

Abstract
Effector trogocytosis between malignant cells and tumor-specific cytotoxic T lymphocytes (CTLs) contributes to immune evasion through antigen loss on target cells and fratricide of antigen-experienced CTLs by other CTLs. The mechanisms regulating these events in tumors remain poorly understood. Here we demonstrate that tumor-derived factors (TDFs) stimulated effector trogocytosis and restricted CTLs’ tumoricidal activity and viability in vitro. TDFs robustly altered the CTL’s lipid profile including depletion of 25-hydroxycholesterol (25HC). 25HC inhibited trogocytosis and prevented CTL’s inactivation and fratricide. Mechanistically, TDFs induced ATF3 transcription factor that suppressed the expression of 25HC-regulating gene - cholesterol 25-hydroxylase (CH25H). Stimulation of trogocytosis in the intratumoral CTL by the ATF3-CH25H axis attenuated anti-tumor immunity, stimulated tumor growth, and impeded the efficacy of chimeric antigen receptor (CAR) T cell adoptive therapy. Through use of armored CAR constructs or pharmacologic agents restoring CH25H expression we reversed these phenotypes and increased the efficacy of immunotherapies.
Keywords: trogocytosis, effector trogocytosis, cancer immunotherapy, tumor microenvironment, cytotoxic T lymphocytes, CD8+ T lymphocytes, tumor-derived factors, chimeric antigen receptor, CAR T cells, ATF3, CH25H, hydroxycholesterol, sumoylation inhibitor, TAK981
INTRODUCTION
Intercellular transfer of biomolecules contributes to organization of collective behavior of cells in a multi-cellular organism. This transfer can be mediated by a number of mechanisms including trogocytosis, which plays an important role in the regulation of immunity and other vital biological processes (Rechavi et al., 2009). Trogocytosis is an evolutionarily conserved process of extraction and subsequent transfer of fragments of cell membrane along with associated proteins and other biomolecules between cells that are in a close contact with each other. As a result of this transfer, a gain or loss of function for either acceptor or donor cells (or both) may occur (Dance, 2019; Rechavi et al., 2009).
The functional consequences of trogocytosis in T cells depend on the context and nature of partner cells. For example, in the course of antigen presentation, a priming T-cell receptor-dependent trogocytosis results in T cells to acquire membrane fragments along with the MHC class I and class II proteins and co-stimulatory and other molecules from the antigen-presenting cells. The effector trogocytosis between activated CD8+ T cells (cytotoxic T lymphocytes, CTLs) and target cells expressing specific antigen leads to a transfer of MHC-antigen complexes onto CTLs (reviewed in (Ahmed et al., 2008; Miyake and Karasuyama, 2021; Nakayama et al., 2021)).
Intriguingly, effector trogocytosis between target malignant cells and therapeutic CD8+ T cells expressing the chimeric antigen receptor (CAR) is implicated in decreased clinical benefits of CTLs (Hamieh et al., 2019). Trogocytosis is associated with decreased viability and activity of chimeric antigen-expressing recipient CTLs due to their exhaustion as well as killing by other CAR CTLs in a process called fratricide. Additionally, the loss of antigen on surviving donor malignant cells masks them from subsequent attack by CAR T cells. These intricate and robust mechanisms help malignant cells to withstand CAR T cell adoptive therapies (Hamieh et al., 2019).
Tumors are adept at utilizing evolutionary conserved processes for evading the immune surveillance. However, it is not clear whether the described above mechanisms are also effective in naturally occurring anti-tumor CTLs. Furthermore, how effector trogocytosis is regulated and how this regulation can be altered during tumor growth is not well understood. Here we present evidence for increased extent of trogocytosis in the tumor microenvironment. Our in vitro studies demonstrate that tumor-derived factors (TDFs) stimulate trogocytosis and decrease viability and activity of anti-tumor CTLs by reprogramming the regulatory axis that involves activating transcription factor-3 (ATF3) and cholesterol 25-hydroxylase (CH25H).
ATF3 is an early stress-responsive gene that could be induced by many factors of tumor microenvironment including hypoxia and nutrient deprivation (reviewed in (Hai et al., 2010). ATF3 has been shown to suppress CH25h expression in macrophages (Gold et al., 2012). Our results in vitro suggest that TDFs-induced ATF3 in CTLs downregulates expression of CH25H. The latter catalyzes monooxygenation of cholesterol into 25-hydroxycholesterol (25HC) (Cyster et al., 2014)), which interferes with trogocytosis stimulated by TDFs. Dysregulation of the ATF3-CH25H axis in the intratumoral natural or CAR-expressing CTLs stimulates effector trogocytosis and undermines viability and anti-tumor activity of these CTLs. Conversely, pharmacological, or genetic restoration of CH25H expression in CTLs inhibits trogocytosis, suppresses tumor growth and increases the efficacy of immunotherapies.
RESULTS
Tumor-derived factors (TDFs) downregulate 25-hydroxycholesterol and stimulate trogocytosis between effector CTL and malignant cells
Effector trogocytosis that transfers antigenic complexes to specific CTLs from malignant cells undermines their killing due to antigen loss and CTL fratricide (Hamieh et al., 2019). Mechanisms regulating the extent of effector trogocytosis are not well understood. We sought to determine how the factors present in the tumor microenvironment may affect the extent of trogocytosis (analyzed as in Figure S1A). To this end, CTLs were exposed to tumor-derived factors (TDFs) produced by MC38 colon adenocarcinoma cells and present within the tumor cell-conditioned media (TCM), as well as purified factors such as prostaglandin E2 (PGE2) and vascular endothelial growth factor (VEGF), which are known to contribute to the immune suppressive properties of tumor microenvironment (Rabinovich et al., 2007). As controls, we used analogous treatment with vehicle, or media conditioned by mouse fibroblasts (FCM) or by intestinal epithelial cells (IECM).
We incubated pretreated OT-I CTLs with OVA-expressing MC38 colon adenocarcinoma cells labeled with DiD lipophilic dye. We detected an increased transfer of DiD from MC38OVA cells onto co-incubated OT-I CTLs after their exposure to TCM, PGE2 or VEGF (Figure 1A, S1B). This transfer was likely a result of trogocytosis because it was notably inhibited by the loss of cell-cell contact due to separation of CTLs and MC38OVA cells in a Transwell setting (Figure S1C). Furthermore, treatment of CTLs with inhibitor of actin polymerization Latrunculin A (previously shown to inhibit trogocytosis, (Hamieh et al., 2019)) or use of parental MC38 cells lacking OVA antigen notably inhibited DiD transfer onto CTLs (Figure S1C–D) indicating that this antigen-dependent transfer stimulated by the tumor-derived factors (TDFs) was a result of trogocytosis.
Figure 1. Tumor-derived factors (TDFs) downregulate 25-hydroxycholesterol and stimulate trogocytosis between effector CTL and malignant cells.

A. Analysis of transfer of DiD dye from DID-labeled OVA-expressing MC38 cells onto co-cultured (for 4 hr) OT-I CD8+ T cells pre-treated with VEGF (50ng/ml), PGE2 (10nM), FCM (fibroblast cell medium), IECM (intestinal epithelial cell medium) or TCM (Tumor conditioned medium) for 8 hr as indicated(n=5).
B. KEGG enrichment analysis of altered pathways in CTLs treated with TCM (compared to FCM) for 8 hr (n=3).
C. Volcano plot of differentially expressed genes in CTLs treated as in panel B.
D. Heatmap of differentially expressed genes in CTLs treated as in panel B (n=3).
E. Heatmap of changes in lipid species that occurred in CTLs treated as in panel B (n=4).
F. Levels of 25HC in CTLs treated as in panel B (n=4).
G. qPCR analysis of Ch25h mRNA expression in CTLs treated with Vehicle, media conditioned by primary mouse intestinal epithelial cells (IECM), or fibroblasts (FCM), or MC38 tumor cells (TCM), or PGE2 (10nM), VEGF (50ng/ml) or MC38 tumor-derived extracellular vesicles (TEVs, 20 μg/mL) for 8 hr (n=4–5).
H. qPCR analysis of Ch25h mRNA expression in CD8+ T cells isolated from MC38 tumors or spleens from naïve or MC38 tumor bearing mice (n=5). Na-sp, spleen from naïve mice; TB-sp, spleen from tumor bearing mice.
I. Pairwise comparison of Ch25h mRNA expression in CD8+ T cells isolated from tumor and spleen of individual mice (n=5).
Data are presented as mean±SEM. Statistical analysis was performed using 2-tailed Students’ t test. n.s, not significant.
See also Figure S1.
To delineate the mechanisms underlying TDF-dependent increase in effector trogocytosis, we profiled gene expression in CTLs treated with TCM. This treatment altered expression of genes involved in several key biological processes including cholesterol metabolism pathway, which was notably suppressed (Figure 1B). Cholesterol controls many crucially important properties of the lipid bilayers of biological membranes such as their compressibility, fluidity, intrinsic curvature and propensity for transfer and fusion (reviewed in (Yang et al., 2016)). Given that these characteristics are instrumental for trogocytosis, we further focused on this pathway.
Among the genes involved in cholesterol metabolism, cholesterol 25-hydroxylase (Ch25h) was the top downregulated gene (Figure S1E). Furthermore, expression of Ch25h was notably decreased among all genes affected by TCM (Figure 1C–D). We focused on CH25H because of its enzymatic role in production of 25HC (Cyster et al., 2014). 25HC is known to alter the fluidity of lipid membranes and to inhibit their fusion (Anggakusuma et al., 2015; Ortiz et al., 2019; Zang et al., 2020), which is essential for incorporating malignant cell membrane fragments into CTL during trogocytosis (Dance, 2019; Rechavi et al., 2009). Intriguingly, the lipidomics profiling of CTLs treated with TCM revealed numerous alterations in the content of specific lipid species (Figure 1E) such as a notable decrease in the levels of hydroxysterols including 25HC (Figure 1E–F).
