

Abstract
Background
Trypanosomatids are parasitic flagellates well known because of some representatives infecting humans, domestic animals, and cultural plants. Many trypanosomatid species bear RNA viruses, which, in the case of human pathogens Leishmania spp., influence the course of the disease. One of the close relatives of leishmaniae, Leptomonas pyrrhocoris, has been previously shown to harbor viruses of the groups not documented in other trypanosomatids. At the same time, this species has a worldwide distribution and high prevalence in the natural populations of its cosmopolitan firebug host. It therefore represents an attractive model to study the diversity of RNA viruses.
Results
We surveyed 106 axenic cultures of L. pyrrhocoris and found that 64 (60%) of these displayed 2–12 double-stranded RNA fragments. The analysis of next-generation sequencing data revealed four viral groups with seven species, of which up to five were simultaneously detected in a single trypanosomatid isolate. Only two of these species, a tombus-like virus and an Ostravirus, were earlier documented in L. pyrrhocoris. In addition, there were four new species of Leishbuviridae, the family encompassing trypanosomatid-specific viruses, and a new species of Qinviridae, the family previously known only from metatranscriptomes of invertebrates. Currently, this is the only qinvirus with an unambiguously determined host. Our phylogenetic inferences suggest reassortment in the tombus-like virus owing to the interaction of different trypanosomatid strains. Two of the new Leishbuviridae members branch early on the phylogenetic tree of this family and display intermediate stages of genomic segment reduction between insect Phenuiviridae and crown Leishbuviridae.
Conclusions
The unprecedented wide range of viruses in one protist species and the simultaneous presence of up to five viral species in a single Leptomonas pyrrhocoris isolate indicate the uniqueness of this flagellate. This is likely determined by the peculiarity of its firebug host, a highly abundant cosmopolitan species with several habits ensuring wide distribution and profuseness of L. pyrrhocoris, as well as its exposure to a wider spectrum of viruses compared to other trypanosomatids combined with a limited ability to transmit these viruses to its relatives. Thus, L. pyrrhocoris represents a suitable model to study the adoption of new viruses and their relationships with a protist host.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12915-023-01687-y.
Keywords: Tombus-like viruses, Ostravirus, Leishbuviridae, Qinviridae, Pyrrhocoris apterus
Background
Parasitic flagellates of the family Trypanosomatidae are a diverse group of protists, some of which are well-known pathogens of humans, domestic animals, and cultural plants [1]. According to their life cycle, they are subdivided into monoxenous (developing in one host) and dixenous (switching between two different hosts) species. Only a few genera, namely phytoparasitic Phytomonas, as well as vertebrate-infecting Trypanosoma and Leishmania sensu lato (i.e., including Porcisia and Endotrypanum) are dixenous, whereas the vast majority of known trypanosomatid lineages are monoxenous parasites of insects [2]. Arguably, the most diverse group within the family is the species-rich subfamily Leishmaniinae, which unites dixenous Leishmania s. l. and their closest monoxenous relatives of the genera Borovskyia, Crithidia, Leptomonas, Lotmaria, Novymonas, and Zelonia [3]. Leptomonas pyrrhocoris is one of the model species of this subfamily. It is an easily cultivable monoxenous parasite of the cosmopolitan and abundant firebug Pyrrhocoris apterus [4], with a genomic sequence assembled to a near-chromosome level [5].
Several species of Leishmania harbor double-stranded Leishmania RNA viruses 1 and 2 (LRV1/2, genus Leishmaniavirus) of the family Totiviridae [6–8]. The distribution of these viruses into the LRV1 and LRV2 clades mirrors the separation of Leishmania into the New and Old World lineages, respectively, indicating a deep co-evolutionary history of leishmaniaviruses and their hosts [9, 10]. The LRV1 in L. guyanensis is responsible for more severe symptoms of mucocutaneous leishmaniasis caused by the virus interference with anti-Leishmania immune response, thereby promoting the survival of the infected macrophages [11–13].
The findings described above have inspired a broad survey of trypanosomatid viromes resulting in the discovery of several new viral groups [14]. These included viruses related to Narnaviridae and the new family Leishbuviridae (LBVs) broadly infecting trypanosomatids, as well as Leptomonas pyrrhocoris tombus-like virus (LeppyrTLV1) and Leptomonas pyrrhocoris Ostravirus (LeppyrOV1) restricted to L. pyrrhocoris. In addition, an endogenous viral element, homologous to the large segment of the LeppyrTLV1, was detected in the genome of L. pyrrhocoris suggesting a long-lasting interaction between the virus and the flagellate [14]. A representative of the trypanosomatid-specific family Leishbuviridae was also documented in a peculiar species of Leishmania—L. martiniquensis (subgenus Mundinia), in which it can modulate macrophage infection [15]. Interestingly, the LRV-related viruses were also found in Blechomonas spp., a divergent clade of monoxenous flea-infecting trypanosomatids, which apparently acquired the virus from Leishmania spp. [16].
Out of all trypanosomatid species studied to date, L. pyrrhocoris represents the most attractive model for studies of the diversity of RNA viruses because of its worldwide distribution, high prevalence in the natural firebug populations, and harboring of unique viruses. In this study, we broadened the scope of known viruses in L. pyrrhocoris by systematically sampling them across Europe. In addition to previously known Ostravirus and tombus-like viruses, we documented new divergent LBVs and a Qin-like virus, the latter being the first detection of this rare viral group in a trypanosomatid host.
Results and discussion
Infection prevalence and detection of double-stranded RNA
Out of 508 dissected firebugs, 374 (74%) were positive for trypanosomatids with prevalence varying between 27 and 100% for different localities. From this material, 106 axenic cultures of L. pyrrhocoris were established, of which 64 (60%) showed the presence of 2–12 double-stranded RNA (dsRNA) bands ranging from 1.3 to 6.2 kb in length (Table 1).
Table 1.
Studied Leptomonas pyrrhocoris isolates: geographic origin and detection of viruses
| Country | City | Trypanosomatid prevalence | Positive for dsRNA/tested | Isolate | Fragments on the gel [kb] | Viral identity | NGS |
|---|---|---|---|---|---|---|---|
| Belarus | Vitebsk | 8/8 (100%) | 1/3 | BY-Vi257 | |||
| BY-Vi260 | 3.5, 2.2 | TLV | Yes | ||||
| BY-Vi262 | |||||||
| Czechia | Benešov | 4/7 (57%) | 0/2 | CZ-Be02 | |||
| CZ-Be04 | |||||||
| Brno | 7/10 (70%) | 2/3 | CZ-Br01 | 3.5, 2.2 | TLV | ||
| CZ-Br02 | 3.5, 2.2 | TLV, OVa, LBV1/2/4a | Yes | ||||
| CZ-Br07 | |||||||
| České Budějovice | 16/25 (64%) | 0/4 | CZ-CB02 | ||||
| CZ-CB03 | |||||||
| CZ-CB13 | |||||||
| CZ-CB16 | |||||||
| Hradec nad Moravicí | 12/16 (75%) | 9/9 | CZ-HM01 | 3.5, 2.2 | TLV, LBV3a | Yes | |
