

GRAPHICAL ABSTRACT:
Allosteric modulators (AMs) that bind allosteric sites can exhibit greater selectivity than the orthosteric ligands and can either enhance agonist-induced receptor activity (termed positive allosteric modulator or PAM), inhibit agonist-induced activity (negative AM or NAM), or have no effect on activity (silent AM or SAM). Until now, it is not clear what the exact effects of AMs are on the orthosteric active site or the allosteric binding pocket(s). In the present work, we collected both the three-dimensional (3D) structures of receptor–orthosteric ligand and receptor–orthosteric ligand–AM complexes of a specific target protein. Using our novel algorithm toolset, molecular complex characterizing system (MCCS), we were able to quantify the key residues in both the orthosteric and allosteric binding sites along with potential changes of the binding pockets. After analyzing 21 pairs of 3D crystal or cryo-electron microscopy (cryo-EM) complexes, including 4 pairs of GPCRs, 5 pairs of ion channels, 11 pairs of enzymes, and 1 pair of transcription factors, we found that the binding of AMs had little impact on both the orthosteric and allosteric binding pockets. In return, given the accurately predicted allosteric binding pocket(s) of a drug target of medicinal interest, we can confidently conduct the virtual screening or lead optimization without concern that the huge conformational change of the pocket could lead to the low accuracy of virtual screening.
Keywords: allosteric modulator, MCCS, drug discovery
INTRODUCTION
Allosteric modulators (AMs) are compounds that bind to a pocket other than the active site(s) of the endogenous ligand(s). AMs are dependent on the existing orthosteric ligands to exert their effects that may instigate a conformational change of the protein structure. Based on their functions, AMs can be classified into three categories, including positive allosteric modulators (PAMs), negative allosteric modulators (NAMs), and silent allosteric modulators (SAMs or neutral allosteric ligands). PAMs increase the activation of agonists of a receptor by raising their affinity or efficacy. NAMs reduce the affinity or the efficacy of agonists of a receptor to decrease the activation of agonists or stabilize the inactive state of the receptor that was bound with antagonists. SAMs have no impact on the activity of orthosteric ligands, but they occupy the allosteric binding sites, resulting in the blocking of the allosteric activities from PAM and NAM.1 Compared to the orthosteric ligands, AMs have the following benefits. Since the modulators only cause an effectiveness when an orthosteric ligand is binding to the protein, there is an “effect ceiling” that makes them safer than orthosteric ligands. The sequences of orthosteric binding sites are highly conserved across a protein family, while the sequences of allosteric binding sites are highly diverse in a receptor family. This feature enables the AMs to possess higher selectivity for different subtypes of a protein family, leading to the lower side effects of drugs.1,2
In the past two decades, studies and publications regarding allosteric mechanisms, pockets, and AMs have grown at a fast pace. Figure 1 shows the related articles in SciFindern climbing from 721 in 2000 to 2153 in 2020. Importantly, 56 AM drugs have been approved by the U.S. Food and Drug Administration (FDA), in which there are 4 for G protein coupled receptors (GPCRs),3–6 40 for ion channels,7–32 and 13 for enzymes (Table S1).33–44 For example, the oldest approved AM in the U.S. is chlordiazepoxide, a benzodiazepine drug approved in 1960 for alcohol withdrawal syndrome and anxiety.45 Chlordiazepoxide is a PAM for the GABA-A receptor that can increase the frequency of GABA-induced chloride channel openings and enhance the binding affinity of GABA in its orthosteric binding site.46,47 Subsequently, several benzodiazepines came on the market in the mid- and late-twentieth century, and the success of benzodiazepines in clinical practice caught the eye of the researchers focused on allosteric drug discovery.9–12,48 In 1998, the first enzyme AM, rifapentine, was approved by the FDA. It is a NAM for bacterial RNA polymerase (RNAP) for the treatment of tuberculosis infection.38 In 2004, the first GPCR AM, cinacalcet, appeared on the market for the treatment of secondary hyperparathyroidism and parathyroid carcinoma.49–53 Cinacalcet is a PAM of the calcium-sensing receptor (CaSR) and has the ability to increase the sensitivity of the calcium-sensing receptor for activation by extracellular calcium.3 The blossoming of AMs in the market makes the development of allosteric drugs an increasingly hot research area.
Figure 1.

Articles regarding “allosteric” in SciFindern from 2000 to 2021.
With X-ray crystallography, transmission electron cryo-microscopy (cryo-EM), and other innovative technologies, more and more three-dimensional (3D) crystal structures complexed with AMs in high resolution have been released. There are 6,772 complexes containing allosteric agents in the Protein Data Bank (https://www.rcsb.org/).54 The allosteric database (http://mdl.shsmu.edu.cn/ASD/) cumulates 82,070 molecules with allosteric modulation features and 538 drugs from preclinical phases to approved status.55 The high quality of 3D complexes of the protein with the modulator allows structural biologists to investigate the binding pocket(s) of receptors, the binding poses of orthosteric ligands and AM(s), and the potential conformational changes of the target protein. For example, Liu et al. reported two crystal structures that included (1) β2 adrenoceptor (ADRB2) binding with orthosteric antagonist carazolol and (2) the complex of ADRB2–carazolol–Cmpd-15(NAM-compound 15). Their studies unveiled the important residues involved in the interactions between Cmpd-15 and ADRB2, the conformational changes caused by the binding of the modulator that stabilized the inactive inward conformation of TM6, and the prevention of coupling to Gs protein caused by the steric clash with Cmpd-15.56 In addition, the high-quality 3D structures of proteins complexed with AMs improve the accuracy of computational experiments and thus facilitate in silico drug discovery.
Dedicated to the development of modulators for GPCRs,57–61 our lab previously compared and analyzed the existing computational methods for detecting the allosteric sites and designing AMs in our review paper.2 We compared six approaches for the prediction of the allosteric binding sites, including sequence-based approaches, structure-based methods, conformational dynamics-based approaches, normal model analysis-based approaches, a combination of conformational dynamics-based and NMA-based approaches, and other allosteric-related approaches. For designing the drug candidates, we classified the strategies into three categories, including pharmacophore models, structure-based virtual screening, and molecular dynamics (MD) simulation. We delineated every method with a detailed introduction, tools, advantages, challenges, and examples of the application study in the article.2 Recently, with a view to deeply investigate the AMs of GPCRs, we collected the available 3D structures of the class A GPCRs with effective interactions between the modulator and receptor, found seven allosteric binding pockets/regions at GPCRs, and analyzed the binding characterization of allosteric binding pockets. In addition, we further predicted the allosteric binding sites at the CB2 receptor as well as the detailed interaction between CB2 and its modulator(s), which will benefit the development of CB2 allosteric drugs. In addition, we recently developed a novel algorithm, molecular complex characterizing system (MCCS), to compute the residue energy contribution for quantifying the binding features or patterns of protein–ligand complexes.62
In this study, we first collected 4-pair complexes in GPCRs, 11 pairs in enzymes, 1 pair in transcription factors, and 5 pairs in ion channels into our dataset. All of these targets play important roles in the nervous system. For example, the β2 adrenergic receptor belongs to the sympathetic nervous system,63,64 glucokinase (GK) gets involved in the neuronal glucose-sensing mechanism,65,66 and ionotropic glutamate receptors, such as AMPA and NMDA receptor (NMDAR), modulate neuronal excitability with the excitatory neurotransmitter, glutamate.67–70 Exploiting our innovative technique, MCCS, we investigated whether the binding of a modulator had an impact on the orthosteric ligand binding via systematically comparing the 3D structure of receptor–ligand with and without a modulator, which would highly facilitate the rational design and development of modulator drug candidates.
RESULTS AND DISCUSSION
Innovation and Application of MCCS.
Our lab has developed an innovative technique, MCCS, a method to quantify the energy contribution of residues involved in the interactions between compounds and protein(s) with high accuracy and high efficiency. MCCS distinguishes itself from molecular dynamics (MD) simulation-based energy decomposition in its ability to generate the residue energy contribution, the binding recognition vector, and vector similarity with reduced time consumption and high accuracy. The detailed protocol of MCCS could be found in our recent publication.62 We also exploited MCCS in several applications or studies. The first study71 was to analyze all of the existing allosteric binding pockets in GPCRs regarding the interactions between important residues and AMs. Moreover, this study also analyzed the allosteric binding site at CB1 and predicted the potential allosteric site(s) for CB2. The second study72 was dedicated to repurposing and combining FDA-approved drugs with high efficiency and also further designing new compounds for the treatment of COVID-19. The latest study73 focused on the binding pockets in the adenosine A2A receptor (AA2AR), especially distinguishing the key residues binding with antagonists from that with agonists, analyzing various statuses of AA2ARs, and investigating selectivity between the AA2AR and adenosine A1A receptor (AA1AR), which could facilitate the rational drug design for treatment involving AA2AR. These studies served as a “proof-of-concept” of MCCS in that it can be applied by other researchers in the field for the design and discovery of functional ligands for a specific target.
Stabilization of Both GPCRs and Orthosteric Ligands by AMs.
For GPCRs, there were four pairs of complexes of either X-ray crystals or cryo-EM structures, including M2 muscarinic acetylcholine receptor (M2 receptor), β2 adrenoceptor, and free fatty acid receptor 1 (FFA1/GPR40). Here, we delineate the results of the M2 receptor and one pair of β2 adrenoceptor structures, and the results of the FFA1 receptor–orthosteric ligand with/without AM and another pair of β2 adrenoceptor structures were similar to those of M2 or β2 receptors (Table 1).
Table 1.
