

Abstract
Class Ia/b cystic fibrosis transmembrane regulator (CFTR) variants cause severe lung disease in 10% of cystic fibrosis (CF) patients and are untreatable with small-molecule pharmaceuticals. Genetic replacement of CFTR offers a cure, but its effectiveness is limited in vivo. We hypothesized that enhancing protein levels (using codon optimization) and/or activity (using gain-of-function variants) of CFTR would more effectively restore function to CF bronchial epithelial cells. Three different variants of the CFTR protein were tested: codon optimized (high codon adaptation index [hCAI]), a gain-of-function (GOF) variant (K978C), and a combination of both (hˆK978C). In human embryonic kidney (HEK293T) cells, initial results showed that hCAI and hˆK978C produced greater than 10-fold more CFTR protein and displayed ∼4-fold greater activity than wild-type (WT) CFTR. However, functionality was profoundly different in CF bronchial epithelial cells. Here, K978C CFTR more potently restored essential epithelial functions (anion transport, airway surface liquid height, and pH) than WT CFTR. hCAI and hˆK978C CFTRs had limited impact because of mislocalization in the cell. These data provide a proof of principle showing that GOF variants may be more effective than codon-optimized forms of CFTR for CF gene therapy.
Video abstract
Keywords: cystic fibrosis, gene therapy, CFTR, airway epithelium, codon optimisation, anion secretion, airway surface liquid, sputum
Graphical abstract
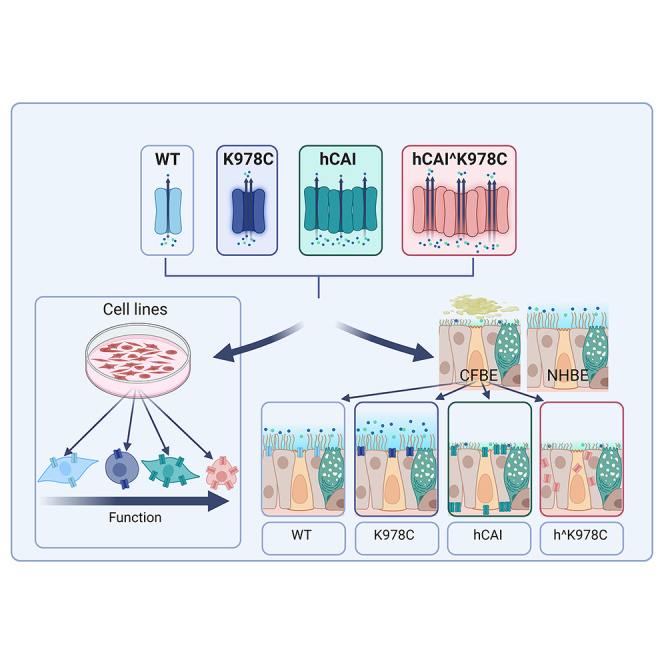
Baines and colleagues show that viral delivery of cystic fibrosis transmembrane regulator (CFTR) gain-of-function variant cDNA (K978C-CFTR) to cystic fibrosis bronchial epithelial (CFBE) cells in the presence of CF sputum was more effective at restoring anion secretion, ASL hydration, and pH than wild-type CFTR.
Introduction
Precision medicine, through pharmacotherapy, has revolutionized the treatment of cystic fibrosis transmembrane regulator (CFTR) variants that exhibit recoverable CFTR protein. Nevertheless, class Ia (no mRNA) and class Ib (no protein) mutations, which are responsible for about 10% of all cystic fibrosis (CF)-causing CFTR variants, remain untreatable.1 Gene therapy and gene editing techniques, while still in their infancy, hold immense promise as a potential cure for all forms of CF, including class I mutations. Proof-of-concept studies have established the feasibility of genetic therapies, but the limited success of CF gene therapy clinical trials highlights the need for further development and understanding of these techniques.
The estimated level of functional endogenous CFTR required to ameliorate severe pulmonary disease in vivo ranges between 5% and 16%,2,3,4,5,6 a level yet to be achieved in clinical trials. In vivo delivery of airway-targeted genetic therapies faces significant challenges because of natural airway barriers, which hinder the delivery and overall potency of genetic therapy formulations.5,7,8 In vitro, where it is easier to manipulate target cells, 10%–60% endogenous expression of CFTR was required to recapitulate normal anion transport function,9,10,11 whereas transduction of only 6%–25% of CF cells with exogenous wild-type (WT) CFTR cDNA generated Cl− transport properties similar to non-CF cultures.12,13 However, none of these studies considered the impact of the CF luminal environment on CFTR and epithelial function; therefore, these estimates may not translate in vivo.
Several innovative strategies are being developed to enhance the potency of gene therapy for CF. For instance, codon optimization of CFTR has been employed to reduce the immunogenicity of the DNA used in gene therapy by depleting cytosine-phosphorothioate-guanine (CpG) motifs, which stimulate the Toll-like receptor 9 (TLR9) inflammatory pathway.14,15,16 Codon optimization also enhances translation of mRNA into protein by substituting rare codons (triplets of DNA bases that code for amino acids less frequently in a specific organism), which can decelerate or even halt the translation process.17,18 Replacement of the rare codons in WT CFTR increases protein production and function dramatically (>10-fold) when expressed in Fisher rat thyroid cells.18 However, research into codon-optimized CFTR function in human airway cells is relatively nascent, with current findings mostly reporting mild changes in CFTR-mediated anion transport, and no data on airway surface liquid (ASL) rehydration are available yet.19 The potential of using gain-of-function (GOF) CFTR mutants as a strategy for improving CF gene therapy remains largely unexplored. Study of CFTR structure and function relationships has identified several such mutations.20,21 One mutation, K978C CFTR, demonstrated a greater than 2-fold higher open probability (Po) in excision patch-clamp studies than WT CFTR.21,22,23 Studies of GOF CFTRs have so far been limited to functional testing in cell lines. However, CF bronchial epithelial (CFBE) cells grown at the air-liquid interface are considered the gold standard for preclinical airway model systems.24,25 In these cells, the function and effectiveness of GOF CFTRs, such as K978C CFTR, for genetic therapy practices is uncharted. K978C was selected over other GOF variants for the unique characteristics of an increased Po while retaining regulation by cyclic AMP (cAMP)/protein kinase A (PKA).21
We hypothesized that use of K978C CFTR, and/or codon-optimized WT or K978C CFTR could improve the performance of CF gene therapy in a physiologically relevant model. We first investigated the protein production and function of our CFTR constructs in HEK293T cells. This provided us with a basic understanding of construct behavior in a reproducible cell line environment. Next, we transitioned our research to primary airway epithelial cells, using non-CF primary human bronchial epithelial (NHBE) cells and CF class 1 (W1282X/R1162X) and F508del genotypes (CFBE cells). We introduced and examined the protein production and localization of the exogenously expressed CFTR constructs, each controlled by a high-activity promoter. In the final stage, we analyzed the function of CFTR constructs in differentiated CFBE cells and compared them with the function of endogenous CFTR from mixed cultures of NHBE and CFBE cells. Importantly, the functional tests were performed in the presence of CF sputum because our previous research indicated that CF sputum inhibits CFTR function and disrupts normal ASL hydration compared with normal lung sputum.26 Our objective was to identify the transduction efficiency required to restore CFTR-dependent anion transport and ASL hydration to that of NHBE cells in the presence of normal lung sputum as a criterion for therapeutic success.26
Results
Expression and function of CFTR cDNAs in HEK293T cells
All CFTR constructs (Figure 1Ai) were transfected into human embryonic kidney (HEK293T) cells to investigate expression and function (Figure 2Aii). All CFTR variants produced protein, visible as mature, complex, glycosylated, ∼170-kDa protein (band C) and immature, core-glycosylated, ∼140-kDa protein (band B) by western blot (Figure 1B). The halide-sensitive (HS) yellow fluorescent protein (YFP) (HS-YFP) protein in co-transfected HEK293T cells was observed at ∼22 kDa. Transfection of HEK293T cells with codon-optimized high codon adaptation index [hCAI] and hˆK978C CFTR resulted in greater than 10-fold more CFTR protein than WT CFTR (14.37 ± 2.2 and 11.21 ± 1.50-fold, respectively; p < 0.05; n = 3) (Figure 1C). There was no significant difference in abundance of K978C compared with WT CFTR protein. An HS YFP fluorescence quenching assay was employed to determine the anion transport function of CFTRs with increased abundance (Figures 1D–1F). There was little decrease of HS YFP fluorescence in the presence of DMSO (vehicle), providing evidence that the K978C GOF mutation did not result in constitutive activity (Figure 1D). Addition of the CFTR activator forskolin to WT-, hCAI CFTR-, and hˆK978C CFTR-transfected HEK293T cells produced a decrease in HS YFP fluorescence (Figure 1E). hˆK978C CFTR displayed a significantly greater maximal rate of I− entry compared with hCAI CFTR (0.3 mM/s and 0.23 mM/s, respectively; p < 0.05; n = 3) and WT CFTR (0.06 mM/s, p < 0.001, n = 3) (Figures 1G and S1). Prior exposure of hCAI CFTR to the potentiator VX770 increased quenching of HS YFP fluorescence (n = 3, p < 0.01), producing a similar maximal rate of I− entry as hˆK978C CFTR (Figures 1F and 1G). The WT maximal rate of iodide entry was also increased greater than 2-fold (0.057 ± 0.011 mM/s to 0.15 ± 0.012 mM/s, p < 0.01, n = 3) (Figure S1), but this remained lower than for hCAI CFTR or hˆK978C CFTR (0.15 mM/s, 0.30 mM/s and 0.32 mM/s, respectively; p < 0.05; n = 3) (Figures 2G and S1).
