

Highlights
-
•
Mutational signatures reflect carcinogenetic pathways.
-
•
Several of mutational signatures provided relevant prognostic information.
-
•
Gene expressions differed remarkably between the low- and high-signature groups.
Keywords: Bladder carcinoma, COSMIC, Mutations, Renal cell carcinoma cancer, Prognosis, Signature, Urothelial cancer
Abstract
Different sources of mutagenesis cause consistently identifiable patterns of mutations and mutational signatures that mirror the various carcinogenetic processes. We used publicly available data from the Cancer Genome Atlas to evaluate the associations between the activity of the mutational signatures and various survival endpoints in six types of urological cancers after adjusting for established prognostic factors. The predictive power of the signatures was evaluated with dynamic area under curve models. In addition, links between mutational signature activities and differences in gene expression patterns were analysed. APOBEC-related signature SBS2 was associated with improved overall survival (OS) and disease-specific survival (DSS) in bladder carcinomas in the multivariate analysis, while clock-like signature SBS1 predicted shortened DSS and progression-free interval (PFI) in clear cell renal cell carcinomas (ccRCC). In papillary renal cell carcinomas (pRCC), SBS45 was a predictor of improved outcomes, and APOBEC-related SBS13 was a predictor of worse outcomes. Gene expression analyses revealed various enriched pathways between the low- and high-signature groups. Interestingly, in both the ccRCC and pRCC cohorts, the genes of several members of the melanoma antigen (MAGE) family were highly upregulated in the signatures, which predicted poor outcomes, and downregulated in signatures, which were associated with improved survival. To summarize, SBS signatures provide substantial prognostic value compared with just the traditional prognostic factors in certain cancer types. APOBEC-related SBS2 and SBS13 seem to provide robust prognostic information for particular urological cancers, maybe driven by the expression of specific groups of genes, including the MAGE gene family.
1. Introduction
Urological cancers are among the most frequently occurring cancers, and they have a significant impact on total cancer mortality worldwide [1]. Their survival rates vary from the excellent long-term disease-specific survival (DSS) rates observed in testicular cancers and local prostate cancers to just a few years of median survival in cases of metastatic urinary bladder and renal cell carcinomas (RCC), despite the introduction of immuno-oncological treatments during the last few years [2,3]. Virtually all urological carcinomas lack any established prognostic factors in addition to the TNM stage and histological subtype. More precise and reproducible prognostic factors would thus be needed to optimize their surgical and oncological treatments, and surveillance.
Mutational signatures are characteristic patterns of mutations that reflect different carcinogenetic pathways [4]. The incidence of these unique mutational patterns in cancer genomes can give insights into, for example, past exposure to carcinogens and defects in DNA repair mechanisms. Single-base substitution (SBS) mutational signatures consist of 53 distinct signatures in the latest version of the Catalogue of Somatic Mutations in Cancer (COSMIC) database [5]. SBS signatures may tell, for example, about the activation of AID/APOBEC cytidine deaminases (signatures SBS2 and SBS13), defects in specific DNA proofreading mechanisms (e.g. SBS10), exposure to specific chemotherapies (e.g. SBS17), or they may be secondary to smoking (e.g. SBS4) [5,6]. The aetiology of some signatures is still unknown, although knowledge is rapidly progressing [5].
The association between COSMIC mutational signatures and outcomes in patients with urological cancers has not yet been studied. We used publicly available data from the Cancer Genome Atlas (TCGA) database to evaluate whether these signatures would be able to show differences in survival endpoints in bladder transitional cell carcinoma (BTCC), clear cell RCC (ccRCC), papillary RCC (pRCC), renal chromophobic carcinoma, prostate adenocarcinoma, and testicular germ cell tumours (TGCT). These studies were complemented by gene expression analysis to gain insight into the potential molecular mechanisms connected to mutational signatures and survival.
2. Materials and methods
2.1. Data
Mutational signature activity data published in [5] were accessed from The International Cancer Genome Consortium (ICGC) data portal: https://dcc.icgc.org/releases/PCAWG/mutational_signatures/Signatures_in_Samples/SP_Signatures_in_Samples. The baseline characteristics for the cohorts used in this study are shown in the Supplementary Tables 1-6. The data comprised whole-genome sequenced tumours from the Pan-Cancer Analysis of Whole-Genomes (PCAWG) consortium and whole-exome sequenced tumours from TCGA. Here we concentrated to TCGA data, where only SBS and ID signatures were available for the samples. Open-access RNA-seq harmonized (aligned to GRCh38 reference genome) gene expression data (HTSeq generated counts) from TCGA were downloaded from the Genomic Data Commons (GDC) portal [8] using the R package TCGAbiolinks v2.16.4 [9].
2.2. Univariate survival analysis
The association between mutational signatures and overall survival (OS) was first tested in a univariate approach utilizing the R packages survival (v3.2–7) [10] and survminer (v0.4.8) [11]. Only TCGA primary tumour samples and patients with available vital status and survival/follow-up time were included. The association between signature activity and OS in each cancer type was analysed according to the following criteria: at least twenty samples had both signature and survival data, there were at least five death events among the patients, and there were at least five samples with nonzero signature activity. The association with survival between low-activity and high-activity tumours was then tested using the log-rank test for the given signature. Low-activity tumours were defined as those with a median or lower activity of the signature within the cancer type, and high-activity tumours were defined as those with above-median activity. For the association between the gene expression of the MAGE family genes and OS, log2-transformed RSEM counts were used, and the subjects were dichotomized (high and low expression) based on the mean gene expression values. Only primary tumour samples were considered. For each analysis, a Kaplan–Meier curve was plotted using the function ggsurvplot from the survminer package.
2.3. Multivariate survival analysis
Multivariate survival analysis was performed for BTCC, ccRCC, pRCC, RCC, prostate adenocarcinoma, and TGCT using TCGA data and the survival and survminer packages. For each cancer type, a Cox proportional hazard regression with multiple variables was fitted, considering all signatures or selected MAGE expressions with nonzero activity in at least 5% of patients within the cancer type and selected clinical variables. For each cancer type, a separate regression model was fitted for each of the following endpoints: OS, DSS, PFI and DFI. The function cox.zph was used to test the proportional hazard assumptions and plot the Schoenfeld residuals for each variable and the combined model. The function coxph was used to run the Cox regression. Forest plots illustrating the hazard ratios of each variable were generated using the function ggforest from the package survminer v0.4.8 [11]. In cases where no uncensored patients (i.e. patients with a qualifying progression event) with a valid value of a clinical variable were available, these variables were either left out, or patients with particular values of that variable were removed. Cross-tables were produced using the R package gtsummary [12].
2.4. Time-dependent area under curve (AUC)
Time-dependent AUC was calculated for each Cox proportional hazard regression with at least one significant COSMIC mutational signature variable. For each model the incident/dynamic AUC was estimated as proposed by Song and Zhou [13] using the survAUC R package across the entire range of endpoint timepoints with one-year intervals. The summary AUC measure (integrated AUC; iAUC) reported is the integral of AUC on the entire time range.
2.5. MAGE gene expressions as predictors of overall survival
Open-access RNA-seq harmonized (aligned to GRCh38 reference genome) gene expression data (HTSeq generated counts) of the The Cancer Genome Atlas (TCGA) Kidney Renal Clear Cell Carcinoma: TCGA-KIRC (ccRCC) and Cervical Kidney renal papillary cell carcinoma: TCGA-KIRP (pRCC) were downloaded from the Genomic Data Commons (GDC) using R package TCGAbiolinks, v. 2.16.4. Gene expression data were quantile normalized using the preprocessCore R package. For ccRCC the following genes from the MAGE family were considered: MAGEB2, MAGEC2, MAGEA4, and MAGEA6 while for pRCC MAGEA3, MAGEA11, MAGEA12, MAGEC1, and MAGEC2. Patients were dichotomized based on median of each MAGE gene expression with patients higher than median classified as High for MAGE.
