al., 2011). Previous evidence suggests that supplementation or increased consumption of omega-3 fatty acids may be beneficial in mood disorders, but additional studies are necessary to more accurately define their clinical efficacy.
7.5. Ketogenic diet
The ketogenic diet is known for its low carbohydrate and high-fat components, which forces the body to use fatty acids as the primary source of energy (DeVivo et al., 1978). Alternatively, the administration of ketone supplements, such as ketone salts or ketone esters, generates rapid and sustained nutritional ketosis (Kovacs et al., 2019). The effects of ketogenic diet or supplements on CNS diseases have not been thoroughly investigated, but ketosis seems to modulate different pathways implicated in psychiatric disorders, including BD (Bostock et al., 2017; Kovacs et al., 2019).
As previously discussed, BD seems to be related to an impaired ability to utilize pyruvate in the oxidative phosphorylation and the Krebs cycle, which results in high levels of pyruvate, derived from glycolysis (Yoshimi et al., 2016b). The increased plasma levels of ketones play an essential role as an alternative energy source by providing acetyl coenzyme A (acetyl-CoA) to the Krebs cycle, and therefore, bypasses the traditional pathway through glycolysis, which seems to be related to its mood stabilization properties (Campbell and Campbell, 2019). Newell et al. (2016) demonstrated that higher levels of ketone bodies, such as beta-hydroxybutyrate, increased the number of mitochondria and the electron transport chain proteins in this organelle, with consequently increased ATP synthesis. Marosi et al. (2016) showed that ketone 3-hydroxybutyrate metabolism increases mitochondrial respiration which drives changes in expression of brain-derived neurotrophic factor (BDNF) in cultured cerebral cortical neurons.
Moreover, ketosis seems to have anti-inflammatory properties and optimizes the cellular antioxidant system by activating nuclear factor erythroid-derived 2-related factor 2 (Nrf2) (Pinto et al., 2018). Furthermore, it has been postulated that the ketogenic diet may regulate neurotransmitters’ function, such as monoamine modulation and enhancement of GABAergic transmission, with consequent anxiolytic effects (Brietzke et al., 2018; Kashiwaya et al., 2013; Rogawski et al., 2016). In addition, some ketone bodies seem to inhibit the vesicular glutamate transporters (Danial et al., 2013), which can act as a buffer under conditions of excess of glutamate, such as acute mood episodes (Jaso et al., 2017). Although ketosis may be a promising treatment for BD, the availability of studies involving the ketogenic diet and BD is limited, and to date, there are no randomized controlled trials published.
8. Conclusion and future directions
Several studies in rodent models and humans suggest and reinforce the mitochondrial dysfunction hypothesis in BD. Mitochondria may present altered morphology and dynamics, as well as decreased metabolism and oxidative stress. Moreover, altered Ca2+ homeostasis and glutamate excitotoxicity likely play a crucial role in apoptosis. Taking together the findings reviewed above it is tempting to suggest that mitochondrial dysfunction is a critical pathological factor in BD that can be intimately linked to a wide range of processes associated with treatment outcomes and disease progression or severity (Fig. 1). The pathophysiology of BD is quite complex and involves several factors, besides mitochondrial dysfunction. Nevertheless, it is worth noting that the mitochondrial hypothesis must be interpreted in light of some limitations since the majority of studies are cross-sectional, which can identify associations but not causal relationships or longitudinal patterns of development, leading to a significant gap in the understanding of the specific role played by mitochondria in the etiology of BD, and whether an abnormal mitochondrial function is a cause or consequence (or even both) of BD subtypes. Furthermore, it is not yet completely understood which mitochondrial mechanism was altered first in addition to the chance that more than one mechanism occurs concurrently during the development of the disease.
9. Literature search strategy
A systematic literature search was performed using databases of Scopus, Embase, PubMed, Cochrane Library, and ISI web of Science from inception to September 2020. Moreover, a hand search was performed on three pre-print databases (medRxiv, bioRxiv, PsyArXiv) in order to find additional articles. Searches were restricted to English language. Results from the databases were merged using EndNote to facilitate the removal of duplicates. Reference lists of studies, review articles and systematic reviews were manually reviewed to identify any additional studies. Key words were grouped using the following Boolean expression and adjusted according to each database: “(bipolar disorder OR bipolar depression OR mood disorders OR mania OR depression) AND (mitochondria OR mitochondrial dysfunction OR energy homeostasis OR cerebral bioenergetics OR mitochondrial DNA OR oxidative stress OR calcium OR redox signaling OR mood stabilizers OR lithium OR mitochondrial agents OR complimentary therapies)”.
Acknowledgments and Disclosures
Translational Psychiatry Program (USA) is funded by a grant from the National Institute of Health/National Institute of Mental Health (1R21MH117636-01A1, to JQ). Center of Excellence on Mood Disorders (USA) is funded by the Pat Rutherford Jr. Chair in Psychiatry, John S. Dunn Foundation and Anne and Don Fizer Foundation Endowment for Depression Research. Translational Psychiatry Laboratory (Brazil) is funded by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (FAPESC), and Instituto Cérebro e Mente. JQ is a 1A CNPq Research Fellow.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Akarsu S, Torun D, Erdem M, Kozan S, Akar H, Uzun O, 2015. Mitochondrial complex I and III mRNA levels in bipolar disorder. J. Affect. Disord 184, 160–163. [DOI] [PubMed] [Google Scholar]
- Andreazza AC, Shao L, Wang JF, Young LT, 2010. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Archiv. Gen. Psychiat 67, 360–368. [DOI] [PubMed] [Google Scholar]
- Andreazza AC, Wang JF, Salmasi F, Shao L, Young LT, 2013. Specific subcellular changes in oxidative stress in prefrontal cortex from patients with bipolar disorder. J. Neurochem 127, 552–561. [DOI] [PubMed] [Google Scholar]
- Axelson D, Goldstein B, Goldstein T, Monk K, Yu H, Hickey MB, Sakolsky D, Diler R, Hafeman D, Merranko J, Iyengar S, Brent D, Kupfer D, Birmaher B, 2015. Diagnostic Precursors to Bipolar Disorder in Offspring of Parents With Bipolar Disorder: A Longitudinal Study. Am. J. Psychiatry 172, 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann RF, Wang Y, Yuan P, Zhou R, Li X, Alesci S, Du J, Manji HK, 2009. Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int. J. Neuropsychopharmacol./Off. Sci. J. Collegium Internationale Neuropsychopharmacol 12, 805–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanza-Martinez V, Fries GR, Colpo GD, Silveira PP, Portella AK, Tabares-Seisdedos R, Kapczinski F, 2011. Therapeutic use of omega-3 fatty acids in bipolar disorder. Expert Rev. Neurother 11, 1029–1047. [DOI] [PubMed] [Google Scholar]
- Beech RD, Lowthert L, Leffert JJ, Mason PN, Taylor MM, Umlauf S, Lin A, Lee JY, Maloney K, Muralidharan A, Lorberg B, Zhao H, Newton SS, Mane S, Epperson CN, Sinha R, Blumberg H, Bhagwagar Z, 2010. Increased peripheral blood expression of electron transport chain genes in bipolar depression. Bipolar Disord 12, 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei E, Salpeas V, Pappa D, Anagnostara C, Alevizos V, Moutsatsou P, 2009. Phosphorylation status of glucocorticoid receptor, heat shock protein 70, cytochrome c and Bax in lymphocytes of euthymic, depressed and manic bipolar patients. Psychoneuroendocrinology 34, 1162–1175. [DOI] [PubMed] [Google Scholar]