Figure 1.

Impact of CFTR cDNAs on protein production and function in transfected HEK293T cells
(A) Illustrations representing each CFTR variant (WT [blue], K978C mutation [purple], high codon adaptation index [hCAI] CFTR [cyan], and hCAI carrying the K978C mutation [hˆK978C, pink]) (i) and schematic of the halide-sensitive (HS) YFP quenching assay (HEK293T cells co-transfected with HS-YFP and CFTR variants) (ii). CFTR activators induce facilitated anion movement through CFTR channels, leading to HS-YFP fluorescence quenching. (B) Western blot of protein extracts from HEK293T cells co-transfected with HS-YFP and CFTR cDNAs. CFTR bands C and B (green) and HS-YFP (red) are indicated. Molecular weight markers (red) are displayed on the left side of the blot. (C) Quantification of CFTR protein abundance, expressed as the mean of (CFTR (bands C + B)/HS-YFP) normalized to the WT for standardization between blots. Data are represented as mean ± SD. Differences were compared by one-way ANOVA followed by Tukey’s post hoc tests. ∗p < 0.05, ∗∗p < 0.01, n = 3. (D–F) HS YFP fluorescence quenching over time in HEK293 cells transfected with HS YFP alone or co-transfected with CFTR cDNAs under different conditions: DMSO (vehicle) (D), forskolin (FSK) (E), and FSK plus the CFTR potentiator VX770 (FSK+VX770) (F), all added at time point 0. (G) The maximal rate of I− influx (Δ[I−]/Δt) is summarized for CFTR cDNA and conditions. Data are represented as mean ± SD along with individual data points. CFTR function was compared by two-way ANOVA followed by Tukey’s post hoc tests. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, n = 3.
Figure 2.

CFTR protein expression and localization in CFBE cells
(A) Representative western blot of soluble protein fraction from differentiated CFBE cell cultures transduced with CFTR cDNAs or GFP alone (as labeled). Blots are probed with anti-CFTR (CFTR 596) and anti-GFP. CFTR bands C and B (green), GFP (red), and molecular weight markers (red) are displayed. (B) Summary of the relative fluorescence units (RFUs) for CFTR bands C and B in relation to the percent final transduction, normalized to the WT for each CFTR cDNA. Data are shown as mean ± SD with individual data points (n = 4–7 from 3 donors). Treatments were compared by two-way ANOVA with Tukey’s post hoc analyses. ∗∗p < 0.01, ∗∗∗p < 0.001. RFU ratios of bands B:C are displayed below the graph. (C) Representative confocal live images (×60, excitation 488 nm/emission 507 nm) of CFBE BMI-1 cells transduced with GFP linked to CFTR cDNAs or GFP alone. Cell blebbing is indicated with white arrows, with a 50-μm scale bar displayed. (D) Schematic detailing the FRAP protocol, indicating the stages of pre-bleach, post-bleach, and end, within the region of interest (ROI), represented by the white square. (E) Kinetics plot showcasing fluorescence (F) change over time in an ROI on the cell periphery. The bleach point is marked with an arrow. (F) Summary of the mobile fraction (Mf) ratio for different GFP-linked CFTRs. Data are shown as mean ± SD with individual data points. Treatments were compared by one-way ANOVA with Tukey’s post hoc analyses. ∗∗p < 0.01, ∗∗∗p < 0.001. (n = 8–13 from 1 CFBE BMI-1 cell donor: F508del/F508del).
Protein expression and localization of CFTR cDNAs in CFBE cells
Transduction of CFBE cells with all CFTR cDNAs produced distinct CFTR proteins, mature (band C, ∼170 kDa) and immature (band B, ∼140 kDa) (Figure 2A). As expected, hCAI CFTR produced the most CFTR protein per transduced cell (p < 0.001, n = 5–7 from 3 donors). However, hˆK978C CFTR produced less CFTR protein than the WT (p < 0.05, n = 4–7 from 3 donors). The abundance of band C was more than 5-fold higher than that of band B (ratio: 5.1–5.2) across all forms of CFTR (p < 0.05, n = 4–7 from 3 donors) except for hˆK978C CFTR (Figure 2B). To address potential protein aggregation often associated with high protein yields, we employed two strategies to the hˆK978C transduced cells: we raised the denaturation temperature of the protein lysate to 80°C from 37°C (Figure S2B) and tested the Triton X-100-insoluble fraction with a stronger detergent, SDS, to detect any trapped protein (Figure S2). These modifications resulted in further reduction of CFTR band intensity, suggesting that the protein was not sequestered in aggregates but indeed expressed at a lower level. The amount of GFP in the cell broadly followed the same pattern, except that GFP for codon-optimized hˆK978C CFTR was higher than for K978C CFTR (p < 0.05, n = 4–7 from 3 donors) (Figure S2D).
We then employed CFTR constructs with GFP fused to the CFTR N terminus to further investigate protein localization in F508del/F508del CFBE cells transduced with BMI-1 (CFBE BMI-1 cells). CFBE BMI-1 cells are functionally similar to primary CFBE cells but have extended proliferation capacity.27 This enabled reproducible acquisition of fluorescence images and fluorescence recovery after photobleaching (FRAP) data across cell passages. WT and K978C CFTR GFP fluorescence was clearly localized to the periphery of the cell, consistent with membrane localization (Figure S2E) hCAI CFTR exhibited increased membrane and intracellular fluorescence (Figures 2C and S2E). hˆK978C CFTR was identified throughout the cell, and blebbing, a characteristic marker of cell apoptosis, was also observed (Figure 2C, white arrows).28 FRAP was used to determine whether the GFP-linked proteins displayed increased free, rapid diffusion characteristic of a non-membrane bound protein (Figures 2D and S2F). hˆK978C CFTR displayed the characteristics of a non-membrane-bound protein, unlike WT, K978C, and hCAI CFTRs (p < 0.001, n = 8–10) (Figures 2E, 2F, and S2F).
Definition of models used to investigate CFTR function in primary CFBE cells
Male (XY) or female (XX) donor CFBE or NHBE cells were isolated from bronchial regions of CF and non-CF lungs, counted, and mixed at 10%, 20%, 50%, and 75% (NHBE:CFBE cells) as represented (Figure 3A). Markers of epithelial differentiation, including cilia (α-tubulin) and F-actin structure associated with tight junctions, visualized by phalloidin, were present in all mixed cultures (only data from the 50:50 mix is shown in Figure 3B). The final percentage of NHBE:CFBE cells in the co-cultures, as analyzed by droplet digital PCR for the amelogenin-X isoform (AMEL-X) or amelogenin-Y isoform (AMEL-Y), was different from the original seeding percentage. This appeared to be donor specific and independent of the CF mutation or whether the donor was male or female (Figure 3C).
