

Abstract

With the resistance increasing to current antimalarial medicines, there is an urgent need to discover new drug targets and to develop new medicines against these targets. We therefore screened the Open Global Health Library of Merck KGaA, Darmstadt, Germany, of 250 compounds against the asexual blood stage of the deadliest malarial parasite Plasmodium falciparum, from which eight inhibitors with low micromolar potency were found. Due to its combined potencies against parasite growth and inhibition of red blood cell invasion, the pyridyl-furan compound OGHL250 was prioritized for further optimization. The potency of the series lead compound (WEHI-518) was improved 250-fold to low nanomolar levels against parasite blood-stage growth. Parasites selected for resistance to a related compound, MMV396797, were also resistant to WEHI-518 as well as KDU731, an inhibitor of the phosphatidylinositol kinase PfPI4KIIIB, suggesting that this kinase is the target of the pyridyl-furan series. Inhibition of PfPI4KIIIB blocks multiple stages of the parasite’s life cycle and other potent inhibitors are currently under preclinical development. MMV396797-resistant parasites possess an E1316D mutation in PfPKI4IIIB that clusters with known resistance mutations of other inhibitors of the kinase. Building upon earlier studies that showed that PfPI4KIIIB inhibitors block the development of the invasive merozoite parasite stage, we show that members of the pyridyl-furan series also block invasion and/or the conversion of merozoites into ring-stage intracellular parasites through inhibition of protein secretion and export into red blood cells.
Keywords: malaria, Plasmodium falciparum, red blood cell invasion, egress, protein trafficking, phosphoinositide phosphate kinase
1.
Malaria is caused by infection with Plasmodium parasites and imposes a substantial global burden with there being an estimated 247 million cases and 619,000 deaths in 2021.1 These estimates are an increase of approximately 10% from previous years, which is believed to be largely because of the SARS-CoV-2 pandemic disrupting health services and increasing resistance to currently available antimalarials. P. falciparum is the species with which most of the malaria disease burden is associated, and of great concern is the emergence of parasite resistance to frontline artemisinin combination therapies (ACTs) around the globe.2−6 This expanding antimalarial resistance highlights an urgent need to discover and develop novel antimalarial medicines that have different mechanisms of action (MoA) from those currently in use, most of which parasites have developed some degree of resistance to. To date, efforts to develop new antimalarials have largely centered on performing phenotypic screens of compound libraries for small molecules that inhibit the growth of the disease-causing asexual blood stage of P. falciparum. This has resulted in the screening of several million compounds and the identification of tens of thousands of inhibitory molecules.7−9
To help prioritize which inhibitory compounds to advance, it is important to know the compound’s MoA. It is also crucial to determine whether the compounds target other stages of the parasite’s lifecycle including the preerythrocytic liver stage and the transmission (sexual) stage. Target identification generally involves growing parasites in the presence of the inhibitors of interest until resistance develops, after which complete genome sequencing of the parasite may reveal mutations in the genes encoding the proteins targeted by the inhibitors. Amplification of genes encoding the target proteins can also be selected to improve resistance through increased protein levels. Resistance selection can take many months and does not always yield resistant parasites. Compounds to which resistance cannot be developed in vitro are said to be irresistible.10,11 Unfortunately, even when resistance can be developed, there is a high likelihood of rediscovering an already well-characterized protein target, for which many inhibitors are already known. Common antimalarial targets often detected by these broad screening methods include PfATP4 and DHODH/cytochrome bc1.12−14 Identifying novel compounds targeting these proteins can be partly mitigated by screening compound libraries with parasites already resistant to a common target such as DHODH/cytochrome bc1 or by performing assays for the biological effects known to be caused by common inhibitors such as those that target PfATP4.12,13
Another way of finding and prioritizing inhibitors with novel MoAs is to perform phenotypic screens for the inhibition of biological phenomena such as egress from old red blood cells (RBCs) and invasion of new RBCs. Such screens of the Medicines for Malaria Venture (MMV) Malaria and Pathogen boxes have identified dozens of compounds that inhibit egress and invasion.15,16 Novel targets identified by these screens include actin and profilin, essential components of the invasion machinery used by the parasite to enter RBCs.17 Egress and invasion inhibitors could be particularly effective compounds since reducing the capacity of invasive merozoite stage parasites to rapidly invade RBCs would leave the merozoites exposed to host immune clearance.18,19
While phenotypic screens for egress and invasion can yield novel targets such as actin and profilin, they are not guaranteed to return solely novel targets.17 Indeed, nearly half of the egress and invasion inhibitors identified in recent screens of the MMV Malaria and Pathogen Boxes were also flagged as inhibitors of PfATP4.12,13,15,16 These have later been determined to be specific inhibitors of egress and not invasion20 and serve to dysregulate sodium ion levels and cellular pH, explaining why late schizont-stage parasites are particularly susceptible to the effects of PfATP4 inhibitors.20,21 Therefore, while egress and invasion screens can also identify inhibitors to well-studied targets, they can also add important additional biological information about the function of targets.
To find inhibitors with novel targets and to reduce the chances of identifying inhibitors of known targets from libraries enriched for antimalarial compounds, we screened the Open Global Health Library (OGHL) of Merck KGaA, Darmstadt, Germany, containing 250 compounds.22 A screen of this library for compounds that reduce the level of growth of P. falciparum parasites identified several inhibitors of low micromolar potency or less. Subsequent egress and invasion inhibition assays indicated half of these compounds were also invasion inhibitors with the pyridyl-furan compound OGHL250 being among the most potent. OGHL250 was selected for further improvement by medicinal chemistry, and the potency of its analogues was greatly increased against parasite growth and invasion. As the pyridyl-furan analogues were structurally related to the compound MMV396797 (M-797), we likewise assayed it against invasion and found it was inhibitory too. Recently, M-797 was shown to block parasite protein secretion and export23 and so we evaluated the pyridyl-furan analogues and found that they too inhibited protein secretion and export. Furthermore, the M-797-resistant parasites were also resistant to the pyridyl-furan analogues that inhibit the enzyme phosphatidylinositol 4-kinase (PfPI4KIIIB), involved in many cellular functions including protein trafficking, suggesting PfPI4KIIIB as the common target. Given this, we believe that the pyridyl-furan series of analogues block protein trafficking and invasion by inhibiting PfPI4KIIIB.
2. Results
2.1. Screening of the OGHL for Inhibitors of P. falciparum Growth Identified Eight Inhibitory Compounds
To screen the OGHL for parasite growth inhibition, P. falciparum parasites transfected with a nanoluciferase (Nluc) reporter protein exported into the RBC compartment were used as these parasites were required for our downstream egress and invasion assays.15,24 The OGHL was diluted to 2 μM in RPMI media and in it were grown young ring-stage Nluc parasites for 72 h during which they passed through 1.5 cell cycles. The biomass of the older trophozoite/schizont-stage parasites was assayed by measuring parasite lactate dehydrogenase (LDH) activity, which indicated seven compounds inhibited growth by >50% relative to a DMSO drug vehicle control (Figure 1A, Table S1). An eighth compound, OGHL236, was slightly above the 50% cutoff and was retained as it was below the cutoff for Nluc inhibition (see next). In parallel, assays measuring Nluc activity were also performed and indicated nine OGHL compounds inhibited growth by >50%, of which eight compounds were common with those identified by the LDH assay (Figure 1B, Table S1).
Figure 1.
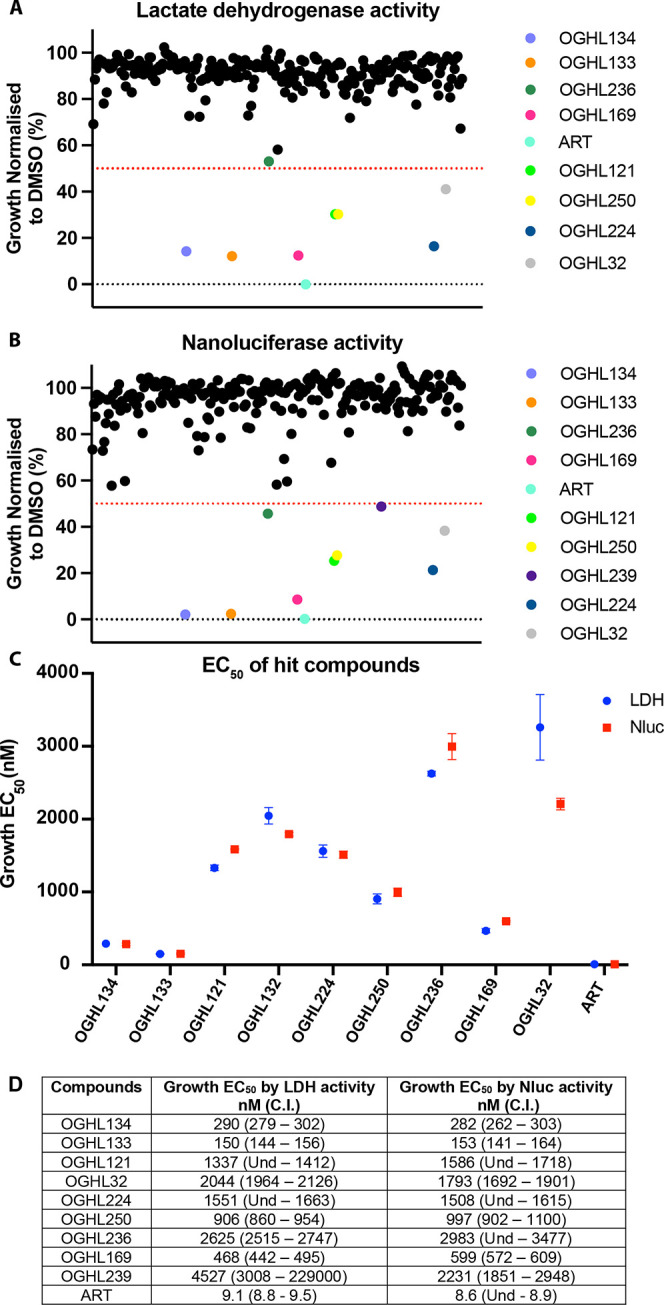
Using P. falciparum parasites expressing an exported nanoluciferase (Nluc) reporter, several Open Global Health Library (OGHL) compounds at 2 μM demonstrated ≥50% blood-stage growth inhibition after a 72 h growth period. (A) Parasite growth after drug treatment was measured using lactate dehydrogenase activity (LDH) and seven compounds inhibited ≥50%. (B) Following measurement of Nluc activity, nine OGHL compounds were found to inhibit growth by ≥50%. All values were normalized to vehicle control (0.1% DMSO) and each dot represents the mean of a compound from one biological repeat of three technical replicates. Dotted lines indicate cutoff values of 50% for growth inhibition. Artemisinin (ART) was used as a positive control at 80 nM. (C, D) Graphical and numerical EC50s of OGHL hit compounds after 72 h growth in serially diluted compounds. Growth was measured by LDH (blue) and Nluc activity (red).