Sixteen Pairs of Complexes Consisted of Either Agonists with PAM or Antagonists with NAM
| receptor | PDB | compounds in PDB | residues with a significant change in the orthosteric pocket | residues rotate in the allosteric pocket when AM was bound | RMSD of the whole protein (Å) | RMSD of the orthosteric binding sites (Å) | RMSD of the allosteric binding pockets (Å) |
|---|---|---|---|---|---|---|---|
| M2 muscarinic acetylcholine receptor | 4MQT | PAM: LY2119620, agonist: iperoxo | NA | Trp422, Phe181 | 0.195 | 0.158 | 0.249 |
| 4MQS | agonist: iperoxo | ||||||
| β2 adrenoceptor | 5X7D | NAM: Cmpd-15, antagonist: carazolol | Asn293 | Arg63, Asp331, Lys267, Phe332 | 0.355 | 0.291 | 0.312 |
| 2RH1 | antagonist: carazolol | ||||||
| 6N48 | PAM: compound-6FA, agonist: BI-167107 | NA | PHE133, TYR141, LEU144, LYS149 | 0.303 | 0.224 | 0.373 | |
| 4LDE | agonist: BI-167107 | ||||||
| FFA1 (GPR40) | 5TZY | PAM: AP8, agonist: MK-8666 | Trp174, Leu186 | LEU190 | 0.990 | 0.656 | 0.631 |
| 5TZR | agonist: MK-8666 | ||||||
| PDK1 | 3HRC | ATP | Ser92, Ser94 | Phe157 | 0.183 | 0.163 | 0.271 |
| 3HRF | PAM:PS48, ATP | ||||||
| 3HRC | ATP | Ser94, Lys111, Glu166, Glu209, Asn210 | Phe157 | 0.297 | 0.536 | 0.626 | |
| 4AW1 | PAM: PS210, ATP | ||||||
| MEK1 (nonphosphorylated MEK1) | 3EQC | NAM: compound-1, ATP-γS | Lys97 | NA | 0.210 | 0.184 | 0.286 |
| 3EQD | ATP-γS | ||||||
| 3EQH | NAM: U0126, ADP | Lys97 | NA | 0.203 | 0.187 | 0.284 | |
| 3EQI | ADP | ||||||
| tyrosine-protein kinase ABL1 | 1OPK | NAM: myristic acid, inhibitor: PD166326 | Ala399 | NA | 0.452 | 0.000 | 0.363 |
| 2G2H | Inhibitor: PD166326 | ||||||
| GK | 3F9M | PAM: TAFMT, agonist: α- d-glucopyranose | NA | Tyr215, Thr65, Pro66 | 0.291 | 0.132 | 0.286 |
| 3IDH | agonist: α-d-glucopyranose | ||||||
| K-ras-gtpase | 4M22 | NAM: acrylamide 16, GDP | Asp30 | Tyr96, Arg68, Gln61, Gln99 | 0.332 | 0.218 | 0.279 |
| 4LDJ | GDP | ||||||
| amine oxidase | 2XFQ | NAM: rasagiline, 2-BFI, FAD | Leu167 | NA | 0.150 | 0.183 | 0.152 |
| 2XFN | 2-BFI, FAD | ||||||
| AMPA GluR2 | 1LBC | PAM: cyclothiazide (CTZ), glutamate | NA | NA | 0.423 | 0.233 | 0.348 |
| 1FTJ | glutamate | ||||||
| AMPA GluR3 | 3M3F | PAM: PEPA, glutamate | Arg515 | NA | 0.522 | 0.343 | 0.369 |
| 3M3K | glutamate | Glu612 | |||||
| NMDAR | 5H8H | PAM: GNE3419, glutamate, glycine | NA | Glu530 (GluN2A), Tyr535 (GluN1) | 0.637 | 0.148 | 0.337 |
| 5H8F | glutamate, glycine | ||||||
| GluK1 | 5MFQ | PAM: BPAM-344, Kainate | NA | NA | 0.443 | 0.233 | 0.221 |
| 4E0X | Kainate |
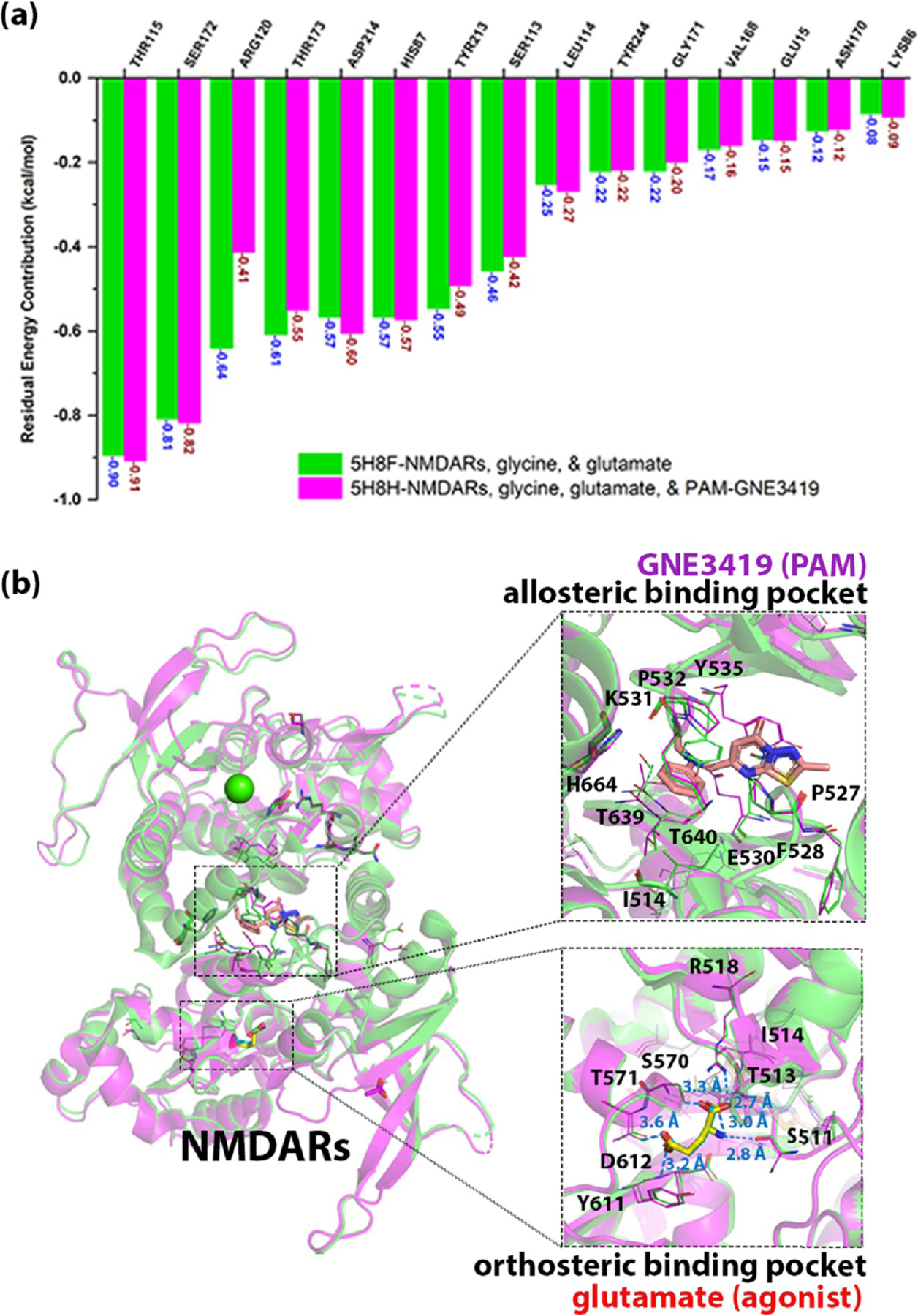
For the M2 receptor, PDB 4MQT is the complex of the M2 muscarinic acetylcholine receptor, PAM-LY2119620, and agonist-iperoxo (Figure 2b, magenta cartoon), while PDB 4MQS is the complex of the M2 muscarinic acetylcholine receptor with iperoxo (Figure 2b, green cartoon). There were no obvious conformational changes for both the orthosteric binding site and the whole structure of M2 when the two structures were aligned in PyMol (Figure 2b). Moreover, in the orthosteric binding pocket of the two structures, there was no residue with a significant change of the energy contribution (Figure 2a), indicating that the interactions between the orthosteric agonist and the receptor were stable after the binding of PAM. Recently, Kruse et al.74 reported the binding affinity of iperoxo with/without LY2119620 using the [3H]- NMS competition binding assay. There was not a significant difference between the pKi values of iperoxo w/o the binding of LY2119620 (the pKi value of iperoxo with the binding of LY2119620 is 8.51 ± 0.04, and the Ki value for iperoxo without LY2119620 is 0.0073 μM, i.e., pKi value = 8.1367). Hence, our results were consistent with the experimental data and indicated that there was no significant conformational change in the orthosteric binding site of the M2 muscarinic acetylcholine receptor when a PAM binds at the allosteric binding site. On the other hand, there were only two residues, Trp422 and Phe181, that endured the conformational change in the allosteric binding pocket along with its energy contribution. When aligning the two structures, the conformation of the Trp422 side chain rotated from the horizontal pose to the vertical pose in the presence of LY2119620, which allowed the binding of LY2119620 to M2 to engage in an aromatic stacking interaction (Figure 2b). The side chain of Phe181 transformed from horizontal to vertical to the PAM, which accommodated the binding of LY2119620.
Figure 2.

Comparison of the complex of M2–iperoxo(agonist) with/without the binding of the positive allosteric modulator (PAM)-LY2119620. (a) Comparison of the binding residues involved in the orthosteric binding pocket between M2–iperoxo (PDB: 4MQS, green bars) and M2–iperoxo-LY2119620 (PDB: 4MQT, magenta bars). (b) Overlap of M2–iperoxo (green cartoon–blue stick) and M2–iperoxo-LY2119620 (magenta cartoon–yellow stick–salmon stick). The residues involved in the binding pockets are shown as thin element–colored lines.