| CZ-HM02 | 3.5, 2.2 | TLV | Yes | ||||
| CZ-HM03 | 3.5, 2.2 | TLV | |||||
| CZ-HM04 | 3.5, 2.2 | TLV | Yes | ||||
| CZ-HM05 | 3.5, 2.2 | TLV | |||||
| CZ-HM06 | 3.5, 2.2 | TLV | Yes | ||||
| CZ-HM07 | 3.5, 2.3 | TLV | |||||
| CZ-HM08 | 3.5, 2.2 | TLV | Yes | ||||
| CZ-HM09 | 3.5, 2.2 | TLV | Yes | ||||
| Ostrava | 24/30 (80%) | 11/13 | CZ-Os00 | 3.5, 2.2 | TLV | ||
| CZ-Os01 | 3.5, 2.2 | TLV | |||||
| CZ-Os02 | 3.5, 2.2 | TLV | |||||
| CZ-Os03 | 3.5, 2.2 | TLV | Yes | ||||
| CZ-Os05 | 3.5, 2.2 | TLV | |||||
| CZ-Os07 | 3.5, 2.2 | TLV | |||||
| CZ-Os08 | 3.5, 2.2 | TLV | |||||
| CZ-Os10 | 3.5, 2.2, 5, 4.2, 4, 3, 2.4, 1.6, 1.3 | TLV, OV | |||||
| CZ-Os11 | 3.5, 2.2 | TLV | Yes | ||||
| CZ-Os12 | 3.5, 2.2 | TLV | |||||
| CZ-Os13 | |||||||
| CZ-Os14 | |||||||
| CZ-Os15 | 3.5, 2.2, 5.0, 4.2, 4.0, 3.0, 2.4, 1.6, 1.3 | TLV, OV | |||||
| Prague | 12/21 (57%) | 1/3 | CZ-Pr02 | ||||
| CZ-Pr14 | 3.5, 2.2, 5.0, 4.2, 4.0, 3.0, 2.4, 1.6, 1.3, 6.2, 1.9, 1.5 | TLV, OV, LBV3 | Yes | ||||
| Cz-Pr24 | |||||||
| Germany | Jena | 3/4 (75%) | 1/1 | DE-Je02 | 3.5, 2.2 | TLV | Yes |
| Hungary | Varbó | 10/16 (62%) | 2/3 | HU-Va04 | 3.5, 2.2 | TLV | Yes |
| HU-Va05 | 6.2, 1.9, 1.5 | LBV3 | Yes | ||||
| HU-Va09 | |||||||
| Italy | Grosseto | 11/28 (39%) | 2/3 | IT-Gr06 | 3.5, 2.2 | TLV | |
| IT-Gr07 | 3.5, 2.2 | TLV | |||||
| IT-Gr10 | |||||||
| Rome | 8/30 (27%) | 6/8 | IT-Ro01 | 3.5, 2.2 | TLV | Yes | |
| IT-Ro02 | 3.5, 2.2 | TLV | |||||
| IT-Ro03 | |||||||
| IT-Ro04 | 3.5, 2.2 | TLV | |||||
| IT-Ro05 | |||||||
| IT-Ro06 | 3.5, 2.2 | TLV | Yes | ||||
| IT-Ro07 | 3.5, 2.2 | TLV | |||||
| IT-Ro08 | 3.5, 2.2 | TLV | |||||
| Lithuania | Vilnius | 8/8 (100%) | 3/3 | LT-Vi06 | 3.5, 2.2 | TLV | Yes |
| LT-Vi08 | 3.5, 2.2 | TLV | Yes | ||||
| LT-Vi09 | 3.5, 2.2, 6.2, 1.9, 1.5 | TLV, LBV3 | Yes | ||||
| Poland | Rzeszów | 18/20 (90%) | 3/6 | PL-Rz05 | |||
| PL-Rz06 | 3.5, 2.2, 5.0, 4.2, 4.0, 3.0, 2.4, 1.6, 1.3, 6.2, 1.9, 1.5 | TLV, OV, LBV3 | Yes | ||||
| PL-Rz11 | |||||||
| PL-Rz12 | |||||||
| PL-Rz13 | 3.5, 2.2 | TLV | Yes | ||||
| PL-Rz18 | 6.2, 1.9, 1.5 | LBV3 | Yes | ||||
| Portugal | Lisbon | 6/13 (46%) | 4/5 | PT-Li02 | 3.5, 2.2 | TLV | |
| PT-Li03 | 3.5, 2.2 | TLV | Yes | ||||
| PT-Li04 | 3.5, 2.2 | TLV | |||||
| PT-Li05 | |||||||
| PT-Li06 | 3.5, 2.2, 6.2, 3.2, 1.8 | TLV, LBV4 | Yes | ||||
| Romania | Luncavița | 2/6 (33%) | 1/1 | RO-Lu01 | 3.5, 2.2 | TLV | |
| Russia | Borisovka | 8/8 (100%) | 0/1 | RU-Bo01 | |||
| Krasnodar | 11/11 (100%) | 1/2 | RU-Kr01 | 3.5, 2.2, 6.2, 1.9, 1.5 | TLV, LBV3, OVa | Yes | |
| RU-Kr02 | |||||||
| Moscow | 14/17 (82%) | 4/4 | RU-Mo01 | 3.5, 2.2 | TLV | Yes | |
| RU-Mo02 | 3.5, 2.2 | TLV | Yes | ||||
| RU-Mo202 | 3.5, 2.2 | TLV | |||||
| RU-Mo203 | 3.5, 2.2 | TLV | Yes | ||||
| Pskov | 92/92 (100%) | 0/3 | RU-Ps01 | ||||
| RU-Ps02 | |||||||
| RU-Ps03 | |||||||
| Suyda | 10/11 (91%) | 1/1 | RU-Su01 | 3.5, 2.2 | TLV | Yes | |
| Serbia | Dimitrovgrad | 12/15 (80%) | 2/4 | SE-Dm02 | |||
| SE-Dm03 | |||||||
| SE-Dm04 | 3.5, 2.2 | TLV | Yes | ||||
| SE-Dm07 | 3.5, 2.2, 6.2 | TLV, LBVb,c | |||||
| Subotica | 10/13 (77%) | 3/7 | SE-Sb01 | ||||
| SE-Sb02 | 3.5, 2.2, 6.2, 1.9, 1.5 | TLV, LBV2/3/4 | Yes | ||||
| SE-Sb03 | |||||||
| SE-Sb04 | |||||||
| SE-Sb06 | 3.5, 2.2 | TLV | |||||
| SE-Sb07 | 3.5, 2.2, 6.2, 1.9, 1.5 | TLV, QINa, LBV2/3/4 | Yes | ||||
| SE-Sb09 | |||||||
| Slovakia | Bratislava | 25/30 (83%) | 4/5 | SK-Br02 | 3.5, 2.2, 6.2 | TLV, LBV2b | Yes |
| SK-Br05 | 3.5, 2.2 | TLV | Yes | ||||
| SK-Br18 | 3.5, 2.2 | TLV | |||||
| SK-Br20 | 3.5, 2.2 | TLV | |||||
| SK-Br24 | |||||||
| Košice | 9/20 (45%) | 0/1 | SKK6 | ||||
| Liptovský Hrádok | 7/10 (70%) | 0/3 | SK-LH01 | ||||
| SK-LH04 | |||||||
| SK-LH06 | |||||||
| Ľubochňa | 18/25 (72%) | 0/4 | SK-Lu01 | ||||
| SK-Lu10 | |||||||
| SK-Lu15 | |||||||
| SK-Lu16 | |||||||
| Ukraine | Zaporizhzhia | 9/14 (64%) | 3/4 | UA-Zp01 | 3.5, 2.2, 5.0, 4.2, 4.0, 3.0, 2.4, 1.6, 1.3 | TLV, OV | Yes |
| UA-Zp02 | 3.5, 2.2 | TLV, OVa, QINa | Yes | ||||
| UA-Zp03 | 3.5, 2.2 | TLV | Yes | ||||
| UA-Zp04 | |||||||
| Total | 374/508 (74%) | 64/106 (60%) | 61 TLV, 13 LBV, 8 OV, 2 QIN | 37 |
aNo fragments detected on the gel
bOnly the large segment detected on the gel
cThe viral species could not be identified in the absence of sequence data
Next-generation sequencing and viral identification
Out of 64 dsRNA-containing isolates, 37 were selected for next-generation sequencing (NGS), with a focus on those displaying complex dsRNA patterns. These included samples from all countries except for Romania. The analysis of the sequence data allowed the identification of 58 viral genomes (Table 1), of which 32 belonged to the previously described LeppyrTLV1, 18 to four new species of the family Leishbuviridae, 6 to LeppyrOV1, and 2 to a new Qin-like virus. Each sequencing library contained 3.5–20 million reads resulting in the coverage ranging from 5 to 104 reads per kilobase per million (RPKM) for an individual viral genomic segment. The vast majority of viral sequences were complete with a few exceptions due to low coverage (the latter sequences were not deposited to GenBank). Complete genomes were assembled for all viruses, except for the clades 1 and 2 of leishbuviruses, in which the middle segments were not found. These glycoprotein-coding segments usually have an order of magnitude lower coverage compared to the large and small segments and highly divergent amino acid sequences, making the search extremely difficult. Fifteen positive isolates contained two to five viruses at once, suggesting coinfections.
Tombus-like virus and Ostravirus
Out of 106 L. pyrrhocoris isolates tested, 8 and 61 displayed dsRNA bands consistent with the presence of LeppyrOV1 and LeppyrTLV1, respectively (Table 1). This was confirmed by the obtained genomic sequences for 6 LeppyrOV1 and 32 LeppyrTLV1 viruses. Sequence similarity to the previously described prototypical viruses [14] was quite high: the minimal nucleotide identity was 93% for tombus-like virus and 95% for Ostravirus. Open reading frames (ORFs) of LeppyrTLV1 and LeppyrOV1 contained 7.6–16.0% and 5.9–11.6% of variable sites at the nucleotide level or 3.5–6.8% and 1.4–12.8% of those at the amino acid level, respectively (Additional file 2: Table S2).
Phylogenetic analysis revealed that LeppyrTLV1 is related to two viruses recently detected in insects: Leuven tombus-like virus 6 from the common wasp Vespula vulgaris [17] and Vai augu virus from the tule mosquito Culex erythrothorax [18] (Fig. 1A). Previously, we proposed that LeppyrTLV1 could have originated from one of the viruses of non-insect invertebrates occasionally serving as food of firebugs [14]. The new data suggest that the ancestral virus could be of insect origin. Of note, it is not always clear, whether the viruses found in metatranscriptomes of insects actually belong to the latter and not to their microbiota, such as trypanosomatids. Indeed, wasps have been recorded as trypanosomatid hosts [19]. Moreover, as predators, they can temporarily contain non-specific parasites acquired from their insect prey. As for the mosquito virus, no trypanosomatids were detected in the Vai augu virus-containing samples [18].
Fig. 1.