Downregulation of Ch25h levels was seen in CTLs exposed to TDFs in vitro (Figure 1G). Accordingly, the intratumoral CTLs expressed less of Ch25h mRNA in comparison with the splenic CTLs (Figure 1H–I). Similar results were obtained in CTLs isolated from tumors/spleens in other syngeneic mouse tumor models including B16F10 melanoma, MH6499c4 pancreatic ductal adenocarcinoma and Hepa1–6 hepatocellular carcinoma (Figure S1F). These results indicate that downregulation of CH25H in CTLs is induced by TDFs in vitro and occurs in the tumor microenvironment.
CH25H is a pivotal regulator of CTL trogocytosis, survival and activity
We next determined the effects of 25HC on CTLs exposed to TDFs in vitro. Stimulation of trogocytosis (assessed by the transfer of OVA antigen complexed with MHC-I from MC38OVA cells onto OT-I CTLs) by TCM was inhibited by 25HC exposure (Figure S2A, 2A). Importantly, 25HC treatment abolished a decrease in viability elicited by TDFs in the antigen-experienced but not antigen-naive CTL (Figure 2B). Furthermore, this treatment also reversed the suppressive effects of TDFs on ability of CTLs to kill MC38OVA target cells (Figure 2C) highlighting the importance of 25HC in the regulation of CTLs viability and function.
Figure 2. CH25H is a pivotal regulator of CTL trogocytosis, survival and activity.

A. Analysis of transfer of OVA-MHC-I complexes from OVA-expressing MC38 cells onto co-cultured (for 4 hr) OT-I CD8+ T cells pre-treated with FCM or IECM or TCM (with or without 25HC, 4μM or GW3965, 2μM) for 8 hr (n=4).
B. Number of live CD8+ CTLs after co-culture experiment described in Panel A (n=4).
C. Luciferase activity-based analysis of lysis of MC38OVA-luc cells incubated with OT-I CD8+ T cells pre-treated with FCM or IECM or TCM (with or without 25HC, 4μM) for 8hr (n=5).
D. A schematic of experiment for assessing the role of Ch25h in viability of CD8+ T cells in vivo (upper panel). Lower panel depicts the flow cytometry analysis of the percentage of CFSE+ CD8+or CFSE− CD8+ T cells isolated from the MC38 or MC38-OVA tumors 24 hr after inoculation.
E. Quantification of viable CFSE+ or CFSE− CD8+ T cells (left) and of transfer of OVA-MHC-I complexes on indicated CTLs (right) from experiment described in Panel D (n=5).
F. Analysis of number of live WT or Ch25h−/− OT-I CD8+ T cells cultured alone or with MC38OVA target cells for indicated time (n=3–5).
G. Analysis of percentage of apoptotic (Annexin V+) T cells treated as in Panel F (n=3–5).
H. Luciferase-based analysis of lysis of MC38OVA-luc cells incubated with WT or CH25H-null OT-I CD8+ T cells at indicated E:T ratios for 4 hr (n=8–10).
I. Flow cytometry analysis of expression of indicated markers on the surface of WT or CH25H-null CD8+ OT-I T cells after co-culture with MC38OVA target cells or MC38 cells (as an antigen-lacking control) for 8 hr (n=5).
J. Quantification of transfer of OVA-MHC-I complexes from MC38OVA target cells on WT or CH25H-null CD8+ OT-I T cells pre-treated or not with 25HC (4μM for 8 hr) (n=5).
K. Numbers of live CTLs from experiment described in Panel J (n=5).
L. Percentage of apoptotic CTLs from experiment described in Panel J (n=5).
M. Luciferase-based analysis of lysis of MC38OVA-luc cells incubated with WT or CH25H-null OT-I CD8+ T cells pre-treated or not with 25HC (4μM for 8hr) (n=5).
Data are presented as mean±SEM. Statistical analysis was performed using 2-tailed Students’ t test (A and C) or 1-way ANOVA with Tukey’s multiple-comparison test (B, E, F, G, H, I, J, K, L and M). n.s, not significant.
See also Figure S2.
Considering that 25HC can directly inhibit fusion of lipid membranes (Anggakusuma et al., 2015; Ortiz et al., 2019; Zang et al., 2020), these effects may explain ability of 25HC to disrupt trogocytosis. However, additional mechanisms such as contribution of 25HC to the activation of the liver X receptor (LXR, as reviewed in (Cyster et al., 2014)) cannot be ruled out. Indeed, treatment of TCM-exposed CD8+ T cells with 25HC upregulated known LXR-stimulated genes such as Abca1, Abcg1, Lbp and Apoe. Similar results were obtained using a bona fide LXR agonist GW3965 (Figure S2B). Importantly, treatment with LXR agonist GW3965 inhibited the extent of trogocytosis (Figure 2A, S2A). These results demonstrate that 25HC in CTLs can contribute to activation of the LXR-dependent pathway and indirectly implicate LXR in the regulation of trogocytosis.
Given that CH25H is downregulated in the intratumoral CTLs (Figure 1H–I, S1F), we sought to determine the importance of this regulation using cells from OT-I mice lacking Ch25h alleles. Knockout of this gene did not significantly affect activation (Figure S2C), proliferation (not shown), exhaustion or apoptosis (Figure S2D) of CD8+ T cells cultured alone. Thus, we assessed the extent of trogocytosis and CTL viability in vivo. To this end, we injected a mixture of selectively labeled WT and CH25H-deficient OT-I CTLs into either control MC38 or antigen-bearing MC38OVA tumors grown in Rag1−/− mice (Figure 2D, S2E). Whereas no difference was detected a day later in MC38 tumors, a notable decrease in numbers and increase in transfer of MHC-I-OVA was seen in Ch25h−/− CTLs that were exposed to OVA antigen in MC38OVA tumors (Figure 2D–E). These results suggest that CH25H protects CTLs from trogocytosis and decrease in viability upon antigen exposure.
We next aimed to recapitulate these phenotypes in vitro. Indeed, knockout of Ch25h notably increased the ability of CTLs to undergo trogocytosis (Figure S2F). Pre-treatment of either WT or Ch25h-deficient cells with 25HC or LXR agonist GW3965 partially decreased the extent of trogocytosis (Figure S2F). This result indicates that LXR activated by 25HC might be involved in the regulation of trogocytosis but not does not rule out the LXR-independent effects of 25HC.
As expected, increased trogocytosis in Ch25h-null cells was antigen-dependent and sensitive to Latrunculin A (Figure S2G–H–I–J). Moreover, when co-cultured with MC38OVA, the increased trogocytosis in Ch25h-deficient CTL was concomitant with decreased viability (Figure 2F), increased apoptosis (Figure 2G) and attenuated tumoricidal activity (Figure 2H). Furthermore, these cells displayed a reduced CD69 activation marker, increased LAG3 and PD-1 exhaustion markers and increased apoptosis (Figure 2I). These results were further supported by experiments, in which adding 25HC to antigen-experienced CTLs reverted the effects of Ch25h ablation on the extent of trogocytosis (Figure 2J), viability (Figure 2K), apoptosis (Figure 2L) and tumoricidal activity (Figure 2M). Collectively, these results suggest that inactivation of CH25H in the tumor microenvironment stimulates trogocytosis and impedes viability and activity of the antigen-experienced CTLs.
Downregulation of CH25H in CTLs attenuates the immune responses and promote tumor growth
Because of downregulation of CH25H in the CTLs isolated from mouse tumors (Figure 1H, S1F) and decreased viability of antigen-experienced CH25H-deficient CTLs (Figure 2F, G, K, L), it is plausible that partial loss of CH25H in the tumor microenvironment may be linked with decrease in tumor-infiltrating lymphocytes. Thus, we next searched for a putative association between the intratumoral expression of CH25H and CD8A, a gene primarily expressed by tumor infiltrating CTLs. Meta analysis of published data (Liu et al., 2020; Park et al., 2019)) revealed a weak correlation between CH25H levels and tumor infiltration by CD8+ T cells and between expression of CH25H and CD8A in tumors from human patients with melanoma, colon and pancreatic cancers (Figure S3A–B). Importantly, low levels of CH25H expression were correlated with poor prognosis manifested by decreased progression-free and overall survival in patients with melanoma (Figure 3A–B). In all, these results indirectly associate CH25H expression with infiltration of human tumors with CTLs and lesser progression of human cancers.
Figure 3. Downregulation of CH25H in CTLs attenuates the immune responses and promote tumor growth.

A. Association between CH25H expression and progress free survival in human melanoma patients.
B. Association between CH25H expression and overall survival in human melanoma patients.
C. Growth of B16F10 melanoma tumors (inoculated s.c. at 1×106) in Ch25hf/f and Ch25hΔCD8Cre mice (left; n=9–10) and survival analysis for tumor-bearing animals (right; n=7–8).
D. Representative images of B16F10 tumor size (left) and weight (right)at day 15 from experiment described in panel C.
E. Immunofluorescence analysis and its quantification for numbers of CD3+CD8+ T cells in MC38 tumors from Ch25hf/f and Ch25hΔCD8Cre mice (n=7–9). Scale bar: 100 μm.
F. Flow cytometry analysis of numbers and percentage of CD3+CD8+ T cells in MC38 tumors and spleens from tumor bearing Ch25hf/f and Ch25hΔCD8Cre mice (n=5).
G. Flow cytometry analysis of percentage of CD69-expressing CTLs from experiment described in Panel F.
H. Flow cytometry analysis of percentage of PD-1-expressing CTLs from experiment described in Panel F.
I. Flow cytometry analysis of percentage of Annexin V-expressing CTLs from experiment described in Panel F.
Data are presented as mean±SEM. Statistical analysis was performed using 2-tailed Students’ t test (D and E ) or 1-way ANOVA with Tukey’s multiple-comparison test (F, G, H, and I ) or 2-way ANOVA with Sidak’s multiple-comparison test (C) or log-rank (Mantel-Cox) test (A, B, and C ). n.s, not significant.
See also Figure S3.
