

Abstract
PURPOSE OF REVIEW:
This article discusses the foundational concepts of genetic treatment strategies employed in neuromuscular medicine, as well as the importance of genetic testing as a requirement for applying gene-based therapy.
RECENT FINDINGS:
Gene therapies have become a reality for several neuromuscular disorders. Exon-skipping and (in Europe) ribosomal read-through approaches are currently available to a subset of patients with Duchenne muscular dystrophy. Microdystrophin gene replacement has shown promise and is nearing the final stages of clinical trials. Numerous gene-based therapies for other muscular dystrophies and congenital myopathies are progressing toward approval as well.
SUMMARY:
Muscular dystrophies and congenital myopathies are a heterogenous group of hereditary muscle disorders. Confirming a diagnosis with genetic testing is not only critical for guiding management, but also an actual prerequisite for current and future gene therapies. Recessive loss-of-function or dominant haploinsufficiency disorders may be treated with gene replacement strategies, whereas dominant negative and toxic gain-of-function disorders are best addressed with a variety of knockdown approaches. It is important to recognize that many therapeutics are mutation specific and will only benefit a subset of individuals with a specific disease.
INTRODUCTION
Gene therapy refers to treatments that involve delivery or modification of genetic material. Genetic-based treatments designed to address the underlying cause of hereditary disorders including muscular dystrophies and congenital myopathies and modify the disease course have not existed until recently. Historically, patients have been provided with aggressive supportive care through multidisciplinary clinics. Genetic treatments for muscular dystrophies and congenital myopathies have been at the forefront of the development of gene-based therapies for years, and several treatments have been approved by the US Food and Drug Administration (FDA).
This article will review the basics of neuromuscular genetics and classes of disease mechanisms, categories of gene therapy strategies and how they address the underlying mechanism, and key examples of current and future gene-based therapies for muscular dystrophies and congenital myopathies. This is an extremely large and rapidly advancing field. This article will not cover specific gene therapies for other neuromuscular disorders such as spinal muscular atrophy, amyotrophic lateral sclerosis, mitochondrial myopathies, and metabolic myopathies; however, we have attempted to write this article with a general approach to gene therapy strategy design, so that the principles may be learned and applied to other disorders. This article will also not review pharmacologic therapies targeting disease mechanisms at the protein level such as enzyme replacement.
CLASSES OF DISEASE MECHANISMS
Muscular dystrophies and congenital myopathies are phenotypically and genotypically heterogenous groups of disorders.1 Hundreds of disease-related genes have been identified for these disorders. Gene-specific therapy approaches may be designed once a causative gene is known and the mechanism of disease is understood enough to be categorized as one of the following (FIGURES 12-1, 12-2, and 12-3).
FIGURE 12-1.
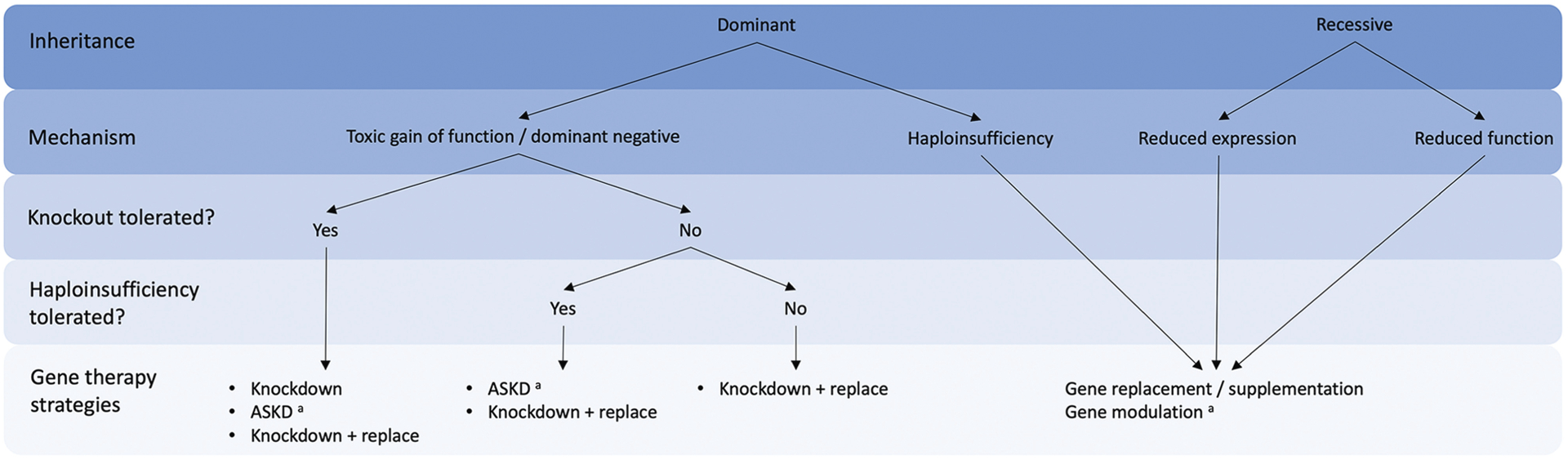
Decision tree to guide selection of gene therapy treatment strategy based on disease mechanism.
ASKD = allele-specific knockdown.
a Mutation-specific therapy.
FIGURE 12-2.
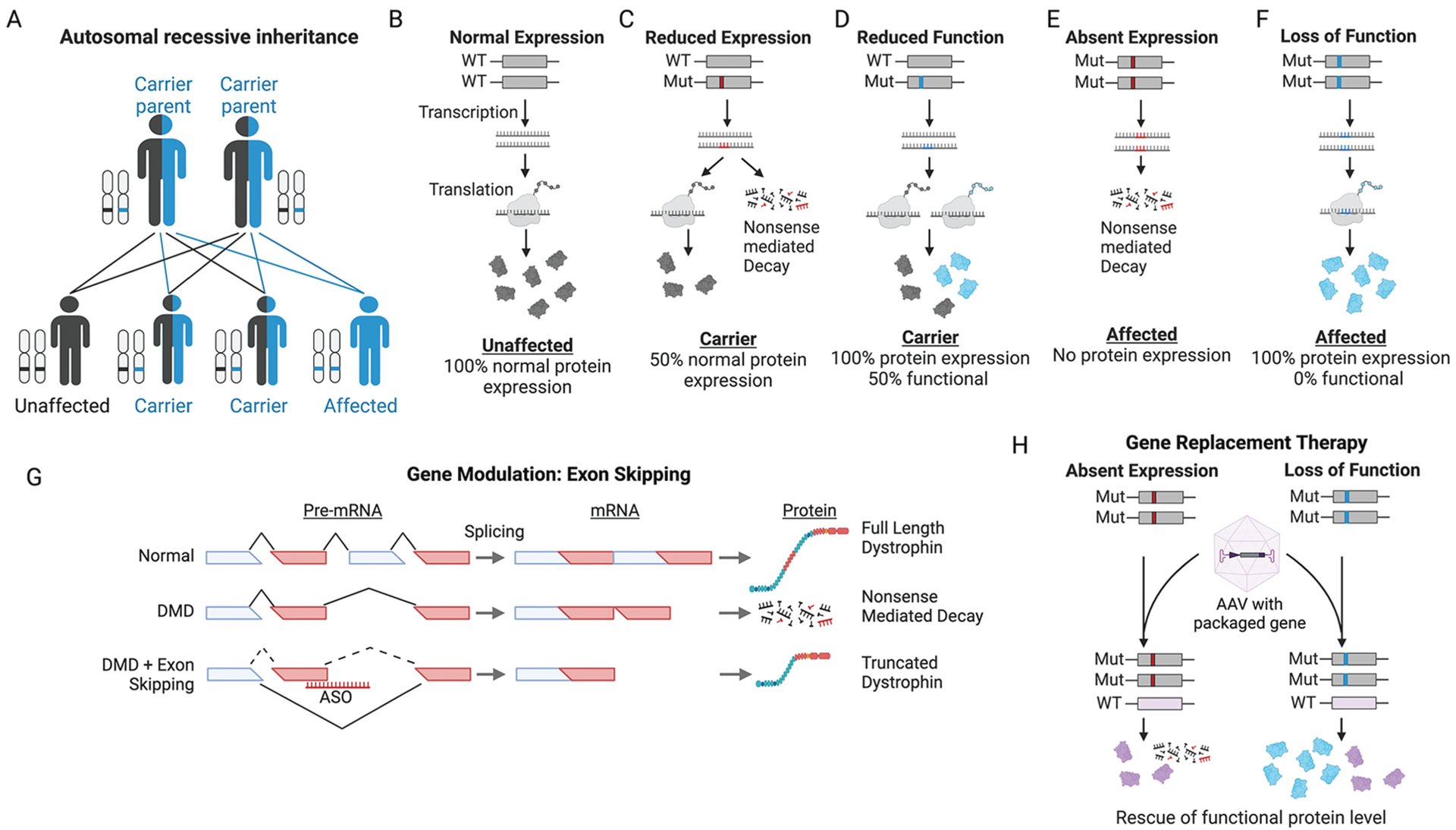
Recessive disease mechanisms and treatment strategies. A, Autosomal recessive inheritance pattern of disease. B-F, Disease mechanisms of recessive disorders. Red represents mutations that reduce expression (eg, nonsense, frameshift). Blue represents mutations that impact function (eg, missense, in-frame deletion). Endogenous functioning proteins are colored gray and nonfunctioning proteins are colored blue. G, Illustration demonstrating mechanism of exon-skipping therapeutic strategy for Duchenne muscular dystrophy (DMD). Individual with DMD contains out-of-frame deletion of an exon, leading to nonsense-mediated decay of transcript, and no production of dystrophin protein. Exon-skipping therapies are achieved via antisense oligonucleotides (ASOs) designed to sterically block splice definition elements, commonly at exon/intron boundaries. In DMD, exons adjacent to the out-of-frame deletion are targeted, resulting in their removal during pre-RNA splicing, in effect enlarging the deletion but restoring the reading frame and resulting in production of a truncated dystrophin protein. H, Illustration demonstrating gene replacement therapy for recessive disorders due to absent expression (red mutations) or loss of function (blue mutations). An adeno-associated virus (AAV) provides a functional copy of the gene (purple). Nonfunctioning proteins are blue, and functional transgenic proteins from AAV are purple.