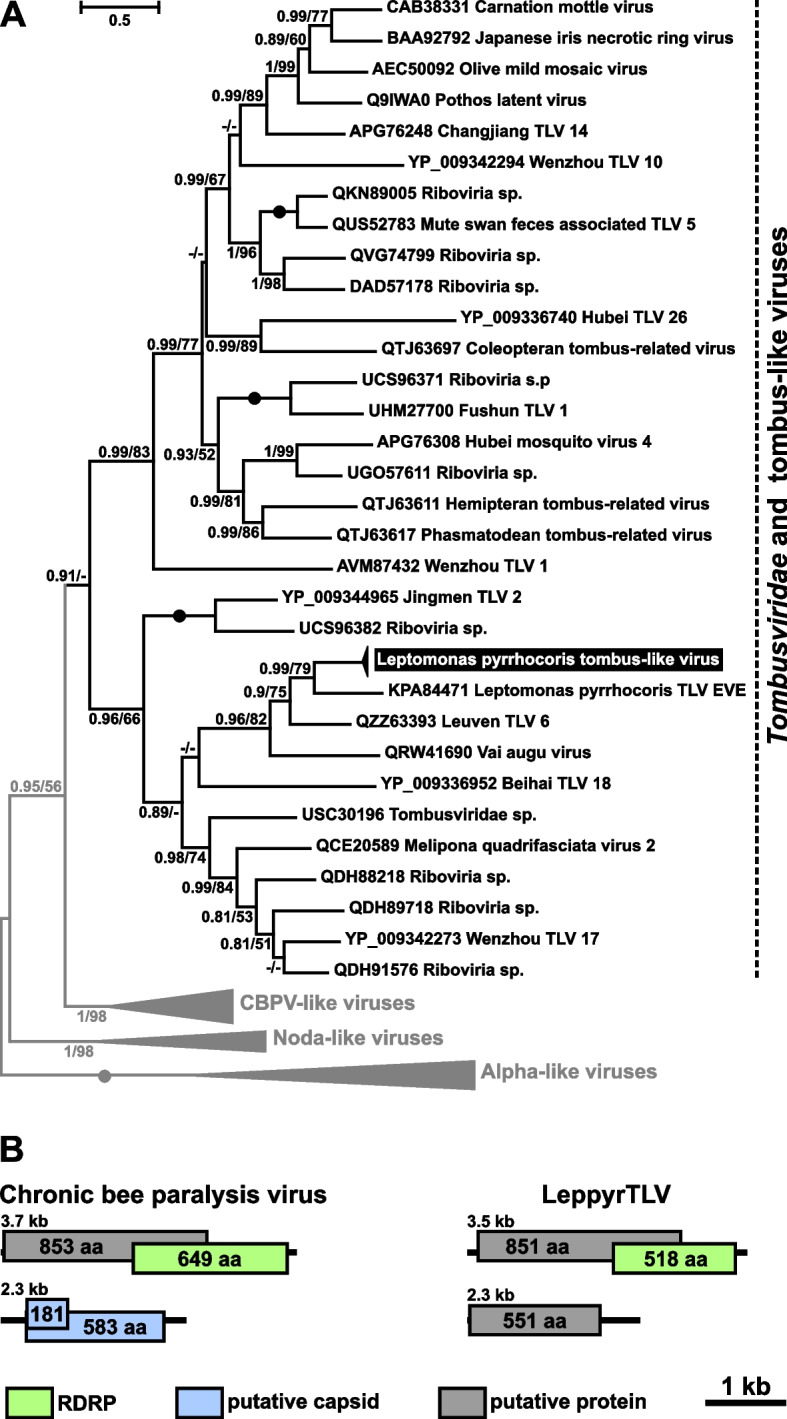
Tombus-like virus of Leptomonas pyrrhocoris. A ML phylogenetic tree based on RDRP amino acid sequences. Numbers at the branches indicate Bayesian posterior probability (PP) and ML bootstrap supports (BS). Only supports PP ≥ 0.8 and BS ≥ 50 are shown, lower values replaced with dashes (-). Circles correspond to maximal statistical support by both methods. The scale bar indicates the number of substitutions per site. Outgroups are shown in gray. Inversed font and background colors indicate the strains studied in this work. B Schemes of the genomic organization of LeppyrTLV1 and chronic bee paralysis virus, the closest available reference species
The genome of LeppyrTLV1 contains two segments (Fig. 1B). The larger one (3.5 kb) carries two overlapping ORFs coding for an unidentified protein and for RNA-dependent RNA polymerase (RDRP). The smaller fragment (2.2 kb) comprises a single ORF for a putative protein. This organization is similar to that of the chronic bee paralysis virus (CBPV), which is the closest relative of LeppyrTLV1 with a known genomic structure. The length of the segments and ORFs are similar in both viruses, but the small segment of CBPV contains an additional small ORF overlapping with the main one and both are predicted to code for capsid proteins [20]. This suggests that the small segment of LeppyrTLV1 can also code for a capsid, although all our attempts to find homology between the proteins of these two viruses failed.
Phylogenetic relationships among individual LeppyrTLV1 viruses were rather poorly resolved, although some clades could be identified (Fig. 2). Importantly, the inferences using different ORFs produced conflicting topologies, and according to likelihood ratio test and Bayes factors analysis, their incompatibility was statistically highly significant (Additional file 3: Table S3). Such topological discordance suggests a reassortment of LeppyrTLV1 genomes, which may occasionally occur during mixed infections by different viral strains and is facilitated by their multi-segment organization. Considering the observed high prevalence of this virus in L. pyrrhocoris (57.5%), such mixed infections are very likely. The lack of strict phylogeographic structure in the inferred trees (i.e., viruses from a single location are not always most closely related to each other) (Fig. 2) suggests some intermixture of viral strains between geographic areas. Since neither LeppyrTLV1 nor its trypanosomatid host L. pyrrhocoris possesses long-lived stages able to travel over long distances themselves, the intermixture must be due to firebugs’ dispersal.
Fig. 2.

ML phylogenetic trees of the three ORFs of LeppyrTLV1 strains based on nucleotide sequences. Numbers at the branches indicate Bayesian posterior probability (PP) and ML bootstrap supports (BS). Only supports PP ≥ 0.8 and BS ≥ 50 are shown, lower values are replaced with dashes. Circles correspond to maximal statistical support by both methods. The scale bar indicates the number of substitutions per site. Some groups are highlighted for easier comparison of topologies
The NGS approach used here allowed identifying a previously overlooked 1.27-kb-long 7th segment of Ostravirus based on the specific 5′-terminal AAAGAAAAAAC sequence (Fig. 3A). Thus, the total length of this viral genome is 21.5 kb, a rather large size for an RNA virus. Confirming our previous assumption on the satellite nature of Ostravirus, it was invariably detected with LeppyrTLV1 [14]. It is surprising that a virus with such a large and complex genome cannot be self-sufficient. Yet, there are similar examples among DNA viruses: the members of Lavidaviridae (also known as virophages) have genome sizes comparable to that of Ostravirus (17–30 kb, 20 predicted ORFs) and can develop in their protist hosts only in the presence of giant viruses of the family Mimiviridae [21]. The relationships between LeppyrTLV1 and LeppyrOV1 appear unbalanced as judged by the ratios of their RPKM values in different samples ranging from nearly equal to over two orders of magnitude preponderance for the former (Additional file 1: Table S1), leading to failure of detection of the latter in the gel (Table 1). It appears plausible that only a minority of cells are infected in the isolates with a very low content of Ostravirus.
Fig. 3.
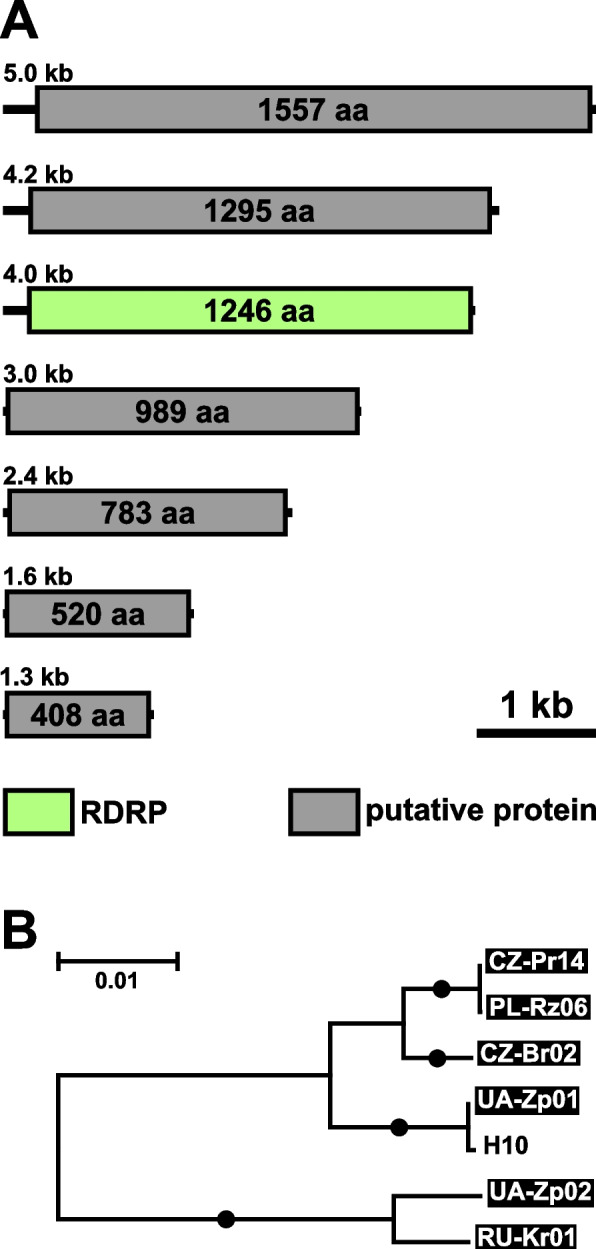
Ostravirus of Leptomonas pyrrhocoris. A Genomic organization of LeppyrOV1. B ML phylogenetic tree of LeppyrOV1 based on the concatenated nucleotide sequences of the seven ORFs. Circles correspond to maximal Bayesian posterior probability and ML bootstrap supports. The scale bar indicates the number of substitutions per site. Inversed font and background colors indicate the strains studied in this work
The phylogenetic tree reconstructed from the concatenated nucleotide alignment of all seven ostraviral ORFs was fully resolved into two major clades containing CZP02 and RUK02 (Czechia and Russia) and CZP01, CZBR02, CZP14, and PLE06 (Czechia, Ukraine, and Russia) with no apparent phylogeographic structure (Fig. 3B).