Driven by these data from human patients with cancer, we sought to determine the importance of expression of CH25H in CTLs for tumor growth. To this end, we crossed Cd8a-Cre and Ch25hf/f mice to generate Ch25hΔCD8 animals (Figure S3C). Whereas no major differences with WT mice was found in spleen or blood of these cells (Figure S3D–E), Ch25hΔCD8 mice supported a notably accelerated growth of B16F10 melanoma tumors (Figure 3C–D). Similar results were obtained in other tumor types including, MC38 (Figure S3F), MH6499c4 (Figure S3G) and Hepa1–6 (Figure S3H). Critically, tumors obtained from Ch25hΔCD8 mice displayed reduced number and a lower frequency of tumor infiltrating CD8+ T cells (Figure 3E–F and S3I–J). Of note, these CH25H-deficient intratumoral (but not splenic) CTLs expressed less CD69 (Figure 3G and S3I–J), more PD-1 (Figure 3H and S3I–J) and exhibited a greater rate of apoptosis (Figure 3I and S3I–J). These results indicate that the expression of CH25H in the intratumoral CTLs plays an important role in their activity and viability. Conversely, inactivation of CH25H in CTLs attenuates the anti-tumor immune responses and promotes tumor growth.
ATF3 regulates effector trogocytosis, activity and viability of CTLs, and tumor growth in a CH25H-dependent manner.
We next sought to delineate the mechanisms underlying downregulation of CH25H in the intratumoral CTLs. Activating transcription factor-3 (ATF3) is a key negative regulator of CH25H expression in macrophages (Gold et al., 2012; Labzin et al., 2015). Chromatin immunoprecipitation assay demonstrated that ATF3 was present on the promoter of the Ch25h gene in mouse OT-I CD8+ T cells and treatment of these cells with TDFs increased this binding (Figure 4A). Importantly, ATF3 is well known as an early stress-responsive gene that could be induced by many factors of tumor microenvironment (reviewed in (Hai et al., 2010). We noted an induction of ATF3 in CTLs treated with media conditioned by MC38 colon adenocarcinoma cells but not by normal intestinal cells or fibroblasts (Figure 1D, S4A). Furthermore, specific TDFs such as PGE2, VEGF and tumor-derived extracellular vesicles were all capable of increasing Atf3 expression in CD8+ T cells in vitro (Figure S4A).
Figure 4. ATF3 regulates effector trogocytosis, activity and viability of CTLs, and tumor growth in a CH25H-dependent manner.
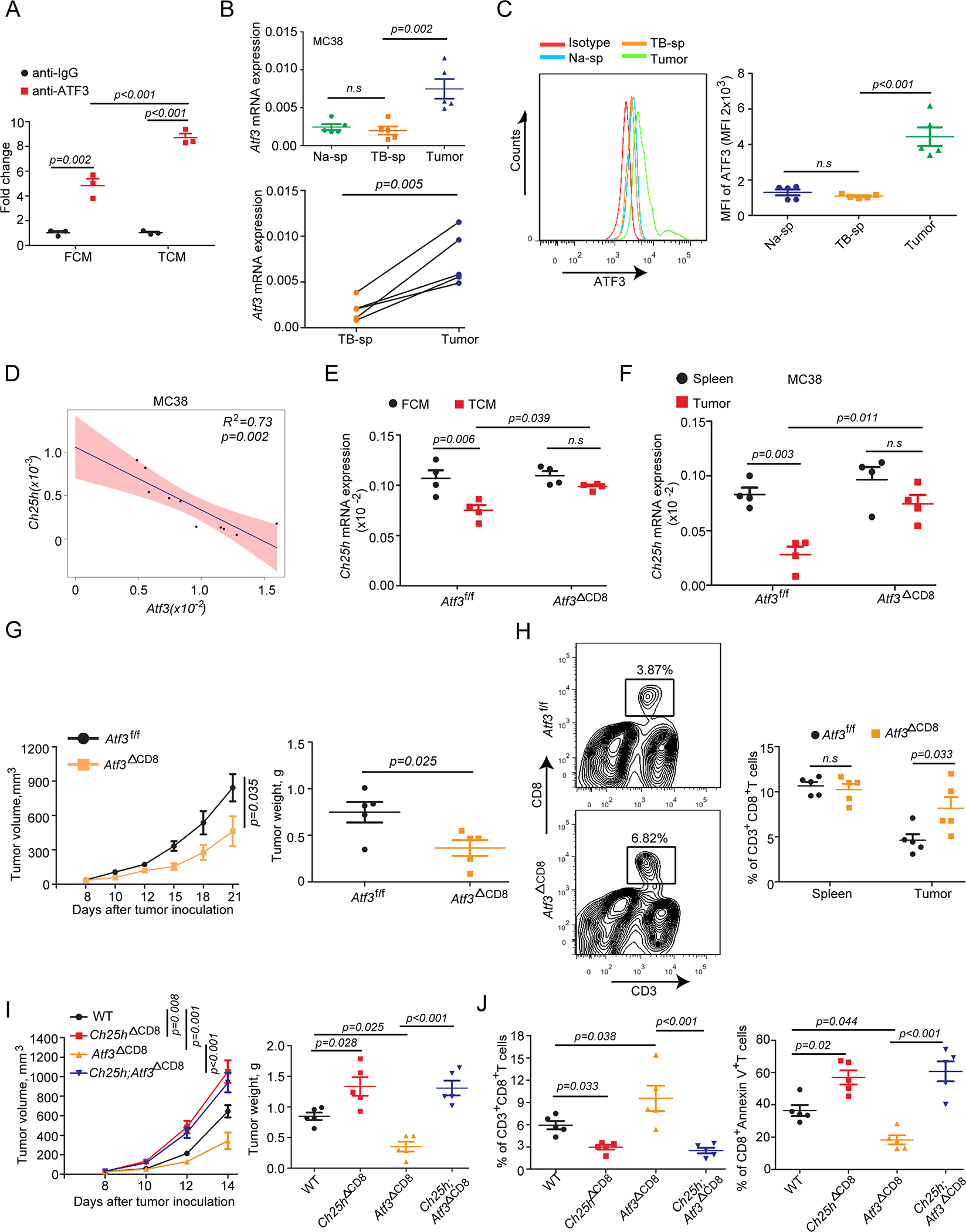
A. ChIP-qPCR analysis of ATF3 binding to Ch25h promoter in OT-I CD8+ T cells treated or not with MC38 TCM for 12 hr (n=3).
B. Overall (upper) and pairwise (below) qPCR analysis of Atf3 mRNA levels in CD8+ T cells from MC38 tumor tissue and spleen of naïve or tumor bearing mice (n=5).
C. Flow cytometry analysis of ATF3 protein level in CD8+ T cells from MC38 tumor tissue and spleen of naïve or tumor bearing mice (n=5).
D. Linear regression analysis of Atf3 and Ch25h mRNA expression in CD8+ T cells isolated from MC38 tumors (n=10).
E. Analysis of Ch25h expression in Atf3f/f or Atf3ΔCD8 CD8+ T cells treated in vitro with TCM or FCM for 8 hr (n=5).
F. Analysis of Ch25h expression in CD8+T cells isolated from MC38 tumor that grew in Atf3f/f or Atf3ΔCD8 mice (n=5).
G. Volume and weight of MC38 s.c. tumors that grew in Atf3f/f or Atf3ΔCD8 mice (n=5).
H. Flow cytometry assay of percentage of CD3+CD8+ T cells in MC38 tumors from Atf3f/f or Atf3ΔCD8 mice (n=5).
I. Volume and weight of B16F10 tumors that grew in WT, Ch25hΔCD8, Atf3ΔCD8 or Ch25h;Atf3ΔCD8 mice (n=5).
J. Percentage of CD3+CD8+ T cells and Annexin V-positive CD3+CD8+ T cells from experiment described in panel I (n=5).
Data are presented as mean±SEM. Statistical analysis was performed using 2-tailed Students’ t test (B, C and G), linear regression analysis (D), 1-way ANOVA with Tukey’s multiple-comparison test (A, E, F, H, I and J) or 2-way ANOVA with Sidak’s multiple-comparison test (G and I). n.s, not significant.
See also Figure S4.
Likewise, analysis of CD8+ T cells isolated from MC38 tumors revealed an increase in ATF3 mRNA and protein levels compared to such cells from spleens of either tumor-bearing or naïve mice (Figure 4B–C). ATF3 induction was also noted in the intratumoral CTLs from B16F10, MH6499c4 and Hepa1–6 tumors (Figure S4B). Importantly, in these models, the expression of Atf3 in the intratumoral CTLs often negatively correlated with Ch25h levels (Figure 4D, S4C). These results are consistent with a hypothesis wherein ATF3 induced in the intratumoral CTLs by TDFs plays an important role in downregulation of CH25H.
To definitively test this possibility, we have generated Atf3ΔCD8 mice, which lacked ATF3 in CD8+ cells but did not differ from Atf3f/f controls in the composition of immune cells in spleen or blood (Figure S4D). Nevertheless, CTL from Atf3ΔCD8 mice did not downregulate CH25H in response to in vitro treatment with TDFs (Figure 4E). Importantly, downregulation of CH25H in the CTLs isolated from MC38 tumors was less pronounced when these tumors grew in Atf3ΔCD8 mice (Figure 4F). Similar results were obtained in other tumor types (Figure S4E) suggesting that ATF3 contributes to decreased expression of CH25H in the intratumoral CTLs.
Examination of tumor volumes from these experiments revealed a significantly slower growth of MC38 tumors in Atf3ΔCD8 mice compared to Atf3f/f controls (Figure 4G). Furthermore, genetic ablation of ATF3 in CTLs increased frequency of these cells within tumors (Figure 4H) and decreased expression of markers of exhaustion and apoptosis on these cells in the tumor microenvironment (Figure S4F). Similar results were found in CTL from B16F10 tumors (Figure 4I–J, S4G–H–I) but not from spleens of B16F10 bearing mice (Figure S4J). Importantly, all phenotypes associated with ablation of ATF3 in CD8+ T cells were reversed upon concurrent ablation of CH25H (Figure 4I–J, S4G–H–I). These results indicate that ATF3-driven downregulation of CH25H in the intratumoral CTLs decreases their viability, suppresses the anti-tumor immune responses, and stimulates tumor growth.