Supplementary compounds of the screening hits and their structures were provided by Merck KGaA Darmstadt, Germany, to enable downstream experiments (Figure S1).22 To derive the EC50 for growth, the nine new OGHL hit compounds were serially diluted and Nluc parasites were grown for 72 h. EC50 values were calculated for both LDH and Nluc assays, and they were in close agreement, except for OGHL239 (Figure 1C,D and Figures S1 and S2). A counter screen was therefore performed using freshly prepared cell lysates of the Nluc parasites. In this screen, compound OGHL239 was observed to strongly inhibit Nluc activity, leaving eight inhibitors with ≥50% growth inhibitory activity (and no specific activity against the Nluc) to be further studied (Figure S3).
2.2. None of the OGHL Growth Inhibitors Affected Egress of P. falciparum Parasites
To measure normal and premature egress of merozoite-stage parasites and their invasion of new RBCs, we employed a previously developed assay (workflow is shown in Figure 2A).15 One of the key events that helps trigger the egress of merozoites from schizonts is the stimulation of protein kinase G (PKG) activity through an increase in cyclic GMP (cGMP) levels.25,26 cGMP-activated PKG triggers a signaling cascade that results in the merozoites releasing proteases and other molecules that help breakdown the RBC so the merozoites can egress and invade new RBCs.27 To prevent premature activation of PKG, phosphodiesterases (PDEs) hydrolyze cGMP, reducing levels of the cyclic nucleotide. PDE inhibitors such as zaprinast lead to a rapid increase in cGMP levels triggering premature activation of PKG and subsequent accelerated egress of immature, poorly invasive merozoites.25,26 Since the OGHL contains many experimental human PDE inhibitors, we assayed Nluc parasites for accelerated egress 30 min after adding the OGHL compounds at 10× EC50 for growth.22 After a 30 min premature egress window, media containing Nluc released from the infected RBCs were harvested and supplemented with the NanoGlo substrate with the bioluminescence measured being proportional to the amount of egress that had occurred.15,26 Relative to egress activator zaprinast, none of the OGHL compounds appeared to accelerate egress, suggesting that they were not inhibitors of P. falciparum PDEs (Figure 2B).
Figure 2.

OGHL hit compounds did not activate egress in the short term (30 min) or inhibit in the longer term (4 h). (A) Workflow summary of how the egress and invasion assay works. (B) Late-stage schizonts were treated with the OGHL compounds for 30 min, and the growth medium was collected to measure Nluc bioluminescence as a marker of egress. None of the OGHL compounds appeared to activate egress. Zaprinast was used as positive control egress stimulator (167% compared with 0.1% DMSO control normalized to 100%), while C1 was an egress inhibitor (24% of DMSO control). Heparin was an invasion inhibitor and had no effect on egress as expected. OGHL compounds were tested at 10× EC50 of growth, and all concentrations specified in parentheses are in μM, except for heparin (in μg/mL). (C) Over a 4 h window, none of the OGHL compounds appeared to significantly inhibit egress, compared to the DMSO control. C1 efficiently blocked normal egress to 38% compared to the 0.1% DMSO control, normalized to 100%. Error bars represent the standard deviation of three biological replicates, each with three technical replicates. Dotted line indicates a cutoff value of 100% for egress activity. Statistical analyses were performed on GraphPad Prism 9 with Welch’s t test between DMSO and C1. **P < 0.01, *P < 0.05. No bar indicates not significant.
To examine the ability of the OGHL inhibitors to inhibit “normal” egress, we next assayed the OGHL inhibitors at 10× EC50 (for growth) for their capacity to inhibit egress over a 4 h window in which most merozoites will have naturally egressed using the same Nluc release assay as used for the premature egress analysis. Briefly, transgenic late schizont-stage parasites exporting Nluc into the RBC compartment were allowed to egress and invade in the presence of 10× EC50 (for growth) of each of the eight OGHL compounds. After the 4 h egress and invasion window, media containing Nluc released from the infected RBCs was harvested and supplemented with NanoGlo substrate as before (Figure 2A). Relative to the control DMSO, the OGHL compounds did not significantly inhibit egress (Figure 2C). The control PKG inhibitor Compound 1 (C1) did however substantially reduce egress as anticipated.15
2.3. Half of the OGHL Inhibitors Reduced P. falciparum Invasion of RBCs
Following the 4 h egress assay, the parasites were treated to remove both the unruptured schizonts and the OGHL compounds leaving the newly invaded ring-stage parasites to continue to grow. The young parasites were grown for a further 24 h in the absence of OGHL compounds, whereupon the older trophozoite-stage parasites were lysed and the Nluc activity quantified as an indicator of the success of invasion or early ring-stage development.15 Compared to the DMSO control, compounds OGHL133 and OGHL224 did not inhibit invasion or early ring-stage development at all (Figure 3). In contrast, compounds OGHL32, OGHL250, OGHL236, and OGHL169 strongly inhibited invasion/early ring-stage development below the 50% cutoff, with compounds OGHL134 and OGHL121 being borderline. Overall, OGHL250 had the strongest combination of low EC50 for invasion and growth; therefore, we decided to optimize this compound for greater potency against P. falciparum followed by efforts to derive the compound’s MoA.
Figure 3.
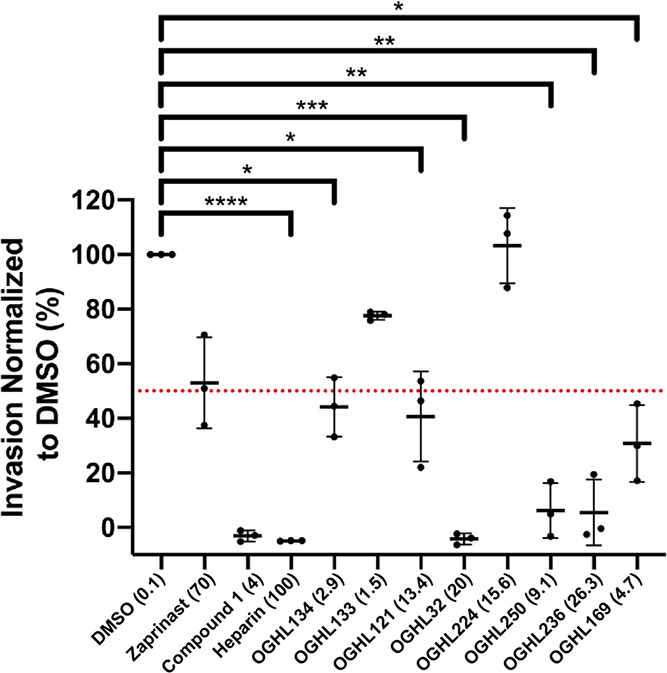
Six of eight OGHL compounds demonstrate varying degrees of invasion inhibition when used at 10× EC50 for growth. When normalized to 0.1% DMSO, the OGHL invasion inhibitors displayed almost complete invasion inhibition for OGHL32 and control compound, heparin, <10% invasion for OGHL250 and OGHL236 and <50% inhibition OGHL134, OGHL121, and OGHL169. All OGHL compounds are in μM, except for heparin (in μg/mL) and DMSO (in %). Error bars represent the standard deviation of three biological replicates, each with three technical replicates. Dotted line indicates a cutoff value of 50% for invasion activity. Statistical analyses were performed on GraphPad Prism 9 with Welch’s t test between DMSO and control compound, heparin, and OGHL compounds. *P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.0001. No bar indicates not significant.
2.4. Optimization of Pyridyl-Furan Parasite Activity
The optimization of the antiparasitic activity of the pyridyl-furan OGHL250 hit chemotype (synthesis outlined in Schemes S1 and S2) began with relocating the endocyclic nitrogen on the isoquinoline ring to the 6-position (1) (Figure 4). This modification resulted in a 10-fold improvement in parasite activity (EC50 = 0.33 μM) while improving the selectivity window against human HepG2 cells (CC50 = 10 μM). The addition of basic functionality by way of an N-methyl piperazine (2) group onto the carboxamide resulted in a 6-fold improvement in parasite activity (EC50 = 0.89 μM) relative to OGHL250. These two modifications were then combined in analogue 3 and resulted in a 2-fold improvement in parasite activity (EC50 0.16 μM) relative to 1. Removal of the methoxy substitution from the benzamide aryl ring (WEHI-518) led to a further 7-fold improvement in activity (EC50 = 0.022 μM).
Figure 4.

Optimized analogues of OGHL250 showing EC50s against blood-stage P. falciparum parasites and CC50 against human HepG2 cells. EC50 data for P. falciparum are averages of three biological replicates, and SD are in brackets. HepG2 toxicity data are averages of three biological replicates and SD in brackets, except for 2 which is n = 1.
The metabolic stability of WEHI-518 was determined by incubation in mouse liver microsomes. It showed that WEHI-518 has low metabolic stability (CLint = 301 μL/min/mg) (Table S2). It was speculated that the methyl substitution on the piperazine of WEHI-518 was susceptible to metabolic N-demethylation. WEHI-212 without the N-methyl group was synthesized and was shown to retain parasite activity (EC50 = 0.058 μM) and improved metabolic stability (CLint = 180 μL/min/mg). This indicated that there are other sites of metabolism in WEHI-518 and WEHI-212 and these are likely to include cytochrome P450-mediated oxidation of one or both pyridyl nitrogens and oxidation of the aryl group. Determination of kinetic aqueous solubility established WEHI-518 possessed high aqueous solubility (160–320 μM) while WEHI-212 was modest (20–40 μM).
