

Abstract
Glioblastoma multiforme (GBM) patients have a poor prognosis. After tumor recurrence statistics suggest an imminent death within 1–4.5 months. Supportive preclinical data, from a rat model, provided the rational for a prototype clinical vaccine preparation, named Gliovac (or ERC 1671) composed of autologous antigens, derived from the patient’s surgically removed tumor tissue, which is administered together with allogeneic antigens from glioma tissue resected from other GBM patients. We now report the first results of the Gliovac treatment for treatment-resistant GBM patients.
Nine (9) recurrent GBM patients, after standard of care treatment, including surgery radio- and chemotherapy temozolomide, and for US patients, also bevacizumab (Avastin™), were treated under a compassionate use/hospital exemption protocol. Gliovac was given intradermally, together with human GM-CSF (Leukine®), and preceded by a regimen of regulatory T cell-depleting, low-dose cyclophosphamide.
Gliovac administration in patients that have failed standard of care therapies showed minimal toxicity and enhanced overall survival (OS). Six-month (26 weeks) survival for the nine Gliovac patients was 100% versus 33% in control group. At week 40, the published overall survival was 10% if recurrent, reoperated patients were not treated. In the Gliovac treated group, the survival at 40 weeks was 77%. Our data suggest that Gliovac has low toxicity and a promising efficacy. A phase II trial has recently been initiated in recurrent, bevacizumab naïve GBM patients (NCT01903330).
Keywords: Glioma, Immunotherapy, Vaccine, Allogeneic, Autologous
1. Introduction
Active immunotherapy against cancer represents an exciting treatment option, involving the stimulation of the patient’s immune system against tumor antigens. However, therapeutic immunization against the most malignant brain tumor – glioblastoma multiforme (GBM) – is a formidable challenge. Although, brain parenchyma infiltrating CD8-positive T cells have been detected in these brain tumors [1,2] and even anecdotal rejection of gliomas following bacterial infection was reported [3], GBM, once established, normally evades immune detection. This is a result of decreased MHC antigen expression and active suppression of local and systemic immune reactions [4]. Apart from tumor-mediated immune suppression the patient’s immune reactivity is further suppressed by both high doses of iatrogenic chemotherapy [5] and corticosteroid treatment. All these factors tend to tilt the balance toward an immune suppressive state [6], as evidenced by significant leucopenia, a decrease in total CD4+ T cells and a functional increase in regulatory T cells.
Glioblastoma mutiforme (GBM) is the most common and most aggressive malignant brain tumor, with a very poor prognosis due to marginally effective standard therapy, involving tumor-debulking surgery, followed by radiotherapy and chemotherapy. This cancer is very difficult to treat and most patients die after tumor recurrence within 12–16 months [7,8]. At the time of tumor recurrence, statistics suggest an imminent death with an average overall survival (OS) of 1–4.5 months [8], depending on the size of the tumor, the Karnofsky performance score (KPS) score, and the tumor localization. In the USA, bevacizumab (Avastin®), a blood vessel growth-inhibiting, anti-angiogenic antibody, is administered as second line of treatment [9], but is not approved by EU authorities. Once the tumor recurs on bevacizumab treatment it is universally fatal with survival times of less than a few weeks [8]. Consequently, novel therapies are highly demanded.
Successful post-operative immunotherapy enabling immune recognition and destruction of residual or recurrent tumor cells would provide an enormous clinical value. Induction of a vaccine-induced immune response by adaptive immune lymphocytes initially requires efficient presentation, by antigen presenting cells, of tumor associated antigens (TAA) (referred to as signal 1) together with co-stimulatory signals (called signal 2). Most TAAs are inherently, poorly antigens and require an adjuvant to break immunological tolerance following proper of induction immune signal 2 [10]. Here we used recombinant granulocyte–macrophage colony stimulating factor (GM-CSF) as an immunological adjuvant, which is able to facilitate both signals 1 and 2 in different types of cancer vaccines [11]. GM-CSF supports dendritic cell (DC) recruitment and development; hence enabling antigen uptake and increasing antigen presentation. In addition, GM-CSF stimulates DC maturation, characterized by expression of co-stimulatory molecules (signal 2), facilitating antigen-presentation for T cells [12]. This cytokine is commonly used to generate DCs for the use in DC cancer vaccines [13]. GM-CSF’s safe pharmacological use in patients is well-established, which makes it attractive and feasible for clinical use in general.
Preclinical efficacy of this immunotherapy approach in an immunocompetent Lewis rat CNS-1 glioma model supported the implementation of this treatment concept in compassionate use for recurrent GBM patients. Here we describe our first clinical data for patients with a KPS score above 60, using this novel immunization approach, consisting of a combined administration of multiple allogeneic and autologous tumor-isolated antigens.
2. Materials and methods
2.1. Treatment scheme
The Gliovac treatment is composed of six cycles of five intradermally administrated treatment doses (Fig. 1). Every dose is composed of both a cellular component and a lysate component, prepared from freshly, surgically removed, GBM tumor tissue, and stored in separate vials.
Fig. 1.