- Belosludtsev KN, Dubinin MV, Belosludtseva NV, Mironova GD, 2019. Mitochondrial Ca2 + Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry (Mosc) 84, 593–607. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Zuk R, Gazawi H, Reshef A, Sheinkman A, Klein E, 1999. Increased mitochondrial complex I activity in platelets of schizophrenic patients. Int. J. Neuropsychopharmacol./Official Scient. J. Collegium Internationale Neuropsychopharmacol 2, 245–253. [DOI] [PubMed] [Google Scholar]
- Berk M, 2009. Neuroprogression: pathways to progressive brain changes in bipolar disorder. Int. J. Neuropsychopharmacol./Off. Scient. J. Collegium Internationale Neuropsychopharmacol 12, 441–445. [DOI] [PubMed] [Google Scholar]
- Berk M, Berk L, Dodd S, Cotton S, Macneil C, Daglas R, Conus P, Bechdolf A, Moylan S, Malhi GS, 2014. Stage managing bipolar disorder. Bipolar Disord 16, 471–477. [DOI] [PubMed] [Google Scholar]
- Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Bush AI, 2008. N-acetyl cysteine for depressive symptoms in bipolar disorder–a double-blind randomized placebo-controlled trial. Biol. Psychiatry 64, 468–475. [DOI] [PubMed] [Google Scholar]
- Berk M, Dean O, Cotton SM, Gama CS, Kapczinski F, Fernandes BS, Kohlmann K, Jeavons S, Hewitt K, Allwang C, Cobb H, Bush AI, Schapkaitz I, Dodd S, Malhi GS, 2011a. The efficacy of N-acetylcysteine as an adjunctive treatment in bipolar depression: an open label trial. J. Affect. Disord 135,389–394. [DOI] [PubMed] [Google Scholar]
- Berk M, Dean OM, Cotton SM, Gama CS, Kapczinski F, Fernandes B, Kohlmann K, Jeavons S, Hewitt K, Moss K, Allwang C, Schapkaitz I, Cobb H, Bush AI, Dodd S, Malhi GS, 2012. Maintenance N-acetyl cysteine treatment for bipolar disorder: a double-blind randomized placebo controlled trial. BMC Med 10, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk M, Kapczinski F, Andreazza AC, Dean OM, Giorlando F, Maes M, Yucel M, Gama CS, Dodd S, Dean B, Magalhaes PV, Amminger P, McGorry P, Malhi GS, 2011b. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev 35, 804–817. [DOI] [PubMed] [Google Scholar]
- Berk M, Turner A, Malhi GS, Ng CH, Cotton SM, Dodd S, Samuni Y, Tanious M, McAulay C, Dowling N, Sarris J, Owen L, Waterdrinker A, Smith D, Dean OM, 2019. A randomised controlled trial of a mitochondrial therapeutic target for bipolar depression: mitochondrial agents, N-acetylcysteine, and placebo. BMC Med 17, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock EC, Kirkby KC, Taylor BV, 2017. The Current Status of the Ketogenic Diet in Psychiatry. Front. Psychiatry 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, Oshino N, Chance B, 1972. The cellular production of hydrogen peroxide. Biochem. J 128, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Sagua R, Parra V, Lopez-Crisosto C, Diaz P, Quest AF, Lavandero S, 2017. Calcium Transport and Signaling in Mitochondria. Compr. Physiol 7, 623–634. [DOI] [PubMed] [Google Scholar]
- Brennan BP, Jensen JE, Hudson JI, Coit CE, Beaulieu A, Pope HG Jr., Renshaw PF, Cohen BM, 2013. A placebo-controlled trial of acetyl-L-carnitine and alpha-lipoic acid in the treatment of bipolar depression. J. Clin. Psychopharmacol 33, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briere JJ, Chretien D, Benit P, Rustin P, 2004. Respiratory chain defects: what do we know for sure about their consequences in vivo? Biochim. Biophys. Acta, Gen. Subj 1659, 172–177. [DOI] [PubMed] [Google Scholar]
- Brietzke E, Mansur RB, Subramaniapillai M, Balanza-Martinez V, Vinberg M, Gonzalez-Pinto A, Rosenblat JD, Ho R, McIntyre RS, 2018. Ketogenic diet as a metabolic therapy for mood disorders: Evidence and developments. Neurosci. Biobehav. Rev 94, 11–16. [DOI] [PubMed] [Google Scholar]
- Brown NC, Andreazza AC, Young LT, 2014. An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res 218, 61–68. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG, 1998. Mitochondria in the life and death of neurons. Essays Biochem 33, 43–52. [DOI] [PubMed] [Google Scholar]
- Burkeen JF, Womac AD, Earnest DJ, Zoran MJ, 2011. Mitochondrial calcium signaling mediates rhythmic extracellular ATP accumulation in suprachiasmatic nucleus astrocytes. J. Neurosci. Off. J. Soc. Neurosci 31, 8432–8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhalter J, Fiumelli H, Allaman I, Chatton JY, Martin JL, 2003. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J. Neurosci. Off. J. Soc. Neurosci 23, 8212–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK, 2016. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IH, Campbell H, 2019. Ketosis and bipolar disorder: controlled analytic study of online reports. BJPsych Open 5, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ, Froimowitz MP, Hassinger LC, Menesale EB, Sargent LW, Logan DJ, Carpenter AE, Cohen BM, 2010. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am. J. Pathol 177, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceylan D, Tuna G, Kirkali G, Tunca Z, Can G, Arat HE, Kant M, Dizdaroglu M, Ozerdem A, 2018. Oxidatively-induced DNA damage and base excision repair in euthymic patients with bipolar disorder. DNA Repair (Amst) 65, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty S, Kabekkodu SP, Singh RP, Thangaraj K, Singh KK, Satyamoorthy K, 2018. Mitochondria in health and disease. Mitochondrion [DOI] [PubMed]
- Chang K, Adleman N, Dienes K, Barnea-Goraly N, Reiss A, Ketter T, 2003. Decreased N-acetylaspartate in children with familial bipolar disorder. Biol. Psychiatry 53, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, Manji HK, 1999. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem 72, 879–882. [DOI] [PubMed] [Google Scholar]
- Chen H, Dzitoyeva S, Manev H, 2012. Effect of valproic acid on mitochondrial epigenetics. Eur. J. Pharmacol 690, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Hou Y, Mattson MP, 2010. Mitochondria and neuroplasticity. ASN Neuro 2, e00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cikankova T, Fisar Z, Bakhouche Y, Luptak M, Hroudova J, 2019. In vitro effects of antipsychotics on mitochondrial respiration. Naunyn-Schmiedeberg’s Arch. Pharmacol 392, 1209–1223. [DOI] [PubMed] [Google Scholar]
- Cikankova T, Sigitova E, Zverova M, Fisar Z, Raboch J, Hroudova J, 2017. Mitochondrial Dysfunctions in Bipolar Disorder: Effect of the Disease and Pharmacotherapy. CNS Neurol. Disord.: Drug Targets 16, 176–186. [DOI] [PubMed] [Google Scholar]
- Clay HB, Sillivan S, Konradi C, 2011. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int. J. Dev. Neurosci 29, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colasanti A, Bugiardini E, Amawi S, Poole OV, Skorupinska I, Skorupinska M, Germain L, Kozyra D, Holmes S, James N, Woodward CE, Quinlivan R, Young AH, Hanna MG, Pitceathly RDS, 2020. Primary mitochondrial diseases increase susceptibility to bipolar affective disorder. J. Neurol. Neurosurg. Psychiatry [DOI] [PubMed]
- Correa C, Amboni G, Assis LC, Martins MR, Kapczinski F, Streck EL, Quevedo J, 2007. Effects of lithium and valproate on hippocampus citrate synthase activity in an animal model of mania. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 31, 887–891. [DOI] [PubMed] [Google Scholar]
- Corson TW, Li PP, Kennedy JL, Macciardi F, Cooke RG, Parikh SV, Warsh JJ, 2001. Association analysis of G-protein beta 3 subunit gene with altered Ca(2+) homeostasis in bipolar disorder. Mol. Psychiatry 6, 125–126. [DOI] [PubMed] [Google Scholar]