In summary of the optimization of the pyridyl-furan chemotype, several key modifications installed in WEHI-518 were made that led to a 250-fold improvement in parasite activity compared to the hit OGHL250. These modifications also inadvertently led to the activity of WEHI-518 against human kinases (IC50 0.22 to >10 μM) (Table S3) compared to potent human kinase activity observed by the hit chemotype.28,29 As a consequence of moderately tuning the kinase activity, the HepG2 selectivity window was also improved. The physicochemical properties installed ensured that WEHI-518 has high aqueous solubility; however, further optimization is required to improve the metabolic stability to a level suitable for in vivo efficacy studies in mouse malaria models.
2.5. Pyridyl-Furan Analogues of OGHL250 Block Parasite Invasion but not Egress
Following development of the pyridyl-furan series, we repeated the 4 h egress and invasion assay as described in Figure 2 with the three lead compounds (OGHL250, WEHI-518, and analogue 3[renamed Compound 3 or C3]), but this time challenged the parasites with a dilution series of the compounds to generate EC50s (Figure 5A–C). As anticipated, WEHI-518 and C3 like OGHL250 did not inhibit parasite egress. In contrast, all the pyridyl-furan compounds blocked invasion/early ring-stage development over 4 h in the same general order of potency as in the 72 h growth assay in which we included the additional antimalarial resistant Dd2 strain (Figure 5E). Generally, the pyridyl-furan compounds inhibited growth more strongly than invasion/early ring-stage development, suggesting that they could target multiple stages of the asexual blood stage cell cycle (Figure 5B).
Figure 5.

M-797 and pyridyl-furan compounds block invasion but not egress during a 4 h assay. (A–D) Dose response curves for 4 h egress and invasion assays for OGHL250, WEHI-518, C3, and M-797, respectively, indicate invasion but not that egress was blocked. Invasion inhibition of purified merozoites indicates all the compounds except for M-797 block RBC invasion. EC50 values (nM) for each type of assay are indicated on the right of each graph. Error bars indicate SD of three biological replicates. (E) EC50 values of 3D7 and Dd2 parasites treated with the compounds for 72 h and EC50s for the 4 h invasion and purified merozoite invasion assays with Nluc parasites are shown for two or three biological replicates. (F) Percentage of invasion of purified merozoites relative to 0.1% DMSO are shown for heparin (100 μg/mL), C1 (4 μM), and CQ (chloroquine, 75 nM) for three biological replicates. (G) Giemsa-stained thin blood smears of P. falciparum parasites after the 4 h egress/invasion assay indicate the pyridyl-furan compounds do not specifically block merozoite invasion as some ring-stage parasites are present. The parasite cultures were treated with DMSO (0.12%), compound 1 (C1; 4 μM), heparin (100 μg/mL), M-797 (4.71 μM), OGHL250 (12 μM), WEHI-518 (W-518, 0.05 μM), and C3 (0.4 μM). Blood smears made after the 4 h drug treatment window (4 h postinvasion [hpi]) and after the 24 hpi show the ring-stage parasites failed to thrive after invasion after treatment with the OGHL250 series and M-797. Cells of interest are labeled with arrows. Size bar = 5 μM. DNI = did not inhibit.
Since we have recently shown that M-797 blocked protein trafficking in parasites and is structurally related to OGHL250 (Figure S4), we decided to determine if M-797 could block egress and invasion/early ring-stage development.23 As RBC invasion requires the trafficking of many invasion proteins to specialized secretory organelles in the merozoite, it was anticipated that M-797 might block invasion.30 Like pyridyl-furans, M-797 did not block egress but did block invasion/early ring-stage development over 4 h with a similar EC50 as 72 h growth inhibition (Figure 5D).
To determine if the pyridyl-furan compounds were disturbing the development of viable merozoites or if they were directly blocking specific invasion processes, schizonts containing merozoites were allowed to mature in the absence of inhibitors. The mature merozoites were then purified and allowed to invade the RBCs in the presence of the compounds. After 30 min, the compounds were removed, and the newly invaded parasites were grown for 24 h to enable quantification of their Nluc levels. We found that OGHL250, WEHI-518, and C3 inhibited invasion/early ring-stage development with EC50 concentrations higher than those found in the 4 h egress and invasion assay (Figure 5A–C,E), indicating that the compounds were not only targeting merozoite development but were also directly blocking invasion/early ring-stage development. M-797 on the other hand did not inhibit merozoite invasion, suggesting that its target was required for merozoite development rather than invasion/early ring-stage development (Figure 5D,E). For merozoite invasion, we also included an invasion-blocking heparin control, which performed as anticipated (Figure 5F).15 The antimalarial chloroquine had no effect on invasion, and the protein kinase G inhibitor C1 also directly inhibited merozoite invasion, which has been noted before with another PKG inhibitor (Figure 5A).15,31
To gain further insights into the compounds’ inhibitory mechanisms, Giemsa-stained blood smears were examined after completion of the 4 h egress and invasion assay (Figure 5G). In DMSO-treated cultures, many healthy, rounded ring-stage parasites were observed, and as anticipated, C1 treatment blocked merozoite egress and heparin blocked invasion (Figure 5G). For M-797, OGHL250, C3, and WEHI-518, small, misshaped ring-stage parasites were observed, suggesting that some invasion had occurred but that conversion into healthy ring-stage parasites was inhibited. By 20 h, the M-797, OGHL250, C3, and WEHI-518-treated parasites had failed to thrive and were noticeably smaller (Figure 5G).
2.6. The Pyridyl-Furan Analogues but not OGHL250 Block Protein Secretion and Export in Parasites
As M-797 had been previously shown to block parasite protein secretion and export into the RBC compartment,23 we next explored the effects of the pyridyl-furans on protein secretion and export. We used a recently developed parasite protein trafficking inhibition assay23 designed to detect inhibitors of protein trafficking both within the parasite and at the specialized parasitophorous vacuole membrane (PVM, the membranous sac containing the parasite within the infected RBC) (Figure 6A). Specifically, we treated parasites with a selection of protein trafficking inhibitors including brefeldin A, torin 2, and M-79723,32,33 (Figure S4). In this assay, young Nluc trophozoite-stage parasites were treated with compounds for 5 h to block secretion into the parasitophorous vacuole (PV) and export into the RBC compartment after which they were differentially lysed in reagents designed to release the contents of the RBC only, RBC and PV or all compartments of the infected RBC (Figure 6A). Lysis of these compartments permitted Nluc to gain access to its substrate and produce a bioluminescent signal proportional to Nluc trapping within that compartment.23 Relative to the DMSO control, brefeldin A, torin 2 and M-797 displayed significantly reduced export into the RBC compartment and therefore greater accumulation in the PV (secreted) and parasite (retained) compartments, as expected for protein trafficking inhibitors (Figure 6B). While OGHL250 did not significantly reduce export of the protein reporter into the RBC compartment, pyridyl-furan analogues WEHI-518 and C3 as well as putative protein trafficking inhibitor KDU73134 behaved similarly to the control export blocking compounds and showed reduced protein export into the RBC compartment with stronger protein trapping at the PV and parasite compartments (Figures 6B and Figure S4).
Figure 6.

PfPI4KIIIB inhibitors reduce protein secretion and export in P. falciparum. (A) Workflow diagram illustrating how the protein export screen works to measure retention of the Nluc reporter in the different compartments of infected RBCs. (B) Percentages of Nluc reporter retained in the parasite, secreted into the PV, or exported into the RBC compartments after 5 h of compound treatment are indicated. Error bars indicate the standard deviations of three biological replicates each of three technical replicates. Compounds used are DMSO (0.1%), brefeldin A (5 μg/mL), torin 2 (10 nM), M-797 (4.8 μM), OGHL250 (3.1 μM), WEHI-518 (200 nM), C3 (1.2 μM), and KDU731 (300 nM). The total Nluc signal relative to DMSO control are indicated by white bars. Welch’s t tests were performed comparing the %Exported signal for each compound versus DMSO using GraphPad PRISM. *p < 0.05, **p < 0.01. (C) Fluorescence microscopy images of parasite infected RBCs treated with the same inhibitory compounds used in panel (B). The cells were probed with rabbit anti-Nluc and an EXP2 mouse monoclonal to locate the Hyp1-Nluc reporter protein and the PVM, respectively. Nuclei were stained with DAPI, and the space bar indicates 4 μm.
Immunofluorescence microscopy of Nluc parasites with an antibody to nanoluciferase confirmed that export into the RBC compartment occurred efficiently in the control DMSO infected RBCs but was inhibited for all compound-treated parasites, except OGHL250 (Figure 6C). All inhibitor treatments except for OGHL250 resulted in the Nluc reporter being trapped inside the parasite’s periphery as denoted by the PVM-staining EXP2 antibody.
2.7. OGHL250 Analogues Act Like PfPI4KIIIB Inhibitors
To identify the protein target and mechanism of action of the pyridyl-furan compounds, we attempted to select for genetic resistance to OGHL250 through repeated rounds of treatment and recovery against engineered “hypermutator” Dd2 parasites that have a higher background error prone DNA replication and are hereafter called Parental parasites.35,36 After three rounds of selection, the DMSO-treated Parental parasites and OGHL250-challenged parasites were subjected to a 72 h growth assay against a dilution series of OGHL250. However, the EC50 was little changed, indicating no resistance could be selected.
In parallel, we selected for resistance to M-797 using the mutator Parental parasites, as we were interested in the protein trafficking target of this compound. The Parental parasites were cycled two times on and off M-797, and resistance to the compound was measured via a 72 h growth assay where four lines with about 16-fold resistance were recovered (Figure 7 and Figure S5A). The M-797-resistant parasite lines were subsequently tested against torin 2 and were not resistant, but they were 8 to 13-fold more resistant to WEHI-518, C3 and KDU731 than the Parental control parasites (Figure 7 and Figure S5B–E). However, the M-797 resistant parasites were not resistant to OGHL250, which was not surprising as OGHL250 did not inhibit protein trafficking, suggesting that its target(s) were not fully shared with the pyridyl-furan analogues, M-797 and KDU731 (Figure 7 and Figure S5F).
Figure 7.

M-797-resistant parasites are resistant to some, but not all, PfPI4KIIIB inhibitors. Four parasite lines selected for resistance to M-797 as well as the nonresistant Parent parasites were grown for 72 h in serially diluted inhibitors, and EC50s for growth inhibition were calculated. The EC50s for three biological replicates and three technical replicates are plotted with bars indicating standard deviations. Y-axis scale represents antilog of drug concentrations in nM.