Figure 3.

Characterization of primary differentiated NHBE:CFBE cell mixed culture and CFBE cell models
(A) Schematic of the NHBE:CFBE cell mixed culture model. CFBE and NHBE cells from male (XY) or female (XX) donors were combined in ratios of 10%, 25%, 50%, and 75% NHBE:CFBE cells and seeded onto semipermeable membranes. (B) Confocal microscope images (maximal projection) of a representative 50% NHBE:CFBE cell, stained with phalloidin (red, F-actin), anti-β-tubulin (yellow, cilia), DAPI (blue, nuclei), and composite image. Scale bars are depicted in the bottom right corner. (C) Relative proportions of AMEL-X and AMEL-Y regions in genomic DNA from three different mixes of male (M) and female (F) NHBE or CF donors post functional analysis, as determined via ddPCR. Data were normalized to values from 100% male or 100% female cultures, and samples were run in duplicate (n = 28). Each data point represents the mean ± SD (some error bars are smaller than the symbol height). (D) Schematic of the lentiviral constructs used for CFTR transduction. (E) Confocal microscope images of GFP CFTR-transduced CFBE cell cultures immunostained with phalloidin (F-actin, red), anti-β-tubulin (cilia, yellow), GFP (transduced cells, green), and composite image with DAPI (blue). (F) Relationship between percent seeded transduced cells and percent final transduced (4 h prior to functional testing) for the different CFTR cDNAs. The ratio of percent final transduced to percent seeded transduced (%T/%S) for each CFTR is given in a table below the graph.
The CFTR variants were introduced into CF cells, specifically W1282X/R1162X or F508del, using a lentiviral vector. These CFTR variants were positioned upstream of a green fluorescent protein (GFP) and were separated by a self-cleaving T2A peptide. This setup allowed efficient analysis of the transduction process (Figure 3D). Transduced CFBE cells maintained CFTR transgene expression for 3–4 weeks, and differentiated ciliated cells were present (Figure 3E). However, the percentages of final transduced cells were reduced from the percentage of seeded transduced cells, indicating a loss of transgene expression over time or a selective pressure against transduced cell survival/proliferation compared with non-transduced cells (Figure 3F). All downstream functional analyses were measured against the final NHBE cell percentage in mixed cultures and the final percentage of CFTR-transduced CFBE cells.
CFTR-dependent anion transport is associated with CFTR in NHBE:CFBE cell co-cultures and with K978C CFTR but not codon-optimized forms of CFTR in transduced CFBE cells
We investigated bioelectric properties of NHBE:CFBE cell co-cultures and transduced CFBE cells in the presence of CF sputum (Figures 4A and 4B). Responses to amiloride, forskolin, CFTR inhibitor 172 (CFTRinh172), and uridine triphosphate (UTP) provided evidence that functional epithelial Na+ channels (ENaCs), CFTR, and calcium-activated chloride channels (CaCCs) were present. The summarized data from all donors and transductions showed that short-circuit current (ΔIsc) in response to CFTRinh172 increased in a linear fashion with the percentage of NHBE cells in the co-culture (coefficient of determination [R2] = 0.91, p < 0.0001) and with the percentage of CF cell transduction of K978C CFTR, WT CFTR, and hCAI CFTRs in transduced CFBE cells (slopes −1.56, −0.75, and −0.12; R2 = 0.95, 0.93, and 0.57; p < 0001; n = 8–12 from 3 donors, respectively) (Figure 4C). Regression analysis indicated that 9.8% transduction of K978C CFTR and 20.4% of WT CFTR would be sufficient to restore CFTR-mediated Isc to that of NHBE cells in the presence of CF sputum (17 μA/cm−2) (Figure 4C). However, 16.7% for K978C CFTR and 34.7% for WT CFTR would be required to achieve Isc in NHBE cells exposed to normal sputum (27.6 μA/cm−2) (indicative of fully restored function, as shown by the dotted lines in Figures 4C and S3A). Similar correlations were observed with basal and forskolin-sensitive Isc (Figures S4A and S4C). However, the percentage of final transduction with hˆK978C CFTR was poorly correlated with these measurements. Further analysis demonstrated that transduction of K978C CFTR amplified CFTRinh172 ΔIsc greater than 2-fold compared with WT CFTR (Figure 4D). hCAI and hˆK978C did not significantly increase CFTRinh172 ΔIsc compared with GFP-only transduced CFBE cells (Figure 4D). Amiloride-sensitive Isc and UTP-stimulated Isc did not correlate with the percentage of NHBE cells (Figures S3C and S3E) or percentage of final transduction with any variants of CFTR in CFBE cells (Figures S4B and S4D), suggesting that this effect was specific to CFTR.
Figure 4.

CFTR function in primary differentiated NHBE:CFBE cell mixed culture and CFBE cell models
(A and B) Example Isc traces from NHBE:CFBE cells and (A) from transduced CFBE cells (B; percent final transduction for individual CFTR variants are given) after incubation with CFS and addition of specific activators and inhibitors of ion transport (as below). (C and D) ΔIsc plotted against percent final NHBE after addition of (C) CFTRinh172 (10 μM) and summarized in (D) as ΔIsc per percent final NHBE or percent transduced cells in the culture (ΔIsc/% final). The dotted horizontal lines in (C) mark ASL height in 100% NHBE cells in the presence of normal lung sputum (healthy) or presence of CFS. (E) TEER/percent final transduced for WT-CFTR and hCAI CFTR. Treatments were compared by unpaired t test. ∗p < 0.05 (n = 8–12 from 3 CF donors). (F) Isc traces from CFBE cells transduced with WT or hCAI CFTRs or GFP alone before and after addition of a basolateral-to-apical Cl− gradient and specific activators and inhibitors of ion transport: amiloride (10 μM), FSK (10 μM), CFTRinh172 (10 μM), and UTP (10 μM). All drugs were added apically with exception of FSK, which was added bilaterally. Direction of chloride concentration gradients (using physiological Cl− and Cl−-free buffers) are indicated, and the duration of application is presented as gray bars. ΔIsc/percent final transduced in response to FSK and CFTRinh172 is given in a table below the graph.
The lack of function associated with hCAI CFTR was counterintuitive considering the increased abundance of membrane-localized protein generated in HEK293T and CFBE cells compared with WT CFTR. However, there is evidence that overexpression of CFTR can result in basolateral expression of CFTR, reducing transepithelial electrical resistance (TEER) and impeding vectorial ion transport under physiological conditions.11 Consistent with this hypothesis, transduction with hCAI CFTR caused a significant decrease in TEER compared with WT CFTR (p < 0.05, n = 8–12 from 3 CF donors) (Figures 4E and S4E). Furthermore, we implemented an artificial basolateral-to-apical Cl− gradient in the Ussing chamber to assess whether hCAI CFTR-transduced CFBE cells were able to facilitate Cl− movement. In the presence of the Cl− gradient, activation of CFTR by addition of forskolin produced a ΔIsc surpassing that of WT CFTR (∼5.8-fold), and this current was CFTR dependent because it was fully inhibited by CFTRinh172 (Figure 4F).
K978C CFTR more effectively restored ASL hydration in CFBE cells than WT CFTR
A crucial aim of CF therapy is to restore hydration to the CF airway. Therefore, we carried out the well-described ASL height assay across the mixed cultures and transduced CF cells in the presence of CF sputum.26 As a benchmark for successful restoration of CFTR function, we again referred to the results obtained from NHBE cells in the presence of normal lung sputum.
ASL height increased with the percentage of transduction of CFBE cells with either K978C CFTR or WT CFTR (1.31 ± 0.53 μm/% final transduced and 0.93 ± 0.50 μm/% final transduced; p < 0.05, n = 6–8 from 3 CF donors, respectively) (Figures 5A, 5B, and S5A; Videos S2, S3, S4, S5, and S6). ASL height did not increase with transduction of codon-optimized forms of CFTR in the presence of CF sputum (hCAI, 0.17 ± 0.22 μm/% final transduced; hˆK978C, 0.04 ± 0.06 μm/% final transduced). Interestingly, in the presence of CF sputum, the percentage of NHBE cells of the co-cultures also had no effect on ASL height (0.08 ± 0.07 μm/% final) (Figure 5B).