- Cotrena C, Branco LD, Kochhann R, Shansis FM, Fonseca RP, 2016. Quality of life, functioning and cognition in bipolar disorder and major depression: A latent profile analysis. Psychiatry Res 241, 289–296. [DOI] [PubMed] [Google Scholar]
- Cui J, Shao L, Young LT, Wang JF, 2007. Role of glutathione in neuroprotective effects of mood stabilizing drugs lithium and valproate. Neuroscience 144, 1447–1453. [DOI] [PubMed] [Google Scholar]
- Cuperfain AB, Zhang ZL, Kennedy JL, Goncalves VF, 2018. The Complex Interaction of Mitochondrial Genetics and Mitochondrial Pathways in Psychiatric Disease. Mol Neuropsychiatry 4, 52–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dager SR, Friedman SD, Parow A, Demopulos C, Stoll AL, Lyoo IK, Dunner DL, Renshaw PF, 2004. Brain metabolic alterations in medication-free patients with bipolar disorder. Arch. Gen. Psychiatry 61, 450–458. [DOI] [PubMed] [Google Scholar]
- Danial NN, Hartman AL, Stafstrom CE, Thio LL, 2013. How does the ketogenic diet work? Four potential mechanisms. J. Child Neurol 28, 1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das TK, Javadzadeh A, Dey A, Sabesan P, Theberge J, Radua J, Palaniyappan L, 2019. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 91, 94–102. [DOI] [PubMed] [Google Scholar]
- de Goede P, Wefers J, Brombacher EC, Schrauwen P, Kalsbeek A, 2018. Circadian rhythms in mitochondrial respiration. J. Mol. Endocrinol 60, R115–R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa RT, Streck EL, Forlenza OV, Brunoni AR, Zanetti MV, Ferreira GK,Diniz BS, Portela LV, Carvalho AF, Zarate CA Jr., Gattaz WF, Machado-Vieira R, 2015. Regulation of leukocyte tricarboxylic acid cycle in drug-naïve Bipolar Disorder. Neurosci. Lett 605, 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa RT, Zarate CA Jr., Zanetti MV, Costa AC, Talib LL, Gattaz WF, Machado-Vieira R, 2014. Oxidative stress in early stage Bipolar Disorder and the association with response to lithium. J. Psychiatr. Res 50, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deicken RF, Pegues MP, Anzalone S, Feiwell R, Soher B, 2003. Lower concentration of hippocampal N-acetylaspartate in familial bipolar I disorder. Am. J. Psychiatry 160, 873–882. [DOI] [PubMed] [Google Scholar]
- DeVivo DC, Leckie MP, Ferrendelli JS, McDougal DB Jr., 1978. Chronic ketosis and cerebral metabolism. Ann. Neurol 3, 331–337. [DOI] [PubMed] [Google Scholar]
- Diler RS, Goldstein TR, Hafeman D, Rooks BT, Sakolsky D, Goldstein BI, Monk K, Hickey MB, Axelson D, Iyengar S, Birmaher B, 2017. Characteristics of depression among offspring at high and low familial risk of bipolar disorder. Bipolar Disord 19, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan AE, Yuksel C, Du F, Chouinard VA, Ongur D, 2018. Brain lactate and pH in schizophrenia and bipolar disorder: a systematic review of findings from magnetic resonance studies. Neuropsychopharmacol. Off. Publicat. Am. Coll. Neuropsychopharmacol 43, 1681–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Stefan G, Horrocks J, Goodday SM, Duffy A, 2019. Investigating the association between anxiety symptoms and mood disorder in high-risk offspring of bipolar parents: a comparison of Joint and Cox models. Int J Bipolar Disord 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos SM, Romeiro CFR, Rodrigues CA, Cerqueira ARL, Monteiro MC, 2019. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxid. Med. Cell Longev 2019, 8409329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F, Yuksel C, Chouinard VA, Huynh P, Ryan K, Cohen BM, Ongur D, 2018. Abnormalities in High-Energy Phosphate Metabolism in First-Episode Bipolar Disorder Measured Using (31)P-Magnetic Resonance Spectroscopy. Biol. Psychiatry 84, 797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubovsky SL, Daurignac E, Leonard KE, 2014. Increased platelet intracellular calcium ion concentration is specific to bipolar disorder. J. Affect. Disord 164, 38–42. [DOI] [PubMed] [Google Scholar]
- Duffy A, Goodday S, Keown-Stoneman C, Grof P, 2019. The Emergent Course of Bipolar Disorder: Observations Over Two Decades From the Canadian High-Risk Offspring Cohort. Am. J. Psychiatry 176, 720–729. [DOI] [PubMed] [Google Scholar]
- Edvardsen J, Torgersen S, Roysamb E, Lygren S, Skre I, Onstad S, Oien PA, 2008. Heritability of bipolar spectrum disorders. Unity or heterogeneity? J. Affect. Disord 106, 229–240. [DOI] [PubMed] [Google Scholar]
- El-Badri SM, Ashton CH, Moore PB, Marsh VR, Ferrier IN, 2001. Electrophysiological and cognitive function in young euthymic patients with bipolar affective disorder. Bipolar Disord 3, 79–87. [DOI] [PubMed] [Google Scholar]
- Ellegaard PK, Licht RW, Nielsen RE, Dean OM, Berk M, Poulsen HE, Mohebbi M, Nielsen CT, 2019. The efficacy of adjunctive N-acetylcysteine in acute bipolar depression: A randomized placebo-controlled study. J. Affect. Disord 245, 1043–1051. [DOI] [PubMed] [Google Scholar]
- Emamghoreishi M, Schlichter L, Li PP, Parikh S, Sen J, Kamble A, Warsh JJ, 1997. High intracellular calcium concentrations in transformed lymphoblasts from subjects with bipolar I disorder. Am. J. Psychiatry 154, 976–982. [DOI] [PubMed] [Google Scholar]
- Eskandari MR, Fard JK, Hosseini MJ, Pourahmad J, 2012. Glutathione mediated reductive activation and mitochondrial dysfunction play key roles in lithium induced oxidative stress and cytotoxicity in liver. Biometals 25, 863–873. [DOI] [PubMed] [Google Scholar]
- Fang D, Yan S, Yu Q, Chen D, Yan SS, 2016. Mfn2 is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/ hiPSC Derived Cortical Neurons. Sci. Rep 6, 31462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feier G, Valvassori SS, Varela RB, Resende WR, Bavaresco DV, Morais MO, Scaini G, Andersen ML, Streck EL, Quevedo J, 2013. Lithium and valproate modulate energy metabolism in an animal model of mania induced by methamphetamine. Pharmacol. Biochem. Behav 103, 589–596. [DOI] [PubMed] [Google Scholar]
- Fernandes BS, Dean OM, Dodd S, Malhi GS, Berk M, 2016. N-Acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J. Clin. Psychiat 77, e457–466. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF, 2000. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J. Neurosci. Off. J. Soc. Neurosci 20, 4389–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J, Scorza FA, 2017. Effects of antiepileptic drugs on mitochondrial functions, morphology, kinetics, biogenesis, and survival. Epilepsy Res 136, 5–11. [DOI] [PubMed] [Google Scholar]
- Fiorentino A, O’Brien NL, Locke DP, McQuillin A, Jarram A, Anjorin A, Kandaswamy R, Curtis D, Blizard RA, Gurling HM, 2014. Analysis of ANK3 and CACNA1C variants identified in bipolar disorder whole genome sequence data. Bipolar Disord 16, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Hamann A, Osiewacz HD, 2012. Mitochondrial quality control: an integrated network of pathways. Trends Biochem. Sci 37, 284–292. [DOI] [PubMed] [Google Scholar]
- Frangou S, Lewis M, McCrone P, 2006. Efficacy of ethyl-eicosapentaenoic acid in bipolar depression: randomised double-blind placebo-controlled study. Br. J. Psychiatry 188, 46–50. [DOI] [PubMed] [Google Scholar]
- Fries GR, Bauer IE, Scaini G, Valvassori SS, Walss-Bass C, Soares JC, Quevedo J, 2019a. Accelerated hippocampal biological aging in bipolar disorder. Bipolar Disord [DOI] [PubMed]
- Fries GR, Bauer IE, Scaini G, Wu MJ, Kazimi IF, Valvassori SS, Zunta-Soares G, Walss-Bass C, Soares JC, Quevedo J, 2017. Accelerated epigenetic aging and mitochondrial DNA copy number in bipolar disorder. Transl. Psychiatry 7, 1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries GR, Pfaffenseller B, Stertz L, Paz AV, Dargel AA, Kunz M, Kapczinski F, 2012. Staging and neuroprogression in bipolar disorder. Curr. Psychiatry Rep 14, 667–675. [DOI] [PubMed] [Google Scholar]