ATF3 and CH25H control trogocytosis and activity of CAR T cells
To directly examine the role of the ATF3-Ch25h regulatory axis in effector trogocytosis as well as viability and function of CTLs, we generated WT, Ch25hΔCD8, Atf3ΔCD8, and Ch25h; Atf3ΔCD8 murine T cells stably transduced with retrovirus for expression of CAR that target human CD19 (hCD19, Figure S5A). Upon incubation with target B16F10-hCD19 cells, we observed trogocytosis manifested by a transfer of hCD19 onto CAR T cells. Similar to results obtained in the OVA-OT-I model (Figure 2), knockout of CH25H stimulated trogocytosis (Figure 5A). Furthermore, under these conditions, CAR T cells lacking CH25H exhibited greater signs of exhaustion (Figure 5B) and apoptosis (Figure 5C) and were less effective in lysis of target B16F10-hCD19 cells (Figure 5D). Importantly, effector trogocytosis was inhibited in ATF3-deficient CAR T cells, whereas these cells were less exhausted/apoptotic (Figure 5B–C) and more effective in killing antigen-expressing malignant cells (Figure 5D). Importantly, all these phenotypes associated with ATF3 deletion were largely reverted by genetic ablation of CH25H (Figure 5A–D).
Figure 5. ATF3 and CH25H control trogocytosis and activity of CAR T cells.

A. Flow cytometry analysis of transfer of CD19 from B16F10-hCD19 target cells onto anti-CD19 CAR T cells (co-incubated for 4 hr) produced from WT, Ch25hΔCD8, Atf3ΔCD8 or Atf3;Ch25h ΔCD8 splenic T cells (n=5).
B. Flow cytometry analysis of the percentage of CD8+Tim3+ anti-CD19-CAR T cells processed as in Panel A (n=5).
C. Flow cytometry analysis of the percentage of CD8+Annexin V+ anti-CD19-CAR T cells processed as in Panel A (n=5).
D. Luciferase-based analysis of lysis of B16F10-hCD19-luc cells incubated with indicated anti-CD19 CAR T cells in vitro (n=5).
E. Percentage of CD19+ CD8+ T cells isolated from B16F10-hCD19 tumors that grew in Rag1−/− mice treated with WT, Ch25hΔCD8, Atf3ΔCD8 or Ch25h;Atf3ΔCD8 anti-CD19 CAR T cells (n=5).
F. Percentage of CD8+ PD-1+ T cells and CD8+Tim3+ T cells isolated from B16F10-hCD19 tumors treated as in Panel E (n=5).
G. Percentage and absolute number of CD8+ anti-CD19 CAR+ T cells infiltrated into B16F10-hCD19 tumors treated as in Panel E (n=5).
H. Volume and weight of B16F10-hCD19 tumor that grew in Rag1−/− mice treated with indicated anti-CD19 CAR T cells (n=5).
I. Survival analysis for mice described in panel H (n=5).
Data are presented as mean±SEM. Statistical analysis was performed using 1-way ANOVA with Tukey’s multiple-comparison test (A, B, C, D, E, F, G and H) or 2-way ANOVA with Sidak’s multiple-comparison test (H) or Kaplan-Meier test (I). n.s, not significant.
See also Figure S5.
These results suggest a critical role for the ATF3-CH25H axis in the regulation of the extent of trogocytosis, viability and activity of CAR T cells in vitro. We further corroborated these results by adoptive transfer of CAR WT, Ch25hΔCD8, Atf3ΔCD8, or Ch25h; Atf3ΔCD8 T cells into Rag1−/− mice bearing B16F10-hCD19 tumors. We then isolated CAR T cells from tumors and analyzed their status. A greater extent of trogocytosis (manifested by frequency of hCD19+ CD8+ T cells) and increase in exhaustion markers were noted in CH25H-null CAR T cells (Figure 5E–F and S5B–C). Whereas the opposite trends were seen in the intratumoral Atf3ΔCD8 CAR T cells, the effects of ATF3 deletion were abolished by additional knockout of CH25H (Figure 5E–F and S5B–C) suggesting an important role for ATF3-CH25H in regulating CAR T viability/activity in vivo.
Indeed, evaluation of TILs in these B16F10-hCD19 tumors revealed a greater infiltration of tumors by ATF3-deficient CAR T cells whereas knockout of CH25H decreased the numbers and frequencies of intratumoral CTLs regardless of the status of ATF3 (Figure 5G). Accordingly, CAR T cells from Atf3ΔCD8 mice exhibited a greater anti-tumor activity as manifested by changes in tumor volume and weight (Figure 5H) and animal survival (Figure 5I). Importantly, knockout of CH25H attenuated the efficacy of CAR T cell therapy in any genetic context tested here (Figure 5H–I). These results characterize the ATF3-CH25H axis as a pivotal regulator of effector trogocytosis and associated changes in CAR T cell viability and efficacy of their anti-tumor effects.
TAK981 sumoylation inhibitor upregulates CH25H, inhibits trogocytosis and augments CAR T cells viability and anti-tumor activities
We next sought a pharmacologic approach to suppressing the effector trogocytosis in order to increase viability and activity of the intratumoral CTLs. Although 25HC worked well in vitro (Figure 2), its suboptimal bioavailability (Li et al., 2017) prompted us to search for a medically relevant small molecule agent that could interfere with ATF3-driven downregulation of CH25H and restore CH25H levels in the intratumoral CTLs. After extensive analysis of literature, we chose to use an inhibitor of the sumoylation pathway because of the following factors: (i) ATF3 directly suppresses transcription of CH25H (Gold et al., 2012) and transcriptional repression is often associated with protein sumoylation (Lyst and Stancheva, 2007); (ii) Furthermore, de-sumoylation of some transcriptional regulators is required for induction of ATF3 (Barysch et al., 2021); (iii) Importantly, sumoylation of ATF3 itself has been implicated in its transcriptional suppressive activities (Wang et al., 2013); and (iv) in addition, activation of innate and adaptive immunity was observed upon genetic (Decque et al., 2016) or pharmacologic (Lightcap et al., 2021) disruption of the sumoylation pathway.
Intriguingly, exposure of activated OT-I CD8+ T cells with TDFs increased the levels of both unmodified and sumoylated ATF3 (Figure S6A). We then tested the ability of selective and potent sumoylation inhibitor TAK981 (Lightcap et al., 2021) to restore the CH25H expression in TDFs-exposed CTLs. Pre-treatment of TDFs-treated CD8+ T cells with TAK981 in vitro partially attenuated the increase in ATF3 mRNA expression and in levels of sumoylated and unmodified ATF3 protein (Figure S6A–B). Furthermore, TAK981 robustly prevented downregulation of CH25H (Figure 6A). We noted that, while TDFs stimulated trogocytosis (assessed by a transfer of the MHC-I-OVA complexes from MC38OVA target tumor cells onto OT-I CTLs), adding TAK981 prevented this stimulation. Importantly, this effect of TAK981 was seen in WT OT-I but not in Ch25h−/− OT-I cells (Figure 6B) indicating that this sumoylation inhibitor can suppress trogocytosis by maintaining the levels of CH25H.
Figure 6. TAK981 sumoylation inhibitor upregulates CH25H, inhibits trogocytosis and augments CAR T viability and anti-tumor activities.

A. qPCR analysis of Ch25h expression in CTLs treated with FCM or TCM in the presence or absence of TAK981 (0.1μM for 8 hr) (n=4).
B. Transfer of OVA-MHC-I complexes from MC38OVA cells onto WT or Ch25h−/− OT-I CTLs pre-treated or not with FCM or TCM (in presence or absence of 0.1μM TAK981) (n=4).
C. Expression of CD69 by the intratumoral CTLs isolated from MC38 tumors that grew in Ch25hf/f or Ch25hΔCD8 mice treated with Vehicle (PBS), anti-PD1 antibody (i.p, 5mg/kg every 4 days) and/or TAK981 (i.v, 15mg/kg once a week) as indicated (n=5).
D. Expression of PD-1 and Annexin V by the intratumoral CTLs from experiments described in Panel C (n=5).
E. Volume of MC38 tumors from experiments described in Panel C (n=5).
F. Weight of MC38 tumors from experiments described in Panel C (n=5).
G. Survival analysis of animals from experiments described in Panel C. Mice were euthanized when tumor volume reached ~2000 mm3 (n=5).
Data are presented as mean±SEM. Statistical analysis was performed using 1-way ANOVA with Tukey’s multiple-comparison test (A, B, C, D, and F) or 2-way ANOVA with Sidak’s multiple-comparison test (E) or Kaplan-Meier test (G). n.s, not significant.
See also Figure S6.
Given that effector trogocytosis of CTLs has been associated with decreased viability and functionality, we examined the effects of TAK981 along with TDFs on these parameters. These treatments did not affect numbers of OT-I CTLs which were not exposed to OVA antigen. However, we noted a decrease in the numbers of WT OT-I CTLs upon their incubation with TCM in the presence of target MC38OVA tumor cells (Figure S6C). Under these conditions, treatment with TAK981 restored the numbers of WT OT-I cells in the culture. Importantly, compared to WT cells, the numbers of co-cultured CH25H-deficient OT-I CTLs were notably lower, and they did not increase in respond to TAK981 treatment (Figure S6C). Likewise, TAK981 restored the tumoricidal activity of WT (but not Ch25h-null) OT-I CTLs against target MC38OVA tumor cells otherwise suppressed by TCM (Figure S6D). These results further suggest that TAK981 prevents trogocytosis and associated decrease in viability and activity of CTLs in an CH25H-dependent manner.
Whereas monotherapy of MC38 tumors in WT mice with TAK981 elicited a modest therapeutic effect, the combination of this agent with anti-PD1 checkpoint inhibitor notably suppressed tumor growth (Lightcap et al., 2021). In Ch25hf/f mice under the same conditions, treatment with TAK981 alone or combined with anti-PD1 increased expression of CD69 and decreased levels of PD-1 and annexin-V on the intratumoral CTLs (Figure 6C–D). We also observed that TAK981+anti-PD1 combination significantly decreased tumor volume and weight and prolonged animal survival (Figure 6E–F–G). Importantly, analysis of intratumoral CD8+ T cells reveal that TAK981 treatment downregulated ATF3 and upregulated CH25H in vivo (Figure S6E). Furthermore, consistent with ability of TAK981 to inhibit trogocytosis (Figure 6A) and apoptosis (Figure 6D), inclusion of this agent into therapeutic regimens also increased infiltration of MC38 tumors with CTLs (Figure S6F). Importantly, these effects were limited to the intratumoral CTLs and were not observed in the spleens from the same animals (Figure S6G).