Mut = mutant; WT = wild type.
Figure created with BioRender.
FIGURE 12-3.
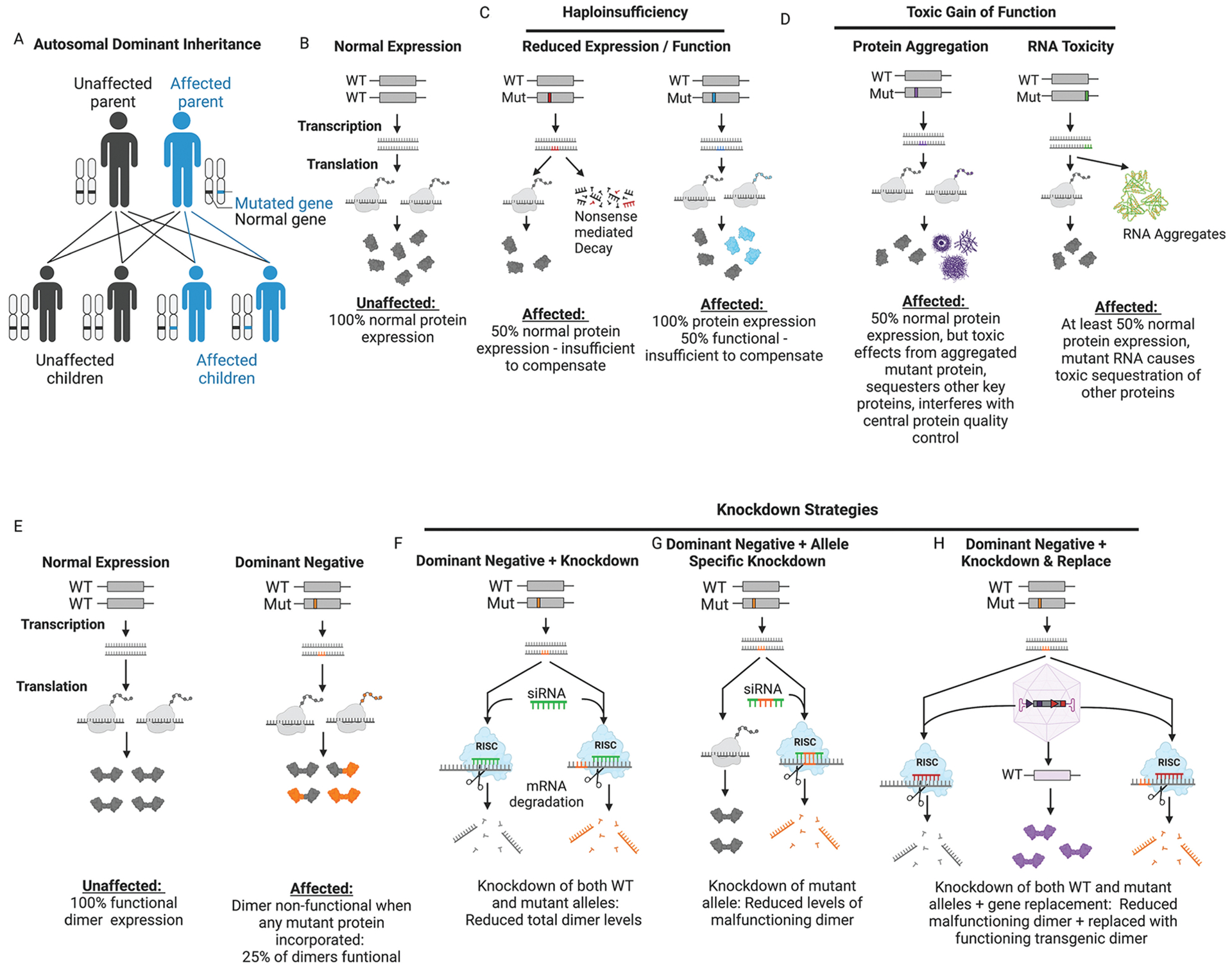
Dominant disease mechanisms and treatment strategies. A, Autosomal dominant inheritance pattern of disease. B-F, Disease mechanisms of dominant disorders. Red represents mutations that reduce expression (eg, nonsense, frameshift). Blue represents mutations that impact function (eg, missense, in-frame deletion). Endogenous functioning proteins are colored gray and nonfunctioning proteins are colored blue. C, In disorders of haploinsufficiency, greater than 50% functioning protein is required to prevent disease. D, Gain-of-function mechanisms refer to those involving increased protein levels (gene duplication or increased stability of mutant protein), hyperactivity of the mutant protein, or misfolding of the mutant protein or RNA, creating aggregates that are toxic when not degraded. E, Dominant negative mechanisms are most easily illustrated in the case of proteins that form dimers or other multimeric structures. Any dimer that contains mutant protein (orange) is rendered nonfunctional. Assuming each allele produces an equal amount of protein with equal stability, only 25% of dimers will be functional. F, Global knockdown of both wild-type and mutant alleles is an ideal treatment strategy for dominant disorders if absence of the gene is tolerated and not deleterious. One method for this is via small inhibitory RNA (siRNA) which causes degradation of the target mRNA. G, Mutant allele-specific knockdown is preferred when knockout is deleterious but haploinsufficiency is tolerated. This can be achieved by designing siRNA (or other antisense knockdown technology) to preferentially bind to the mutant allele. H, In cases where neither knockout nor haploinsufficiency are tolerated, a knockdown-and-replace approach may be required. This can be achieved via viral delivery of interference RNA (red) targeting the affected gene and simutaneous gene replacement using a codon-optimized transgene (purple) to avoid knockdown.
Mut = mutant; RISC = RNA-induced silencing complex; WT = wild type.
Figure created with BioRender.
- Recessive inheritance
- Reduced expression
- Loss of function with preserved expression
- Dominant inheritance
- Toxic gain of function
- Dominant negative
- Haploinsufficiency
Of note, disorders associated with mitochondrial DNA mutations are not included in this classification, but they deserve their own discussion. These classifications are generalizations and are an oversimplification, especially for many dominant disorders with complex disease processes which may involve multiple mechanisms2; however, they are useful for learning and conceptualization.
It is also important to mention X-linked disorders. They commonly manifest with a recessive mechanism of disease as in Duchenne muscular dystrophy (DMD), which primarily affects males due to lack of a second X chromosome. However, some X-linked myopathies such as those associated with FHL1 can behave in a dominant manner, affecting both males and females (FIGURE 12-1).
Recessive disorders require both copies of a gene to be faulty, resulting in reduced expression or impaired function of the encoded protein (FIGURES 12-2A to 12-2F). An example is Pompe disease, which is due to recessive mutations in GAA which encodes acid alpha-1,4-glucosidase, a lysosomal protein involved in glycogen breakdown.3 Disease results from either reduced expression of alpha-1,4-glucosidase or production of a protein with impaired function. The end result from either type of mutation is reduced enzymatic activity, which can be measured in tissues such as white blood cells.3 The consequences of reduced enzymatic activity can also be seen in muscle biopsies from patients, which demonstrate glycogen accumulation. Enzymatic activity in blood and glycogen levels in tissue can both be considered as biomarkers of the disease, and can even be used to monitor response to treatment.3 Other examples, albeit X-linked, are Duchenne and Becker muscular dystrophies.4 Duchenne muscular dystrophy results from DMD mutations causing complete loss of dystrophin production, whereas Becker muscular dystrophy results from mutations causing impaired function or reduced levels of dystrophin. Gene therapy approaches for recessive disorders are aimed at replacing or restoring lost function (FIGURES 12-1, 12-2G, and 12-2H).