2.6. Differential expression analysis
The HTseq count matrix was filtered to include only subjects with available mutational signature information. Additionally, data from only primary tumour samples were considered. Duplicated samples (different aliquots of the same patient) were also excluded. Samples (subjects) were dichotomized into low and high mutational signature groups based on the population median mutational signature activity. Finally, the count matrix was filtered to exclude genes with fewer than 10 reads in total across all samples. Data normalization and differential expression analysis were performed using the R package DESeq2 [14] by contrasting gene expression in the high versus the low group for each cancer type and mutational signature combination. Differentially expressed genes were tested on the null hypothesis that the log2-fold change between groups was equal to 0 (lfc0). Significant genes were identified as those with an adjusted p-value of < 0.05 and an absolute log2-fold change of > 1.
2.7. Gene ontology enrichment
Overrepresentation tests of differentially expressed genes in gene ontology, biological processes, molecular function, and cellular component terms were performed using the R package clusterProfiler [15,16]. Hypergeometric test p-values were corrected for multiple testing using the Benjamini–Hochberg multiple testing adjustment. Terms with an adjusted p-value of < 0.05 were considered significantly enriched. Enriched terms were further visualized using the R package clusterProfiler.
3. Results
3.1. COSMIC mutational signatures and survival
First, we studied whether specific mutational signatures were associated with survival endpoints (overall survival (OS), DSS, disease-free interval (DFI), or progression-free interval (PFI)) in the six studied cancer types from the TCGA cohort. We performed Cox proportional hazard analyses, which are reported in more detail in Table 1.
Table 1.
Summary of Cox proportional hazard (CoxPH) analyses from TCGA data. The clinical variables used in the models included age, gender (if applicable), tissue, or organ of origin, and primary diagnosis. American Joint Committee on Cancer (AJCC) staging was also included, except for prostate adenocarcinoma, where this variable was replaced with two Gleason score groups: (1) a Gleason score of < 7 and 7 (3 + 4) and (2) a Gleason score of > 7 and 7 (4 + 3).
| Cancer type | Endpoint | Signatures eligible for the analysis | Excluded variables / variable levels | Variables with rejected proportional hazards assumptions | Signatures with p-value < 0.05 from the CoxPH test |
|---|---|---|---|---|---|
| Bladder transitional cell carcinoma | OS | SBS1, SBS2, SBS3, SBS5, SBS13 | AJCC pathologic: stage I samples, Primary diagnosis: Carcinoma, Papillary adenocarcinoma and Squamous cell carcinoma samples, Tissue: Bladder neck and Ureteric orifice samples | SBS2 (Improved survival) | |
| DSS | SBS1, SBS2, SBS3, SBS5, SBS13 | Same as above | SBS2 (Improved survival) | ||
| DFI | SBS1, SBS2, SBS3, SBS5, SBS13 | Tissue or organ of origin and AJCC pathologic stage and signature SBS3 not included in the model | SBS2, SBS5, SBS13 | ||
| PFI | SBS1, SBS2, SBS3, SBS5, SBS13 | AJCC pathologic: stage I samples, Primary diagnosis: Carcinoma, Papillary adenocarcinoma and Squamous cell carcinoma samples, Tissue: Bladder neck and Ureteric orifice samples | SBS1 | SBS2 (Improved survival) | |
| Clear cell renal cell carcinoma | OS | SBS1, SBS5, SBS40, SBS45, SBS52 | Tissue or organ of origin and Primary diagnosis not included in the model | Age | |
| DSS | SBS1, SBS5, SBS40, SBS45, SBS52 | Same as above | SBS40 | SBS1 (Decreased survival) | |
| DFI | SBS1, SBS5, SBS40, SBS45, SBS52 | Tissue or organ of origin, Primary diagnosis and SBS45 not included in the model, AJCC pathologic stage IV samples | |||
| PFI | SBS1, SBS5, SBS40, SBS45, SBS52 | Tissue or organ of origin and Primary diagnosis not included in the model | SBS45 | SBS1 (Decreased survival) | |
| Renal papillary cell carcinoma | OS | SBS1, SBS2, SBS5, SBS13, SBS45 | Same as above | AJCC pathologic stage, gender, SBS1 | SBS45 (Improved survival) |
| DSS | SBS1, SBS2, SBS5, SBS13, SBS45 | Same as above | AJCC pathologic stage | SBS45 (Improved survival) | |
| DFI | SBS1, SBS2, SBS5, SBS13, SBS45 | Tissue or organ of origin and Primary diagnosis not included in the model, AJCC pathologic stage IV samples | AJCC pathologic stage | SBS13 (Decreased survival) | |
| PFI | SBS1, SBS2, SBS5, SBS13, SBS45 | Tissue or organ of origin and Primary diagnosis not included in the model | AJCC pathologic stage, SBS1, SBS5 | SBS13 (Decreased survival) | |
| Renal chromophobic carcinoma | OS | SBS1, SBS5, SBS17a, SBS46 | Tissue or organ of origin and Primary diagnosis not included in the model, AJCC pathologic stage and signatures SBS5 and SBS46 also as model did not converge | ||
| DSS | SBS1, SBS5, SBS17a, SBS46 | Tissue or organ of origin and Primary diagnosis not included in the model, AJCC pathologic stage and signatures SBS5, SBS17a and SBS46 also as model did not converge | |||
| DFI | SBS1, SBS5, SBS17a, SBS46 | Tissue or organ of origin and Primary diagnosis not included in the model, AJCC pathologic stage and all signatures as model did not converge | |||
| PFI | SBS1, SBS5, SBS17a, SBS46 | Tissue or organ of origin and Primary diagnosis not included in the model, AJCC pathologic stage and signatures SBS5, SBS17a and SBS46 also as model did not converge | |||
| Prostate adenocarcinoma | OS | SBS1, SBS5, SBS10b, SBS15, SBS40, SBS45 | Tissue or organ of origin not included; samples with primary diagnosis ‘Adenocarcinoma with mixed subtypes’ or ‘Mucinous adenocarcinoma’, signatures SBS10b and SBS15 due to inflated coefficients | SBS40, SBS45 (Decreased survival) | |
| DSS | SBS1, SBS5, SBS10b, SBS15, SBS40, SBS45 | Tissue or organ of origin not included; samples with primary diagnosis ‘Adenocarcinoma with mixed subtypes’ or ‘Mucinous adenocarcinoma’, Gleason group and signatures SBS10b and SBS15 due to inflated coefficients | SBS5, SBS40 (Decreased survival) | ||
| DFI | SBS1, SBS5, SBS10b, SBS15, SBS40, SBS45 | Tissue or organ of origin not included; samples with primary diagnosis ‘Adenocarcinoma with mixed subtypes’ or ‘Mucinous adenocarcinoma’ | |||
| PFI | SBS1, SBS5, SBS10b, SBS15, SBS40, SBS45 | Same as above | |||
| Testicular germ cell tumours | OS | SBS1, SBS2, SBS5, SBS7a, SBS13, SBS15, SBS19, SBS24, SBS42, SBS44 | Tissue or organ of origin not included, ‘AJCC pathologic stage’ subtypes combined into 3 stages, ‘Primary diagnosis’ categories with few samples combined into ‘Other’ group, signatures SBS2, SBS7a, SBS13, SBS15, SBS19, SBS24 and SBS44 due to inflated coefficients | ||
| DSS | SBS1, SBS2, SBS5, SBS7a, SBS13, SBS15, SBS19, SBS24, SBS42, SBS44 | Tissue or organ of origin not included, ‘AJCC pathologic stage’ and ‘Primary diagnosis’ and signatures SBS1, SBS2, SBS7a, SBS13, SBS15, SBS19, SBS24 and SBS44 due to inflated coefficients | |||
| DFI | SBS1, SBS2, SBS5, SBS7a, SBS13, SBS15, SBS19, SBS24, SBS42, SBS44 | Tissue or organ of origin not included, ‘AJCC pathologic stage’ due to inflated coefficients | SBS44 | ||
| PFI | SBS1, SBS2, SBS5, SBS7a, SBS13, SBS15, SBS19, SBS24, SBS42, SBS44 | Primary diagnosis, SBS2 |
OS = overall survival; DSS = disease-specific survival; DFI = disease-free interval; PFI = progression-free interval; SBS = single-base substitution.