2.8. Parasites Resistant to M-797 Have a Mutation in Their PfPI4KIIIB Gene
As torin 2 analogue NCATS-SM3710 is a known inhibitor of PfPI4KIIB (PF3D7_0509800) and KDU731 inhibits the PI4K enzyme of Cryptosporidium spp., we predicted that M-797, WEHI-518, and C3 might also be PI4KIIIB inhibitors (Figure S4).34,37 PfPI4KIIIB (PF3D7_0509800) is 1559 aa in size, and the last three-quarters of the protein is predicted to comprise a phosphatidylinositol 4-kinase beta domain in which resistance mutations to other PI4K inhibitors have been found (Figure 8A). As a first step to address this, M-797, OGHL250, and WEHI-518 were assayed with the previously described 3H-hypoxanthine incorporation assay against a parasite line resistant to MMV390048 that has a S743T mutation in PI4KIIIB (Figure S4).38 Compared to the parental Dd2 parasites, the S743T parasites were 2-fold and 4-fold more resistant to M-797 and WEHI-518, respectively, but not OGHL250 (Table S4).
Figure 8.

M-797 resistance mutation in pfpi4kiiib gene clusters with resistance mutations to other inhibitory compounds. (A, Top) Diagram of whole pfpi4kiiib gene showing exons (green) and introns (dashed line), location of PCR amplifed gene fragments, and all known resistance mutations. Image made with SnapGene. An expanded region of pfpi4kiiib indicated with dashed lines shows a cluster of resistance mutations. (B) Homology model of the Pf catalytic domain of PI4K (PF3D7_0509800) was built using the PDB 3ENE as previously described by McNamara (2013).39 MMV390048 (green) was docked using the CLC Drug Discovery workbench. M-797 (cyan) was overlaid using the X-ray structure of this chemotype bound to human PI4KIIIB (PDB 4WAG).40 Mutations conferring drug resistance are shown: E1316D (gold) for M-797; A1319 V (orange) for MMV390048; G1309 V and Y1342F (magenta) for UCT943; S1320L, Y1356F (yellow) for BQR695; V1357G (gray) for NCATS-SM3710; H1484Y (purple) for KAI407. (C) Surface presentation showing a closeup of the ligand binding site with nearby amino acids shown.
To determine if the M-797-resistant parasites have a mutation in their pfpi4kiiib gene, genomic DNA was extracted from the four M-797 resistant lines as well as the Parental parasites. The PI4K domain of PF3D7_0509800 was amplified as a series of 5 overlapping fragments by PCR from one resistant line and the Parental line and compared to pi4kiiib of Dd2 (Figure 8A). In the resistant line but not the Parental line, a GAA-GAC mutation was found that encoded an E1316D mutation in the PCR4 fragment near other known drug resistance mutations (Figure 8A). PCR4 fragments from two clones of each of the other three resistant lines were also sequenced, and two of the other lines also had the same GAA-GAC mutation. The remaining line contained a different GAA-GAT mutation at the same position, which also encoded the same E1316D mutation. The occurrence of different nucleotide mutations that were independently derived but produced the same amino acid change made us confident that resistance to M-797 was due to this mutation.
To determine if the various resistance mutations mapped closely to each other in the predicted 3D structure, a homology model of the P. falciparum catalytic domain of PI4KIIIB was built using the PDB 3ENE as previously described by McNamara (2013).39 M-797 was predicted to overlap with MMV390048 when overlaid using the X-ray structure of this chemotype bound to human PI4KIIIB (Figure 8B,C).40 Mutations conferring drug resistance are shown: E1316D for M-797, A1319 V for MMV390048, G1309 V and Y1342F for UCT943, S1320L and Y1356F for BQR695, V1357G for NCATS-SM3710 and H1484Y for KAI407 all appear to be near each other (Figure 8 and Figure S4). Interestingly, the MMV390048 S743T resistance mutation is located some distance upstream from the other mutations. These parasites are, however, known to be resistant to UCT943, which in turn has its own resistance mutation at Y1342F located near the M-797 resistance mutation E1316D. Alpha-fold structures of PfPI4KIIIB indicate that S743 could project toward the predicted binding sites of the inhibitors providing a mechanism for the resistance of S743T parasites against MMV390048 and other inhibitors (Figure S6).41−43
2.9. The Inhibitors Block Recombinant PvPI4KB as Well as Parasite Transmission
To support that the putative PfPI4KIIIB inhibitors were active against this enzyme, the compounds were assayed against recombinant PvPI4Kβ as per ref (44) and were found to inhibit kinase activity generally in proportion to their growth inhibition of cultured blood stage parasites (Table S5).
To explore transmission inhibition activity of the compounds, they were assayed for their ability to block the transformation of male and female gametocytes into gametes with the potency of WEHI-518 > C3 > M-797 (Figure S8).45 The compounds exhibited slightly greater activity against female over male gametes, which has been previously observed (Figure S8).44
3. Discussion
Here, we screened the 250 compound OGHL library of Merck KGaA Darmstadt, Germany, for novel inhibitors of merozoite egress and invasion by first performing a 72 h growth inhibitory screen against the asexual blood stage of P. falciparum. This resulted in eight compounds being identified with low micromolar potency. This subset was further screened and although none were observed to inhibit egress, four strongly inhibited parasite invasion of human RBCs. Compound OGHL250 was selected for further optimization as it had a strong combination of growth and invasion inhibition. A 250-fold improvement in growth inhibition was achieved with the series-leading WEHI-518 compound, but high metabolic turnover in liver microsomes precluded further evaluation in the Plasmodium berghei mouse model of malaria. WEHI-212 was subsequently developed to limit metabolic turnover, and although this was reduced, it was not significant enough to consider evaluation in P. berghei. WEHI-518 has some mild inhibitory activity against human IKKe and Syk kinases, and this will need to be reduced along with improvements in the metabolic stability of the series before further evaluation in mouse parasite models can occur.
Attempts to identify the target of OGHL250 through the selection of parasite resistance to the compound were unsuccessful. OGHL250 is a known inhibitor of human spleen tyrosine kinase (Syk) for which inhibitory compounds have been developed to treat several diseases.46 Given OGHL250’s modest potency against parasite growth and poor inhibition of protein export, the compound could have multiple targets in P. falciparum including one or more of the parasite’s many kinases.47 Activity against multiple parasite proteins would decrease the chances of recovering multiple resistance mutations in a single parasite, which may have been why resistance to OGHL250 was not recovered.
Resistance was however recovered to the protein export blocking M-797 compound whose core structurally resembles that of the OGHL250 series, prompting us to evaluate the resistance of these parasites to WEHI-518 and C3. As the M-797-resistant parasites were also resistant to known PfPI4KIIIB inhibitor KDU731, we sequenced the pfpi4kiiib gene in our resistant parasites recovering an E1316D mutation, which was located near several other pfpi4kiiib drug resistance mutations.37−39,41 Examination of a homology structure of PfPI4KIIIB indicated that the resistance mutations clustered closely to the binding sites of MMV390048 and M-797 predicted by in silico docking, providing a rationale for how the E1316D resistance mutation could reduce the affinity of OGHL250-like compounds for the kinase.39 The M-797 resistant parasites were refractory to the pyridyl-furan lead compounds WEHI-518 and C3 but not to OGHL250, indicating that during the development of the lead compounds, their specificity for PfPI4KIIIB increased while activity against other proteins targeted by OGHL250 decreased.
During preparation of this manuscript, we became aware that human mTOR inhibitor sapanisertib also inhibits parasite PfPI4KIIIB and PfPKG (Figure S4).44 Parasites selected for resistance to sapanisertib were found to contain the same E1316D mutation as our M-797 resistant parasites, supporting our finding that PfPI4KIIIB is very likely the main target of M-797. Unlike sapanisertib, M-797 is unlikely to be a major inhibitory target for PfPKG as M-797 did not inhibit egress like known PfPKG inhibitor compound 1, which was used here as a positive control in our egress inhibition experiments (Figure 2).
Several inhibitory lead compounds to PfPI4KIIIB have already been developed, some of which have recently undergone phase 1 clinical trials.48,49 The treatment of schizonts with the PfPI4KIIIB inhibitor KAI407, closely related to KDU691, has been previously observed to produce poorly invasive merozoites.39 This could be due to defective plasma membrane ingression in the schizont, disrupting the proper formation of membranes around the developing merozoites. To investigate this further, we examined Giemsa-stained smears of inhibitor-treated parasites after the 4 h invasion assay and found many young, malformed rings that failed to grow properly even 20 h after compound removal. This indicates that the pyridyl-furan compounds could also be inhibiting the conversion of newly invaded merozoites into ring-stage parasites.
In contrast to developing merozoites, fully formed merozoites mechanically released from their old RBCs could efficiently invade new RBCs in the presence of KAI407.39 We found that this was also the case for M-797 and contrasts with our observations for OGHL250, WEHI-518, and C3 where purified merozoites appeared to poorly invade RBCs in the presence of these compounds. Based on our observations of Giemsa-stained smears of merozoites that were allowed to egress and invade over a 4 h window, some of the purified merozoites may have invaded but failed to differentiate into rings. It is possible that OGHL250, WEHI-518, and C3 could block important protein trafficking events essential for RBC invasion such as the secretion of the merozoites’ microneme or rhoptry organelles. It is also likely that the compounds inhibited membrane and protein trafficking events required for the differentiation of newly invaded merozoites into rings. If these events depend upon PfPI4KIIIB, then it is unknown why KAI407 and M-797 did not inhibit invasion. It is possible that OGHL250, WEHI-518, and C3 may have targets additional to PfPI4KIIIB, which could explain why their EC50s for the 4 h and purified merozoite invasion assays are higher than for growth.