Leishbuviridae
The family Leishbuviridae (previously, Leishbunyaviridae) is a speciose group of negative-sense single-stranded RNA (-ssRNA) viruses commonly infecting various lineages of trypanosomatids [14–16]. Here, we identified four novel representatives of this family in L. pyrrhocoris, which has not been previously recorded as a host of viruses from this group [14]. Interestingly, these viruses, named LeppyrLBV1–LeppyrLBV4 (for Leptomonas pyrrhocoris leishbuvirus 1 to 4), are not closely related to LBVs from other members of the subfamily Leishmaniinae (Fig. 4). Two new members of LBVs, namely LeppyrLBV1 and LeppyrLBV2, represent sister taxa with their closest relative being the virus from a plant-infecting trypanosomatid Phytomonas sp. TCC231. Some viral sequences previously reported from insect metatranscriptomes [22] proved to be nested within trypanosomatid LBVs (Fig. 4) suggesting that their hosts are also these flagellates. Indeed, one of these is a Huangshi Humpbacked Fly virus, identified in a fly, which, as we previously revealed, harbored trypanosomatids [14].
Fig. 4.

ML phylogenetic tree of Leishbuviridae based on RDRP amino acid sequences. Numbers at the branches indicate Bayesian posterior probability (PP) and ML bootstrap supports (BS). Only supports PP ≥ 0.8 and BS ≥ 50 are shown, lower values are replaced with dashes. Circles correspond to maximal statistical support by both methods. The scale bar indicates the number of substitutions per site. The double-crossed branch is at 50% of the original length. Outgroup is shown in gray. Inversed font and background colors indicate the strains studied in this work. Single asterisk marks viruses detected in trypanosomatid cultures, and two asterisks indicate viruses found in metatranscriptomes along with trypanosomatids (detected previously in [14] or reported in a corresponding NCBI Sequence Read Archive record in the “Taxonomy Analysis” section)
Similarly to other Bunyavirales, LBVs have three genomic segments: large (L), medium (M), and small (S) encoding the cap-snatching-endonuclease/RDRP complex, the glycoprotein, and the nucleoprotein, respectively [23]. We have previously demonstrated that the M and S segments of LBVs are much reduced in size as compared to those of the insect viruses from the sister family Phenuiviridae [14]. In LeppyrLBV1 and LeppyrLBV2, the S segment is 0.8 kb long, while the M segment could be identified neither on the gel nor in the NGS data (in the “crown” LBVs the respective sizes are 0.7–1.0 kb and 1.1–1.4 kb). Intriguingly, LeppyrLBV4, the most early-diverging representative of L. pyrrhocoris LBVs, contains the 2.9-kb-long M and 1.0-kb-long S segments (Fig. 5A, Additional file 1: Table S1), which are close in length to those found in Gouleako virus (Goukovirus gouleakoense) of the family Phenuiviridae (3.2 and 1.1 kb, respectively) [24]. In addition, clade 3 contains viruses with a 1.9-kb-long M segment (still longer than in LBVs described before) and a 1.5-kb-long S segment, which is even longer than that in all other related viruses considered here (Fig. 5A, Additional file 1: Table S1). The analysis of the proteins encoded in these segments (nucleocapsid and glycoprotein for M and S, respectively) revealed a clearer trend of gradual length reduction in the evolution of Leishbuviridae (Fig. 5B).
Fig. 5.

Genome variability of leishbuviruses from Leptomonas pyrrhocoris. A Schemes of genome organization of LeppyrLBVs and the reference species for the families Leishbuviridae and Phenuiviridae—Leishmania martiniquensis leishbuvirus 1 and Gouleako virus (Goukovirus gouleakoense), respectively. B Juxtaposition of schematic phylogeny and lengths of predicted nucleocapsid protein and glycoprotein demonstrating the gradual reduction of the latter in the evolution of Leishbuviridae
In one of the analyzed isolates, PL-Rz06, two distinct L segments of LeppyrLBV3 showing only 89.8% identity were assembled, while the minimal identity among all available sequences was 89.2%. A phylogenetic analysis of the nucleotide sequences of these segments demonstrated their association with two different clades within LeppyrLBV3 (Additional file 4: Fig. S1). This indicates the presence of two different strains of this virus in one isolate. Interestingly, this was not observable for the other two segments, apparently due to their lower variability (99.8% and 96.3% minimal identity for the M and S segments, respectively), which is counterintuitive considering that at the interspecific level, they are so divergent that it is sometimes difficult to reveal homology between them [14].
All bunyaviral segments have specific terminal complementary sequences (TCSs), which bring together both ends of genomic RNA forming a double-stranded “panhandle.” This facilitates RNA-RDRP interaction necessary for transcription, replication, and packaging of genomic segments into a virion. The TCS is a stretch of 20 to 30 complementary nucleotides interrupted by kinks or bulges [25]. In LeppyrLBVs, there are differences in sequence and shape of the TCSs not only between viruses, but also between the L, M, and S segments in different clades (Additional file 5: Fig. S2).
Showing the highest similarity of panhandles both in the primary and secondary structures, LBV4 represents an exception among these viruses. The most unusual is the L segment of LBV3, which contains a complex structure “disfiguring” the panhandle: a multi-branched loop, a big bulge, and a short hairpin (Additional file 5: Fig. S2). Such variability of TCSs revealed here is surprising. In Bunyavirales, the last eight nucleotides of TCS are conserved and family-specific [23]. For the L segment of the family Phenuiviridae, as well as LBV4 and all previously reported leishbuviruses, which form the crown group of LBVs in phylogenetic trees, this sequence is invariably ACACAAAG. The two sister species, LeppyrLBV1 and LeppyrLBV2, have a different sequence—AAG(A)AACA, while the related Phytomonas sp. TCC231 LBV1 has the canonical one [14]. In LBV3, that sequence is disrupted by the abovementioned complex structure (Additional file 5: Fig. S2).
Thus, the LBVs of L. pyrrhocoris represent both the missing link between the ancestral insect-infecting bunyaviruses similar to extant Phenuiviridae, as well as display significantly divergent TCSs. The basal phylogenetic position of L. pyrrhocoris LBVs can be explained by a restricted host range of L. pyrrhocoris, which infects predominantly firebugs [4, 26]. This resulted in the evolutionary preservation of the ancestral forms with the full-length M and S segments. The main driving force behind LBV diversification within L. pyrrhocoris appears to be a change in TCS, as clades 2, 3, and 4 deviate from the ancestral ACACAAAG form. The reasons for such changes, which independently occurred in three out of four L. pyrrhocoris LBVs, are obscure but may be related to the high prevalence and diversity of other viruses in this trypanosomatid species.
Qin-like virus
Two highly similar viruses detected in L. pyrrhocoris isolates from Ukraine and Serbia were identified as representatives of a single species of Qinviridae, a family of -ssRNA viruses recently discovered in metatranscriptomes of invertebrates [22]. It is considered that their genome consists of two segments, of which the first is 5–6.5 kb in size and encodes a large RDRP domain-containing protein, while the second one (in most members) is reported to be 1.6–1.9 kb long and code for a single hypothetical protein. Phylogenetically, Qinviridae along with a few other groups of bipartite viruses are close to non-segmented Mononegavirales and are united with them into the subphylum Haploviricotina, as opposed to multipartite Polyploviricotina [27]. The Leptomonas pyrrhocoris Qin-like virus (LeppyrQLV1) detected in this work falls into a clade together with viruses from metatranscriptomes of mosquitoes belonging to the genera Aedes and Culex, while other more distantly related representatives of the family were revealed in other arthropods and a nematode [28–30] (Fig. 6A). Notably, Qinviridae-specific reads in those metatranscriptomes had low abundance suggesting that these viruses infected rather microbiota than arthropods [28, 29].
Fig. 6.