Given that protein sumoylation affects numerous biologically important processes (Geiss-Friedlander and Melchior, 2007), these phenotypes cannot be attributed entirely to the effects of TAK981 on the levels of CH25H and on the extent of effector trogocytosis. However, ablation of CH25H in CD8+ T cells rendered these cells and their host mice much less sensitive to effects of TAK981, anti-PD1 and their combination (Figure 6C–D–E–F–G). These results are consistent with our hypothesis that, albeit not essential, the prevention of CH25H downregulation in the intratumoral CTLs represents an important mechanism of action for anti-cancer therapies that involve TAK981.
Armored CAR designed to re-express CH25H inhibit trogocytosis and increase therapeutic efficacy
To counteract downregulation of CH25H in the intratumoral CTLs, we re-designed previously described anti-MESO and anti-CD19 CARs (Milone et al., 2009) to enable the co-expression of CH25H (Figure 7A). The ability of mouse CTLs expressing conventional anti-MESO CAR T to kill target EM-Meso-GFP-Luc cells (Figure S7A) was notably attenuated by knockout of CH25H but was restored with treatment with 25HC in vitro (Figure S7B). Experiments comparing CTLs harboring either conventional CAR or CAR armored with CH25H (Figure S7C) revealed that re-expression of CH25H notably inhibited the extent of trogocytosis (Figure 7B), and increased viability (Figure 7C) and tumoricidal activity (Figure 7D) of CAR T cells. Accordingly, treatment of NSG mice harboring EM-Meso-GFP-Luc tumors (Figure 7E) with CTLs harboring conventional or CH25H-expressing anti-MESO CAR revealed that expression of CH25H significantly increased the efficacy of this adoptive cell therapy as seen from assessment of tumor volume and weight and animal survival (Figure 7F and S7D). Under these conditions, T cells harboring CH25H-expressing armored CAR displayed increased numbers in tumors and blood compared to control CAR T cells (Figure 7G and S7E).
Figure 7. CARs designed to re-express CH25H inhibit trogocytosis and increase therapeutic efficacy.

A. Design of anti-Meso-CAR, anti-Meso-Ch25h CAR, anti-CD19 CAR and anti-CD19-Ch25h CAR constructs.
B. Transfer of MESO from EM-Meso-GFP-Luc cells onto WT or CH25H-null anti-MESO or anti-MESO-Ch25h CAR T cells co-cultured for 4 hr (n=5).
C. Numbers of live WT or CH25H-null anti-MESO or anti-MESO-Ch25h CAR T cells after co-culture with EM-Meso-GFP-Luc target cells for 8 hr (n=5).
D. Luciferase-based analysis of killing of EM-Meso-GFP-Luc target cells by WT or CH25H-null anti-MESO or anti-MESO-Ch25h CAR T cells (n=5).
E. A schematic of experiment for comparing the efficacy of anti-Meso-CAR and anti-MESO-Ch25h T cells in vivo.
F. Volume of EM-Meso-GFP-Luc tumors (inoculated s.c. at 1×106) in NSG mice treated with vehicle, Meso-CAR T cells or Meso-Ch25h CAR T cells as indicated in Panel E (n=6). Kaplan-Meier survival analysis of survival of tumor–bearing mice (euthanized when tumors reached 1000 mm3) is shown on the right (n=7).
G. Quantification of percentage (left) and absolute numbers (right) of CD3+CD8+ T cells (i.e. CAR T cells) in EM-Meso-GFP-Luc tumors (upper panels) and blood (bottom panels) from NSG mice treated as in Panel E (n=5).
H. Volume of B16F10-hCD19 tumors (inoculated s.c. at 0.3×106) growing in Rag1−/− mice treated with vehicle or indicated CAR T cells generated from WT or Ch25h−/− splenocytes using anti-CD19 CAR or anti-CD19-Ch25h CAR constructs as indicated (upper panel). Kaplan-Meier survival analysis of survival of tumor–bearing mice (euthanized when tumors reached 1000 mm3) is shown on the bottom (n=5).
I. A schematic of experiment for comparing the efficacy of human anti-CD19 and anti-CD19-Ch25h CAR T cells in the model of NALM6 acute lymphoblastic leukemia in NSG mice.
J. Numbers of indicated CAR T cells in blood of NSG mice at day 17, 24 and 31 after injection of NALM6 leukemic cells (n=6).
K. Numbers of NALM6 cell in blood of NSG mice at day 17, 24 and 31 after injection of NALM6 leukemic cells (n=5–6).
L. Survival analysis of NALM6 leukemia cells-bearing mice treated with PBS or indicated CAR T cells (n=5).
Data are presented as mean±SEM. Statistical analysis was performed using 1-way ANOVA with Tukey’s multiple-comparison test (B, C and D) or 2-way ANOVA with Sidak’s multiple-comparison test (F, H and L) or Kaplan-Meier test (F, H and L) or 2-tailed Students’ t test (G, J and K). n.s, not significant.
See also Figure S7.
To corroborate these data, we used another set of CARs designed against human CD19 (Figure 7A, S7F–G). When tested in vitro, CAR T cells that co-express CH25H exhibited a greater killing activity (Figure S7H), and decreased trogocytosis manifested by transfer of antigen onto CTLs (Figure S7I) or by disappearance of antigen from target tumor cells (Figure S7J). CH25H-expressing CAR T cells also displayed lesser exhaustion (Figure S7K), and improved viability (Figure S7L). Furthermore, when tested in vivo, CAR T cells expressing CH25H demonstrated a greater therapeutic efficacy against B16F10-hCD19 tumors (Figure 7H, S7M), better infiltrated these tumors (Figure S7N), and expressed lower levels of markers of exhaustion and apoptosis (Figure S7O).
We next tested the effects of co-expressing CH25H on the efficacy of adoptive transfer of human CAR T cells harboring anti-CD19 CAR in the model of NALM6 acute B cell leukemia (Figure 7I). Under these conditions, CAR T cells expressing CH25H better persisted in the host (Figure 7J) and exhibited a greater anti-tumor efficacy as manifested by decreased number of leukemic cells in the blood (Figure 7K) and prolonged animal survival (Figure 7L). Collectively, these data argue that re-expression of CH25H in adoptively transferred CAR T cells protects them from trogocytosis and associated exhaustion and death and increases their tumoricidal activity and therapeutic potential.
DISCUSSION
Data presented herein reveal that activation of ATF3 and ensuing suppression of CH25H in the tumor microenvironment stimulate the extent of effector trogocytosis between tumor-specific CTLs and malignant cells. These events also limit viability and activity of the intratumoral CTLs thereby stimulating tumor growth and eliciting resistance against adoptive cell transfer therapies including CAR T cell-based regimens. Maintaining expression of CH25H by administration of TAK981 sumoylation inhibitor or co-expressing CH25H along with CAR leads to restricted trogocytosis, increased survival and activity of CTLs and improved efficacy of CAR T cell treatment. It is, however, important to note that TDFs elicited diverse and numerous changes in CTLs’ gene expression profile as well as distinct alterations in levels of lipid species (Figure 1). Given that ATF3 is an early stress-responsive gene that could be induced by many factors of tumor microenvironment (reviewed in (Hai et al., 2010)), it is plausible that many redundant factors besides PGE2 and VEGF are capable of downregulating CH25H and promoting trogocytosis. Furthermore, although our data clearly implicate the ATF3-CH25H regulatory axis in TDFs-stimulated trogocytosis, we should not rule out additional potentially important mechanisms by which trogocytosis can be upregulated in the tumor microenvironment.
Results of studies utilizing TAK981 suggest that protein sumoylation may contribute to both induction of ATF3 and to ATF3-dependent suppression of CH25H. Although sumoylation can occur on ATF3 itself (Wang et al., 2013), it is very likely that disrupted sumoylation of other important regulators contributes to ability of TAK981 to restore CH25H expression and inhibit trogocytosis. Additional studies are warranted to unravel these potentially interesting and important mechanisms that may lead to identification of additional targets and pharmacologic agents that could counteract trogocytosis associated with downregulation of CH25H in CTLs.
Our data further suggest a mechanism by which CH25H inhibits trogocytosis. Although inactivation of CH25H can channel cholesterol monooxygenation toward formation of other biologically and immunologically active oxysterols (such as 27-hydroxycholesterol (Baek et al., 2017), we think that negative regulation of trogocytosis by CH25H is conferred by production of 25HC. Indeed, we observed a decrease in 25HC in CTLs exposed to the TDFs (Figure 1). Furthermore, current studies demonstrate a direct inhibitory effect of 25HC on trogocytosis and associated defects in viability and activity of CTLs (Figure 2). Furthermore, 25HC is known to inhibit lipid membrane fusion (Anggakusuma et al., 2015; Zang et al., 2020), which must happen for incorporating malignant cell membrane fragments into CTL during trogocytosis. These activities of 25HC are also implicated in suppression of uptake of the tumor-derived extracellular vesicles (Lu et al., 2021; Ortiz et al., 2019), which were proposed to contribute to the process of transfer of T cell receptor-containing complexes (Choudhuri et al., 2014). Importantly, 25-hydroxylation along with other modifications generates ligands for the LXR pathway (Cyster et al., 2014). Inhibition of trogocytosis in T cells treated with LXR agonist is suggestive of a role for the LXR pathway – potentially downstream of the ATF3-CH25H axis. Finally, 25HC also acts as a negative regulator of the cell membrane cholesterol (Kandutsch and Chen, 1975); and reduction of cholesterol levels in CD8+ T cells prevents their exhaustion in the tumor microenvironment (Ma et al., 2019).
