

Abstract
The late-stage functionalization of peptides and proteins holds significant promise for drug discovery and facilitates bioorthogonal chemistry. This selective functionalization leads to innovative advances in in vitro and in vivo biological research. However, it is a challenging endeavor to selectively target a certain amino acid or position in the presence of other residues containing reactive groups. Biocatalysis has emerged as a powerful tool for selective, efficient, and economical modifications of molecules. Enzymes that have the ability to modify multiple complex substrates or selectively install nonnative handles have wide applications. Herein, we highlight enzymes with broad substrate tolerance that have been demonstrated to modify a specific amino acid residue in simple or complex peptides and/or proteins at late-stage. The different substrates accepted by these enzymes are mentioned together with the reported downstream bioorthogonal reactions that have benefited from the enzymatic selective modifications.
Keywords: Amino acids, Biocatalysis, Bioorthogonal reactions, C–H activation, Enzyme catalysis
Graphical Abstract
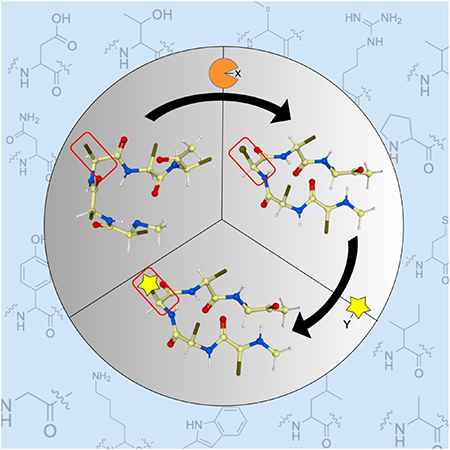
1. Introduction
Late-stage functionalization (LSF) has gained popularity in the last few years compared to early-stage bond activation and there are excellent reviews that discuss the general concept of LSFs.[1–3] LSF allows the direct installation of chemical functionalities on molecules leading to the generation of compound libraries without the need for de novo synthesis requiring multiple synthetic approaches with tirelessly work-up and purification steps for different derivatives. Within this context, site-selective LSF of peptides and proteins has attracted great attention.[4–6] Installation of chemical moieties on peptides and proteins expands their chemical space, alters their biological activities or receptor selectivity, and/or fine-tunes their physical and chemical properties. Furthermore, LSF allows the attachment of drugs to antibodies or proteins for targeted drug delivery and can facilitate bioorthogonal chemical reactions (Figure 1).[7–9] The latter are covalent organic reactions that are performed under aqueous and mild biological conditions to modify biomolecules with exogenous functional groups including fluorophores or affinity tags. These bioorthogonal reactions facilitate the study and manipulation of peptides via imaging and mapping of their cellular localization.[10,11] The most common bioorthogonal reactions utilize azide or alkyne handles which have the advantage of being small in size and uncharged with minimal effect on the properties of the targeted molecule.[12] The installed azides/alkynes can then be modified by copper-catalyzed or copper-free click chemistry via affinity handles or fluorescent dyes enabling visualization or enrichment of the desired molecules (Figure 1). Another popular bioorthogonal process is the inverse electron demand Diels–Alder (IEDDA) reaction that relies on the interaction between tetrazines and electron-rich alkenes (Figure 1). Thus, LSF paves the way for success in drug discovery and development and enhances biological research.
Figure 1.
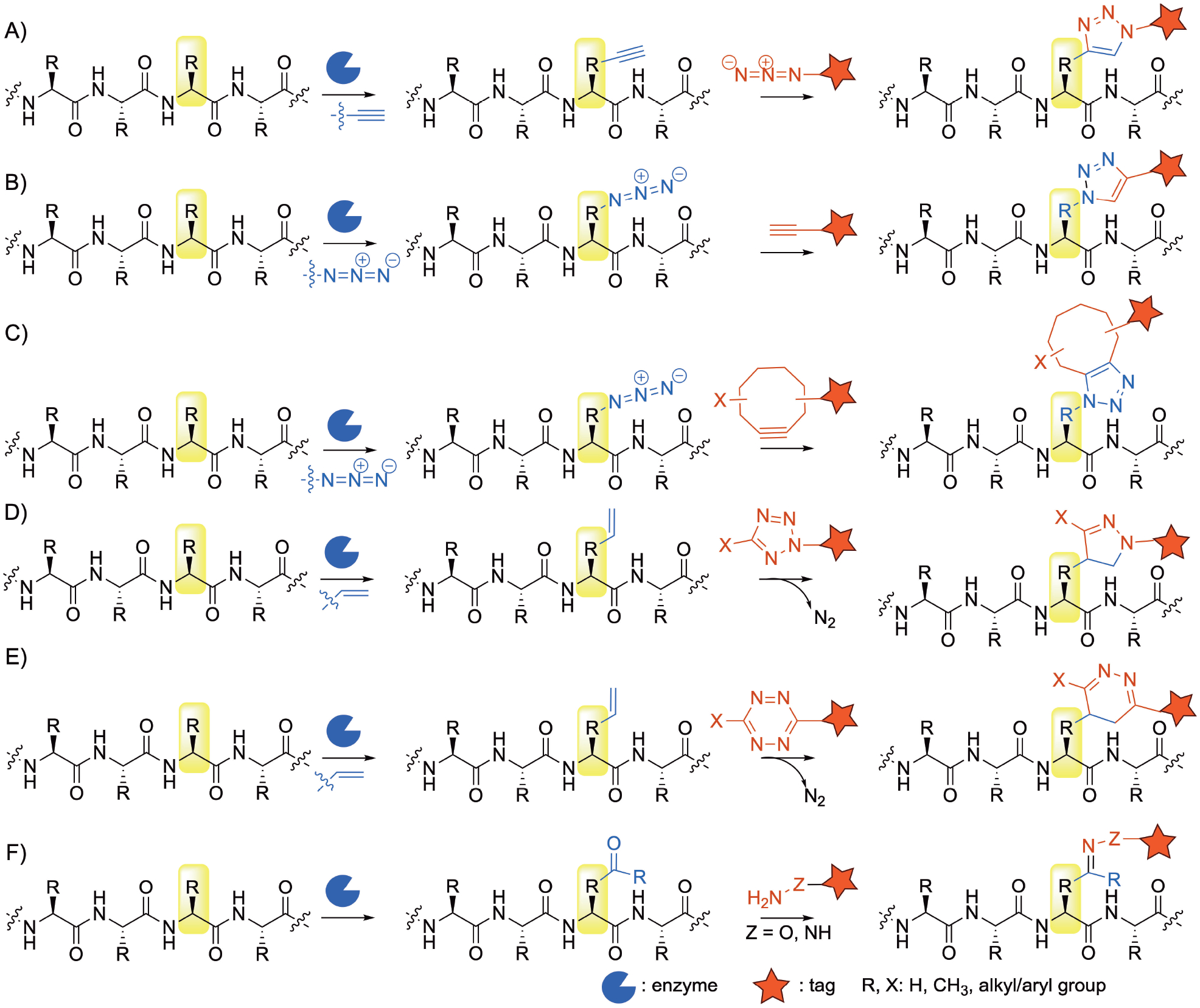
Examples of labelling and tagging reactions performed on peptides after enzymatic modifications. A) and B) copper catalyzed azide-alkyne cycloaddition (CuAAC); C) Strain promoted azide-alkyne cycloaddition (SPAAC); D) tetrazole alkene/alkyne cycloaddition; E) inverse electron-demand Diels–Alders (IEDDA) reaction; F) carbonyl-dependent oxime/hydrazone ligation reactions.
Site-selective LSF of peptides is associated with challenges to maintain specificity due to the presence of i) similar reactive groups that are prone to modifications; ii) multiple C–H bonds with similar chemical environments impacting dissociation energy; iii) stereocenters that are liable for racemization; iv) requirement of mild or physiological conditions when dealing with unstable peptides or biomolecules or the need to perform bioorthogonal reactions. Nevertheless, several groups have developed chemical,[13–16] enzymatic,[17] or photocatalytic[18] approaches to achieve LSF of peptides that have been reviewed extensively.
Enzymes are present in all walks of life to perform functions for the producing organisms. Some are responsible for protein posttranslational modifications or involved in signaling pathways while others are encoded in the biosynthetic gene clusters of natural products.[19,20] They have been shown to catalyze reactions in nature that would be difficult to achieve using traditional synthetic methods. Enzymes perform their reactions under physiological pH, temperature, and pressure in aqueous conditions in a regio-, chemo-, and stereoselective manner and thus can be exploited and repurposed to achieve desired reactions as precise and efficient biological catalysts. Wild-type enzymes or engineered variants can be overproduced in heterologous hosts and used in in vitro reactions. Many enzymes have shown broad substrate flexibility enabling them to be utilized to functionalize nonnative substrates. Because of their selectivity, they do not require protection/deprotection steps or the use of toxic solvents or transition metals.[21] These advantages make enzymes catalyze the synthesis of variable steps in an efficient way, in fewer steps, more scalable fashion, and at a lower cost. Moreover and owing to their reactivity under physiological reaction conditions, enzymes can be used to achieve bioorthogonal chemical reactions.[22] Thus, it seems logical to leverage the machinery that Nature has developed to achieve peptide LSF.
The use of enzymes to achieve LSF of peptides requires the availability of studies that demonstrate their activity and amino acid residue specificity, as well as their substrate scope and whether it needs a recognition sequence. Different enzymes have been shown to be selective in modifying a specific amino acid present in a complex peptide or protein despite the presence of multiple functional groups (Table 1). While enzymes are known to be specific to their substrates, several have been reported to have broad substrate tolerance allowing them to functionalize nonnative peptides in a regio- and chemoselective manner. In this Review, we highlight the enzymes reported to modify small or complex peptides and/or proteins that target specific canonical amino acid residues or specific to one or both of the termini and have relaxed substrate flexibility enabling them to modify nonnative peptides. We also highlight any reported nonnative substrates and the applications that utilized the selective enzymatic modifications.
Table 1.
Amino acid residue-specific modifying enzymes mentioned in this Review and their native functions. Nonnative substrates and downstream bioorthogonal reactions are mentioned in detail in the text and figures.
| Amino acid residue | Modifying Enzyme | Native function |
|---|---|---|
| Ala | N/A | N/A |
| Arg | protein Arg methyltransferases | mono- and dimethylation of the guanidino group |
| 2-oxoglutarate oxygenases YcfD | oxidation of the Arg Cβ carbon | |
| Asp/Asn | peptide Asp ligases/Asn endopeptidases | ligation and/or hydroylsis of Asp/Asn-containing peptides, some catalyze macrocyclization |
| Cys | farnesyltransferases/geranylgeranyltransferases | transfer of farnesyl/geranylgeranyl group to Cys |
| formyl-generating enzymes | converts Cys thiol group to formaldehyde | |
| cyclodehydratases YcaO family | cyclodehydration and formation of thiazoline ring | |
| LanM enzymes | formation of thioether link with dehydrated Ser/Thr | |
| Glu | N/A | N/A |
| Gln | transglutaminases | ligation with Lys-containing peptides or compounds with primary amines conversion of Gln to Glu |
| Gly | N-myristoyltransferases | transfer of myristoyl group to the N-terminus of Gly |
| sortase A | hydroylsis and ligation of peptides containing Thr-Gly | |
| His | 2-oxoglutarate oxygenases (NO66 and MINA53) | hydroxylation of the His Cβ carbon |
| LimF prenyltransferase | C-prenylation of the imidazole ring | |
| Ile | N/A | N/A |
| Leu | α-ketoglutarate dioxygenase, Mur15 | Cβ-hydroxylation of Leu in muraymycins-type peptides |
| Lys | transglutaminases | ligation with Gln-containing peptides |
| N-myristoyltransferases | transfer of myristoyl group to the ε-NH2 of Lys at the N-terminus | |
| Lys acyltransferases | transfer of acetyl group to the Lys ε-NH2 | |
| Lys methyltransferases | transfer of methyl to the Lys ε-NH2 | |
| biotin ligase | transfer of biotin to the Lys ε-NH2 | |
| lipoic acid ligases LplA | transfer of lipoyl group to the Lys ε-NH2 | |
| Met | Met aminopeptidases | cleavage of the Met at N-terminus of peptides |
| Phe | N/A | N/A |
| Pro | tyrosinase abTyr | attaches Tyr to N-Pro and oxidizes it to O-quinones |
| Ser | formyl-generating enzymes | converts Ser side chain to formaldehyde |
| Ser/Thr | O-linked N-acetylglucosamine transferase | transfers Glc-NAc to OH side chain |
| prenyltransferase TolF | transfers prenyl group to OH side chain | |
| cyclodehydratases YcaO | forms oxazoline and methyloxazoline rings | |
| LanM enzymes | dehydration of Ser/Thr before forming thioether link with Cys side chain | |
| Thr | flavin transferase ApbE | transfers a flavin group to the Thr hydroxy group |
| Trp | flavin-dependent Trp halogen- ases | installs halogen on the C-2 |
| indole prenyltransferases | transfers prenyl groups to the Trp indole | |
| hydroxylase BhaQ | converts the benzene ring of Trp to o-hydroxy-p-quinone | |
| C-mannosyltransferse | catalyzes C-mannosyl glycosylation to the Trp indole ring | |
| Tyr | tyrosinase/catechol O-meth- yltransferase | alkoxylate a Tyr group |
| F-prenyltransferases | transfer C5 and C10 prenyl moieties to the O-Tyr | |
| glycosyltransferase GtfE | transfers sugars to Tyr in vancomycins and teicoplanin-related peptides | |
| Val | N/A | N/A |
| N-terminus | N-methyltransferases (Mtfadbv and Mtfapek) | catalyze N-terminus methylation of teicoplanin-like heptapeptides |
| protease PatA | cleaves peptides following a specific recognition sequence | |
| ligase subtiligase | ligation between the N at the N-terminus of a peptide and an ester/thioester peptide | |
| Met and Pro aminopeptidases | excision of Met and Pro in proteins containing Met-Pro-Cys at the N-terminus to expose reactive Cys | |
| C-terminus | tubulin tyrosine ligase | recognizes a tubulin tag sequence at the C-terminus of a protein and installs Tyr-derivatives |
| N-/C-terminus | dual prenyl and meth- yltransferase AgeMTPT | transfers prenyl and methyl group to the N- and C-terminus of peptides, respectively |
| ligase trypsiligase | hydrolyzes peptides with the Tyr-Arg-His tag at the N- or C-terminus between Tyr and Arg and ligates a tag with a guanidinophenyl ester or Arg-His moiety | |
| Cyclization | ligase Omniligase-1 | ligation and cyclization of peptides |
| cyclodehydratase YcaO (PatG) | N–C macrocyclization | |
| macrocyclase PsnB | cyclization between Thr and Glu | |
| macrocyclase PCY1 | macrocyclization of peptides with recognition sequence | |
| cytochrome P450 OxyA-C | catalyze oxidative crosslinking | |
| subtiligase | macrocyclization of peptides containing 12–31 amino acids |
2. Alanine
No enzyme with broad substrate flexibility targeting Ala in peptides was reported.
3. Arginine
Protein Arg methyltransferases (PRMTs) play a major role in cellular events including signal transduction, protein interactions, and gene transcription in prokaryotes and eukaryotes. They catalyze mono- (type III) or asymmetric (type I) or symmetric (type II) dimethylation of the guanidino side chain of arginine in peptides and proteins using S-adenosyl-l-methionine (SAM)[23] (Figure 2A) with reports of successful switch of methylation pattern via rational design.[24] In addition to the type of PRMT, the recognition sequence might also affect the methylation pattern.[25] There are nine families of PRMTs that differ in their Arg-containing recognition sequence preferences and specificities. Many PRMTs recognize the RGG motifs,[26] some prefer RxR surrounded by Lys residues,[27] while others modify Arg in a PGM-rich environment[28] with several exceptions.[29] Yet, PRMTs also differ in their promiscuity towards accepting peptides or proteins. PRMTs were also found to accept several SAM analogs. For example, N-mustard analogs functionalized with azides and alkynes (Figure 2i) were used to probe the Arg in the peptide AcH4–21, Ac-SGRGKGGKGLGKGGAKRHRKV[30] and more complex proteins (Figure 2A).[31] The installed azide and alkyne groups were conjugated with biotin via copper-catalyzed click chemistry to enable visualization and isolation of histone H4.
Figure 2.
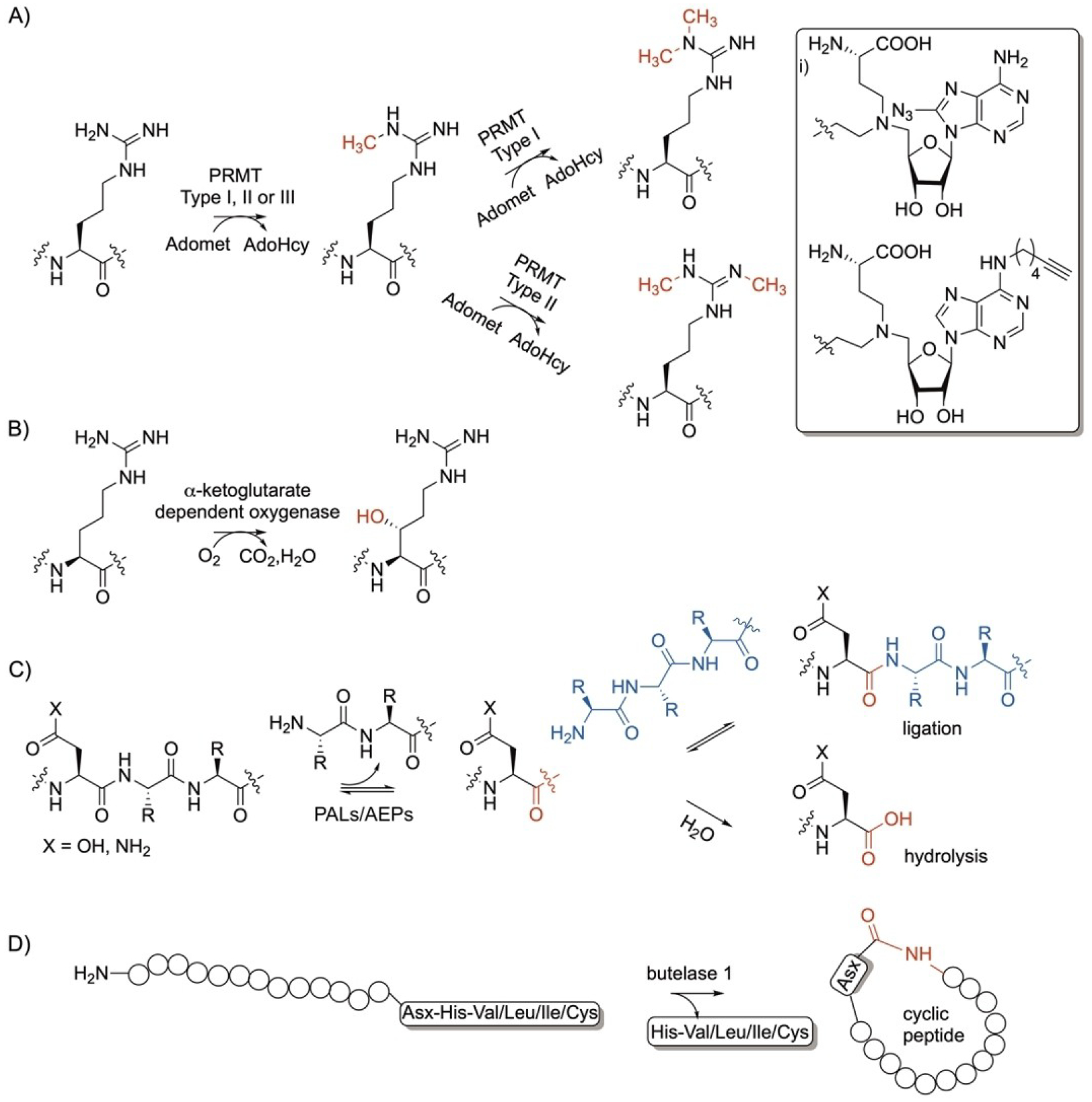
Arg and Asx-specific enzymatic modifications. A) protein Arg methyltransferases (PRMTs) and B) α-ketoglutarate dependent oxygenase; C) peptide Asp ligases (PALs)/ Asn endopeptidases (AEPs) and D) butelase 1. i) Nonnative moieties used to install azide and terminal alkyne groups by PRMTs.