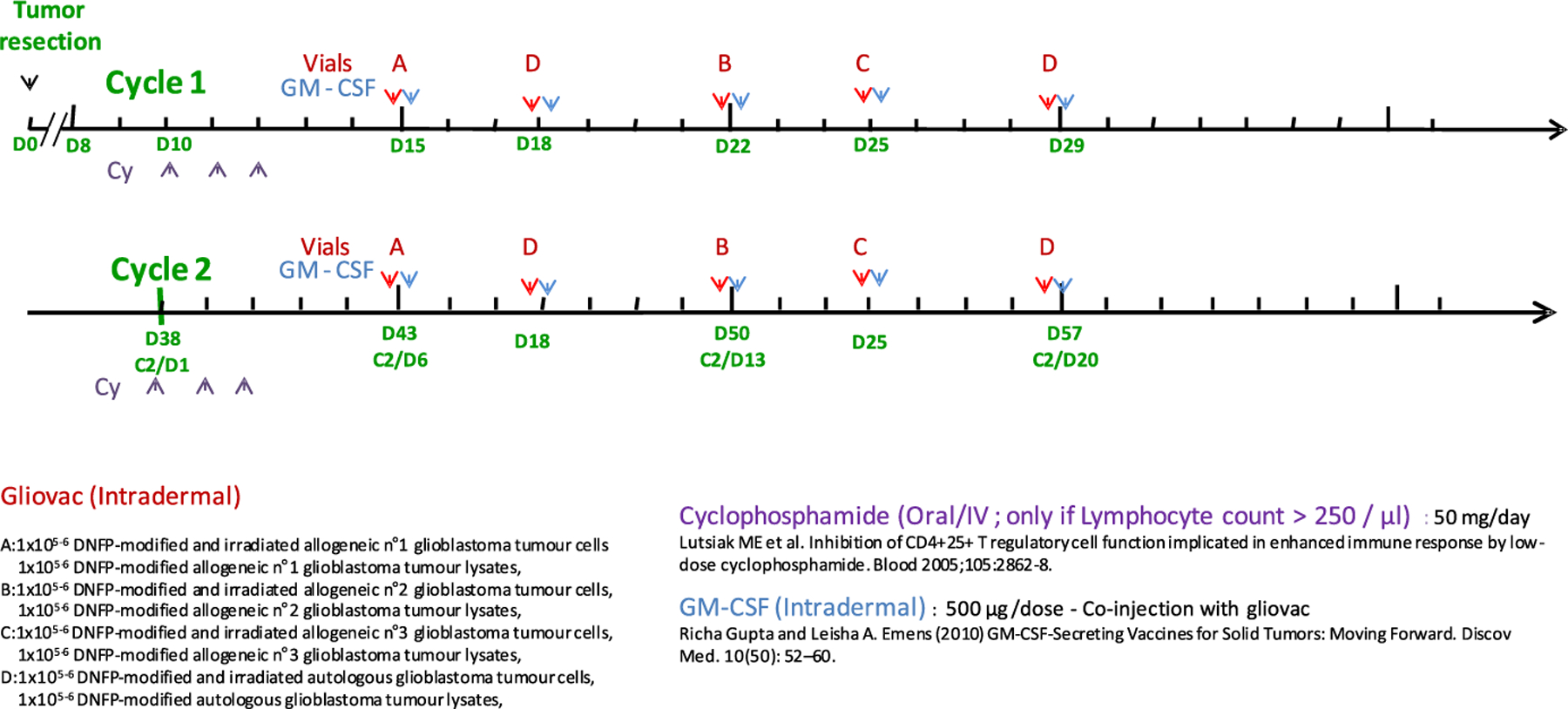
Time line of Gliovac treatment administration. The tumor resection is considered as day 0 (D0). The Gliovac is administered in repeated cycles. Ten days after surgery, the patient receives low-dose cyclophosphamide (Cy) for three consecutive doses (day 10–12: D10–D12; purple arrows) in order to reduce immune inhibitory immune cells, such as regulatory T cells [14]. The first immunization with an allogeneic tumor antigen-preparation, in conjunction with GM-CSF, is given on day 15 (D15). Subsequent immunizations, given at a 3–4 day intervals, consist of the patient-derived autologous antigens, two distinct allogeneic antigen-preparations, and a final autologous antigen preparation – all in combination with GM-CSF. The patient is left in rest for 1 week and a new cycle (cycle 2) restart with cyclophosphamide for three consecutive doses from D38 followed by immunization Gliovac treatment. This treatment has been repeated for six cycles.
The cell vial contains 250 μl of a suspension of 1 × 105–1 × 106 irradiated DNFB-modified tumor cells, and the lysate vial contains 250 μl of the equivalent of a lysate of 1 × 105–1 × 106 irradiated DNFB-modified tumor cells. In the schedule (Fig. 1) the allogenic Gliovac A, B, and C product doses are prepared from three different glioblastoma tumor donors, while autologous Gliovac D dose is derived from the patient’s tumor.
Gliovac treatment is administered together with GM-CSF (Leukine®) as adjuvant following the oral administration of a low dose of cyclophosphamide for 3 days (Endoxan®). The treatment scheme of two cycles is depicted in Fig. 1. The six treatment cycles were repeated every 28 days.
2.2. Vaccine production
The Gliovac product has been manufactured, under GMP approved aseptic conditions, from surgically removed GBM tissues. The tumor tissues were received and released by a tissue bank of human body material, after testing for absence of viral infections, including HIV, HBC, HCV, CMV, HTLV, and also Syphilis. After coding by a suitable anonymization procedure, they were sent in temperature-controlled conditions, to the GMP manufacturing site, immediately after the surgery. The cells were isolated by mechanical dissection and washed in Earl’s balanced salt solution (EBSS) medium. Isolated cells were counted and haptenized with 1-fluoro 2,4-dinitrofluorobenzene (DNFB), to improve immunogenicity. The total amount of haptinized cells was collected and divided in two equal parts. One part of cells was preserved for freezing in a sucrose medium, one part was lysed by osmotic shock. Both, the solutions of the cells and the lysates were irradiated with 25 Gray of gamma radiation to make the cells replication incompetent as a result of DNA damage. All preparations were stored at −80 °C.
2.3. Patient characteristics
Eligible adult patients, with histologically confirmed WHO grade IV malignant glioma and documented treatment failure to standard of care treatment (SOC), including surgery followed by concomitant chemotherapy plus radiotherapy with TMZ, and bevacizumab (Avastin™) in second line of treatment for one of them. All the patients presented a relapse of glioblastoma. Included patients are patients with an operable tumor mass since the treatment is composed, in part, of autologous tumor cells and lysates. Patient surgery was generally limited by the localization of the tumor (≤95% of the total tumor mass). Primary end points collected for each individual patient were toxicity, while secondary end points were median overall survival (OS) and radiographic responses. A total of nine (9) patients, presenting a KPS score of >60, enrolled, four on a phase 0/I protocol at the Cliniques of South Luxembourg-Belgium, one on compassionate/single patient IND protocol at UC Irvine Medical Center, one from Vilnius Hospital (Lithuania), one from the University Hospital Saarland, Homburg (Germany), and two from the Foundation Center for Epilepsy and neurological Diseases (FIRE) (Colombia). Median age was 48 years, with five female and four male patients. The average KPS was 80 (60–100). In Europe, treatments were performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. US patients were treated under IRB-approved protocols. All patients (or their guardians; if applicable) signed an informed consent form prior to their inclusion, and each treatment was approved by the hospital’s ethical committee. Patient demographics are shown in Table 1.