The high prevalence of SBS2 signatures in BTCC was associated in multivariate analysis with improved PFI (Hazard Ratio (HR) 0.42, 95% Confidence Interval (CI) 0.24–0.73), DSS (HR 0.34, 95% CI 0.16–0.70), and OS (HR 0.48, 95% CI 0.26–0.86; see Figure 1). The iAUC values in the Cox model were 0.6659 for PFI, 0.6956 for DSS and 0.6619 for OS (Figure 2.). Adding mutational signatures to the model improved especially the prognostic value of PFI when compared to using only the traditional prognostic factors. The prognostic value of the SBS2 signature also remained separate in the stage I–III and stage IV cohorts (Figure 3).
Fig. 1.

Cox proportional hazards regression analysis results for bladder transitional cell carcinoma. In these models, nonmetastatic disease and high SBS2 prevalence were associated with better OS (A) and DSS (B). A high number of SBS2 signatures also predicted a better PFI (C). AIC = Akaike Information Criterion; AJCC = American Joint Committee on Cancer; NOS = not otherwise specified; SBS = single-base substitution.
Fig. 2.
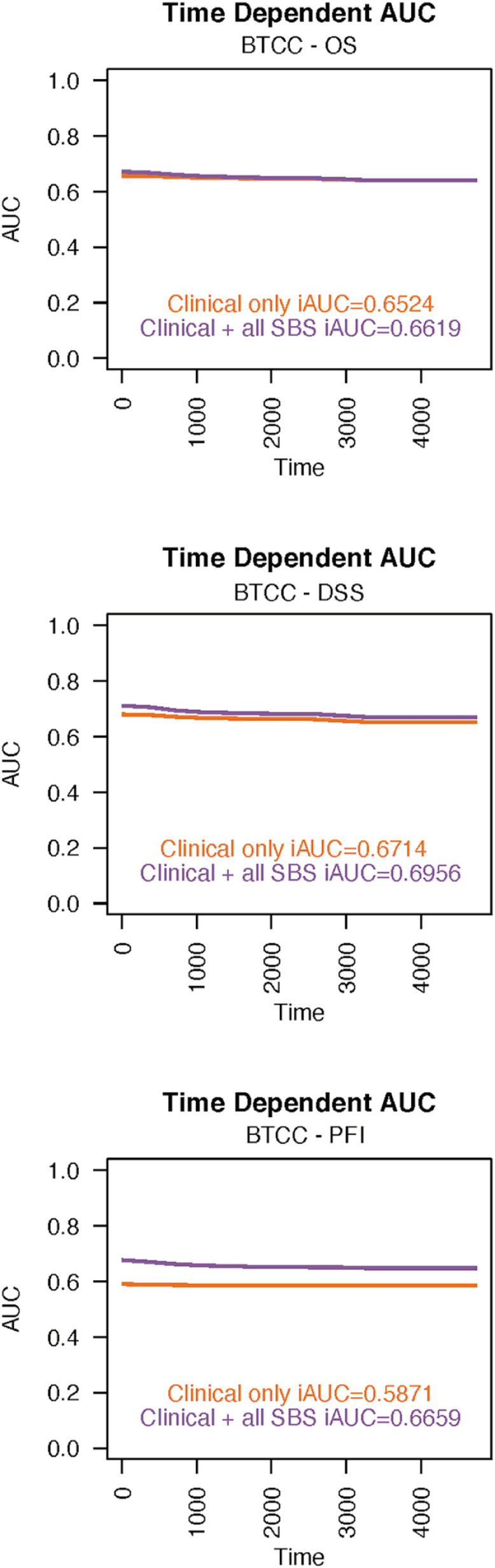
The time-dependent integrated areas under the curves (iAUC) values in bladder transitional cell carcinoma for progression-free interval (A), for disease-specific survival (B) and for overall survival (C). SBS = single-base substitution.
Fig. 3.

Kaplan–Meier curves showing that the prognostic value of SBS2 in terms of OS was observed separately in both stage I–III (left) and stage IV (right) bladder transitional cell carcinomas. The coloured areas represent 95% confidence intervals.
In ccRCC, the high prevalence of the SBS1 signature was associated with decreased DFI (HR 7.69, 95% CI 1.45–40.80) and PFI (HR 1.90, 95% CI 1.19–3.00; see Figure 4). The iAUC values were 0.8139 and 0.7773, respectively (Figure 5). There was a notable increase in the prognostic power of the DFI endpoint, when mutational signatures were added to the model apart from clinical prognostic factors.
Fig. 4.

In patients with ccRCC, the high activity of SBS1 was associated with a worse DFI (A) and PFI (B). Stage was still the most predominant prognostic factor in both cases. AJCC = American Joint Committee on Cancer; SBS = single-base substitution.
Fig. 5.

The time-dependent integrated areas under the curves show (iAUC) values in ccRCC for progression-free interval (A) and for disease-free interval (B). SBS = single-base substitution.
Again, in pRCC, both SBS13 and SBS45 signatures were significantly associated with survival endpoints after adjusting for traditional prognostic factors (Figure 6). A high number of SBS13 signatures was associated with worse DFI (HR 4.00, 95% CI 1.20–13.30) and PFI (HR 3.15, 95% CI 1.30–7.60), while the presence of SBS45 was linked to improvements in the clinically more significant endpoints DSS (HR 0.27; 95% CI 0.083–0.85) and OS (HR 0.39; 95% CI 0.154–0.97). The predictive power analyzed for the Cox model with ROC showed iAUC value of 0.7367 for DFI, 0.7625 for PFI, 0.9259 for DSS and 0.7824 for OS, with substantial increase in the iAUC value especially with the OS endpoint (Figure 7).
Fig. 6.

In pRCC patients, a high number of SBS45 signatures predicted longer OS (A) and DSS (B). Again, a high number of SBS13 signatures were associated with worse DFI (C) and PFI (D). AIC = Akaike information criterion; AJCC = American Joint Committee on Cancer; SBS = single-base substitution.
Fig. 7.

The integrated areas under the curves (iAUC) values in the pRCC cohort demonstrate the predictive power over time for disease-free interval (A), progression-free interval (B), disease-specific survival (C) and overall survival (D). SBS = single-base substitution.
SBS5, SBS40, and SBS45 were found to be significant predictors of DSS and OS in prostate adenocarcinoma but with very wide CIs (Figure 8). Specifically, the high prevalence of SBS40 predicted both decreased DSS (HR 98.73; 95% CI 3.47–2810.71) and OS (HR 8.42; 95% CI 1.243–57.10). The SBS5 signature was associated with shorter DSS (HR 24.27; 95% CI 1.75–335.59) and SBS45 with shorter OS (HR 17.02; 95% CI 2.606–111.20). Adding mutational signatures to the traditional clinical prognostic factors yielded a lot more accurate predictive model, with all SBS iAUC values of 0.9104 for DSS and 0.8347 for OS (Figure 9).
Fig. 8.

The high numbers of SBS40 and SBS45 signatures were predictors of poor OS (A) and SBS5 and SBS45 of DSS (B) in patients with prostate adenocarcinoma in Cox multivariate analysis. AIC = Akaike Information Criterion; AJCC = American Joint Committee on Cancer; NOS = not otherwise specified; SBS = single-base substitution.
Fig. 9.

The integrated areas under the curves (iAUC) values in the prostate adenocarcinoma cohort demonstrate the predictive power over time for disease-specific survival (A) and overall survival (B). SBS = single-base substitution.
No significant associations between survival and COSMIC mutational signatures were found in renal chromophobe carcinoma, or TGCT after adjusting for traditional prognostic factors.
3.2. Mutational signatures and gene expression analysis
Next, we identified putative links between mutational signature activity (low activity vs. high activity) and gene expression patterns based on TCGA data. To elucidate the potential drivers of the observed survival differences at the gene expression level, these analyses were conducted only for cancer types and signatures, in which statistically significant survival differences were observed in multivariate analyses.