The microbial 2-oxoglutarate oxygenase, YcfD, is a ribosomal hydroxylase in E. coli that recognizes the Arg residue in Arg-containing peptides and proteins. YcfD was shown to hydroxylate the Arg Cβ with a stereochemistry of 2S, 3R in a series of peptides ranging from hexa- to hexadecaptides, in addition to proteins using oxygen (Figure 2B).[32]
4. Aspartate and Asparagine
Peptide Asparginyl Ligases (PALs) and Asn endopeptideases (AEPs) are cysteine proteases that catalyze ligation/hydrolysis of Asp and Asn-containing peptides.[33–35] These enzymes are responsible for the formation of the plant macrocyclic peptides, cyclotides that play a major role as antimicrobials and insecticidals. The enzymes form a thioester-linked acyl-enzyme intermediate which facilitates nucleophilic attack by the nitrogen of an N-terminus peptide or by water leading to ligation or hydrolysis, respectively (Figure 2C).[33,36] Examples of enzymes that favor ligation include butelase 1, VyPAL2, and OaAEP1b while those that favor hydroylsis include OaAEP1, butelase-2, MCoAEP1, and MCoAEP2.[37] The reactions are reversible and the product can be resued in the reverse directions. Some enzymes favor one reaction over the other which is proposed to be determined by their sequences. Presence of the marker ligase activity (MLA) or ligase-activity determinants (LAD) motifs can suggest that the enzyme favors ligation reactions. The presence of a truncated version of MLA or presence of a distant Gly gatekeeper residue in the MLA indicates ligase activty.[33,35] In addition, LAD1 where Trp/Tyr at the first position and Val/Ile/Cys/Ala at the second position with Gly/Ala at the first position of LAD2 favors ligation.[34] Structural analysis of enzymes that favored ligation has identified 16 key covarying residues and introduction of these residues into an enzyme switched the catalysis to favor ligation instead of hydrolysis.[35] Enzymes that favor hydroylsis recognize Asp or Asn followed by two amino acid residues at the C-terminus of a protein. The addition of a large excess of one of the ligation substrates can shift the reaction equilibrum towards ligation. Ligation reactions can also be favored when performed at slightly acidic pH.[38] The ligation reaction was recently made irreversible by designing the acyl donor peptide to have Gln attached to Asn and coupling the reaction with glutaminyl cyclase which quenches the Gln amide in a lactam.[39] Structure-guided protein engineering efforts have shown the capability to convert OaAEP1 and other enzymes with endopeptidase activity to efficient PALs.[40–42] Another approach to drive the equilibrium of the reaction towards ligation activity was performed using 2-formyl phenylboronic acid to react with the released dipeptide forming inert thiazolidine derivatives.[43] Recently, MCoAEP2 was found to act as an efficient PAL and AEP with broad substrate specificity.[44]
One example that belongs to this class is butelase 1. The enzyme was discovered from the tropical plant Bunga telang (Clitoria ternatea), family Fabaceae.[45] The native substrate of butelase 1 is the linear 31-residue kB1-NHV with a Asn-His-Val at the C-terminus sequence. It catalyzes the Asn ligation to the N-terminus of the polypepitde and subsequent amide bond formation and intramolecular cyclization (Figure 2D). Butelase 1 was found to be promiscuous that it can accept non-native peptides with Asn or Asp (Asx) at the C-terminus and either Leu, Ile, Val or Cys as the second amino acid after Asx (Figure 2D).[45] Butelase 1 has ligation, cyclization, and transamidation activities with high catalytic efficiencies and no apparent protease activity at neutral pH. Butelase 1 is able to accept d-amino acids[46] and able to catalyze ligation and intermolecular cyclization between multiple peptides ranging in size between 10 to more than 200 amino acids.[47] The enzyme has reversible activity which has been overcome by the use of high peptide to enzyme ratio or the use of thiodepsipeptides.[48] Butelase 1 enabled the N-terminus labelling with functional groups such as biotin. Immobilization of butelase 1 as well as other PALs allows enzyme recycling and increases their stability and half-life enabling high enzyme concentration to be used which enhances PAL’s applications in basic and translational research.[49]
5. Cysteine
Mammalian and yeast protein farnesyltransferase (FTase) and geranylgeranyltransferase (GGTase) enzymes play major roles in all walks of life and are essential for the proper cellular activity and localization of several proteins. Interfering with the role of these enzymes has demonstrated roles in human health and diseases.[50–52] FTases and GGTases recognize a Cys in a C-terminus sequence called the CaaX box, where C is Cys, followed by aliphatic amino acids and an amino acid residue X that determines specificity towards FTases or GGTases.[50] FTases and GGTases transfer a C15 or C20 isoprenoid units, from farnesyl and geranylgeranyl pyrophosphates (FPP and GGPP) respectively, to the thiol side chain of Cys in peptides and proteins (Figure 3A).[53–56] These enzymes are promiscuous towards the peptide recognition sequence as they can also prenylate Cys in a tri- or pentapeptide sequence, Cxx or CxxxX located at the C-terminus of proteins.[57–60] They recognize standalone 3–5 amino acid residues in peptides naturally present or genetically-encoded in proteins. FTases and GGTases are also promiscuous towards their isoprenoid substrates and have shown to accept other non FPP and GGPP substrates (Figure 3i). For example, FTases accepted a library of anilinogeranyl[61,62] and phenoxygeranyl[62] diphosphate derivatives as well as branched FPP analogs.[61] FTases also accepted other FPP analogs with carbonyl groups,[63–65] photoswitchable azobenzene,[66] terminal azide,[67] alkyne,[65,67] or trans-cyclooctene[68] (Figure 3i) which allows them to undergo downstream oxime, hydrazone, cycloaddition, and tetrazine ligation reactions. FTases have facilitated protein tagging and conjugation to small[65,69] and complex[70,71] molecules in vitro and in vivo and enabled downstream click and Staudinger reactions specific to the installed handles.
Figure 3.
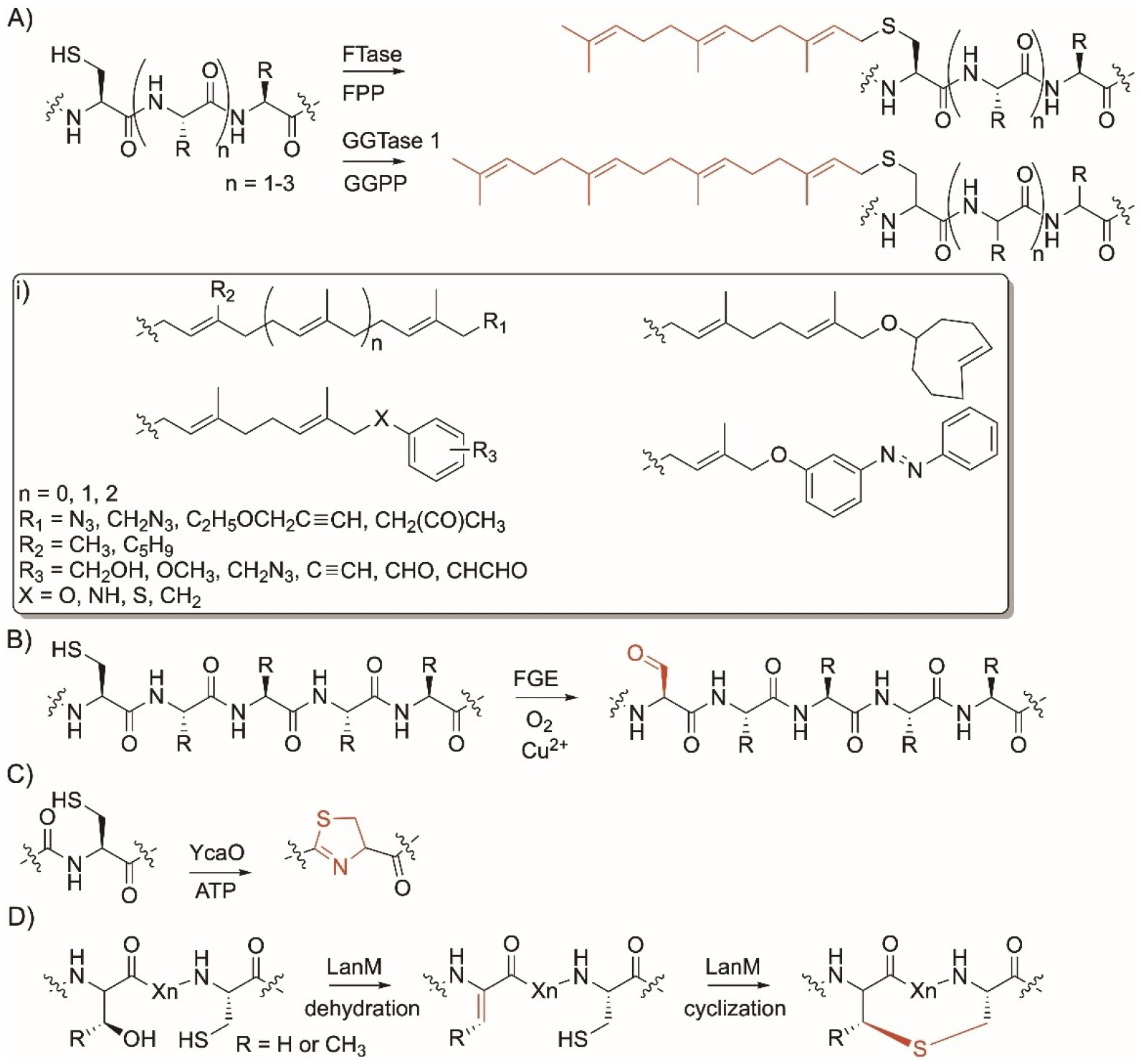
Cys-specific enzyme modifications. A) Reaction of farnesyl and geranylgeranyl transferases (FTases and GGTases, respectively); FPP and GGPP, farnesyl and geranylgeranyl pyrophosphate; B) reaction of aerobic formylglycine generating enzymes; C) reaction of cyclodehydratase YcaO superfamily; D) reaction of LanM enzymes. i) Nonnative moieties installed by FTases.
Aerobic formylglycine-generating enzymes (FGEs) are present in prokaryotic and eukaryotic organisms. They recognize Cys in the N- or C-terminus sequence CxPxR and introduce Cα-formylglycine (FGly) into proteins.[72,73] Aerobic FGEs catalyze the conversion of the thiol side chain of cysteine to an electrophilic aldehyde-bearing group via copper and oxygen-dependent catalysis (Figure 3B).[74–76] Their native function is to activate type-I sulfatases via oxidation and subsequent hydrolysis and their deficiency is associated with several diseases.[77,78] Different bacterial FGEs were found to be promiscuous in recognizing their Cys-containing peptide sequence and modified protein with the tag Cxxxx.[79] FGEs’ ability to install a bioorthogonal aldehyde functional group have allowed them to mediate ligation reactions[80] including trapped-Knoevenagel ligation[81] using a thiopyrazolone-maytansine and aminooxy-maytansine compounds or undergo a hydazino-iso-Pictet-Spengler[82] or reaction with hydrazide- or aminooxy-functionalized reagents.[83] They have been used to conjugate to small molecules as well as antibodies and enabled in vivo and cell-surface labelling and binding with fluorophore, polyethylene glycol, affinity tags, and glycans.[72,82–88] Engineering of the theromstable Thermomonospora curvata FGE enzyme (C187A and Y273F) improved catalytic efficiency 40-fold.[89]
YcaO superfamily enzymes are present in cyanobacteria and other bacteria. They are cyclodehydratases that catalyze the formation of thiazoline rings in Cys-containing ribosomal precursor peptides (Figure 3C).[90] Examples of YcaO enzymes that modify Cys include TruD,[91] PatD,[91] LynD,[92] 1SrpC,[93] and MprC.[94] YcaO enzymes have an N-terminus motif called RRE that binds to the recognition sequence (RS) elements in ribosomal substrate precursor peptides. The substrate peptide is designed such that its C-terminus contains a Cys-containing 7–8 amino acid core peptide sandwiched between RSs that act as a guide for YcaO enzyme activities. Different YcaO enzymes can bind to different RSs leading to formation of thiazolines.[95] The N-terminus of the substrate has a region that acts as a leader peptide usually with a L(S/T)EEXL sequence motif recognized by YcaO and was thought to be essential for activity. YcaO enzymes are promiscuous allowing them to incorporate different 5-membered rings into different peptides. Cys-containing peptides with the full or minimal leader sequence have been incorporated with thiazoline opening the door for introduction of new and active moieties into peptides.[96] PatD/TruD were also used to generate selenoazoles and selenoazoline-containing cyanobactins when using peptides where Cys was replaced with selenocystenine.[97] Engineered enzymes with minimal leader sequence facilitated the synthesis of new peptides. For example, the Schmidt group has generated swap PatD and TruD mutant constructs to identify the residues responsible for chemoselectivity between Cys and other residues. Their efforts have led to that development of a covalent protein TruD fused with RS1 capable of modifying Cys-containing large and small peptides.[98] An in vitro translation system that combines genetic code reprogramming and a YcaO enzyme was developed to convert Cys-containing precursor peptides to thiazoline, expanding the repertoire of thiazoline-containing artificial peptides.[99]
Enzymes that belong to the LanM family such as ProcM[100] from the cyanobacterium Prochlorococcus sp. and SyncM[101] from the cyanobacterium Synechococcus sp. are bifunctional enzymes involved in the biosynthetic gene clusters (BGCs) of the cyclic ribosomal peptides, class II lanthipeptides. In addition to dehydrating Ser/Thr, they catalyze the reaction between the thiol side chain of Cys and a dehydrated Ser or Thr via nucleophilic attack with the subsequent formation of lanthionine or 3-methyllanthionine, respectively (Figure 3D). LanM enzymes have low substrate specificity which allowed them to synthesize a diverse library of >80 lanthionine-containing polycyclic peptides.[102]
6. Glutamate
No enzyme with broad substrate flexibility targeting Glu in peptides was reported.
7. Glutamine
Microbial and mammalian transglutaminases (MTGase) catalyze the amide bond formation between the ɛ-amino group of Lys or a molecule with a primary amine and the γ-carboxyamine of Gln (Figure 4A).[103] They can also deamidate a protein Gln residue in the presence of water to form Glu (Figure 4B). Microbial transglutaminases have broader applications than mammalian ones because they have lower molecular weight, lower substrate specificities and do not rely on calcium and guanosine-5′- triphosphate.[104] They are used in food production to elongate shelf-life and improve quality. They can tolerate moderately higher temperatures and several organic solvents which have broadened their applications in industry.[105] The enzyme recognizes Gln in a sequence of LLQG called Q-tag and recognizes lysine in a MKHKGS sequence known as the K tag.
Figure 4.
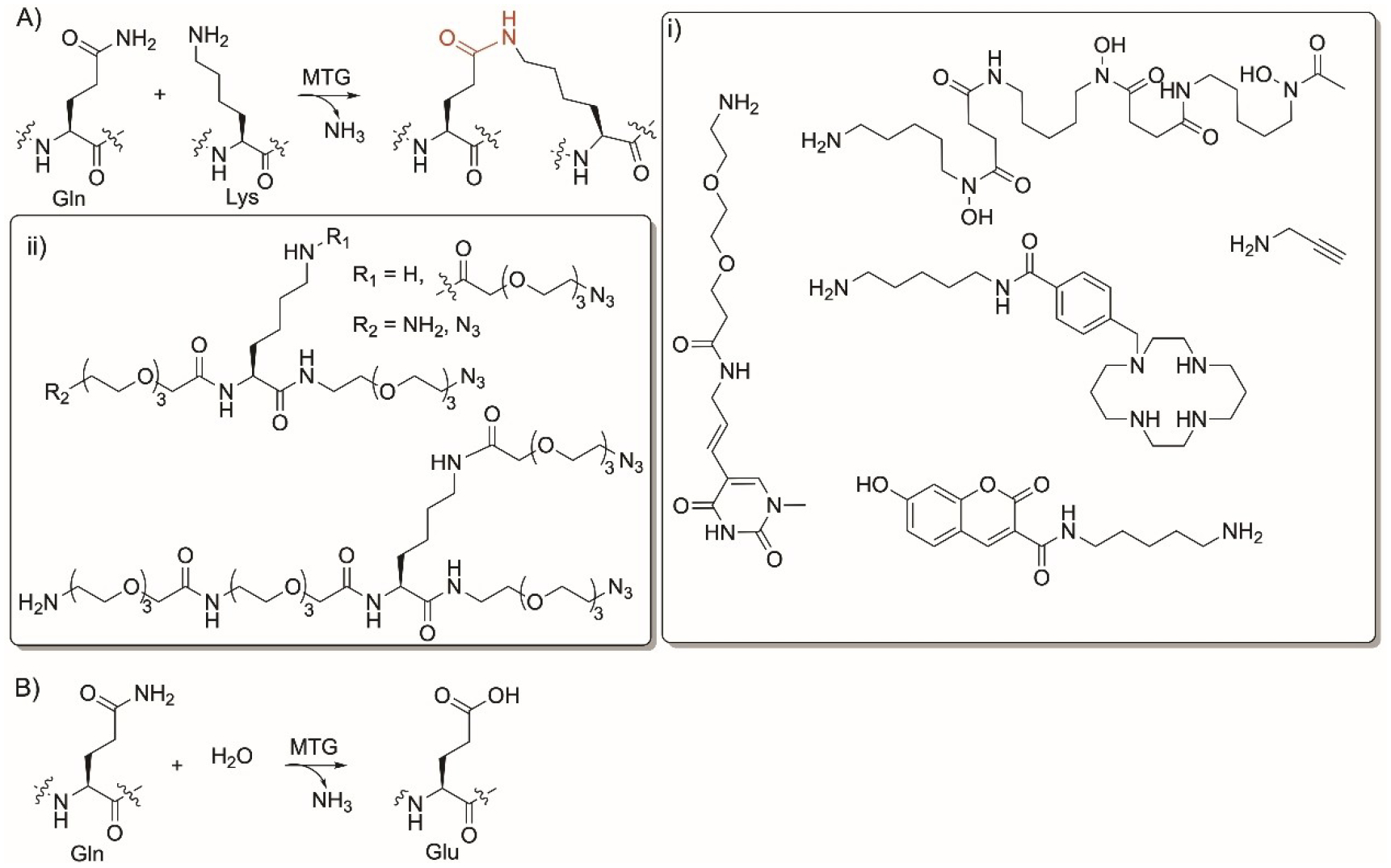
Gln-specific enzyme modifications. Reaction of microbial and mammalian transglutaminase (MTG) enzyme to A) ligate peptides and B) convert Gln to Glu. i) nonnative substrates accepted by MTGs to label Gln-containing peptides using alkyne- and azide-amine conjugation; ii) nonnative substrates used for PEGylation of Gln in peptides.
However, members of this class have shown broad substrate flexibility and can recognize Gln in the Q-tag as hexapeptides or as an introduced tag in peptides. Several MTGs have shown low substrate specificities towards their amine substrates enabling primary amines to crosslink to the Gln residue (Figure 4i). MTG has found several applications in protein and peptide labelling.[106] Different functional groups were added to proteins and peptides to act as bioorthogonal functional groups such as triazoles, tetrazines, terminal alkyne, and triphosphine (Figure 4i).[107,108] MTGs were used to facilitate protein lipidation,[106] PEGylation (Figure 4ii)[109,110] and labelling with oligonucleotides,[111,112] propargyl,[108,113] as well as fluorescence and biotin moieties.[114–117]
8. Glycine
N-myristoyltransferase (NMT) enzyme is ubiquitous in eukaryotes and is important for many biological processes such as immunity, tumorogenesis, and cellular differentiation.[118] NMT catalyzes the covalent attachment of a saturated C14 carbon chain, myristic acid, from a CoA-myristoyl thioester co-substrate to the nitrogen of a Gly residue at the N-terminus of a protein (Figure 5A).[119] NMT acylates Gly in the recognition sequence GXXXS/T(K) (Figure 5A). The reaction occurs via a nucleophilic attack of the N-terminus amine onto the carbonyl carbon of the myristoyl-CoA. While it has been accepted that the lack of side chain in Gly enables free rotation and facilitates the reaction, the free amino group of N-terminus Lys has also been shown to be modified by NMT.[120] ω-Azido fatty acids were used as alternative substrates (Figure 5i) by NMT allowing Staudinger ligation with phosphine-biotin.[121–123] This enabled visualization by streptavidin blotting and expands the bioorthogonal applications of NMT-catalyzed reactions.
Figure 5.