Table 1.
Patient information. For each patient are indicated: age (gender), HLA (autologous), HLA of allogeneic donors (in bold overlapping HLA), number of cycles, efficacy of the treatment in terms of changes in tumor mass, if dead on week 40, toxicity observed (ERY, local erythema; HA, headache; NA, not applicable), overall survival (OS) from the relapse detection. UN, unavailable.
| Patient number | Age (gender) | HLA autologous | Known HLA allogeneic received | Cycles received | Efficacy | Dead | Toxicity observed | OS from relapse |
|---|---|---|---|---|---|---|---|---|
| 1 | 61 (F) | A 02/03 B 15/44 C 03/05 |
A 1/2/3/24/26/32/33 B 7/8/18/35/37/40/44/50/51 C 2/3/6/7 |
5 cycles 4 cycles |
Tumor regression Stable 17 wk Stable 28 wk |
Yes | ERY ERY + HA |
28 w >40 w |
| 2 | 65 (M) | A 11/26 B 44/52 C 14/12 |
A 1/2/3/24/32/33 B 7/8/15/18/35/37/40/44 C 2/3/4/5/6/7/12 |
4 cycles | Stable | ERY + HA | >40 w | |
| 3 | 47 (M) | UN | A 2/24/26/32/33 B 7/35/37/40/44/50/51 C 2/3/4/6/7 |
|||||
| 4 | 64 (F) | A 25/31 B 18/51 C 04/12 |
A 1/2/3/11/24/26 B 7/8/13/15/18/35/37/40/44/52 C 2/3/4/5/6/7/12/14 |
5 cycles | Stable 30 wk | ERY | >40 w | |
| 5 | 50 (F) | UN | A 1/2/3/11/24/26/30 B 8/13/15/18/35/44/52 C 3/4/5/6/7/12/14 |
3 cycles | Stable 26 wk Disease progression |
Yes | NA | 35 w |
| 6 | 52 (M) | UN | A 1/2/11/24 B 8/13/18/35/44 C 4/5/6/7 |
6 cycles | Tumor regression | NA | >40 w | |
| 7 | 57 (F) | A 03/68 B 27/35 C 04/07 |
A 1/2/3/11/24 B 8/13/15/18/35/44 C 3/4/5/6/7/ |
6 cycles | Stable | ERY | >40 w | |
| 8 | 28 (M) | A 02/24 B 07/49 C 07/– |
A 1/2/3/11/26/29/30/68 B 7/8/13/27/35/44/51/52 C 4/6/7/12/14/16 |
6 cycles | Stable | NA | >40 w | |
| 9 | 27 (F) | A 23/24 B 35/− C 04/− |
A 3/11/26/29/30/68 B 7/13/27/35/44/51/52 C 4/6/7/12/14/16 |
6 cycles | Stable | NA | >40 w |
2.4. Immunomodulators and potentiators
Cyclophosphamide (CY; CalBiochem, 239785) was given at 50 mg/dose.
Human-GM-CSF (Leukine®) was purchased as an Escherichia coli expression product from Bayer HealthCare Pharmaceuticals (Seattle, WA, USA), and administered intradermally, with the vaccine, at 500 μg/dose diluted in 500 μl of water for injection (WFI).
2.5. Statistics
For software for the statistical analysis of the patient data was GraphPad Prism 5.03 for Windows. Median Overall-survival (OS-time between recurrence and death) was determined. The impact on OS of the treatment received (with or without GLIOVAC) was analyzed. For the univariate analysis of potential prognostic factors, time-to-event distributions of the patients were constructed using Kaplan–Meier plots and P values were obtained using log-rank tests. Significance was set at P < 0.05.
3. Results
3.1. Rat model
In a syngeneic, immunocompetent Lewis rat CNS-1 model we noted complete tumor regression (six out of six animals) only in the group of animals that received the vaccine (antigens from syngeneic and allogeneic cells) in conjunction with GM-CSF and cyclophosphamide (CY) pre-treatment (data not shown). In the control groups, some delay in measurable tumor growth was observed, relative to the untreated control groups (zero out of six animals showed tumor growth reduction). In the control groups, receiving CY only (tumor growth delay was noted in three out of six animals), in the CY plus GM-CSF group (one out of six animals showed tumor growth delay), while in the CY plus vaccine group three out of six animals showed growth delay.
3.2. Clinical findings
GBM patients have a very poor prognosis. Upon relapse, the overall survival depends of a multitude of factors, however, majority of the patients face imminent death after 1–4.5 months at best [8].
Encouraging findings in the preclinical rat model provided the rational and scientific basis to investigate the safety and efficacy of this immunotherapeutic concept, named Gliovac (or ERC1671), in individual GBM relapsing patients, with no remaining treatment options, under compassionate use/hospital exemption conditions. Gliovac is an immunotherapy based on (allo)immune response triggering following non-syngeneic tumor antigen (cells and lysates) injection/transplantation, reflecting the preclinical approach described in CNS-1 Lewis rats. During each immunization cycle, the immune effector response is triggered by breaking tolerance to the patient’s tumor antigens upon administration of allogeneic (non-self) DNFP-modified tumor antigens, at the first injection (Gliovac A), and subsequent focusing of the triggered immune reaction toward the patient’s tumor antigens, upon administration of patient-derived autologous tumor antigens (Gliovac D) (Fig. 1). This is followed by two additional (booster) injections of allogeneic antigen preparations (Gliovac B and C) and a final injection of Gliovac D. The immunizations are preceded by a short regimen of low-dose, metronomic cyclophosphamide (CY) [14], which depletes immune inhibitory immune cells. Each immunization with tumor antigens is accompanied by a co-injection of GM-CSF [11,12].