Figure 5.

K978C CFTR more efficiently restores ASL height and pH to differentiated CFBE cells than WT CFTR
(A) Representative XZ images of airway surface liquid (ASL) labeled with dextran (red layer) overlying transduced CFBE cells following incubation with CFS. Transduced cells express cytosolic GFP (green). (B) ASL height, after exposure to CFS and stimulation with vasoactive intestinal peptide (VIP), plotted against percent final NHBE (in NHBE:CFBE cell mixtures) or percent final transduced CFBE cells (for CFTR variants). Dotted lines show linear regression for each CFTR variant (n = 8–11 from 3 CF donors). The dotted horizontal line marks ASL height in 100% NHBE cells in the presence of normal lung sputum. Labeled arrows show percent final transduction of K978C CFTR required to reach this value. (C) Summary of ASL height in μm per percent final NHBE or percent final transduced CFBE cells in culture (μm/percent final) after exposure to CFS and VIP. Data are shown as mean ± SD with individual data points (n = 6–13 from 3 CF donors and 6 non-CF donors). (D) ASL height measured in transduced cells exposed to CFS or NHBE exposed to normal lung sputum before (−) or after (+) treatment with VIP. Data are shown as mean ± SD with individual data points (n = 6–15 from 3 CF donors and 6 non-CF donors). Mixed-effects analysis with Sidak’s post hoc analyses was used to compare −VIP with +VIP. †††p < 0.001. (E) ASL pH per final percent of NHBE or CFTR transduction in culture (ΔpH/percent final). Data are shown as mean ± SD with individual data points for n = 8–11 from 3 CF donors. Treatments were compared by two-way ANOVA with Tukey’s post hoc analyses; significantly increased as compared to CFBE cells: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The change in ASL height was greatest with transduction of K978C CFTR at ∼1.6-fold more than WT CFTR (Figure 5C). While we observed a trend toward an increase in ASL height following stimulation of CFTR with vasoactive intestinal peptide (VIP), this increase did not reach statistical significance under any condition when CF sputum was present (Figures 5D and S5B).26 Only NHBE cells in the presence of normal lung sputum produced a significant increase in ASL height following VIP stimulation (from 8.51 ± 3.27 μm to 16.51 ± 9.41 μm, p < 0.001, n = 9).
ASL pH levels were restored to physiological levels (7.35) in a similar manner, with 13.0% final transduction of K978C CFTR (n = 8 from 3 donors) and 18.7% WT CFTR (n = 11 from 3 donors) (Figures 5E, S5C, and S5D), suggesting that HCO3− secretion through CFTR was also facilitated. Interestingly, hCAI CFTR and hˆK978C CFTR also appeared to increase ASL pH levels compared with GFP-only-transduced CFBE cells, albeit less effectively than K978C or WT CFTR.
Discussion
The aim of this research was to determine whether use of codon-optimized or GOF forms of CFTR could improve the efficacy of CF gene therapy in a physiologically relevant airway model that included CF sputum.
As reported previously, in HEK293T cells, CFTR protein abundance from codon-optimized cDNAs was greatly increased compared with WT-CFTR.18 Replacement of lysine with cysteine in K978C CFTR and hˆK978C CFTR decreased protein expression, similar to that reported for G551D/K978C compared with G551D expression in HEK293T cells.21 The decreased CFTR expression in K978C CFTR and hˆK978C CFTR may be linked to altered cellular homeostasis because of their enhanced activity, potentially impacting protein synthesis or degradation. Dysregulated ion transport or protein folding can induce cellular stress and alter protein levels.28,29,30 Further research is needed to clarify this relationship. The halide transport function of the CFTR variants corelated with protein abundance and the presence of the K978C mutation. K978C is hypothesized to destabilize the inactive state of CFTR and lead to a 2-fold increase in Po of the channel.21,31 Interestingly, pre-treatment of hCAI CFTR with the CFTR activator VX770 increased activity to the same level as hˆK978C CFTR. VX770 is predicted to induce a conformational change to stabilize the open state of CFTR,23,32 increasing Po of WT CFTR by ∼2-fold,33,34 a phenomenon we replicated using the HS YFP quenching assay.35 Thus, our results support that K978C CFTR produces a similar outcome as CFTR potentiation by VX770.
In CFBE cells and in the presence of CF sputum, K978C CFTR was much more effective than other forms of CFTR we studied. K978C CFTR produced the largest CFTRinh172-sensitive Isc, increased ASL height, and more effectively restored normal ASL pH levels than WT CFTR, implying that K978C CFTR better facilitated Cl− and HCO3− secretion from airway cells. Most notably, we show that K978C CFTR transduction of CFBE cells in the presence of CF sputum restored ASL heights to those observed in NHBE cells in the presence of normal lung sputum, a desirable aim for genetic therapy research.
In our study, we used a lentivirus load designed to transduce less than 40% of cells, maximizing the likelihood of single transgene incorporation per transduced cell. This allowed a more effective comparison of the CFTR variants under consideration in our study. We did not measure mRNA levels; thus, variations in transgene expression might exist, depending on the site of genomic integration. However, this effect appears to be limited, as evidenced by the tight correlation of functional activity with the increase in the percentage of transduced cells.
We provide evidence to support the importance of considering use of CF sputum in pre-clinical models for CF genetic therapy. In the presence of CF sputum, there was a tight linear relationship between CFTRinh172-sensitive Isc and percentage of NHBE cells (endogenous CFTR expression) using the more accurate droplet digital PCR (ddPCR) methodology as opposed to initial seeding density. These data contrast findings where CFTR-mediated Cl− secretion plateaued with 50%–75% CFTR expression or less in other mixed-culture methods, interpreted as being limited by transporter-driven basolateral Cl− entry.9,10,11 We have shown previously that the presence of CF sputum compared with normal sputum lowered CFTR-dependent Isc in NHBE cell cultures.26 Thus, CFTRinh172-sensitive Isc may not be limited by basolateral Cl− under this circumstance. Our previous work also showed that exposure to CF sputum abrogated hydration of the ASL and responses to VIP compared with normal lung sputum.26 The lack of change in ASL height with percentage of NHBE cells suggests that endogenous CFTR function alone may not be sufficient to efficiently rehydrate the ASL in the CF luminal environment.
Our data demonstrate that, in the presence of CF sputum, approximately 10% final transduction of cells with K978C CFTR was sufficient to restore ASL hydration to normal levels (NHBE cells in the presence of normal sputum). This is approximately half that required for WT CFTR, in line with the previously reported difference in Po.9 Full restoration of “normal function” in vitro has been described with less than 25% transduction of cells with WT CFTR.12,13 It is important to highlight, however, that these estimates and interpretation of “normal” function apply to airway epithelial cells that were not exposed to sputum.
In the context of in vivo studies, it is suggested that only 10% of fully functional CFTR transcripts might be enough to prevent development of lung disease.2 While there are limitations when accurately modeling endogenous and exogenous CFTR expression in vitro, our data suggest a promising potential to achieve the desired levels of CFTR function in a CF scenario with K978C CFTR.