PfPI4KIIIB phosphorylates phosphatidylinositol to produce phosphatidylinositol 4-phosphate. Although phosphoinositides only make up about 1% of membrane phospholipids, they play important roles in many cellular processes such as signal transduction, membrane trafficking, and protein transport.50 Apart from position 4, the inositol headgroup can be phosphorylated singly and multiply at several other positions by different phosphoinositol kinases to produce up to seven types of phosphoinositol phosphate (PIPs). Through enrichment at different organellar membranes and locations, the PIPs act as a code to regulate membrane-dependent cellular functions. Recent investigation of PIPs in P. falciparum using protein reporters that recognize the different PIP types indicated that PI4P is found at the Golgi apparatus and plasma membrane, PI3P concentrates at the apicoplast, plasma membrane, and food vacuole membranes, and PI5P is found at the plasma membrane, nuclear membrane, and possibly the transitional ER in schizonts.50 The multiply phosphorylated PIP species PI(3,4)P2 and PI(3,4,5)P3 are cytosolic, and PI(4,5)P2 is in the PM and possibly cytostome, a structure that endocytoses the RBC cytosol. PI(3,5)P2 was not detected, indicating that Plasmodium may not utilize the full complement of PIP types.50 In developing merozoites, PI4P is found in the plasma membrane and an internal punctate structure. In mature, free merozoites, PI4P disappears from the plasma membrane but is retained within an internal structure that does not appear to overlap with the microneme or rhoptry organelles that release proteins involved in invasion.50 While the PI4P produced by PfPI4KIIIB appears to play a role in merozoite development, it is unclear what role PI4P might play in invasion. Recently however, the protein PfPH2, which binds to several PIP species, has been shown to play a role in the secretion of a subset of micronemes potentially linking PI4P to invasion.51
Although our data indicate that PfPI4KIIIB is important for successful invasion, it is possible that PI4P may not be required for merozoite penetration of the RBC but for downstream events where the internalized merozoite must rapidly differentiate into an amoeboid ring-stage parasite over several minutes.52 This would involve disassembling many internal membranous structures, such as the inner membrane complex and apical organelles, as well as the secretion and export of parasite proteins into the RBC compartment to remodel the host cell for subsequent rapid growth, replication, and immune evasion.53−55 Metamorphosis of the extracellular merozoite into an intraerythrocytic form that feeds and replicates would require extensive reorganization of membranous structures and proteins, and this is likely heavily dependent on PI4P and the other PIPs.
In conclusion, we have identified the P. falciparum-killing compound OGHL250, from the OGHL of Merck KGaA, Darmstadt, Germany, that also appears to inhibit merozoite invasion of RBCs. From OGHL250, the highly potent analogues WEHI-518 and C3 were produced that likely target PfPI4KIIIB, an enzyme required to produce PI4P, which has an important role in vesicular membrane and protein trafficking from the ER to the plasma membrane via the Golgi. PI4P can also be converted into PI4,5P2 and PI3,4,5P3, which may also have important invasion-related functions.50 M-797 resistant parasites containing an E1316D mutation in PfPI4KIIIB are also resistant to WEHI-518 and C3 but not to OGHL250, which could be targeting another kinase involved in invasion. The E1316D mutation clusters with several other resistance mutations to PfPI4KIIIB inhibitors, some of which are undergoing clinical trials in humans such as MMV390048.48 PfPI4KIIIB is an excellent target with inhibitors blocking multiple parasite lifecycle stages.38,39 While WEHI-518 and C3 are as potent as other leading PfPI4KIIIB inhibitors, they are metabolically unstable and not suitable for further clinical development. Despite this, our work does shed light on the mechanism of action of this important future drug target.
4. Materials and Methods
4.1. Compounds
All compounds (zaprinast, heparin, artemisinin, chloroquine, brefeldin A, and torin 2) were sourced from Merck KGaA, Darmstadt, Germany, unless otherwise stated. C1 (4-[2-(4-fluorphenyl)-5-(1-methylpiperidine-4-yl)-1H-pyrrol-3-yl]pyridine) and KDU731 were made according to published protocols.34,56 WR99210 was purchased from Jacobus Pharmaceutical Company.
4.2. In Vitro Culturing of P. falciparum and Parasite Strains
Parasites were cultured in human red blood cells (Australian Red Cross Blood Bank) at 4% hematocrit in RPMI media (Sigma-Aldrich, UK), supplemented with 0.2% NaHCO3 (Thermo Scientific, Australia), 0.25% Albumax II (Gibco, New Zealand), 0.37 mM hypoxanthine (Sigma, USA), 25 mM HEPES (Gibco, USA), and 31.25 mg/L gentamicin (Gibco, USA) (cRPMI) and incubated at 37 °C with 1% O2, 5% CO2, and 94% N2. The parasite lines used in this study includes the 3D7 African wild-type strain parasite transfected with a Nanoluciferase reporter proteins targeted to the RBC compartment (Hyp1-Nluc)24 and Dd2 strain parasite with a mutated exonuclease domain within the DNA polymerase δ (Dd2-Polδ).35 Hyp1-Nluc was cultivated under 2.5 nM WR99210 (Jacobus Pharmaceutical Company) selection to maintain episomal expression of the Hyp1-Nluc gene.57
To produce synchronous parasites for protein export and invasion assays, they were first synchronized with 5% sorbitol to enrich for ring-stage parasites.58 The parasite were grown until late schizonts and layered over a 67% Percoll density gradient buffer in cRPMI (Percoll, 10 mM NaH2PO4, 143 mM NaCl (GE Healthcare Bio- Sciences, Sweden)) and centrifuged (1500g/15 min).59 The dark-colored synchronous schizont layer was removed and used for the downstream experiments.
4.3. Screening of the Open Global Health Library for P. falciparum Growth Inhibitors
The assay was conducted on sorbitol-synchronized ring-stage Hyp1-Nluc parasites at 2% hematocrit and 0.3% parasitemia in a 96-well U-bottom plate (Thermo Scientific, Australia) for 72 h in the presence of 2 μM of the 250 compound OGHL. The plate was subsequently stored at −80 °C and thawed at room temperature for 4 h to lyse parasites. Parasite growth was quantified through colorimetric measurement of parasite lactate dehydrogenase activity (LDH).60,61 To measure parasite Nluc activity, 5 μL of lysed parasite culture was dispensed into a 96-well white luminescent plate, prior to the addition of 45 μL of 1× NanoGlo Lysis buffer to lyse the infected RBCs (iRBCs), releasing Nluc. The lysis buffer contained a 1:4000 NanoGlo (furimazine) substrate (Promega, USA), to generate a bioluminescence signal through substrate hydrolysis.62 The total Nluc signal was then measured with a CLARIOstar multimode plate reader (BMG Labtech).
4.4. Nluc Counter Screen
The assay was adapted from15 where Hyp1-Nluc schizonts at 1% hematocrit and 1–2% parasitaemia were lysed in 1x NanoGlo buffer (Promega, USA) within a 96-well flat-bottom plate and parasite lysates were added to compounds and incubated for 10 min at 37 °C. Nluc activity was measured as above. Nluc activity detected in the presence of compounds was normalized to the vehicle control (0.1% DMSO).
30 min Egress Assay
The assay is adapted from ref (15) and conducted on Hyp1-Nluc schizonts at early- to midstages (36–40 hpi) isolated via Percoll purification, at 3–5% parasitaemia at 1% hematocrit in a 96-well U-bottom plate with a 30 min drug-treatment and incubation period at 37 °C. The supernatant was removed for Nluc activity detection as above. The assay included a background control for egress comprising of culture media of Percoll-purified cultures kept at 4 °C during the 30 min incubation period, which accounted for the potential Nluc signal from spontaneous lysis or cell leakage. The background control was subtracted from the egress values and normalized relative to the vehicle control (0.1% DMSO).
4 h Egress/Invasion Assay
The assay was performed as per ref (15) with background controls subtracted from both the egress and invasion values and normalized relative to the vehicle control (0.1% DMSO).15
72 h Growth Inhibition Assay with Dd2 Parasites and the MMV390048-Resistant Dd2 with S743T Mutation in PI4KIIIB (Swiss TPH)
The testing was performed with the modified [3H]-hypoxanthine incorporation assay, as previously reported.63
Chemistry
See the Supplementary Experimental and Chemistry sections.
In Vitro Drug Resistance Selection of P. falciparum Dd2-Polδ
Dd2-Polδ mutant parasites were used for in vitro drug resistance selection to increase the likelihood of selection of random mutations for drug resistance accelerating the selection time.35 Dd2-Polδ mixed-stage parasite cultures at 5% parasitemia and 4% hematocrit were dispensed into each well of a six-well flat-bottom plate (Thermo Scientific, Australia). Five cultures were treated with 10× the growth EC50 of OGHL250 (9.1 μM) and M-797 (4.8 μM), and one was treated with the vehicle control (DMSO) at the same final volume used in the compound-treated wells. Parasite cultures were exposed to the compound until they began to die, and the drug was removed. The drug treatment was repeated when the parasite population had regrown, and this drug-cycling process was repeated 2−5 times. Following this, 72 h growth assays in the presence of inhibitory compounds were performed to determine if the parasites had developed resistance. Clonal parasite lines were derived from resistant populations by limiting dilution and retested for resistance before isolation of genomic DNA.
Known and Putative PfPI4KIIIB Inhibitors Block Protein Export in P. falciparum
Trophozoite stage 20–24 h post-invasion (hpi), P. falciparum parasites expressing a Nluc reporter were treated with PfPI4KIIIB inhibitory compounds for 5 h before being assayed for inhibition of Nluc reporter secretion and export as previously described.23
Sequence Analysis of PfPI4KIIIB Gene Sequence from Resistant Parasites
Genomic DNA was isolated from two clones resistant to M-797and two parental clones. Most of the pfpki4kiiib was amplified as five overlapping PCR fragments using Phusion DNA polymerase (Thermo) as per the manufacturer’s instruction. Primer sequences are listed in Table S6, and as only PCR fragment 4 was found to contain a mutation in the resistant parasites, two clones each from another three resistant populations were ligated into the pJet 1.2 plasmid (Thermo) and were sequenced. Sequence data was aligned an analyzed in SnapGene.
Invasion Inhibition Assay with Purified Merozoites
Merozoites were grown and purified, inhibitor-treated, and assayed for invasion as described elsewhere.15
PfPI4KIIIB Kinase Inhibition Assay
The compounds used here were assayed against recombinant PvPI4Kβ (PVX_098050) kinase with inhibitory activity detected using the ADP-Glo kinase assay kit (Promega).44
Transmission blocking assay
The transmission blocking activity of M-797, WEHI-518, and C3 was evaluated by incubating the compounds against stage V gametocytes and evaluating their transition to male gametes by counting exflagellation centers and female gametes by counting anti-Pfs25 IgG parasites as per.45
Acknowledgments
We are grateful to Merck KGaA Darmstadt, Germany, for provision of the Open Global Helath Library and to Sven Lindemann for supplying additional compound material. We thank Lifeblood Biological Resources for providing blood and Medicine for Malaria Venture for facilitating the testing of three compounds in the hypoxanthine incorporation assay. We are extrememly thankful to Kelly Chibale, Lauren Arendse and Nicolaas Salomane from the Holistic Drug Discovery and Development (H3D) Centre, University of Cape Town for testing compounds on recombinant parasite PvPI4KB. We are grateful to Trent Ashton from Walter and Eliza Hall Institute for providing a sample of M-797. We are grateful for Dr Kate Jarman and Katie Loi from Walter and Eliza Hall Institute for 3D7 LDH and HepG2 assay support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.3c00138.