It is plausible that suppressing CH25H expression and stimulating effector trogocytosis might represent an evolutionarily important mechanism that attenuates the function of immune system. Such mechanisms may plausibly play a protective role to minimize tissue damage during inflammation and T cell-driven autoimmune reactions. If that is true, it would not be the first time, when such protective mechanism is appropriated by a growing tumor to evade the T cell immunity and perhaps even develop a resistance to therapies that involve adoptive transfer of CTLs.
The trogocytosis-driven loss of antigen on target malignant cells that persist following an encounter with CTL (Hamieh et al., 2019) is likely temporary and reversible. Nevertheless, these events provide cancer cells with a window of opportunity for evading future killing through numerous mechanisms including downregulation of MHC-I (Jhunjhunwala et al., 2021), immunoediting and loss of antigen expression (Zheng et al., 2019), production of additional immune-suppressive ligands and substances (e.g. PD-L1, adenosine, tumor-derived extracellular vesicles, (Becker et al., 2013; Gajewski et al., 2006; Whiteside, 2016)) and other mechanisms. Restricting trogocytosis by re-expression of CH25H in CTLs and production of 25HC is expected to close this temporal window and prevent resistance to adoptive cell transfer therapies.
Besides masking malignant cells through decreasing antigen density, trogocytosis also affects CTLs themselves. A number of consequences ensue for an effector CTL that did not manage to kill target malignant cell and, instead, engaged in trogocytosis. Among these consequences are exposure to other CTLs and subsequent fratricide as well as trogocytosis-associated exhaustion described for both CAR T and natural effector CD8+ T cells (Hamieh et al., 2019; Huang et al., 1999). It is likely that both consequences richly contribute to reduced killing capacity of CTLs in vitro and decreased efficacy of these cells in the therapeutic settings. Future studies are warranted to understand how trogocytosis affects the killing activities of a CTL coupled with a target malignant cell. Furthermore, at the present time we cannot entirely rule out that changes in CTLs viability and activity are indirect and perhaps propensity for trogocytosis may reflect other ATF3/CH25H-dependent mechanisms that occur in parallel but not conferred by trogocytosis.
Regardless of the mechanism, current data suggest that restricting trogocytosis via inducing CH25H expression using small molecule agents or CAR constructs engineered to re-express CH25H in CTLs should lessen the time and chance for malignant cells to undergo immunoediting and generate the immune-suppressive tumor microenvironment. This strategy should increase the efficacy of anti-cancer therapies involving adoptive transfer of CTLs such as CAR T cells (as demonstrated in Figure 7).
25HC robustly inhibits trogocytosis in vitro, however, this oxysterol has limited stability and suboptimal bioavailability (Li et al., 2017) complicating its use in vivo. Presented here data reveal that sumoylation inhibitor TAK981 can decrease the extent of trogocytosis (Figure 6). This agent acts to re-activate the anti-tumor immunity (Lightcap et al., 2021) and is currently investigated in several oncology clinical trials (including NCT03648372, NCT04074330, NCT04776018 and NCT04381650). Our current data suggest that the efficacy of TAK981 relates to its ability to prevent downregulation of CH25H expression in the CD8+ T cells (Figure 6). While clinical testing of this agent and its combinations with different types of immune therapies is underway, it is important to stratify these efforts and focus on scenarios where trogocytosis in CTLs acts to promote tumor growth. Otherwise, indiscriminate suppression of trogocytosis may negate the benefits of trogocytosis-mediated cross-priming (Hudrisier et al., 2005; Uzana et al., 2015; Zhang et al., 2008) and of trogocytosis-dependent anti-tumor effects elicited by neutrophils or macrophages (Horner et al., 2007; Stasilojc et al., 2016; Velmurugan et al., 2016). Besides these considerations, protein sumoylation affect numerous important cellular processes and continuous use of sumoylation inhibitors may complicate the efforts to manage oncological disease due to toxicity.
Given these considerations, it would be more attractive to restrict the inhibition of trogocytosis to CTLs. Engineering human CAR T cells that either lack ATF3 (for example, via CRISPR/Cas9-driven knockout) or re-express CH25H would represent such strategy. However, ATF3 is a key transcription factors, which regulates many important genes, and whose effects on tumor growth and progression are complex (Hai et al., 2010). Thus, we preferred to design a third generation armored CAR that co-expresses CH25H to guard against its downregulation in the tumor microenvironment. Our study demonstrates a proof of principle that such approach works. Expression of CH25H within CAR indeed protects CAR T cells from trogocytosis and associated decrease in survival and activity and increases therapeutic efficacy of mouse and human CAR T cells (Figure 7). Future clinical studies will determine the utility of this CAR design in patients with solid tumors and leukemia.
Limitations of Study
Presented here data demonstrate the importance of the ATF3-CH25H regulatory axis in stimulation of the effector trogocytosis in the tumor microenvironment. Consistent with previous reports regarding the consequences of effector trogocytosis in B cell leukemia (Hamieh et al., 2019), our results obtained in several solid tumor models reveal that trogocytosis is associated with decreased viability and impaired activity of CTLs. Accordingly, our results demonstrate the importance of activation of the ATF3-CH25H pathway in restricting the capacity of anti-tumor effects elicited by endogenous and CAR-bearing CTLs. These data support a model where induction of ATF3 and suppression of CH25H in the intratumoral CTLs stimulate trogocytosis and undermine activities and viability of these CTLs. However, additional ATF3-CH25H-dependent and independent mechanisms related to the antigen exposure are likely to contribute to inactivation and demise of the trogocytosis-experienced CTLs in addition to trogocytosis itself. In addition, further studies are required to elucidate putative LXR-dependent and LXR-independent mechanisms by which 25HC restricts the ability of CTLs to undergo trogocytosis. Furthermore, given that isolation of apoptosis-prone trogocytosis-experienced CTLs from human tumors is increasingly challenging, a proper assessment of the extent of effector trogocytosis in human cancers and association of these events with infiltration of CD8+ cytotoxic lymphocytes and with tumor growth and progression requires future technological advances and additional studies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Serge Y. Fuchs (syfuchs@upenn.edu).
Materials availability statement
Mouse lines and constructs generated in this study can be requested from the Lead Contact and obtained under appropriate MTA or/and license from the University of Pennsylvania.
Data and code availability
Bulk RNA-seq data have been deposited at GEO (accession number: GSE190702) and are publicly available as of the date of publication. Accession numbers are also listed in the key resources table. Original western blot images and all raw data used to create the graphs can be found in Data S1 (contains uncropped scans of Western blots and original data related to Figures 1–7 and S1–S4, S6 and S7)
No original code was generated for all data in this study.
Any additional information required to analyze the data reported in this study is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti- mouse CD8-APC/Cy7 | Biolegend | Cat# 100714; RRID: AB_312753 |
| anti-mouse CD69-PE | Biolegend | Cat# 104507; RRID: AB_313110 |
| anti-mouse PD-1-BV605 | BioLegend | Cat# 135219; RRID: AB_11125371 |
| anti-mouse LAG3-PE/Cy7 | BioLegend | Cat# 125226; RRID: AB_2715764 |
| anti-mouse CD366-BV421 | BioLegend | Cat# 134019; RRID: AB_2814028 |
| anti-mouse Annexin V-FITC | BioLegend | Cat# 640906; RRID: AB_2561292 |
| anti-mouse CD45-APC/Cy7 | BioLegend | Cat# 157203; RRID: AB_2876533 |
| anti-mouse CD3-PE | BioLegend | Cat# 100206; RRID: AB_312663 |
| anti-mouse CD8-AF700 | BioLegend | Cat# 100730; RRID: AB_493703 |
| anti-mouse CD69-BV421 | BioLgend | Cat# 104545; RRID: AB_2686969 |
| anti-mouse PD-1-PE/Cy7 | BioLegend | Cat# 109110; RRID: AB_572017 |
| anti-mouse LAG3-BV650 | BioLegend | Cat# 125227; RRID: AB_2687209 |
| Anti-mouse Annexin V-APC | BioLegend | Cat# 640941; RRID: AB_2616657 |
| anti-hCD19-APC | Biolegend | Cat# 392504; RRID: AB_2728416 |
| anti-human CD19 CAR-PE | Acrobiosystems | Cat#FM3-HPY53 |
| anti-human CD8-PE/Cy7 | BioLegend | Cat# 344750; RRID: AB_2687201 |
| anti-mouse CD4-APC/Cy7 | BioLegend | Cat# 100414; RRID: AB_312699 |
| anti-mouse CD19-PE | BioLegend | Cat# 115507; RRID: AB_313642 |
| anti-mouse NK1.1-APC | BioLegend | Cat# 108709; RRID: AB_313396 |
| Biotin anti-mouse CD3 | BioLegend | Cat# 100243; RRID: AB_2563946 |