Our evidence indicated that protein expression and localization of codon-optimized CFTR in CFBE cells was disrupted, leading to a lack of vectorial Cl− transport. FRAP can be used to determine the rates of local protein turnover, identify mobile fractions, and demonstrate exchange between cellular compartments or lack thereof in live cells.36 In this assay, hˆK978C CFTR displayed free, rapid diffusion, providing evidence that the protein was not membrane bound. hCAI CFTR was present in a membrane-associated state but likely mislocalized, as evidenced by a reduction in TEER and the requirement of an artificial Cl− gradient to demonstrate function. Such overexpression of CFTR has been shown to perturb polarization of epithelial cells, localization of membrane proteins, and membrane potential, all of which compromise vectorial Cl− transport.37,38 For example, CFTR under control of a CMV promoter generated basolateral CFTR.11 Furthermore, codon optimization can affect subsequent mRNA secondary structure, co-translational protein folding, and interaction with noncoding RNAs, which are involved in regulation of CFTR gene expression, none of which was explored in this study.39,40,41 Interestingly, the codon-optimized variants of CFTR did restore ASL pH levels but at higher percentages of cellular transduction rates than the WT or K978C, a phenomenon also captured in other work.19 Explanations for this include increased paracellular basolateral-to-apical movement of HCO3− because of decreased TEER or increased HCO3− secretion via other channels that interact with CFTR; for example, SLC26 transporters.19,42 Exclusive use of a high-activity viral spleen focus forming virus (SSFV) promoter is a notable constraint of our study. As a result, further investigation is required to discern whether use of lower-activity promoters that are translatable to in vivo study might improve the ability of hCAI CFTR and hˆK978C CFTR to restore critical epithelial characteristics in CFBE cells. These include the phosphoglycerate kinase (PGK) and elongation factor 1α (EF1α) promoters, both of which have been employed to drive the expression of codon-optimized CFTR in CFBE cells with some success.19
While this study provided evidence of CFTR expression in transduced cells (Figure 3E), we did not focus on which cell types expressed the CFTRs. Lentiviral transduction does not impede differentiation of airway progenitor cells, and all differentiated airway cell types are capable of expressing the introduced transgene.43 Ciliated and secretory cells are the most numerous cells among NHBE/CFBE cells. Ionocytes are rare (0.5%–1.0%) but express the highest levels of CFTR, although the relative contribution of each cell type to epithelial anion transport is still under debate.44,45,46,47 Given the cellular transduction efficiencies we observed, functional restoration is likely driven by cell types other than ionocytes. How individual cell transduction modifies CFTR function is a compelling direction for future research to refine genetic therapy strategies for CF.
In conclusion, we provided a proof of principle showing that GOF variant K978C CFTR was more effective at restoring anion secretion, ASL hydration, and pH levels to CFBE cells than WT CFTR. However, we found that increasing CFTR protein using codon optimization did not translate to better function in CFBE cells. Importantly, K987C CFTR was able to restore function to CFBE cells even in the presence of CF sputum. These data provide a proof of principle showing that GOF variants may be more effective than codon-optimized forms of CFTR for CF gene therapy.
Materials and methods
Primary human bronchial epithelial cell culture
CFBE and NHBE cells were obtained with ethics approval from the University of North Carolina at Chapel Hill Biomedical Review Board (protocol 03 1396). Cells were cultured on permeable supports and maintained at air-liquid interface (ALI) in modified bronchial epithelial growth medium for 21–28 days.48 Co-cultures were generated by mixing NHBE and CFBE cells of different sexes (XX or XY) at 10%, 25%, 50%, and 75% (non-CF:CF). Cultures were incubated with 20 μL apically applied CF sputum 4 h prior to functional experimentation as described previously.26 Donor demographics are provided in Table S1. CFBE BMI-1 cells were generously supplied by Prof. Stephen Hart (University College London, Institute of Child Health).27
Sputum preparation
Airway sputum samples were obtained as described in the University of North Carolina protocol.49 An ultrasonic nebulizer was filled with 30 mL of 5% hypertonic saline for a 12-min inhalation period. After the 12-min inhalation period, subjects underwent a cleansing procedure: gargling and rinsing the mouth with water, scraping and clearing the back of the throat (to avoid inclusion of non-airway fluid samples), and blowing the nose. The subjects were asked to deliver a chesty cough and expectorated the secretions into a sterile specimen jar. This was capped and placed on ice and stored at −80°C. The demographics of the induced sputum donors are provided in Table S2 and reported in Woodall et al., 2021.26
For each experiment, unrefined sputum samples were thawed on ice and centrifuged at 4,000 × g for 20 min to remove cells, bacteria, and macromolecules. The supernatants were pooled for all downstream experimentation.
ddPCR
ddPCR using primers for AMEL-X) and AMEL-Y and probes with FAM and VIC reporter dyes, respectively (AB-Bioscience, UK), was performed as described previously.50 Primer sequences are provided in Table S6. FAM- and VIC-positive droplets from each well of the PCR plate were measured and analyzed using the associated software QuantaSoft. These data are presented normalized with exclusively male cultures (n = 10) set at 0% and female (n = 10) set at 100%.
Immunohistochemistry
ALI cultures were fixed in 4% paraformaldehyde (PFA), permeabilized (0.1% Triton X-100 in PBS), and blocked with blocking buffer (Table S5) prior to overnight incubation at 4°C with primary antiserum (α-tubulin, GFP), followed by visualization with Alexa Fluor 568 or Alexa Fluor 488. Membranes were counterstained with phalloidin (4 μg/mL) and DAPI (2 μg/mL) for 30 min at room temperature. Images were captured using an SP8 confocal microscope with LAS AF (Leica) acquisition software (antiserum and fluorophore dilutions and catalog numbers can be found in Tables S3 and S4).
Electrophysiological measurements
Transepithelial ion transport was measured using the Ussing chamber technique using symmetrical buffers (Table S5) and the following drugs: amiloride (apical, 100 μM) to inhibit the ENaC, forskolin (bilaterally, 10 μM) to stimulate CFTR, CFTRinh172 (apical, 10 μM) to inhibit CFTR, and UTP (apical, 100 μM) to stimulate CaCCs, as described previously.26 For Cl− gradient studies, the apical chamber was replaced with Cl−-free Ussing buffer (Table S5). Data were analyzed using Acquire and Analysis (v.1.2) software (Physiologic Instruments). All drugs/chemicals were obtained from Sigma-Aldrich.
ASL height/pH measurements
ASL exposed to CF sputum (CFS) or normal lung sputum (20 μL) were labeled with 0.5 mg/mL of 10-kDa dextran-tetramethyl rhodamine (Life Technologies, USA). Images were obtained before and 60 min after addition of basolateral VIP (100 nM) to induce CFTR-mediated secretion by using a Leica SP8 confocal microscope with a ×63/1.3 numerical aperture (NA) glycerol immersion lens in XZ-scanning mode as described previously.26,51 ASL pH was measured using Alexa Fluor 647 dextran (10 μM) and pH-sensitive pHrodo red dextran (10 μM) in 20 μL CFS. After 60-min incubation at 37°C in air +5% CO2, excitation/emission at 562/592nm and 650/668nm was measured. Apical pH was calculated as the fluorescence ratio pHrodo red dextran:Alexa Fluor 647 dextran less background fluorescence from non-labeled ASL, and the results were aligned to a standard curve generated from controls of known pH (pH 6.0–7.5) as described previously.52
CFTR codon optimization
The WT-CFTR construct, generously supplied by Stephen Hart, contained the native codons derived from a cDNA clone (https://www.ncbi.nlm.nih.gov/nuccore/M28668.1) encoding human CFTR. The codon-optimized versions of CFTR were generously supplied by David Mueller, in which the codons were nearly entirely derived using codons with hCAI53 values (from www.jcat.de) and named hCAI by the laboratory team that designed it (Department of Biochemistry and Molecular Biology, Rosalind Franklin University).18
Site-directed mutagenesis
Primers containing the nucleotide sequence to alter WT CFTR with the K978C mutation (K978C) and hCAI with the K978C mutation (hˆK978C), or for T2A mutagenesis were designed using SnapGene software (Table S6) and synthesized by Sigma-Aldrich.
Site-directed mutagenesis was carried out on an Eppendorf Mastercycler Pro Thermal Cycler using Phusion Hot Start II High-Fidelity PCR Master Mix, following the manufacturer’s protocol. The PCR-amplified DNA products were confirmed by sequencing (GENEWIZ; https://www.genewiz.com).