- Fries GR, Vasconcelos-Moreno MP, Gubert C, Santos B.T.M.Q.d., da Rosa ALST, Eisele B, Sartori J, Pfaffenseller B, Kapczinski F, Kauer-Sant’Anna M, 2014. Early apoptosis in peripheral blood mononuclear cells from patients with bipolar disorder. J. Affect. Disorders 152–154, 474–477. [DOI] [PubMed]
- Fries GR, Walss-Bass C, Bauer ME, Teixeira AL, 2019b. Revisiting inflammation in bipolar disorder. Pharmacol. Biochem. Behav 177, 12–19. [DOI] [PubMed] [Google Scholar]
- Fries GR, Zamzow MJ, Andrews T, Pink O, Scaini G, Quevedo J, 2020. Accelerated aging in bipolar disorder: A comprehensive review of molecular findings and their clinical implications. Neurosci. Biobehav. Rev 112, 107–116. [DOI] [PubMed] [Google Scholar]
- Fusar-Poli L, Surace T, Vanella A, Meo V, Patania F, Furnari R, Signorelli MS, Aguglia E, 2019. The effect of adjunctive nutraceuticals in bipolar disorder: A systematic review of randomized placebo-controlled trials. J. Affect. Disord 252, 334–349. [DOI] [PubMed] [Google Scholar]
- Gama CS, Kunz M, Magalhaes PV, Kapczinski F, 2013. Staging and neuroprogression in bipolar disorder: a systematic review of the literature. Rev. Bras Psiquiatr 35, 70–74. [DOI] [PubMed] [Google Scholar]
- Geoffroy PA, 2018. Clock Genes and Light Signaling Alterations in Bipolar Disorder: When the Biological Clock Is Off. Biol. Psychiatry 84, 775–777. [DOI] [PubMed] [Google Scholar]
- Geoffroy PA, Etain B, Lajnef M, Zerdazi EH, Brichant-Petitjean C, Heilbronner U, Hou L, Degenhardt F, Rietschel M, McMahon FJ, Schulze TG, Jamain S, Marie-Claire C, Bellivier F, 2016. Circadian genes and lithium response in bipolar disorders: associations with PPARGC1A (PGC-1alpha) and RORA. Genes Brain Behav 15, 660–668. [DOI] [PubMed] [Google Scholar]
- Giorgi C, Marchi S, Pinton P, 2018. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol 19, 713–730. [DOI] [PubMed] [Google Scholar]
- Giorgi C, Romagnoli A, Pinton P, Rizzuto R, 2008. Ca2 + signaling, mitochondria and cell death. Curr. Mol. Med 8, 119–130. [DOI] [PubMed] [Google Scholar]
- Giorgio V, Guo L, Bassot C, Petronilli V, Bernardi P, 2018. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 70, 56–63. [DOI] [PubMed] [Google Scholar]
- Gorlach A, Bertram K, Hudecova S, Krizanova O, 2015. Calcium and ROS: A mutual interplay. Redox Biol 6, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II, Laursen TM, Bertelsen A, Mortensen PB, 2010. Severe mental disorders in offspring with 2 psychiatrically ill parents. Arch. Gen. Psychiatry 67, 252–257. [DOI] [PubMed] [Google Scholar]
- Gould TD, Manji HK, 2005. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacol. Off. Publicat. Am. College Neuropsychopharmacol 30, 1223–1237. [DOI] [PubMed] [Google Scholar]
- Grande I, Berk M, Birmaher B, Vieta E, 2016. Bipolar disorder. Lancet 387, 1561–1572. [DOI] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME, 2008. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. [DOI] [PubMed] [Google Scholar]
- Gubert C, Stertz L, Pfaffenseller B, Panizzutti BS, Rezin GT, Massuda R, Streck EL, Gama CS, Kapczinski F, Kunz M, 2013. Mitochondrial activity and oxidative stress markers in peripheral blood mononuclear cells of patients with bipolar disorder, schizophrenia, and healthy subjects. J. Psychiatr. Res 47, 1396–1402. [DOI] [PubMed] [Google Scholar]
- Gurvich N, Klein PS, 2002. Lithium and valproic acid: parallels and contrasts in diverse signaling contexts. Pharmacol. Ther 96, 45–66. [DOI] [PubMed] [Google Scholar]
- Hager K, Kenklies M, McAfoose J, Engel J, Munch G, 2007. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease–a 48 months follow-up analysis. J. Neural Transm Suppl. 189–193. [DOI] [PubMed]
- Hahn CG, Gomez G, Restrepo D, Friedman E, Josiassen R, Pribitkin EA, Lowry LD, Gallop RJ, Rawson NE, 2005. Aberrant intracellular calcium signaling in olfactory neurons from patients with bipolar disorder. Am. J. Psychiatry 162, 616–618. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Hall N, Mould A, Al-Juffali N, Tunbridge EM, 2019. Cellular calcium in bipolar disorder: systematic review and meta-analysis. Mol. Psychiatry [DOI] [PMC free article] [PubMed]
- Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM, 2002. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43, 1173–1179. [DOI] [PubMed] [Google Scholar]
- Hemmer W, Wallimann T, 1993. Functional aspects of creatine kinase in brain. Dev. Neurosci 15, 249–260. [DOI] [PubMed] [Google Scholar]
- Holper L, Ben-Shachar D, Mann JJ, 2019. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacol. Off. Publicat. Am. Coll. Neuropsychopharmacol 44, 837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough C, Lu SJ, Davis CL, Chuang DM, Post RM, 1999. Elevated basal and thapsigargin-stimulated intracellular calcium of platelets and lymphocytes from bipolar affective disorder patients measured by a fluorometric microassay. Biol. Psychiatry 46, 247–255. [DOI] [PubMed] [Google Scholar]
- Hroudova J, Fisar Z, 2011. Connectivity between mitochondrial functions and psychiatric disorders. Psychiatry Clin. Neurosci 65, 130–141. [DOI] [PubMed] [Google Scholar]
- Huzayyin AA, Andreazza AC, Turecki G, Cruceanu C, Rouleau GA, Alda M, Young LT, 2014. Decreased global methylation in patients with bipolar disorder who respond to lithium. Int. J. Neuropsychopharmacol. Off. Scient. J. Colleg. Internationale Neuropsychopharmacol 17, 561–569. [DOI] [PubMed] [Google Scholar]
- Isobe Y, Hida H, Nishino H, 2011. Circadian rhythm of metabolic oscillation in suprachiasmatic nucleus depends on the mitochondrial oxidation state, reflected by cytochrome C oxidase and lactate dehydrogenase. J. Neurosci. Res 89, 929–935. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Kakiuchi C, Bundo M, Ikeda K, Kato T, 2004. Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol. Psychiatry 9, 406–416. [DOI] [PubMed] [Google Scholar]
- Jacobi D, Liu S, Burkewitz K, Kory N, Knudsen NH, Alexander RK, Unluturk U, Li X, Kong X, Hyde AL, Gangl MR, Mair WB, Lee CH, 2015. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab 22, 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafarian I, Eskandari MR, Mashayekhi V, Ahadpour M, Hosseini MJ, 2013. Toxicity of valproic acid in isolated rat liver mitochondria. Toxicol. Mech. Methods 23, 617–623. [DOI] [PubMed] [Google Scholar]
- Jaso BA, Niciu MJ, Iadarola ND, Lally N, Richards EM, Park M, Ballard ED, Nugent AC, Machado-Vieira R, Zarate CA, 2017. Therapeutic Modulation of Glutamate Receptors in Major Depressive Disorder. Curr. Neuropharmacol 15, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan SD, Lamia KA, 2013. AMPK at the crossroads of circadian clocks and metabolism. Mol. Cell. Endocrinol 366, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadenbach B, Ramzan R, Wen L, Vogt S, 2010. New extension of the Mitchell Theory for oxidative phosphorylation in mitochondria of living organisms. Biochim. Biophys. Acta, Gen. Subj 1800, 205–212. [DOI] [PubMed] [Google Scholar]
- Kapczinski F, Dal-Pizzol F, Teixeira AL, Magalhaes PV, Kauer-Sant’Anna M, Klamt F, Pasquali MA, Quevedo J, Gama CS, Post R, 2010. A systemic toxicity index developed to assess peripheral changes in mood episodes. Mol. Psychiatry 15, 784–786. [DOI] [PubMed] [Google Scholar]
- Kapczinski F, Vieta E, Andreazza AC, Frey BN, Gomes FA, Tramontina J, Kauer-Sant’anna M, Grassi-Oliveira R, Post RM, 2008. Allostatic load in bipolar disorder: implications for pathophysiology and treatment. Neurosci. Biobehav. Rev 32, 675–692. [DOI] [PubMed] [Google Scholar]
- Kasahara T, Kato T, 2018. What Can Mitochondrial DNA Analysis Tell Us About Mood Disorders? Biol. Psychiatry 83, 731–738. [DOI] [PubMed] [Google Scholar]