For the β2 adrenoceptor, PDB 5X7D is the complex of the β2 adrenoceptor, compound 15 (Cmpd-15, NAM), and the orthosteric antagonist carazolol, while PDB 2RH1 is the complex of the β2 adrenoceptor binding with carazolol. In the orthosteric binding site (Figure 3a), most of the energy contributions of the residues were identical in the two structures, except for Asn293. The total free-energy contribution of Asn293 reduced significantly when the NAM was bound to the receptor. To be more specific, the steric force of Asn293 decreased greatly after the binding of Cmpd-15. Aligning the two structures showed that the position of Asn293 shifted for about 0.9 Å along with the slightly inward movement of TM6 caused by the NAM that stabilized the inactive conformation of the receptor (Figure 3b). It is worth noting that Asn293 is an important residue for interactions with both agonists and antagonists. A recent report by Wieland et al.75 about the site-directed mutagenesis studies of Asn293 replaced by Leu supports that Asn293 is crucial for stereospecificity and intrinsic activity of agonists in their interactions with the receptor. Hanson et al.76 reported a structure containing β2 adrenoceptor and timolol (PDB code: 3D4S), and their results showed that Asn293 played an important role in the interaction between the antagonist and β2 adrenoceptor. Our computational results agreed with the experimental data and further unveiled the detailed energetic change of residues. Finally, we also investigated the detailed interactions between NAM-Cmpd-15 and its surrounding residues. Most of the allosteric binding residues did not endure significant conformational changes, including Leu64, Asn69, Ala271, Thr274, Tyr326, and Ser329. However, four residues, Arg63, Asp331, Lys267, and Phe332, rotated to either accommodate or interact with Cmpd-15.
Figure 3.

Comparison of the complex of β2–carazolol(antagonist) with/without the binding of the negative allosteric modulator (NAM)-Cmpd-15. (a) Comparison of the binding residues involved in the orthosteric binding pocket between β2–carazolol (PDB: 2RH1, green bars) and β2–carazolol–Cmpd-15 (PDB: 5X7D, magenta bars). (b) Alignment of β2–carazolol (green cartoon–blue stick) and β2–carazolol–Cmpd-15 (magenta cartoon–yellow stick–orange stick). The residues involved in the binding pockets are shown as thin element–colored lines. The red arrow shows the inward movement of TM6.
Stable Conformations and Interaction of the Enzymes–Orthosteric Ligand by AMs.
There were 11 pairs of enzymes collected in the present study, and the receptors included 3-phosphoinositide-dependent protein kinase 1 (PDK1), dual specificity mitogen-activated protein kinase 1 (MEK1), tyrosine-protein kinase ABL1, glucokinase (GK), K-Ras GTPase, acetylcholinesterase, amine oxidase [flavin-containing] B (MAO-B), and mitochondrial glutamate dehydrogenase 1 (GDH 1). The results of 11 pairs of enzymes were similar in terms of the number of binding residues with a significant conformational change (Tables 1 and 2).
Table 2.
Five Pairs of Complexes Comprised of Either Agonists with NAM or Antagonists with PAM
| receptor | PDB | compounds in PDB | residues with a significant change in orthosteric pocket | residues rotate in allosteric pocket when AM was bound | RMSD values for protein structures | RMSD values for orthosteric binding sites | RMSD values for allosteric binding pockets |
|---|---|---|---|---|---|---|---|
| tyrosine-protein kinase ABL1 | 3PYY | PAM: DPH, inhibitor: imatinib | His361 | NA | 0.340 | 0.287 | 0.363 |
| 2HYY | inhibitor: imatinib | Asp381 | |||||
| acetylcholinesterase | 5HF9 | PAM: HI-6, inhibitor: DEP | Phe295 | Trp286 | 0.277 | 0.132 | 0.188 |
| 5HF5 | inhibitor: DEP | ||||||
| GDH 1 | 3ETG | NAM: GW5074, glutamic acid, GTP, NADPH | Val378 Lys114 Lys90 Met111 |
Lys143 | 0.291 | 0.214 | 0.188 |
| 6DHQ | glutamic acid, GTP, NADPH | ||||||
| NMDAR | 5H8N | NAM: compound 6, glutamate, glycine | NA | Glu530 (GluN2A), Arg639 (GluN1) | 0.260 | 0.163 | 0.338 |
| 5H8F | glutamate, glycine | ||||||
| AR | 2YHD | NAM: 4-(2,3-dihydro-1H-perimidin-2-yl) benzene-1,2-diol, TES | NA | Met734 | 0.253 | 0.207 | 0.232 |
| 2AM9 | TES |
Taking GK as an example, PDB 3F9M is the complex of GK, substrate α-d-glucopyranose, and PAM-TAFMT, while PDB 3IDH is the complex of GK and α-d-glucopyranose. As shown in Figure 4a, the computed results of the orthosteric binding pocket in the two structures were almost identical, and there were no residues undergoing conformational change after the PAM was bound (Figure 4b). In addition, the residues involved in the allosteric pocket of PAM also did not endure significant conformation change, except for Tyr215 rotating toward the pocket center, and Thr65 as well as Pro66 moving along with the loop when the PAM was bound to the pocket (Figure 4b). Moreover, studies for site-directed mutagenesis of Tyr215 to Ala and the natural variant of Thr65 to Ile support that both residues play important roles in the interaction between GK and PAM. To be more specific, Y215A and T65I both raised the affinity of the glucose binding and the glucokinase activity, which was measured by the catalytic efficiency.77–79
Figure 4.

Comparison of the complex of GK–α-d-glucopyranose(agonist) with/without the binding of the positive allosteric modulator (PAM)- TAFMT. (a) Comparison of the binding residues involved in the orthosteric binding pocket between GK–α-d-glucopyranose (PDB:3IDH, green bars) and GK–α-d-glucopyranose(agonist)–TAFMT (PDB: 3F9M, magenta bars). (b) Alignment of GK–α-d-glucopyranose (green cartoon–blue stick) and GK–α-d-glucopyranose(agonist)–TAFMT (magenta cartoon–yellow stick–salmon stick). The residues involved in the binding pockets are shown as thin element–colored lines.
Taking MEK1 as another example, PDB 3EQC is the complex of MEK1, ATP-γS, and NAM-compound-1, while PDB 3EQD is the complex of MEK1 and ATP-γS. Based on the literatures, MEK1 in the two complexes is in an inactive state.80–83 By comparing the binding characterization of the orthosteric pocket in 3EQC with 3EQD, only one binding residue, Lys97, endured significant change with respect to the total free-energy contribution (Figure 5a). To be more specific, when the NAM was bound to the complex of MEK1–ATP-γS, the total free energy and the repulsion contribution of Lys97 sharply decreased. According to the literature, Lys97 plays an important role in the binding of ATP and inhibitors and is also a key residue in distinguishing the active and inactive states of MEK1.82–90 Aligning PDB 3EQC and PDB 3EQD, we found that the important residues in the orthosteric binding pocket in the two proteins are nearly overlapped (Figure 5b). The ribose and triphosphate groups in the ATP-γS rotated slightly. Lys97 on the β sheets near the triphosphate group of ATP-γS was slightly rotating and shifting (about 0.3 Å with Cα as reference) along with the movement of β sheets. Similarly, we also found that the binding residues involved in the allosteric active site kept stable in the structure, as shown in Figure 5b; only a few residues, such as Leu215 and Val211, shifted along with the movement of the activation loop.
Figure 5.

Comparison of the complex of MEK1–ATP-γS(agonist) with/without the binding of the negative allosteric modulator (NAM)- compound-1. (a) Comparison of the binding residues involved in the orthosteric binding pocket between MEK1–ATP-γS (PDB: 3EQD, green bars) and MEK1–ATP-γS–compound-1 (PDB: 3EQC, magenta bars). (b) Alignment MEK1–ATP-γS (green cartoon–blue stick) and MEK1–ATP-γS–compound-1 (magenta cartoon–yellow stick–orange stick). The residues involved in the binding pockets are shown as thin element–colored lines.
Stability in Ion Channels and Transcription Factor.
There were five pairs of complexes that belonged to ligand-gated ion channels in the present study, including a pair of the AMPA receptor 2 (GluR2), a pair of the AMPA receptor 3 (GluR3), two pairs of NMDA receptors (NMDARs), and a pair of glutamate receptor, ionotropic, kainate 1 (GluK1). The results of five pairs of ion channels were similar, in which there were only zero to two binding residues in the orthosteric and allosteric sites undergoing significantly conformational change when an AM was bound to the receptors (Tables 1 and 2).