Among the BTCC cohort, between SBS-low and SBS-high subjects, 550 genes were differentially expressed (absolute log2-fold change of > 1 and an adjusted p-value of < 0.05) (Supplementary Figure 1). The most downregulated gene in the SBS2-high group was delta-like 1 homologue, with 9.65-fold (95% CI 1.38; p = 1.9*10−8) expression in the SBS2-low group in patients with BTCC (Table 2). In the BTCC cohort, somatostatin was among the most downregulated genes in the SBS2 group.
Table 2.
Examples of the differences in gene expression between the low- and high-signature cohorts. Renal chromophobic carcinoma and TCGT are not included in the table since there were no associations with survival outcomes in these cohorts in the multivariate analysis.
| Cancer | Signature | Differentially expressed genes between low-signature and high-signature groups | Examples of differentially expressed genes between low-signature and high-signature groups (fold-change, adjusted p-value) | Examples of enriched GO terms |
|---|---|---|---|---|
| Bladder transitional cell carcinoma | SBS2 (association with increased survival) | 550 | Delta-like noncanonical Notch ligand 1 (9.65-fold downregulation, p = 1.9*10−9) Somatostatin (7.70-fold downregulation, p = 8.4*10−8) |
Humoral immune response, regulation of metal ion transport |
| Clear cell renal cell carcinoma | SBS1 (association with decreased survival) | 455 | Melanoma antigen-B2 (10.1-fold upregulation, p = 1.8*10−5) Melanoma antigen-C2 (7.52-fold upregulation, p = 0.00078) Melanoma antigen A4 (6.82-fold upregulation, p = 0.019) Uromodulin (4.68-fold upregulation, p = 4.7*10−5) Melanoma antigen A6 (4.38-fold upregulation, p = 0.048) |
Antigen binding, immunoglobulin genes, complement, T-cell/B-cell response related pathway upregulated |
| Papillary renal cell carcinoma | SBS13 (association with decreased survival) | 842 | Melanoma antigen-C2 (69.7-fold upregulation, p = 3.2*10−8) Melanoma antigen-C1 27.7-fold upregulation, p = 4.4*10−5) Melanoma antigen-A11 (12.2-fold upregulation, p = 0.013) Melanoma antigen-A3 (11.3-fold upregulation, p = 0.011) Melanoma antigen-A12 (10.3-fold upregulation, p = 0.000088) BUB1 (2.05-fold upregulation, p = 7.8*10−7) Survivin (2.2-fold upregulation, p = 3.1*10−7) Kallikrein-related peptidase 3 (9.45-fold downregulation, p = 7.9*10−8) Kallikrein 1 (7.69-fold downregulation, p = 1*10−10) Kallikrein-related peptidase 2 (5.36-fold downregulation, p = 0.00017) |
Mitosis, cell division, nuclear segregation |
| Papillary renal cell carcinoma | SBS45 (association with increased survival) | 337 | Melanoma antigen-A3 (27.6-fold downregulation, p = 0.00011) Melanoma antigen-C2 (12.1-fold downregulation, p = 0.041) Melanoma antigen-C1 (7.98-fold downregulation, p = 0.042) Kallikrein-1 (5.00-fold upregulation, p = 3.9*10−6) |
Several pathways related to ion channel activity downregulated |
| Prostate adenocarcinoma | SBS5 (association with decreased survival) | 144 | Cytochrome c oxidase subunit VIa polypeptide 1 (COX6A1) (3.03-fold downregulation, p = 0.002) Cytochrome c oxidase subunit 7B2 (5.23-fold upregulation, p = 0.01) |
Related to vascular system |
| SBS40 (association with decreased survival) | 56 | Prostate and testis expressed 1 (5.77-fold upregulation, p = 2.7*10−10) | Sperm function and spermatid development | |
| SBS45 (association with decreased survival) | 108 | Myosin light chain 3 (3.83-fold upregulation, p = 0.00034) Actin alpha 1, skeletal muscle (2.96-fold upregulation, p = 0.046) Troponin C1, slow skeletal, and cardiac type (2.62-fold upregulation, p = 0.016) Troponin I1, slow skeletal type 2.37-fold upregulation, p = 0.042) |
Several pathways related skeletal muscles |
GO = gene ontology; SBS = single-base substitution.
In the ccRCC group, 455 genes were differentially expressed between SBS1-low and SBS1-high subjects. Of these genes, 343 were upregulated in the SBS1-high group, and 90 were immunoglobulin-related genes. The 22 most upregulated genes in the SBS1-high group included four members of the melanoma antigen (MAGE) family: MAGE-B2 (10.1-fold expression, 95% CI 1.54, p = 0.000018), MAGE-C2 (7.52-fold expression, 95% CI 1.56, p = 0.00078), MAGE-A4 (6.81-fold expression, 95% CI 1.75, p = 0.018), and MAGE-A6 (4.38-fold expression, 95% CI 1.64, p = 0.048). The high expressions of MAGE-B2, MAGE-C2, MAGE-A4, and MAGE-A6 were all associated with poor OS (Figure 10). We also studied if MAGE-B2, MAGE-C2, MAGE-A4 or MAGE-A6 gene expression could provide prognostic power over the traditional prognostic factors in the ccRCC cohort, but the additional prognostic value of the gene expression was very limited (Figure 11A).
Fig. 10.

The high expressions of the MAGE-B2, MAGE-C2, MAGE-A4, and MAGE-A6 genes were associated with worse overall survival in patients with clear cell renal cell carcinoma (ccRCC) in univariate analysis. These genes were significantly upregulated in patients with a high SBS1 signature (poor prognosis signature) in the ccRCC cohort.
Fig. 11.

The expression of genes MAGE-B2, MAGE-C2, MAGE-A4 or MAGE-A6 provided only minor additional prognostic value over the traditional prognostic (clinical) values in the ccRCC cohort (A). In pRCC, the expression of MAGE-A3, MAGE-A11, MAGE-A12, MAGE-C1, and MAGE-C2 genes was a better predictor when compared only to the traditional prognostic factors. This model also almost reached the prognostic level of mutational signatures and traditional prognostic factors combined (B).
As another potentially interesting gene, encoding uromodulin (UMOD, Tamm–Horsfall protein), was 4.69-fold upregulated (95% CI 1.31, p = 4.7*10−6) in SBS1-high subjects in the ccRCC cohort. The five most enriched biological processes in SBS1-high patients were all related to immunoglobulins and immune responses (Supplementary Figure 2).
High SBS45 levels, which were associated with improved DSS and OS in pRCC patients, were associated with 337 differentially expressed genes, of which 312 were downregulated in high SBS45-high patients compared to SBS45-low pRCC patients. MAGE-A3 was the most downregulated gene in the SBS45-high group (27.6-fold change, 95% CI 1.90; p = 0.00011) in the pRCC cohort, and the 10 most downregulated genes also included MAGE family members MAGEC2 and MAGEC1, with 12.1-fold change (95% CI 2.08, p = 0.041) and 7.98-fold (95% CI 1.84, p = 0.041) downregulations, respectively. Kallikrein-1 was the second most upregulated gene in the SBS45-high group in the pRCC cohort, with a 5.00-fold increase (95% CI 1.32, p = 3.9*10−6) compared to the SBS45-low group in the same cohort. The most enriched biological processes within the differentially expressed genes were associated predominantly with processes such as epidermis development, keratinocyte differentiation, and keratinization (Supplementary Figure 3). High SBS45 prevalence was also connected with several genes related to ion transport in patients with pRCC (Figure 12A).
Fig. 12.

Top twenty significantly enriched (adjusted p-value of < 0.05) gene ontology molecular function terms in differentially expressed genes between SBS45-low and SBS45-high (A) and SBS13-low and SBS13-high (B) patients with papillary renal cell carcinoma.