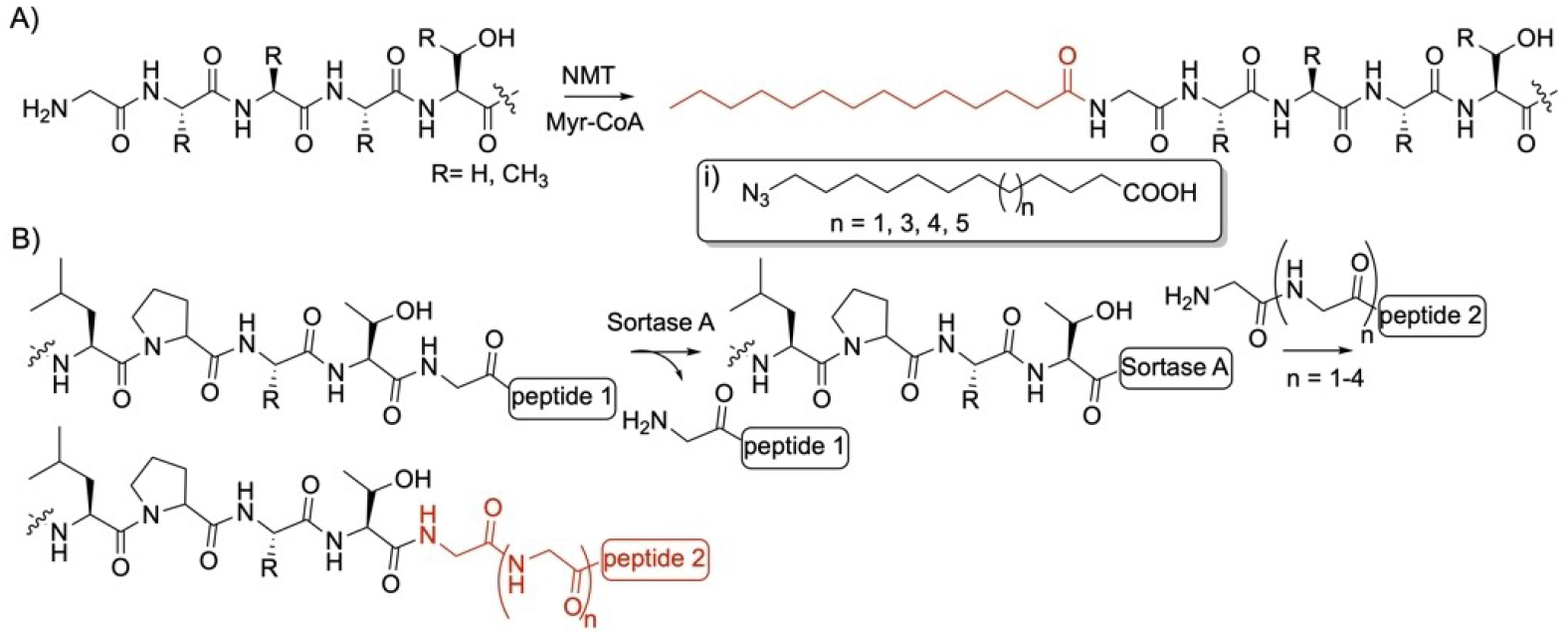
Gly-specific enzyme modifications. Reactions of A) N-myristoyltransferase (NMT) and B) sortase A enzymes. i) ω-azido fatty acids used as nonnative substrates by NMT.
Sortase A (SrtA) is a Ca2+-dependent transpeptidase found at the plasma membrane of Gram-positive bacteria to anchor surface proteins to the cell wall envelope. It recognizes the motif LPxTG in the C-terminus of a protein and through its Cys residue, forms a Thr-Cys thioester bond which leads to cleavage at the Thr residue and release of the Gly-containing peptide downstream of the Thr (Figure 5B).[124] The sortase-bound thioester can be ligated to the N-terminus amino group of a 2–5 Gly-containing peptide (Figure 5B). Other homologs of SrtA in other Gram-positive bacteria ligate proteins with motifs containing Ala or Ser in place of Gly.[124] Efforts have succeeded to discover and engineer enzymes with lower Km improving their catalytic activity.[125] A directed sortase evolution strategy has been used to generate variants with up to 22-fold increase in catalytic efficiency compared to the wild-type enzyme. SrtA was also utilized to probe intracellular contact using a PEG-lipid conjugate to a triglycine peptide followed by fluorescence labelling.[126] It has been shown to have broad substrate tolerance allowing it to modify peptidoglycan as well as its precursor lipid II.[127] SrtA has found applications in protein modifications and immobilization on surfaces and synthesis of protein polymer conjugates.[128]
9. Histidine
The human 2-oxoglutarate oxygenases, NO66 and MINA53, are linked to cancer and other diseases.[129] They are homologs to the Arg-specific, YcfD mentioned in section 3 (Figure 2B). These 2-oxoglutarate oxygenases hydroxylate the Cβ of His in a series of His-containing 19- and 20-mer peptides using O2 (Figure 6A).[32] The stereochemistry of the generated hydroxylated derivative is 2S, 3S. Both enzymes were able to hydroxylate a specific His residue in the ribosomal proteins, Rpl8 and Rpl27a, respectively.
Figure 6.
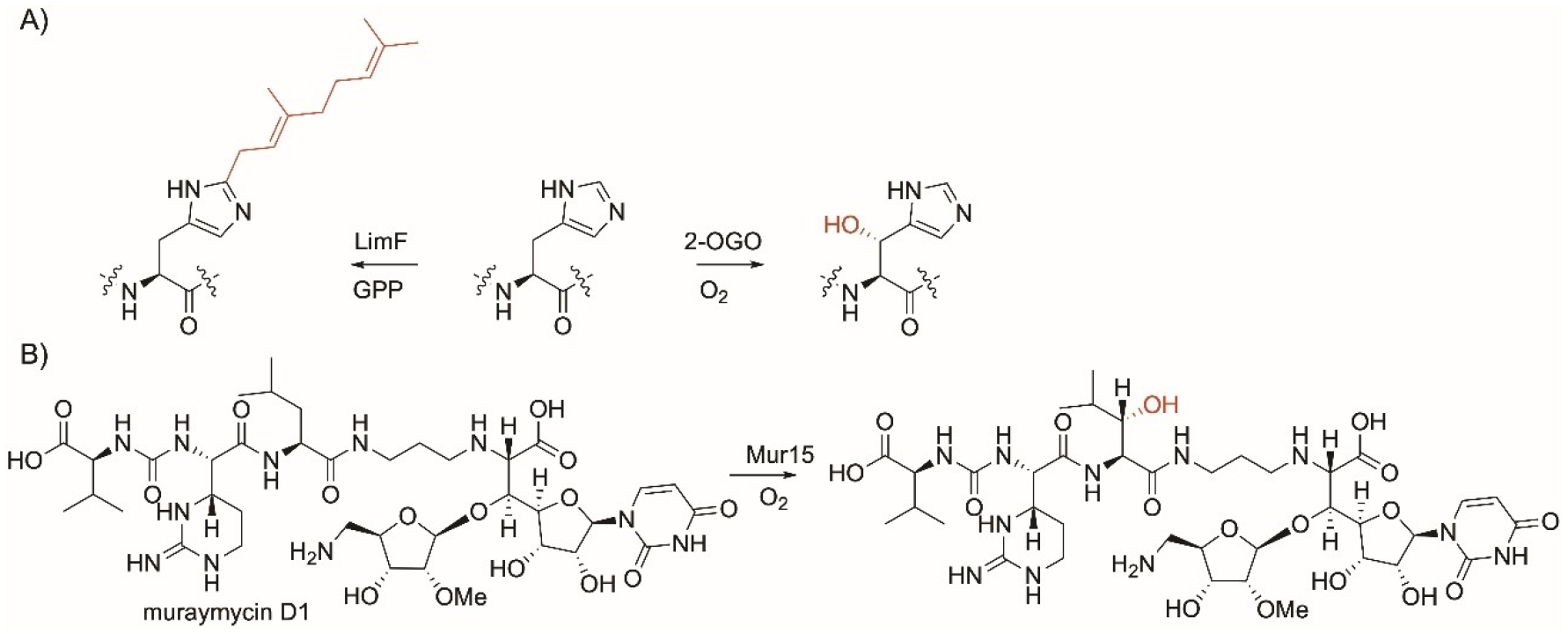
His and Leu residues-specific enzyme modifications. A) His-specific reactions showing 2-oxoglutarate oxygenase and LimF prenyltransferase; B) Leu-specific reaction showing the α-ketoglutarate dioxygenase enzyme, Mur15.
LimF in an enzyme involved in a cyanobactin BGC from the cyanobacteria Limnothrix sp.[130] LimF catalyzes C-1′ normal prenylation and the addition of a C10 geranyl group onto the C2 position of the imidazole side chain of His present in linear and thioether cyclic peptides. The enzyme was shown to geranylate the His-containing FDA-approved leuprorelin and cimetidine. Two LimF residues, His172 and Glu54, were found to be essential for the recognition and catalysis of the imidazole ring of His. LimF can also catalyze the Tyr-O-geranylation as a secondary function as indicated by the lower catalytic efficiency.
10. Isoleucine
No enzyme with broad substrate flexibility targeting Ile in peptides was reported.
11. Leucine
The hydroxylase Mur15 is encoded in the Streptomyces biosynthetic gene clusters responsible for the formation of the antibiotics muraymycins. It is a non-heme, Fe(II)-dependent α-ketoglutarate dioxygenase enzyme. Functional characterization showed that Mur15 catalyzes the late-stage Cβ-hydroxylation of Leu in Muraymycin D1 (Figure 6B). The enzyme was found to be promiscuous and able to hydroxylate Leu in other structurally-related peptides such as muraymycins B2, B6, A1, D2, and the deaminoribose of muraymycin D1.[131]
12. Lysine
Lysine is subjected to numerous enzymatic modifications due to the reactivity of the primary amine group.[132] In addition to MTGs mentioned in section 7 (Figure 4A) and NMT mentioned in section 8 (Figure 5A), other enzymes are specific to Lys. Lysine acetyl- or acytransferases (KATs, mark writers) are present in all walks of life and play a major role in protein posttranslational modifications. They modify histones and other proteins and play a role in many cellular processes such as DNA repair, regulation and transcription and have been involved in diseases.[133] Examples of KATs include GCN5, HAT1, PCAF, p300, CBP,[134] and the bacterial HBO1.[135] KATs catalyze the transfer of an acyl group from acyl coenzyme A (Ac-CoA) onto the ε-amino group of a Lys residue in a protein (Figure 7).[136,137] KATs are promiscuous towards their substrates and can use different acyl CoAs including fluoro,[138] chloro,[139] and other[140] substituted acetyl and benzoyl[135] substrates (Figure 7i), which allows labelling of and hijacking the KAT reactions. KATs were used to introduce alkynyl-acetyl and azidopropionyl groups (Figure 7i) to install bioorthogonal alkyne and azido groups respectively, onto lysine which enabled copper-catalyzed azide–alkyne cycloaddition and subsequent labelling with biotin tag.[141,142] This promiscuity facilitated the development of fluorescence-based assay for detection of KATs.[143] KATs also achieved Lys crotonylation (Kcr) facilitating reaction with carboxylic acid-containing bioorthogonal phosphines.[144] Engineering of KATs expanded the substrate profiles and enabled diverse acyl coenzyme A with terminal alkyne or azides to be accepted.[145,146] Recently, a platform based on thiol recycling was used to introduce bioorthogonal handles and isotopic labels into KAT.[147] Lysine deacetylases (KDACs, mark erasers) catalyze the reverse reaction of KATs and remove acetyl groups from Lys and together with KATs, fine-tunes the Lys acylation in cells. When combined with KAT, KDAC could unravel the biological effects of acetylation/deacetylation in living cells and the development of targeted therapeutics.[148]
Figure 7.
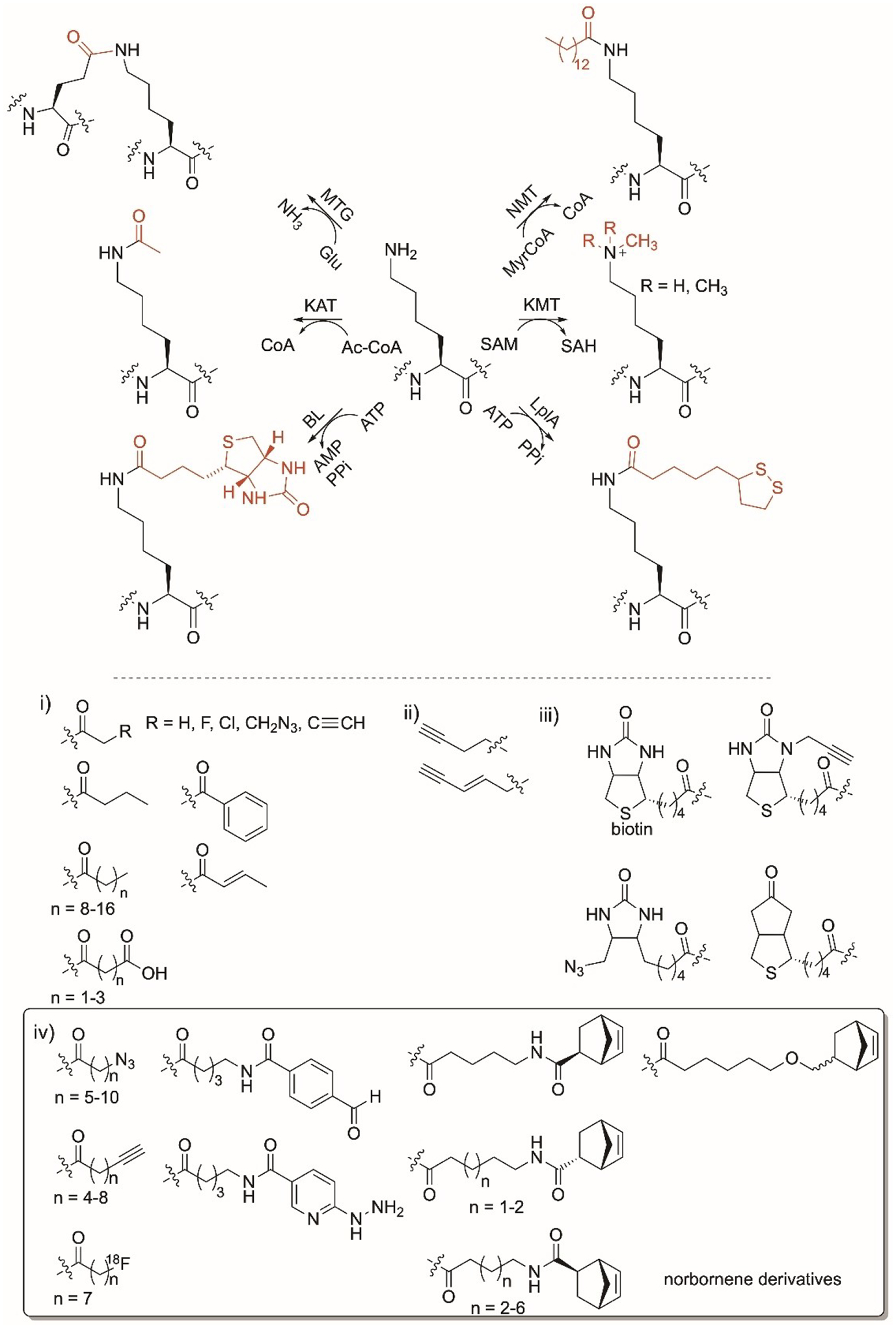
Lys-specific enzyme modifications. Reactions of transglutaminase (MTG), N-myristoyltransferase (NMT), lysine acyltransferase (KAT), lysine methyltransferase (KMT), biotin ligase (BL) and lipoic acid ligase (LplA) enzymes. Nonnative moieties installed by i) KAT; ii) KMT; iii) BL; iv) LplA ligase are shown. Norbornene substrates with >10% conversions are drawn.
Lysine methyltransferases (KMTs) play major roles in mammalian cell regulation as well as histone methylation and regulation of chromatin structure and gene expression and mutations in KMT are associated with cancer.[149] They transfer a methyl group from S-adenosylmethionine (SAM) to Lys in a peptide or a protein (Figure 7).[150] KMTs have been reported to cause mono-, di-, or trimethylation on the side chain of Lys. They are substrate specific, causing them to be an avenue for drug design as specific Lys residues can be targeted. Analogs of SAM have been engineered to replace the sulfur with selenium and replace the methyl group with bioorthogonal groups using chemical- and enzymatic-based approaches (Figure 7ii).[151–153] Installing propargyl on Lys enabled copper catalyzed azide-alkyne click chemistry.[154,155] Assays have been developed to detect KMT in vitro that rely on radio-[156] or fluorescence-[157] labelling.
Biotin ligase (BL) enzymes are present in prokaryotes and eukaryotes. They recognize Lys in a 15 amino acid sequence GLNDIFEAQKIEWHE, known as the acceptor peptide, at the N- or C-terminus of a protein.[17,158] BLs such as phBL, BirA, AaBL, and StBL catalyze the transfer of biotin to the Lys ɛ-amino group in an ATP-dependent manner (Figure 7). The Lys-containing acceptor domain can be appended to proteins of interest to facilitate biotinylation by these enzymes. BLs have been used to label proteins in cells allowing for strong fluoresence.[159,160] Biotin-derivatives carrying ketone, azide, and propargyl (Figure 7iii) were accepted by different BLs to site-specifically introduce phosphine-FLAG peptide onto proteins.[158,161] Methods that rely on wild-type and engineered BLs coupled with biotin were used to study protein-protein interactions in vitro and in vivo by fusing the first protein of interest to a BL and the second appended to an acceptor peptide and using streptavidin to measure biotinylation.[162–164]
Lipoic acid ligase (LplA) recognizes the GFEIDKVWYDLDA acceptor peptide sequence in a protein and catalyzes the ATP-dependent transfer of a lipoate group to the Lys side chain (Figure 7).[17] Engineering of microbial LplA enabled the attachment of lipoic acid analogs with terminal azido group (Figure 7iv) enabling conjugation with cyclooctene derivatives and fluorescent labelling.[165,166] Installation of a fluorooctanoyl moiety enabled protein radiofluorination[167] while the use of norbornene derivatives led to the formation of tetrazine fluorophores (Figure 7iv).[168] LplA variants catalyzed the installation of chemical handles including hydrazine and aldehyde aryl analogs (Figure 7iv) that enabled fluoresce labelling and Diels-Alder reaction with inverse electron demand.[169–171]
13. Methionine
Methionine aminopeptidases (MetAPs) are present in prokaryotes and eukaryotes. They catalyze the N-terminus Met excision of most proteins and peptides when the second amino acid is not bulky such as Gly, Ala, Ser, Cys, Pro, and with lower efficiencies if the second amino acid is Thr or Val (Figure 8A).[172,173]
Figure 8.