Table of growth inhibition assay data (XLSX)
Synthetic route to 3, WEHI-518, and WEHI-212; tables of growth inhibition assay data, aqueous solubility and in vitro metabolism, human kinase activity, summary of growth inhibitor activity, and PCR primers; structures of P. falciparum growth inhibitory compounds from the OGHL and compounds mentioned in the text, dose–response curves, Nluc activity counter screen, growth of M-797 resistant parasite lines, ALPHA-fold model of Pf PI4KIIIB, and inhibition assay (PDF)
Author Contributions
D.L., W.N., O.L., B.S., A.B., M.D., H.B., and .PG. conceived this research and designed the experiments. D.L., O.L., Z.R., K.M., C.S., S.W., and M.T.F. conducted the experiments and generated the figures. P.G. wrote the manuscript. All authors edited the manuscript. B.S., B.C., and P.G. provided funding. All authors have approved the submitted manuscript.
This work was supported by funding from the Victorian Operational Infrastructure Support Program received by the Burnet Institute. Funding was provided by the National Health and Medical Research Council (grant numbers 1185354, 2001073 and 119780521).
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organisation World Malaria Report 2021. 2021.
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P. J.; Lindegardh N.; Socheat D.; White N. J. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009, 361 (5), 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann C.; van Loon W.; Habarugira F.; Tacoli C.; Jager J. C.; Savelsberg D.; Nshimiyimana F.; Rwamugema E.; Mbarushimana D.; Ndoli J.; Sendegeya A.; Bayingana C.; Mockenhaupt F. P. Increase in Kelch 13 Polymorphisms in Plasmodium falciparum, Southern Rwanda. Emerg Infect Dis 2021, 27 (1), 294–296. 10.3201/eid2701.203527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N.; Yamauchi M.; Morikawa R.; Hombhanje F.; Mita T. Increase in the proportion of Plasmodium falciparum with kelch13 C580Y mutation and decline in pfcrt and pfmdr1 mutant alleles in Papua New Guinea. Malar. J. 2021, 20 (1), 410. 10.1186/s12936-021-03933-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautu-Gumal D.; Razook Z.; Koleala T.; Nate E.; McEwen S.; Timbi D.; Hetzel M. W.; Lavu E.; Tefuarani N.; Makita L.; Kazura J.; Mueller I.; Pomat W.; Laman M.; Robinson L. J.; Barry A. E. Surveillance of molecular markers of Plasmodium falciparum artemisinin resistance (kelch13 mutations) in Papua New Guinea between 2016 and 2018. Int. J. Parasitol. Drugs Drug Resist 2021, 16, 188–193. 10.1016/j.ijpddr.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward K. E.; Fidock D. A.; Bridgford J. L. Plasmodium falciparum resistance to artemisinin-based combination therapies. Curr. Opin Microbiol. 2022, 69, 102193 10.1016/j.mib.2022.102193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465 (7296), 305–310. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jiménez-Díaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; Mazouni F. E.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; DeRisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. Chemical genetics of Plasmodium falciparum. Nature 2010, 465 (7296), 311–315. 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister S.; Plouffe D. M.; Kuhen K. L.; Bonamy G. M.; Wu T.; Barnes S. W.; Bopp S. E.; Borboa R.; Bright A. T.; Che J.; Cohen S.; Dharia N. V.; Gagaring K.; Gettayacamin M.; Gordon P.; Groessl T.; Kato N.; Lee M. C.; McNamara C. W.; Fidock D. A.; Nagle A.; Nam T. G.; Richmond W.; Roland J.; Rottmann M.; Zhou B.; Froissard P.; Glynne R. J.; Mazier D.; Sattabongkot J.; Schultz P. G.; Tuntland T.; Walker J. R.; Zhou Y.; Chatterjee A.; Diagana T. T.; Winzeler E. A. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334 (6061), 1372–1377. 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte B.; Ottilie S.; Plater A.; Campo B.; Dechering K. J.; Gamo F. J.; Goldberg D. E.; Istvan E. S.; Lee M.; Lukens A. K.; McNamara C. W.; Niles J. C.; Okombo J.; Pasaje C. F. A.; Siegel M. G.; Wirth D.; Wyllie S.; Fidock D. A.; Baragana B.; Winzeler E. A.; Gilbert I. H. Prioritization of Molecular Targets for Antimalarial Drug Discovery. ACS Infect. Dis. 2021, 7 (10), 2764–2776. 10.1021/acsinfecdis.1c00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okombo J.; Kanai M.; Deni I.; Fidock D. A. Genomic and Genetic Approaches to Studying Antimalarial Drug Resistance and Plasmodium Biology. Trends Parasitol. 2021, 37 (6), 476–492. 10.1016/j.pt.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehane A. M.; Ridgway M. C.; Baker E.; Kirk K. Diverse chemotypes disrupt ion homeostasis in the Malaria parasite. Mol. Microbiol. 2014, 94 (2), 327–339. 10.1111/mmi.12765. [DOI] [PubMed] [Google Scholar]
- Dennis A. S. M.; Rosling J. E. O.; Lehane A. M.; Kirk K. Diverse antimalarials from whole-cell phenotypic screens disrupt malaria parasite ion and volume homeostasis. Sci. Rep. 2018, 8 (1), 8795. 10.1038/s41598-018-26819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Voorhis W. C.; Adams J. H.; Adelfio R.; Ahyong V.; Akabas M. H.; Alano P.; Alday A.; Alemán Resto Y.; Alsibaee A.; Alzualde A.; Andrews K. T.; Avery S. V.; Avery V. M.; Ayong L.; Baker M.; Baker S.; Ben Mamoun C.; Bhatia S.; Bickle Q.; Bounaadja L.; Bowling T.; Bosch J.; Boucher L. E.; Boyom F. F.; Brea J.; Brennan M.; Burton A.; Caffrey C. R.; Camarda G.; Carrasquilla M.; Carter D.; Belen Cassera M.; Chih-Chien Cheng K.; Chindaudomsate W.; Chubb A.; Colon B. L.; Colón-López D. D.; Corbett Y.; Crowther G. J.; Cowan N.; D’Alessandro S.; Le Dang N.; Delves M.; DeRisi J. L.; Du A. Y.; Duffy S.; Abd El-Salam El-Sayed S.; Ferdig M. T.; Fernández Robledo J. A.; Fidock D. A.; Florent I.; Fokou P. V. T.; Galstian A.; Gamo F. J.; Gokool S.; Gold B.; Golub T.; Goldgof G. M.; Guha R.; Guiguemde W. A.; Gural N.; Guy R. K.; Hansen M. A. E.; Hanson K. K.; Hemphill A.; Hooft van Huijsduijnen R.; Horii T.; Horrocks P.; Hughes T. B.; Huston C.; Igarashi I.; Ingram-Sieber K.; Itoe M. A.; Jadhav A.; Naranuntarat Jensen A.; Jensen L. T.; Jiang R. H. Y.; Kaiser A.; Keiser J.; Ketas T.; Kicka S.; Kim S.; Kirk K.; Kumar V. P.; Kyle D. E.; Lafuente M. J.; Landfear S.; Lee N.; Lee S.; Lehane A. M.; Li F.; Little D.; Liu L.; Llinás M.; Loza M. I.; Lubar A.; Lucantoni L.; Lucet I.; Maes L.; Mancama D.; Mansour N. R.; March S.; McGowan S.; Medina Vera I.; Meister S.; Mercer L.; Mestres J.; Mfopa A. N.; Misra R. N.; Moon S.; Moore J. P.; Morais Rodrigues da Costa F.; Müller J.; Muriana A.; Nakazawa Hewitt S.; Nare B.; Nathan C.; Narraidoo N.; Nawaratna S.; Ojo K. K.; Ortiz D.; Panic G.; Papadatos G.; Parapini S.; Patra K.; Pham N.; Prats S.; Plouffe D. M.; Poulsen S. A.; Pradhan A.; Quevedo C.; Quinn R. J.; Rice C. A.; Abdo Rizk M.; Ruecker A.; St. Onge R.; Salgado Ferreira R.; Samra J.; Robinett N. G.; Schlecht U.; Schmitt M.; Silva Villela F.; Silvestrini F.; Sinden R.; Smith D. A.; Soldati T.; Spitzmüller A.; Stamm S. M.; Sullivan D. J.; Sullivan W.; Suresh S.; Suzuki B. M.; Suzuki Y.; Swamidass S. J.; Taramelli D.; Tchokouaha L. R. Y.; Theron A.; Thomas D.; Tonissen K. F.; Townson S.; Tripathi A. K.; Trofimov V.; Udenze K. O.; Ullah I.; Vallieres C.; Vigil E.; Vinetz J. M.; Voong Vinh P.; Vu H.; Watanabe N. a.; Weatherby K.; White P. M.; Wilks A. F.; Winzeler E. A.; Wojcik E.; Wree M.; Wu W.; Yokoyama N.; Zollo P. H. A.; Abla N.; Blasco B.; Burrows J.; Laleu B.; Leroy D.; Spangenberg T.; Wells T.; Willis P. A.; Phillips M. A. Open Source Drug Discovery with the Malaria Box Compound Collection for Neglected Diseases and Beyond. PLoS Pathog. 2016, 12 (7), e1005763 10.1371/journal.ppat.1005763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dans M. G.; Weiss G. E.; Wilson D. W.; Sleebs B. E.; Crabb B. S.; de Koning-Ward T. F.; Gilson P. R. Screening the Medicines for Malaria Venture Pathogen Box for invasion and egress inhibitors of the blood stage of Plasmodium falciparum reveals several inhibitory compounds. Int. J. Parasitol. 2020, 50 (3), 235–252. 10.1016/j.ijpara.2020.01.002. [DOI] [PubMed] [Google Scholar]