- Kashiwaya Y, Bergman C, Lee JH, Wan R, King MT, Mughal MR, Okun E, Clarke K, Mattson MP, Veech RL, 2013. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka M, Matoba N, Sawada T, Kazuno AA, Ishiwata M, Fujii K, Matsuo K, Takata A, Kato T, 2016. Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations. Mol. Psychiatry 21, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, 2005. Mitochondrial dysfunction in bipolar disorder: from 31P-magnetic resonance spectroscopic findings to their molecular mechanisms. Int. Rev. Neurobiol 63, 21–40. [DOI] [PubMed] [Google Scholar]
- Kato T, 2008. Role of mitochondrial DNA in calcium signaling abnormality in bipolar disorder. Cell Calcium 44, 92–102. [DOI] [PubMed] [Google Scholar]
- Kato T, 2011. Mitochondrial dysfunction and bipolar disorder. Curr. Top Behav. Neurosci 5, 187–200. [DOI] [PubMed] [Google Scholar]
- Kato T, Kato N, 2000. Mitochondrial dysfunction in bipolar disorder. Bipolar Disord 2, 180–190. [DOI] [PubMed] [Google Scholar]
- Kato T, Kunugi H, Nanko S, Kato N, 2001. Mitochondrial DNA polymorphisms in bipolar disorder. J. Affect. Disord 62, 151–164. [DOI] [PubMed] [Google Scholar]
- Kato T, Murashita J, Kamiya A, Shioiri T, Kato N, Inubushi T, 1998. Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity. Eur. Arch. Psychiatry Clin. Neurosci 248, 301–306. [DOI] [PubMed] [Google Scholar]
- Kato T, Takahashi S, Shioiri T, Inubushi T, 1992. Brain phosphorous metabolism in depressive disorders detected by phosphorus-31 magnetic resonance spectroscopy. J. Affect. Disord 26, 223–230. [DOI] [PubMed] [Google Scholar]
- Kato T, Takahashi S, Shioiri T, Murashita J, Hamakawa H, Inubushi T, 1994. Reduction of brain phosphocreatine in bipolar II disorder detected by phosphorus-31 magnetic resonance spectroscopy. J. Affect. Disord 31, 125–133. [DOI] [PubMed] [Google Scholar]
- Kazuno AA, Munakata K, Kato N, Kato T, 2008. Mitochondrial DNA-dependent effects of valproate on mitochondrial calcium levels in transmitochondrial cybrids. The Int. J. Neuropsychopharmacol. Off. Scient. J. Collegium Internationale Neuropsychopharmacol 11, 71–78. [DOI] [PubMed] [Google Scholar]
- Kazuno AA, Munakata K, Mori K, Nanko S, Kunugi H, Nakamura K, Mori N, Yamada K, Yoshikawa T, Kato N, Kato T, 2009. Mitochondrial DNA haplogroup analysis in patients with bipolar disorder. Am. J. Med. Genet. Part B, Neuropsychiat. Genetics: Off. Publicat. Int. Soc. Psychiatr. Genetics 150B, 243–247. [DOI] [PubMed] [Google Scholar]
- Kim HK, Chen W, Andreazza AC, 2015. The Potential Role of the NLRP3 Inflammasome as a Link between Mitochondrial Complex I Dysfunction and Inflammation in Bipolar Disorder. Neural. Plast 2015, 408136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HW, Rapoport SI, Rao JS, 2010. Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol. Disease 37, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Vadodaria KC, Lenkei Z, Kato T, Gage FH, Marchetto MC, Santos R, 2019. Mitochondria, Metabolism, and Redox Mechanisms in Psychiatric Disorders. Antioxid. Redox Signal 31, 275–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Ko HH, Han ES, Lee CS, 2007. Lamotrigine inhibition of rotenone- or 1-methyl-4-phenylpyridinium-induced mitochondrial damage and cell death. Brain Res. Bull 71, 633–640. [DOI] [PubMed] [Google Scholar]
- Knezevic V, Nedic A, 2013. Influence of misdiagnosis on the course of bipolar disorder. Eur. Rev. Med. Pharmacol. Sci 17, 1542–1545. [PubMed] [Google Scholar]
- Knorr U, Simonsen AH, Roos P, Weimann A, Henriksen T, Christensen EM, Vinberg M, Mikkelsen RL, Kirkegaard T, Jensen RN, Akhoj M, Forman J, Poulsen HE, Hasselbalch SG, Kessing LV, 2019. Cerebrospinal fluid oxidative stress metabolites in patients with bipolar disorder and healthy controls: a longitudinal case-control study. Transl. Psychiatry 9, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koene S, Kozicz TL, Rodenburg RJ, Verhaak CM, de Vries MC, Wortmann S, van de Heuvel L, Smeitink JA, Morava E, 2009. Major depression in adolescent children consecutively diagnosed with mitochondrial disorder. J. Affect. Disord 114, 327–332. [DOI] [PubMed] [Google Scholar]
- Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S, 2004. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch. Gen. Psychiatry 61, 300–308. [DOI] [PubMed] [Google Scholar]
- Kornmann B, 2014. Quality control in mitochondria: use it, break it, fix it, trash it. F1000Prime Rep 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs Z, D’Agostino DP, Diamond D, Kindy MS, Rogers C, Ari C, 2019. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 10, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang H, Duong A, Jeong H, Zachos K, Andreazza AC, 2018. Lactate in bipolar disorder: A systematic review and meta-analysis. Psychiatry Clin. Neurosci 72, 546–555. [DOI] [PubMed] [Google Scholar]
- Leboyer M, Kupfer DJ, 2010. Bipolar disorder: new perspectives in health care and prevention. J. Clin. Psych 71, 1689–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaz G, Genova ML, 2010. Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid. Redox Signal 12, 961–1008. [DOI] [PubMed] [Google Scholar]
- Levenson JC, Axelson DA, Merranko J, Angulo M, Goldstein TR, Mullin BC, Goldstein BI, Brent DA, Diler R, Hickey MB, Monk K, Sakolsky D, Kupfer DJ, Birmaher B, 2015. Differences in sleep disturbances among offspring of parents with and without bipolar disorder: association with conversion to bipolar disorder. Bipolar Disord 17, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Bijur GN, Jope RS, 2002. Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection . Bipolar Disord 4, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li S, Liu T, Borjigin J, Lin JD, 2007. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature 447, 477–481. [DOI] [PubMed] [Google Scholar]
- Liu J, 2008. The effects and mechanisms of mitochondrial nutrient alpha-lipoic acid on improving age-associated mitochondrial and cognitive dysfunction: an overview. Neurochem. Res 33, 194–203. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rosenthal RE, Starke-Reed P, Fiskum G, 1993. Inhibition of postcardiac arrest brain protein oxidation by acetyl-L-carnitine. Free Radic. Biol. Med 15, 667–670. [DOI] [PubMed] [Google Scholar]
- Lopes-Borges J, Valvassori SS, Varela RB, Tonin PT, Vieira JS, Goncalves CL, Streck EL, Quevedo J, 2015. Histone deacetylase inhibitors reverse manic-like behaviors and protect the rat brain from energetic metabolic alterations induced by ouabain. Pharmacol. Biochem. Behav 128, 89–95. [DOI] [PubMed] [Google Scholar]
- Ly CV, Verstreken P, 2006. Mitochondria at the synapse. Neuroscientist 12, 291–299. [DOI] [PubMed] [Google Scholar]
- Lyoo IK, Kong SW, Sung SM, Hirashima F, Parow A, Hennen J, Cohen BM, Renshaw PF, 2003. Multinuclear magnetic resonance spectroscopy of high-energy phosphate metabolites in human brain following oral supplementation of creatine-monohydrate. Psychiatry Res 123, 87–100. [DOI] [PubMed] [Google Scholar]
- Lyoo IK, Sung YH, Dager SR, Friedman SD, Lee JY, Kim SJ, Kim N, Dunner DL, Renshaw PF, 2006. Regional cerebral cortical thinning in bipolar disorder. Bipolar Disord 8, 65–74. [DOI] [PubMed] [Google Scholar]
- MacDonald ML, Naydenov A, Chu M, Matzilevich D, Konradi C, 2006. Decrease in creatine kinase messenger RNA expression in the hippocampus and dorsolateral prefrontal cortex in bipolar disorder. Bipolar Disord 8, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Andreazza AC, Viale CI, Zanatto V, Cereser V Jr., da Silva Vargas R, Kapczinski F, Portela LV, Souza DO, Salvador M, Gentil V, 2007. Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: a possible role for lithium antioxidant effects. Neurosci. Lett 421, 33–36. [DOI] [PubMed] [Google Scholar]