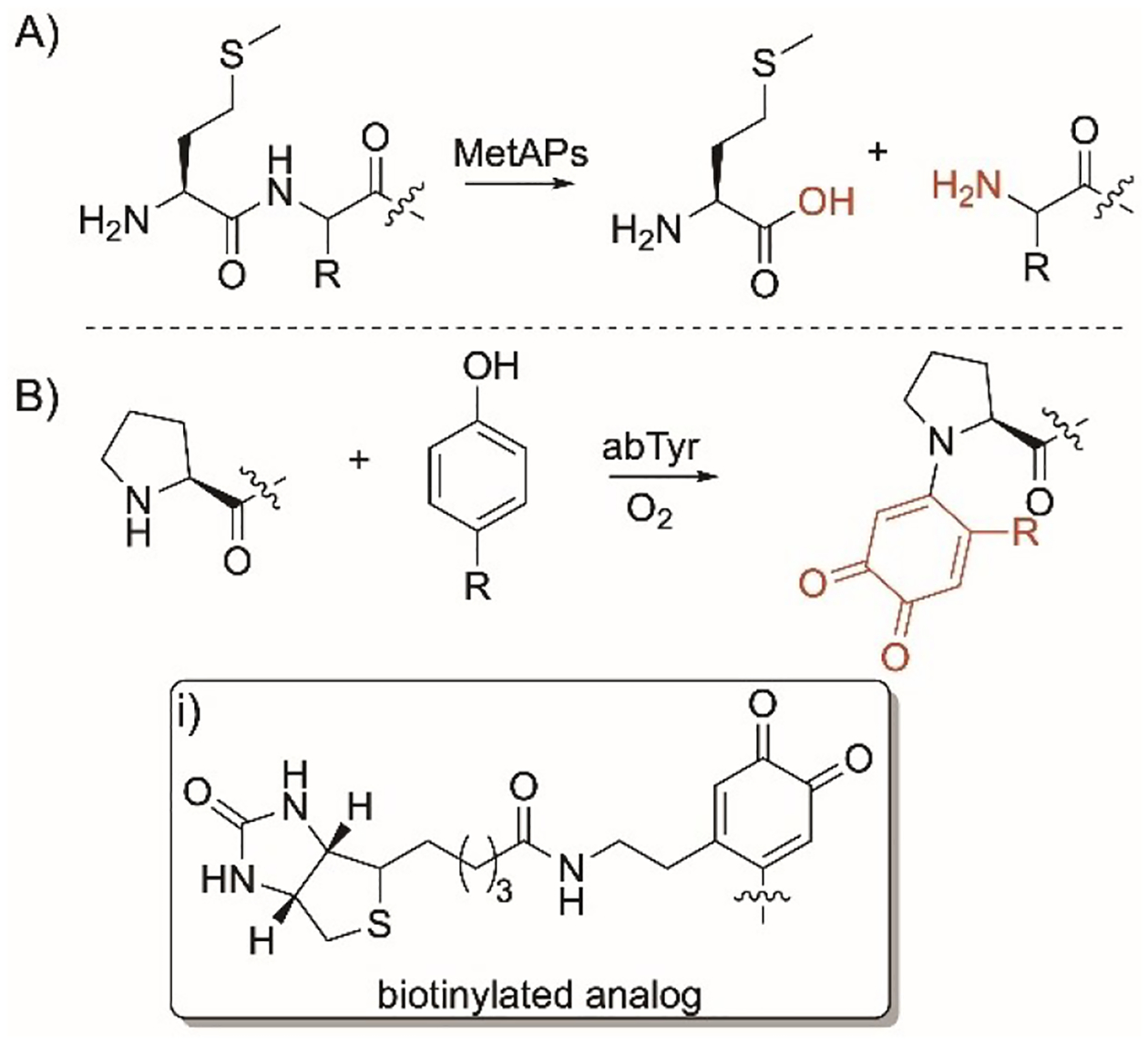
Met and Pro-specific modifications. Reactions of A) Met and B) Pro-specific enzymatic reactions. i) biotinylated group installed by abTyr.
14. Phenylalanine
No enzyme with broad substrate flexibility targeting Phe in peptides was reported.
15. Proline
The tyrosinase abTyr is a 120 kDa tetrameric enzyme that was isolated from the mushroom Agaricus bisporus.[174,175] It selectively modifies N-terminus Pro in proteins. It selectively by installing o-quinone intermediates on the nitrogen ring of Pro by reacting with phenols or catechols under aerobic conditions (Figure 8B). The desired phenols and catechols could be prepared by reacting N-hydroxysuccinamide and tyramine to form highly reactive intermediates that readily couple with the N-terminus Pro. This approach enabled the installation of o-quinone intermediates and the site selective biotinylation (Figure 8i) onto enzymes such as creatine phosphokinase and aldolase, self-assembled bacteriophage MS2 viral capsid, enzymes, and a chitin binding domain.[175] This approach showed more regioselectivity than alternative chemical methods.
16. Serine
In addition to oxygen-dependent FGE mentioned in section 5 with Cys (Figure 3B), anaerobic FGEs such as the prokaryotic AtsB from Methanosarcina mazei, can also cause Ser residue-specific modification. AtsB converts the hydroxy side chain of Ser to aldehyde using S-adenosyl methionine in an oxygen-independent manner (Figure 9A).[72,176] AtsB is an iron-sulfur protein that uses redox chemistry to generate the formylglycine moiety. In vitro experiments require the presence of strong reducing agents as sodium dithionite. AtsB modifies Ser in a minimal motif SXA/PXR in small peptides and large proteins which facilitated ligation with a HIPS-CF fluoroscent conjugate showing the application of this system in bioorthogonal chemistry.[176]
Figure 9.
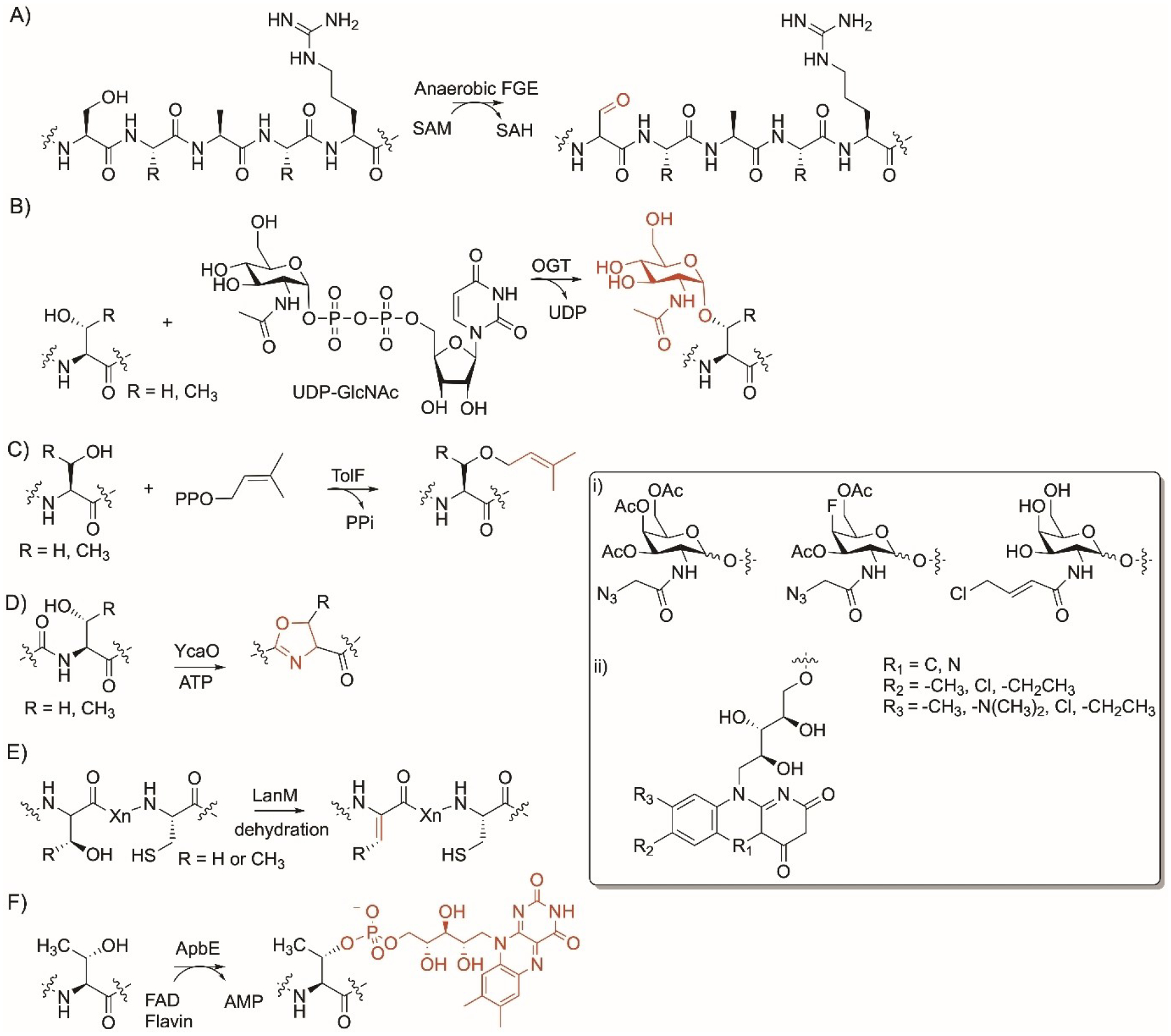
Ser and Thr residue-specific enzyme modifications. Reactions of A) anaerobic formylglycine generating enzyme (FGE); B) O-linked N-acetylglucosamine transferase (OGT); C) TolF prenyltransferase; D) cyclodehydaratase YcaO; E) ApbE flavin transferase. Nonnative groups accepted by the enzymes. i) OGTs and ii) ApbE.
17. Serine and Threonine
O-GlcNAcylation occurs in mammals, yeasts, bacteria, and viruses and are involved in several biological processes including the regulation of signal transduction, cell cycle, transcription, and protein synthesis and degradation with reports that abnormal patterns can lead to diseases in human.[177] O-linked N-acetylglucosamine transferase (OGT) is an enzyme that catalyzes the transfer of O-linked β-N-acetylglucosamine from the glycosyl donor uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc) to the hydroxy side chain of Ser and Thr in proteins (Figure 9B).[178] OGT is characterized by an N-terminus tetratricopeptide repeat (TPR) structural Asn-rich motifs and C-terminus two catalytic domains that form an active site for substrate binding. The TPR motif is folded into a super α-helix and is thought to generate unique binding sites. When these binding sites are occupied, a conformational change occurs to enable the target peptide or proteins to access the active site. Although there is not a strict typical recognition sequence, OGT is known to modify Ser and Thr residues that are flanked with Pro and β-branched amino acids to restrict flexibility.[179,180] In addition to large proteins, OGT also modified Ser-containing small peptides ranging from 9 to 26 amino acids.[180,181] OGT also accepted N-allylchloride and N-azido acetyl analog of UDP-GlcNAc which enabled colorimetric detection after treatment with phosphine-FLAG as well as a 4-fluoro-azido UDP-GlcNAc analog (Figure 9i).[181–183] This shows that OGT has substrate flexibility towards peptides and proteins as well as the UDP-sugar donors.
The prenyltransferase TolF is encoded in the gene cluster responsible for the formation of hexapeptide thiazoline-containing tolypamide from the cyanobacterium Tolypothrix sp.[184] It catalyzes the prenylation reaction of the hydroxyl group of Thr residue of the mature hexapeptide using dimethylallylpyrophosphate (DMAPP) (Figure 9C). In addition to Thr, TolF was found to prenylate Ser with less preference compared to Thr. TolF catalyzes only C-1′ normal (forward) prenylation and has a strict requirement for DMAPP (Figure 9C). The authors narrowed down a gate keeper residue Met232 that might result in accepting larger donors. Nevertheless, TolF was found to have relaxed substrate flexibility towards their acceptors and prenylate Thr- and Ser-containing short peptides.[184]
Similar to Cys in section 5 (Figure 3C), YcaO enzymes catalyze a synthetically challenging reaction via the introduction of a five membered ring into peptides. YcaO members as PatD modify Ser and Thr and convert them into oxazoline and methyloxazoline, respectively using ATP (Figure 9D).[185] Although most YcaO enzymes have a preference to modify Cys, several enzymes modify Ser and Thr at equal level or higher such as MprC[94] and TruD.[98]
As mentioned in section 5 (Figure 3D), LanM enzymes are bifunctional and can dehydrate Ser and Thr and form dehydroalanine and dehydrobutyrine, respectively, before forming thiol ether bridge.[100,101] These enzymes have low substrate specificity and can modify Ser/Thr present in different positions in peptides (Figure 9E).
18. Threonine
ApbE homologs are found in bacteria and are cofactors for the Na+-translocating NADH:quinone oxidoreductase.[186] It is Mg2+-dependent and transfers flavin from FMN to the Thr residues in the sequence DxxxxAT/S via a phosphoester covalent link (Figure 9F).[187] This sequence, called flavin tag, was used to label proteins with the flavin chromo- and fluorophore by genetically encoding the flavin tags at the N- or C-terminus or the loop region. This method facilitates purification and detection due to the fluorescence and yellow color of the flavin-labelled proteins.[187] The presence of FAD synthetase improves the efficiency of flavin incorporation and was used to label alcohol dehydrogenase, maltose-binding protein, and ubiquitinin-related SUMO.[188] ApbE accepts FMN-derivatives as well as nicotinamide and the bright-red, roseoflavin compound (Figure 9ii).
19. Tryptophan
Flavin-dependent tryptophan halogenases (FDHs) catalyze the installation of a halogen onto a Trp residue within a peptide leading to functionalization of the indole C–H bond (Figure 10).[189,190] Examples of FDHs include the microbial enzymes ThaI, PyrH, SttH, RebH, and PrnA. ThaI was found to halogenate short and long peptides without the need for carrier proteins.[189] The reaction relies on FADH2, oxygen, and halide salt while the presence of flavin reductase, PrnF, and excess NADH or phosphite dehydrogenase for NADH regeneration avoids cofactor build up and increases conversion (Figure 10). ThaI also halogenates d-trp-containing peptides including a derivative of the integrin ligand cilengitide. PyrH and SttH were reported to halogenate Trp in peptides containing 2–5 amino acid residues.[190] These 2–5 amino acids Trp-containing tags were attached to target proteins at the C-terminus and subsequently halogenated by wild-type PyrH as well as a more efficient engineered enzyme (PyrH_Q160N) enabling the introduction of new physicochemical properties to the protein.[190]
Figure 10.
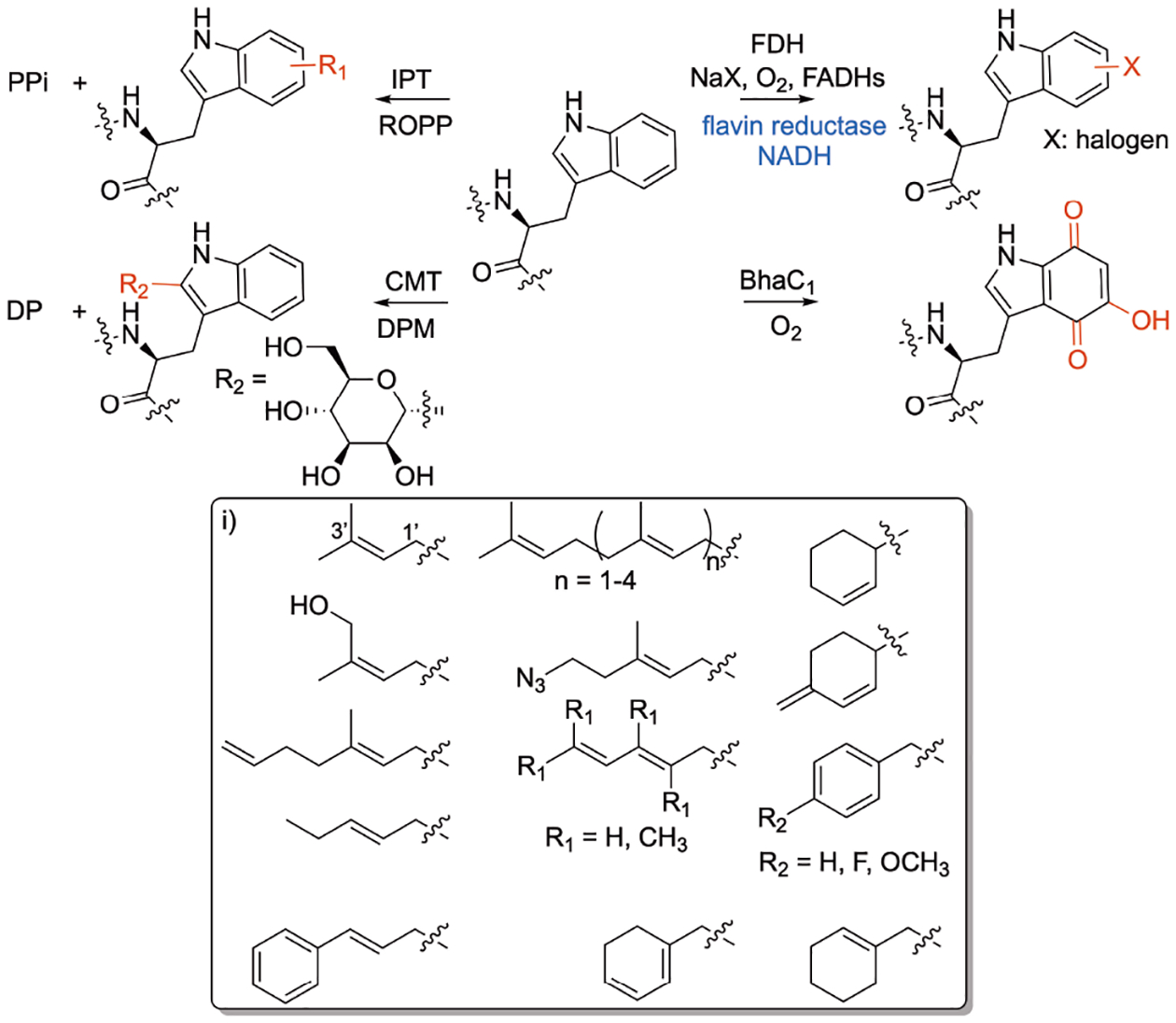
Trp residue-specific enzyme modifications. Reactions of flavin-dependent transferase (FDH), indoleprenyltransferases (IPTs), o-hydroxy-p-quinone-forming enzyme BhaC1, and C-mannosyltransferase (CMT). i) Non-native chemical moieties installed by IPTs on peptides at late-stage. DPM, dolichylphosphate mannose; DP, dolichylphosphate.
Indole prenyltransferases (IPTs) are enzymes present in many bacterial and fungal biosynthetic gene clusters. They catalyze the transfer of isoprenoid moieties into the indole moiety of Trp and other indole-containing compounds (Figure 10).[191,192] IPTs are dual substrate enzymes where they accommodate the acceptor, usually Trp or indole-derived molecule, and a donor isoprenoid diphosphates such as DMAPP, GPP, FPP, GGPP, and GFPP. Our lab[193,194] and others[195–197] have shown that IPTs have a wide substrate flexibility towards acceptor and donor substrates.[198] The release of the diphosphate moiety from donors generates a carbocation that is stabilized by positive charge delocalization.[199] This creates an electron deficient carbon at either C1′ or C3′ of the donor substrate leading to normal (forward) or reverse prenylation, respectively. IPTs differ in the degree of their substrate flexibility where several were found to accept non-native and synthetic pyrophosphates with multiple functionalities including alkyl, allylic, benzylic, anilino, and phenoxy geranyl analogs with aromatic or heteroaromatic moieties or terminal alkyne or azide groups (Figure 10i).[195,196,198,200] IPTs are also promiscuous towards their acceptor donors enabling them to modify Trp and Trp-containing-peptides. Our lab developed a chemoenzymatic approach to screen different IPTs with a library of Trp-containing peptides. We utilized this approach to develop regiospecific analogs of the FDA-approved calcium-dependent daptomycin antibiotic selectively modified at the Trp moiety.[193] The modified analogs exhibited improved antimicrobial properties against daptomycin-sensitive and daptomycin-resistant strains. Most importantly, in contrast to the parent daptomycin, the developed analogs did not rely on calcium for activity.[193] Moreover, our lab synthesized a substrate carrying a hydroxy-bearing allyl group (Figure 10ii). We selectively installed this moiety directly on the indole ring of Trp in Trp-containing peptides carrying 2–13 amino acids.[194] Recently, the Houssen group utilized AcyF coupled with diphosphate donors carrying a cyclohexene moiety to selectively modify Trp in a 10-mer peptide at the N1 position of the indole.[201] The installed handles were used to label the Trp moiety of the peptide with tetrazine–fluorescein via inverse-electron-demand Diels–Alder click reactions.
The regiospecificity (position of modification) of IPTs seems to be controlled by key catalytic residues such as Lys,[202] Gln,[203] Glu,[204] or His.[205] Three-dimensional structural analysis and engineering of IPTs have revealed insights into the promiscuity of several members of this class. The size of the active site as well as the presence and absence of specific residues control substrate specificity.[198,204,206] In addition, rational design of IPTs has led to the generation of variants with increased promiscuity,[207] change of regiospecificity,[203,205,208] or altered donor preference.[209] Mutation of the gateway residue from a bulky residue to Gly has allowed incorporation of larger donors.[209] Alteration of certain key residues has switched the donor preferences.[208,210,211] IPTs show great applications in late-stage functionalization and the conversion of C–H to new C–C and C–N bonds directly on the indole ring. When coupled with diphosphate donors, IPTs could serve as potential tools for orthogonal chemistry, labelling, and conjugation of peptides and proteins as well as the synthesis of therapeutically-important compounds.