Dominant disorders are caused by only one defective copy of a gene (FIGURE 12-3A).2 Therefore, individuals have one normal and one mutated allele. This mutant allele can be inherited from an affected parent, or it can occur sporadically (de novo) from a new mutation developing in a patient without a family history of the disorder. At least three generalized molecular mechanisms exist that can result in dominantly inherited disorders (FIGURES 12-3B to 12-3E).Gene therapy approaches for dominantly inherited disorders are mostly aimed at knocking down or reducing levels of the mutant mRNA, but with some exceptions (FIGURE 12-3F to 12-3H).5
Haploinsufficiency
Haploinsufficiency refers to disorders where mutations cause only half the amount of functional protein to be produced, and this amount of protein is insufficient for normal cellular function (FIGURE 12-3C). An example of haploinsufficiency is facioscapulohumeral muscular dystrophy type 2, which is due to heterozygous loss-of-function mutations in SMCHD1, a methyltransferase that is required to prevent inappropriate expression of toxic DUX4.6 Haploinsufficiency disorders can in theory be addressed by any treatment that increases levels of functioning protein. One such approach would be gene replacement therapy (FIGURE 12-1).
Gain of Function
A gain-of-function mechanism results from mutations that either increase the protein’s activity, prolong its stability and thereby increase its effect in the cell, or cause the protein (or RNA) to gain some additional toxic function, unrelated to its given role (FIGURE 12-3D).2 One example of a muscle disease with a likely toxic gain-of-function mechanism is due to mutations in the Z-disc protein myotilin.7 Point mutations in this structural protein cause a vacuolar myopathy with Z-disc abnormalities and prominent aggregates known as spheroid bodies.7 Myotilin mutations cause it to misfold and become insoluble, thereby reducing its turnover.8 The prominent aggregates in muscle contain not only myotilin, but also other sarcomeric proteins like desmin, presumably due to its critical role in facilitating protein-protein interactions of the skeletal muscle cytoskeleton.9 Additionally, absence of myotilin does not cause abnormalities in mouse skeletal muscle, arguing against a dominant negative or haploinsufficiency mechanism.10 Treatments for myotilinopathies have centered around knockdown of the mutant allele (FIGURE 12-3G).11 Another example of a disorder with a toxic gain-of-function mechanism is myotonic dystrophy type 1 (DM1).12 It is caused by an expansion of CTG repeats within the 3’ untranslated region of the DMPK gene. This expansion results in production of toxic DMPK RNA, which causes aberrant alternative splicing of many unrelated genes, some of which include MBLN1, CLCN1, and TWNK.12 It is the downstream alteration of these bystander proteins that results in many of the clinical features observed in DM1. Several treatments for DM1 are in development. To address the toxic gain of function, many of these therapies aim to knockdown or reduce levels of DMPK RNA.12
Dominant Negative
A dominant negative mechanism results from mutations that negate the activity of the functioning allele (FIGURE 12-3E).2 This is often seen with proteins that multimerize, where each protein complex that the mutant protein is a part of is rendered nonfunctional (FIGURE 12-3E). For example, if a protein of interest forms a dimer, and if equal amounts of normal and mutant proteins are assumed, only 25% of dimers would be expected to be normal, and the remaining 75% would be nonfunctional due to the presence of at least one mutant protein (FIGURE 12-3E).2 Collagen VI–related dystrophies may result from dominant negative mutations in the COL6 genes, resulting in collagen subunits that disrupt multimerization to form collagen VI microfibrils.13 Absence of collagen VI is also not tolerated, as evidenced by recessive loss-of-function mutations causing disease as well.13 Selective knockdown of just the mutant allele is an ideal treatment strategy for dominant collagen VI–related dystrophies and other disorders with dominant negative mechanisms, as it avoids the potential damaging effects of complete knockdown (FIGURES 12-1, 12-3F, and 12-3G).14 In fact, allele-specific knockdown appears to have therapeutic potential in preclinical studies for dominantly inherited collagen VI–related dystrophies as well as dynamin-2–related centronuclear myopathy.15
GENETIC TREATMENT STRATEGIES EMPLOYED IN NEUROMUSCULAR MEDICINE
Several categories of gene-based treatment strategies exist, but the strategy optimally suited to treat a specific disease largely depends on the disorder’s mechanistic category (FIGURE 12-1). Genetic testing is the necessary step to having effective gene therapies for muscular dystrophies and congenital myopathies (CASE 12-1).
CASE 12-1.
A 34-year-old woman presented for an initial neuromuscular clinic appointment with 3 years of progressive weakness and a prior diagnosis of limb-girdle muscular dystrophy (LGMD). Sixteen years earlier, the patient reported being admitted to the hospital for “muscle breakdown” after presenting to the emergency department with chest pain. She reported follow-up with a rheumatologist and having a deltoid muscle biopsy that suggested muscular dystrophy. Over the past 15 years she had seen neurologists for fatigue but had maintained activities of daily living. She had been offered genetic testing but had been reluctant to pursue it because of insurance concerns and having been told that no treatment was available and genetic testing would not change management. Following the delivery of her first child 3 years ago, she noticed increased weakness and difficulty carrying her son.
On neurologic examination the patient was noted to have mild proximal weakness and used her arms to push up from a chair. Her creatine kinase level was 2180 U/L. Electrodiagnostic testing confirmed a myopathy.
After undergoing genetic counseling, the patient was evaluated for hereditary muscle diseases via panel-based genetic testing, which revealed heterozygous pathogenic variants in CAPN3 (c.1468C>T, p.Arg490Tyr & c.IVS11+1G>C, splice site mutation). Due to her clinical presentation, the lack of family history (suggesting a recessive pattern of inheritance), and the autosomal recessive mutations in CAPN3, a diagnosis of autosomal recessive LGMD type 1 (LGMDR1) was established. The patient kept a personal copy of her genetic test results in a file cabinet at her home. The patient was counseled that her son and any subsequent children would be carriers of a pathogenic variant of CAPN3 but it would not manifest with weakness.
The patient registered with the Muscular Dystrophy Association and a patient advocacy group. She now participates in an LGMD-specific natural history and biomarker study aimed at clinical trial readiness for LGMDR1.
COMMENT
This case illustrates a patient with an established genetic etiology to her weakness and the importance of reengaging patients with potential genetic causes of muscle disease. While a gene therapy for LGMDR1 is currently not available, establishing the diagnosis is critical, not only for research purposes, but as a required prerequisite to receive future genetic-based treatments.
Gene therapies can be first broken into two categories: disease-gene targeting or nondisease-gene targeting.
Therapeutics targeting the disease gene (the gene that results in disease when mutated) may function at the DNA or RNA level. General categories include gene replacement, gene modulation, gene correction, and gene knockdown (FIGURES 12-2G, 12-2H, and 12-3F to 12-3H).16 Recessive loss-of-function and dominant haploinsufficiency disorders are the primary target of gene replacement and repair strategies. Gene replacement provides a functional copy of the mutated gene, whereas gene modulation modifies the mutation for functional benefit (eg, exon skipping, stop codon read-through) (FIGURES 12-2G and 12-2H).16
Knockdown strategies (allele- or nonallele-specific) are the primary approach for dominantly inherited disorders due to either toxic gain-of-function or dominant negative mechanisms (FIGURES 12-3F to 12-3H).5 And lastly, gene correction via CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) and other technologies such as prime editing have the potential to completely fix the disease-causing mutation, and could in theory apply to any hereditary disorder, regardless of mechanism.17
Nondisease gene–targeting therapies can involve modifier genes or disease gene homologs. Disease-modifying genes do not cause the disease themselves but alter the severity of disease. Modifier genes are targeted in order to modulate or correct downstream events that are either specific to a particular disease or common to many disorders of muscle, such as atrophy or fibrosis. Modulation of disease gene homologs (ie, utrophin) can in theory provide therapeutic benefit by compensating for the mutated disease gene in cases of loss-of-function disorders.18
Gene Replacement Therapies
Recessive loss-of-function as well as dominant haploinsufficiency disorders can be treated with gene replacement therapies (FIGURES 12-1 and 12-2H). Gene replacement compensates for a missing or faulty gene by providing an additional copy; this involves delivering a functional version of the gene to a person’s cells. While some other gene therapy approaches are dependent on patients having a specific type of mutation (eg, deletion of certain exons, nonsense mutation causing premature stop codon), gene replacement can apply to any mutation causing loss of function or expression.