Lentivirus construction and cell transduction
Second-generation lentiviral transfer constructs contained the SFFV promoter upstream of GFP and CFTR. In some constructs, the transgene was separated by a self-cleaving T2A peptide. The construct, packaging plasmid (pMD2.G), and envelope plasmid (pCMVR8.74) were packaged into HEK293T cells via co-transfection with Lipofectamine 2000 (Thermo Fisher Scientific).54 The day before transduction, 2.5 × 105 target cells were seeded onto a collagen-coated 6-well plate. Opti-MEM supplemented with 8 μg Polybrene was mixed with the desired volume of harvested lentivirus per well. For viral titer definition, volumes of 10 μL, 5 μL, 2.5 μL, and 1.25 μL of lentivirus suspension were added to this mix. Cells were washed once with PBS prior to addition of the transduction mixture. Cells were incubated for 6 h, after which the transduction mix was aspirated and replaced with 2 mL of growth medium. Viral titer was measured by quantifying GFP-positive cells with flow cytometry on an Attune NxT 2 and analyzed with FlowJo software.
Following the methodology suggested by Charrier et al.,55 we employed virus suspensions that transduced fewer than 40% of CFBE cells from each donor (MOI < 0.4). This approach was chosen to ensure integration of a single vector copy per cell, translating to incorporation of one transgene per cell.
Titer and quantification of transduction efficiency
For definition of the final percentage of cells expressing the transgene (percent final transduced), cultures were analyzed 4 h prior to functional assays. Cultures were first washed three times with PBS to remove residual phenol red in growth medium and submerged in Ussing buffer for the duration of microscopy analysis. At least 10 images were taken from each well with a Cytation 5 Cell Imaging Multi-Mode Reader at 4× magnification.
The fluorescent signal and transmitted light were imaged for each well. Images were subject to analysis on ImageJ with a macro designed specifically for the purpose of quantifying the fluorescent:non-fluorescent cell percentage. The macro converted transmitted light images and fluorescence images from the same XY position to binary masks and allowed accurate identification of the fluorescent signal and non-fluorescent cells. Dead cells were discarded by a size and granulation threshold. Transduction efficiency was calculated with the following equation, where n is the number of images taken per well:
The results produced from this method were initially verified by flow cytometry analysis of a 1.5 × 105 cell sample from the respective wells.
HS YFP quenching assay
HEK293T cells were transfected with 100 ng pcDNA3.1_YFP-H148Q/I152L (HS YFP)56 and CFTR cDNAs (WT, K978C, hCAI, and hˆK978C) (Figure 2A) using Lipofectamine (Thermo Fisher Scientific). 24 h post transfection, the medium was replaced with 100 μL standard buffer (Table S5). HS YFP fluorescence in the cells was measured using ImageXpress as described previously57 with a 20× objective and 472/520-nm excitation/emission filters at 28°C. Images were acquired every 2 s for 150 s. At 20 s, I− in standard buffer was automatically dispensed into the extracellular medium so that the final concentration of I− in the extracellular medium was 100 mM. At 40 s, forskolin was added at a final concentration of 10 μM while keeping the concentrations of the other components unchanged. Images were analyzed using ImageJ (http://rsbweb.nih.gov/ij/). Iodide binding to HS YFP decreases fluorescence (F), thus F/Fmax (f) is used to quantify I− entry, where the concentration of iodide in the cells was defined as [I−]in = KI ((1-f) / f).35,57,58 The binding affinity of I− to HS YFP, KI, was set to 1.9 mM.35,56,57,58 CFTR activity was quantified as the maximal rate of I− entry into cells (mM/s).57,59
Western blot
Cells were lysed in NP-40 lysis buffer (Table S5), incubated on ice for 20 min, and then centrifuged at 15,000 × g for 20 min. Protein concentration was determined by Pierce BCA Protein Assay Kit, and 20 μg protein was denatured with 2.5 μL LDS sample buffer and 1 μL reducing agent (NuPAGE) at 37°C for 30 min. Samples were resolved on NuPAGE 4%–12% BisTris protein gels with mass standards of 10–250 kDa (LI-COR). Proteins were transferred to Immobilon-FL polyvinylidene fluoride (PVDF) membranes (Millipore). Membranes were blocked in Odyssey Blocking Buffer (LI-COR Biosciences), immunostained with anti-CFTR or anti-α-tubulin followed by IRDye 800CW goat anti-mouse immunoglobulin G (IgG; LI-COR Biosciences), visualized, and quantified on an Odyssey IR Imager (LI-COR Biosciences) (Tables S3 and S4).
FRAP
GFP-linked CFTR-transduced CFBE BMI-1 cells were imaged on a Leica SP5 inverted confocal microscope 63×/1.3 glycerol as described previously.60 Regions of interest (ROIs) of 20 pixels were selected as the point of highest fluorescence of individual cells. The mean fluorescence of 3 scan iterations (∼1 s per iteration) were acquired. ROIs were using 100% transmission of the 488-nm-wavelength laser, and FRAP was measured for ∼70 scan iterations. The fluorescence values were normalized to the initial pre-bleach value (1) and the value immediately post bleach (0). The mobile fraction (Mf) was calculated using the equation .
Pre-FRAP images were used to obtain mean fluorescence values from the intracellular region of the cell. At least 10 ROIs were measured for each cell. Ratios of mean intracellular to mean peripheral (2.5 μm from and including the most distal point of fluorescence of each cell) fluorescence (relative fluorescence units [RFUs]) were obtained using NIS-Elements software (Figure S2C).
Statistics
Normally distributed data were analyzed using ANOVA followed by Tukey’s test or unpaired t test with Welch’s correction. Paired t tests were applied to samples from the same donor but subject to different treatments. Non-parametric equivalents (Mann-Whitney test, Kruskal-Wallis test with Dunn’s multiple comparisons test) were used when data were not normally distributed. Data are shown as individual points and/or mean ± standard deviation. For linear regression, the R2 and whether the slope significantly differed from zero are presented. ns indicates no significant difference. Significant differences are indicated as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data analyses were performed using GraphPad Prism v.9.1.0.
Data and code availability
The experimental data that support the findings of this study are available in Figshare with the identifiers https://doi.org/10.6084/m9.figshare.23915565.v1, https://doi.org/10.6084/m9.figshare.23907711.v1, and https://doi.org/10.6084/m9.figshare.23907726.v1. Additional image sets are available upon request from the corresponding author.
Acknowledgments
This work was funded by Cystic Fibrosis Trust Project SRC 006. Provision of cells and media was supported by TARRAN17GO from the Cystic Fibrosis Foundation, BOUCHE15RO from the Cystic Fibrosis Foundation, and P30 DK065988 from the NIH (USA). We thank the CF SRC team for advice and input, particularly Ileana Guerrini (ICH) for help with developing the lentiviral constructs. We thank Emily Langron (UCL) for advice and direction with the halide-sensitive YFP assay. We thank Wei Wang (UAB) for sharing knowledge on K978C CFTR. We thank the University of North Carolina (UNC) Cystic Fibrosis Center Tissue Core (Director: Scott Randell) for providing cells, media, and expert advice. In addition, we thank R.L., Michael Chua, Lolita Radet, Saira Ahmad, Patrick Moore, Megan Webster, Ozge Beyazcicek, Eric Scott, and Maria Sassano for help and support at UNC. BioRender was used to create images included in this manuscript.
Author contributions
D.B. and M.W. conceived the idea. M.W. and H.A. carried out the experimental work. R.T. and R.L. provided resources and supervised M.W. S.P., P.V., and J.C. provided resources and helped perform assays. D.B. and S.H. supervised M.W. and edited the manuscript. D.B. and M.W. carried out analyses and wrote and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.08.006.
A video abstract is available at https://doi.org/10.1016/j.omtm.2023.08.006#mmc2.