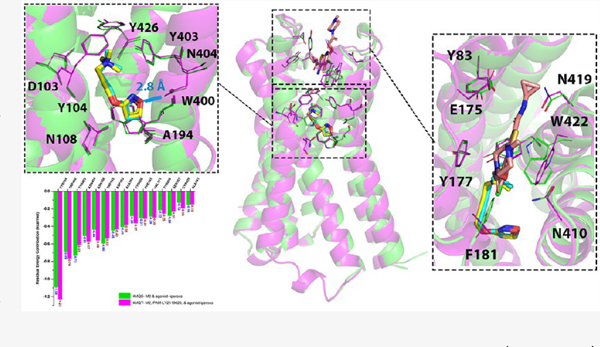
Taking NMDARs as examples, we utilized two pairs of NMDARs in this study, including PDB 5H8H–5H8F and PDB 5H8N–5H8F. NMDARs are the complex of heterotetramers consisting of two NMDA 1 (GluN1) and typically two NMDR 2 (GluN2) subunits.91 Activation of NMDARs requires glycine/D-serine, glutamate, and membrane depolarization.92,93 PDB 5H8F is the complex of GluN1–GluN2A–glycine–glutamate. PDB 5H8N is the complex of GluN1–GluN2A–glycine–glutamate–NAM-compound 6. PDB 5H8H is the complex of GluN1–GluN2A–glycine–glutamate–PAM-GNE3419. GNE3419 has been reported to have an impact on the interactions between glutamate and NMDARs.94 By comparing the glutamate binding sites within each pair, the conformational change and energy contribution of the key binding residues in the orthosteric pockets were similar to each other (Figures 6 and 7), indicating that the binding of PAM and NAM can stabilize the conformations of both ion channels and the orthosteric ligands. Only a few residues underwent significant change in the allosteric binding pocket when an AM was bound. Specifically, when the NAM was bound to the pocket, Glu530 (GluN2A) rotated away from and Arg639 (GluN1) approached toward the NAM, while when the PAM was bound to the pocket, Glu530 (GluN2A) and Tyr535 (GluN1) rotated away from the PAM to accommodate the binding of the compound.
Figure 6.
Comparison of the complex of NMDARs–glycine/glutamate with/without the binding of PAM-GNE3419. (a) Energy contributions of the binding residues in the orthosteric binding pocket of NMDARs–glycine/glutamate (PDB: 5H8F) are highlighted in green bars, while those of NMDARs–glycine/glutamate–GNE3419 (PAM, PDB: 5H8H) are highlighted in magenta bars. (b) Alignment of NMDARs–glycine/glutamate (green cartoon–blue stick) and NMDARs–glycine/glutamate–(PAM) GNE3419 (magenta cartoon–yellow stick–salmon stick). The residues involved in the binding pockets are shown as thin element–colored lines.
Figure 7.

Comparison of the complex of NMDARs–glycine/glutamate with/without the binding of NAM-compound 6. (a) Energy contributions of the binding residues in the orthosteric binding pocket of NMDARs–glycine/glutamate (PDB: 5H8F) are highlighted in green bars, while those of NMDARs–glycine/glutamate–compounds 6 (NAM, PDB: 5H8N) are highlighted in magenta bars. (b) Alignment of NMDARs–glycine/glutamate (green cartoon–blue stick) and NMDARs–glycine/glutamate–(NAM) compound 6 (magenta cartoon–yellow stick–orange stick). The residues involved in the binding pockets are shown as thin element–colored lines.
The androgen receptor (AR) is the only transcription factor collected in this study. PDB 2YHD is the complex of AR, agonist testosterone (TES), and NAM 4-(2,3-dihydro-1H-perimidin-2-yl) benzene-1,2-diol, while PDB 2AM9 is the complex of AR and TES. By comparing the TES binding pocket in the two complexes, we found that the total free-energy contribution of each important residue was similar (Figure 8a), yet the energy composition of Arg752 changed (Figure S1). When the NAM was bound, the repulsion force dropped and the strength of hydrogen bonds in Arg752 declined. The result was consistent with the observation of aligning the two 3D structures, as shown in Figure 8b. The binding of NAM only caused slight conformational change except for the outward movement of helix-10 to approach the helix 9. The side chain of Arg752 twisted about 20° when the NAM was bound to AR. In addition, the allosteric binding residues did not endure huge conformational changes except for Met734 rotating away from the pocket center when the NAM occupied the pocket.
Figure 8.

Comparison of the complex of AR–testosterone (TES) with/without the binding of the negative allosteric modulator (NAM)-4-(2,3-dihydro-1H-perimidin-2-yl) benzene-1,2-diol. (a) Energy contributions of the binding residues of AR–testosterone (TES) (PDB: 2AM9) are highlighted in green bars, while the energy contributions of the binding residues of AR–testosterone (TES)–NAM (PDB: 2YHD) are highlighted in magenta bars. (b) Alignment AR–testosterone (TES) (green cartoon–blue stick) and AR–testosterone (TES)–NAM (magenta cartoon–yellow stick–orange stick). The residues involved in the binding pockets are shown as thin element–colored lines.
Comparison of Orthosteric- and Allosteric-Binding Sites between the 21 Pairs X-ray Crystal or Cryo-EM Complexes.
According to the pharmacological features of the ligands in complexes, 21 pairs could be divided into 2 groups that were (1) agonist with PAM and antagonist with NAM (Table 1) and (2) agonist with NAM and antagonist with PAM (Table 2). In the first group with 16 pairs of structures, AMs have little impact on the conformation of the orthosteric binding pocket. To be more specific, most of them only have one or two binding residues undergoing significant conformational change after an AM was bound to the receptors. On the other hand, in the second group with five pairs of complexes, we also found that the binding of AM did not lead to a significant change in the interaction between orthosteric compounds and the protein. Specifically, four of them with no more than two important residues and one pair with four residues underwent significant change.
Moreover, aligning the complexes within each pair showed that the structure around the allosteric binding sites did not endure significant conformational change when an AM was bound to the pocket: most of the important residues only shifted mildly when the α-helixes or β-sheets that were around the allosteric pocket moved slightly. However, in some of the allosteric binding sites, a few residues that possessed a large side chain may have either rotated away from or approached the allosteric binding pocket to accommodate the binding of AMs. Based on the findings above, we observed that the binding of AM had little impact on the conformation of both the orthosteric- and allosteric binding pockets.
To further validate our results, we calculated the root-mean-square deviation (RMSD) value within each pair of complexes to compare the whole protein structures, orthosteric binding sites, and allosteric binding pockets. As shown in Tables 1 and 2, all of the RMSD values are smaller than 1 Å, indicating that the conformation of the protein structures and their pockets did not undergo significantly conformational change when an AM was bound to a specific protein.
In addition, most of the rotated residues in allosteric pockets play important roles in the binding of AM. Take the M2 receptor as an example, with in silico site mutagenesis of W422A, the normalized total binding free energy between the residues and LY2119620 dropped from −7.484 to −6.161 kcal/mol. However, the residues in the AM pocket that did not undergo significant conformational change did not mean they are not important for the AM binding. The calculation with MCCS can identify the important residues in AM pockets. Take Y177 in the M2 receptor as another example for residues without undergoing significant rotation during the binding of AM; the in silico results showed that Y177 in the PDB 4MQT structure contributed binding free energy of −1.555 kcal/mol for the interaction with LY2119620, while the Y177A mutant contributed free energy of −0.637 kcal/mol and lead to the reduction of total binding free energy from −7.484 to −6.774 kcal/mol. Our predictions were consistent with recent studies, e.g., Valant et al.95 reported the binding affinity of NAM gallamine for Y177A mutant drop with an 18.621-fold change of the Ki value, and Gregory et al.96 published the Ki value for the binding of PAM 77-LH-28-1 for the Y177A mutant increase with a 3.165-fold change.
Molecular Dynamics Simulation Studies.
Molecular dynamic simulation studies were conducted to investigate the stability and dynamics of the PDB structures collected in this study. Here, we selected two pairs of complexes: one is PDB 4MQS–4MQT consisting of the M2 receptor, agonist-iperoxo, and PAM-LY2119620; another is PDB 2RH1-5X7D comprised of the β2 adrenoceptor, antagonist carazolol, and NAM Cmpd-15.
As shown in Figure 9a,b, the complex of PDB 4MQS and PDB 4MQT was stable during the simulation with the RMSD values of the receptor, the agonist, and the PAM in each complex were all under 5 Å. Specifically, the M2 receptor without the PAM was stable in the active state with the RMSD value around 2 Å, while the M2 receptor with the binding of PAM was more stable with the RMSD value around 1.5 Å, indicating that the binding of PAM stabilized the M2 receptor in an active state. Figure S2a,b shows the overlay of the first and the last frames of the simulation, portraying the dynamic conformational variation of the receptors and ligands during the simulation time. To evaluate that the binding of PAM does not lead to a significant conformational change of the orthosteric binding pocket, we aligned the last frames of the simulation of PDB 4MQS and 4MQT (Figure S2c). The residues in the orthosteric site moved along with the helixes where they were located but did not undergo a significant conformational change.
Figure 9.