- Maciukiewicz M, Dmitrzak-Weglarz M, Pawlak J, Leszczynska-Rodziewicz A, Zaremba D, Skibinska M, Hauser J, 2014. Analysis of genetic association and epistasis interactions between circadian clock genes and symptom dimensions of bipolar affective disorder. Chronobiol. Int 31, 770–778. [DOI] [PubMed] [Google Scholar]
- Magalhaes PV, Dean OM, Bush AI, Copolov DL, Malhi GS, Kohlmann K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Berk M, 2011. N-acetyl cysteine add-on treatment for bipolar II disorder: a subgroup analysis of a randomized placebo-controlled trial. J. Affect. Disord 129, 317–320. [DOI] [PubMed] [Google Scholar]
- Magalhaes PV, Dean OM, Bush AI, Copolov DL, Malhi GS, Kohlmann K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Berk M, 2013. A preliminary investigation on the efficacy of N-acetyl cysteine for mania or hypomania. Austral. New Zealand J. Psychiatry 47, 564–568. [DOI] [PubMed] [Google Scholar]
- Manelis A, Ladouceur CD, Graur S, Monk K, Bonar LK, Hickey MB, Dwojak AC, Axelson D, Goldstein BI, Goldstein TR, Bebko G, Bertocci MA, Gill MK, Birmaher B, Phillips ML, 2016. Altered functioning of reward circuitry in youth offspring of parents with bipolar disorder. Psychol. Med 46, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji HK, Henter ID, Zarate CA Jr., 2011. Bipolar disorder: a neurobiological synthesis. Curr Top Behav Neurosci 5, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P, 2018. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. [DOI] [PubMed] [Google Scholar]
- Marosi K, Kim SW, Moehl K, Scheibye-Knudsen M, Cheng A, Cutler R, Camandola S, Mattson MP, 2016. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem 139, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marpegan L, Swanstrom AE, Chung K, Simon T, Haydon PG, Khan SK, Liu AC, Herzog ED, Beaule C, 2011. Circadian regulation of ATP release in astrocytes. The J. Neurosci. Off. J. Soc. Neurosci 31, 8342–8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer IC, Schippel P, Volz HP, 2009. Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord 11, 515–522. [DOI] [PubMed] [Google Scholar]
- McCarthy MJ, Nievergelt CM, Kelsoe JR, Welsh DK, 2012. A survey of genomic studies supports association of circadian clock genes with bipolar disorder spectrum illnesses and lithium response. PLoS ONE 7, e32091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MJ, Wei H, Marnoy Z, Darvish RM, McPhie DL, Cohen BM, Welsh DK, 2013. Genetic and clinical factors predict lithium’s effects on PER2 gene expression rhythms in cells from bipolar disorder patients. Transl. Psychiatry 3, e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A, 2003. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch. Gen. Psychiatry 60, 497–502. [DOI] [PubMed] [Google Scholar]
- Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, Zheng Y, Diffenderfer KE, Zhang J, Soltani S, Eames T, Schafer ST, Boyer L, Marchetto MC, Nurnberger JI, Calabrese JR, Odegaard KJ, McCarthy MJ, Zandi PP, Alda M, Nievergelt CM, Pharmacogenomics of Bipolar Disorder, S., Mi S, Brennand KJ, Kelsoe JR, Gage FH, Yao J, 2015. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhiet V, Boudebesse C, Bellivier F, Drouot X, Henry C, Leboyer M, Etain B, 2014. Circadian abnormalities as markers of susceptibility in bipolar disorders. Front. Biosci. (Schol Ed) 6, 120–137. [DOI] [PubMed] [Google Scholar]
- Molz P, Schroder N, 2017. Potential Therapeutic Effects of Lipoic Acid on Memory Deficits Related to Aging and Neurodegeneration. Front. Pharmacol 8, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK, 2000. Lithium-induced increase in human brain grey matter. Lancet 356, 1241–1242. [DOI] [PubMed] [Google Scholar]
- Morava E, Gardeitchik T, Kozicz T, de Boer L, Koene S, de Vries MC, McFarland R, Roobol T, Rodenburg RJ, Verhaak CM, 2010. Depressive behaviour in children diagnosed with a mitochondrial disorder. Mitochondrion 10, 528–533. [DOI] [PubMed] [Google Scholar]
- Moutsatsou P, Tsoporis JN, Salpeas V, Bei E, Alevizos B, Anagnostara C, Izhar S, Proteau G, Rizos E, Hatziagelaki E, Toumpoulis IK, Rizos IK, Parker TG, 2014. Peripheral blood lymphocytes from patients with bipolar disorder demonstrate apoptosis and differential regulation of advanced glycation end products and S100B. Clin. Chem. Lab. Med 52, 999–1007. [DOI] [PubMed] [Google Scholar]
- Munakata K, Iwamoto K, Bundo M, Kato T, 2005. Mitochondrial DNA 3243A>G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol. Psychiatry 57, 525–532. [DOI] [PubMed] [Google Scholar]
- Munakata K, Tanaka M, Mori K, Washizuka S, Yoneda M, Tajima O, Akiyama T, Nanko S, Kunugi H, Tadokoro K, Ozaki N, Inada T, Sakamoto K, Fukunaga T, Iijima Y, Iwata N, Tatsumi M, Yamada K, Yoshikawa T, Kato T, 2004. Mitochondrial DNA 3644T–>C mutation associated with bipolar disorder. Genomics 84, 1041–1050. [DOI] [PubMed] [Google Scholar]
- Muneer A, 2016. Staging Models in Bipolar Disorder: A Systematic Review of the Literature. Clin. Psychopharmacol. Neurosci 14, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkholm K, Peijs L, Vinberg M, Kessing LV, 2015. A composite peripheral blood gene expression measure as a potential diagnostic biomarker in bipolar disorder. Transl. Psychiatry 5, e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BL, Stoll AL, Harris PQ, Ravichandran C, Babb SM, Carlezon WA Jr., Cohen BM, 2012. Omega-3 fatty acid treatment, with or without cytidine, fails to show therapeutic properties in bipolar disorder: a double-blind, randomized add-on clinical trial. J. Clin. Psychopharmacol 32, 699–703. [DOI] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P, 2009. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM, 2011. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol 12, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov AV, MacDonald ML, Ongur D, Konradi C, 2007. Differences in lymphocyte electron transport gene expression levels between subjects with bipolar disorder and normal controls in response to glucose deprivation stress. Arch. Gen. Psychiatry 64, 555–564. [DOI] [PubMed] [Google Scholar]
- Newell C, Shutt TE, Ahn Y, Hittel DS, Khan A, Rho JM, Shearer J, 2016. Tissue Specific Impacts of a Ketogenic Diet on Mitochondrial Dynamics in the BTBR(T+tf/j) Mouse. Front. Physiol 7, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Hough CJ, Chuang DM, 1998. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. PNAS 95, 2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Gorman E, Beutner G, Wallimann T, Brdiczka D, 1996. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim. Biophys. Acta, Gen. Subj 1276, 161–170. [DOI] [PubMed] [Google Scholar]
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B, 2007. Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922. [DOI] [PubMed] [Google Scholar]
- Palacios HH, Yendluri BB, Parvathaneni K, Shadlinski VB, Obrenovich ME, Leszek J, Gokhman D, Gasiorowski K, Bragin V, Aliev G, 2011. Mitochondrion-specific antioxidants as drug treatments for Alzheimer disease. CNS Neurol. Disord.: Drug Targets 10, 149–162. [DOI] [PubMed] [Google Scholar]
- Panchal K, Tiwari AK, 2019. Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 47, 151–173. [DOI] [PubMed] [Google Scholar]
- Paraskevi M, James NT, Vasileios S, Ekaterini B, Basel A, Chrysoula A, Shehla I, Gerald P, Emmanouil R, Erifili H, Ioannis KT, Ioannis KR, Thomas GP, 2014. Peripheral blood lymphocytes from patients with bipolar disorder demonstrate apoptosis and differential regulation of advanced glycation end products and S100B. Clin. Chem. Laborat. Med. (CCLM) 52, 999–1007. [DOI] [PubMed] [Google Scholar]