BhaC1 is an enzyme encoded in the biosynthesis of pyrroloiminoquinone-type natural product in Alkalihalobacillus halodurans.[212] The enzyme installs three hydroxy groups onto the benzene ring of Trp which readily oxidizes to o-hydroxy-p-quinone moiety (Figure 10).[213] The enzyme has substrate flexibility towards its 20 mer peptide substrate as long as it ends with Ala-Trp. The first nine residues are important for enzyme recognition. This broad substrate flexibility was applied to maltose binding protein, resulting in a handle that can subsequently be used for bioconjugation. Peptide amino-acyl tRNA ligases were used to attach the recognition sequence into proteins allowing the formation of an electrophilic o-hydroxy-p-quinone handle and subsequent labelling of targeted proteins.[213]
Trp C-mannosyltransferase (CMT) enzymes are involved in function, transport, and folding of secretory and transmembrane proteins.[214] The C. elegans DPY-19 is a Trp CMT glycosylating enzyme. It catalyzes the C–C bond formation by transferring mannose from the dolichylphosphate mannose to the C2 indole position of first Trp in a WxxW/C sequon in proteins and peptides (Figure 10). Structural data of DPY-19 indicate that the mechanism of C-glucosylation proceeds through electrophilic aromatic substitution and that Glu71 seems to act as the catalytic base.[215]
20. Tyrosine
Members of the F prenyltransferase family, TruF1 (trunkamide), LynF (aestuaramide), and PagF (prenylagaramide) in the cyanobacterial ribosomal peptide pathways catalyze electrophilic alkylation of dimethylallyl moiety onto the hydroxy group of Tyr residues in peptides (Figure 11).[216] They have broad substrate tolerance and do not require a recognition sequence on the precursor peptide. They can achieve normal and reverse O-prenylation on the mature linear or cyclic peptides containing d- and l-Tyr up to 16 amino acids. The broad substrate specificity of this family was attributed to the truncation of the α/β barrel-fold observed in the F-PTs compared to other PTs. Most PTs have an occluded base due to the presence of a long α-helix or extended aromatic residue loop at the enzyme base. In contrast, the TruF superfamily has an open base resulting in a solvent-exposed channel.[216] The acceptor peptide binds at the base of the active site forming a deep encapsulated hydrophobic cavity that shields the allylic carbocation from solvent. These enzymes can catalyze Tyr in a cyclic peptide where PagF and LynF modify Tyr in the normal and reverse prenylation, respectively while TruF1 has a preference to catalyze Ser/Thr over Tyr. Similarly, PirF (piricyclamide pathway) can catalyze the addition of a C10 geranyl chain from GPP onto Tyr-containing linear and cyclic peptides (Figure 11).[217] The specificity of a C10 carbon chain of PirF was attributed to a presence of a less bulky amino acid residue Gly221 compared to other F-PT enzymes.[218] Moreover and as mentioned in section 9, LimF can catalyze O-geranylation as a secondary preference.[130] Noteworthy, the donor specificity of PagF towards C5 was switched to a C10 geranyl moiety upon a single amino acid switch of Phe220 to Ala or Gly. The broad substrate tolerance of the cyanobactin prenyltransferases towards acceptor substrates suggests they might have applications in biotechnology.[219]
Figure 11.
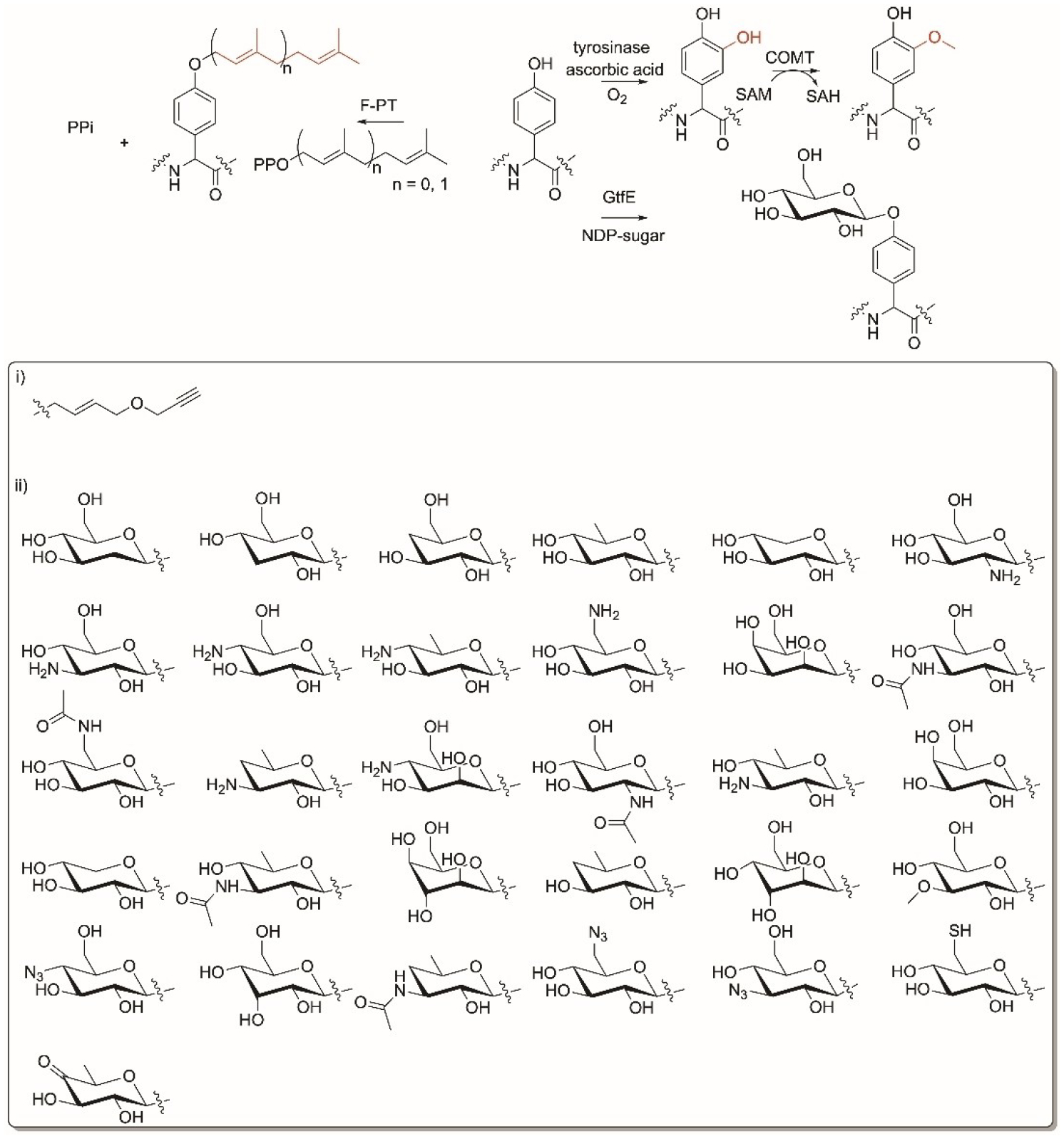
Tyr residue-specific enzyme modifications. Reactions of F-prenyltransferases, enzymatic cascade of tyrsoniase/catechol-O-methyltransferase (COMT) and glycosyltransferases (GT), GtfE. i) nonnative groups installed by COMT on the tyrosinase-introduced hydroxy group, ii) nonnative sugars accepted by GtfE.
The Micklefield group used an enzymatic cascade of two enzymes to selectively alkoxylate Tyr in peptides (Figure 11).[220] The cascade consists of a mushroom tyrosinase that introduces a hydroxyl group onto the aromatic ring to from a catechol and a mammalian catechol-O-methyltransferase (COMT) that methylates the installed hydroxy group using SAM (Figure 11). This tyrosinase/COMT dual system is selective for Tyr residues and has broad substrate tolerance. The system was used to alkoxylate and introduce a bioorthogonal terminal alkyne group using S-4-propargyloxybut-2-enyl analogue of AdoMet (Figure 11i) onto hexapeptides, goserin, oxytocin as well as small proteins such as tyrosine- N- and C-terminus inserted BtrI.[220]
Leloir glycosyltransferases (GTs) catalyze the attachment of monosaccharides onto acceptor molecules. Of those, the vancomycin aglycon GT from Streptomyces, GtfE, has shown broad substrate tolerance towards cyclic aglycone peptides as well as sugar donors (Figure 11).[221,222] GtfE was used to generate a library of vancomycin and teicoplanin antibiotics onto the O-Tyr residue when combined with different nucleotide diphospho sugars (NDP-sugars) (Figure 11ii). The glycoylated peptides were subjected to downstream chemoselective Huisgen 1,3-dipolar cycloaddition of azide installed on the sugar and alkynes to give the corresponding 1,2,3-triazoles in the presence of Cu+.[223,224] This reaction allowed the tagging of glycosylated peptides with fluorescent labels when reacting the azide with modified alkynes, dabcyl, and dansyl cadavarine propargyl imide.[223] The reversibility of GT-catalyzed reactions and development of efficient enzymes such as the OleD Loki variant, facilitated the formation of sugar nucleotides from aromatic carriers that can be coupled to the glycosylation of peptides aglycons.[225,226]
21. Valine
No enzyme with broad substrate flexibility targeting Val in peptides was reported.
22. N-Terminus Modifications
Several enzymes exist that modify peptide/protein N-terminus. The N-methyltransferases, Mtfadbv and Mtfapek, are found in the gene clusters of the glycopeptides A40926 and pekiskomycin, respectively.[227] Both enzymes catalyze N-terminus methylation of teicoplanin-like heptapetides bound or not bound to the NRPS (Figure 12A). Bicyclic peptides of teicoplanin aglycone were favored over linear and monocyclic intermediates. The enzymes were also promiscuous in attaching allyl and p-vinylbenzyl groups when using the corresponding Adomet substrates.
Figure 12.
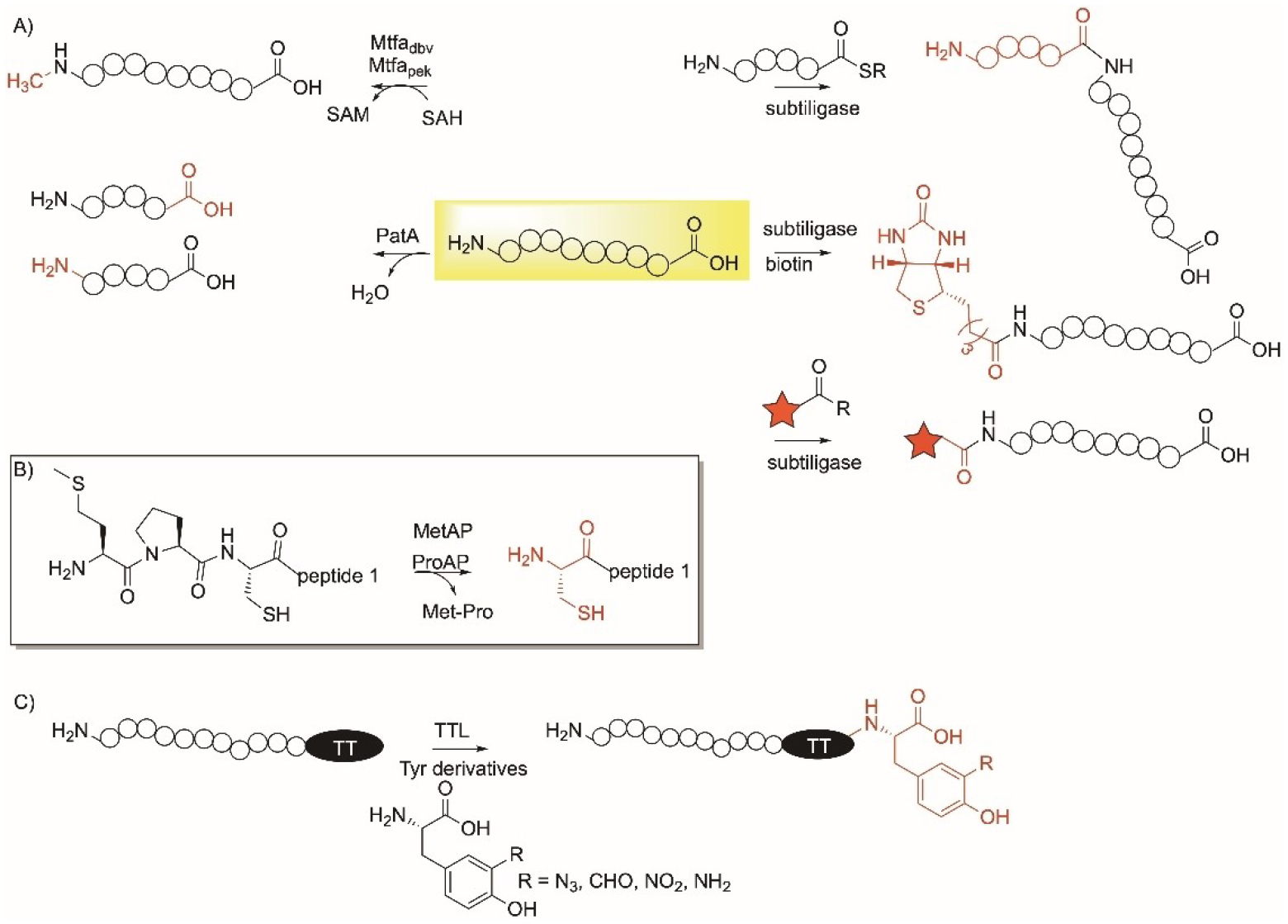
Enzymatic modifications specific to the N- and C-terminus of peptides and proteins showing A) the reactions of N-methyltransferases, Mtfadbv and Mtfapek, subtiligase, PatA protease and subtiligase-peptide ligation, biotinylation, and labelling; B) reaction of using methionine and proline aminopeptidase (MetAP and ProAP) enzymes in a N-terminus Met-Pro-Cys to expose Cys; C) reaction of α-tubulin tyrosine ligase (TTL) on the C-terminus of peptides/proteins containing tubulin tag (TT).
PatA enzyme from the patellamide pathway is a protease that can be used to catalyze N-terminus proteolysis (Figure 12A).[228] PatA is selective and able to directly cleave a peptide following a recognition sequence, including GVDAX, AVLAX, or GVEPX. The cleavage of the amide bond is not impacted by the methylation of nitrogen residues in the protein.
Subtiligase is a peptide ligase enzyme engineered from the promiscuous Bacillus serine protease, subtilisin.[229] Subtilisin was engineered to reduce the hydrolase activity and increase the ligase by creating a S221C/P225A subtilisin variant.[230,231] Subtiligase catalyzes a ligation reaction between the nitrogen of the N-terminus of a peptide and an ester/thioester peptide (Figure 12A). Furthermore, subtiligase catalyzed the attachment of other ester/thioester handles and the use of thioesters was found to be more efficient than ester peptides or substrates (Figure 12A).[232] A proteome-derived peptide libraries approach involving proteomic identification of ligation sites (PILS) led to the discovery of a family of 72 subtiligase mutants and identify their specificities.[231] Subtiligase variants-catalyzed reactions have enabled peptide and protein synthesis as well as bioconjugation with fluorescent tags and the formation of antibody-drug conjugates.
As mentioned in section 13, Met aminopeptidase (MetAP) is used to cleave Met from N-terminus peptides and proteins (Figure 13A). MetAP was coupled with Pro aminopeptidases, to cleave Met and Pro from an N-terminus protein.[233] Both enzymes were used to generate an N-terminus Cys protein when applied to peptides and proteins with the Met-Pro-Cys sequence at the N-terminus (Figure 12B). The method was advantageous due to the presence of Pro that protects Cys from undergoing many other reactions with endogenous enzymes. Once the proline was removed, downstream chemical labelling of the N-terminus Cys allowed ligation with a cell penetrating peptide and fluorescein. This was demonstrated using a variant of the Ras-binding domain of c-Raf, RBDV, but shows promise in eukaryotic proteins as well.
Figure 13.
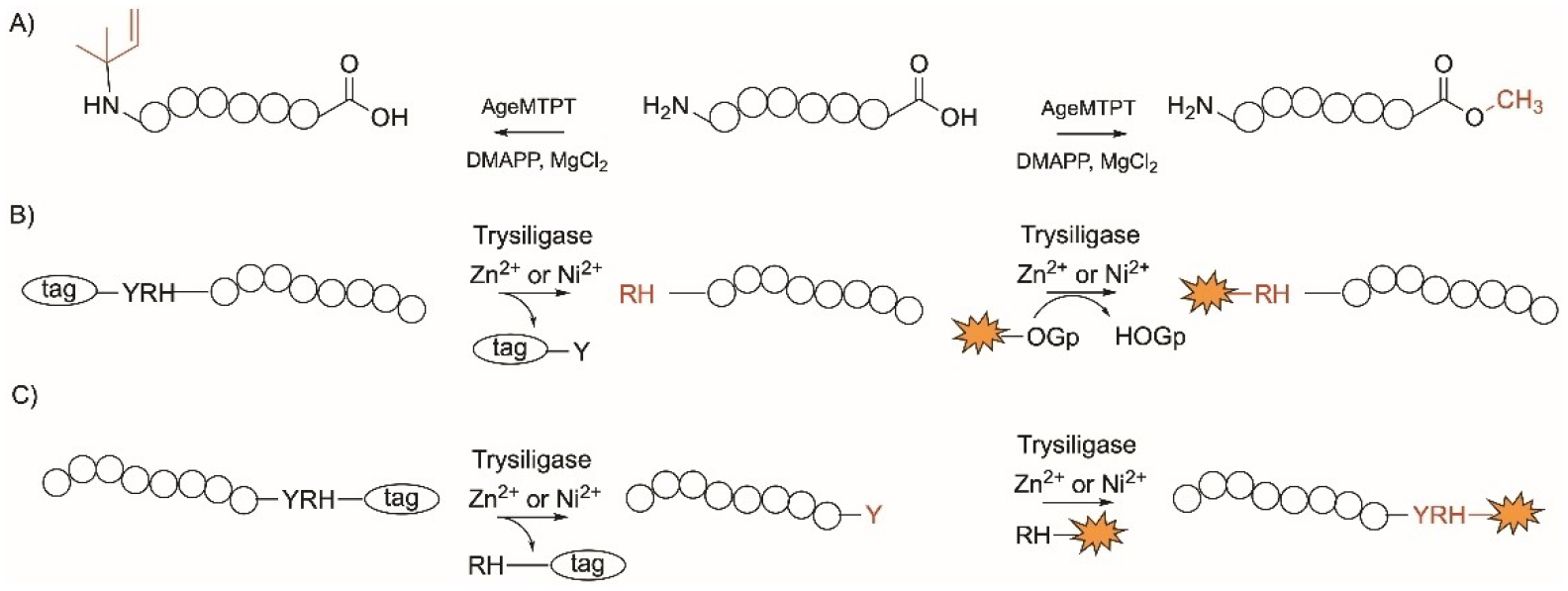
Enzymatic modifications specific to the N- and C-terminus of peptides and proteins showing A) the dual action of AgeMTPT; B) and C) Trypsiligase-mediatied cleavage specific to the Tyr-Arg-His at the B) N-terminus and C) C-terminus and attachment of desired tag. Number of circles does not represent number of residues.