As described above, SBS13 was associated with worse outcomes for pRCC patients. Upon closer investigation of different gene expressions between high and low SBS13 phenotype individuals, 842 genes were differentially expressed between the SBS13 groups, and 744 of them were upregulated. For example, various enzyme activities belong to the most enriched gene ontology molecular function terms describing the differentially expressed genes between SBS13 low and high patient groups (Figure 12B). In contrast with the list of the most upregulated genes in the SBS45-high pRCC cohort, there was a significant upregulation of several MAGE family members in the SBS13-high pRCC cohort: MAGE-C2 (69.7-fold upregulation, 95% CI 2.154, p = 5.1*10-6), MAGE-C1 (27.7-fold upregulation, 95% CI 1.93, p = 4.4*10-5), MAGE-A11 (12.2-fold upregulation, 95% CI 2.03, p = 0.013), MAGE-A3 (11.3-fold upregulation, 95% CI 1.97, p = 0.011) and MAGE-A12 (10.3-fold upregulation, 95% CI 1.70, p = 0.00088) in the SBS13-high group. The high expressions of MAGE-A3, MAGE-C1, and MAGE-C2 were all associated with poor OS (Figure 13). When studied in the Cox model, the expression of MAGE-A3, MAGE-A11, MAGE-A12, MAGE-C1, and MAGE-C2 genes was a better predictor than just the traditional prognostic factors, and this model almost reached the prognostic level of mutational signatures and traditional prognostic factors combined (Figure 11B). Again, kallikrein-related peptidase 3, kallikrein 1, and kallikrein-related peptidase 2 were among the most downregulated genes in the SBS13-high group. In particular, the processes linked to mitosis and nuclear segregation were upregulated in the patients with the worst prognoses, i.e. those with a high SBS13 prevalence (Supplementary Figure 4). Among those, BUB1 and survivin (BIRC5) upregulation were identified as genes of specific interest.
Fig. 13.

The high expressions of the MAGE-A3, MAGE-A11, MAGE-A12, MAGE-C1, and MAGE-C2 genes were associated with worse overall survival in patients with papillary renal cell carcinoma (pRCC) in univariate analysis. These genes were significantly upregulated in patients with a high SBS13 signature (poor prognosis signature) in the pRCC cohort.
In patients with prostate adenocarcinoma, the SBS45 signature was associated with the enrichment of several pathways related to skeletal muscle development and regulation (Supplementary Figure 5). Again, between SBS40-high and SBS40-low patients, differentially expressed genes especially included those having functions in sperm and spermatid capacitation and function and in antibacterial and antimicrobial humoral responses (Supplementary Figure 6). Instead, SBS5 was associated with several regulators of blood pressure and vasoconstriction (Supplementary Figure 7). Mutational signatures were compared with clinical and pathological parameters, and these results are shown in Supplementary Tables 7–12.
3.3. Mutational burden
As expected, age-related mutational signatures SBS1 and SBS5, in particular, correlated with the total number of mutations (Supplementary Figure 8). The correlation between the individual signatures and the mutational burden was most frequently observed in the BTCC cohort, where SBS1, SBS2, SBS5, and SBS13 were associated with the total mutation count.
Mutation rate of MAGE family genes across urological cancers is shown in Supplementary Figure 9. Highest mutation rates were detected in the BTCC cohort, while mutations in MAGE genes were infrequent in other cohorts.
4. Discussion
This is the first study to assess the prognostic significance of COSMIC mutational signatures in urological cancers. One of the main findings was the association of a high number of SBS2 signatures with improved PFI, DSS, and OS in BTCC. In particular, PFI, and DSS have been considered reliable endpoints in this TCGA cohort [7]. The effect of SBS2 on DSS was of a similar magnitude as the traditionally with the most powerful predictor of outcome and stage in the multivariate analysis (HR 0.34, 95% CI 0.16–0.70). Notably, SBS2 activity was not associated with the traditional prognostic factors, and a survival benefit was observed in stage I–III and stage IV BTCCs. The SBS2 signature is formed due to APOBEC deamination of cytosine to uracil, and the APOBEC family of enzymes is a frequent source of hypermutation in most urothelial carcinomas [17]. While uracil subsequently enters the replication process uncorrected and pairs with A, this yields C-to-T mutations, which are characteristic of SBS2 [5,18,19]. Indeed, SBS2 is one of the most well-defined mutational signatures, and it is often accompanied by SBS13. Logically, with the hypermutator role of APOBEC, we observed a strict correlation between the number of SBS2 signatures and the total mutational burden in BTCC.
Recent data suggest that the APOBEC-driven subgroups of high SBS2 and SBS13 would form the largest subgroup of metastatic urothelial carcinomas and that SBS2 and SBS13 would be the major features of radiotherapy-associated BTCC [20,21]. APOBEC-related mutations also seem to drive the aggressiveness of non-muscle invasive BTCC and are associated with tobacco smoking, being an early, a critical event in bladder carcinogenesis [22], [23], [24], [25]. Patients with APOBEC-related mutations have doubled OS rates compared to patients with APOBEC-low BTCC [22]. The APOBEC-A-high SBS5 phenotype was linked to the absence of recurrences in a series of 62 high-grade, T1 non-muscle-invasive bladder cancers [26]. Non-COSMIC-based APOBEC mutational signatures have also been linked to improved outcomes in patients with advanced urothelial cancers, with most patients having BTCC [27]. To summarize, our results of a favourable prognosis in patients with a high number of SBS2 signatures are in line with the previously reported results regarding the behaviours of less aggressive cancers in APOBEC-high BTCC. Our results complete the previous reports by using COSMIC database, reporting the prognostic value from all stages, and using also additional endpoints than used in the previous studies. In gene expression analysis, we observed notable upregulation of, for example, delta-like noncanonical Notch ligand 1-gene expression in the SBS2-low group (worsened prognosis) in BTCC patients. This gene has been previously linked to a poor prognosis in a variety of different cancers [28], but has remained unstudied in bladder cancers in vivo.
In the ccRCC cohort, the high prevalence of the SBS1 signature provided additional prognostic information over staging in terms of decreased DFI and PFI. In particular, PFI has been considered a reliable endpoint in ccRCCs in TCGA data, with data containing 162 progression events in 362 evaluable patients [7]. SBS1 signatures are produced after the deamination of 5-methylcytosine to thymine, generating G:T mismatches in double-stranded DNA and further C-to-T substitutions [5]. The burden of SBS1 is associated with age in virtually all cancer types examined [29], but we still do not observe an association with OS in the ccRCC cohort.
The mutational load of ccRCCs is among the most common of all cancer types, and ccRCCs are consequently among the most immunologically active carcinomas. In our gene expression analysis, immune-related pathways were activated in SBS1-high (poor prognosis) subjects in the ccRCC cohort. Among these, 90 immunoglobulin genes were upregulated in patients with a high SBS1 signature. Several of these immunoglobulin genes have been associated with survival in ccRCC [30]. In line with our results, immune cell infiltration and enhanced immunological response are established adverse prognostic factors in ccRCC [31], [32], [33]. There is no previous literature on COSMIC signatures and prognosis in ccRCCs, and neither has the relationship between SBS1 and immune cell activation been previously evaluated in RCC. In primary and metastatic cutaneous melanomas, SBS1 activity was associated with lower activity of cytotoxic T and NK cells [34].