3.2.1. Patient selection
From January 2012 to July 2014, nine adult patients with recurrent glioblastoma were treated under Institutional review board (IRB)-approved protocols at the Clinique du Sud Luxembourg, Arlon, Belgium, University of California, Irvine, CA, USA, Universitäts Klinikum Homburg, UKS, Germany, from Vilnius Hospital (Lithuania) and the Foundation Center for Epilepsy and Neurological Diseases (FIRE) (Colombia). All these patients were previously treated with standard care, including surgery followed by concomitant radiotherapy and chemotherapy with TMZ, and for US patient, bevacizumab (Avastin™) as a second line of treatment. All the patients presented with recurrent, treatment resistant tumors. Only patients with an operable tumor mass were included in this protocol, since the treatment is composed, in part, of autologous tumor cells and lysates. Patient surgery, however, was generally limited due to the localization of the tumor (≤95% of the total tumor mass).
Primary data collected were toxicity, while secondary endpoints were median overall survival (OS) and radiographic response.
3.2.2. Clinical safety
The most common toxicities observed were mild and transient: two out of nine patients developed grade 2 headaches, and four showed grade 2 local erythema at the injection site. The local skin reactions (induration, erythema and ulceration) are not surprising for a local immune reaction following intradermal administration [15]. In fact, these local reactions indicate the development of immune responses. The diameters of the observed erythema’s were between 1 and 3 cm, which however, were not observed in all patients. Hence, no clear correlation between efficacy and erythema response can be concluded (as yet). Also, other observed mild systemic reactions, including self-limiting fever and chills, represent expected outcomes related to the intended immune stimulation [16]. The treatment did not trigger other serious adverse events.
3.2.3. Clinical efficacy – Radiology data
Magnetic resonance imaging (MRI) of the brain with and without contrast was used to evaluate the tumor response to treatment – using the RANO criteria [17]. Significant responses were seen on imaging – which, suggest that Gliovac/ERC1671 shows efficacy within our clinical settings, as illustrated for two patients in more detail in the case report (Box 1 supplementary info), visible in Figs. 5A–C and 6, online only. Clear imaging results were also noted in the MRIs of most other patients (Figs. 2, 3, and 6). One patient showed multifocal GBM, with multiple tumors (Fig. 2, left panel), which all showed remarkable reduction after one treatment cycle (Fig. 2, right panel). Another patient showed a noteworthy reduction in tumor load, visible at the end of cycle 1 (Fig. 3, left panel), after the second treatment period (Fig. 3, right panel).
Fig. 2.
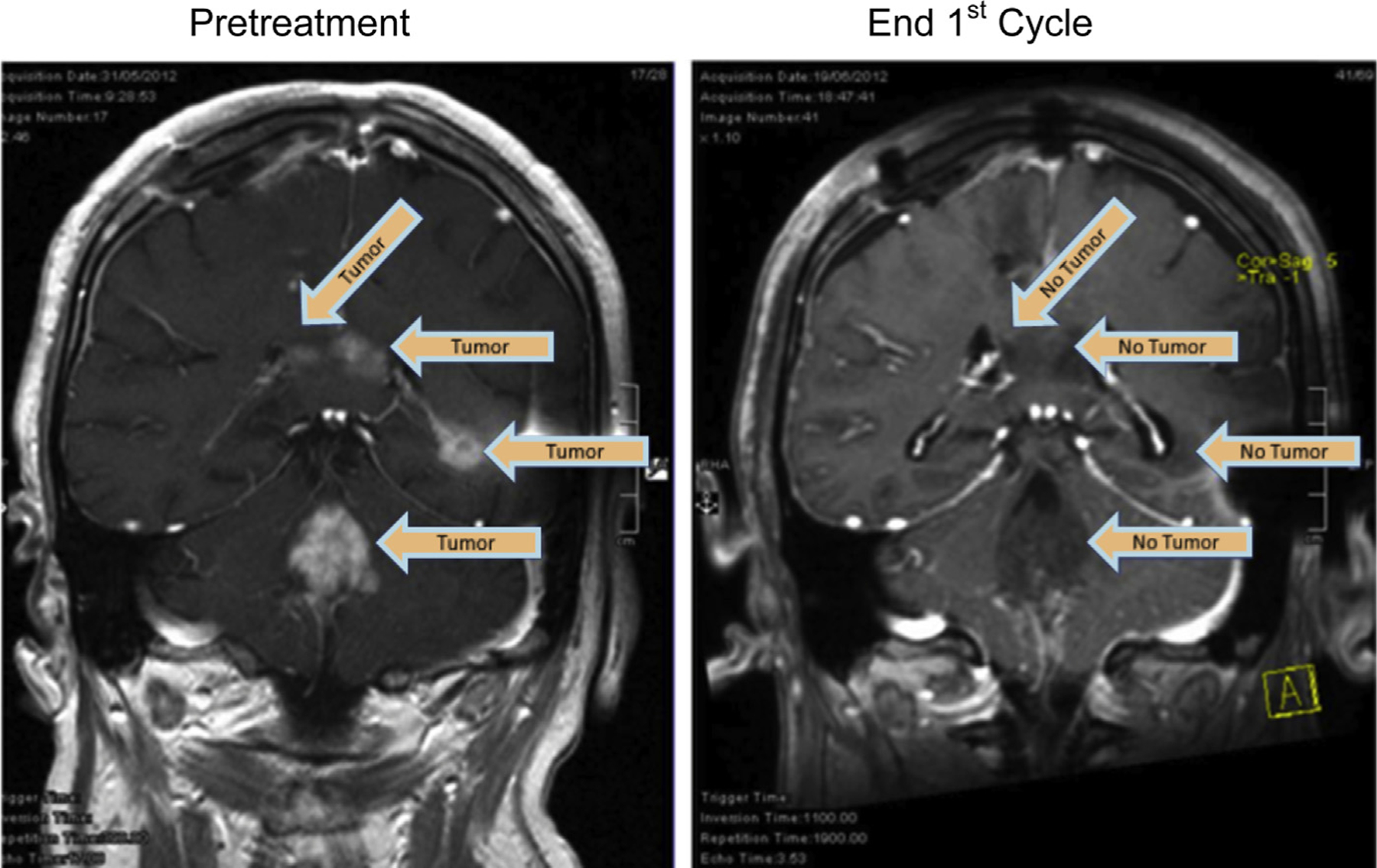
MRI scans of a patient made on 31 May 2012 (left, pretreatment), versus June 19, 2012 (right) following one treatment cycle (MRI scan, coronal view; end 1st cycle). Arrows indicate the locations of tumor tissue contrast staining.