The approach may seem simple at first, but many important considerations go into developing a successful gene replacement strategy. Key areas of consideration include mechanism of delivery to tissue, target tissue for gene expression, potential toxicity, and evading the human immune system. Gene replacement has been successfully translated to the clinic for spinal muscular atrophy.19 DMD gene replacement for Duchenne muscular dystrophy is currently being studied in phase 3 clinical trials, and may be available in the near future. Several other gene replacement therapies are in development for recessive limb-girdle muscular dystrophies and congenital myopathies.
Much of what is known about gene therapy has been learned through the development and advancement of a treatment for Duchenne muscular dystrophy. Previously, adenoviral vectors were used to deliver the replacement gene, but they had strong immunogenic effects, which were fatal in some cases.20 Adeno-associated viruses (AAVs)—smaller, less immunogenic, replication-defective viruses that are not known to cause disease—are now used to deliver the replacement gene. These clinically administered AAVs are unable to use the host cell machinery to replicate. Additionally, their genomes largely persist as an episome (an extrachromosomal, nonintegrated, circular DNA molecule) within transduced cells, the importance of which is highlighted by lentiviral vectors that were previously used but can lead to malignancy following integration of their genome into the host’s.20 AAV capsids have been identified that preferentially target certain cell types.20 Additional specificity is achieved by selecting a promoter that expresses the transgene only in tissues of interest.20 In DMD, a muscle-specific promoter is used that is active in all muscle types including the heart and diaphragm. One limitation of AAVs is their limited packaging capacity of approximately 4.7 kilobases (kb).20 DMD is the largest gene in the genome at roughly 2.4 million bases long, with approximately 14 kb of coding sequence. To circumvent this issue, a microdystrophin transgene capable of fitting within an AAV was designed. This microdystrophin transgene was based on a patient with Becker muscular dystrophy with a large deletion, involving approximately 46% of DMD, but only a mild phenotype.21
Additional barriers to gene therapy involve the immune system. Some patients have preexisting neutralizing antibodies to AAVs within the natural environment, causing clearance of the vectors before they can transduce cells.22 Also, successfully transduced cells will load AAV capsid proteins onto major histocompatibility complex class 1 molecules, which then present them to cytotoxic CD8+ T cells, causing elimination of transduced cells.22 Another issue is development of an immune response against the newly expressed transgenic protein in an individual with a recessive disorder from loss of expression.22 This has clearly been demonstrated in the micro-dystrophin gene transfer trials for DMD, and tends to occur in patients with N-terminal DMD deletions overlapping with parts of the micro-dystrophin transgene. Another issue is the theoretical need for redosing as it is currently unknown how long these treatments may last. The immune response that develops following initial treatment may be a barrier to repeat dosing.22 Hepatotoxicity is a common adverse event associated with AAV gene therapy,22 and four children died of complications related to liver failure during a clinical trial when treated with MTM1 gene replacement therapy for X-linked myotubular myopathy.23
Gene Modulation
Gene modulation refers to treatment strategies that alter the disease gene for therapeutic benefit. They do not provide a replacement copy nor do they aim to knock down the disease gene. These types of treatments are commonly mutation specific, meaning they cannot be widely applied to all individuals with a certain disorder, regardless of their exact mutation (FIGURE 12-1).
Exon skipping using antisense oligonucleotides for the treatment of Duchenne muscular dystrophy is a key example (FIGURE 12-2G).24
Antisense oligonucleotides are small, single-stranded sequences of nucleic acid containing a variety of modifications to the backbone to improve stability against nucleases (FIGURE 12-4).25 Antisense oligonucleotides are designed to be complementary to a sequence of interest. When antisense oligonucleotides bind to their target RNA, they can induce several changes,26 which include RNA destruction via ribonuclease H (RNase H) recruitment or RNA modification to cause changes in splicing, impair translation, or alter RNA stability (FIGURES 12-4C to 12-4G).26 RNase H–mediated destruction of RNA will be discussed further in the section on knockdown gene therapies. When antisense oligonucleotides are designed to bind intron/exon junctions or other splicing elements, they physically block the binding of splicing factors, resulting in altered splicing of pre-mRNA to either include or exclude targeted exons.26 Antisense oligonucleotides can also be designed to sterically block translation by binding the 5’ untranslated region of a target mRNA, or reduce mRNA stability by targeting polyadenylation sites.26
FIGURE 12-4.
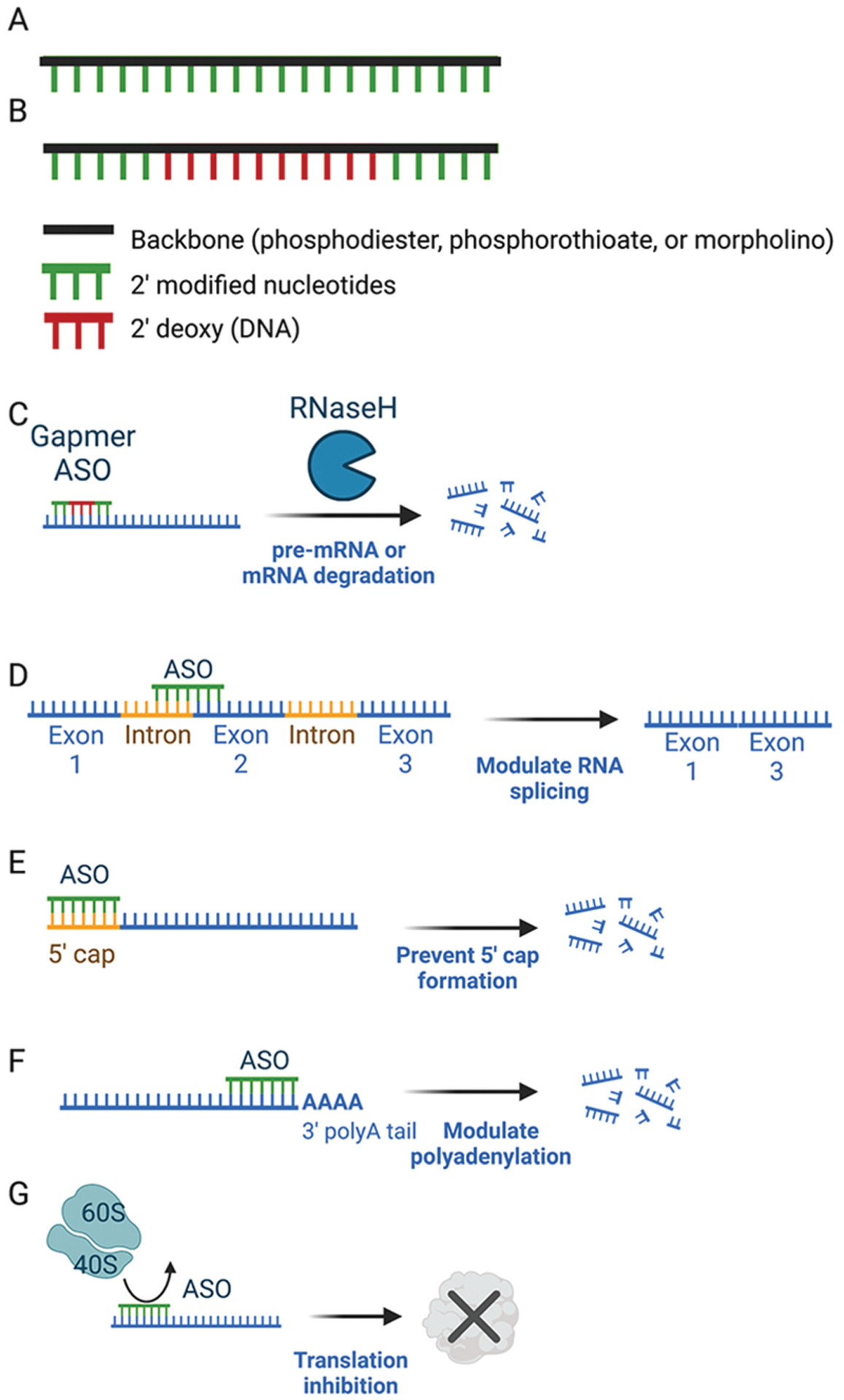
Oligonucleotide mechanisms. A, Example of 2’ modified antisense oligonucleotide (ASO) that does not support ribonuclease H (RNase H) activity but could be used for steric blockade of target sequences. The 2’-sugar modification is used along the entire length of the ASO. B, Example of gapmer ASO that supports RNase H activity. The middle section of the ASO contains 2’ unmodified nucleotides which allow for RNase H cleavage in the central region of the ASO. The outer portions of the ASO with 2’ modified nucleotides have increased target binding affinity to RNA. Representative mechanisms of action are shown for gapmers causing RNA degradation via RNAse H (C), steric-blocking ASOs modulating splicing (D), blocking 5’ cap formation (E), modulating polyadenyulation (F), or preventing initiation of protein translation (G).