Supplemental information
References
- 1.Wilschanski M. Class 1 CF mutations. Front. Pharmacol. 2012;3:117. doi: 10.3389/fphar.2012.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu C.S., Trapnell B.C., Curristin S.M., Cutting G.R., Crystal R.G. Extensive posttranscriptional deletion of the coding sequences for part of nucleotide-binding fold 1 in respiratory epithelial mRNA transcripts of the cystic fibrosis transmembrane conductance regulator gene is not associated with the clinical manifestations of cystic fibrosis. J. Clin. Invest. 1992;90:785–790. doi: 10.1172/JCI115952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Char J.E., Wolfe M.H., Cho H.J., Park I.H., Jeong J.H., Frisbee E., Dunn C., Davies Z., Milla C., Moss R.B., et al. A little CFTR goes a long way: CFTR-dependent sweat secretion from G551D and R117H-5T cystic fibrosis subjects taking ivacaftor. PLoS One. 2014;9:e88564. doi: 10.1371/journal.pone.0088564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooney A.L., Abou Alaiwa M.H., Shah V.S., Bouzek D.C., Stroik M.R., Powers L.S., Gansemer N.D., Meyerholz D.K., Welsh M.J., Stoltz D.A., et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight. 2016;1:e88730. doi: 10.1172/jci.insight.88730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maule G., Arosio D., Cereseto A. Gene Therapy for Cystic Fibrosis: Progress and Challenges of Genome Editing. Int. J. Mol. Sci. 2020;21:3903–3913. doi: 10.3390/ijms21113903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramalho A.S., Beck S., Meyer M., Penque D., Cutting G.R., Amaral M.D. Five Percent of Normal Cystic Fibrosis Transmembrane Conductance Regulator mRNA Ameliorates the Severity of Pulmonary Disease in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2002;27:619–627. doi: 10.1165/rcmb.2001-0004OC. [DOI] [PubMed] [Google Scholar]
- 7.King N.E., Suzuki S., Barillà C., Hawkins F.J., Randell S.H., Reynolds S.D., Stripp B.R., Davis B.R. Correction of Airway Stem Cells: Genome Editing Approaches for the Treatment of Cystic Fibrosis. Hum. Gene Ther. 2020;31:956–972. doi: 10.1089/hum.2020.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooney A.L., McCray P.B., Sinn P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes. 2018;9:538. doi: 10.3390/genes9110538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah V.S., Ernst S., Tang X.X., Karp P.H., Parker C.P., Ostedgaard L.S., Welsh M.J. Relationships among CFTR expression, HCO3- secretion, and host defense may inform gene- and cell-based cystic fibrosis therapies. Proc. Natl. Acad. Sci. USA. 2016;113:5382–5387. doi: 10.1073/pnas.1604905113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dannhoffer L., Blouquit-Laye S., Regnier A., Chinet T. Functional Properties of Mixed Cystic Fibrosis and Normal Bronchial Epithelial Cell Cultures. Am. J. Respir. Cell Mol. Biol. 2009;40:717–723. doi: 10.1165/rcmb.2008-0018OC. [DOI] [PubMed] [Google Scholar]
- 11.Farmen S.L., Karp P.H., Ng P., Palmer D.J., Koehler D.R., Hu J., Beaudet A.L., Zabner J., Welsh M.J. Gene transfer of CFTR to airway epithelia: low levels of expression are sufficient to correct Cl − transport and overexpression can generate basolateral CFTR. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;289:L1123–L1130. doi: 10.1152/ajplung.00049.2005. [DOI] [PubMed] [Google Scholar]
- 12.Johnson L.G., Olsen J.C., Sarkadi B., Moore K.L., Swanstrom R., Boucher R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L., Button B., Gabriel S.E., Burkett S., Yan Y., Skiadopoulos M.H., Dang Y.L., Vogel L.N., McKay T., Mengos A., et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7:e1000155. doi: 10.1371/journal.pbio.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alton E.W.F.W., Armstrong D.K., Ashby D., Bayfield K.J., Bilton D., Bloomfield E.V., Boyd A.C., Brand J., Buchan R., Calcedo R., et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015;3:684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Padegimas L., Kowalczyk T.H., Adams S., Gedeon C.R., Oette S.M., Dines K., Hyatt S.L., Sesenoglu-Laird O., Tyr O., Moen R.C., Cooper M.J. Optimization of hCFTR lung expression in mice using DNA nanoparticles. Mol. Ther. 2012;20:63–72. doi: 10.1038/mt.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yew N.S., Wang K.X., Przybylska M., Bagley R.G., Stedman M., Marshall J., Scheule R.K., Cheng S.H. Contribution of plasmid DNA to inflammation in the lung after administration of cationic lipid:pDNA complexes. Hum. Gene Ther. 1999;10:223–234. doi: 10.1089/10430349950019011. [DOI] [PubMed] [Google Scholar]
- 17.Bartoszewski R., Króliczewski J., Piotrowski A., Jasiecka A.J., Bartoszewska S., Vecchio-Pagan B., Fu L., Sobolewska A., Matalon S., Cutting G.R., et al. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell. Mol. Biol. Lett. 2016;21:23. doi: 10.1186/s11658-016-0025-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah K., Cheng Y., Hahn B., Bridges R., Bradbury N.A., Mueller D.M. Synonymous codon usage affects the expression of wild type and F508del CFTR. J. Mol. Biol. 2015;427:1464–1479. doi: 10.1016/j.jmb.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marquez Loza L.I., Cooney A.L., Dong Q., Randak C.O., Rivella S., Sinn P.L., McCray P.B. Increased CFTR expression and function from an optimized lentiviral vector for cystic fibrosis gene therapy. Mol. Ther. Methods Clin. Dev. 2021;21:94–106. doi: 10.1016/j.omtm.2021.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linsdell P. Anion conductance selectivity mechanism of the CFTR chloride channel. Biochim. Biophys. Acta. 2016;1858:740–747. doi: 10.1016/j.bbamem.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Wang W., Wu J., Bernard K., Li G., Wang G., Bevensee M.O., Kirk K.L. ATP-independent CFTR channel gating and allosteric modulation by phosphorylation. Proc. Natl. Acad. Sci. USA. 2010;107:3888–3893. doi: 10.1073/pnas.0913001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jih K.Y., Hwang T.C. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc. Natl. Acad. Sci. USA. 2013;110:4404–4409. doi: 10.1073/pnas.1215982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeh H.I., Qiu L., Sohma Y., Conrath K., Zou X., Hwang T.C. Identifying the molecular target sites for CFTR potentiators GLPG1837 and VX-770. J. Gen. Physiol. 2019;151:912–928. doi: 10.1085/jgp.201912360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodall M.N.J., Masonou T., Case K.M., Smith C.M. Human models for COVID-19 research. J. Physiol. 2021;599:4255–4267. doi: 10.1113/JP281499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Awatade N.T., Wong S.L., Hewson C.K., Fawcett L.K., Kicic A., Jaffe A., Waters S.A. Human Primary Epithelial Cell Models: Promising Tools in the Era of Cystic Fibrosis Personalized Medicine. Front. Pharmacol. 2018;9:1429. doi: 10.3389/fphar.2018.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodall M., Reidel B., Kesimer M., Tarran R., Baines D.L. Culture with apically applied healthy or disease sputum alters the airway surface liquid proteome and ion transport across human bronchial epithelial cells. Am. J. Physiol. Cell Physiol. 2021;321:C954–C963. doi: 10.1152/ajpcell.00234.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munye M.M., Shoemark A., Hirst R.A., Delhove J.M., Sharp T.V., McKay T.R., O’Callaghan C., Baines D.L., Howe S.J., Hart S.L. BMI-1 extends proliferative potential of human bronchial epithelial cells while retaining their mucociliary differentiation capacity. Am. J. Physiol. Lung Cell Mol. Physiol. 2017;312:L258–L267. doi: 10.1152/ajplung.00471.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charras G.T. A short history of blebbing. J. Microsc. 2008;231:466–478. doi: 10.1111/j.1365-2818.2008.02059.x. [DOI] [PubMed] [Google Scholar]
- 29.Rutkowski D.T., Kaufman R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Miura G. Death by ions. Nat. Chem. Biol. 2014;10:795. [Google Scholar]