Fig. 3.
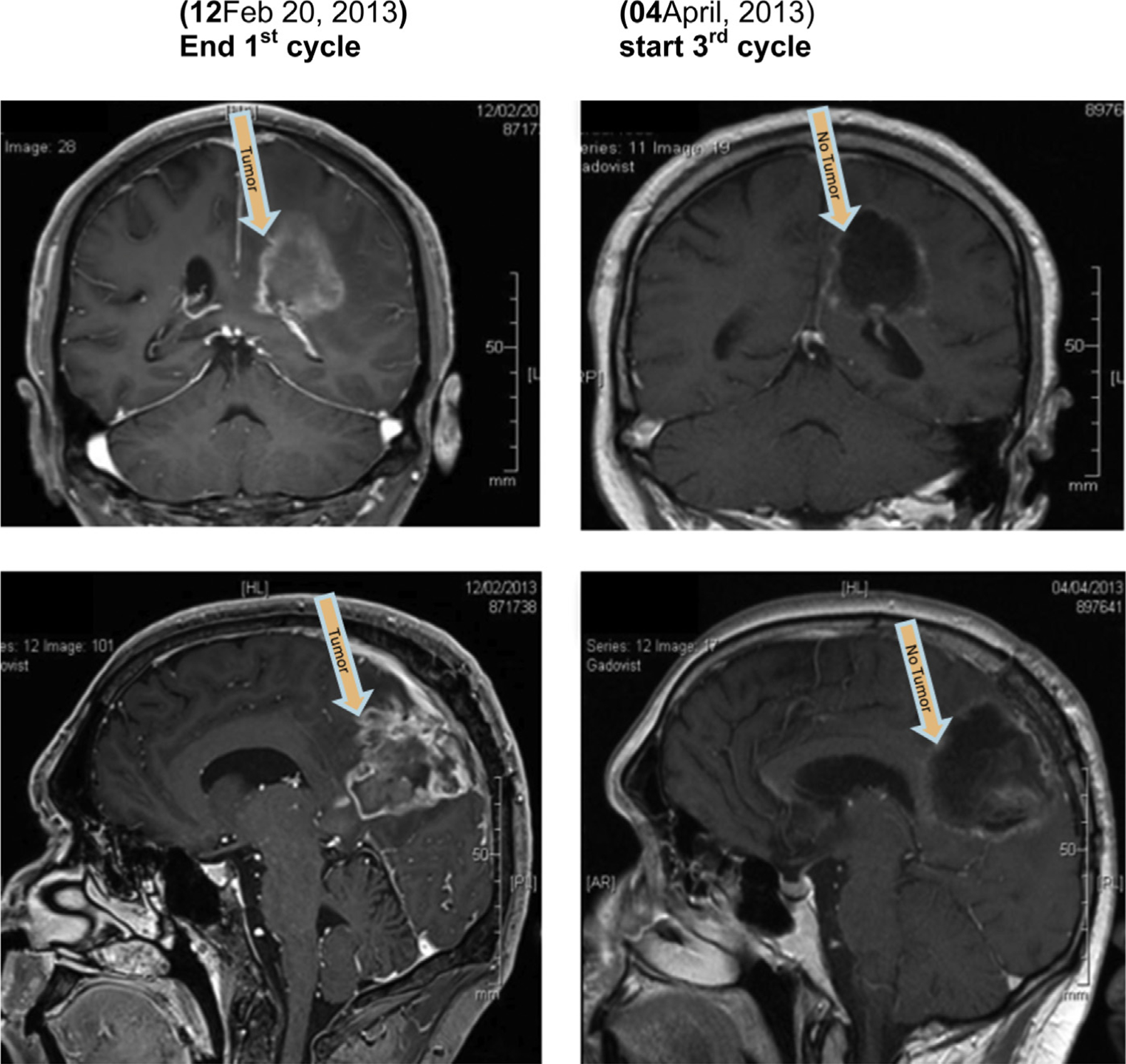
MRI scans of a patient made on February 12, 2013 (left) versus April 04, 2013 (right), following one additional treatment cycle (MRI scan, coronal [top] view and sagittal view [bottom]).
All the patients with a KPS of >60, when treated with Gliovac, responded to the treatment by a stabilization of the tumor, and, at 40 weeks post recurrence a prolongation of survival for about 30 weeks (at 77% survival) was observed versus historic untreated control patients (10 weeks).
3.2.4. Clinical efficacy – Overall survival
Of the nine patients, all had complete follow-up (until week 40). Patients’ mean age was 48 (range from 27 to 63) years. The mean Karnofsky performance score (KPS) 1 week after the surgery at the time of recurrence was 80 (range from 60 to 100). Our data were compared to the published survival data of reoperated, untreated (KPS >60) patients receiving standard care [18].
The rates of overall survival (OS) achieved by Gliovac/ERC1671 treatment are significantly increased (p = 0.0001), relative to those reported historically for patients after surgery for recurrent GBM in a recent retrospective analysis published by Barker et al. [18] (Fig. 4). Fig. 4 shows the comparison between Gliovac treated patients (n = 9; solid line) and control patients (n = 39; dashed line), both with KPS scores ranging between 60–100. Data were recorded until week 40 after reoperation. Six-month (26 weeks) survival for the nine Gliovac patients was 100% versus 33% in control group. At week 40, the published overall survival was 10% if patients were not treated. In the Gliovac treated group, the survival at 40 weeks was 77%. The statistic analysis clearly indicates a significant effect of Gliovac on the survival of recurrent patient (p < 0.0001).
Fig. 4.