Figure created with BioRender.
In the case of Duchenne muscular dystrophy, many patients have deletions or duplications that result in out-of-frame mutations, causing nonsense-mediated decay and no dystrophin production. The milder Becker muscular dystrophy is commonly caused by in-frame DMD deletions, producing a truncated, semifunctional dystrophin protein. This is an oversimplification as many exceptions to the “reading-frame rule” exist, and it should therefore not be used in isolation to predict phenotypic outcomes. The aim of exon-skipping therapy is to convert an out-of-frame DMD mutation into an in-frame BMD mutation.24 This is achieved by using antisense oligonucleotides to skip an additional exon, enlarging the size of the mutation in the case of deletions, but restoring the disrupted reading frame, avoiding nonsense-mediated decay, and resulting in production of a truncated, partly functional dystrophin protein.24 These therapies do not work for all individuals with DMD as they are mutation specific. Several FDA-approved exon-skipping therapies exist for a subset of individuals with DMD. Eteplirsen, an exon 51–skipping drug, is predicted to restore the reading frame for 13% of all patients with DMD.27 Golodirsen and viltolarsen, both exon 53–skipping drugs, would address 8% of patients, and lastly, casimersen, an exon 45–skipping drug, is predicted to address mutations in another 8% of boys.27 To determine if a boy with DMD is a candidate for one of these therapies, genetic testing must be performed to identify their exact mutation (FIGURE 12-5) (CASE 12-2).
FIGURE 12-5.
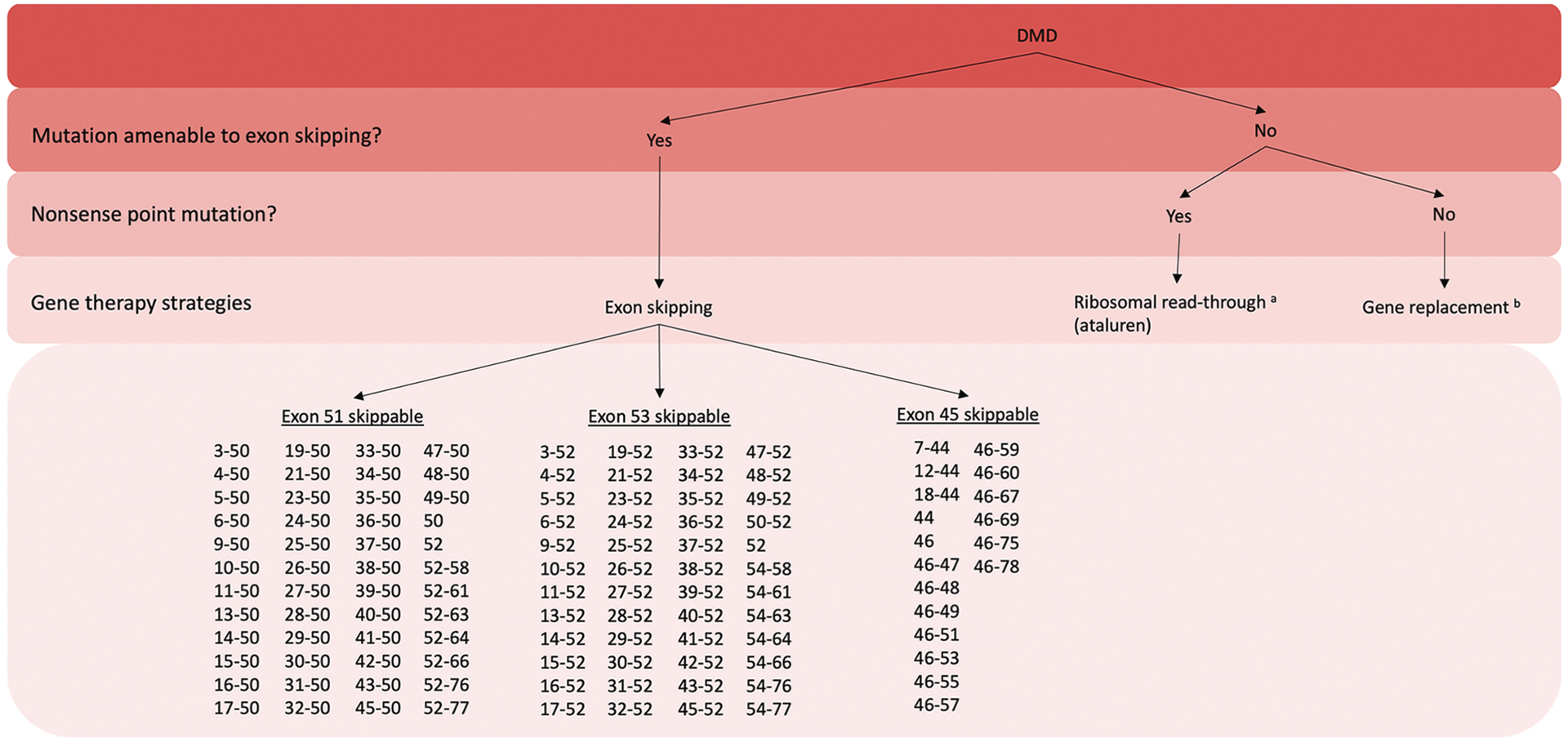
Flowchart demonstrating several gene therapy treatment options for boys with Duchenne muscular dystrophy (DMD). All currently available gene therapies are mutation specific.
a Only available in Europe.
b Not clinically available as of October 18, 2022.
Figure created with BioRender.
CASE 12-2.
A 5-year-old boy with motor delay presented to a neuromuscular clinic. He walked independently at age 19 months. His running was slow and climbing stairs one foot after another was difficult. No family history of nerve or muscle problems was reported.
Examination demonstrated large calves, toe walking, pronounced lordosis, difficulty standing up from the ground and the presence of Gowers sign, and slow running.
Laboratory testing showed a creatine kinase level of 9646 U/L. Genetic testing was performed for a presumed diagnosis of Duchenne muscular dystrophy. The patient’s test was “positive” with a deletion of DMD exons 48 to 50. This deletion is treatable with eteplirsen, an exon 51–skipping antisense oligonucleotide, which he was started on along with oral corticosteroids.
COMMENT
This case illustrates a boy with Duchenne muscular dystrophy with a mutation amenable to exon 51–skipping therapy. Exon-skipping therapies are mechanistically targeted toward specific DMD mutations and are therefore beneficial to only a subset of individuals.
Another example of gene-modulation therapy for DMD is ataluren. This drug promotes ribosomal read-through of nonsense mutations and is therefore only applicable to boys with nonsense point mutations in DMD, which is approximately 10% to 15% of all patients with DMD.24 Bypassing the nonsense mutation allows translation to continue and produces a functioning dystrophin protein (CASE 12-3). This compound is approved for use in Europe, but has not been approved by the FDA.24 Gene-modulation therapies clearly highlight the importance of knowing an individual’s exact mutation, as it can significantly change management (CASE 12-4) (FIGURE 12-5).
CASE 12-3.
A 30-month-old boy initially presented for neurologic evaluation with motor delay. He started walking at 16 months. No other relevant medical or family history was reported.
Examination at age 30 months revealed a waddling gait and Gowers sign. Creatine kinase level was 11,341 U/L. Genetic testing was performed for presumed Duchenne muscular dystrophy but identified no deletion or duplication. Full DNA sequencing of DMD from peripheral blood identified a nonsense mutation (c.10141C>T) within exon 70 resulting in a premature stop codon p.(Arg3381X), presumably leading to nonsense-mediated decay of DMD transcript and no dystrophin production. Review of the Leiden Open Variation Database revealed that all prior reports of this mutation have been associated with a Duchenne muscular dystrophy phenotype.
At 6 years old, the patient was started on prednisone 0.75 mg/kg/d and vitamin D. At 8 years old, he had a waddling gait, lordosis, a positive Gowers sign, proximal muscle weakness, and was unable to run. He also had short stature, calf pseudohypertrophy, and tight heel cords. His family had questions about future therapies. Ataluren and micro-dystrophin gene replacement clinical trials were discussed. The family was provided with additional information regarding ongoing studies at clinicaltrials.gov, and referred to patient advocacy groups with updated information on clinical studies.