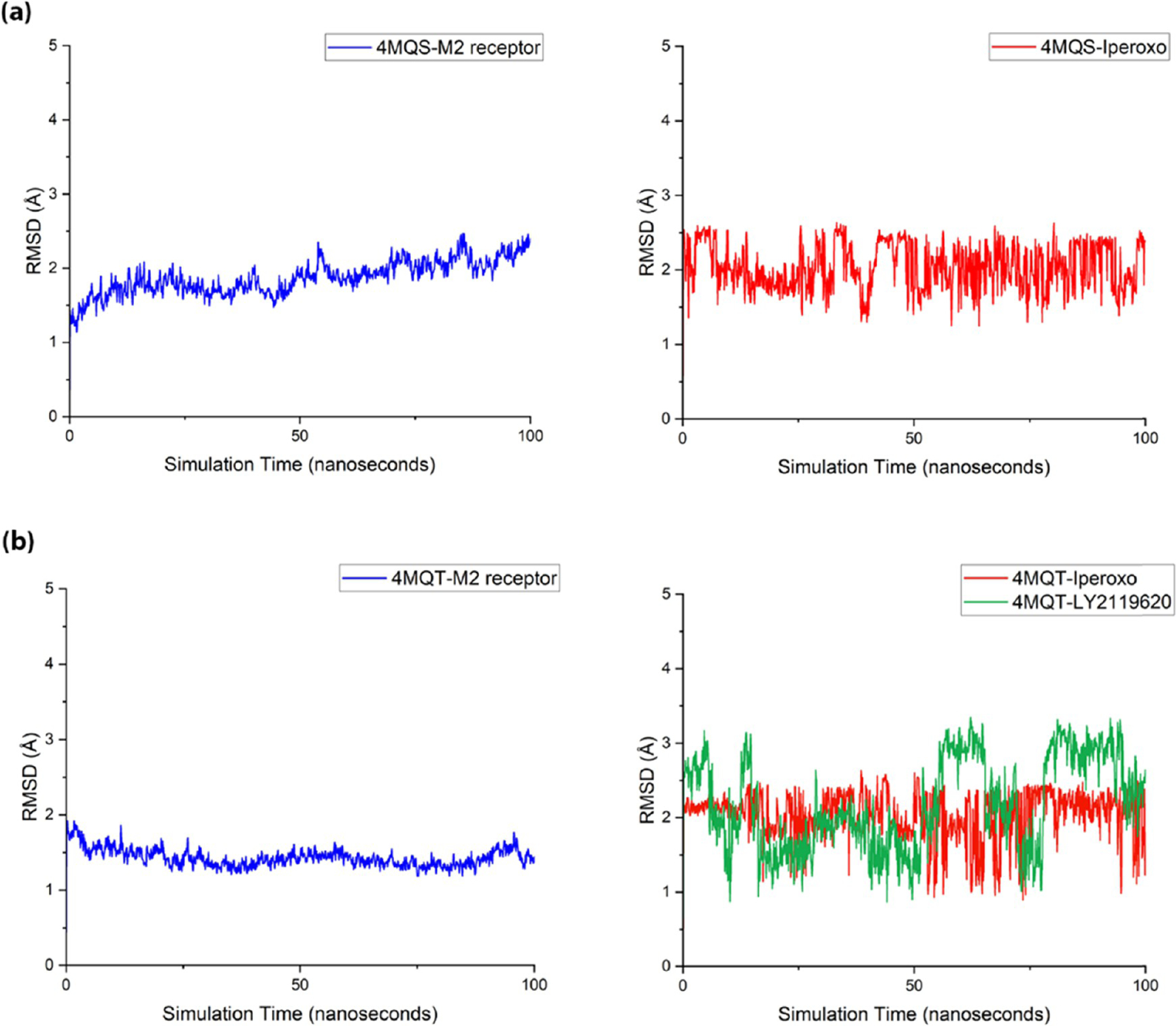
MD simulation studies of PDB 4MQT-4MQS structures. The time course of RMSD of mainchain atoms of the M2 receptor (black), heavy atoms of agonist-iperoxo (red), and PAM-LY2119620 (blue). (a) PDB 4MQS and (b) PDB 4MQT.
Figure S2c also shows the G protein binding site of the M2 receptor with the alignment of the last frames of the simulation for the receptor w/o PAM as well as the cryo-EM structure of the M2 receptor complexed with the same agonist, PAM, and plus, the G protein subunits (PDB: 6OIK). The intracellular portion of TMs 3,5,6,7 and helix 8 in both complex w/o PAM moved outward during the simulation, leaving a larger space for the insertion of the G protein α subunit. Specifically, the intracellular part of TMs 5,6,7 and helix 8 in the complex with PAM rotated and twisted much away from the center of the G protein binding site than that of the complex without PAM during the simulation course. PDB 6OIK showed that after coupling with G protein, TMs 5,6,7 and helix 8 moved inward to accommodate the α5 helix of G protein. The alignment of the three structures indicates that the binding of PAM allows the M2 receptor to couple with G protein more easily with a larger space at the G protein binding pocket, while the binding of G protein may lead to the reduction of the binding site volume for tighter interaction.
For PDB 2RH1-5X7D, the NAM stabilized the receptor at the inactive state, which was delineated by Figure 10a,b. During the simulation, the receptor in PDB 2RH1 was stable with the RMSD value oscillating around 2–3 Å, while the protein in PDB 5X7D was more stable than that in PDB 2RH1, with the RMSD value fluctuating around 1.5–2.5 Å in the first 40 ns and around 1.5–2 Å for the last 60 ns. The alignment of the first and the last frames of the simulation of the two complexes in Figure S3a,b delineates the structural fluctuations of the β2 adrenoceptor, antagonist, and NAM during the simulation course. Figure S3c shows the alignment of the last frame of the simulation of the receptor w/o NAM, which demonstrated that the binding of NAM did not result in a significant conformational change in the orthosteric binding site.
Figure 10.

MD simulation studies of PDB 5X7D–2RH1 structures. The time course of RMSD of mainchain atoms of the β2 adrenoceptor (blue), heavy atoms of antagonist carazolol (red), and NAM Cmpd-15 (green). (a) PDB 2RH1 and (b) PDB 5X7D.
METHODS AND MATERIALS
Protein–Ligand Complexes.
The X-ray crystal and cryo-EM structures were collected from the Protein Data Bank (https://www.rcsb.org).97 Two different complexes of any target protein were used in our work, including the structure of the protein coupled with the orthosteric ligand and the allosteric modulator as well as the structure of protein bound with the orthosteric ligand only. The orthosteric ligand in these two kinds of complexes should be the same.
Molecular Complex Characterizing System (MCCS).
MCCS62 was applied to prepare the complexes and calculate the residue energy contribution. It can analyze the binding recognition between receptors and ligands by calculating the energy contribution of each individual residue and the corresponding energy terms, such as hydrogen bonding, hydrophobic force, repulsion, etc.
Figure 11 shows the workflow of the MCCS. After we input the PDB structure, Chimera (version 1.15)98 was first applied to repair the residues with an incomplete side chain. To be more specific, Chimera first scanned the entire protein structures and reported the residues with missing parts. Then, the truncated side chains were replaced by a complete side chain of the same residue type with the Dunbrack rotamer library.99 Next, we split the complex into the ligand and the protein PDB Files. VEGA100 was applied to both ligand and protein files to add the polar hydrogens, Vina force field, and Gasteiger charges. Moreover, VEGA would define rotatable bonds for ligand files. The format of the output files was transformed from PDB into PDBQT by VEGA. In addition, PROPKA (version 3.4)101,102 was applied to predict the pKa values of ligands and generated a PKA format. When the computed pKa values of ligands were greater than or equal to the given pH (7.4 by default), MCCS would protonate the tertiary (3°) amine in the molecules. The PDBQT files of the protein and the ligand together with the PKA file of the ligand form the input of the next step in MCCS scoring and docking with jdock (version 2.2.3c, https://github.com/stcmz/jdock).
Figure 11.

Workflow of MCCS. Blue portion is the preparation steps. The green part is the performance of jdock. The orange part is the function of MCCSX.
As a variant and successor of idock,103 jdock is a docking and scoring program that can predict the binding pose of a compound within a complex and calculate the total binding free energy as well as energy contribution of each residue, which is involved in the interaction between a ligand and a protein. The binding affinity of a ligand can then be predicted with the calculated total binding free energy.104,105 Adopting the same five-term scoring function (gauss1, gauss2, repulsion, hydrophobic, and hbonding) invented by AutoDock Vina,106,107 jdock can generate a vector of residue free energy from the conformation either predicted by the Monte Carlo-based docking algorithm or determined by X-ray crystallography or cryo-EM. Those energy terms are related to the distance between two interacting atoms and the van der Waals radii of the interacting atoms.
In this study, we applied the scoring function of jdock to analyze the binding features of 21 pairs of X-ray crystal and cryo-EM structures, in which the scores of all receptor–ligand atom pairs were directly calculated and summed to form the overall score. The scoring function in jdock can generate nine binding recognition vectors for a given receptor–ligand complex, including (1) Gauss (Gauss1 + Gauss2), (2) Gauss1, (3) Gauss2, (4) repulsion, (5) steric (Gauss1 + Gauss2 + repulsion), (6) hydrogen bonding, (7) hydrophobic, (8) nonsteric (hydrogen bonding + hydrophobic), and (9) residue energy contribution. More details can be found in our previous publication.62
Vector or Pattern Similarity Calculated by MCCS.
The mccsx (version 1.4.1, https://github.com/stcmz/mccsx), another part of the MCCS implementation, was applied to quantify the similarity between two vectors generated by jdock using the Pearson correlation coefficient (PCC). The row vectors and/or column vectors in the resulting similarity matrix were clustered using the Farthest Point Algorithm (i.e., the Voor Hees Algorithm) with cosine distance and/or Pearson’s distance. Eventually, based on the energy values (scaled in the same way as the Vina scoring function) of residues in the binding pocket, the complexes were clustered in the way that similar proteins with similar ligand conformation were grouped together.
Alignment of the Structures and Calculation of RMSD.
Alignment of the structures within each pair and the root-mean-square deviation (RMSD) value for the protein structure and binding pockets in each pair were calculated by the “align” command in PyMol, which was suitable for the two protein structures with similar sequences. Orthosteric- and allosteric binding pockets consisted of residues around the ligands within 8 Å, which was the cutoff value for the minimal distance to generate interaction between two atoms and was adopted in the algorithms of jdock in MCCS and Autodock Vina.106
In Silico Site Mutagenesis.
In silico site mutagenesis was conducted with the Mutagenesis Wizard in PyMol. The conformation of the mutated residues was selected when the ligands were bound to the receptors.
Molecular Dynamics (MD) Simulation.