- Passos IC, Mwangi B, Vieta E, Berk M, Kapczinski F, 2016. Areas of controversy in neuroprogression in bipolar disorder. Acta Psychiatr. Scand 134, 91–103. [DOI] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J, 2013. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Wallace DC, 2018. Mitochondrial Etiology of Neuropsychiatric Disorders. Biol. Psychiatry 83, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perova T, Wasserman MJ, Li PP, Warsh JJ, 2008. Hyperactive intracellular calcium dynamics in B lymphoblasts from patients with bipolar I disorder. The Int. J. Neuropsychopharmacol. Off. Scient. J. Collegium Internationale Neuropsychopharmacologicum 11, 185–196. [DOI] [PubMed] [Google Scholar]
- Pettegrew JW, Levine J, Gershon S, Stanley JA, Servan-Schreiber D, Panchalingam K, McClure RJ, 2002. 31P-MRS study of acetyl-L-carnitine treatment in geriatric depression: preliminary results. Bipolar Disord 4, 61–66. [DOI] [PubMed] [Google Scholar]
- Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, Benatti S, Gibellini L, Cotichini R, Stazi MA, Trenti T, Franceschi C, Cossarizza A, 2014. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol 44, 1552–1562. [DOI] [PubMed] [Google Scholar]
- Pinto JV, Passos IC, Librenza-Garcia D, Marcon G, Schneider MA, Conte JH, da Silva JPA, Lima LP, Quincozes-Santos A, Kauer-Sant Anna M, Kapczinski F, 2018. Neuron-glia Interaction as a Possible Pathophysiological Mechanism of Bipolar Disorder. Curr. Neuropharmacol 16, 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post RM, Fleming J, Kapczinski F, 2012. Neurobiological correlates of illness progression in the recurrent affective disorders. J. Psychiatr. Res 46, 561–573. [DOI] [PubMed] [Google Scholar]
- Raffa M, Barhoumi S, Atig F, Fendri C, Kerkeni A, Mechri A, 2012. Reduced antioxidant defense systems in schizophrenia and bipolar I disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 39, 371–375. [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, Takahashi JS, Imai S, Bass J, 2009. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasic D, Hajek T, Alda M, Uher R, 2014. Risk of mental illness in offspring of parents with schizophrenia, bipolar disorder, and major depressive disorder: a meta-analysis of family high-risk studies. Schizophr. Bull 40, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG, 1999. Redox signaling: hydrogen peroxide as intracellular messenger. Exp. Mol. Med 31, 53–59. [DOI] [PubMed] [Google Scholar]
- Rhoads DM, Umbach AL, Subbaiah CC, Siedow JN, 2006. Mitochondrial reactive oxygen species. Contribution to oxidative stress and interorganellar signaling. Plant Physiol 141, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C, 2012. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol 13, 566–578. [DOI] [PubMed] [Google Scholar]
- Robinson LJ, Ferrier IN, 2006. Evolution of cognitive impairment in bipolar disorder: a systematic review of cross-sectional evidence. Bipolar Disord 8, 103–116. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Loscher W, Rho JM, 2016. Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harb. Perspect. Med 6. [DOI] [PMC free article] [PubMed]
- Roitman S, Green T, Osher Y, Karni N, Levine J, 2007. Creatine monohydrate in resistant depression: a preliminary study. Bipolar Disord 9, 754–758. [DOI] [PubMed] [Google Scholar]
- Rosa AR, Singh N, Whitaker E, de Brito M, Lewis AM, Vieta E, Churchill GC, Geddes JR, Goodwin GM, 2014. Altered plasma glutathione levels in bipolar disorder indicates higher oxidative stress; a possible risk factor for illness onset despite normal brain-derived neurotrophic factor (BDNF) levels. Psychol. Med 44, 2409–2418. [DOI] [PubMed] [Google Scholar]
- Sahar S, Nin V, Barbosa MT, Chini EN, Sassone-Corsi P, 2011. Altered behavioral and metabolic circadian rhythms in mice with disrupted NAD+ oscillation. Aging (Albany NY) 3, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi A, Gholamifar E, Naserzadeh P, Hosseini MJ, Pourahmad J, 2017. Toxicity of lithium on isolated heart mitochondria and cardiomyocyte: A justification for its cardiotoxic adverse effect. J. Biochem. Mol. Toxicol 31. [DOI] [PubMed]
- Samuni Y, Goldstein S, Dean OM, Berk M, 2013. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta, Gen. Subj 1830, 4117–4129. [DOI] [PubMed] [Google Scholar]
- Sanz A, Caro P, Gomez J, Barja G, 2006. Testing the vicious cycle theory of mitochondrial ROS production: effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. J. Bioenerg. Biomembr 38, 121–127. [DOI] [PubMed] [Google Scholar]
- Sarris J, Mischoulon D, Schweitzer I, 2011. Adjunctive nutraceuticals with standard pharmacotherapies in bipolar disorder: a systematic review of clinical trials. Bipolar Disord 13, 454–465. [DOI] [PubMed] [Google Scholar]
- Scafidi S, Fiskum G, Lindauer SL, Bamford P, Shi D, Hopkins I, McKenna MC, 2010a. Metabolism of acetyl-L-carnitine for energy and neurotransmitter synthesis in the immature rat brain. J. Neurochem 114, 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi S, Racz J, Hazelton J, McKenna MC, Fiskum G, 2010b. Neuroprotection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Dev. Neurosci 32, 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaini G, Barichello T, Fries GR, Kennon EA, Andrews T, Nix BR, Zunta-Soares G, Valvassori SS, Soares JC, Quevedo J, 2019. TSPO upregulation in bipolar disorder and concomitant downregulation of mitophagic proteins and NLRP3 inflammasome activation. Neuropsychopharmacol. Off. Publicat. Am. College Neuropsychopharmacol 44, 1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaini G, Fries GR, Valvassori SS, Zeni CP, Zunta-Soares G, Berk M, Soares JC, Quevedo J, 2017a. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl. Psychiatry 7, e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaini G, Fries GR, Valvassori SS, Zeni CP, Zunta-Soares G, Berk M, Soares JC, Quevedo J, 2017b. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl. Psychiatry 7, e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaini G, Rezin GT, Carvalho AF, Streck EL, Berk M, Quevedo J, 2016. Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev 68, 694–713. [DOI] [PubMed] [Google Scholar]
- Scaini G, Valvassori SS, Diaz AP, Lima CN, Benevenuto D, Fries GR, Quevedo J, 2020. Neurobiology of bipolar disorders: a review of genetic components, signaling pathways, biochemical changes, and neuroimaging findings. Braz. J Psychiatry [DOI] [PMC free article] [PubMed]
- Schmitt K, Grimm A, Dallmann R, Oettinghaus B, Restelli LM, Witzig M, Ishihara N, Mihara K, Ripperger JA, Albrecht U, Frank S, Brown SA, Eckert A, 2018. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab 27 (657–666), e655. [DOI] [PubMed] [Google Scholar]
- Schneider MR, DelBello MP, McNamara RK, Strakowski SM, Adler CM, 2012. Neuroprogression in bipolar disorder. Bipolar Disord 14, 356–374. [DOI] [PubMed] [Google Scholar]
- Schulmann A, Ryu E, Goncalves V, Rollins B, Christiansen M, Frye MA, Biernacka J, Vawter MP, 2019. Novel Complex Interactions between Mitochondrial and Nuclear DNA in Schizophrenia and Bipolar Disorder. Mol Neuropsychiatry 5, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scola G, Kim HK, Young LT, Andreazza AC, 2013. A fresh look at complex I in microarray data: clues to understanding disease-specific mitochondrial alterations in bipolar disorder. Biol. Psychiatry 73, e4–5. [DOI] [PubMed] [Google Scholar]
- Scola G, Kim HK, Young LT, Salvador M, Andreazza AC, 2014. Lithium reduces the effects of rotenone-induced complex I dysfunction on DNA methylation and hydroxymethylation in rat cortical primary neurons. Psychopharmacology 231, 4189–4198. [DOI] [PubMed] [Google Scholar]
- Shalbuyeva N, Brustovetsky T, Brustovetsky N, 2007. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J. Biol. Chem 282, 18057–18068. [DOI] [PubMed] [Google Scholar]