COMMENT
This case illustrates a boy with Duchenne muscular dystrophy due to a nonsense point mutation. This mutation is not treatable with any exon-skipping therapies currently available in the United States. In Europe, this boy could be treated with ataluren, a compound that causes ribosomal read-through of premature stop codons. In the future, if microdystrophin gene replacement proves efficacious, many more individuals with Duchenne muscular dystrophy could be treated.
CASE 12-4.
A 4-year-old boy presented to his pediatrician because of difficulty running and large calves. He did not walk until age 17 months. The mother of the boy had a brother with Duchenne muscular dystrophy (DMD) who had similar calves as the child, required a wheelchair by age 10, and died at age 23. Examination revealed large calves and proximal weakness of the arms and legs. Creatine kinase level was 16,385 U/L.
He was referred to a neuromuscular clinic where these findings were confirmed on exam. Duchenne muscular dystrophy was suspected and a genetic analysis of the DMD gene revealed an out-of-frame duplication of exon 2. Sixty percent of boys with this mutation have a DMD phenotype as in this patient, 31% have a Becker muscular dystrophy–like phenotype, and 9% have an intermediate phenotype.28 Prednisone was started at 0.75 mg/kg/d.
The family wanted to know if any gene therapies were available to treat their son. Based on his mutation, the neuromuscular specialist informed them of two trials the patient may qualify for: a microdystrophin gene replacement trial, and an exon-skipping trial using an adeno-associated virus (AAV) to treat exon 2 duplications.29
COMMENT
This boy had Duchenne muscular dystrophy due to an out-of-frame duplication of exon 2.This case again highlights the importance of confirming a genetic diagnosis. Although no gene therapies are currently approved for individuals with exon 2 duplications, many therapies are in development, including micro-dystrophin gene replacement and exon 2 skipping using an AAV-based approach. The exon 2–skipping approach is especially notable as it is capable of producing full-length normal dystrophin by skipping just one of the duplicated exons.
Ataluren is a drug capable of causing ribosomes to read through premature stop codons. It is available in Europe for boys with nonsense point mutations.
Knockdown
Knockdown strategies are ideally suited for treating dominantly inherited disorders (FIGURES 12-1 and 12-3). These strategies have lagged behind gene replacement strategies and are largely in preclinical stages. The various tools used in knockdown approaches, how to decide between the various knockdown strategies (allele versus nonallele specific, or knockdown and replace), and some preclinical examples in muscular dystrophies and congenital myopathies will be discussed.
The RNA interference (RNAi) pathway within cells is commonly utilized for knockdown strategies (FIGURE 12-6). Briefly, endogenous, small noncoding RNAs called microRNAs (miRNAs) bind to coding mRNAs with complementary sequences via Watson-Crick base pairing, and cause gene silencing via translational inhibition or transcript degradation (FIGURE 12-6).5 Naturally occurring miRNAs can be engineered so their sequence is complementary to any gene of interest, resulting in knockdown of the target. Similar to miRNA molecules, small interfering RNAs (siRNA) are short RNA duplexes that are synthesized in vitro and can bypass several RNAi processing steps miRNAs must go through before being capable of gene silencing (FIGURE 12-6).5 Both siRNA and miRNA can be used to therapeutically knockdown a target. Several factors are important to consider when deciding between an siRNA- or miRNA-based approach. Sustained knockdown of a gene requires repeated siRNA administration as they are degraded over time. However, miRNAs can be transcribed within a cell by placing their sequence downstream of a promoter, allowing for sustained gene silencing.5 Tissue-specific miRNA expression can be guided by promoter selection. Additional tissue specificity can be achieved by delivering the miRNA via viral vector with a tropism for certain cell types.20 Without modification, siRNAs will be distributed systemically, unless injected locally into the affected tissue. Tissue-specific targeting of systemically delivered siRNA is an area of active research.25 One common approach is to conjugate siRNA to a ligand (eg, antibody, protein, carbohydrate) that interacts with surface receptors on specific cell types.25
FIGURE 12-6.
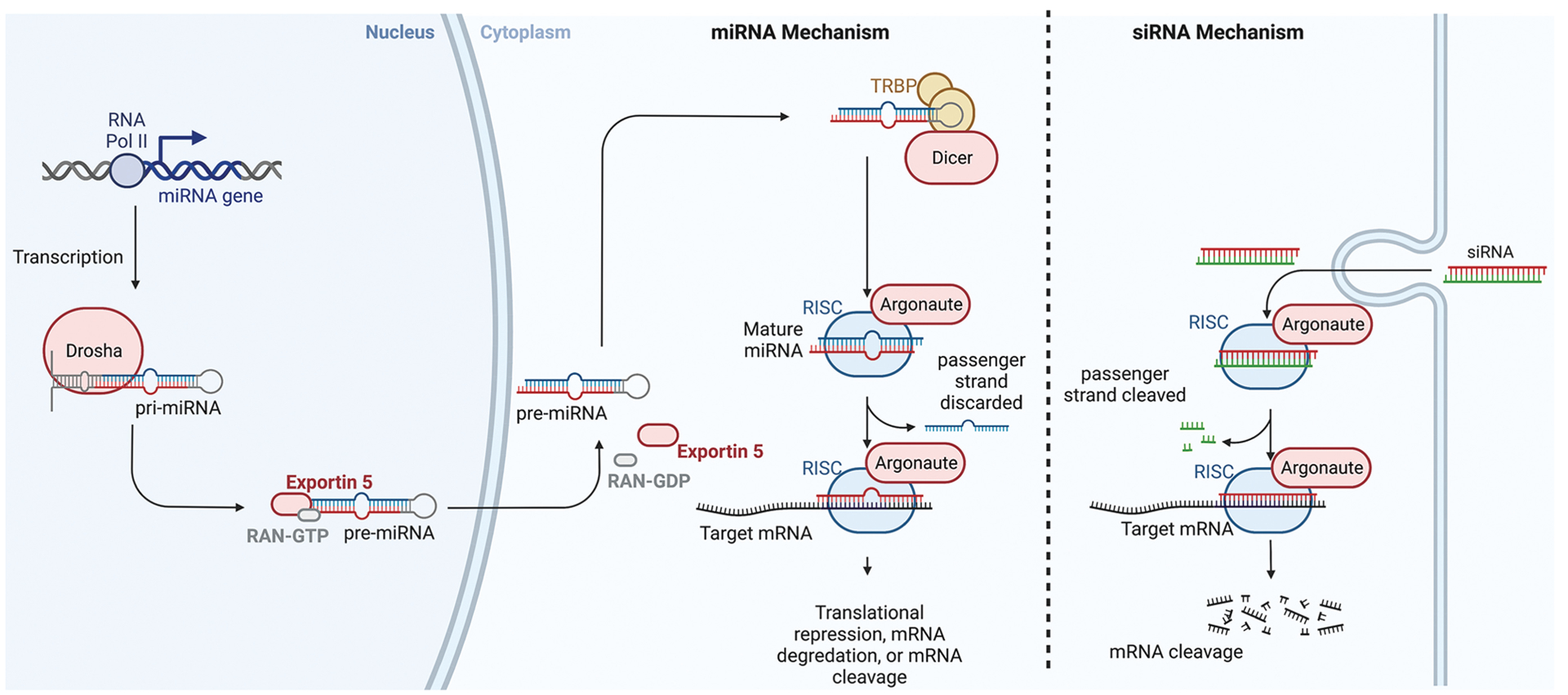
Inteference RNA pathways. Molecular mechanisms of microRNA (miRNA) and small inhibitory RNA (siRNA). miRNA transcription occurs in the nucleus via RNA polymerase II, which generates pre-miRNA. This is cleaved by drosha to form pre-miRNA. Pre-miRNA is transported to the cytoplasm via exportin-5 where it is cleaved by dicer to form miRNA. miRNA is then loaded into the RNA-induced silencing complex (RISC) and the passenger strand is discarded. The remaining miRNA guide strand directs the RISC to the target mRNA via complementary base pairing, resulting in either prevention of translation, mRNA degradation, or mRNA cleavage. siRNA can be artificially introduced. Once inside the cell it is loaded into the RISC, where Argonaute cleaves the passenger strand. The guide strand then directs the active RISC to the target mRNA. Full complementary base pairing between the siRNA and the target mRNA results in mRNA cleavage.
RAN-GDP/-GTP = guanosine-diphosphate/-triphospate–bound Ras-related nuclear protein; RNA Pol II = RNA polymerase II; TRBP = trans-activation response RNA-binding protein
Figure created with BioRender.