- 31.Wang W., Roessler B.C., Kirk K.L. An electrostatic interaction at the tetrahelix bundle promotes phosphorylation-dependent cystic fibrosis transmembrane conductance regulator (CFTR) channel opening. J. Biol. Chem. 2014;289:30364–30378. doi: 10.1074/jbc.M114.595710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langron E., Prins S., Vergani P. Potentiation of the cystic fibrosis transmembrane conductance regulator by VX-770 involves stabilization of the pre-hydro. Br. J. Pharmacol. 2018;175:3990–4002. doi: 10.1111/bph.14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Goor F., Hadida S., Grootenhuis P.D.J., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A., et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu H., Burton B., Huang C.J., Worley J., Cao D., Johnson J.P., Urrutia A., Joubran J., Seepersaud S., Sussky K., et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012;11:237–245. doi: 10.1016/j.jcf.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Galietta L.J., Springsteel M.F., Eda M., Niedzinski E.J., By K., Haddadin M.J., Kurth M.J., Nantz M.H., Verkman A.S. Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J. Biol. Chem. 2001;276:19723–19728. doi: 10.1074/jbc.M101892200. [DOI] [PubMed] [Google Scholar]
- 36.Bancaud A., Huet S., Rabut G., Ellenberg J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, Photoactivation, Photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010;2010:pdb.top90. doi: 10.1101/pdb.top90. [DOI] [PubMed] [Google Scholar]
- 37.Mohammad-Panah R., Demolombe S., Riochet D., Leblais V., Loussouarn G., Pollard H., Baró I., Escande D. Hyperexpression of recombinant CFTR in heterologous cells alters its physiological properties. Am. J. Physiol. 1998;274:C310–C318. doi: 10.1152/ajpcell.1998.274.2.C310. [DOI] [PubMed] [Google Scholar]
- 38.Ye L., Chan S., Chow Y.-H., Tsui L.-C., Hu J. Regulated Expression of the Human CFTR Gene in Epithelial Cells. Mol. Ther. 2001;3:723–733. doi: 10.1006/mthe.2001.0314. [DOI] [PubMed] [Google Scholar]
- 39.Mauro V.P. Codon Optimization in the Production of Recombinant Biotherapeutics: Potential Risks and Considerations. BioDrugs. 2018;32:69–81. doi: 10.1007/s40259-018-0261-x. [DOI] [PubMed] [Google Scholar]
- 40.Glasgow A.M.A., de Santi C., Greene C.M. Non-coding RNA in cystic fibrosis. Biochem. Soc. Trans. 2018;46:619–630. doi: 10.1042/BST20170469. [DOI] [PubMed] [Google Scholar]
- 41.Kim S.J., Yoon J.S., Shishido H., Yang Z., Rooney L.A., Barral J.M., Skach W.R. Translational tuning optimizes nascent protein folding in cells. Science. 2015;348:444–448. doi: 10.1126/science.aaa3974. [DOI] [PubMed] [Google Scholar]
- 42.Garnett J.P., Hickman E., Burrows R., Hegyi P., Tiszlavicz L., Cuthbert A.W., Fong P., Gray M.A. Novel role for pendrin in orchestrating bicarbonate secretion in cystic fibrosis transmembrane conductance regulator (CFTR)-expressing airway serous cells. J. Biol. Chem. 2011;286:41069–41082. doi: 10.1074/jbc.M111.266734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooney A.L., Thurman A.L., McCray P.B., Pezzulo A.A., Sinn P.L. Lentiviral vectors transduce lung stem cells without disrupting plasticity. Mol. Ther. Nucleic Acids. 2021;25:293–301. doi: 10.1016/j.omtn.2021.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montoro D.T., Haber A.L., Biton M., Vinarsky V., Lin B., Birket S.E., Yuan F., Chen S., Leung H.M., Villoria J., et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018;560:319–324. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scudieri P., Musante I., Venturini A., Guidone D., Genovese M., Cresta F., Caci E., Palleschi A., Poeta M., Santamaria F., et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells. 2020;9:2090. doi: 10.3390/cells9092090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plasschaert L.W., Žilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., Klein A.M., Jaffe A.B. A single cell atlas of the tracheal epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018;560:377–381. doi: 10.1038/s41586-018-0394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato Y., Kim D., Turner M.J., Luo Y., Zaidi S.S.Z., Thomas D.Y., Hanrahan J.W. 2023. Ionocyte-Specific Regulation of CFTR. [DOI] [PubMed] [Google Scholar]
- 48.Fulcher M.L., Randell S.H. Human Nasal and Tracheo-Bronchial Respiratory Epithelial Cell Culture. Methods Mol. Biol. 2013;945:109–121. doi: 10.1007/978-1-62703-125-7_8. [DOI] [PubMed] [Google Scholar]
- 49.Alexis N.E., Hu S.C., Zeman K., Alter T., Bennett W.D. Induced sputum derives from the central airways: Confirmation using a radiolabeled aerosol bolus delivery technique. Am. J. Respir. Crit. Care Med. 2001;164:1964–1970. doi: 10.1164/ajrccm.164.10.2104051. [DOI] [PubMed] [Google Scholar]
- 50.George D., Czech J., John B., Yu M., Jennings L.J. Detection and quantification of chimerism by droplet digital PCR. Chimerism. 2013;4:102–108. doi: 10.4161/chim.25400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi H.-C., Kim C.S.K., Tarran R. Automated acquisition and analysis of airway surface liquid height by confocal microscopy. Am. J. Physiol. Lung Cell Mol. Physiol. 2015;309:L109–L118. doi: 10.1152/ajplung.00027.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garland A.L., Walton W.G., Coakley R.D., Tan C.D., Gilmore R.C., Hobbs C.A., Tripathy A., Clunes L.A., Bencharit S., Stutts M.J., et al. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc. Natl. Acad. Sci. USA. 2013;110:15973–15978. doi: 10.1073/pnas.1311999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grote A., Hiller K., Scheer M., Münch R., Nörtemann B., Hempel D.C., Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33:W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vink C.A., Counsell J.R., Perocheau D.P., Karda R., Buckley S.M.K., Brugman M.H., Galla M., Schambach A., McKay T.R., Waddington S.N., Howe S.J. Eliminating HIV-1 Packaging Sequences from Lentiviral Vector Proviruses Enhances Safety and Expedites Gene Transfer for Gene Therapy. Mol. Ther. 2017;25:1790–1804. doi: 10.1016/j.ymthe.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charrier S., Ferrand M., Zerbato M., Précigout G., Viornery A., Bucher-Laurent S., Benkhelifa-Ziyyat S., Merten O.W., Perea J., Galy A. Quantification of lentiviral vector copy numbers in individual hematopoietic colony-forming cells shows vector dose-dependent effects on the frequency and level of transduction. Gene Ther. 2011;18:479–487. doi: 10.1038/gt.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galietta L.J., Haggie P.M., Verkman A.S. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001;499:220–224. doi: 10.1016/s0014-5793(01)02561-3. [DOI] [PubMed] [Google Scholar]
- 57.Langron E., Simone M.I., Delalande C.M.S., Reymond J.L., Selwood D.L., Vergani P. Improved fluorescence assays to measure the defects associated with F508del-CFTR allow identification of new active compounds. Br. J. Pharmacol. 2017;174:525–539. doi: 10.1111/bph.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prins S., Langron E., Hastings C., Hill E.J., Stefan A.C., Griffin L.D., Vergani P. Fluorescence assay for simultaneous quantification of CFTR ion-channel function and plasma membrane proximity. J. Biol. Chem. 2020;295:16529–16544. doi: 10.1074/jbc.RA120.014061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pedemonte N., Zegarra-Moran O., Galietta L.J.V. High-throughput screening of libraries of compounds to identify CFTR modulators. Methods Mol. Biol. 2011;741:13–21. doi: 10.1007/978-1-61779-117-8_2. [DOI] [PubMed] [Google Scholar]
- 60.Woodall M., Jacob J., Kalsi K.K., Schroeder V., Davis E., Kenyon B., Khan I., Garnett J.P., Tarran R., Baines D.L. E-cigarette constituents propylene glycol and vegetable glycerin decrease glucose uptake and its metabolism in airway epithelial cells in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 2020;319:L957–L967. doi: 10.1152/ajplung.00123.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental data that support the findings of this study are available in Figshare with the identifiers https://doi.org/10.6084/m9.figshare.23915565.v1, https://doi.org/10.6084/m9.figshare.23907711.v1, and https://doi.org/10.6084/m9.figshare.23907726.v1. Additional image sets are available upon request from the corresponding author.