- Shao L, Young LT, Wang JF, 2005. Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol. Psychiatry 58, 879–884. [DOI] [PubMed] [Google Scholar]
- Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y, 2011. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet 20, 4605–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wittke-Thompson JK, Badner JA, Hattori E, Potash JB, Willour VL, McMahon FJ, Gershon ES, Liu C, 2008. Clock genes may influence bipolar disorder susceptibility and dysfunctional circadian rhythm. Am. J. M.ed. Genet. Part B, Neuropsychiatr. Genet. Off. Publicat. Int. Soc. Psychiat. Genet 147B, 1047–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak S, Honig A, Christophe A, Maes M, Helsdingen RW, De Vriese SA, Riedel WJ, 2004. Lower high-density lipoprotein cholesterol and increased omega-6 polyunsaturated fatty acids in first-degree relatives of bipolar patients. Psychol. Med 34, 103–112. [DOI] [PubMed] [Google Scholar]
- Soczynska JK, Kennedy SH, Chow CS, Woldeyohannes HO, Konarski JZ, McIntyre RS, 2008. Acetyl-L-carnitine and alpha-lipoic acid: possible neurotherapeutic agents for mood disorders? Expert Opin. Invest. Drugs 17, 827–843. [DOI] [PubMed] [Google Scholar]
- Stoll AL, Severus WE, Freeman MP, Rueter S, Zboyan HA, Diamond E, Cress KK, Marangell LB, 1999. Omega 3 fatty acids in bipolar disorder: a preliminary double-blind, placebo-controlled trial. Arch. Gen. Psychiatry 56, 407–412. [DOI] [PubMed] [Google Scholar]
- Strakowski SM, DelBello MP, Zimmerman ME, Getz GE, Mills NP, Ret J, Shear P, Adler CM, 2002. Ventricular and periventricular structural volumes in first-versus multiple-episode bipolar disorder. Am. J. Psychiatry 159, 1841–1847. [DOI] [PubMed] [Google Scholar]
- Streck EL, Goncalves CL, Furlanetto CB, Scaini G, Dal-Pizzol F, Quevedo J, 2014. Mitochondria and the central nervous system: searching for a pathophysiological basis of psychiatric disorders. Rev Bras Psiquiatr 36, 156–167. [DOI] [PubMed] [Google Scholar]
- Sun X, Wang JF, Tseng M, Young LT, 2006. Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J. Psychiat. Neurosci. JPN 31, 189–196. [PMC free article] [PubMed] [Google Scholar]
- Taanman JW, 1999. The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta, Gen. Subj 1410, 103–123. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL, 2000. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol 279, L1005–1028. [DOI] [PubMed] [Google Scholar]
- Toniolo RA, Silva M, Fernandes FBF, Amaral J, Dias RDS, Lafer B, 2018. A randomized, double-blind, placebo-controlled, proof-of-concept trial of creatine monohydrate as adjunctive treatment for bipolar depression. J. Neural Transm. (Vienna) 125, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvassori SS, Bavaresco DV, Feier G, Cechinel-Recco K, Steckert AV, Varela RB, Borges C, Carvalho-Silva M, Gomes LM, Streck EL, Quevedo J, 2018. Increased oxidative stress in the mitochondria isolated from lymphocytes of bipolar disorder patients during depressive episodes. Psychiatry Res 264, 192–201. [DOI] [PubMed] [Google Scholar]
- Valvassori SS, Rezin GT, Ferreira CL, Moretti M, Goncalves CL, Cardoso MR, Streck EL, Kapczinski F, Quevedo J, 2010. Effects of mood stabilizers on mitochondrial respiratory chain activity in brain of rats treated with d-amphetamine. J. Psychiatr. Res 44, 903–909. [DOI] [PubMed] [Google Scholar]
- Velot C, Srere PA, 2000. Reversible transdominant inhibition of a metabolic pathway. In vivo evidence of interaction between two sequential tricarboxylic acid cycle enzymes in yeast. J. Biol. Chem 275, 12926–12933. [DOI] [PubMed] [Google Scholar]
- Wasserman MJ, Corson TW, Sibony D, Cooke RG, Parikh SV, Pennefather PS, Li PP, Warsh JJ, 2004. Chronic lithium treatment attenuates intracellular calcium mobilization. Neuropsychopharmacol. Off. Publicat. Am. Coll. Neuropsychopharmacol 29, 759–769. [DOI] [PubMed] [Google Scholar]
- Winsberg ME, Sachs N, Tate DL, Adalsteinsson E, Spielman D, Ketter TA, 2000. Decreased dorsolateral prefrontal N-acetyl aspartate in bipolar disorder. Biol. Psychiatry 47, 475–481. [DOI] [PubMed] [Google Scholar]
- Wu G, Zhu Q, Zeng J, Gu X, Miao Y, Xu W, Lv T, Song Y, 2019. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-kappaB. J. Thorac Dis 11, 4816–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaki N, Otsuka I, Numata S, Yanagi M, Mouri K, Okazaki S, Boku S, Horai T, Ohmori T, Shirakawa O, Sora I, Hishimoto A, 2018. Mitochondrial DNA copy number of peripheral blood in bipolar disorder: The present study and a meta-analysis. Psychiatry Res 269, 115–117. [DOI] [PubMed] [Google Scholar]
- Yang S, Van Dongen HP, Wang K, Berrettini W, Bucan M, 2009. Assessment of circadian function in fibroblasts of patients with bipolar disorder. Mol. Psychiatry 14, 143–155. [DOI] [PubMed] [Google Scholar]
- Yoon IS, Li PP, Siu KP, Kennedy JL, Cooke RG, Parikh SV, Warsh JJ, 2001a. Altered IMPA2 gene expression and calcium homeostasis in bipolar disorder. Mol. Psychiatry 6, 678–683. [DOI] [PubMed] [Google Scholar]
- Yoon IS, Li PP, Siu KP, Kennedy JL, Macciardi F, Cooke RG, Parikh SV, Warsh JJ, 2001b. Altered TRPC7 gene expression in bipolar-I disorder. Biol. Psychiatry 50, 620–626. [DOI] [PubMed] [Google Scholar]
- Yoshimi N, Futamura T, Bergen SE, Iwayama Y, Ishima T, Sellgren C, Ekman CJ, Jakobsson J, Palsson E, Kakumoto K, Ohgi Y, Yoshikawa T, Landen M, Hashimoto K, 2016a. Cerebrospinal fluid metabolomics identifies a key role of isocitrate dehydrogenase in bipolar disorder: evidence in support of mitochondrial dysfunction hypothesis. Mol. Psychiatry [DOI] [PMC free article] [PubMed]
- Yoshimi N, Futamura T, Bergen SE, Iwayama Y, Ishima T, Sellgren C, Ekman CJ, Jakobsson J, Palsson E, Kakumoto K, Ohgi Y, Yoshikawa T, Landen M, Hashimoto K, 2016b. Cerebrospinal fluid metabolomics identifies a key role of isocitrate dehydrogenase in bipolar disorder: evidence in support of mitochondrial dysfunction hypothesis. Mol. Psychiatry 21, 1504–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuksel C, Du F, Ravichandran C, Goldbach JR, Thida T, Lin P, Dora B, Gelda J, O’Connor L, Sehovic S, Gruber S, Ongur D, Cohen BM, 2015. Abnormal high-energy phosphate molecule metabolism during regional brain activation in patients with bipolar disorder. Mol. Psychiatry 20, 1079–1084. [DOI] [PubMed] [Google Scholar]
- Yuzefovych LV, Pastukh VM, Ruchko MV, Simmons JD, Richards WO, Rachek LI, 2019. Plasma mitochondrial DNA is elevated in obese type 2 diabetes mellitus patients and correlates positively with insulin resistance. PLoS ONE 14, e0222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanardi R, Smeraldi E, 2006. A double-blind, randomised, controlled clinical trial of acetyl-L-carnitine vs. amisulpride in the treatment of dysthymia. Europ. Neuropsychopharmacol. J. Eur. College Neuropsychopharmacol 16, 281–287. [DOI] [PubMed] [Google Scholar]
- Zarate CA Jr., Singh J, Manji HK, 2006. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol. Psychiatry 59, 1006–1020. [DOI] [PubMed] [Google Scholar]
- Zarate CA Jr., Tohen M, Land M, Cavanagh S, 2000. Functional impairment and cognition in bipolar disorder. Psychiatr. Q 71, 309–329. [DOI] [PubMed] [Google Scholar]
- Zavodnik IB, 2016. Mitochondria, calcium homeostasis and calcium signaling. Biomed Khim 62, 311–317. [DOI] [PubMed] [Google Scholar]
- Zverova M, Hroudova J, Fisar Z, Hansikova H, Kalisova L, Kitzlerova E, Lambertova A, Raboch J, 2019. Disturbances of mitochondrial parameters to distinguish patients with depressive episode of bipolar disorder and major depressive disorder. Neuropsychiatr. Dis. Treat 15, 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]