Another important point to consider is target specificity. SiRNAs result in degradation of a specific mRNA when near-perfect complementarity exists. MiRNAs, on the other hand, result in knockdown of numerous targets as translational inhibition only requires partial sequence complementarity.5 MiRNA shuttles are a hybrid of miRNA and siRNA, where the miRNA backbone is used to deliver an siRNA sequence targeting a specific gene of interest. This approach combines the benefits of both strategies: the sustained expression and tissue specificity of miRNA and the target specificity of siRNA.5
Another commonly utilized class of compounds used for gene silencing is antisense oligonucleotides. As discussed earlier, antisense oligonucleotides are single-stranded oligonucleotides that have undergone chemical modifications to their backbone to improve stability within cells and increase their half-life (FIGURE 12-4).25 Antisense oligonucleotides are designed to bind to target RNA by Watson-Crick base pairing. Depending on their structure, antisense oligonucleotides can be used to degrade an RNA target via the RNAse H pathway, or sterically block its target sequence, resulting in either inhibition of translation or modulation of pre-mRNA splicing.26 Gapmers, antisense oligonucleotides with chemically modified molecules at their ends and a gap of unmodified DNA in the center, are used to recruit RNAse H and promote target degradation (FIGURES 12-4B and 12-4C).26 Gapmers are able to degrade both mRNA and pre-mRNA as RNAse H localizes in both the cytoplasm and nucleus of cells.25 This is a key difference from siRNA and miRNA strategies which target only mRNA, as the RNAi machinery is strictly cytoplasmic (FIGURE 12-6).25 Nongapmer antisense oligonucleotides do not recruit RNAse H, but are capable of target knockdown by sterically blocking sequences that are key for initiating protein translation (FIGURES 12-4A and 12-4D to 12-4G).26 Similar to siRNA, antisense oligonucleotides are chemically synthesized, systemically or locally delivered, and can be modified by ligand conjugation or delivered via modified lipid nanoparticles to target specific tissues. Careful consideration of all the previously mentioned factors can help with choosing an antisense-oligonucleotide– or RNAi-mediated approach to knockdown.
Another important consideration for treatment of dominantly inherited disorders is whether allele or nonallele-specific knockdown is indicated (FIGURES 12-1, 12-3F, and 12-3G). This can be determined based on whether or not knockout or haploinsufficiency of the affected gene is tolerated. If complete knockout or less than 50% of gene expression is not detrimental, then nonallele-specific knockdown is the most simple and straightforward approach (FIGURE 12-1). This is the case for facioscapulohumeral muscular dystrophy and DUX4 knockdown, myofibrillar myopathy due to myotilin mutations, and DM1. However, if knockout is deleterious and haploinsufficiency is tolerated, an allele-specific strategy is required in order to selectively silence the mutation and avoid toxic effects of global knockdown (FIGURE 12-1). Examples include genes where disease may arise from either autosomal dominant or recessive mutations, such as RYR1, LMNA, TTN, COL6, and many others. Allele-specific knockdown is technically more difficult but feasible, even if the two transcripts differ by only one base pair, and is achieved by designing RNAi or antisense-oligonucleotide sequences that preferentially bind with the mutant allele.5 Several preclinical studies are pursuing this strategy and have shown promise in collagen VI–related myopathies, centronuclear myopathy due to DNM2 mutations, and several dominantly inherited cardiomyopathy syndromes.30
If both knockout and haploinsufficiency are deleterious, or allele-specific knockdown is not achievable, a dual gene therapy strategy combining nonallele-specific knockdown with gene replacement would be required (FIGURES 12-1 and 12-3H).5 Gene replacement in this case would require a codon-optimized transgene to avoid being silenced by the RNAi or antisense oligonucleotide treatment. This approach has proved beneficial for oculopharyngeal muscular dystrophy in preclinical mouse studies and in theory could be applied to any of the previously mentioned disease categories.31
Knockdown treatments are not yet clinically available for dominantly inherited myopathies. Several studies are currently in preclinical development for disorders such as facioscapulohumeral muscular dystrophy and centronuclear myopathy due to DNM2 mutations. Knockdown strategies are currently a reality for other dominantly inherited neuromuscular disorders such as TTR-related amyloidosis and on the horizon for amyotrophic lateral sclerosis due to SOD1 mutations.32
Gene Correction
One area of gene therapy that is worth mentioning but is not yet clinically ready is gene correction. Technologies such as CRISPR/Cas9 use double-strand DNA breaks and are capable of completely fixing disease-causing mutations.17 More recently developed technologies such as base editing and prime editing do not require double-strand breaks, and have exciting potential as well.33 The details of their mechanisms are beyond the scope of this article. As with other gene therapies, delivery of these editing tools to muscle is a major hurdle, as are immunogenicity and target specificity. These technologies are capable of causing permanent changes at the DNA level; therefore, their specificity is of utmost importance to avoid potentially harmful, permanent, off-target effects. Some of these tools work better in dividing cells, whereas others function ideally in terminally differentiated tissues.17 Skeletal muscle and quiescent satellite cells are nondividing tissues, making them difficult targets for gene correction via CRISPR/Cas9 due to low levels of the endogenous homology-directed repair machinery required for repairing a mutation.17 These are some of the many hurdles to be surmounted before gene correction becomes a reality.
CONCLUSION
Gene therapies are progressing towards reality for muscular dystrophies and congenital myopathies. Key strategies include gene replacement for recessive loss-of-function or dominant haploinsufficiency disorders and gene knockdown for dominant negative or toxic gain-of-function disorders. A confirmed genetic diagnosis is a prerequisite for gene-based therapies. While some treatment approaches may be applied to all individuals with a certain disorder (gene replacement), many gene therapies are mutation specific and apply only to a subset of individuals with a certain disease (exon skipping, allele-specific knockdown). Mutation-specific therapeutics further highlight the importance of obtaining a confirmed genetic diagnosis in individuals with muscular dystrophy or congenital myopathy.
KEY POINTS.
Genetic diseases are inherited in a recessive, dominant, or mitochondrial pattern.
Recessive disorders result from mutations causing reduced expression or loss of function despite preserved expression.
Dominant disorders result from several mechanisms including haploinsufficiency, dominant negative, or toxic gain of function.
Recessive disorders may be treated via gene therapies aimed at replacing or restoring lost function.
Dominant disorders are primarily treated via knockdown gene therapy approaches.
Dominant disorders due to haploinsufficiency, where 50% of functional protein is deleterious, may be addressed via gene replacement therapies.
Dominant toxic gain-of-function disorders result from mutations increasing a protein’s activity or stability, or by imparting an additional toxic function.
Dominant negative mechanisms commonly occur in proteins that multimerize and result from the mutant allele negating the function or activity of the normal allele.
Allele-specific knockdown is an ideal treatment approach for dominantly inherited disorders if haploinsufficiency is not deleterious.
KEY POINTS.
Gene therapies may target the disease gene or nondisease genes.
Gene therapies may be categorized as gene replacement, modulation, correction, or knockdown.
Nondisease genes may be targeted for therapeutic benefit when they are involved in downstream portions of the pathomechanism (eg, fibrosis, atrophy), or are homologs of the disease gene (ie, utrophin).
Gene replacement strategies such as microdystrophin for Duchenne muscular dystrophy are capable of addressing any DMD mutation.
Adeno-associated viruses are the primary vector used for gene therapy. Their immunogenicity is tolerable, their genomes are largely nonintegrative, and they have modifiable capsids that determine tissue tropism.
The immune system is a major barrier to gene therapy due to preexisting antibodies to adeno-associated viruses (AAVs) and clearance of transduced cells by cytotoxic CD8+ T cells after AAV capsid proteins are presented on major histocompatibility complex class I surface molecules.
KEY POINTS.
Hepatotoxicity is a major concern with adeno-associated virus–mediated gene therapies.
Exon skipping for Duchenne muscular dystrophy is a key example of a gene-modulation therapy where the disease gene is not replaced or knocked down, but instead is altered for therapeutic benefit.
Antisense oligonucleotides are capable of gene knockdown via ribonuclease H- mediated destruction of RNA or by sterically blocking protein translation initiation. In exon skipping, antisense oligonucleotides modify pre-mRNA splicing by sterically blocking exon/intron definition elements.
Exon-skipping therapies are available for a subset of boys with Duchenne muscular dystrophy. Their mutations must be put back into frame by skipping exon 51, 53, or 45.
KEY POINTS.
Cytoplasmic RNA interference pathways in cells are utilized for knockdown approaches with microRNA or small inhibitory RNA. Antisense-oligonucleotide gapmers use the nuclear and cytoplasmic RNase H pathway and are therefore capable of targeting both mRNA and pre-mRNA.
Knockdown strategies for dominantly inherited disorders may require allele specificity if haploinsufficiency is deleterious.