| Alexa Fluor-594 streptavidin | BioLegend | N/A |
| Alexa Fluor-488 anti-mouse CD8 | BioLegend | Cat# 100723; RRID: AB_389304 |
| Rabbit anti-mouse ATF3 antibody | Cell Signaling | Cat#18665S |
| Anti-mouse SUMO1 | Invitrogen | Cat# 33–2400; RRID: AB_2533109 |
| VeriBlot for IP Detection Reagent HRP | Abcam | N/A |
| Goat anti-mouse HRP-conjugated antibody | Cell Signaling | Cat# 7076S |
| Rat anti-mouse β-tubulin antibody | Cell Signaling | Cat# 2146 |
| PE-anti-mouse H-2Kb bounds to SIINFEKL antibody | BioLegend | Cat# 141603; RRID: AB_10897938 |
| Recombinant Anti-Mesothelin antibody | Abcam | Cat# ab196235 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Healthy human peripheral T cells | Human Immunology Core at the University of Pennsylvania | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Prostaglandin E2 | Sigma | Cat# P0409 |
| DMSO | Sigma | Cat# 20–139 |
| VEGF165 | Sigma | Cat# V5765 |
| 1,1’-dioctadecyl-3,3,3’,3’tetramethylindodicarbocyanine | Biotium | Cat# 60014 |
| Latrunculin A | Sigma | Cat# L5163 |
| 25-hydroxycholesterol | Sigma | Cat# H1015 |
| GW3965 | Sigma | Cat# G6295 |
| Golgi stop | BD | Cat# BDB554724 |
| Transwell Chambers | Millipore | Cat# CLS3422–48EA |
| DNase I | Roche | Cat# 10104159001 |
| Collagenase D | Roche | Cat# 11088882001 |
| Percoll | Sigma | Cat# 17–0891-01 |
| Lymphoprep™ | Stem Cell | Cat# 07851 |
| Recombinant human IL-2 | Sigma | Cat# I0523 |
| Ovalbumin (257–264) chicken | Sigma | Cat# S7951 |
| Anti-PD-1 | Bio X cell | Cat# BE0146 Clone: RMP1–14 |
| CFSE | Biolegend | Cat# 79898 |
| Dynabeads™ Mouse T-Activator CD3/CD28 | Gibco | Cat# 11453D |
| Dynabeads™ Human T-Activator CD3/CD28 | Gibco | Cat# 11132D |
| CountBright™ Absolute Counting Beads | Invitrogen | Cat# C36950 |
| Fetal Bovine Serum | Hyclone | Cat# SH30071.03 |
| High-Capacity RNA-to-cDNA™ Kit | Applied Biosystems | Cat# 4387406 |
| Gibco™ L-Glutamine | Gibco | Cat# 25–030-081 |
| Intracellular Fix & Perm Buffer set | eBioscience | Cat# 88–8824 |
| Lipofectamin2000 | Invitrogen | Cat# 52887 |
| Retronectin | Takara | Cat# T100B |
| RBC lysis buffer | Biolegend | Cat# 420302 |
| Critical commercial assays | ||
| ONE-Glo™ Luciferase Assay System | Promega | Cat# E6120 |
| Power SYBR Green PCR Master Mix | Thermo Fisher | Cat# 4367659 |
| SimpleChIP Plus Enzymatic Chromatin IP Kit | Cell Signaling | Cat# 9005S |
| RNeasy Kits | Qiagen | Cat# 74004 |
| EasySep™ Mouse CD8+ T Cell Isolation Kit | Stem Cell | Cat# 19853 |
| EasySep™ Mouse T Cell Isolation Kit | Stem Cell | Cat# 19851 |
| EasySep™ Mouse CD4+ T Cell Isolation Kit | Stem Cell | Cat# 19852 |
| Deposited data | ||
| Bulk RNA-sequence data | This paper | GEO: GSE190702 |
| Raw data | This paper | Data S1 |
| Experimental models: Cell lines | ||
| Human: Phoenix-ECO | ATCC | Cat# CRL-3214™ |
| Mouse: MC38 | ATCC | N/A |
| Mouse: MC38 OVA | Cancer Cell, 2017, 31: 194–207 | N/A |
| Mouse: MC38-OVA-Luciferase | Nat Cancer 2022 May 30. doi: 10.1038/s43018–022- 00383–0 | N/A |
| Mouse: Hepa1–6 | ATCC | Cat# CRL-1830™ |
| Mouse: B16F10 | ATCC | Cat# CRL-6475™ |
| Mouse: MH6499c4 | Immunity 49, 178–193 | N/A |
| Human: NALM6 | ATCC | Cat# CRL-3273™ |
| Human: EM-Meso-GFP-Luciferase | Oncoimmunology, 2018, VOL. 7, NO. 3, e1395997 | N/A |
| Mouse: NIH 3T3 | ATCC | Cat# CRL-1658™ |
| Experimental models: Organisms/strains | ||
| Mouse: WT C57BL/6 | Jackson Laboratory | Cat# 000664 |
| Mouse: Ch25h−/− C57BL/6 | Jackson Laboratory | Cat# 016263 |
| Mouse: Ch25hΔCD8 C57BL/6 | This paper | N/A |
| Mouse: Atf3ΔCD8 C57BL/6 | This paper | N/A |
| Mouse: OT1 C57BL/6 | Jackson Laboratory | Cat# 003831 |
| Mouse: Rag1−/− C57BL/6J | Jackson Laboratory | Cat# 002216 |
| Mouse: NSG NOD/ShiLtJ | Jackson Laboratory | Cat# 005557 |
| Oligonucleotides | ||
| Primers for q-PCR, see table S1 | This paper | N/A |
| Recombinant DNA (plasmid) | ||
| Lenti-p-EF1α-Meso-scFV | 10.1073/pnas.0813101106. | N/A |
| Lenti-p-EF1α-Meso-scFV-T2A-Ch25h | This paper | N/A |
| Retro-p-CD19-scFV-BBZ-MSGV | 10.1016/j.cell.2021.08.004. | N/A |
| Retro-p-CD19-scFV-BBZ-T2A-Ch25h MSGV | This paper | N/A |
| Software and algorithms | ||
| Prism 9 | GraphPad Prism9 | https://www.graphpad.com/ |
| FlowJo 7.6 version | FlowJo | https://www.flowjo.com/ |
| Image J | Image J | https://imagej.nih.gov/ij/ |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study approvals
Human T cells were previously collected from healthy female and male donors (of age of 18–55 years) by the Human Immunology Core at the University of Pennsylvania under informed consent and could not be directly or indirectly linked to individual human subjects. Use of these cells was approved for by IRB of the University of Pennsylvania. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and were carried out in accordance with the IACUC guidelines. All animals were kept in SPF (specific pathogen-free) facility accordance with American Association for Laboratory Animal Science guidelines. All mice had water ad libitum and were fed with regular food. Littermates from different cages were randomly assigned into different group and were exposed to the same environment.
Animal studies
NSG, WT, Ch25h−/−, Rag1−/−, Cd8 Cre and OT-I mice were purchased from Jackson Laboratory and used for study directly after importing into facility. The conditional Ch25h allele was created by flanking the single exon of the Ch25h gene with the loxP sites inserted into the non-conservative regions (Lu et al., 2021). OT-I WT and OT-I Ch25h−/− mice were generated by crossing OT-I mice with WT or Ch25h−/− mice. Atf3f/f mice were previously described (Wolford et al., 2013) and kindly provided by Dr. Tsonwin Hai (Ohio State University). Ch25hΔCD8 or Atf3ΔCD8 or Ch25h;Atf3ΔCD8 mice were generated by intercrossing Cd8-Cre mice with conditional Ch25hf/f or Atf3f/f mice.
All animal experiments were performed using both male and female littermates (mated in-house) of 6–8 weeks of age. All mice except NSG were in the C57BL/6J background. Mice were housed in a specific pathogen free (SPF) condition (12 hr light/12 hr dark cycle, temperature 20–25°C) and had free access to water and chow (ANIMAL SPECIALTIES AND PROVISIONS, Lab diet 5010). Animal health status was routinely checked by qualified veterinarians.
Cell culture and conditioned media
Human 293T cell and B16F10 cell were purchased from ATCC. NALM6 acute lymphoblastic leukemia (from Michael C. Milone, University of Pennsylvania), MC38 colon adenocarcinoma (from Susan Ostrand-Rosenberg, University of Maryland), B16F10-hCD19 melanoma (B16F10 melanoma stably expressing human CD19 - from Andy Minn, University of Pennsylvania), human EM-Meso-GFP-Luc cell line (from Edmund Moon, University of Pennsylvania), MH6499c4 pancreatic ductal adenocarcinoma (from Ben Stanger, University of Pennsylvania) and Hepa1–6 hepatocellular carcinoma (from Malay Haldar, University of Pennsylvania) were kindly gifted. MC38-OVA and MC-38OVA-luc cells were previously described (Katlinski et al., 2017; Zhang et al., 2022). B16F10-hCD19-luc melanoma cells were generated by stable expression of luciferase in B16F10-hCD19 cells. 293T cell and all cancer cells were cultured at 37°C with 5% CO2 in DMEM including 10% heat-inactivated Fetal Bovine Serum (FBS), 100 U/ml penicillin-streptomycin and L-glutamine. Phoenix packaging cells (from ATCC) were maintained at 37°C with 5% CO2 in RPMI-1640 including 10% heat-inactivated Fetal Bovine Serum (FBS), 50 U/ml penicillin-streptomycin and L-glutamine. Tumor-condition medium (TCM) and NIH3T3 fibroblast-conditioned media (FCM) was prepared as previously reported (Gui et al., 2020). Mouse intestinal epithelial cells were obtained and cultured as previously described (Katlinskaya et al., 2016). Briefly, they were scraped gently using bladder and cultured in Advanced DMEM/F12 medium containing 10mM HEPES, 1XPenicillin-Streptomycin, 2 mM GlutaMAX, 1XN2 supplement, 50 ng/ml mEGF, and 1 mg/ml Rspondin for 12 hr. After that, complementary medium was replaced with serum-free DMEM/F12 medium for 24 hr and the conditioned medium was collected.
METHOD DETAILS
CAR T cells: vector generation, T cell isolation, expansion, and CAR T cells generation
The second generation anti-MESO CAR lentiviral construct, which included mCherry reporter, CD8 hinge, meso scFv, 4–1BB and h28 signaling domains (described in (Milone et al., 2009)). was a gift from Michael C. Milone (University of Pennsylvania). For generation of the anti-Meso-Ch25h CAR plasmid, a cassette encoding a 54 nucleotide self-cleaving peptide (T2A) positioned 5’ of Ch25h was commercially manufactured (Genscript, NJ) with 5’ AvrII and 3’ SalI restriction sites flanking the sequence and inserted into this vector as shown in Figure 7A. Similar strategy was employed to modify the anti-CD19 CAR retrovirus backbone consisting of a CD8 hinge, CD19 scFv, 4–1BB and h28 signaling domains (obtained from Andy J. Minn, University of Pennsylvania). The final constructs were verified using AvrII and SalI restriction digests and Sanger sequencing of the T2A-Ch25h insert using the final anti-MESO-Ch25h or anti-CD19-Ch25h constructs as the templates.