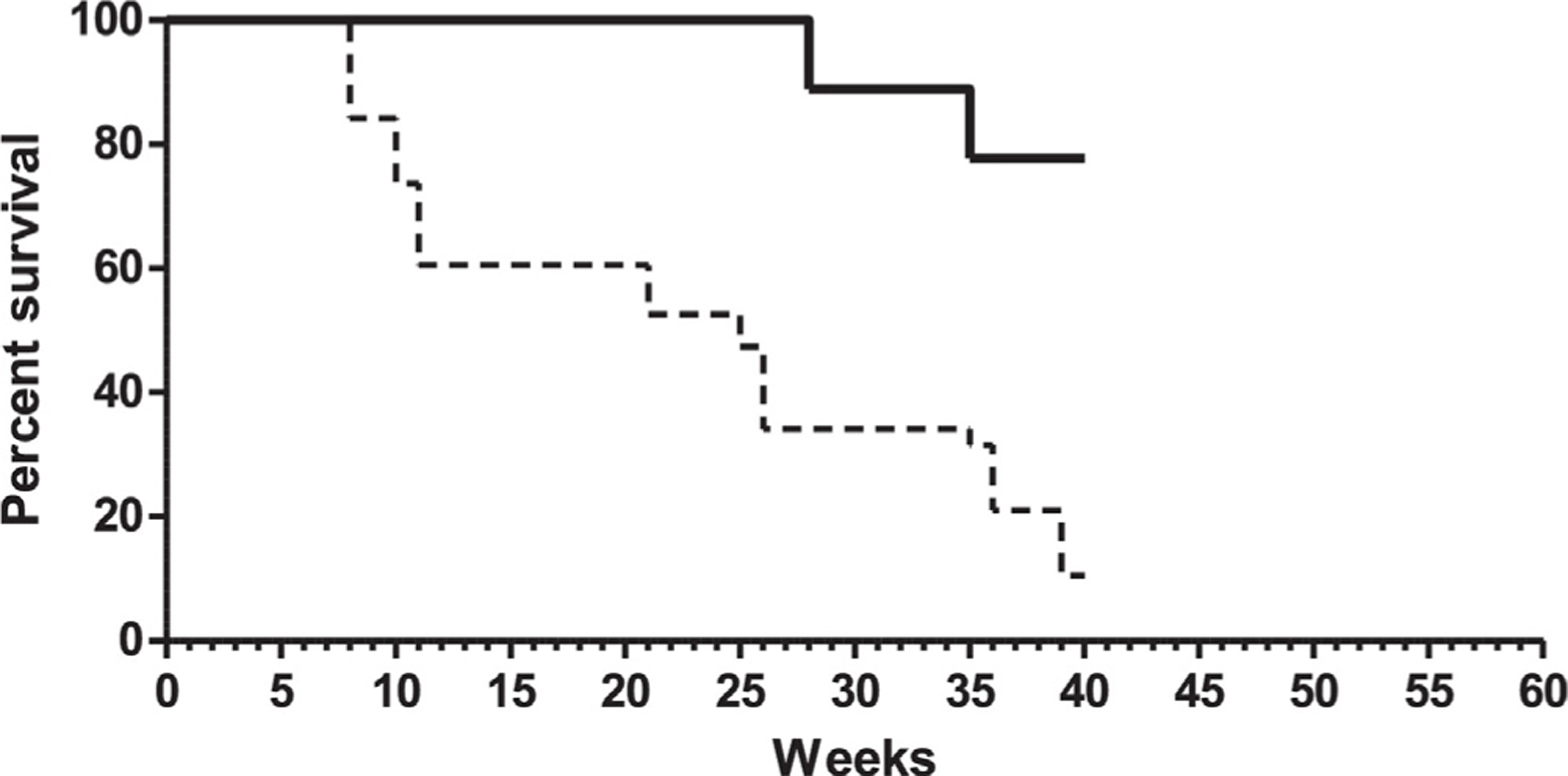
Overall survival of patients treated with Gliovac (n = 9; solid line) versus overall survival of published control patients (dashed line). Data of control patients are extracted from Fig. 3A of the publication [18].
All patients with a KPS score between 60 and 100, treated with Gliovac/ERC 1671 were still alive at week 28 (about 7 months) (Fig. 4, solid line). One patient showed after three cycles of vaccination complete tumor regression, which was observed after an average of 8 (4–12) weeks. It should be noted that for most of the recurrent Gliovac/ERC1671 patients included in this report the surgery achieved only limited, subtotal resection, due to the vital brain area and critical location of the tumor, which is a negative prognostic factor relative to complete resection [19].
For one patient, further histological analysis of residual tumor biopsies showed that the Gliovac treatment corresponds with infiltration of activated macrophages (CD68-positive), CD4+ and CD8+ T cells, and strongly reduced viable tumor growth (Ki67 staining; data not shown) [20]. These observations sustain the efficacy of immune effector response-induction and local immune infiltration in the tumor bed.
Those first results in man are highly encouraging, despite the late stage of disease, the resistance to standard therapeutic treatment, and the incomplete tumor resection by surgery. However, future clinical trials require strict selection criteria, limiting the extent of disease to patients who do not have multifocal or leptomeningeal disease [8,21]. Potential improvements should also address the timing of Gliovac administration after the initial diagnosis, e.g., treatment before immunosuppressive chemotherapy.
3.3. Conclusive interpretation of clinical results
Current data suggest that even in advanced stages of disease, the Gliovac treatment increases overall survival of recurrent, treatment resistant GBM patients. These encouraging clinical case study results, from relapsing GBM patients in a compassionate use program, provided support to FDA authorities (FDA) to approve the investigation of the product in a phase II, randomized, double blinded clinical trial, comparing the product’s safety and efficacy in combination with bevacizumab with bevacizumab in combination with placebo treatment in GBM patients who have failed temozolomide.
4. Discussion
The present study shows that Gliovac (or ERC 1671) immunotherapy is safe and potentially effective in treatment-resistant GBM patients. At 40 weeks post recurrence this approach prolonged the observed 77% survival among relapsing glioblastoma patients with an increase in survival of about 5-month (30 weeks) relative to historic controls (10 weeks) [18].
Our clinical protocol has been designed based on supportive proof-of-concept data observed in a CNS-1 glioma model in Lewis rats. In the rat model we observed tumor regression, visible as a reduction in tumor growth rate after about 2 weeks of initiation of immunotherapy, using allogenic and syngeneic antigens from glioma cell lines, when administered together with GM-CSF as immunological adjuvant, eventually resulting in non-detectable tumor volumes. This anti-tumor response resulted in immunological memory, since the majority of animals that controlled the first tumor, also rejected a secondary tumor without noticeable tumor growth. All animals were pretreated with a low-dose CY in order to deplete the immunosuppressive regulatory T cells [22].