Several MAGE genes were upregulated in SBS1-high tumours in the ccRCC cohort. MAGE genes are highly conserved in all eukaryotes, and they play a crucial role in adaptation against environmental stress [35]. For example, MAGE-B2, which was the most upregulated gene in the SBS1-high ccRCC cohort, promotes stress tolerance, giving a growth advantage to cancer cells, and its high expression has been implicated in tumour growth and progression in various cancers in vitro but has not been studied previously in RCCs [35], [36], [37], [38]. MAGE-C2, with the observed 7.4-fold upregulation in SBS1 ccRCCs, is a cancer cell-specific regulator of TRIM28, and its high expression is associated with poor survival in various malignancies, including bladder and prostate carcinomas; however, the role of MAGE-C2 has not yet been evaluated in RCCs [39], [40], [41], [42]. Other highly induced MAGE genes in the SBS1-high group in the ccRCC cohort included MAGE-A4 and MAGE-A6. In particular, MAGE-A6 seems to have significant pro-oncogenic roles [43,44], whereas the role of MAGE-A4 is less well understood. The high RNA levels of MAGE-B2, MAGE-A4, and MAGE-A6 were related to a dismal prognosis. A single potentially interesting gene, UMOD, was > 4.5-fold upregulated in SBS1-high subjects in the patients with ccRCC. For a small set of samples, UMOD RNA expression has been reported as one of the most upregulated transcripts in ccRCCs compared to benign renal tissue [45]. Based on our ccRCC results, it is possible that the dismal prognosis for SBS1-high patients may be driven by the upregulation of the immune system and MAGE family genes.
Of all renal cancers, pRCC accounts for approximately 15% and is genetically and morphologically distinct from other RCCs [46]. With the exception of staging, reliable prognostic factors are virtually absent in pRCC. According to our analyses, a high number of SBS13 signatures was associated with worse DFI and PFI in the TCGA pRCC cohort. SBS1 and SBS13 have also been attributed to the activity of AID/APOBEC family of cytidine deaminases [5,47]. However, while high APOBEC activity has been constantly linked to improved outcomes in BTCC, there are no previous data on the role of APOBEC in pRCC. In line with our results, high SBS13 activity is associated with a higher risk of postresection recurrence in non-small-cell lung cancers [48] and with radioiodine treatment resistance in papillary thyroid carcinomas [49]. We found that SBS13 activity was associated with the upregulation of 744 genes in pRCC, which were mainly linked to mitosis, cell division, and nuclear segregation. Notably, several MAGE family members were upregulated in pRCC in the SBS13 high cohort. The most upregulated of all genes in this cohort was MAGE-C2, which was upregulated 70-fold in the SBS13 high group in the pRCC cohort. In addition, MAGE-C1, MAGE-A11, MAGE-A3, and MAGE-A12 were among the most upregulated genes, with at least a 10-fold increase in the SBS13 high group.
In urological cancers, MAGE-A11 is best known for its protooncogenic role in prostate cancer [50]. Although MAGE family members in RCCs have rarely been studied, there is evidence that a specific single-nucleotide polymorphism of MAGE-A11 provides prognostic information on OS in RCCs in materials of mixed histologies [51]. The MAGE-A3 gene shares a 96% sequence similarity with more well-characterized prooncogenic MAGE-A6 [52]. MAGE-C1 is highly expressed in several cancers and has prognostic relevance in multiple myeloma and breast cancers [53,54]. The possible biological relationship between APOBEC enzymes and MAGE family members or kallikreins remains unstudied. However, it is known that the expression of MAGE-A genes is regulated by DNA methylation [55], which could be modified by APOBEC enzymes.
The high prevalence of SBS45 signatures was associated with improved OS and DSS in pRCCs, and in the time-dependent SBS45-including model the iAUC value was as high as 0.93 for DSS. Adding mutational signatures to the model with only traditional clinical variables increased especially the predictive power of OS analysis in the pRCC cohort. Among the SBS45-high patients, MAGE-A3, MAGE-C1, and MAGE-C2 were among the most downregulated genes, and kallikrein 1 was highly upregulated in the SBS45-high patients. The function of these genes, and the respective proteins with tumour-suppressive functions, are well-characterised in prostate carcinogenesis, for example, but no previous literature exists on their role in RCCs [56,57]. The high RNA expression of MAGE-A3, MAGE-C1, and MAGE-C2 individually associated with dismal survival rates in pRCC. According to the ROC analysis, gene expression of the five selected MAGE genes almost reached the prognostic significance of mutational signatures and traditional prognostic factors combined and exceeded substantially the prognostic power of that of just the traditional prognostic factors. In clinical practice, using gene expression analysis could be more feasible prognostic tool than mutational signatures requiring whole-exome sequencing (WES). It is worth noting that the SBS45 signature may be a sequencing artefact due to the use of 8-oxoguanine during sequencing [58].
In patients with prostate cancer, SBS5, SBS40, and SBS45 were associated with worse OS and DSS. There were very wide CIs in these analyses due to the limited number of events in this disease, which has a generally favourable prognosis. In addition, only PFI and DFI have been considered reliable endpoints in prostate cancer, while OS and DSS should be used with caution [7]. Thus, this study could not reveal any usable prognostic factors for use in patients with prostate cancer. We propose that the prognostic value of mutational signatures should be evaluated in the future, preferably in local high-risk or metastasized prostate cancers.
Despite urological TCGA cohorts containing a substantial number of patients with comprehensive clinical parameters, the major limitation of this study was the use of only a single dataset. WES is currently performed only to a minority of cancer patients, which limits the immediate implementation of the WES-based prognostic tools. As discussed above and by [7], the sample sizes in different TCGA cohorts vary considerably, and the usability of different survival endpoints is diverse. As TCGA contains only exome data, not all mutations in the human genome are covered. This is also a likely reason for our final analyses, including only SBSs, but not the other types of mutational signatures. Furthermore, mutations occurring at noncoding regulatory regions, including promoters, distal genomic enhancers, and silencer elements, might play crucial roles in gene expression levels. As the current study was not able to evaluate if the observed changes in gene expressions were drivers or passengers, this topic requires functional studies in the future. No significant associations between survival and COSMIC mutational signatures were found in chromophobe RCCs or in TGCT after adjusting for the traditional prognostic factors. This is due, at least partly, to the small number of events in both datasets.
Conclusions
Several COSMIC signatures seem to provide prognostic information in urological cancers, and a significant proportion of the prognostic signatures are related to the activation of APOBEC enzymes. Based on the iAUC values, adding mutational signatures to Cox models seem to provide best prognostic information in pRCC with the iAUC values up to 0.93. The prognostic relevance was worst in BTCC, although mutational signatures are able to provide additional prognostic information also for BTCC patients over traditional prognostic factors. We presented a novel finding of the prominent upregulation of the genes belonging to the MAGE family in the groups where the signature predicted worse survival (SBS1 in ccRCC and SBS13 in pRCC) and the respective downregulation of MAGE genes in signature group with improved survival (SBS45 in pRCC). With using only selected MAGE genes we were also able to provide prognostic information near to that of mutational signatures and clinical variables combined. This suggests that MAGE genes could be key drivers of the current results and deserve further functional studies.
Funding
No reportable funding was used to conduct this study.
Author contributions
PK drafted the manuscript. All authors participated in the planning the study, evaluating the results and writing the final versions of the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank Genevia Technologies for their help in statistical analyses and in composing the figures and tables. Open access funded by Helsinki University Library.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neo.2023.100933.
Appendix. Supplementary materials
Data availability
The datasets used and analyzed are available on reasonable request.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Motzer RJ, Tannir NM, McDermott DF, Burotto M, Choueiri TK. Conditional survival and 5-year follow-up in CheckMate 214: first-line nivolumab + ipilimumab (N+I) versus sunitinib (S) in advanced renal cell carcinoma (aRCC) Ann. Oncol. 2021;32:S678. [Google Scholar]
- 3.Lenis AT, Lec PM, Chamie K, Mshs MD. Bladder cancer: a review. JAMA. 2020 Nov 17;324(19):1980–1991. doi: 10.1001/jama.2020.17598. [DOI] [PubMed] [Google Scholar]
- 4.Alexandrov LB, Stratton MR. The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014;24:52–60. doi: 10.1016/j.gde.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koh G, Degasperi A, Zou X, Momen S, Nik-Zainal S. Mutational signatures: emerging concepts, caveats and clinical applications. Nat. Rev. Cancer. 2021;21:619–637. doi: 10.1038/s41568-021-00377-7. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V, Cancer Genome Atlas Research Network. Hu H. An integrated TCGA pan-cancer clinical data resource to drive high quality survival outcome analytics. Cell. 2018;173(2):400–416. doi: 10.1016/j.cell.2018.02.052. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016 Sep 22;375(12):1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, Ceccarelli M, Bontempi G, Noushmehr H. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic. Acids. Res. 2016 May 5;44(8):e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Therneau, T. A package for survival analysis. In R Package Version 3.2–7. 2020. https://CRAN.R-project.org/package=survival.