- Subramanian G.; Belekar M. A.; Shukla A.; Tong J. X.; Sinha A.; Chu T. T. T.; Kulkarni A. S.; Preiser P. R.; Reddy D. S.; Tan K. S. W.; Shanmugam D.; Chandramohanadas R.; Sullivan W. J. Targeted Phenotypic Screening in Plasmodium falciparum and Toxoplasma gondii Reveals Novel Modes of Action of Medicines for Malaria Venture Malaria Box Molecules. mSphere 2018, 3 (1), e00534 10.1128/mSphere.00534-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dans M. G.; Piirainen H.; Nguyen W.; Khurana S.; Mehra S.; Razook Z.; Geoghegan N. D.; Dawson A. T.; Das S.; Parkyn Schneider M.; Jonsdottir T. K.; Gabriela M.; Gancheva M. R.; Tonkin C. J.; Mollard V.; Goodman C. D.; McFadden G. I.; Wilson D. W.; Rogers K. L.; Barry A. E.; Crabb B. S.; de Koning-Ward T. F.; Sleebs B. E.; Kursula I.; Gilson P. R.; Kim K. Sulfonylpiperazine compounds prevent Plasmodium falciparum invasion of red blood cells through interference with actin-1/profilin dynamics. PLOS Biol. 2023, 21, e3002066 10.1371/journal.pbio.3002066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G. E.; Crabb B. S.; Gilson P. R. Overlaying Molecular and Temporal Aspects of Malaria Parasite Invasion. Trends Parasitol. 2016, 32 (4), 284–295. 10.1016/j.pt.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Weiss G. E.; Gilson P. R.; Taechalertpaisarn T.; Tham W. H.; de Jong N. W.; Harvey K. L.; Fowkes F. J.; Barlow P. N.; Rayner J. C.; Wright G. J.; Cowman A. F.; Crabb B. S. Revealing the sequence and resulting cellular morphology of receptor-ligand interactions during Plasmodium falciparum invasion of erythrocytes. PLoS Pathog 2015, 11 (2), e1004670 10.1371/journal.ppat.1004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C. B. G.; Dans M. G.; Jonsdottir T. K.; Crabb B. S.; Gilson P. R. PfATP4 inhibitors in the Medicines for Malaria Venture Malaria Box and Pathogen Box block the schizont-to-ring transition by inhibiting egress rather than invasion. Front. Cell. Infect. Microbiol. 2022, 12, 1060202. 10.3389/fcimb.2022.1060202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M.; McNamara C.; Yeung B. K. S.; Lee M. C. S.; Zou B.; Russell B.; Seitz P.; Plouffe D. M.; Dharia N. V.; Tan J.; Cohen S. B.; Spencer K. R.; González-Páez G. E.; Lakshminarayana S. B.; Goh A.; Suwanarusk R.; Jegla T.; Schmitt E. K.; Beck H. P.; Brun R.; Nosten F.; Renia L.; Dartois V.; Keller T. H.; Fidock D. A.; Winzeler E. A.; Diagana T. T. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329 (5996), 1175–80. 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merck Healthcare KGaA Open Global Health Library URL. https://www.merckgroup.com/en/research/open-innovation/biopharma-open-innovation-portal/open-global-health-library.html.
- Looker O.; Dans M. G.; Bullen H. E.; Sleebs B. E.; Crabb B. S.; Gilson P. R. The Medicines for Malaria Venture Malaria Box contains inhibitors of protein secretion in Plasmodium falciparum blood stage parasites. Traffic 2022, 23 (9), 442–461. 10.1111/tra.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo M. F.; Nie C. Q.; Elsworth B.; Charnaud S. C.; Sanders P. R.; Crabb B. S.; Gilson P. R. Plasmodium falciparum transfected with ultra bright NanoLuc luciferase offers high sensitivity detection for the screening of growth and cellular trafficking inhibitors. PLoS One 2014, 9 (11), e112571 10.1371/journal.pone.0112571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. R.; Hackett F.; Strath M.; Penzo M.; Withers-Martinez C.; Baker D. A.; Blackman M. J. Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog. 2013, 9 (5), e1003344 10.1371/journal.ppat.1003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard B. L.; Harvey K. L.; Stewart R. J.; Azevedo M. F.; Crabb B. S.; Jennings I. G.; Sanders P. R.; Manallack D. T.; Thompson P. E.; Tonkin C. J.; Gilson P. R. Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites. ACS Chem. Biol. 2015, 10 (4), 1145–1154. 10.1021/cb501004q. [DOI] [PubMed] [Google Scholar]
- Baker D. A.; Drought L. G.; Flueck C.; Nofal S. D.; Patel A.; Penzo M.; Walker E. M. Cyclic nucleotide signalling in malaria parasites. Open Biol. 2017, 7 (12), 170213 10.1098/rsob.170213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf L.; Ross T.; Deutsch C.. Furopyridine derivatives. WO Patent 2,013,124,025, 2013.
- Eggenweiler H. M.; Hoelzamann D. D.; Dorsch D.. Furo [3,2-b]-and thieno [3,2-b] pyridine derivatives as TBK1 and IKK inhibitors. 2013. [Google Scholar]
- Burns A. L.; Dans M. G.; Balbin J. M.; de Koning-Ward T. F.; Gilson P. R.; Beeson J. G.; Boyle M. J.; Wilson D. W. Targeting malaria parasite invasion of red blood cells as an antimalarial strategy. FEMS Microbiol Rev. 2019, 43 (3), 223–238. 10.1093/femsre/fuz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. M.; Solyakov L.; Bottrill A. R.; Flueck C.; Siddiqui F. A.; Singh S.; Mistry S.; Viskaduraki M.; Lee K.; Hopp C. S.; Chitnis C. E.; Doerig C.; Moon R. W.; Green J. L.; Holder A. A.; Baker D. A.; Tobin A. B. Phosphoproteomics reveals malaria parasite Protein Kinase G as a signalling hub regulating egress and invasion. Nat. Commun. 2015, 6, 7285. 10.1038/ncomms8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benting J.; Mattei D.; Lingelbach K. Brefeldin A inhibits transport of the glycophorin-binding protein from Plasmodium falciparum into the host erythrocyte. Biochem. J. 1994, 300, 821–826. 10.1042/bj3000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson K. K.; Ressurreição A. S.; Buchholz K.; Prudêncio M.; Herman-Ornelas J. D.; Rebelo M.; Beatty W. L.; Wirth D. F.; Hänscheid T.; Moreira R.; Marti M.; Mota M. M. Torins are potent antimalarials that block replenishment of Plasmodium liver stage parasitophorous vacuole membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (30), E2838–E2847. 10.1073/pnas.1306097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U. H.; Vinayak S.; Zambriski J. A.; Chao A. T.; Sy T.; Noble C. G.; Bonamy G. M. C.; Kondreddi R. R.; Zou B.; Gedeck P.; Brooks C. F.; Herbert G. T.; Sateriale A.; Tandel J.; Noh S.; Lakshminarayana S. B.; Lim S. H.; Goodman L. B.; Bodenreider C.; Feng G.; Zhang L.; Blasco F.; Wagner J.; Leong F. J.; Striepen B.; Diagana T. T. A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 2017, 546 (7658), 376–380. 10.1038/nature22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuümpornsin K.; Kochakarn T.; eo T.; Luth M.; Pearson R.; Hoshizaki J.; Mok S.; Park H.; Uhlemann A.-C.; Cubel S.; Franco V.; Gomez-Lorenzo M.; Gamo F.; Winzeler E.; Fidock D.; Chookajorn T.; Lee M. Generation of a mutator parasite to drive resistome discovery in Plasmodium falciparum. Nat. Commun. 2022, 14 (1), 3059. 10.1101/2022.08.23.504974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu D.; Pei J. V.; Rosling J. E. O.; Thathy V.; Li D.; Xue Y.; Tanner J. D.; Penington J. S.; Aw Y. T. V.; Aw J. Y. H.; Xu G.; Tripathi A. K.; Gnadig N. F.; Yeo T.; Fairhurst K. J.; Stokes B. H.; Murithi J. M.; Kumpornsin K.; Hasemer H.; Dennis A. S. M.; Ridgway M. C.; Schmitt E. K.; Straimer J.; Papenfuss A. T.; Lee M. C. S.; Corry B.; Sinnis P.; Fidock D. A.; van Dooren G. G.; Kirk K.; Lehane A. M. A G358S mutation in the Plasmodium falciparum Na(+) pump PfATP4 confers clinically-relevant resistance to cipargamin. Nat. Commun. 2022, 13 (1), 5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan K.; Ziniel P.; Li H.; Huang X.; Hupalo D.; Gombakomba N.; Guerrero S. M.; Dotrang T.; Lu X.; Caridha D.; Sternberg A. R.; Hughes E.; Sun W.; Bargieri D. Y.; Roepe P. D.; Sciotti R. J.; Wilkerson M. D.; Dalgard C. L.; Tawa G. J.; Wang A. Q.; Xu X.; Zheng W.; Sanderson P. E.; Huang W.; Williamson K. C. Torin 2 Derivative, NCATS-SM3710, Has Potent Multistage Antimalarial Activity through Inhibition of P. falciparum Phosphatidylinositol 4-Kinase (Pf PI4KIIIbeta). ACS Pharmacol. Transl. Sci. 2020, 3 (5), 948–964. 10.1021/acsptsci.0c00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet T.; Le Manach C.; Cabrera D. G.; Younis Y.; Henrich P. P.; Abraham T. S.; Lee M. C. S.; Basak R.; Ghidelli-Disse S.; Lafuente-Monasterio M. J.; Bantscheff M.; Ruecker A.; Blagborough A. M.; Zakutansky S. E.; Zeeman A. M.; White K. L.; Shackleford D. M.; Mannila J.; Morizzi J.; Scheurer C.; Angulo-Barturen I.; Martínez M. S.; Ferrer S.; Sanz L. M.; Gamo F. J.; Reader J.; Botha M.; Dechering K. J.; Sauerwein R. W.; Tungtaeng A.; Vanachayangkul P.; Lim C. S.; Burrows J.; Witty M. J.; Marsh K. C.; Bodenreider C.; Rochford R.; Solapure S. M.; Jiménez-Díaz M. B.; Wittlin S.; Charman S. A.; Donini C.; Campo B.; Birkholtz L. M.; Hanson K. K.; Drewes G.; Kocken C. H. M.; Delves M. J.; Leroy D.; Fidock D. A.; Waterson D.; Street L. J.; Chibale K. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci. Transl. Med. 2017, 9 (387), eaad9735 10.1126/scitranslmed.aad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara C. W.; Lee M. C.; Lim C. S.; Lim S. H.; Roland J.; Nagle A.; Simon O.; Yeung B. K.; Chatterjee A. K.; McCormack S. L.; Manary M. J.; Zeeman A. M.; Dechering K. J.; Kumar T. R.; Henrich P. P.; Gagaring K.; Ibanez M.; Kato N.; Kuhen K. L.; Fischli C.; Rottmann M.; Plouffe D. M.; Bursulaya B.; Meister S.; Rameh L.; Trappe J.; Haasen D.; Timmerman M.; Sauerwein R. W.; Suwanarusk R.; Russell B.; Renia L.; Nosten F.; Tully D. C.; Kocken C. H.; Glynne R. J.; Bodenreider C.; Fidock D. A.; Diagana T. T.; Winzeler E. A. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 2013, 504 (7479), 248–253. 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejdrova I.; Chalupska D.; Kogler M.; Sala M.; Plackova P.; Baumlova A.; Hrebabecky H.; Prochazkova E.; Dejmek M.; Guillon R.; Strunin D.; Weber J.; Lee G.; Birkus G.; Mertlikova-Kaiserova H.; Boura E.; Nencka R. Highly Selective Phosphatidylinositol 4-Kinase IIIbeta Inhibitors and Structural Insight into Their Mode of Action. J. Med. Chem. 2015, 58 (9), 3767–3793. 10.1021/acs.jmedchem.5b00499. [DOI] [PubMed] [Google Scholar]