Qin-like virus of Leptomonas pyrrhocoris. A ML phylogenetic tree of Qinviridae based on RDRP amino acid sequences. Numbers at the branches indicate Bayesian posterior probability (PP) and ML bootstrap supports (BS). Only supports PP ≥ 0.8 and BS ≥ 50 are shown, lower values are replaced with dashes. Circles correspond to maximal statistical support by both methods. The scale bar indicates the number of substitutions per site. Outgroup is shown in gray. Inversed font and background colors indicate the strains studied in this work. B genomic organization of LeppyrQLV1 and its closest available reference—Wilkie qin-like virus
Here, we documented the first representative of the family Qinviridae, for which the host specificity is unambiguously established. Similarly to other members of this group, its genome consists of two segments: 5.3 and 2.2 kb long (Fig. 6B). The large one encodes a 1679-aa-long protein with the RDRP domain. The small segment contains two ORFs in one frame coding for hypothetical proteins of 375 and 231 amino acids (aa) separated by a stop codon. This is distinct from the typical genome architecture for the family as exemplified by the closely related Wilkie qin-like virus, the small segment of which is shorter (1.6 kb) and comprises only a single ORF similar in size (407 aa) to the first one in LeppyrQLV1 (Fig. 6B). This longer ORF of LeppyrQLV1 yielded a few homologs from related meta-transcriptomic qinviruses, while nothing was retrieved for the shorter one from GenBank and UniClust20 databases. The larger hypothetical protein was predicted to be cytosolic and to contain two glycosylation sites, which is in line with an assumption that it is a nucleocapsid/matrix protein. In the shorter hypothetical protein, a single glycosylation site was identified and two transmembrane helices were predicted near the C-terminus, suggesting that it can be a surface glycoprotein. Interestingly, the distantly related Collembolan qin-related virus has been predicted to have these two proteins as well, but each of them resides in an individual genomic segment and their sequences are longer (623 and 430 aa for nucleocapsid and glycoprotein, respectively) [27].
Analysis of virus distribution heterogeneity within isolates
The presence of more than one viral species in some isolates of L. pyrrhocoris raised a question of whether they represent mixtures of cells with different infection statuses. To address this question, we selected three cultures, in which two to four viruses had been detected: Lt-Vi09 (with LeppyrTLV1 and LeppyrLBV3), Pt-Li06 (with LeppyrTLV1 and LeppyrLBV3), and SE-Sb02 (with LeppyrTLV1, and LeppyrLBV2-LBV4). For each of these isolates, eight clones were analyzed by gel electrophoresis of dsRNAs (Additional file 6: Fig. S3).
In the Lt-Vi09 and PT-Li09 isolates, which bore mixtures of two viruses, individual clones displayed all possible variants of viral distribution. They could be virus-free, contain one of two viruses, or both of them at once. All analyzed clones of the SE-Sb02 isolate harbored LeppyrLBVs, of which we could reliably identify only LeppyrLBV2 and LeppyrLBV3 in some samples displaying the bands for the characteristic S segments (Additional file 6: Fig. S3). The absence of LeppyrTLV1 in the clones of SE-Sb02 is explained by the significantly lower proportion of this virus in the original uncloned isolate as compared to Lt-Vi09 and PT-Li09 (Additional file 6: Fig. S3).
The obtained results confirm our hypothesis on the mixed nature of L. pyrrhocoris isolates, being proxies of individual micropopulations of this species in the firebugs’ gut. It is very likely that with such a high parasite prevalence (up to 100% in some populations of Pyrrhocoris apterus) (Table 1), many individuals can be repeatedly infected by flagellates with different patterns of viral presence. Another important conclusion drawn from these findings is that two different viruses can coexist in a single cell. This resonates with our inferences suggesting coinfections of a single trypanosomatid cell by two strains of LeppyrTLV1 (see above).
Interestingly, some isolates can contain virus-free cells along with virus-bearing ones. Considering that the cultures underwent multiple passages, during which the cells had chances to become homogeneous in terms of viral infection status, two mutually non-exclusive explanations could be proposed: (1) some flagellates lose viruses due to inefficient segregation during cell division (vertical transmission) and/or (2) the horizontal transmission between cells is limited (e.g., due to insusceptibility of some cells or unsuitability of in vitro conditions for the viral exchange). It has been reported previously that viruses in trypanosomatid cultures can be stably preserved upon decades of continuous passaging or be depleted up to a complete loss [14]. The heterogeneity of cultures that we detected can explain the change in total viral load in the culture over time without the necessity to assume viral loss: the virus-free cells can overgrow the virus-bearing ones if they better fit to the in vitro conditions.
Little is known about the majority of the viral groups discussed here, but at least in LBVs, regular enveloped virions with glycoprotein spikes similar to those of Phenuiviridae have been detected by electron microscopy [14, 15]. Therefore, some viruses of L. pyrrhocoris can probably be shared through virion shedding. Another likely transmission route involves the exchange of extracellular vesicles, as demonstrated for LRV in Leishmania spp. [31, 32]. Finally, a direct cytoplasm contact occurring during cell mating can represent one more transmission route. Although the presence of genetic exchange requiring such a mechanism has not been so far demonstrated in L. pyrrhocoris, this is known to occur in its close relatives Crithidia bombi and Leishmania spp. [33–36].
Conclusions
Our survey of RNA viruses in L. pyrrhocoris revealed seven viruses belonging to four groups. Only two out of these—LeppyrTLV1 (the most prevalent virus, present in 95.3% of all positive isolates or 57.5% of the tested ones) and LeppyrOV1 (12.5% and 7.5% prevalence, respectively)—have been previously detected in this flagellate. In addition, we discovered four new species of the family Leishbuviridae (20.3% and 12.3% prevalence, respectively), whose representatives have been previously documented in other trypanosomatids, and a new species of Qinviridae (3.1% and 1.9% prevalence, respectively), the family so far known only from the metatranscriptomes of invertebrates. Such a wide range of viruses, and the simultaneous presence of up to five different viruses in a single isolate of L. pyrrhocoris, are unprecedented among trypanosomatids. The uniqueness of this flagellate is determined by its peculiar host, the firebug P. apterus. This insect has a nearly cosmopolitan distribution and high abundance, ensuring the same for its parasites. Moreover, the gregarious lifestyle, coprophagy, and cannibalism of firebugs [37, 38] stipulate a very high (up to 100%) prevalence of L. pyrrhocoris [4]. This, in turn, creates conditions for repeated infections of the same host by parasites, which can represent different strains. Although most individuals of firebugs are short-winged and therefore flightless, there is also a long-winged (macropterous) morph, which can sometimes fly and is highly mobile in any case. This morph is considered to play the main role in the dispersal of this species [37, 39]. The active dispersal of firebugs creates conditions for mixing parasites from different host populations and, subsequently, mixing viruses, resulting in coinfection by different viral strains and species.
Importantly, P. apterus is polyphagous and, in addition to plant seeds, feeds on corpses of various invertebrates [37]. Thus, L. pyrrhocoris is exposed in the host intestine to a much wider spectrum of viruses than other trypanosomatids. After multiple attempts, some of these viruses can become adapted to this flagellate and eventually perform a host switch [40]. The representatives of Leishbuviridae, which are typical for trypanosomatids, could be adopted directly from other flagellates getting into the firebugs’ gut from the corpses of other insects. Conversely, L. pyrrhocoris probably cannot share its viruses with other trypanosomatids, since only this species and Blastocrithidia papi [41] are known to infect firebugs. In addition, the pungent defensive secretions of P. apterus repel the overwhelming majority of predators [37, 42], thereby restricting the chances of L. pyrrhocoris to “share” its viruses with trypanosomatids of other insects.
The multiplicity and high abundance of viruses in L. pyrrhocoris suggest that they may play a role in the relationship between this trypanosomatid and its firebug host. Although, to the best of our knowledge, this question has not been addressed so far for any trypanosomatid—insect pair, we can speculate based on the available information about the immune system in insects. In contrast to the majority of monoxenous trypanosomatids, L. pyrrhocoris was detected not only in the gut, but also in the hemolymph and salivary glands [43, 44]. The viruses released from infected flagellates via shedding, exocytosis, or following cell death can be perceived by the host immune system. The Toll pathway, known to play an important role in the biology of LRV-infected Leishmania spp., is also used by insects in antiviral and antimicrobial defense [45, 46]. Therefore, released viruses may trigger this pathway. The following activation of defense mechanisms such as recruitment of hemocytes to the infection sites (also including the gut) and melanization [47, 48] may lead to harsher conditions for the parasites, especially in the secondary (extraintestinal) infection sites. While frequent gut infections do not seem to be affected by viruses (Table 1), this may not be the case in rather rare infections of hemolymph and salivary glands [49].
The effects of viruses described here can be very diverse. Indeed, even closely related viral species can have different levels of integration into the cellular processes of their trypanosomatid hosts. In a recent experimental study, the removal of LRV1 from Leishmania guyanensis had no significant effect on the growth and transcription profile of the latter, whereas ablation of LRV2 from L. major resulted in a decreased proliferation rate and conspicuous stress effect as judged by changes in the gene expression [50].
In sum, the peculiar biology of the insect host makes L. pyrrhocoris a unique “hoarder” of viruses collected from various sources. We propose that this flagellate is a good model to study the adoption of new viruses and their relationships with a protist host. Since our study concerned only European isolates, the global diversity of viruses in this trypanosomatid is likely to be significantly higher.
Methods
Collection, cultivation, and identification of isolates
The screening for the presence of dsRNA included 106 trypanosomatid isolates established from field samples of 508 firebugs (Pyrrhocoris apterus) from 13 European countries (Table 1). Insects were dissected and analyzed for the presence of trypanosomatids as described previously [51]. Cultured parasites were maintained in the Brain Heart Infusion medium (Sigma-Aldrich/Merck, St. Louis, USA) supplemented with 10 µg/ml of hemin (Jena Bioscience, Jena, Germany), 10% fetal bovine serum (FBS), 500 units/ml of penicillin, and 0.5 mg/ml of streptomycin (all from Thermo Fisher Scientific, Waltham, USA). DNA for the species validation was isolated from 5 × 107 cells using the Qiagen DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany). All isolates were confirmed to be axenic L. pyrrhocoris by 18S rRNA and glycosomal glyceraldehyde phosphate dehydrogenase gene sequence analyses as described previously [52].