23. C-Terminus Modifications
Tubulin tyrosine ligase (TTL) is a mammalian enzyme that catalyzes the posttranslational addition of l-Tyr to α-tubulin and is involved in tubulin hemostasis.[234] It recognizes a Tub-tag, a 14 amino acid sequence, VDSVEGEGEEEGEE, at the C-terminus of proteins and installs Tyr or Tyr-derivatives to the carboxylic acid (Figure 12C).[235] The enzyme is promiscuous towards tyrosine derivatives including those carrying azide and formyl moieties (Figure 12C). TTL was used to install tyrosine with unique chemical bioorthogonal handles onto proteins that contain proper recognition motifs.[236,237] The Tub-tag labelling approach was applied on a GFP-specific nanobody that was labelled with Tyr-azide moiety which was used to conjugate to a dibenzocyclooctyne (DBCO)-biotin derivative via azide-alkyne cycloaddition. The azide analog was also used to employ bioorthogonal reactions including Staudinger ligation and Staudinger-phosphite reaction while the addition of formyl-Tyr enabled oxime- and hydrazone-forming reactions. These reactions were useful to incorporate fluorophores and biotin.[235]
24. N- and C-Terminus Modifications
AgeMTPT is an enzyme encoded in the cyanobacterial aeruginosamide B peptide biosynthetic gene cluster.[238] It catalyzes the dual isoprenylation and methyl esterification at the N- and C-termini, respectively protecting both termini of peptides (Figure 13A). AgeMTPT was found to modify the peptide with sequence FXPPSYD, as well as peptides with amino acids ranging from 6–10.[239] The enzyme was also used to modify Leu- enkephalin and prenylate SAM. Mutation of the Tyr263 to Leu led to an enzyme with a faster catalysis and broader substrate tolerance while Glu215 to Gly allowed a geranyl group to be utilized and installed onto the N-terminus of peptides.[239]
Trypsiligase is a K60E/N143H/E151H/D189K engineered variant of the human protease, trypsin that depends on divalent cations such as Zn2+ or Ni2+ for activity.[240] The mutations lead to an enzyme with reduced proteolytic and increased ligation and acyl transfer activity. Proteins of interest are attached to an affinity tag with the recognition tripeptide sequence YRH at the N- or the C-terminus and cleaves between the Tyr and Arg residues (Figure 13B, 13C). Trypsiligase then installs the substrate mimetics, acyl 4-guanidinophenyl esters (OGps) linked to fluoresence or labelling groups enabling the coupling of peptidic and nonpeptidic acyl moieties onto the protein of interest. A click bioorthogonal counterpart can be added to label the protein (Figure 13B, 13C).[241] This approach has been used to install fluorescence group and affinity label biotin when linked to OGps in proteins and living cells[242] such as the human Cyclophilin 18,[240,243] Pin1,[243] the Fab fragments anti-Her2 and anti-TNFα,[243] CD 147 and the EGF receptor.[242]
25. Cyclization
Omniligase-1 is an engineered version of the serine protease subtilisin BPN′ allowing it to have a higher ligase compared to the hydroylsis activity.[244,245] It catalyzes the head-to-tail cyclization of linear peptides carrying a glycolate ester at the C-terminus introduced during peptide synthesis (Figure 14). Omniligase cyclizes peptides of amino acids with a preferred sequence of SY-(SG)n-FSKL-OCam-L (where n=2–6, Ocam, O-carboxamidomethyl ester), yet the enzyme can cyclize larger peptides such as the 39-amio acid exenatide.[246] With peptides of 12 amino acids or less, the enzyme forms cyclic multimers. Omniligase-1 can also be used to introduce isopeptide and PEG moieties as well as d- and l-amino acids (Figure 14).[245] Omniligase-1 also converted a linear peptide to a tricyclic peptide when reacted with tris(bromomethyl)benzene and was used for the synthesis of the Cys knot-containing macrocyclic peptides, cyclotides by achieving one-pot cyclization and oxidative folding (Figure 14).[245]
Figure 14.
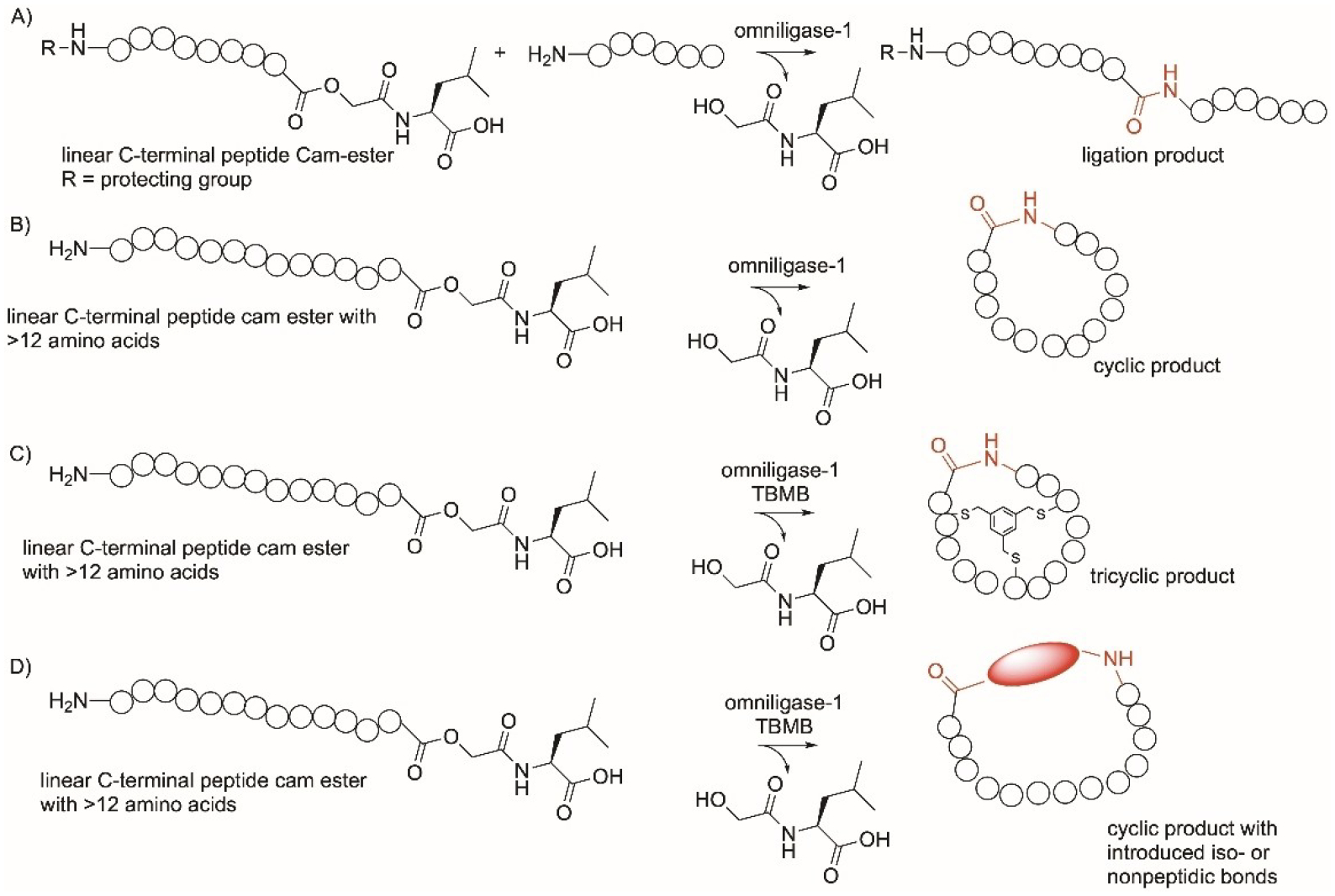
Omniligase-mediated cyclization reactions on proteins. TBMB, tris(bromomethyl)benzene.
Peptide macrocyclization remains a challenge in chemistry.[247] In addition to YcaO mentioned in section 5 (Figure 3C) and 17 (Figure 9D) for the modification of Cys, Ser, and Thr-containing peptides, cyanobactin macrocyclases are promiscuous subtilisin-like serine proteases that are present in the pathways of cyanobacterial peptides.[248,249] Examples include PatG, TruG, and PagG from the patellamide,[250,251] trunkamide,[252] and prenylagaramide[253] pathways, respectively. The PatG-related enzymes catalyze the N–C macrocyclization of short linear peptides composed of proteinogenic and non-proteinogenic amino acids, attached to a C-terminus recognition sequence that ranges from 3–7 amino acids and is different among G enzymes (Figure 15A).[254,255] They favor peptide sequences with azolines and with less extent, Pro. In particular, PagG is a highly promiscuous macrocyclase that recognizes the sequence FAGDDAE and cyclizes the upstream core sequence of a heptamer that contains a Pro at its terminus. A library of 114 peptide sequences was tested where every amino acid of the non-Pro amino acid was replaced with a different proteinogenic amino acid showed that 81% of the peptides were cyclized by PagG.[253]
Figure 15.
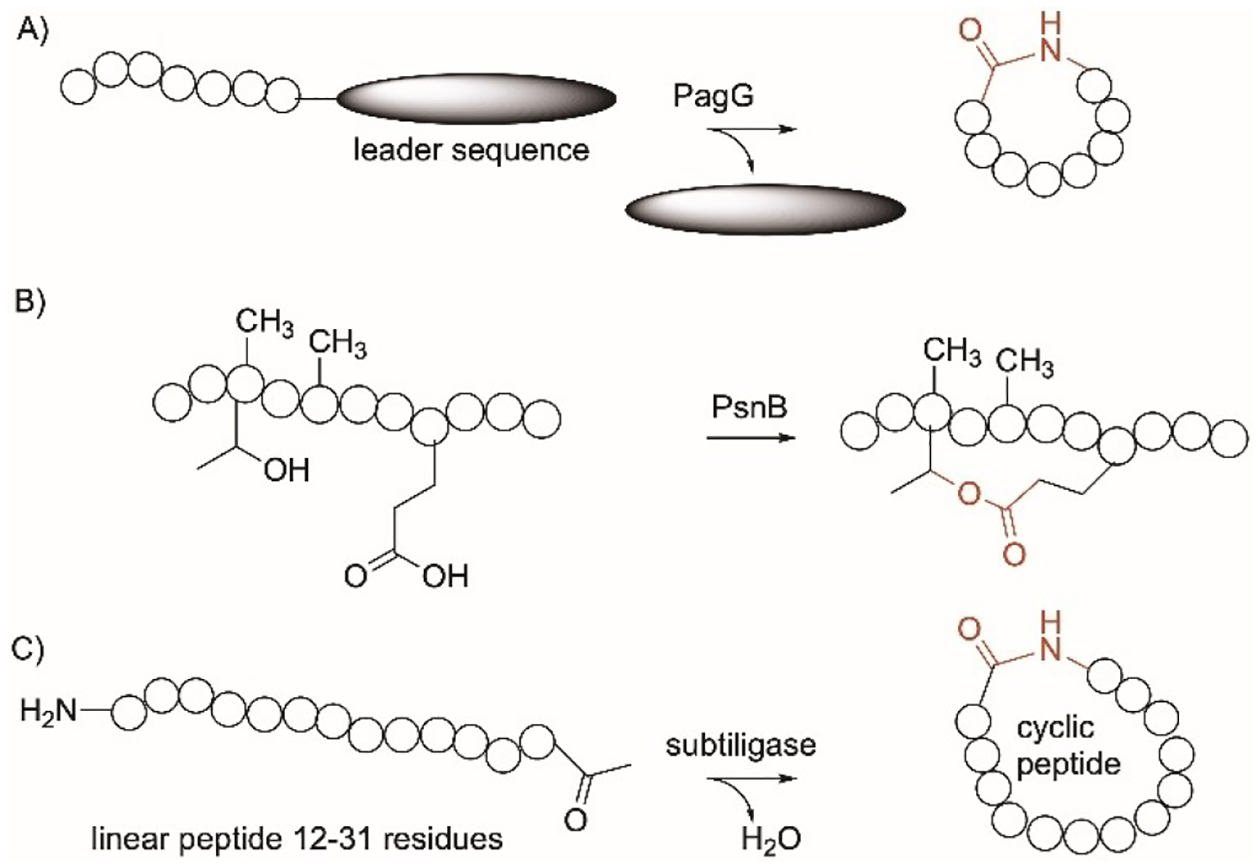
Cyclization-enzymes specific on proteins. A) PagG protease; B) PsnB; C) subtiligase.
PsnB[256] is an ATP-grasp ligase from the cyanobactin plesiocin pathway while PCY1,[257] peptide cyclase 1, is a serine protease from the plant orbitide pathway. PsnB forms a bond between Thr and Glu in N-methylated peptides to form a cyclic compound with a ω-ester linkage (Figure 15B). PsnB was used to synthesize the antiprotease microviridin-like bicyclic peptide, plesiocin from a linear peptide and ATP by performing eight esterification reactions.[256] PsnB is proposed to expand the chemical diversity of peptides due to the possible formation of variable crosslinks and different topological connectivities. PCY1 recognizes peptide sequences with FQA and catalyze the macrocyclization of N-methylated core peptides. In contrast to PsnB, PCY1 requires cleavage of the recognition sequence from the C-terminus before cyclization.[228] PCY1 can be useful in creating cyclic N-methylated peptides, useful for drug development.
OxyA–C are cytochrome P450 enzymes from the glycopeptide antibiotics pathways. They catalyze the oxidative crosslinking between amino acids based on the vancomycin template. These enzymes have moderate substrate tolerance and accept different PCP-bound peptides especially when altering amino acids at the N-terminus.[258]
Subtiligase was mentioned in section 22 (Figure 12A) which was used to ligate peptides or moieties to the N-terminus. Subtiligase can also form cyclic peptides when used with 12–31 mer linear peptides (Figure 15C).[229] Using this enzyme to achieve peptide cyclization does not rely on concentration and does not require peptide protection.
26. Conclusions
LSF of peptides and proteins using wild-type and engineered enzymes represent a practical tool for selective modification of complex peptides and proteins (Table 1). The specificity and selectivity of enzymes can complement the flexibility of synthetic chemistry in achieving peptide LSF. Our aim in this Review was to show that biocatalysis can contribute significantly to the field of residue-specific peptide modification. The examples represented here highlight promiscuous enzymes that achieve functionalization of peptides/proteins at late- or near-late stage in an amino acid residue-specific manner or at one or both of the peptide termini (Table 1). The advantages of peptide functionalization include i) altering the biological activities and receptor binding; ii) fine-tuning the physical and chemical properties; iii) enabling downstream bioorthogonal chemical reactions compatible with biological components and selective to the installed handles such as azide-alkyne cycloaddition, Staudinger ligation, Staudinger-phosphite reaction, carbonyl-based oxime- and hydrazone-forming reactions, as well as tetrazine and tetrazole ligation (Figure 1). As mentioned above, several groups have utilized this to attach fluorescence or biotin tags or ligate to other peptides or moieties to achieve peptide visualization and tagging.
The precision and efficiency offered by biocatalysis have caused an increase in the utilization of enzymes in academia and industry. Immobilization approaches have been shown to improve enzyme properties and enable recycling which in turn reduces the time, cost, and effort of exploiting enzymes. Yet, several challenges limit the wide application of biocatalysis in peptide LSF. First, the number of enzymes functionally and structurally characterized is low compared to what is available in nature. This is in part due to inability of some enzymes to be produced or become properly folded in heterologous hosts or under laboratory conditions. This could also be attributed to undefined or inaccessible native substrates. Second, the number of enzymes that were tested against different non-native substrates to determine full substrate panel is extremely low. Third, different enzymes require cofactors or coenzymes for activity that may not be defined. Fourth, lack of enzymes to catalyze the desired reaction, modify the required amino acid residue, or accommodate the targeted peptide or protein. Fifth, many chemists are reluctant to integrate enzymes in their reactions.
The enzymes presented here (Table 1) have shown peptide/protein promiscuity and represent the potential for further substrate tolerance broadening through rational design or directed evolution. There is no doubt that we need to grow our library of amino acid residue-specific enzymes that have broad substrate tolerance. New biocatalysts can be discovered through genome and metagenome mining, enzyme characterization, and three-dimensional structural analysis. Within this context, there has been an enormous progress in the characterization of the three-dimensional structure methods of enzymes with over 204,000 enzyme structures deposited on the protein databank (Protein Data Bank, accessed 05/2023) which can help in structural-guided functional characterization of enzymes[259,260] and identification of their mechanisms.[205]
Amidst the technological advances in DNA sequencing, bioinformatics, and enzyme discovery, combined with machine-learning[261] and the development of high-throughput robotics,[262] we are confident that the repertoire of residue-specific enzymes will expand and that more enzymes will be available for selective peptide and protein LSF. There has also been significant advancement in the engineering of enzymes through rationale design[260,263] or directed evolution.[262,264] This opens the door to modify enzyme’s activity, selectivity, and substrate scope, expanding their capability as powerful tools in peptide modifications.[265] This will greatly impact on drug discovery and biological research bringing in further exciting developments in these rapidly evolving areas.
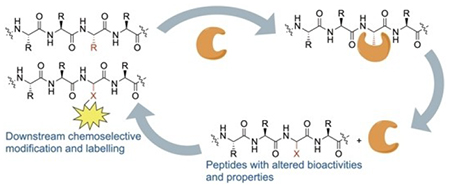
Biocatalysis is a powerful tool to achieve selective modifications on peptides with strong potential in the development of pharmaceuticals and in biological research. This review focuses on amino acid residue-specific enzymes with broad substrate tolerance as well as their nonnative substrates. These enzymes have enabled the modification of a wide range of peptides and proteins at late-stage.
Acknowledgements
This work was supported in part by NIH Award Number R03AI168826 (S. I. E.) and Chapman University School of Pharmacy (CUSP).
Biographies

Ashley Alexander is currently an undergraduate student at Chapman University. She is in pursuit of a BS degree in Biochemistry and Molecular Biology. Her research interests center on drug development using chemoenzymatic approaches.

Sherif Elshahawi is an Assistant Professor at Chapman University School of Pharmacy. He earned his Bachelor’s degree in Pharmacy from Cairo University (2000), Master’s in Pharmacognosy from the University of Mississippi (2006) and PhD in Biochemistry and Molecular Biology from Oregon Health & Science University (2012). His research interests focus on applying tools from natural product chemistry, biocatalysis, and biosynthesis towards drug discovery and development.