- Brunschwig C.; Lawrence N.; Taylor D.; Abay E.; Njoroge M.; Basarab G. S.; Le Manach C.; Paquet T.; Cabrera D. G.; Nchinda A. T.; de Kock C.; Wiesner L.; Denti P.; Waterson D.; Blasco B.; Leroy D.; Witty M. J.; Donini C.; Duffy J.; Wittlin S.; White K. L.; Charman S. A.; Jimenez-Diaz M. B.; Angulo-Barturen I.; Herreros E.; Gamo F. J.; Rochford R.; Mancama D.; Coetzer T. L.; van der Watt M. E.; Reader J.; Birkholtz L. M.; Marsh K. C.; Solapure S. M.; Burke J. E.; McPhail J. A.; Vanaerschot M.; Fidock D. A.; Fish P. V.; Siegl P.; Smith D. A.; Wirjanata G.; Noviyanti R.; Price R. N.; Marfurt J.; Silue K. D.; Street L. J.; Chibale K. UCT943, a Next-Generation Plasmodium falciparum PI4K Inhibitor Preclinical Candidate for the Treatment of Malaria. Antimicrob. Agents Chemother. 2018, 62 (9), e00012 10.1128/aac.00012-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Zidek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M.; Anyango S.; Deshpande M.; Nair S.; Natassia C.; Yordanova G.; Yuan D.; Stroe O.; Wood G.; Laydon A.; Zidek A.; Green T.; Tunyasuvunakool K.; Petersen S.; Jumper J.; Clancy E.; Green R.; Vora A.; Lutfi M.; Figurnov M.; Cowie A.; Hobbs N.; Kohli P.; Kleywegt G.; Birney E.; Hassabis D.; Velankar S. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50 (D1), D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendse L. B.; Murithi J. M.; Qahash T.; Pasaje C. F. A.; Godoy L. C.; Dey S.; Gibhard L.; Ghidelli-Disse S.; Drewes G.; Bantscheff M.; Lafuente-Monasterio M. J.; Fienberg S.; Wambua L.; Gachuhi S.; Coertzen D.; van der Watt M.; Reader J.; Aswat A. S.; Erlank E.; Venter N.; Mittal N.; Luth M. R.; Ottilie S.; Winzeler E. A.; Koekemoer L. L.; Birkholtz L. M.; Niles J. C.; Llinas M.; Fidock D. A.; Chibale K. The anticancer human mTOR inhibitor sapanisertib potently inhibits multiple Plasmodium kinases and life cycle stages. Sci. Transl Med. 2022, 14 (667), eabo7219 10.1126/scitranslmed.abo7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delves M. J.; Straschil U.; Ruecker A.; Miguel-Blanco C.; Marques S.; Dufour A. C.; Baum J.; Sinden R. E. Routine in vitro culture of P. falciparum gametocytes to evaluate novel transmission-blocking interventions. Nat. Protoc. 2016, 11 (9), 1668–1680. 10.1038/nprot.2016.096. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Liu R.; Li M.; Liu P. The spleen tyrosine kinase (SYK): A crucial therapeutic target for diverse liver diseases. Heliyon 2022, 8 (12), e12130 10.1016/j.heliyon.2022.e12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyakov L.; Halbert J.; Alam M. M.; Semblat J. P.; Dorin-Semblat D.; Reininger L.; Bottrill A. R.; Mistry S.; Abdi A.; Fennell C.; Holland Z.; Demarta C.; Bouza Y.; Sicard A.; Nivez M. P.; Eschenlauer S.; Lama T.; Thomas D. C.; Sharma P.; Agarwal S.; Kern S.; Pradel G.; Graciotti M.; Tobin A. B.; Doerig C. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2011, 2, 565. 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- McCarthy J. S.; Donini C.; Chalon S.; Woodford J.; Marquart L.; Collins K. A.; Rozenberg F. D.; Fidock D. A.; Cherkaoui-Rbati M. H.; Gobeau N.; Mohrle J. J. A Phase 1, Placebo-controlled, Randomized, Single Ascending Dose Study and a Volunteer Infection Study to Characterize the Safety, Pharmacokinetics, and Antimalarial Activity of the Plasmodium Phosphatidylinositol 4-Kinase Inhibitor MMV390048. Clin. Infect. Dis. 2020, 71 (10), e657–e664. 10.1093/cid/ciaa368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinxadi P.; Donini C.; Johnstone H.; Langdon G.; Wiesner L.; Allen E.; Duparc S.; Chalon S.; McCarthy J. S.; Lorch U.; Chibale K.; Mohrle J.; Barnes K. I. Safety, Tolerability, Pharmacokinetics, and Antimalarial Activity of the Novel Plasmodium Phosphatidylinositol 4-Kinase Inhibitor MMV390048 in Healthy Volunteers. Antimicrob. Agents Chemother. 2020, 64 (4), e01896 10.1128/aac.01896-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimzadeh Z.; Mukherjee A.; Richard D. A map of the subcellular distribution of phosphoinositides in the erythrocytic cycle of the malaria parasite Plasmodium falciparum. Int. J. Parasitol. 2018, 48 (1), 13–25. 10.1016/j.ijpara.2017.08.015. [DOI] [PubMed] [Google Scholar]
- Ebrahimzadeh Z.; Mukherjee A.; Crochetière M.; Sergerie A.; Amiar S.; Thompson L. A.; Gagnon D.; Gaumond D.; Stahelin R. V.; Dacks J. B.; Richard D. A pan-apicomplexan phosphoinositide-binding protein acts in malarial microneme exocytosis. EMBO Rep. 2019, 20 (6), e47102 10.15252/embr.201847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson P. R.; Crabb B. S. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int. J. Parasitol. 2009, 39 (1), 91–96. 10.1016/j.ijpara.2008.09.007. [DOI] [PubMed] [Google Scholar]
- de Koning-Ward T. F.; Gilson P. R.; Boddey J. A.; Rug M.; Smith B. J.; Papenfuss A. T.; Sanders P. R.; Lundie R. J.; Maier A. G.; Cowman A. F.; Crabb B. S. A newly discovered protein export machine in malaria parasites. Nature 2009, 459 (7249), 945–949. 10.1038/nature08104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsworth B.; Crabb B. S.; Gilson P. R. Protein export in malaria parasites: an update. Cell. Microbiol. 2014, 16 (3), 355–363. 10.1111/cmi.12261. [DOI] [PubMed] [Google Scholar]
- Jonsdottir T. K.; Gabriela M.; Crabb B. S.; F. de Koning-Ward T.; Gilson P. R. Defining the Essential Exportome of the Malaria Parasite. Trends Parasitol. 2021, 37 (7), 664–675. 10.1016/j.pt.2021.04.009. [DOI] [PubMed] [Google Scholar]
- Gurnett A. M.; Liberator P. A.; Dulski P. M.; Salowe S. P.; Donald R. G.; Anderson J. W.; Wiltsie J.; Diaz C. A.; Harris G.; Chang B.; Darkin-Rattray S. J.; Nare B.; Crumley T.; Blum P. S.; Misura A. S.; Tamas T.; Sardana M. K.; Yuan J.; Biftu T.; Schmatz D. M. Purification and molecular characterization of cGMP-dependent protein kinase from Apicomplexan parasites. A novel chemotherapeutic target. J. Biol. Chem. 2002, 277 (18), 15913–15922. 10.1074/jbc.M108393200. [DOI] [PubMed] [Google Scholar]
- Fidock D. A.; Wellems T. E. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (20), 10931–10936. 10.1073/pnas.94.20.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambros C.; Vanderberg J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65 (3), 418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- Rivadeneira E. M.; Wasserman M.; Espinal C. T. Separation and concentration of schizonts of Plasmodium falciparum by Percoll gradients. J. Protozool. 1983, 30 (2), 367–370. 10.1111/j.1550-7408.1983.tb02932.x. [DOI] [PubMed] [Google Scholar]
- Makler M. T.; Hinrichs D. J. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop Med. Hyg. 1993, 48 (2), 205–210. 10.4269/ajtmh.1993.48.205. [DOI] [PubMed] [Google Scholar]
- Charnaud S. C.; Jonsdottir T. K.; Sanders P. R.; Bullen H. E.; Dickerman B. K.; Kouskousis B.; Palmer C. S.; Pietrzak H. M.; Laumaea A. E.; Erazo A. B.; McHugh E.; Tilley L.; Crabb B. S.; Gilson P. R. Spatial organization of protein export in malaria parasite blood stages. Traffic 2018, 19 (8), 605–623. 10.1111/tra.12577. [DOI] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7 (11), 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder C.; Chollet J.; Santo-Tomas J.; Scheurer C.; Wittlin S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp. Parasitol. 2007, 115 (3), 296–300. 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.