The rationale of the Gliovac prototype vaccine is to evoke oligoclonal, partly allo-specific, immune induction, using a broad set of tumor antigens, derived from freshly resected whole tumor tissue. This will reduce the chance of immune escape, which is more likely to occur when using a single-antigen-targeted immunotherapy. The vaccine is composed of autologous antigens, derived from the patient’s surgically removed tumor tissue, which is administered in conjunction with antigens from glioma tumor tissue that was surgically removed from allogenic donor patients. This allogenic tumor material provides an additional source of antigens that can be stored in a tissue bank for “off-the shelf” use. The allogenic TAAs may display partial HLA-matching with the patient. The mismatching HLA molecules serve to trigger and enhance an allo-immune response. In this first in man study, partial HLA mismatch information is available (Table 1), but the role of HLA mismatches in effectiveness will have to be evaluated in a stringent clinical trial currently ongoing (NCT01903330). Relevant unique or shared TAAs overexpressed by tumor cells are present among thousands of irrelevant immunotolerant non-tumor associated antigens. A multivalent vaccine will prevent or minimize escape of residual tumor cells, due to antigenic loss, or active MHC down-regulation. In addition, a tumor antigen mixture is preferred above monovalent synthetic peptides, because of their restricted use in patients with defined HLA types only.
We used GM-CSF as an immune adjuvant, which is known to augment immune responses against protein of peptide based vaccines [23], as well as to tumor cell vaccines genetically engineered to secrete GM-CSF [24]. This cytokine has been used as a hematopoietic growth factor in patients undergoing chemotherapy, and is well-tolerated [25]. When administered in the skin it recruits and activates antigen-presenting cells, including epidermal Langerhans cells [26] Moreover, GM-CSF showed positive effects relative to other cytokines, in preclinical rat and mouse glioma vaccine studies [27,28].
Immunological protection against gliomas has been ascribed to cell-mediated immune reactions involving cytolytic CD8+ T lymphocytes [29]. Depletion of these cells has demonstrated their critical role in vaccine-mediated antitumor immunity [30]. These observations are in line with the histological results from sequentially taken tumor autopsy specimens. Biopsy specimens of a Gliovac treated patient showed local immune infiltration in the tumor bed, consisting of abundant activated macrophages (CD68), as well as CD4 and CD8 T cells. This immunohistological staining was associated with a strongly reduced viable tumor growth index, as evidenced by reduced Ki67 positive cells [20]. Although, in general, tumor-specific immune response monitoring and a clear relationship with clinical outcome has proven difficult for tumor vaccines, it will be of interest to investigate in detail the contribution of particular lymphocyte populations to protective antitumor efficacy of Gliovac – for example, by monitoring of the number and function of both peripheral blood regulatory T cells and interferon-γ-producing CD8 T cells specific for prototype glioma antigens.
Clinical studies using cell-based vaccination, employing a broad set of tumor antigens, have been carried out before. Some used autologous cells, e.g., M-vax [31], or allogeneic cells, e.g., Canvaxin [32], or autologous lysates, e.g., oxidized tumor cell lysate (OC-L), or allogeneic lysates, such as Melacine [33]. Although safe, in phase I and II clinical trials, these products failed to provide convincing statistical evidence of positive immunological and clinical outcome. The innovative aspect of the Gliovac is to combine all elements (autologous and allogeneic, cells and lysates) in order to trigger strong polyclonal immune reactions. Autologous components contain patient-specific antigen, while allogeneic components are able to induce an allo-immune reaction. This strategy enables triggering of an immune response against a broad array of tumor antigens, including tolerance breaking allo-immune reactivity, -a classical allograft-directed immune response-, typical for non-matching major histocompatibility between the injected graft cells and antigens and the host. The allogenic part of the Gliovac treatment contains antigens from GBM tumors from allogeneic donor patients, that overlap with specific tumor antigens in the patient.
The observed safety and promising clinical results of Gliovac in the compassionate use program, lead the US authorities (FDA) to approve the development of a phase II clinical trial registered under number (NCT01903330), which is currently enrolling patients.
Supplementary Material
Acknowledgments
We thank Drs. Joachim Oertel, and David Breuskin at the Klinik fur Neurochirurgie, Universitaetsklinikum des Saarlandes, Homburg, Germany, and Dr. Adas Darinskas, Center of Oncosurgery, Institute of Oncology, Vilnius University, Vilnius, Lithuania, for excellent support to treat patients with Gliovac.
Financial disclosure
Epitopoietic Research Corporation (ERC) provided financial and material support for this work and was involved in all stages of this work.