An additional strategy for dominantly inherited disorders with toxic gain-of-function or dominant negative mechanisms involves a combination of knockdown with gene replacement, where the transgene has been codon optimized to avoid knockdown.
RELATIONSHIP DISCLOSURE:
Dr Findlay has received personal compensation in the range of $500 to $4999 for serving as a consultant for Atheneum, Guidepoint, RiverVest and Triangle Insights Group. The institution of Dr Findlay has received research support from the American Academy of Neurology, the American Society for Gene and Cell Therapy, the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital, the LGMD-1D DNAJB6 Foundation and International Registry, and the National Institutes of Health. Dr Weihl has received personal compensation in the range of $500 to $4999 for serving as a consultant or on a scientific advisory or data safety monitoring board for Abata Therapeutics, Acceleron Pharma, Casma Therapeutics, and Sarepta Therapeutics.
Footnotes
UNLABELED USE OF PRODUCTS/INVESTIGATIONAL USE DISCLOSURE:
Drs Findlay and Weihl report no disclosure.
USEFUL WEBSITES
ClinicalTrials.gov is a registry of clinical trials. It is run by the United States National Library of Medicine at the National Institutes of Health. It is a helpful resource for physicians to answer patient questions regarding available trials, their locations, and contact information.
CUREDUCHENNE
CureDuchenne is a nonprofit organization. This specific webpage lists DMD mutations that are treatable with exon-skipping therapies.
cureduchenne.org/cure/exon-skipping
LEIDEN MUSCULAR DYSTROPHY PAGES
Leiden University Medical Center maintains this database of DMD mutations. There are many exceptions to the “reading-frame rule.” This website is useful for looking up clinical phenotypes (Duchenne versus Becker muscular dystrophy) for specific DMD mutations.
REFERENCES
- 1.Dowling JJ, Weihl CC, Spencer MJ. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat Rev Mol Cell Biol 2021; 22(11):713–732. doi: 10.1038/s41580-021-00389-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veitia RA, Caburet S, Birchler JA. Mechanisms of Mendelian dominance. Clin Genet 2018;93(3): 419–428. doi: 10.1111/cge.13107 [DOI] [PubMed] [Google Scholar]
- 3.Kohler L, Puertollano R, Raben N. Pompe disease: from basic science to therapy. Neurotherapeutics 2018;15(4):928–942. doi: 10.1007/s13311-018-0655-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waldrop MA, Flanigan KM. Update in Duchenne and Becker muscular dystrophy. Curr Opin Neurol 2019;32(5):722–727. doi: 10.1097/WCO.0000000000000739 [DOI] [PubMed] [Google Scholar]
- 5.Harper SQ. RNAi therapy for dominant muscular dystrophies and other myopathies. In: Duan D, Mendell JR, editors. Muscle gene therapy. 2nd ed. Cham: Springer, 2019:491–507. [Google Scholar]
- 6.Jia FF, Drew AP, Nicholson GA, et al. Facioscapulohumeral muscular dystrophy type 2: an update on the clinical, genetic, and molecular findings. Neuromuscul Disord 2021;31(11):1101–1112. doi: 10.1016/j.nmd.2021.09.010 [DOI] [PubMed] [Google Scholar]
- 7.Selcen D, Engel AG. Myofibrillar myopathies. Handb Clin Neurol 2011;101:143–154. doi: 10.1016/B978-0-08-045031-5.00011-6 [DOI] [PubMed] [Google Scholar]
- 8.von Nandelstadh P, Soliymani R, Baumann M, Carpen O. Analysis of myotilin turnover provides mechanistic insight into the role of myotilinopathy-causing mutations. Biochem J 2011;436(1):113–121. doi: 10.1042/BJ20101672 [DOI] [PubMed] [Google Scholar]
- 9.Maerkens A, Olivé M, Schreiner A, et al. New insights into the protein aggregation pathology in myotilinopathy by combined proteomic and immunolocalization analyses. Acta Neuropathol Commun 2016;4:8. doi: 10.1186/s40478-016-0280-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moza M, Mologni L, Trokovic R, et al. Targeted deletion of the muscular dystrophy gene myotilin does not perturb muscle structure or function in mice. Mol Cell Biol 2007;27:(1)244–252. doi: 10.1128/MCB.00561-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Wallace LM, Garwick-Coppens SE, et al. RNAi-mediated gene silencing of mutant myotilin improves myopathy in LGMD1A mice. Mol Ther Nucleic Acids 2014;3(4):e160. doi: 10.1038/mtna.2014.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson NE. Myotonic muscular dystrophies. Continuum (Minneap Minn) 2019;25(6):1682–1695. doi: 10.1212/CON.0000000000000793 [DOI] [PubMed] [Google Scholar]
- 13.Bönnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 2011;7(7):379–390. doi: 10.1038/nrneurol.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolduc V, Zou Y, Ko D, Bönnemann CG. siRNA-mediated allele-specific silencing of a COL6A3 mutation in a cellular model of dominant ullrich muscular dystrophy. Mol Ther Nucleic Acids 2014;3(2):e147. doi: 10.1038/mtna.2013.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trochet D, Prudhon B, Beuvin M, et al. Allele-specific silencing therapy for dynamin 2-related dominant centronuclear myopathy. Embo Mol Med 2018;10(2):239–253. doi: 10.15252/emmm.201707988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duan D Considerations on preclinical neuromuscular disease gene therapy studies. In: Duan D, Mendell JR, editors. Muscle gene therapy. 2nd ed. Cham: Springer, 2019:291–326. [Google Scholar]
- 17.Chemello F, Bassel-Duby R, Olson EN. Correction of muscular dystrophies by CRISPR gene editing. J Clin Invest 2020;130(6):2766–2776. doi: 10.1172/JCI136873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan D Micro-utrophin therapy for Duchenne muscular dystrophy. Mol Ther 2019;27(11): 1872–1874. doi: 10.1016/j.ymthe.2019.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 2017;377(18):1713–1722. doi: 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- 20.Bulcha JT, Wang Y, Ma H, et al. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther 2021;6(1):53. doi: 10.1038/s41392-021-00487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendell JR, Sahenk Z, Lehman K, et al. Assessment of systemic delivery of rAAVrh74. MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol 2020;77(9):1122–1131. doi: 10.1001/jamaneurol.2020.1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev 2017;8:87–104. doi: 10.1016/j.omtm.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morales L, Gambhir Y, Bennett J, Stedman HH. Broader implications of progressive liver dysfunction and lethal sepsis in two boys following systemic high-dose AAV. Mol Ther 2020;28(8): 1753–1755. doi: 10.1016/j.ymthe.2020.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol 2019;15(7):373–386. doi: 10.1038/s41582-019-0203-3 [DOI] [PubMed] [Google Scholar]
- 25.Hammond SM, Aartsma-Rus A, Alves S, et al. Delivery of oligonucleotide-based therapeutics: challenges and opportunities. EMBO Mol Med 2021;13(4):e13243. doi: 10.15252/emmm.202013243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoch KM, Miller TM. Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 2017;94(6):1056–1070. doi: 10.1016/j.neuron.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dzierlega K, Yokota T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther 2020;27(9): 407–416. doi: 10.1038/s41434-020-0156-6 [DOI] [PubMed] [Google Scholar]
- 28.Zambon AA, Waldrop MA, Alles R, et al. Phenotypic spectrum of dystrophinopathy due to Duchenne muscular dystrophy exon 2 duplications. Neurology 2022;98(7):e730–e738. doi: 10.1212/WNL.0000000000013246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simmons TR, Vetter TA, Huang N, et al. Preclinical dose-escalation studies establish a therapeutic range for U7snRNA-mediated DMD exon 2 skipping. Mol Ther Methods Clin Dev 2021;21:325–340. doi: 10.1016/j.omtm.2021.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaleta-Rivera K, Dainis A, Ribeiro AJS, et al. Allele-specific silencing ameliorates restrictive cardiomyopathy attributable to a human myosin regulatory light chain mutation. Circulation 2019; 140(9):765–778. doi: 10.1161/CIRCULATIONAHA.118.036965 [DOI] [PubMed] [Google Scholar]
- 31.Abu-Baker A, Kharma N, Perreault J, et al. RNA-based therapy utilizing oculopharyngeal muscular dystrophy transcript knockdown and replacement. Mol Ther Nucleic Acids 2019;15: 12–25. doi: 10.1016/j.omtn.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med 2020;383(2):109–119. doi: 10.1056/NEJMoa2003715 [DOI] [PubMed] [Google Scholar]
- 33.Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019; 576(7785):149–157. doi: 10.1038/s41586-019-1711-4 [DOI] [PMC free article] [PubMed] [Google Scholar]