RNA isolation, dsRNA purification, and next-generation sequencing
Total RNA was isolated from 5 × 108 to 1 × 109 cells using the TRI Reagent (Molecular Research Center, Cincinnati, USA) as described elsewhere [53]. For the initial screening, 50 µg of total RNA was treated with DNase I/S1 nuclease enzyme mix [54, 55]. The resulting dsRNA was resolved on 0.8% agarose gel and post-stained with Midori green dye (Nippon Genetic Europe, Düren, Germany). For next-generation sequencing (NGS), 400 µg of total RNA was digested with DNase I/S1 nuclease enzyme mix and purified using the Zymoclean Gel RNA recovery kit (Zymo Research, Irvine, USA). The RiboMinus libraries were sequenced using Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) at Macrogen Inc. (Amsterdam, The Netherlands) or the Institute of Applied Biotechnologies (Olomouc, Czechia).
Virus genome assembly and search
Reads were trimmed using Trimmomatic v. 0.40 [56] (ILLUMINACLIP:TruSeq3-PE-2.fa:2:20:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50) and assembled de novo with Trinity v. 2.13.2 [57]. The mapping and sorting of reads were performed using Bowtie 2 v. 2.4.4 [58] and SAMtools v.1.13 [59], respectively, with default settings. The read per kilobase per million (RPKM) values for each sample were calculated using a custom awk script from the “per base” coverage file generated by BEDTools v. 2.30.0 [60]. Contigs containing viral genes were identified by BLASTN (BLAST + v. 2.12 [61]) and BLASTX (DIAMOND v. 2.0.2 [62]) searches against UniClust50 database. Nucleocapsid proteins of divergent leishbuviruses were found by HHblits search [63] against a custom-built sequence profile of nucleocapsids from Leishbuviridae and Phenuiviridae. Additional viral ORFs were identified using the following criteria: (i) contig length corresponding to that of the dsRNA band on the gel, (ii) contig coverage correlating with the relative brightness of the dsRNA band as compared to an identified contig/band pair, and (iii) the presence of specific viral terminal sequences. The analysis of putative Qin-like virus glycoprotein was performed with web-based tools: CCTOP [64] for transmembrane helices, NetNGlyc v. 1.0 [65] for N-linked glycosylation sites, and SignalP v. 6.0 [66] for signal peptides. Assembled viral fragments were submitted to GenBank under the accession numbers OP722764 – OP722922.
Phylogenetic inferences
For LBVs, the dataset of RNA-dependent RNA polymerase (RDRP) proteins was taken from [15]. Homologs of divergent LBVs were added by running BLASTP [61] with the respective sequences against the non-redundant (nr) database of GenBank. The sequences were aligned in MAFFT v. 7.490 [67] using the G-INS-i algorithm with a maximum of 1000 iterations and trimmed in TrimAl v. 1.4 [68] with “automated1” algorithm resulting in 1139 amino acid positions. The maximum likelihood (ML) phylogenetic tree was inferred in IQ-Tree v. 2.2.0 under the automatically selected LG + I + G4 + F substitution model and branch supports estimated using 1000 thorough bootstrap replicates [69]. The tree was rooted according to the topologies obtained in the previous studies using wider taxonomic datasets [14, 16]. Bayesian inference of phylogeny was performed in MrBayes v. 3.2.7 run for 1 million generations under the same substitution model as above, sampling every 250th generation and all other parameters set by default [70]. Posterior probability values from Bayesian analysis were overlaid onto the ML tree topology with bootstrap supports.
A similar approach was applied to novel Qin-like viruses. The sequences of RDRP homologs were retrieved from the nr database using PSI-BLAST (4 iterations), and only Qin-like viruses and their closest relatives—Yue-like viruses (serving as outgroup)—were taken for further analysis. Sequences were aligned in MAFFT using the E-INS-i algorithm with default settings and trimmed as above. The resulting alignment of 1047 amino acid positions was analyzed as above with the only difference being that the Bayesian analysis was run for 1.5 million generations.
For the inference of the phylogenetic position of LeppyrTLV1, the amino acid RDRP sequences of 32 tombus-like viruses and 32 outgroups, belonging to chronic bee paralysis virus (CBPV)-like viruses, Alpha-like viruses, and Noda-like viruses were retrieved from the GenBank, and processed as for Qin-like viruses, resulting in a 255-amino acid (aa)-long alignment. The ML and Bayesian inferences were performed as above except for the latter analysis which was run for 4 million generations.
Phylogenetic relationships between the strains of LeppyrTLV1 were inferred using sequences obtained in this work and those for the isolates H10 and F165 published previously [14]. Analyses were performed separately for each of the three ORFs using automatically selected nucleotide substitution models with partitioning by codon position in IQ-Tree and MrBayes with rate multiplier unlinked across partitions. The significance of the discordance between individual ORF topologies was assessed using the likelihood ratio test and Bayes factors [71]. For that, phylogenetic inferences with linked and unlinked topologies for the three ORFs were performed using nucleotide and amino acid substitution models in both IQ-Tree and MrBayes. The marginal likelihoods for the Bayes factors were estimated using the stepping stone method [70]. The p-values for the χ2 statistic obtained in LRT analysis were calculated using an online tool at https://goodcalculators.com/chi-square-calculator/.
For the reconstruction of phylogenetic relationships between ostraviruses, the sequences of all seven ORFs were concatenated after processing as for the qinviruses using a custom bash script. The ML and Bayesian inferences were performed without partitioning under the GTR + F + I model and other details as for LBVs.
Analysis of virus distribution heterogeneity within isolates
Considering that the isolates of Leptomonas pyrrhocoris obtained in this work could represent mixtures of cells differing in viral infection status, we performed an additional screening of viruses on the clonal level. For that, we selected three isolates that showed the presence of more than one virus on the gel: Lt-Vi09, PT-Li06, and Se-Sb02. They were cloned by serial dilution in microtiter plates (0.2 cells per well) and eight clones in each case were screened by gel electrophoresis of dsRNA preparations as described above.
Supplementary Information
Additional file 1: Table S1. Summary of the NGS data obtained for the viral RNAs of the studied trypanosomatid isolates.
Additional file 2: Table S2. Sequence variation in LeppyrTLV1 and LeppyrOV1.
Additional file 3: Table S3. Analysis of the incompatibility of tree topologies for different ORFs in LeppyrTLV1.
Additional file 4: Fig. S1. Phylogenetic inference of relationships between the RDRP nucleotide sequences of LeppyrLBV3. Numbers at the branches indicate Bayesian posterior probability (PP) and ML bootstrap supports (BS), respectively. Only bootstrap supports BS ≥ 50 are shown, lower values replaced with dashes (-). Circles correspond to maximal statistical support by both methods. The scale bar indicates the number of substitutions per site.
Additional file 5: Fig. S2. Terminal complementary sequences in LBVs of Leptomonas pyrrhocoris. Primary structures and/or alignments are shown on the left, secondary structures are on the right. The panhandle-distorting complex structure containing a multi-branched loop, a big bulge, and a short hairpin is outlined.
Additional file 6: Fig. S3. Analysis of virus distribution heterogeneity within isolates. Note that not all fragments for a virus (see graphic legend) can be always detected on the gel.
Acknowledgements
We thank the members of our laboratories for stimulating the discussions. Computational resources were provided by the e-INFRA CZ project (ID:90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic.
Abbreviations
- BS
Bootstrap support
- CBPV
Chronic bee paralysis virus
- CQRV
Collembolan qin-related virus
- dsRNA
Double-stranded RNA
- L (segment)
Large
- LBV(s)
Leishbuvirus(es), Leishbuviridae
- LeppyrLBV
Leptomonas pyrrhocoris Leishbuvirus
- LeppyrOV1
Leptomonas pyrrhocoris Ostravirus 1
- LeppyrTLV1
Leptomonas pyrrhocoris tombus-like virus 1
- LRV
Leishmania RNA virus
- M (segment)
Medium
- ML
Maximum likelihood
- NGS
Next-generation sequencing
- ORF
Open reading frames
- PP
Posterior probability
- RDRP
RNA-dependent RNA polymerase
- RPKM
Read per kilobase per million
- S (segment)
Small
- -ssRNA
Negative-sense single-stranded RNA
- TCSs
Terminal complementary sequences
- TLV
Tombus-like virus(es)
Authors’ contributions
AYK and VY conceived the study and coordinated the work. DHM, JV, TY, JŠ, MUD, KZ, JL, ES, SS, AYK, MNM, AOF, and VY collected and/or dissected the firebugs and established the trypanosomatid cultures. JR, AM, AYK, and AJ identified the isolates. DHM and DK isolated and analyzed the dsRNAs. DK performed the cloning experiments. DG assembled the viral genomes from NGS data. DG and AYK completed the phylogenetic inferences. DG and AYK prepared the illustrations. DG, AYK, and VY wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Grant Agency of Czech Republic 20-22689S to V.Y. The computational resources used in this work were funded by the European Regional Development Funds CZ.02.1.01/16_019/0000759 to VY, AYK, JV, and JL as well as the EU’s Operational Program “Just Transition” CZ.10.03.01/00/22_003/0000003 LERCO to VY. The funders had no role in the study design, data collection and interpretation, or the decision to submit this work for publication.