- 11.Kassambara, A, Kosinski, M, Biecek, P. Survminer: drawing survival curves using ‘ggplot2’. R Package Version 0.4.8. 2020. https://CRAN.R-project.org/package=survminer.
- 12.Sjoberg DD, Whiting K, Curry M, Lavery JA, Larmarange J. Reproducible summary tables with the gtsummary package. R. J. 2021; 2021;13:570–580. [Google Scholar]
- 13.Song X, Zhou X-H. A semiparametric approach for the covariate specific ROC curve with survival outcome. Stat. Sin. 2008;18:947–965. [Google Scholar]
- 14.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, x Fu, Liu S, Bo X, Yu G. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb) 2021;2(3) doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glaser AP, Fantini D, Shilatifard A, Schaeffer EM, Meeks JJ. The evolving genomic landscape of urothelial carcinoma. Nat. Rev. Urol. 2017 Feb 7;14(4):215–229. doi: 10.1038/nrurol.2017.11. [DOI] [PubMed] [Google Scholar]
- 18.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, Brinkman AB, Morganella S, Aure MR, Lingjærde OC, Langerød A, Ringnér M, Ahn SM, Boyault S, Brock JE, Broeks A, Butler A, Desmedt C, Dirix L, Dronov S, Fatima A, Foekens JA, Gerstung M, Hooijer GK, Jang SJ, Jones DR, Kim HY, King TA, Krishnamurthy S, Lee HJ, Lee JY, Li Y, McLaren S, Menzies A, Mustonen V, O'Meara S, Pauporté I, Pivot X, Purdie CA, Raine K, Ramakrishnan K, Rodríguez-González FG, Romieu G, Sieuwerts AM, Simpson PT, Shepherd R, Stebbings L, Stefansson OA, Teague J, Tommasi S, Treilleux I, Van den Eynden GG, Vermeulen P, Vincent-Salomon A, Yates L, Caldas C, van't Veer L, Tutt A, Knappskog S, Tan BK, Jonkers J, Borg Å, Ueno NT, Sotiriou C, Viari A, Futreal PA, Campbell PJ, Span PN, Van Laere S, Lakhani SR, Eyfjord JE, Thompson AM, Birney E, Stunnenberg HG, van de Vijver MJ, Martens JW, Børresen-Dale AL, Richardson AL, Kong G, Thomas G, Stratton MR. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;2(534):47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, Kim J, Kwiatkowski DJ, Fargo DC, Mieczkowski PA, Getz G, Gordenin DA. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015 Sep;47(9):1067–1072. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakauma-González JA, Rijnders M, van Riet J, van der Heijden MS, Voortman J, Cuppen E, Mehra N, van Wilpe S, Oosting SF, Rijstenberg LL, Westgeest HM, Zwarthoff EC, de Wit R, van der Veldt AAM, van de Werken HJG, Lolkema MPJ, Boormans JL. Comprehensive molecular characterization reveals genomic and transcriptomic subtypes of metastatic urothelial carcinoma. Eur. Urol. 2022 Apr;81(4):331–336. doi: 10.1016/j.eururo.2022.01.026. [DOI] [PubMed] [Google Scholar]
- 21.Mossanen M, Carvalho FLF, Muralidhar V, Preston MA, Reardon B, Conway JR, Curran C, Freeman D, Sha S, Sonpavde G, Hirsch M, Kibel AS, Van Allen EM, Mouw KW. Genomic features of muscle-invasive bladder cancer arising after prostate radiotherapy. Eur. Urol. 2022 May;81(5):466–473. doi: 10.1016/j.eururo.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171:540–556. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Akbani A, Creighton CJ, Lerner SP, Weinstein JN, Getz G. Invasive bladder cancer: genomic insights and therapeutic promise. Clin. Cancer Res. 2015;21:4514–4524. doi: 10.1158/1078-0432.CCR-14-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindskrog SV, Prip F, Lamy P, Taber A, Groeneveld CS, Birkenkamp-Demtröder K, Jensen JB, Strandgaard T, Nordentoft I, Christensen E, Sokac M, Birkbak NJ, Maretty L, Hermann GG, Petersen AC, Weyerer V, Grimm MO, Horstmann M, Sjödahl G, Höglund M, Steiniche T, Mogensen K, de Reyniès A, Nawroth R, Jordan B, Lin X, Dragicevic D, Ward DG, Goel A, Hurst CD, Raman JD, Warrick JI, Segersten U, Sikic D, van Kessel KEM, Maurer T, Meeks JJ, DeGraff DJ, Bryan RT, Knowles MA, Simic T, Hartmann A, Zwarthoff EC, Malmström PU, Malats N, Real FX, Dyrskjøt L. An integrated multi-omics analysis identifies prognostic molecular subtypes of non-muscle-invasive bladder cancer. Nat. Commun. 2021 Apr 16;12(1):2301. doi: 10.1038/s41467-021-22465-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koutros S, Rao N, Moore LE, Nickerson ML, Lee D, Zhu B, Pardo LA, Baris D, Schwenn M, Johnson A, Jones K, Garcia-Closas M, Prokunina-Olsson L, Silverman DT, Rothman N, Dean M. Targeted deep sequencing of bladder tumors reveals novel associations between cancer gene mutations and mutational signatures with major risk factors. Clin. Cancer Res. 2021 Jul 1;27(13):3725–3733. doi: 10.1158/1078-0432.CCR-20-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellmunt J, Kim J, Reardon B, Perera-Bel J, Orsola A, Rodriguez-Vida A, Wankowicz SA, Bowden M, Barletta JA, Morote J, de Torres I, Juanpere N, Lloreta-Trull J, Hernandez S, Mouw KW, Taplin ME, Cejas P, Long HW, Van Allen EM, Getz G, Kwiatkowski DJ. Genomic predictors of good outcome, recurrence, or progression in high-grade T1 non-muscle-invasive bladder cancer. Cancer Res. 2020 Oct 15;80(20):4476–4486. doi: 10.1158/0008-5472.CAN-20-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Natesan D, Zhang L, Martell HJ, Jindal T, Devine P, Stohr B, Espinosa-Mendez C, Grenert J, Van Ziffle J, Joseph N, Umetsu S, Onodera C, Turski M, Chan E, Desai A, Aggarwal R, Wong A, Porten S, Chou J, Friedlander T, Fong L, Small EJ, Sweet-Cordero A, Koshkin VS. APOBEC mutational signature and tumor mutational burden as predictors of clinical outcomes and treatment response in patients with advanced urothelial cancer. Front. Oncol. 2022 Mar 7;12 doi: 10.3389/fonc.2022.816706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pittaway JFH, Lipsos C, Mariniello K, Guasti L. The role of delta-like non-canonical Notch ligand 1 (DLK1) in cancer. Endocr. Relat. Cancer. 2021 Oct 15;28(12):R271–R287. doi: 10.1530/ERC-21-0208. [DOI] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia ZN, Wang XY, Cai LC, Jian WG, Zhang C. IGLL5 is correlated with tumour-infiltrating immune cells in clear cell renal cell carcinoma. FEBS Open Bio. 2021 Mar;11(3):898–910. doi: 10.1002/2211-5463.13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X, Yang J, Chen Y. Identification of a four immune-related gene signature based on an immunogenomic landscape analysis of clear cell renal cell carcinoma. J. Cell. Physiol. 2020;235(12):9834–9850. doi: 10.1002/jcp.29796. [DOI] [PubMed] [Google Scholar]
- 32.Diaz-Montero CM, Rini BI, Finke JH. The immunology of renal cell carcinoma. Nat. Rev. Nephrol. 2020;16(12):721–735. doi: 10.1038/s41581-020-0316-3. [DOI] [PubMed] [Google Scholar]