Two pairs of complexes, PDB 4MQS–4MQT and PDB 2RH1-5X7D, were set up for MD simulation. Each complex consisted of a receptor and an orthosteric ligand w/o an AM, which were put into a 0.15 M NaCl solution with a cubic water box, 300 POPC lipid molecules, and about 20,809 TIP3P water molecules.108 CHARMM-GUI Online Toolkit109 (https://charmm-gui.org) was applied to add POPC lipids and prepared the bilayer membrane where the membrane protein embedded with the size about 128 Å × 110 Å × 128 Å. The protein was modeled with the AMBER ff14SB force field.110 The partial atomic charges of ligands were derived via the semiempirical with bond charge correction (AM1-BCC) method.111,112 The residue topologies for ligands were prepared with the ANTECHAMBER module. The other force field parameters were obtained from GAFF in AMBER16.112
The MD simulations were conducted with the PMEMD.mpi and PMEMD.cuda modules in the AMBER18113–115 package. The system was first minimized by several steps to avoid possible steric clashes. Subsequently, each system was gradually heated from 0 to 300 K during the heating stage and maintained at 300 K during the following equilibrium and production stages with a time step of 2 fs. The constant temperature and pressure ensembles were maintained with a periodic boundary condition. The pressure was set at 1 atm and controlled by the anisotropic (x-, y-, z-) pressure scaling protocol with a pressure relaxation time of 1 ps. The temperature was regulated using Langevin dynamics with a collision frequency of 2 ps−1.116,117 The particle mesh Ewald (PME) method118,119 was adopted to handle long-range electrostatics and a 10 Å cutoff was set to treat real-space interactions. All covalent bonds involving hydrogen atoms were constrained with the SHAKE algorithm.120 Each system was subject to a 100 ns MD simulation and the trajectory of simulated systems was saved every 100 ps.
CONCLUSIONS
When an AM was bound to a target protein complexed with an orthosteric ligand, it stabilizes the complex of protein–ligand into an active, inactive, or intermediate state and may lead to a mild conformational change of the whole protein structure. However, there was no evidence regarding the influences of the binding of AMs on the orthosteric binding pocket and the allosteric ones. In the present study, we exploited an innovative technique, MCCS, which was developed by our lab and can characterize the interactions between compounds and proteins to analyze 21 pairs of 3D crystal or cryo-EM complexes of protein–ligand–AM, including 4 pairs of GPCRs, 5 pairs of ion channels, 11 pairs of enzymes, and 1 pair of transcription factor. The results demonstrated that the binding of AM has few impacts on the orthosteric binding site based on the little change of either conformation or energy contribution of the residues involved in the interactions between the protein and the orthosteric ligand. Moreover, the binding of AMs did not cause a significant conformational change of allosteric binding pocket(s), and only a few residues with a large side chain may rotate away or approach toward the AMs. MD simulation studies supported that the structures of the protein and ligands are stable on the multi-ns time scale. Therefore, if the allosteric binding pocket(s) was predicted accurately, we may conduct virtual screening as well as lead compound-based optimization for the AMs. In summary, our study can facilitate the rational design and development of allosteric drug candidates.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge the funding support to the Xie laboratory from the NIH NIDA (R01DA052329 and P30 DA035778A1).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.1c00749.
Detailed energy contributions of important binding residues in the orthosteric pocket of AR, the overlay of structures in MD simulation studies, AM drugs approved by U.S. FDA (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.1c00749
Notes
The authors declare no competing financial interest.
Contributor Information
Chih-Jung Chen, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States; National Center of Excellence for Computational Drug Abuse Research, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Chen Jiang, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States; National Center of Excellence for Computational Drug Abuse Research, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Jiayi Yuan, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States; National Center of Excellence for Computational Drug Abuse Research, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Maozi Chen, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States; National Center of Excellence for Computational Drug Abuse Research, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Jacob Cuyler, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States; National Center of Excellence for Computational Drug Abuse Research, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Xiang-Qun Xie, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States; National Center of Excellence for Computational Drug Abuse Research, Drug Discovery Institute, and Departments of Computational Biology and Structural Biology, School of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Zhiwei Feng, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States; National Center of Excellence for Computational Drug Abuse Research, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
REFERENCES
- (1).He X; Ni D; Lu S; Zhang J Characteristics of Allosteric Proteins, Sites, and Modulators. In Protein Allostery in Drug Discovery; Zhang J; Nussinov R, Eds.; Springer: Singapore, 2019; Vol. 1163, pp 107–139. [DOI] [PubMed] [Google Scholar]
- (2).Feng Z; Hu G; Ma S; Xie XQ Computational Advances for the Development of Allosteric Modulators and Bitopic Ligands in G Protein-Coupled Receptors. AAPS J 2015, 17, 1080–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jacobsen SE; Gether U; Bräuner-Osborne H Investigating the molecular mechanism of positive and negative allosteric modulators in the calcium-sensing receptor dimer. Sci. Rep 2017, 7, No. 46355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wold EA; Chen J; Cunningham KA; Zhou J Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem 2019, 62, 88–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Springthorpe B; Bailey A; Barton P; Birkinshaw TN; Bonnert RV; Brown RC; Chapman D; Dixon J; Guile SD; Humphries RG; Hunt SF; Ince F; Ingall AH; Kirk IP; Leeson PD; Leff P; Lewis RJ; Martin BP; McGinnity DF; Mortimore MP; Paine SW; Pairaudeau G; Patel A; Rigby AJ; Riley RJ; Teobald BJ; Tomlinson W; Webborn PJ; Willis PA From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg. Med. Chem. Lett 2007, 17, 6013–6018. [DOI] [PubMed] [Google Scholar]
- (6).Garcia-Perez J; Rueda P; Staropoli I; Kellenberger E; Alcami J; Arenzana-Seisdedos F; Lagane B New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J. Biol. Chem 2011, 286, 4978–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Waugh DJ; Gaivin RJ; Damron DS; Murray PA; Perez DM Binding, partial agonism, and potentiation of alpha(1)- adrenergic receptor function by benzodiazepines: A potential site of allosteric modulation. J. Pharmacol. Exp. Ther 1999, 291, 1164–1171. [PubMed] [Google Scholar]
- (8).Morlock EV; Czajkowski C Different residues in the GABAA receptor benzodiazepine binding pocket mediate benzodiazepine efficacy and binding. Mol. Pharmacol 2011, 80, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sigel E Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr. Top. Med. Chem 2002, 2, 833–839. [DOI] [PubMed] [Google Scholar]
- (10).Sigel E; Steinmann ME Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem 2012, 287, 40224–40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhu S; Noviello CM; Teng J; Walsh RM Jr.; Kim JJ; Hibbs RE Structure of a human synaptic GABA(A) receptor. Nature 2018, 559, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Möhler H; Fritschy JM; Rudolph U A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther 2002, 300, 2–8. [DOI] [PubMed] [Google Scholar]
- (13).Riss J; Cloyd J; Gates J; Collins S Benzodiazepines in epilepsy: pharmacology and pharmacokinetics. Acta Neurol. Scand 2008, 118, 69–86. [DOI] [PubMed] [Google Scholar]
- (14).Quirk JC; Nisenbaum ES Multiple molecular determinants for allosteric modulation of alternatively spliced AMPA receptors. J. Neurosci 2003, 23, 10953–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Desai MA; Burnett JP; Ornstein PL; Schoepp DD Cyclothiazide acts at a site on the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor complex that does not recognize competitive or noncompetitive AMPA receptor antagonists. J. Pharmacol. Exp. Ther 1995, 272, 38–43. [PubMed] [Google Scholar]
- (16).Feng HJ; Jounaidi Y; Haburcak M; Yang X; Forman SA Etomidate produces similar allosteric modulation in α1β3δ and α1β3γ2L GABA(A) receptors. Br. J. Pharmacol 2014, 171, 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vinkers CH; Olivier B; Hanania T; Min W; Schreiber R; Hopkins SC; Campbell U; Paterson N Discriminative stimulus properties of GABAA receptor positive allosteric modulators TPA023, ocinaplon and NG2-73 in rats trained to discriminate chlordiazepoxide or zolpidem. Eur. J. Pharmacol 2011, 668, 190–193. [DOI] [PubMed] [Google Scholar]