Availability of data and materials
The sequences obtained in this study are available from the GenBank under the accession numbers OP722764 – OP722922.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Diego H. Macedo and Danyil Grybchuk contributed equally to this work.
Contributor Information
Vyacheslav Yurchenko, Email: vyacheslav.yurchenko@osu.cz.
Alexei Yu. Kostygov, Email: kostygov@gmail.com
References
- 1.Maslov DA, Opperdoes FR, Kostygov AY, Hashimi H, Lukeš J, Yurchenko V. Recent advances in trypanosomatid research: genome organization, expression, metabolism, taxonomy and evolution. Parasitology. 2019;146(1):1–27. doi: 10.1017/S0031182018000951. [DOI] [PubMed] [Google Scholar]
- 2.Kostygov AY, Karnkowska A, Votýpka J, Tashyreva D, Maciszewski K, Yurchenko V, Lukeš J. Euglenozoa: taxonomy, diversity and ecology, symbioses and viruses. Open Biol. 2021;11(3):200407. doi: 10.1098/rsob.200407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kostygov AY, Yurchenko V. Revised classification of the subfamily Leishmaniinae (Trypanosomatidae) Folia Parasitol. 2017;64:020. doi: 10.14411/fp.2017.020. [DOI] [PubMed] [Google Scholar]
- 4.Votýpka J, Klepetková H, Yurchenko VY, Horák A, Lukeš J, Maslov DA. Cosmopolitan distribution of a trypanosomatid Leptomonas pyrrhocoris. Protist. 2012;163(4):616–631. doi: 10.1016/j.protis.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Flegontov P, Butenko A, Firsov S, Kraeva N, Eliáš M, Field MC, Filatov D, Flegontova O, Gerasimov ES, Hlaváčová J, et al. Genome of Leptomonas pyrrhocoris: a high-quality reference for monoxenous trypanosomatids and new insights into evolution of Leishmania. Sci Rep. 2016;6:23704. doi: 10.1038/srep23704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stuart KD, Weeks R, Guilbride L, Myler PJ. Molecular organization of Leishmania RNA virus 1. Proc Natl Acad Sci U S A. 1992;89(18):8596–8600. doi: 10.1073/pnas.89.18.8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zangger H, Hailu A, Desponds C, Lye LF, Akopyants NS, Dobson DE, Ronet C, Ghalib H, Beverley SM, Fasel N. Leishmania aethiopica field isolates bearing an endosymbiontic dsRNA virus induce pro-inflammatory cytokine response. PLoS Negl Trop Dis. 2014;8(4):e2836. doi: 10.1371/journal.pntd.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klocek D, Grybchuk D, Tichá L, Votýpka J, Volf P, Kostygov AY, Yurchenko V. Evolution of RNA viruses in trypanosomatids: new insights from the analysis of Sauroleishmania. Parasitol Res. 2023. 10.1007/s00436-023-07928-x. [DOI] [PMC free article] [PubMed]
- 9.Cantanhêde LM, Mata-Somarribas C, Chourabi K, Pereira da Silva G, Dias das Chagas B, de Oliveira RPL, Cortes Boite M, Cupolillo E. The maze pathway of coevolution: a critical review over the Leishmania and its endosymbiotic history. Genes. 2021;12(5):657. doi: 10.3390/genes12050657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostygov AY, Grybchuk D, Kleschenko Y, Chistyakov DS, Lukashev AN, Gerasimov ES, Yurchenko V. Analyses of Leishmania-LRV co-phylogenetic patterns and evolutionary variability of viral proteins. Viruses. 2021;13(11):2305. doi: 10.3390/v13112305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ives A, Ronet C, Prevel F, Ruzzante G, Fuertes-Marraco S, Schutz F, Zangger H, Revaz-Breton M, Lye LF, Hickerson SM, et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science. 2011;331(6018):775–778. doi: 10.1126/science.1199326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eren RO, Reverte M, Rossi M, Hartley MA, Castiglioni P, Prevel F, Martin R, Desponds C, Lye LF, Drexler SK, et al. Mammalian innate immune response to a Leishmania-resident RNA virus increases macrophage survival to promote parasite persistence. Cell Host Microbe. 2016;20(3):318–328. doi: 10.1016/j.chom.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartley MA, Bourreau E, Rossi M, Castiglioni P, Eren RO, Prevel F, Couppie P, Hickerson SM, Launois P, Beverley SM, et al. Leishmaniavirus-dependent metastatic leishmaniasis is prevented by blocking IL-17A. PLoS Pathog. 2016;12(9):e1005852. doi: 10.1371/journal.ppat.1005852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grybchuk D, Akopyants NS, Kostygov AY, Konovalovas A, Lye LF, Dobson DE, Zangger H, Fasel N, Butenko A, Frolov AO, et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc Natl Acad Sci U S A. 2018;115(3):E506–515. doi: 10.1073/pnas.1717806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grybchuk D, Macedo DH, Kleschenko Y, Kraeva N, Lukashev AN, Bates PA, Kulich P, Leštinová T, Volf P, Kostygov AY, et al. The first non-LRV RNA virus in Leishmania. Viruses. 2020;12(2):168. doi: 10.3390/v12020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grybchuk D, Kostygov AY, Macedo DH, Votýpka J, Lukeš J, Yurchenko V. RNA viruses in Blechomonas (Trypanosomatidae) and evolution of Leishmaniavirus. mBio. 2018;9(5):e01932–01918. doi: 10.1128/mBio.01932-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remnant EJ, Baty JW, Bulgarella M, Dobelmann J, Quinn O, Gruber MAM, Lester PJ. A diverse viral community from predatory wasps in their native and invaded range, with a new virus infectious to honey bees. Viruses. 2021;13(8):1431. doi: 10.3390/v13081431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Batson J, Dudas G, Haas-Stapleton E, Kistler AL, Li LM, Logan P, Ratnasiri K, Retallack H. Single mosquito metatranscriptomics identifies vectors, emerging pathogens and reservoirs in one assay. eLife. 2021;10:e68353. doi: 10.7554/eLife.68353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Podlipaev SA. Catalogue of world fauna of Trypanosomatidae (Protozoa), vol. 144. Leningrad: Zoologicheskii Institut AN SSSR; 1990. [Google Scholar]
- 20.Olivier V, Blanchard P, Chaouch S, Lallemand P, Schurr F, Celle O, Dubois E, Tordo N, Thiery R, Houlgatte R, et al. Molecular characterisation and phylogenetic analysis of chronic bee paralysis virus, a honey bee virus. Virus Res. 2008;132(1–2):59–68. doi: 10.1016/j.virusres.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 21.Duponchel S, Fischer MG. Viva lavidaviruses! Five features of virophages that parasitize giant DNA viruses. PLoS Pathog. 2019;15(3):e1007592. doi: 10.1371/journal.ppat.1007592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, Qin XC, Li J, Cao JP, Eden JS, et al. Redefining the invertebrate RNA virosphere. Nature. 2016;540(7634):539–543. doi: 10.1038/nature20167. [DOI] [PubMed] [Google Scholar]
- 23.Ferron F, Weber F, de la Torre JC, Reguera J. Transcription and replication mechanisms of Bunyaviridae and Arenaviridae L proteins. Virus Res. 2017;234:118–134. doi: 10.1016/j.virusres.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marklewitz M, Handrick S, Grasse W, Kurth A, Lukashev A, Drosten C, Ellerbrok H, Leendertz FH, Pauli G, Junglen S. Gouleako virus isolated from West African mosquitoes constitutes a proposed novel genus in the family Bunyaviridae. J Virol. 2011;85(17):9227–9234. doi: 10.1128/JVI.00230-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren F, Zhou M, Deng F, Wang H, Ning YJ. Combinatorial minigenome systems for emerging banyangviruses reveal viral reassortment potential and importance of a protruding nucleotide in genome “panhandle” for promoter activity and reassortment. Front Microbiol. 2020;11:599. doi: 10.3389/fmicb.2020.00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Votýpka J, Kment P, Kriegová E, Vermeij MJA, Keeling PJ, Yurchenko V, Lukeš J. High prevalence and endemism of trypanosomatids on a small Caribbean island. J Eukaryot Microbiol. 2019;66(4):600–607. doi: 10.1111/jeu.12704. [DOI] [PubMed] [Google Scholar]
- 27.Käfer S, Paraskevopoulou S, Zirkel F, Wieseke N, Donath A, Petersen M, Jones TC, Liu S, Zhou X, Middendorf M, et al. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS Pathog. 2019;15(12):e1008224. doi: 10.1371/journal.ppat.1008224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi M, Neville P, Nicholson J, Eden JS, Imrie A, Holmes EC. High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in western Australia. J Virol. 2017;91(17):e00680–00617. doi: 10.1128/JVI.00680-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pettersson JH, Shi M, Eden JS, Holmes EC, Hesson JC. Meta-transcriptomic comparison of the RNA viromes of the mosquito vectors Culex pipiens and Culex torrentium in northern Europe. Viruses. 2019;11(11):1033. doi: 10.3390/v11111033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams SH, Levy A, Yates RA, Somaweera N, Neville PJ, Nicholson J, Lindsay MDA, Mackenzie JS, Jain K, Imrie A, et al. The diversity and distribution of viruses associated with Culex annulirostris mosquitoes from the Kimberley region of Western Australia. Viruses. 2020;12(7):717. doi: 10.3390/v12070717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atayde VD, da Silva Lira Filho A, Chaparro V, Zimmermann A, Martel C, Jaramillo M, Olivier M. Exploitation of the Leishmania exosomal pathway by Leishmania RNA virus 1. Nat Microbiol. 2019;4:714–723. doi: 10.1038/s41564-018-0352-y. [DOI] [PubMed] [Google Scholar]