Footnotes
Conflict of interest
Only authors affiliated to ERC, including VS, CP, LD, DP, ThC, and AS, received financial support, either as personal consulting fees, employment, shares, or honoraria.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.vaccine.2015.03.095
References
- [1].Yang I, Tihan T, Han SJ, Wrensch MR, Wiencke J, Sughrue ME, et al. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci 2010;17(11):1381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schneider J, Hofman FM, Apuzzo ML, Hinton DR. Cytokines and immunoregulatory molecules in malignant glial neoplasms. J Neurosurg 1992;7(2):265–73. [DOI] [PubMed] [Google Scholar]
- [3].Bowles AP, Perkins E. Long-term remission of malignant brain tumors after intracranial infection. A report of four cases. Neurosurgery 1999;44(3):636–42. [DOI] [PubMed] [Google Scholar]
- [4].Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 2006;66(6):3294–302. [DOI] [PubMed] [Google Scholar]
- [5].Weir GM, Liwski RS, Mansour M. Immune modulation by chemotherapy to enhance cancer vaccines. Cancers 2011;3:3114–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fadul CE, Fisher JL, Gui J, Hampton TH, Côté AL, Ernstoff MS. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. NeuroOncolology 2011;13(4):393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352(10):987–96. [DOI] [PubMed] [Google Scholar]
- [8].Park JK, Hodges T, Arko L, Shen M, Dello Iacono D, McNabb A, et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J Clin Oncol 2010;28(24):3838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chamberlain MC. Bevacizumab for the treatment of recurrent glioblastoma. Clin Med Insights Oncol 2011;5:117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schijns VE, Lavelle EC. Trends in vaccine adjuvants. Expert Rev Vaccines 2011;10(4):539–50. [DOI] [PubMed] [Google Scholar]
- [11].Chang DZ, Lomazow W, Joy Somberg C, Stan R, Perales MA. Granulocyte–macrophage colony stimulating factor: an adjuvant for cancer vaccines. Hematology 2004;9(3):207–15. [DOI] [PubMed] [Google Scholar]
- [12].Min L, Mohammad Isa SA, Shuai W, Piang CB, Nih FW, Kotaka M, et al. Cutting edge: granulocyte–macrophage colony-stimulating factor is the major CD8+ T cell-derived licensing factor for dendritic cell activation. J Immunol 2010;184(9):4625–9. [DOI] [PubMed] [Google Scholar]
- [13].Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol 2003;15:138–47. [DOI] [PubMed] [Google Scholar]
- [14].Daenen LG, Shaked Y, Man S, Xu P, Voest EE, Hoffman RM, et al. Low-dose metronomic cyclophosphamide combined with vascular disrupting therapy induces potent anti-tumor activity in preclinical human tumor xenograft. Models Mol Cancer Ther 2009;8(10):2872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gidudu J, Kohl KS, Halperin S, Hammer SJ, Heath PT, Hennig R, et al. A local reaction at or near injection site: case definition and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine 2008;26:6800–13. [DOI] [PubMed] [Google Scholar]
- [16].Khasraw M, Holodny A, Goldlust SA, DeAngelis LM. Intracranial hemorrhage in patients with cancer treated with bevacizumab: the memorial Sloan-Kettering experience. Ann Oncol 2012;23(2):458–63. [DOI] [PubMed] [Google Scholar]
- [17].Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 2010;28(11):1963–72. [DOI] [PubMed] [Google Scholar]
- [18].Barker FG, Chang SM, Gutin PH, Malec MK, McDermott MW, Prados MD, et al. Neurosurgery 1998;42:709–20. [DOI] [PubMed] [Google Scholar]
- [19].Stummer W, Reulen H-J, Meinel T, Pichlmeier U, Schumacher W, Tonn JC, et al. Extent of resection and survival in glioblastoma multiforme: identification of and adjustment for bias. Neurosurgery 2008;62:564–76. [DOI] [PubMed] [Google Scholar]
- [20].Bota DA, Alexandru-Abrams D, Pretto C, Hofman FM, Chen TC, Fu B, et al. Use of ERC-1671 Vaccine in a Patient with Recurrent Glioblastoma Multiforme after Progression during Bevacizumab Therapy: First Published Report. Perm J 2015. Mar 1, 10.7812/TPP/14-042 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chamberlain MC, Johnston SK. Salvage therapy with single agent benvacizumab for recurrent glioblastoma. J Neurooncol 2010;105(3):523–30. [DOI] [PubMed] [Google Scholar]
- [22].Rozados VR, Mainetti LE, Rico MJ, Zacarías Fluck MF, Matar P, Scharovsky OG. The immune response and the therapeutic effect of metronomic chemotherapy with cyclophosphamide. Oncol Res 2010;18(11–12):601–5. [DOI] [PubMed] [Google Scholar]
- [23].Disis ML, Bernhard H, Shiota FM, Hand SL, Gralow JR, Huseby ES, et al. Granulocyte–macrophage colony-stimulating factor: an effective adjuvant for protein and peptide-based vaccines. Blood 1996;88(1):202–10. [PubMed] [Google Scholar]
- [24].Dranoff G GM-CSF-based cancer vaccines. Immunol Rev 2002;188:147–54. [DOI] [PubMed] [Google Scholar]
- [25].Armitage JO. The use of granulocyte–macrophage colony-stimulating factor in bone marrow transplantation. Semin Hematol 1992;29(4 Suppl 3):14–8. [PubMed] [Google Scholar]
- [26].Kaplan G, Walsh G, Guido LS, Meyn P, Burkhardt RA, Abalos RM, et al. Novel responses of human skin to intradermal recombinant granulocyte/macrophage-colony-stimulating factor: Langerhans cell recruitment, keratinocyte growth, and enhanced wound healing. J Exp Med 1992;175(6): 1717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Herrlinger U, Aulwurm S, Strik H, Weit S, Naumann U, Weller M. MIP-1alpha antagonizes the effect of a GM-CSF-enhanced subcutaneous vaccine in a mouse glioma model. J Neurooncol 2004;66(1–2):147–54. [DOI] [PubMed] [Google Scholar]
- [28].Soiffer R, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger JC, et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte–macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc Natl Acad Sci U S A 1998;95(22):13141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ghulam Muhammad AK, Candolfi M, King GD, Yagiz K, Foulad D, Mineharu Y, et al. Antiglioma immunological memory in response to conditional cytotoxic/immune-stimulatory gene therapy: humoral and cellular immunity lead to tumor regression. Clin Cancer Res 2009;15(19):6113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Barcia C Jr, Gómez A, Gallego-Sanchez JM, Perez-Vallés A, Castro M, Lowenstein PR, et al. Infiltrating CTLs in human glioblastoma establish immunological synapses with tumorigenic cells. Am J Pathol 2009;175(2): 786–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Berd D, Sato T, Maguire HC, Kairys J Jr, Mastrangelo MJ. Immunopharmacologic analysis of an autologous, hapten-modified human melanoma vaccine. J Clin Oncol 2004;22(3):403–15. [DOI] [PubMed] [Google Scholar]
- [32].Hsueh EC, Morton DL. Antigen-based immunotherapy of melanoma: canvaxin therapeutic polyvalent cancer vaccine. Semin Cancer Biol 2003;13(6):401–7. [DOI] [PubMed] [Google Scholar]
- [33].Sosman JA, Sondak VK. Melacine: an allogeneic melanoma tumor cell lysate vaccine. Expert Rev Vaccines 2003;2(3):353–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.