- (18).Richter G; Liao VWY; Ahring PK; Chebib M The Z-Drugs Zolpidem, Zaleplon, and Eszopiclone Have Varying Actions on Human GABA (A) Receptors Containing γ1, γ2, and γ3 Subunits. Front. Neurosci 2020, 14, No. 599812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jayakar SS; Zhou X; Chiara DC; Dostalova Z; Savechenkov PY; Bruzik KS; Dailey WP; Miller KW; Eckenhoff RG; Cohen JB Multiple propofol-binding sites in a γ-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J Biol Chem 2014, 289 (40), 27456–27468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liang Q; Anderson WD; Jones ST; Souza CS; Hosoume JM; Treptow W; Covarrubias M Positive Allosteric Modulation of Kv Channels by Sevoflurane: Insights into the Structural Basis of Inhaled Anesthetic Action. PLoS One 2015, 10, No. e0143363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mapelli J; Gandolfi D; Giuliani E; Casali S; Congi L; Barbieri A; D’Angelo E; Bigiani A The effects of the general anesthetic sevoflurane on neurotransmission: an experimental and computational study. Sci. Rep 2021, 11, No. 4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ahmed AH; Oswald RE Piracetam defines a new binding site for allosteric modulators of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors. J. Med. Chem 2010, 53, 2197–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Quirk JC; Nisenbaum ES Multiple molecular determinants for allosteric modulation of alternatively spliced AMPA receptors. J. Neurosci 2003, 23, 10953–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rogawski MA; Hanada T Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist. Acta Neurol. Scand 2013, 127, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schoemaker H; Boles RG; Roeske WR; Yamamura HI Allosteric modulation by diltiazem and verapamil of [3H]nitrendipine binding to calcium channel sites in rat brain. Proc. West Pharmacol. Soc 1983, 26, 219–224. [PubMed] [Google Scholar]
- (26).Holck M; Fischli W; Hengartner U Effects of temperature and allosteric modulators on [3H] nitrendipine binding: methods for detecting potential Ca2+ channel blockers. J. Recept. Res 1984, 4, 557–569. [DOI] [PubMed] [Google Scholar]
- (27).Tang L; Gamal El-Din TM; Lenaeus MJ; Zheng N; Catterall W Structural Basis for Diltiazem Block of a Voltage-gated Ca2+ Channel. Mol. Pharmacol 2019, 96, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Porzig H; Becker C Potential-dependent allosteric modulation of 1,4-dihydropyridine binding by d-(cis)-diltiazem and (+/−)-verapamil in living cardiac cells. Mol. Pharmacol 1988, 34, 172–179. [PubMed] [Google Scholar]
- (29).Guzman JN; Ilijic E; Yang B; Sanchez-Padilla J; Wokosin D; Galtieri D; Kondapalli J; Schumacker PT; Surmeier DJ Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Invest 2018, 128, 2266–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vallés AS; Garbus I; Antollini SS; Barrantes FJ A novel agonist effect on the nicotinic acetylcholine receptor exerted by the anticonvulsive drug Lamotrigine. Biochim. Biophys. Acta, Biomembr 2008, 1778, 2395–2404. [DOI] [PubMed] [Google Scholar]
- (31).Twyman RE; Rogers CJ; Macdonald RL Differential regulation of gamma-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann. Neurol 1989, 25, 213–220. [DOI] [PubMed] [Google Scholar]
- (32).Krause RM; Buisson B; Bertrand S; Corringer PJ; Galzi JL; Changeux JP; Bertrand D Ivermectin: a positive allosteric effector of the alpha7 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol 1998, 53, 283–294. [DOI] [PubMed] [Google Scholar]
- (33).Häberle J Role of carglumic acid in the treatment of acute hyperammonemia due to N-acetylglutamate synthase deficiency. Ther. Clin. Risk Manage 2011, 7, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gilmartin AG; Bleam MR; Groy A; Moss KG; Minthorn EA; Kulkarni SG; Rominger CM; Erskine S; Fisher KE; Yang J; Zappacosta F; Annan R; Sutton D; Laquerre SG GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res 2011, 17, 989–1000. [DOI] [PubMed] [Google Scholar]
- (35).Chiu MI; Katz H; Berlin V RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc. Natl. Acad. Sci. U.S.A 1994, 91, 12574–12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen J; Yang J; Sun X; Wang Z; Cheng X; Lu W; Cai X; Hu C; Shen X; Cao P Allosteric inhibitor remotely modulates the conformation of the orthestric pockets in mutant IDH2/R140Q. Sci. Rep 2017, 7, No. 16458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Schauer GD; Huber KD; Leuba SH; Sluis-Cremer N Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence. Nucleic Acids Res 2014, 42, 11687–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Artsimovitch I; Vassylyeva MN; Svetlov D; Svetlov V; Perederina A; Igarashi N; Matsugaki N; Wakatsuki S; Tahirov TH; Vassylyev DG Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell 2005, 122, 351–363. [DOI] [PubMed] [Google Scholar]
- (39).Changeux J-P; Christopoulos A Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102. [DOI] [PubMed] [Google Scholar]
- (40).Chiarini F; Lonetti A; Teti G; Orsini E; Bressanin D; Cappellini A; Ricci F; Tazzari PL; Ognibene A; Falconi M; Pagliaro P; Iacobucci I; Martinelli G; Amadori S; McCubrey JA; Martelli AM A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget 2012, 3, 1615–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yang L; Manithody C; Walston TD; Cooper ST; Rezaie AR Thrombomodulin enhances the reactivity of thrombin with protein C inhibitor by providing both a binding site for the serpin and allosterically modulating the activity of thrombin. J. Biol. Chem 2003, 278, 37465–37470. [DOI] [PubMed] [Google Scholar]
- (42).Cheng Y; Tian H Current Development Status of MEK Inhibitors. Molecules 2017, 22, No. 1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cho HS; Mason K; Ramyar KX; Stanley AM; Gabelli SB; Denney DW Jr.; Leahy DJ Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [DOI] [PubMed] [Google Scholar]
- (44).Franklin MC; Carey KD; Vajdos FF; Leahy DJ; de Vos AM; Sliwkowski MX Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [DOI] [PubMed] [Google Scholar]
- (45).Wick JY The history of benzodiazepines. Consult. Pharm 2013, 28, 538–548. [DOI] [PubMed] [Google Scholar]
- (46).Skerritt JH; Johnston GA Enhancement of GABA binding by benzodiazepines and related anxiolytics. Eur. J. Pharmacol 1983, 89, 193–198. [DOI] [PubMed] [Google Scholar]
- (47).Study RE; Barker JL Cellular mechanisms of benzodiazepine action. JAMA 1982, 247, 2147–2151. [PubMed] [Google Scholar]
- (48).Olsen RW; Yang J; King RG; Dilber A; Stauber GB; Ransom RW Barbiturate and benzodiazepine modulation of GABA receptor binding and function. Life Sci 1986, 39, 1969–1976. [DOI] [PubMed] [Google Scholar]
- (49).de Francisco AL Cinacalcet HCl: a novel therapeutic for hyperparathyroidism. Expert Opin. Pharmacother 2005, 6, 441–452. [DOI] [PubMed] [Google Scholar]
- (50).Moe SM; Cunningham J; Bommer J; Adler S; Rosansky SJ; Urena-Torres P; Albizem MB; Guo MD; Zani VJ; Goodman WG; Sprague SM Long-term treatment of secondary hyperparathyroidism with the calcimimetic cinacalcet HCl. Nephrol., Dial., Transplant 2005, 20, 2186–2193. [DOI] [PubMed] [Google Scholar]
- (51).Cunningham J Management of secondary hyperparathyroidism. Ther. Apheresis Dial 2005, 9, S35–S40. [DOI] [PubMed] [Google Scholar]
- (52).Eriguchi R; Umakoshi J; Tominaga Y; Sato Y Successful treatment of inoperable recurrent secondary hyperparathyroidism with cinacalcet HCl. NDT Plus 2008, 1, 218–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Meola M; Petrucci I; Barsotti G Long-term treatment with cinacalcet and conventional therapy reduces parathyroid hyperplasia in severe secondary hyperparathyroidism. Nephrol., Dial., Transplant 2008, 24, 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Goodsell DS; Zardecki C; Di Costanzo L; Duarte JM; Hudson BP; Persikova I; Segura J; Shao C; Voigt M; Westbrook JD; Young JY; Burley SK RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Sci 2020, 29, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Huang Z; Zhu L; Cao Y; Wu G; Liu X; Chen Y; Wang Q; Shi T; Zhao Y; Wang Y; Li W; Li Y; Chen H; Chen G; Zhang J ASD: a comprehensive database of allosteric proteins and modulators. Nucleic Acids Res 2011, 39, D663–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Liu X; Ahn S; Kahsai AW; Meng KC; Latorraca NR; Pani B; Venkatakrishnan AJ; Masoudi A; Weis WI; Dror RO; Chen X; Lefkowitz RJ; Kobilka BK Mechanism of intracellular allosteric β(2)AR antagonist revealed by X-ray crystal structure. Nature 2017, 548, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Feng Z; Ma S; Hu G; Xie X-Q Allosteric Binding Site and Activation Mechanism of Class C G-Protein Coupled Receptors: Metabotropic Glutamate Receptor Family. AAPS J 2015, 17, 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hou T; Bian Y; McGuire T; Xie X-Q Integrated multiclass classification and prediction of GPCR allosteric modulators by machine learning intelligence. Biomolecules 2021, 11, No. 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bian Y; Feng Z; Yang P; Xie X-Q Integrated In Silico Fragment-Based Drug Design: Case Study with Allosteric Modulators on Metabotropic Glutamate Receptor 5. AAPS J 2017, 19, 1235–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Feng Z; Alqarni MH; Yang P; Tong Q; Chowdhury A; Wang L; Xie X-Q Modeling, Molecular Dynamics Simulation, and Mutation Validation for Structure of Cannabinoid Receptor 2 Based on Known Crystal Structures of GPCRs. J. Chem. Inf. Model 2014, 54, 2483–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Bian Y; Jing Y; Wang L; Ma S; Jun JJ; Xie X-Q Prediction of Orthosteric and Allosteric Regulations on Cannabinoid Receptors Using Supervised Machine Learning Classifiers. Mol. Pharmaceutics 2019, 16, 2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Chen M; Feng Z; Wang S; Lin W; Xie XQ MCCS, a novel characterization method for protein-ligand complex. Briefings Bioinf 2021, 22, No. bbaa239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Molinoff PB Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs 1984, 28, 1–15. [DOI] [PubMed] [Google Scholar]
- (64).Triposkiadis F; Karayannis G; Giamouzis G; Skoularigis J; Louridas G; Butler J The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol 2009, 54, 1747–1762. [DOI] [PubMed] [Google Scholar]