- 33.Shen H, Liu J, Chen S, Ma X, Ying Y, Li J, Wang W, Wang X, Xie L. Prognostic value of tumor-associated macrophages in clear cell renal cell carcinoma: a systematic review and meta-analysis. Front. Oncol. 2021 Apr 26;11 doi: 10.3389/fonc.2021.657318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roufas C, Georgakopoulos-Soares I, Zaravinos A. Distinct genomic features across cytolytic subgroups in skin melanoma. Cancer Immunol. Immunother. 2021 Nov;70(11):3137–3154. doi: 10.1007/s00262-021-02918-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai Z, Zhao Y, Yang X, Wang L, Yin X, Chen Y, Lu J. A novel prognostic ferroptosis-related long noncoding RNA signature in clear cell renal cell carcinoma. J. Oncol. 2022 Feb 22;2022 doi: 10.1155/2022/6304824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peche LY, Ladelfa MF, Toledo MF, Mano M, Laiseca JE, Schneider C, Monte M. Human MAGEB2 protein expression enhances E2F transcriptional activity, cell proliferation, and resistance to Ribotoxic stress. J. Biol. Chem. 2015 Dec 4;290(49):29652–29662. doi: 10.1074/jbc.M115.671982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pattani KM, Soudry E, Glazer CA, Ochs MF, Wang H, Schussel J, Sun W, Hennessey P, Mydlarz W, Loyo M, Demokan S, Smith IM, Califano JA. MAGEB2 is activated by promoter demethylation in head and neck squamous cell carcinoma. PLoS One. 2012;7(9):e45534. doi: 10.1371/journal.pone.0045534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamada R, Takahashi A, Torigoe T, Morita R, Tamura Y, Tsukahara T, Kanaseki T, Kubo T, Watarai K, Kondo T, Hirohashi Y, Sato N. Preferential expression of cancer/testis genes in cancer stem-like cells: proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens. 2013 Jun;81(6):428–434. doi: 10.1111/tan.12113. [DOI] [PubMed] [Google Scholar]
- 39.von Boehmer L, Keller L, Mortezavi A, Provenzano M, Sais G, Hermanns T, Sulser T, Jungbluth AA, Old LJ, Kristiansen G, van den Broek M, Moch H, Knuth A, Wild PJ. MAGE-C2/CT10 protein expression is an independent predictor of recurrence in prostate cancer. PLoS One. 2011;6(7):e21366. doi: 10.1371/journal.pone.0021366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hodgson A, Jungbluth AA, Katabi N, Xu B, Downes MR. Evaluation of cancer testis antigen (CT10, PRAME) and MHC I expression in high-grade urothelial carcinoma of the bladder. Virchows. Arch. 2020 Apr;476(4):535–542. doi: 10.1007/s00428-019-02661-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Q, Xu WT, Shalieer T. Pilot study on MAGE-C2 as a potential biomarker for triple-negative breast cancer. Dis. Markers. 2016;2016 doi: 10.1155/2016/2325987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen X, Wang L, Liu J, Huang L, Yang L, Gao Q, Shi X, Li J, Li F, Zhang Z, Zhao S, Zhang B, Van der Bruggen P, Zhang Y. Expression and prognostic relevance of MAGE-A3 and MAGE-C2 in non-small cell lung cancer. Oncol. Lett. 2017 Mar;13(3):1609–1618. doi: 10.3892/ol.2017.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, Potts PR. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell. 2015;160(25679763):715–728. doi: 10.1016/j.cell.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang MAGE-A, MAGE-B, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in mage-positive cell lines. Cancer Res. 2007;67(17942928):9954–9962. doi: 10.1158/0008-5472.CAN-07-1478. [DOI] [PubMed] [Google Scholar]
- 45.Eikrem OS, Strauss P, Beisland C, Scherer A, Landolt L, Flatberg A, Leh S, Beisvag V, Skogstrand T, Hjelle K, Shresta A, Marti HP. Development and confirmation of potential gene classifiers of human clear cell renal cell carcinoma using next-generation RNA sequencing. Scand. J. Urol. 2016 Dec;50(6):452–462. doi: 10.1080/21681805.2016.1238007. [DOI] [PubMed] [Google Scholar]
- 46.Mendhiratta N, Muraki P, Sisk AE, Jr., Shuch B. Papillary renal cell carcinoma: review. Urol. Oncol. 2021 Jun;39(6):327–337. doi: 10.1016/j.urolonc.2021.04.013. [DOI] [PubMed] [Google Scholar]
- 47.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerød A, Oslo Breast Cancer Consortium (OSBREAC) Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van 't Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Børresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caso R, Sanchez-Vega F, Tan KS, Mastrogiacomo B, Zhou J, Jones GD, Nguyen B, Schultz N, Connolly JG, Brandt WS, Bott MJ, Rocco G, Molena D, Isbell JM, Liu Y, Mayo MW, Adusumilli PS, Travis WD, Jones DR. The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma. J. Thorac. Oncol. 2020 Dec;15(12):1844–1856. doi: 10.1016/j.jtho.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siraj S, Masoodi T, Siraj AK, Azam S, Qadri Z, Parvathareddy SK, Bu R, Siddiqui KS, Al-Sobhi SS, AlDawish M, Al-Kuraya KS. APOBEC SBS13 mutational signature—a novel predictor of radioactive iodine refractory papillary thyroid carcinoma. Cancers (Basel) 2022 Mar 21;14(6):1584. doi: 10.3390/cancers14061584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willett YM. Evolution of melanoma antigen-A11 (MAGEA11) during primate phylogeny. J. Mol. Evol. 2018;86(29574604):240–253. doi: 10.1007/s00239-018-9838-8. [DOI] [PubMed] [Google Scholar]
- 51.Su S, Gu Q, Xu A, Shen S, Liu S, Zhang C, Miao C, Qin C, Liu B, Wang Z. Genetic variations in MAGE-A11 predict the risk and survival of renal cell cancer. J. Cancer. 2019 Aug 27;10(20):4860–4865. doi: 10.7150/jca.32675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pineda CT, Potts PR. Oncogenic MAGEA-TRIM28 ubiquitin ligase downregulates autophagy by ubiquitinating and degrading AMPK in cancer. Autophagy. 2015;11(5):844–846. doi: 10.1080/15548627.2015.1034420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hou S, Sang M, Zhao L, Hou R, Shan B. The expression of MAGE-C1 and MAGE-C2 in breast cancer and their clinical significance. Am. J. Surg. 2016 Jan;211(1):142–151. doi: 10.1016/j.amjsurg.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 54.Pabst C, Zustin J, Jacobsen F, Luetkens T, Kröger N, Schilling G, Bokemeyer C, Sauter G, Atanackovic D, Marx A. Expression and prognostic relevance of MAGE-C1/CT7 and MAGE-C2/CT10 in osteolytic lesions of patients with multiple myeloma. Exp. Mol. Pathol. 2010 Oct;89(2):175–181. doi: 10.1016/j.yexmp.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 55.Colemon A, Harris TM, Ramanathan S. DNA hypomethylation drives changes in MAGE-A gene expression resulting in alteration of proliferative status of cells. Genes Environ. 2020;42:24–36. doi: 10.1186/s41021-020-00162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hassan S, Blick T, Thompson EW, Williams ED. Diversity of epithelial-mesenchymal phenotypes in circulating tumour cells from prostate cancer patient-derived xenograft models. Cancers (Basel) 2021 Jun 1;13(11):2750. doi: 10.3390/cancers13112750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Z, Wu H, Zhou H, Gu Y, Bai Y, Yu S, An R, Qi J. Identification of potential key genes and high-frequency mutant genes in prostate cancer by using RNA-Seq data. Oncol. Lett. 2018 Apr;15(4):4550–4556. doi: 10.3892/ol.2018.7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, Kim S, Gabriel SB, Lander ES, Fisher S, Getz G. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic. Acids. Res. 2013 Apr 1;41(6):e6. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and analyzed are available on reasonable request.