Footnotes
Conflict of Interests
The authors declare no conflict of interest.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- [1].Romero E, Jones BS, Hogg BN, Rué Casamajo A, Hayes MA, Flitsch SL, Turner NJ, Schnepel C, Angew. Chem. Int. Ed 2021, 60, 16824–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lasso JD, Castillo-Pazos DJ, Li C-J, Chem. Soc. Rev 2021, 50, 10955–10982. [DOI] [PubMed] [Google Scholar]
- [3].Guillemard L, Kaplaneris N, Ackermann L, Johansson MJ, Nat. Chem. Rev 2021, 5, 522–545. [DOI] [PubMed] [Google Scholar]
- [4].Wang W, Lorion MM, Shah J, Kapdi AR, Ackermann L, Angew. Chem. Int. Ed 2018, 57, 14700–14717. [DOI] [PubMed] [Google Scholar]
- [5].Gruß H, Sewald N, Chemistry 2020, 26, 5328–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Allouche EMD, Grinhagena E, Waser J, Angew. Chem. Int. Ed 2022, 61, e202112287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Parker CG, Pratt MR, Cell 2020, 180, 605–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rigolot V, Biot C, Lion C, Angew. Chem. Int. Ed 2021, 60, 23084–23105. [DOI] [PubMed] [Google Scholar]
- [9].Mitry MMA, Greco F, Osborn HMI, Chemistry 2023, 29, e202203942. [DOI] [PubMed] [Google Scholar]
- [10].Porte K, Riberaud M, Châtre R, Audisio D, Papot S, Taran F, ChemBioChem 2021, 22, 100–113. [DOI] [PubMed] [Google Scholar]
- [11].Bird RE, Lemmel SA, Yu X, Zhou QA, Bioconjugate Chem 2021, 32, 2457–2479. [DOI] [PubMed] [Google Scholar]
- [12].Stump B, ChemBioChem 2022, 23, e202200016. [DOI] [PubMed] [Google Scholar]
- [13].Hoyt EA, Cal PMSD, Oliveira BL, Bernardes GJL, Nat. Chem. Rev 2019, 3, 147–171. [Google Scholar]
- [14].Isenegger PG, Davis BG, J. Am. Chem. Soc 2019, 141, 8005–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Krall N, da Cruz FP, Boutureira O, Bernardes GJL, Nat. Chem 2016, 8, 103–113. [DOI] [PubMed] [Google Scholar]
- [16].deGruyter JN, Malins LR, Baran PS, Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Y, Park K-Y, Suazo KF, Distefano MD, Chem. Soc. Rev 2018, 47, 9106–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bellotti P, Huang H-M, Faber T, Glorius F, Chem. Rev 2023, DOI 10.1021/acs.chemrev.2c00478. [DOI] [PubMed] [Google Scholar]
- [19].Walsh CT, Nat. Prod. Rep 2023, 40, 326–386. [DOI] [PubMed] [Google Scholar]
- [20].Clinger JA, Zhang Y, Liu Y, Miller MD, Hall RE, Van Lanen SG, Phillips GN, Thorson JS, Elshahawi SI, ACS Chem. Biol 2021, 16, 2816–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhan B-B, Jiang M-X, Shi B-F, Chem. Commun 2020, 56, 13950–13958. [DOI] [PubMed] [Google Scholar]
- [22].Jalali E, Thorson JS, Curr. Opin. Biotechnol 2021, 69, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Blanc RS, Richard S, Mol. Cell 2017, 65, 8–24. [DOI] [PubMed] [Google Scholar]
- [24].Gui S, Gathiaka S, Li J, Qu J, Acevedo O, Hevel JM, J. Biol. Chem 2014, 289, 9320–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gui S, Wooderchak-Donahue WL, Zang T, Chen D, Daly MP, Zhou ZS, Hevel JM, Biochemistry 2013, 52, 199–209. [DOI] [PubMed] [Google Scholar]
- [26].Hamey JJ, Rakow S, Bouchard C, Senst JM, Kolb P, Bauer U-M, Wilkins MR, Hart-Smith G, FEBS J 2021, 288, 5668–5691. [DOI] [PubMed] [Google Scholar]
- [27].Feng Y, Maity R, Whitelegge JP, Hadjikyriacou A, Li Z, Zurita-Lopez C, Al-Hadid Q, Clark AT, Bedford MT, Masson J-Y, Clarke SG, J. Biol. Chem 2013, 288, 37010–37025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shishkova E, Zeng H, Liu F, Kwiecien NW, Hebert AS, Coon JJ, Xu W, Nat. Commun 2017, 8, 15571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tewary SK, Zheng YG, Ho M-C, Cell. Mol. Life Sci 2019, 76, 2917–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hymbaugh Bergman SJ, Comstock LR, Bioorg. Med. Chem 2015, 23, 5050–5055. [DOI] [PubMed] [Google Scholar]
- [31].Hymbaugh SJ, Pecor LM, Tracy CM, Comstock LR, ChemBioChem 2019, 20, 379–384. [DOI] [PubMed] [Google Scholar]
- [32].Ge W, Wolf A, Feng T, Ho C, Sekirnik R, Zayer A, Granatino N, Cockman ME, Loenarz C, Loik ND, Hardy AP, Claridge TDW, Hamed RB, Chowdhury R, Gong L, Robinson CV, Trudgian DC, Jiang M, Mackeen MM, Mccullagh JS, Gordiyenko Y, Thalhammer A, Yamamoto A, Yang M, Liu-Yi P, Zhang Z, Schmidt-Zachmann M, Kessler BM, Ratcliffe PJ, Preston GM, Coleman ML, Schofield CJ, Nat. Chem. Biol 2012, 8, 960–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rehm FBH, Tyler TJ, Xie J, Yap K, Durek T, Craik DJ, ChemBioChem 2021, 22, 2079–2086. [DOI] [PubMed] [Google Scholar]
- [34].Hemu X, El Sahili A, Hu S, Wong K, Chen Y, Wong YH, Zhang X, Serra A, Goh BC, Darwis DA, Chen MW, Sze SK, Liu C-F, Lescar J, Tam JP, Proc. Natl. Acad. Sci. USA 2019, 116, 11737–11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jackson MA, Gilding EK, Shafee T, Harris KS, Kaas Q, Poon S, Yap K, Jia H, Guarino R, Chan LY, Durek T, Anderson MA, Craik DJ, Nat. Commun 2018, 9, 2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Harris KS, Durek T, Kaas Q, Poth AG, Gilding EK, Conlan BF, Saska I, Daly NL, van der Weerden NL, Craik DJ, Anderson MA, Nat. Commun 2015, 6, 10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tang TMS, Luk LYP, Org. Biomol. Chem 2021, 19, 5048–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang D, Wang Z, Hu S, Balamkundu S, To J, Zhang X, Lescar J, Tam JP, Liu C-F, J. Am. Chem. Soc 2021, 143, 8704–8712. [DOI] [PubMed] [Google Scholar]
- [39].Xia Y, Li F, Zhang X, Balamkundu S, Tang F, Hu S, Lescar J, Tam JP, Liu C-F, J. Am. Chem. Soc 2023, 145, 6838–6844. [DOI] [PubMed] [Google Scholar]
- [40].Yang R, Wong YH, Nguyen GKT, Tam JP, Lescar J, Wu B, J. Am. Chem. Soc 2017, 139, 5351–5358. [DOI] [PubMed] [Google Scholar]
- [41].Rehm FBH, Harmand TJ, Yap K, Durek T, Craik DJ, Ploegh HL, J. Am. Chem. Soc 2019, 141, 17388–17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hemu X, El Sahili A, Hu S, Zhang X, Serra A, Goh BC, Darwis DA, Chen MW, Sze SK, Liu C, Lescar J, Tam JP, ACS Catal 2020, 10, 8825–8834. [Google Scholar]
- [43].Tang TMS, Cardella D, Lander AJ, Li X, Escudero JS, Tsai Y-H, Luk LYP, Chem. Sci 2020, 11, 5881–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Du J, Yap K, Chan LY, Rehm FBH, Looi FY, Poth AG, Gilding EK, Kaas Q, Durek T, Craik DJ, Nat. Commun 2020, 11, 1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nguyen GKT, Wang S, Qiu Y, Hemu X, Lian Y, Tam JP, Nat. Chem. Biol 2014, 10, 732–738. [DOI] [PubMed] [Google Scholar]
- [46].Nguyen GKT, Hemu X, Quek J-P, Tam JP, Angew. Chem. Int. Ed 2016, 55, 12802–12806. [DOI] [PubMed] [Google Scholar]
- [47].Nguyen GKT, Qiu Y, Cao Y, Hemu X, Liu C-F, Tam JP, Nat. Protoc 2016, 11, 1977–1988. [DOI] [PubMed] [Google Scholar]
- [48].Nguyen GKT, Cao Y, Wang W, Liu CF, Tam JP, Angew. Chem. Int. Ed 2015, 54, 15694–15698. [DOI] [PubMed] [Google Scholar]
- [49].Hemu X, To J, Zhang X, Tam JP, J. Org. Chem 2020, 85, 1504–1512. [DOI] [PubMed] [Google Scholar]
- [50].Palsuledesai CC, Distefano MD, ACS Chem. Biol 2015, 10, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Suazo KF, Schaber C, Palsuledesai CC, Odom John AR, Distefano MD, Sci. Rep 2016, 6, 38615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chang H-Y, Cheng T-H, Wang AH-J, IUBMB Life 2021, 73, 40–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang Y-C, Distefano MD, Chem. Commun 2012, 48, 8228–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Krzysiak AJ, Scott SA, Hicks KA, Fierke CA, Gibbs RA, Bioorg. Med. Chem. Lett 2007, 17, 5548–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Krzysiak AJ, Aditya AV, Hougland JL, Fierke CA, Gibbs RA, Bioorg. Med. Chem. Lett 2010, 20, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].London N, Lamphear CL, Hougland JL, Fierke CA, Schueler-Furman O, PLoS Comput. Biol 2011, 7, e1002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang Y-C, Dozier JK, Beese LS, Distefano MD, ACS Chem. Biol 2014, 9, 1726–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schey GL, Buttery PH, Hildebrandt ER, Novak SX, Schmidt WK, Hougland JL, Distefano MD, Int. J. Mol. Sci 2021, 22, 12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ashok S, Hildebrandt ER, Ruiz CS, Hardgrove DS, Coreno DW, Schmidt WK, Hougland JL, Biochemistry 2020, 59, 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Blanden MJ, Suazo KF, Hildebrandt ER, Hardgrove DS, Patel M, Saunders WP, Distefano MD, Schmidt WK, Hougland JL, J. Biol. Chem 2018, 293, 2770–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Troutman JM, Subramanian T, Andres DA, Spielmann HP, Biochemistry 2007, 46, 11310–11321. [DOI] [PubMed] [Google Scholar]
- [62].Subramanian T, Liu S, Troutman JM, Andres DA, Spielmann HP, ChemBioChem 2008, 9, 2872–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lee J, Choi H-J, Yun M, Kang Y, Jung J-E, Ryu Y, Kim TY, Cha Y, Cho H-S, Min J-J, Chung C-W, Kim H-S, Angew. Chem. Int. Ed 2015, 54, 12020–12024. [DOI] [PubMed] [Google Scholar]
- [64].Rashidian M, Song JM, Pricer RE, Distefano MD, J. Am. Chem. Soc 2012, 134, 8455–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rashidian M, Kumarapperuma SC, Gabrielse K, Fegan A, Wagner CR, Distefano MD, J. Am. Chem. Soc 2013, 135, 16388–16396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Morstein J, Bader T, Cardillo AL, Schackmann J, Ashok S, Hougland JL, Hrycyna CA, Trauner DH, Distefano MD, ACS Chem. Biol 2022, 17, 2945–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Labadie GR, Viswanathan R, Poulter CD, J. Org. Chem 2007, 72, 9291–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wollack JW, Monson BJ, Dozier JK, Dalluge JJ, Poss K, Hilderbrand SA, Distefano MD, Chem. Biol. Drug Des 2014, 84, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K, Nat. Biotechnol 2003, 21, 86–89. [DOI] [PubMed] [Google Scholar]
- [70].Seo J, Lee S, Poulter CD, J. Am. Chem. Soc 2013, 135, 8973–8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yeo JE, Wickramaratne S, Khatwani S, Wang Y-C, Vervacke J, Distefano MD, Tretyakova NY, ACS Chem. Biol 2014, 9, 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Krüger T, Dierks T, Sewald N, Biol. Chem 2019, 400, 289–297. [DOI] [PubMed] [Google Scholar]
- [73].Appel MJ, Bertozzi CR, ACS Chem. Biol 2015, 10, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wu Y, Zhao C, Su Y, Shaik S, Lai W, Angew. Chem. Int. Ed 2023, 62, e202212053. [DOI] [PubMed] [Google Scholar]
- [75].Appel MJ, Meier KK, Lafrance-Vanasse J, Lim H, Tsai C-L, Hedman B, Hodgson KO, Tainer JA, Solomon EI, Bertozzi CR, Proc. Natl. Acad. Sci. USA 2019, 116, 5370–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Knop M, Engi P, Lemnaru R, Seebeck FP, ChemBioChem 2015, 16, 2147–2150. [DOI] [PubMed] [Google Scholar]
- [77].Mashima R, Nakanishi M, Int. J. Mol. Sci 2022, 23, 8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Asiimwe N, Mazid MFA, Jeong YT, Lee J, Lee J-S, RSC Adv 2022, 12, 18884–18888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Rush JS, Bertozzi CR, J. Am. Chem. Soc 2008, 130, 12240–12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Janson N, Krüger T, Karsten L, Boschanski M, Dierks T, Müller KM, Sewald N, ChemBioChem 2020, 21, 3580–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kudirka R, Barfield RM, McFarland J, Albers AE, de Hart GW, Drake PM, Holder PG, Banas S, Jones LC, Garofalo AW, Rabuka D, Chem. Biol 2015, 22, 293–298. [DOI] [PubMed] [Google Scholar]
- [82].Boschanski M, Krüger T, Karsten L, Falck G, Alam S, Gerlach M, Müller B, Müller KM, Sewald N, Dierks T, Bioconjugate Chem 2021, 32, 1167–1174. [DOI] [PubMed] [Google Scholar]
- [83].Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR, Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Carrico IS, Carlson BL, Bertozzi CR, Nat. Chem. Biol 2007, 3, 321–322. [DOI] [PubMed] [Google Scholar]
- [85].Drake PM, Albers AE, Baker J, Banas S, Barfield RM, Bhat AS, de Hart GW, Garofalo AW, Holder P, Jones LC, Kudirka R, McFarland J, Zmolek W, Rabuka D, Bioconjugate Chem 2014, 25, 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rupniewski I, Rabuka D, Enzyme-Mediat. Ligation Methods, Springer, New York, NY, 2019, p. 63. [DOI] [PubMed] [Google Scholar]
- [87].Krüger T, Weiland S, Falck G, Gerlach M, Boschanski M, Alam S, Müller KM, Dierks T, Sewald N, Angew. Chem. Int. Ed 2018, 57, 7245–7249. [DOI] [PubMed] [Google Scholar]
- [88].Smith EL, Giddens JP, Iavarone AT, Godula K, Wang L-X, Bertozzi CR, Bioconjugate Chem 2014, 25, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Knop M, Lemnaru R, Seebeck FP, ChemBioChem 2017, 18, 1755–1761. [DOI] [PubMed] [Google Scholar]
- [90].Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, Mitchell DA, Chem. Rev 2017, 117, 5389–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].McIntosh JA, Donia MS, Schmidt EW, J. Am. Chem. Soc 2010, 132, 4089–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Koehnke J, Mann G, Bent AF, Ludewig H, Shirran S, Botting C, Lebl T, Houssen WE, Jaspars M, Naismith JH, Nat. Chem. Biol 2015, 11, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Nguyen NA, Lin Z, Mohanty I, Garg N, Schmidt EW, Agarwal V, J. Am. Chem. Soc 2021, 143, 10221–10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Nguyen NA, Cong Y, Hurrell RC, Arias N, Garg N, Puri AW, Schmidt EW, Agarwal V, ACS Chem. Biol 2022, 17, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA, Nat. Chem. Biol 2015, 11, 564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Goto Y, Ito Y, Kato Y, Tsunoda S, Suga H, Chem. Biol 2014, 21, 766–774. [DOI] [PubMed] [Google Scholar]
- [97].Koehnke J, Morawitz F, Bent AF, Houssen WE, Shirran SL, Fuszard MA, Smellie IA, Botting CH, Smith MCM, Jaspars M, Naismith JH, ChemBioChem 2013, 14, 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gu W, Zheng Y, Pogorelov T, Nair SK, Schmidt EW, ACS Chem. Biol 2022, 17, 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Goto Y, Suga H, ChemBioChem 2020, 21, 84–87. [DOI] [PubMed] [Google Scholar]
- [100].Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, van der Donk WA, Proc. Natl. Acad. Sci. USA 2010, 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Arias-Orozco P, Yi Y, Ruijne F, Cebrián R, Kuipers OP, ACS Synth. Biol 2023, 12, 164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Le T, van der Donk WA, Trends Chem 2021, 3, 266–278. [Google Scholar]
- [103].Deweid L, Avrutina O, Kolmar H, Biol. Chem 2019, 400, 257–274. [DOI] [PubMed] [Google Scholar]
- [104].Strop P, Bioconjugate Chem 2014, 25, 855–862. [DOI] [PubMed] [Google Scholar]
- [105].Fuchsbauer H-L, FEBS J 2022, 289, 4680–4703. [DOI] [PubMed] [Google Scholar]
- [106].Abe H, Goto M, Kamiya N, Chemistry 2011, 17, 14004–14008. [DOI] [PubMed] [Google Scholar]
- [107].Rachel NM, Pelletier JN, Chem. Commun 2016, 52, 2541–2544. [DOI] [PubMed] [Google Scholar]
- [108].Oteng-Pabi SK, Pardin C, Stoica M, Keillor JW, Chem. Commun 2014, 50, 6604–6606. [DOI] [PubMed] [Google Scholar]
- [109].Braun AC, Gutmann M, Mueller TD, Lühmann T, Meinel L, J. Controlled Release 2018, 279, 17–28. [DOI] [PubMed] [Google Scholar]
- [110].Spolaore B, Raboni S, Satwekar AA, Grigoletto A, Mero A, Montagner IM, Rosato A, Pasut G, Fontana A, Bioconjugate Chem 2016, 27, 2695–2706. [DOI] [PubMed] [Google Scholar]
- [111].Kitaoka M, Tsuruda Y, Tanaka Y, Goto M, Mitsumori M, Hayashi K, Hiraishi Y, Miyawaki K, Noji S, Kamiya N, Chemistry 2011, 17, 5387–5392. [DOI] [PubMed] [Google Scholar]
- [112].Takahara M, Wakabayashi R, Minamihata K, Goto M, Kamiya N, Bioconjugate Chem 2017, 28, 2954–2961. [DOI] [PubMed] [Google Scholar]
- [113].Gnaccarini C, Ben-Tahar W, Mulani A, Roy I, Lubell WD, Pelletier JN, Keillor JW, Org. Biomol. Chem 2012, 10, 5258–5265. [DOI] [PubMed] [Google Scholar]
- [114].Guy J, Castonguay R, Pineda NBC-R, Jacquier V, Caron K, Michnick SW, Keillor JW, Mol. BioSyst 2010, 6, 976–987. [DOI] [PubMed] [Google Scholar]
- [115].Oteng-Pabi SK, Clouthier CM, Keillor JW, PLoS One 2018, 13, e0197956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Jeger S, Zimmermann K, Blanc A, Grünberg J, Honer M, Hunziker P, Struthers H, Schibli R, Angew. Chem. Int. Ed 2010, 49, 9995–9997. [DOI] [PubMed] [Google Scholar]
- [117].Anami Y, Xiong W, Gui X, Deng M, Zhang CC, Zhang N, An Z, Tsuchikama K, Org. Biomol. Chem 2017, 15, 5635–5642. [DOI] [PubMed] [Google Scholar]
- [118].Kosciuk T, Lin H, ACS Chem. Biol 2020, 15, 1747–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Rosen CB, Francis MB, Nat. Chem. Biol 2017, 13, 697–705. [DOI] [PubMed] [Google Scholar]
- [120].Dian C, Pérez-Dorado I, Rivière F, Asensio T, Legrand P, Ritzefeld M, Shen M, Cota E, Meinnel T, Tate EW, Giglione C, Nat. Commun 2020, 11, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Su D, Kosciuk T, Yang M, Price IR, Lin H, ACS Catal 2021, 11, 14877–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Heal WP, Wright MH, Thinon E, Tate EW, Nat. Protoc 2012, 7, 105–117. [DOI] [PubMed] [Google Scholar]
- [123].Hang HC, Geutjes E-J, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL, J. Am. Chem. Soc 2007, 129, 2744–2745. [DOI] [PubMed] [Google Scholar]
- [124].Freund C, Schwarzer D, ChemBioChem 2021, 22, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Zou Z, Mate DM, Rübsam K, Jakob F, Schwaneberg U, ACS Comb. Sci 2018, 20, 203–211. [DOI] [PubMed] [Google Scholar]
- [126].Yamaguchi S, Ikeda R, Umeda Y, Kosaka T, Yamahira S, Okamoto A, ChemBioChem 2022, 23, e202200474. [DOI] [PubMed] [Google Scholar]
- [127].Apostolos AJ, Kelly JJ, Ongwae GM, Pires MM, ChemBioChem 2022, 23, e202200412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kumari P, Bowmik S, Paul SK, Biswas B, Banerjee SK, Murty US, Ravichandiran V, Mohan U, Biotechnol. Bioeng 2021, 118, 4577–4589. [DOI] [PubMed] [Google Scholar]
- [129].Bundred JR, Hendrix E, Coleman ML, Cell. Mol. Life Sci 2018, 75, 4093–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zhang Y, Hamada K, Nguyen DT, Inoue S, Satake M, Kobayashi S, Okada C, Ogata K, Okada M, Sengoku T, Goto Y, Suga H, Nat. Catal 2022, 5, 682–693. [Google Scholar]
- [131].Cui Z, Nguyen H, Bhardwaj M, Wang X, Büschleb M, Lemke A, Schütz C, Rohrbacher C, Junghanns P, Koppermann S, Ducho C, Thorson JS, Van Lanen SG, J. Am. Chem. Soc 2021, 143, 19425–19437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Wang ZA, Cole PA, Cell Chem. Biol 2020, 27, 953–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Blasl A-T, Schulze S, Qin C, Graf LG, Vogt R, Lammers M, Biol. Chem 2022, 403, 151–194. [DOI] [PubMed] [Google Scholar]