- (65).De Backer I; Hussain SS; Bloom SR; Gardiner JV Insights into the role of neuronal glucokinase. Am. J. Physiol.: Endocrinol. Metab 2016, 311, E42–E55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Iynedjian PB Molecular physiology of mammalian glucokinase. Cell. Mol. Life Sci 2009, 66, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Platt SR The role of glutamate in central nervous system health and disease–a review. Vet. J 2007, 173, 278–286. [DOI] [PubMed] [Google Scholar]
- (68).Limatola C Neurotrophic effects of AMPA. Cerebellum 2004, 3, 2–10. [DOI] [PubMed] [Google Scholar]
- (69).Zhou Q; Sheng M NMDA receptors in nervous system diseases. Neuropharmacology 2013, 74, 69–75. [DOI] [PubMed] [Google Scholar]
- (70).Zanetti L; Regoni M; Ratti E; Valtorta F; Sassone J Presynaptic AMPA Receptors in Health and Disease. Cells 2021, 10, No. 2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Feng Z; Liang T; Wang S; Chen M; Hou T; Zhao J; Chen H; Zhou Y; Xie XQ Binding Characterization of GPCRs-Modulator by Molecular Complex Characterizing System (MCCS). ACS Chem. Neurosci 2020, 11, 3333–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Feng Z; Chen M; Xue Y; Liang T; Chen H; Zhou Y; Nolin TD; Smith RB; Xie XQ MCCS: a novel recognition pattern-based method for fast track discovery of anti-SARS-CoV-2 drugs. Briefings Bioinf 2021, 22, 946–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Cheng J; Chen M; Wang S; Liang T; Chen H; Chen CJ; Feng Z; Xie XQ Binding Characterization of Agonists and Antagonists by MCCS: A Case Study from Adenosine A(2A) Receptor. ACS Chem. Neurosci 2021, 12, 1606–1620. [DOI] [PubMed] [Google Scholar]
- (74).Kruse AC; Ring AM; Manglik A; Hu J; Hu K; Eitel K; Hübner H; Pardon E; Valant C; Sexton PM; Christopoulos A; Felder CC; Gmeiner P; Steyaert J; Weis WI; Garcia KC; Wess J; Kobilka BK Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wieland K; Zuurmond HM; Krasel C; Ijzerman AP; Lohse MJ Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta 2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 9276–9281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Hanson MA; Cherezov V; Griffith MT; Roth CB; Jaakola VP; Chien EY; Velasquez J; Kuhn P; Stevens RC A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Gloyn AL; Noordam K; Willemsen MA; Ellard S; Lam WW; Campbell IW; Midgley P; Shiota C; Buettger C; Magnuson MA; Matschinsky FM; Hattersley AT Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes 2003, 52, 2433–2440. [DOI] [PubMed] [Google Scholar]
- (78).Heredia VV; Carlson TJ; Garcia E; Sun S Biochemical basis of glucokinase activation and the regulation by glucokinase regulatory protein in naturally occurring mutations. J. Biol. Chem 2006, 281, 40201–40207. [DOI] [PubMed] [Google Scholar]
- (79).Martínez R; Gutierrez-Nogués Á; Fernández-Ramos C; Velayos T; Vela A; Navas M; Castaño L Heterogeneity in phenotype of hyperinsulinism caused by activating glucokinase mutations: a novel mutation and its functional characterization. Clin. Endocrinol 2017, 86, 778–783. [DOI] [PubMed] [Google Scholar]
- (80).Wu PK; Park JI MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol 2015, 42, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Harding A; Giles N; Burgess A; Hancock JF; Gabrielli BG Mechanism of Mitosis-specific Activation of MEK1*. J. Biol. Chem 2003, 278, 16747–16754. [DOI] [PubMed] [Google Scholar]
- (82).Kornev AP; Haste NM; Taylor SS; Eyck LF Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Fischmann TO; Smith CK; Mayhood TW; Myers JE; Reichert P; Mannarino A; Carr D; Zhu H; Wong J; Yang R-S; Le HV; Madison VS Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry 2009, 48, 2661–2674. [DOI] [PubMed] [Google Scholar]
- (84).Ohren JF; Chen H; Pavlovsky A; Whitehead C; Zhang E; Kuffa P; Yan C; McConnell P; Spessard C; Banotai C; Mueller WT; Delaney A; Omer C; Sebolt-Leopold J; Dudley DT; Leung IK; Flamme C; Warmus J; Kaufman M; Barrett S; Tecle H; Hasemann CA Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol 2004, 11, 1192–1197. [DOI] [PubMed] [Google Scholar]
- (85).Spicer JA; Rewcastle GW; Kaufman MD; Black SL; Plummer MS; Denny WA; Quin J 3rd; Shahripour AB; Barrett SD; Whitehead CE; Milbank JB; Ohren JF; Gowan RC; Omer C; Camp HS; Esmaeil N; Moore K; Sebolt-Leopold JS; Pryzbranowski S; Merriman RL; Ortwine DF; Warmus JS; Flamme CM; Pavlovsky AG; Tecle H 4-anilino-5-carboxamido-2-pyridone derivatives as noncompetitive inhibitors of mitogen-activated protein kinase kinase. J. Med. Chem 2007, 50, 5090–5102. [DOI] [PubMed] [Google Scholar]
- (86).Warmus JS; Flamme C; Zhang LY; Barrett S; Bridges A; Chen H; Gowan R; Kaufman M; Sebolt-Leopold J; Leopold W; Merriman R; Ohren J; Pavlovsky A; Przybranowski S; Tecle H; Valik H; Whitehead C; Zhang E 2-Alkylamino- and alkoxy-substituted 2-amino-1,3,4-oxadiazoles-O-Alkyl benzohydroxamate esters replacements retain the desired inhibition and selectivity against MEK (MAP ERK kinase). Bioorg. Med. Chem. Lett 2008, 18, 6171–6174. [DOI] [PubMed] [Google Scholar]
- (87).Tecle H; Shao J; Li Y; Kothe M; Kazmirski S; Penzotti J; Ding YH; Ohren J; Moshinsky D; Coli R; Jhawar N; Bora E; Jacques-O’Hagan S; Wu J Beyond the MEK-pocket: can current MEK kinase inhibitors be utilized to synthesize novel type III NCKIs? Does the MEK-pocket exist in kinases other than MEK? Bioorg. Med. Chem. Lett 2009, 19, 226–229. [DOI] [PubMed] [Google Scholar]
- (88).Iverson C; Larson G; Lai C; Yeh LT; Dadson C; Weingarten P; Appleby T; Vo T; Maderna A; Vernier JM; Hamatake R; Miner JN; Quart B RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res 2009, 69, 6839–6847. [DOI] [PubMed] [Google Scholar]
- (89).Dong Q; Dougan DR; Gong X; Halkowycz P; Jin B; Kanouni T; O’Connell SM; Scorah N; Shi L; Wallace MB; Zhou F Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett 2011, 21, 1315–1319. [DOI] [PubMed] [Google Scholar]
- (90).Wallace MB; Adams ME; Kanouni T; Mol CD; Dougan DR; Feher VA; O’Connell SM; Shi L; Halkowycz P; Dong Q Structure-based design and synthesis of pyrrole derivatives as MEK inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 4156–4158. [DOI] [PubMed] [Google Scholar]
- (91).Salussolia CL; Prodromou ML; Borker P; Wollmuth LP Arrangement of subunits in functional NMDA receptors. J. Neurosci 2011, 31, 11295–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Guo H; Camargo LM; Yeboah F; Digan ME; Niu H; Pan Y; Reiling S; Soler-Llavina G; Weihofen WA; Wang HR; Shanker YG; Stams T; Bill A A NMDA-receptor calcium influx assay sensitive to stimulation by glutamate and glycine/D-serine. Sci. Rep. 2017, 7, No. 11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Bonaccorso C; Micale N; Ettari R; Grasso S; Zappalà M Glutamate binding-site ligands of NMDA receptors. Curr. Med. Chem 2011, 18, 5483–5506. [DOI] [PubMed] [Google Scholar]
- (94).Hackos DH; Lupardus PJ; Grand T; Chen Y; Wang TM; Reynen P; Gustafson A; Wallweber HJ; Volgraf M; Sellers BD; Schwarz JB; Paoletti P; Sheng M; Zhou Q; Hanson JE Positive Allosteric Modulators of GluN2A-Containing NMDARs with Distinct Modes of Action and Impacts on Circuit Function. Neuron 2016, 89, 983–999. [DOI] [PubMed] [Google Scholar]
- (95).Valant C; Gregory KJ; Hall NE; Scammells PJ; Lew MJ; Sexton PM; Christopoulos A A novel mechanism of G protein-coupled receptor functional selectivity. Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J. Biol. Chem 2008, 283, 29312–29321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Gregory KJ; Hall NE; Tobin AB; Sexton PM; Christopoulos A Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J. Biol. Chem 2010, 285, 7459–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (99).Shapovalov MV; Dunbrack RL Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Pedretti A; Villa L; Vistoli G VEGA – An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput.-Aided Mol. Des 2004, 18, 167–173. [DOI] [PubMed] [Google Scholar]
- (101).Olsson MHM; Søndergaard CR; Rostkowski M; Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
- (102).Søndergaard CR; Olsson MHM; Rostkowski M; Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- (103).Li H; Leung K; Wong M-H In Idock: A Multithreaded Virtual Screening Tool for Flexible Ligand Docking, 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), 2012.
- (104).Wan S; Bhati AP; Zasada SJ; Coveney PV Rapid, accurate, precise and reproducible ligand–protein binding free energy prediction. Interface Focus 2020, 10, No. 20200007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Hall R; Dixon T; Dickson A On Calculating Free Energy Differences Using Ensembles of Transition Paths. Front. Mol. Biosci 2020, 7, No. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Trott O; Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (109).Jo S; Kim T; Im W Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2007, 2, No. e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Jakalian A; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- (112).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (113).Götz AW; Williamson MJ; Xu D; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Salomon-Ferrer R; Götz AW; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- (115).Case DA; Betz RM; Cerutti DS; Cheatham TE III; Darden TA; Duke RE et al. AMBER 2016; University of California: San Francisco, 2016. [Google Scholar]
- (116).Loncharich RJ; Brooks BR; Pastor RW Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [DOI] [PubMed] [Google Scholar]
- (117).Izaguirre JA; Catarello DP; Wozniak JM; Skeel RD Langevin stabilization of molecular dynamics. J. Chem. Phys 2001, 114, 2090–2098. [Google Scholar]
- (118).Darden T; York D; Pedersen L Particle mesh Ewald: An N· log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (119).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (120).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.