- 32.Lafleur A, Olivier M. Viral endosymbiotic infection of protozoan parasites: how it influences the development of cutaneous leishmaniasis. PLoS Pathog. 2022;18(11):e1010910. doi: 10.1371/journal.ppat.1010910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmid-Hempel R, Salathe R, Tognazzo M, Schmid-Hempel P. Genetic exchange and emergence of novel strains in directly transmitted trypanosomatids. Infect Genet Evol. 2011;11(3):564–571. doi: 10.1016/j.meegid.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Akopyants NS, Kimblin N, Secundino N, Patrick R, Peters N, Lawyer P, Dobson DE, Beverley SM, Sacks DL. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science. 2009;324(5924):265–268. doi: 10.1126/science.1169464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glans H, Lind Karlberg M, Advani R, Bradley M, Alm E, Andersson B, Downing T. High genome plasticity and frequent genetic exchange in Leishmania tropica isolates from Afghanistan, Iran and Syria. PLoS Negl Trop Dis. 2021;15(12):e0010110. doi: 10.1371/journal.pntd.0010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Telittchenko R, Descoteaux A. Study on the occurrence of genetic exchange among parasites of the Leishmania mexicana complex. Front Cell Infect Microbiol. 2020;10:607253. doi: 10.3389/fcimb.2020.607253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Socha R. Pyrrhocoris apterus (Heteroptera) - an experimental-model species: a review. Eur J Entomol. 1993;90(3):241–286. [Google Scholar]
- 38.Frolov AO, Kostygov AY, Yurchenko V. Development of monoxenous trypanosomatids and phytomonads in insects. Trends Parasitol. 2021;37(6):538–551. doi: 10.1016/j.pt.2021.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Socha R, Zemek R. Wing morph-related differences in the walking pattern and dispersal in a flightless bug, Pyrrhocoris apterus (Heteroptera) Oikos. 2003;100(1):35–42. [Google Scholar]
- 40.Parrish CR, Holmes EC, Morens DM, Park EC, Burke DS, Calisher CH, Laughlin CA, Saif LJ, Daszak P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev. 2008;72(3):457–470. doi: 10.1128/MMBR.00004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frolov AO, Malysheva MN, Ganyukova AI, Yurchenko V, Kostygov AY. Life cycle of Blastocrithidia papi sp. n. (Kinetoplastea, Trypanosomatidae) in Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae) Eur J Protistol. 2017;57:85–98. doi: 10.1016/j.ejop.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Farine JP, Bonnard O, Brossut R, Le Quere JL. Chemistry of defensive secretions in nymphs and adults of fire bug, Pyrrhocoris apterus L. (Heteroptera, Pyrrhocoridae) J Chem Ecol. 1992;18(10):1673–1682. doi: 10.1007/BF02751094. [DOI] [PubMed] [Google Scholar]
- 43.Butenko A, Vieira TDS, Frolov AO, Opperdoes FR, Soares RP, Kostygov AY, Lukeš J, Yurchenko V. Leptomonas pyrrhocoris: genomic insight into parasite’s physiology. Curr Genomics. 2018;19(2):150–156. doi: 10.2174/1389202918666170815143331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zotta G. Sur un flagellé du type Herpetomonas chez Pyrrhocoris apterus. Ann Sci Univ Jassy. 1912;7:211–223. [Google Scholar]
- 45.Nakamoto M, Moy RH, Xu J, Bambina S, Yasunaga A, Shelly SS, Gold B, Cherry S. Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity. 2012;36(4):658–667. doi: 10.1016/j.immuni.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He YJ, Lu G, Qi YH, Zhang Y, Zhang XD, Huang HJ, Zhuo JC, Sun ZT, Yan F, Chen JP, et al. Activation of Toll immune pathway in an insect vector induced by a plant virus. Front Immunol. 2020;11:613957. doi: 10.3389/fimmu.2020.613957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang L, Goldsmith MR, Xia Q. Advances in the arms race between silkworm and Baculovirus. Front Immunol. 2021;12:628151. doi: 10.3389/fimmu.2021.628151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leite THJF, Ferreira AGA, Imler JL, Marques JT. Distinct roles of hemocytes at different stages of infection by dengue and Zika viruses in Aedes aegypti mosquitoes. Front Immunol. 2021;12:660873. doi: 10.3389/fimmu.2021.660873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frolov AO, Malysheva MN, Kostygov AY. Homoxenous trypanosomatids from true bugs Pyrrhocoris apterus (L.) in the north of the Pskov region. Parazitologiia. 2014;48(6):461–471 . [PubMed] [Google Scholar]
- 50.Saura A, Zakharova A, Klocek D, Gerasimov ES, Butenko A, Macedo DH, Servienė E, Zagirova D, Meshcheryakova A, Rogozin IB, et al. Elimination of LRVs elicits different responses in Leishmania spp. mSphere. 2022;7(4):e0033522. doi: 10.1128/msphere.00335-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yurchenko V, Kostygov A, Havlová J, Grybchuk-Ieremenko A, Ševčíková T, Lukeš J, Ševčík J, Votýpka J. Diversity of trypanosomatids in cockroaches and the description of Herpetomonas tarakana sp. n. J Eukaryot Microbiol. 2016;63(2):198–209. doi: 10.1111/jeu.12268. [DOI] [PubMed] [Google Scholar]
- 52.Ishemgulova A, Butenko A, Kortisova L, Boucinha C, Grybchuk-Ieremenko A, Morelli KA, Tesarova M, Kraeva N, Grybchuk D, Panek T, et al. Molecular mechanisms of thermal resistance of the insect trypanosomatid Crithidia thermophila. PLoS One. 2017;12(3):e0174165. doi: 10.1371/journal.pone.0174165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chomczyński P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 54.Isorce N, Fasel N. Viral double-stranded RNA detection by DNase I and nuclease S1 digestions in Leishmania parasites. Bio Protoc. 2020;10(9):e3598. doi: 10.21769/BioProtoc.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zangger H, Ronet C, Desponds C, Kuhlmann FM, Robinson J, Hartley MA, Prevel F, Castiglioni P, Pratlong F, Bastien P, et al. Detection of Leishmania RNA virus in Leishmania parasites. PLoS Negl Trop Dis. 2013;7(1):e2006. doi: 10.1371/journal.pntd.0002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez-Gonzalez RH, Bonnal R, Caccamo M, Maclean D. Bio-SAMtools: ruby bindings for SAMtools, a library for accessing BAM files containing high-throughput sequence alignments. Source Code Biol Med. 2012;7(1):6. doi: 10.1186/1751-0473-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan AR. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47:11.12.11–11.12.34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18(4):366–368. doi: 10.1038/s41592-021-01101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger SJ, Söding J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20(1):473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobson L, Reményi I, Tusnády GE. CCTOP: a consensus constrained TOPology prediction web server. Nucleic Acids Res. 2015;43(W1):W408–W412. doi: 10.1093/nar/gkv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput. 2002;7:310–22. [PubMed]
- 66.Teufel F, Almagro Armenteros JJ, Johansen AR, Gislason MH, Pihl SI, Tsirigos KD, Winther O, Brunak S, von Heijne G, Nielsen H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol. 2022;40(7):1023–1025. doi: 10.1038/s41587-021-01156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Capella-Gutiérrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37(5):1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kass RE, Raftery AE. Bayes factors. J Am Statist Assoc. 1995;90(430):773–795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Summary of the NGS data obtained for the viral RNAs of the studied trypanosomatid isolates.
Additional file 2: Table S2. Sequence variation in LeppyrTLV1 and LeppyrOV1.
Additional file 3: Table S3. Analysis of the incompatibility of tree topologies for different ORFs in LeppyrTLV1.
Additional file 4: Fig. S1. Phylogenetic inference of relationships between the RDRP nucleotide sequences of LeppyrLBV3. Numbers at the branches indicate Bayesian posterior probability (PP) and ML bootstrap supports (BS), respectively. Only bootstrap supports BS ≥ 50 are shown, lower values replaced with dashes (-). Circles correspond to maximal statistical support by both methods. The scale bar indicates the number of substitutions per site.
Additional file 5: Fig. S2. Terminal complementary sequences in LBVs of Leptomonas pyrrhocoris. Primary structures and/or alignments are shown on the left, secondary structures are on the right. The panhandle-distorting complex structure containing a multi-branched loop, a big bulge, and a short hairpin is outlined.
Additional file 6: Fig. S3. Analysis of virus distribution heterogeneity within isolates. Note that not all fragments for a virus (see graphic legend) can be always detected on the gel.
Data Availability Statement
The sequences obtained in this study are available from the GenBank under the accession numbers OP722764 – OP722922.