- [134].He M, Han Z, Liu L, Zheng YG, Angew. Chem. Int. Ed 2018, 57, 1162–1184. [DOI] [PubMed] [Google Scholar]
- [135].Tan D, Wei W, Han Z, Ren X, Yan C, Qi S, Song X, Zheng YG, Wong J, Huang H, iScience 2022, 25, 105443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Andrews FH, Strahl BD, Kutateladze TG, Nat. Chem. Biol 2016, 12, 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Talibov VO, Fabini E, FitzGerald EA, Tedesco D, Cederfeldt D, Talu MJ, Rachman MM, Mihalic F, Manoni E, Naldi M, Sanese P, Forte G, Lepore Signorile M, Barril X, Simone C, Bartolini M, Dobritzsch D, Del Rio A, Danielson UH, ChemBioChem 2021, 22, 1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lyu Z, Zhao Y, Buuh ZY, Gorman N, Goldman AR, Islam MS, Tang H-Y, Wang RE, J. Am. Chem. Soc 2021, 143, 1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Yu M, de Carvalho LPS, Sun G, Blanchard JS, J. Am. Chem. Soc 2006, 128, 15356–15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Simon RP, Rumpf T, Linkuviene V, Matulis D, Akhtar A, Jung M, J. Med. Chem 2019, 62, 2582–2597. [DOI] [PubMed] [Google Scholar]
- [141].Yang Y-Y, Ascano JM, Hang HC, J. Am. Chem. Soc 2010, 132, 3640–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Han Z, Chou C, Yang X, Bartlett MG, Zheng YG, ACS Chem. Biol 2017, 12, 1547–1555. [DOI] [PubMed] [Google Scholar]
- [143].Han Z, Luan Y, Zheng YG, ChemBioChem 2015, 16, 2605–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Bos J, Muir TW, J. Am. Chem. Soc 2018, 140, 4757–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Yang C, Mi J, Feng Y, Ngo L, Gao T, Yan L, Zheng YG, J. Am. Chem. Soc 2013, 135, 7791–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Song J, Han Z, Zheng YG, Curr. Protoc 2022, 2, e497. [DOI] [PubMed] [Google Scholar]
- [147].Schnepel C, Pérez LR, Yu Y, Angelastro A, Heath RS, Lubberink M, Falcioni F, Mulholland K, Hayes MA, Turner NJ, Flitsch SL, Nat. Catal 2023, 6, 89–99. [Google Scholar]
- [148].Xiao R, Zhao L, Ma H, Liu Q, Qin H, Luo X, Xuan W, ChemBioChem 2022, 23, e202100551. [DOI] [PubMed] [Google Scholar]
- [149].Wang MY, Liow P, Guzman MIT, Qi J, ACS Chem. Biol 2022, 17, 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Del Rizzo PA, Trievel RC, Biochim. Biophys. Acta Gene Regul. Mech 2014, 1839, 1404–1415. [DOI] [PubMed] [Google Scholar]
- [151].Huber TD, Clinger JA, Liu Y, Xu W, Miller MD, Jr Phillips GN, Thorson JS, ACS Chem. Biol 2020, 15, 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Michailidou F, Klöcker N, Cornelissen NV, Singh RK, Peters A, Ovcharenko A, Kümmel D, Rentmeister A, Angew. Chem. Int. Ed 2021, 60, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Bothwell IR, Luo M, Org. Lett 2014, 16, 3056–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Binda O, Boyce M, Rush JS, Palaniappan KK, Bertozzi CR, Gozani O, ChemBioChem 2011, 12, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Willnow S, Martin M, Lüscher B, Weinhold E, ChemBioChem 2012, 13, 1167–1173. [DOI] [PubMed] [Google Scholar]
- [156].Idigo NJ, Voigt P, Histone Methyltransferases Methods Protoc, Springer US, New York, NY, 2022, p. 43. [DOI] [PubMed] [Google Scholar]
- [157].Florea M, Kudithipudi S, Rei A, González-Álvarez MJ, Jeltsch A, Nau WM, Chemistry 2012, 18, 3521–3528. [DOI] [PubMed] [Google Scholar]
- [158].Chen I, Howarth M, Lin W, Ting AY, Nat. Methods 2005, 2, 99–104. [DOI] [PubMed] [Google Scholar]
- [159].Santos-Barriopedro I, van Mierlo G, Vermeulen M, Nat. Commun 2021, 12, 5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Sueda S, Yoneda S, Hayashi H, ChemBioChem 2011, 12, 1367–1375. [DOI] [PubMed] [Google Scholar]
- [161].Slavoff SA, Chen I, Choi Y-A, Ting AY, J. Am. Chem. Soc 2008, 130, 1160–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Fernández-Suárez M, Chen TS, Ting AY, J. Am. Chem. Soc 2008, 130, 9251–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Roux KJ, Kim DI, Raida M, Burke B, J. Cell Biol 2012, 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ, Mol. Biol. Cell 2016, 27, 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Fernández-Suárez M, Baruah H, Martínez-Hernández L, Xie KT, Baskin JM, Bertozzi CR, Ting AY, Nat. Biotechnol 2007, 25, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Liu DS, Tangpeerachaikul A, Selvaraj R, Taylor MT, Fox JM, Ting AY, J. Am. Chem. Soc 2012, 134, 792–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Drake CR, Sevillano N, Truillet C, Craik CS, VanBrocklin HF, Evans MJ, ACS Chem. Biol 2016, 11, 1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Best M, Degen A, Baalmann M, Schmidt TT, Wombacher R, ChemBioChem 2015, 16, 1158–1162. [DOI] [PubMed] [Google Scholar]
- [169].Cohen JD, Zou P, Ting AY, ChemBioChem 2012, 13, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Baalmann M, Best M, Wombacher R, Noncanonical Amino Acids Methods Protoc, Springer, New York, NY, 2018, p. 365. [Google Scholar]
- [171].Schirer A, Rouch A, Marcheteau E, Stojko J, Landron Sophie, Jeantet E, Fould B, Ferry G, Boutin J, Mol. Biol. Rep 2022, 49, 149–161. [DOI] [PubMed] [Google Scholar]
- [172].Wiltschi B, Merkel L, Budisa N, ChemBioChem 2009, 10, 217–220. [DOI] [PubMed] [Google Scholar]
- [173].Frottin F, Martinez A, Peynot P, Mitra S, Holz RC, Giglione C, Meinnel T, Mol. Cell. Proteomics 2006, 5, 2336–2349. [DOI] [PubMed] [Google Scholar]
- [174].Kjærsgaard NL, Nielsen TB, Gothelf KV, ChemBioChem 2022, 23, e202200245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Maza JC, Bader DLV, Xiao L, Marmelstein AM, Brauer DD, ElSohly AM, Smith MJ, Krska SW, Parish CA, Francis MB, J. Am. Chem. Soc 2019, 141, 3885–3892. [DOI] [PubMed] [Google Scholar]
- [176].Krüger T, Weiland S, Boschanski M, Sinha PK, Falck G, Müller KM, Dierks T, Sewald N, ChemBioChem 2019, 20, 2074–2078. [DOI] [PubMed] [Google Scholar]
- [177].Hu C-W, Worth M, Li H, Jiang J, ChemBioChem 2019, 20, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Ju Kim E, ChemBioChem 2020, 21, 3026–3035. [DOI] [PubMed] [Google Scholar]
- [179].Yang X, Qian K, Nat. Rev. Mol. Cell Biol 2017, 18, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Pathak S, Alonso J, Schimpl M, Rafie K, Blair DE, Borodkin VS, Schüttelkopf AW, Albarbarawi O, van Aalten DMF, Nat. Struct. Mol. Biol 2015, 22, 744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Leavy TM, Bertozzi CR, Bioorg. Med. Chem. Lett 2007, 17, 3851–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Hu C-W, Worth M, Fan D, Li B, Li H, Lu L, Zhong X, Lin Z, Wei L, Ge Y, Li L, Jiang J, Nat. Chem. Biol 2017, 13, 1267–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Jackson EG, Cutolo G, Yang B, Yarravarapu N, Burns MWN, Bineva-Todd G, Roustan C, Thoden JB, Lin-Jones HM, van Kuppevelt TH, Holden HM, Schumann B, Kohler JJ, Woo CM, Pratt MR, ACS Chem. Biol 2022, 17, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Purushothaman M, Sarkar S, Morita M, Gugger M, Schmidt EW, Morinaka BI, Angew. Chem. Int. Ed 2021, 60, 8460–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Sardar D, Lin Z, Schmidt EW, Chem. Biol 2015, 22, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Bertsova YV, Fadeeva MS, Kostyrko VA, Serebryakova MV, Baykov AA, Bogachev AV, J. Biol. Chem 2013, 288, 14276–14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Tong Y, Lee M, Drenth J, Fraaije MW, Bioconjugate Chem 2021, 32, 1559–1563. [DOI] [PubMed] [Google Scholar]
- [188].Tong Y, Loonstra MR, Fraaije MW, ChemBioChem 2022, 23, e202200144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Schnepel C, Moritzer A-C, Gäfe S, Montua N, Minges H, Nieß A, Niemann HH, Sewald N, ChemBioChem 2023, 24, e202200569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Sana B, Ke D, Li EHY, Ho T, Seayad J, Duong HA, Ghadessy FJ, Biomol. Eng 2022, 12, 1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Walsh CT, ACS Chem. Biol 2014, 9, 2718–2728. [DOI] [PubMed] [Google Scholar]
- [192].Mori T, J. Nat. Med 2020, 74, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Mupparapu N, Lin Y-HC, Kim TH, Elshahawi SI, Chemistry 2021, 27, 4176–4182. [DOI] [PubMed] [Google Scholar]
- [194].Mupparapu N, Brewster L, Ostrom KF, Elshahawi SI, Chemistry 2022, 28, e202104614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Liebhold M, Xie X, Li S-M, Org. Lett 2012, 14, 4882–4885. [DOI] [PubMed] [Google Scholar]
- [196].Liebhold M, Li S-M, Org. Lett 2013, 15, 5834–5837. [DOI] [PubMed] [Google Scholar]
- [197].Scull EM, Bandari C, Johnson BP, Gardner ED, Tonelli M, You J, Cichewicz RH, Singh S, Appl. Microbiol. Biotechnol 2020, 104, 7853–7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Elshahawi SI, Cao H, Shaaban KA, Ponomareva LV, Subramanian T, Farman ML, Spielmann HP, Phillips GN, Thorson JS, Singh S, Nat. Chem. Biol 2017, 13, 366–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Tanner ME, Nat. Prod. Rep 2014, 32, 88–101. [DOI] [PubMed] [Google Scholar]
- [200].Gardner ED, Dimas DA, Finneran MC, Brown SM, Burgett AW, Singh S, Catalysts 2020, 10, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Colombano A, Dalponte L, Dall’Angelo S, Clemente C, Idress M, Ghazal A, Houssen WE, Angew. Chem. Int. Ed 2023, 62, e202215979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Metzger U, Schall C, Zocher G, Unsöld I, Stec E, Li S-M, Heide L, Stehle T, Proc. Natl. Acad. Sci. USA 2009, 106, 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Ostertag E, Zheng L, Broger K, Stehle T, Li S-M, Zocher G, J. Mol. Biol 2021, 433, 166726. [DOI] [PubMed] [Google Scholar]
- [204].Eaton SA, Ronnebaum TA, Roose BW, Christianson DW, Biochemistry 2022, 61, 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Aoun AR, Mupparapu N, Nguyen DN, Ho Kim T, Nguyen CM, Pan Z, Elshahawi SI, ChemCatChem 2023, 15, e202300423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Roose BW, Christianson DW, Biochemistry 2019, 58, 3232–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Eggbauer B, Schrittwieser JH, Kerschbaumer B, Macheroux P, Kroutil W, ChemBioChem 2022, 23, e202200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Mori T, Zhang L, Awakawa T, Hoshino S, Okada M, Morita H, Abe I, Nat. Commun 2016, 7, 10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Liao G, Mai P, Fan J, Zocher G, Stehle T, Li S-M, Org. Lett 2018, 20, 7201–7205. [DOI] [PubMed] [Google Scholar]
- [210].Mai P, Zocher G, Stehle T, Li S-M, Org. Biomol. Chem 2018, 16, 7461–7469. [DOI] [PubMed] [Google Scholar]
- [211].Shen Y, Zhang L, Yang M, Shi T, Li Y, Li L, Yu Y, Deng H, Lin H-W, Zhou Y, ACS Chem. Biol 2023, 18, 123–133. [DOI] [PubMed] [Google Scholar]
- [212].Daniels PN, Lee H, Splain RA, Ting CP, Zhu L, Zhao X, Moore BS, van der Donk WA, Nat. Chem 2022, 14, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Daniels PN, van der Donk WA, Biochemistry 2023, 62, 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H, Nat. Rev. Mol. Cell Biol 2020, 21, 729–749. [DOI] [PubMed] [Google Scholar]
- [215].Bloch JS, John A, Mao R, Mukherjee S, Boilevin J, Irobalieva RN, Darbre T, Scott NE, Reymond J-L, Kossiakoff AA, Goddard-Borger ED, Locher KP, Nat. Chem. Biol 2023, 19, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Hao Y, Pierce E, Roe D, Morita M, McIntosh JA, Agarwal V, Cheatham TE, Schmidt EW, Nair SK, Proc. Natl. Acad. Sci. USA 2016, 113, 14037–14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Morita M, Hao Y, Jokela JK, Sardar D, Lin Z, Sivonen K, Nair SK, Schmidt EW, J. Am. Chem. Soc 2018, 140, 6044–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Estrada P, Morita M, Hao Y, Schmidt EW, Nair SK, J. Am. Chem. Soc 2018, 140, 8124–8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Zheng Y, Cong Y, Schmidt EW, Nair SK, Acc. Chem. Res 2022, 55, 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Struck A-W, Bennett MR, Shepherd SA, Law BJC, Zhuo Y, Wong LS, Micklefield J, J. Am. Chem. Soc 2016, 138, 3038–3045. [DOI] [PubMed] [Google Scholar]
- [221].Losey HC, Jiang J, Biggins JB, Oberthür M, Ye X-Y, Dong SD, Kahne D, Thorson JS, Walsh CT, Chem. Biol 2002, 9, 1305–1314. [DOI] [PubMed] [Google Scholar]
- [222].Losey HC, Peczuh MW, Chen Z, Eggert US, Dong SD, Pelczer I, Kahne D, Walsh CT, Biochemistry 2001, 40, 4745–4755. [DOI] [PubMed] [Google Scholar]
- [223].Fu X, Albermann C, Jiang J, Liao J, Zhang C, Thorson JS, Nat. Biotechnol 2003, 21, 1467–1469. [DOI] [PubMed] [Google Scholar]
- [224].Fu X, Albermann C, Zhang C, Thorson JS, Org. Lett 2005, 7, 1513–1515. [DOI] [PubMed] [Google Scholar]
- [225].Peltier-Pain P, Marchillo K, Zhou M, Andes DR, Thorson JS, Org. Lett 2012, 14, 5086–5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Gantt RW, Peltier-Pain P, Singh S, Zhou M, Thorson JS, Proc. Natl. Acad. Sci. USA 2013, 110, 7648–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Brieke C, Yim G, Peschke M, Wright GD, Cryle MJ, Chem. Commun 2016, 52, 13679–13682. [DOI] [PubMed] [Google Scholar]
- [228].Sarkar S, Gu W, Schmidt EW, ACS Chem. Biol 2022, 17, 2165–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Weeks AM, Wells JA, Chem. Rev 2020, 120, 3127–3160. [DOI] [PubMed] [Google Scholar]
- [230].Weeks AM, Wells JA, Curr. Protoc. Chem. Biol 2020, 12, e79. [DOI] [PubMed] [Google Scholar]
- [231].Weeks AM, Wells JA, Nat. Chem. Biol 2018, 14, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Tan X, Yang R, Liu C-F, Org. Lett 2018, 20, 6691–6694. [DOI] [PubMed] [Google Scholar]
- [233].Hempfling JP, Sekera ER, Sarkar A, Hummon AB, Pei D, J. Am. Chem. Soc 2022, 144, 21763–21771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Nieuwenhuis J, Brummelkamp TR, Trends Cell Biol 2019, 29, 80–92. [DOI] [PubMed] [Google Scholar]
- [235].Schumacher D, Helma J, Mann FA, Pichler G, Natale F, Krause E, Cardoso MC, Hackenberger CPR, Leonhardt H, Angew. Chem. Int. Ed 2015, 54, 13787–13791. [DOI] [PubMed] [Google Scholar]
- [236].Gerlach M, Stoschek T, Leonhardt H, Hackenberger CPR, Schumacher D, Helma J, Enzyme-Mediat. Ligation Methods, Springer, New York, NY, 2019, p. 327. [DOI] [PubMed] [Google Scholar]
- [237].Schumacher D, Leonhardt H, Hackenberger CPR, Helma J, Bioconjugation Methods Protoc, Springer, New York, NY, 2019, p. 167. [Google Scholar]
- [238].Sardar D, Hao Y, Lin Z, Morita M, Nair SK, Schmidt EW, J. Am. Chem. Soc 2017, 139, 2884–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Cong Y, Scesa PD, Schmidt EW, ACS Synth. Biol 2022, 11, 3699–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Liebscher S, Schöpfel M, Aumüller T, Sharkhuukhen A, Pech A, Höss E, Parthier C, Jahreis G, Stubbs MT, Bordusa F, Angew. Chem. Int. Ed 2014, 53, 3024–3028. [DOI] [PubMed] [Google Scholar]
- [241].Liebscher S, Bordusa F, Enzyme-Mediat. Ligation Methods, Springer, New York, NY, 2019, pp. 111–133. [Google Scholar]
- [242].Liebscher S, Mathea S, Aumüller T, Pech A, Bordusa F, ChemBioChem 2021, 22, 1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Meyer C, Liebscher S, Bordusa F, Bioconjugate Chem 2016, 27, 47–53. [DOI] [PubMed] [Google Scholar]
- [244].Toplak A, Nuijens T, Quaedflieg PJLM, Wu B, Janssen DB, Adv. Synth. Catal 2016, 358, 2140–2147. [Google Scholar]
- [245].Schmidt M, Toplak A, Quaedflieg PJLM, Ippel H, Richelle GJJ, Hackeng TM, van Maarseveen JH, Nuijens T, Adv. Synth. Catal 2017, 359, 2050–2055. [Google Scholar]
- [246].Schmidt M, Nuijens T, Enzyme-Mediat. Ligation Methods, Springer, New York, NY, 2019, p. 43. [Google Scholar]
- [247].Wills R, Adebomi V, Raj M, ChemBioChem 2021, 22, 52–62. [DOI] [PubMed] [Google Scholar]
- [248].Gu W, Dong S-H, Sarkar S, Nair SK, Schmidt EW, Methods Enzymol 2018, 604, 113–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Houssen WE, Methods Mol. Biol. Clifton NJ 2019, 2012, 193–210. [DOI] [PubMed] [Google Scholar]
- [250].Koehnke J, Bent A, Houssen WE, Zollman D, Morawitz F, Shirran S, Vendome J, Nneoyiegbe AF, Trembleau L, Botting CH, Smith MCM, Jaspars M, Naismith JH, Nat. Struct. Mol. Biol 2012, 19, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J, Proc. Natl. Acad. Sci. USA 2005, 102, 7315–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Ruffner DE, Schmidt EW, Heemstra JR, ACS Synth. Biol 2015, 4, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Sarkar S, Gu W, Schmidt EW, ACS Catal 2020, 10, 7146–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].McIntosh JA, Robertson CR, Agarwal V, Nair SK, Bulaj GW, Schmidt EW, J. Am. Chem. Soc 2010, 132, 15499–15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Oueis E, Nardone B, Jaspars M, Westwood NJ, Naismith JH, ChemistryOpen 2016, 6, 11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Lee H, Park Y, Kim S, Biochemistry 2017, 56, 4927–4930. [DOI] [PubMed] [Google Scholar]
- [257].Barber CJS, Pujara PT, Reed DW, Chiwocha S, Zhang H, Covello PS, J. Biol. Chem 2013, 288, 12500–12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Ho YTC, Schittenhelm RB, Iftime D, Stegmann E, Tailhades J, Cryle MJ, ChemBioChem 2023, 24, e202200686. [DOI] [PubMed] [Google Scholar]
- [259].Xu Q, Mengin-Lecreulx D, Patin D, Grant JC, Chiu H-J, Jaroszewski L, Knuth MW, Godzik A, Lesley SA, Elsliger M-A, Deacon AM, Wilson IA, Structure 2014, 22, 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Elshahawi SI, Ramelot TA, Seetharaman J, Chen J, Singh S, Yang Y, Pederson K, Kharel MK, Xiao R, Lew S, Yennamalli RM, Miller MD, Wang F, Tong L, Montelione GT, Kennedy MA, Bingman CA, Zhu H, Phillips GN, Thorson JS, ACS Chem. Biol 2014, 9, 2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Yang KK, Wu Z, Arnold FH, Nat. Methods 2019, 16, 687–694. [DOI] [PubMed] [Google Scholar]
- [262].Pyser JB, Chakrabarty S, Romero EO, Narayan ARH, ACS Cent. Sci 2021, 7, 1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Wang J-B, Li G, Reetz MT, Chem. Commun 2017, 53, 3916–3928. [DOI] [PubMed] [Google Scholar]
- [264].Arnold FH, Angew. Chem. Int. Ed 2018, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Bell EL, Finnigan W, France SP, Green AP, Hayes MA, Hepworth LJ, Lovelock SL, Niikura H, Osuna S, Romero E, Ryan KS, Turner NJ, Flitsch SL, Nat. Rev. Methods Primer 2021, 1, 1–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.