Mouse T cells or CD8+ T cells were isolated from spleen of different genetic mice with T cell or CD8+ T cell isolation kit. Human T cells were isolated from heathy male and female donors with age of 18–55 by the Human Immunology Core at the University of Pennsylvania and used for study immediately after isolation. T cells were stimulated with Dynabeads Mouse T-Activator CD3/CD28 beads or Dynabeads Human T-Activator CD3/CD28 beads for 48 hr and were separated from beads prior to transduction. Indicated retroviruses were added into retronectin (20 μg/ml) pe-coated 24-well plate and spun down at 1000g for 1 hr. Activated T cells were transduced with viruses at MOI=3000 for 48 hr. Transduction efficiency, expression of anti-MESO and anti-CD19 CARs and expression of Ch25h were assessed using flow cytometry and quantitative RT-PCR (qPCR) analysis, respectively.
Cytotoxicity, Viability and Exhaustion assays
OT-I-WT or -Ch25h−/− splenocytes were in vitro stimulated with OVA peptide (0.5 μg/ml for 48 hr) followed by treatment with vehicle or TDFs in the presence or absence of 25HC (4μM) for 8 hr. Then, these differentially treated-CTLs were co-cultured with target MC38OVA cells expressing luciferase at indicated E/T ratio for 4 hr. For CAR T cells setting, anti-MESO or anti-CD19 CAR T cells were generated and treated with Vehicle or 25HC at 4μM for 8 hr. After that, CAR T cells were co-cultured with target EM-Meso-GFP-Luc or B16F10-hCD19-luc cells at indicated E/T ratio for 4 hr followed by the luciferase-based cytotoxicity assay.
For the luciferase-based cytotoxicity assay (described in detail in (Zhang et al., 2022)), target cells alone were set as negative control with spontaneous death RLU and target cells lysed with water were set as positive control with maximal killing RLU. After 4 hr co-culture, 100 μl of luciferase substrate (Promega Bright-Glo) was added to each well containing CTLs and target cells. After 10 min incubation, luminescence was measured using the EnVision (PerkinElmer) plate reader. The percentage of killing was calculated using the formula:
For the viability and exhaustion assay, we co-cultured OT-I or CAR T CTLs with respective target cells for 2, 4, 8 or 16 hr, washed with PBS and analyzed by flow cytometry using appropriate antibodies such as: anti-CD8-APC/Cy7, anti-CD69-PE, anti-PD-1-BV605, anti-LAG3-PE/Cy7, anti-CD366-BV421 and anti-Annexin V-FITC. Following staining, cells were washed with FACS buffer and mixed with counting beads followed by flow cytometry analysis.
Flow cytometry of tumors or spleens, immunofluorescence, immunoprecipitation and immunoblotting.
For immunoprofiling of solid tumors, tumor tissue was dissected and digested with 1 mg/ml Collagenase D (Roche,) plus with 100 μg/ml DNase I (Roche) in RPMI medium with 2% FBS for 1 hr with continuous agitation at 37 °C. Digestion mixture was passed through 100 μm cell strainer to prepare single cell suspension and washed with PBS supplemented with 2mM EDTA and 1% FBS. Single cells were stained with cell surface antibodies: anti-CD45-APC/Cy7, anti-CD3-PE, anti-CD8-AF700, anti- CD69-BV421, anti-PD-1-PE/Cy7, anti-LAG3-BV650, anti-CD336-BV421 and Annexin V-APC. Following staining, cells were washed with PBS and subjected to flow cytometry analysis.
For leukemia model, mouse blood was collected through retro-orbital bleeding and red cells were removed using RBC lysis buffer. After washes with PBS, cells were stained with anti-hCD19, anti-human CD19 CAR, anti-human CD8 on ice for 30 min. Following staining, cells were washed with PBS and subjected to flow cytometry analysis.
For spleen immune profile assay, spleens were ground up and passed through 40 μm cell strainer. Red cells were removed using RBC buffer. After that, single cells were incubated with anti-CD8-AF700, anti-CD4, anti-CD19, anti-NK1.1, anti-CD69, anti-PD-1 and anti-Annexin V antibodies.
For immunofluorescence analyses, the samples were prepared as previous described (Lu et al., 2021) and the Biotin anti-mouse CD3 antibody (1:100), Alexa Fluor-594 streptavidin antibody 1:500), Alexa Fluor-488 anti-mouse (1:100) were used as indicated.
For analysis of ATF3 sumoylation, OT-I WT T cells were isolated and stimulated with OVA peptide (0.5 μg/ml for 48 hr) followed by treatment with Vehicle or TCM in the presence or absence of TAK981 (0.1μM) for 12 hr. Cells were lysed, and 1 mg of pre-cleared protein lysates from each sample were taken into immunoprecipitation using rabbit anti-mouse ATF3 antibody. Immunoprecipitation was carried out as previously described (Huangfu et al., 2012) and resulting precipitates were analyzed by immunoblot using primary antibodies against SUMO1 (3μg/ml), ATF3 and secondary antibodies (VeriBlot for IP Detection Reagent HRP, 1:200), and goat anti-mouse HRP-conjugated antibody (1:5000). Loading levels in initial samples was controlled by direct immunoblot using antibody against β-tubulin (1:1000).
Flow cytometry analysis of trogocytosis
OT-I and CAR T settings were used to assess the extent of trogocytosis. In OT-I settings, splenocytes isolated from OT-I WT (Ch25h+/+) or OT-I Ch25h−/− mice were stimulated with OVA peptide (0.5 μg/ml for 48 hr) followed by treatment with Vehicle, IECM, PGE2 (10nM), VEGF165 (50 ng/ml) or TCM in the presence or absence of 25HC (4μM) or GW3965 (2μM) for 8 hr as indicated. For DiD labeling, MC38 or MC38OVA cells were stained with 0.3μM DiD (1,1′-dioctadecyl-3,3,3′,3′- tetramethylindodicarbocyanine,) at 37oC for 10 min followed by PBS washes for 3 times. A total of 1×105 treated-CTLs were co-cultured with DiD-labeled target cells in 96-well plates at a 5:1 of ratio for indicated times. After co-culture, cells were washed with PBS and stained with anti-CD8-APC/Cy7, PE-anti-mouse H-2Kb bounds to SIINFEKL antibody on ice for 30 min. Following staining, cells were washed with FACS buffer and subjected to flow cytometry. Trogocytosis was tested by the acquisition of either DiD or OVA-MHC-I complexes by the CD8+ CTLs.
In CAR T cell setting, a total of 1×105 CAR T cells were co-cultured with target cells at a 1:2 ratio for 4 hr. After co-culture, cells were washed with PBS and stained with anti-CD8-APC/Cy7, anti-CD19 CAR and anti-hCD19 on the ice for 30 min. Following staining, cells were washed with FACS buffer and subjected to flow cytometry. Trogocytosis was tested by the acquisition of either MESO or hCD19 antigens by the CAR T cells and the loss of the CD19 antigen on target tumor cells.
For the inhibition of trogocytosis, CTLs or CAR T cells were pre-treated with vehicle of latrunculin A (1μM) at 37°C for 20 min before co-incubation with target cells. In the Transwell setting, parental or DiD-labeled MC38 were seeded into the upper portion of Transwell chambers and T cells were seeded on the bottom these plates.
Quantitative Real-time PCR
Total of RNA of CD8+ T cells which was treated with Vehicle or TCM or isolated from tumor tissue was extracted using RNA isolation kit. Concentration of RNA was measured by nanodrop2000 and the mRNA expression of Ch25h and Atf3 were tested using real-time PCR. Primer sequences are provided in Table S1.
Chromatin immunoprecipitation (ChIP) analysis
OT-I WT T cells were isolated from spleen of OT-I WT mice and stimulated with OVA (0.5μg/ml) for 48 hr followed by treated with FCM or TCM for 12hr. T cells were harvested and the Immunoprecipitation (IP) was conducted following instruction from SimpleChIP Plus Enzymatic Chromatin IP Kit. ATF3 antibody used for the IP was Rabbit anti-mouse ATF3 antibody (1:50). The ChIP-qPCR for the Ch25h promoter used the following primers: forward 5-TAGCAGCCCATGCTGAGACTATGT-3 and reverse primer, 5 TTCTTTAGCAGGGAAAGGGAGGTG-3.
Tumorigenesis studies
For syngeneic subcutaneous tumor model, B16F10 (1×106), MC38 (1×106), MH6499c4 (1×106) and Hepa1–6 (2×106) were s.c inoculated into right flank of mice and tumor size was measured every other day using caliper. Tumor volume was calculated as width x width x length x 0.5 and mice survival were tracked until tumor volume reached ~1000 mm3.
Combination therapy and CAR T therapies
For the therapeutic combination of TAK981 with PD-1, MC38 (1×106) cells were suspended into 100 μl PBS and inoculated into the right flank of Ch25hf/f and Ch25hΔCD8 mice at day 0. TAK981 was dissolved in 20% hydroxypropyl beta-cyclodextrin and formulated as previously described (Lightcap et al., 2021). Vehicle or anti-PD-1 (i.p, 5mg/kg every 4 days) or TAK981 (i.v, 15mg/kg, twice a week) or combination were injected into tumor-bearing mice when the tumor volume reached ~50–80 mm3. Tumor tissues were collected at day 25 for immune profiling analysis. For the mice survival analysis, mice were euthanized when tumor volume reached.
For anti-MESO/anti-MESO-Ch25h CAR T therapy, human EM-Meso-GFP-Luc cells (s.c., 1×106) were inoculated into the right flank of NSG mice at day 0. Human anti-MESO-CAR or anti-MESO-Ch25h CAR T cells (generated as described above) were i.v. injected into tumor bearing mice at dose of 1×106/mouse at days 16, 23, 30 and 37; PBS was used as control. Tumor tissue was collected at day 45 for immune profiling. For the survival analysis, mice were euthanized when tumor volume reached ~1000 mm3.
For anti-CD19/anti-CD19-Ch25h CAR T therapy, B16F10-hCD19 cells (s.c, 0.3×106) were inoculated into the right flank of Rag1−/− mice at day 0. Mouse anti-CD19 CAR or anti-CD19-Ch25h CAR T cells were generated and i.v. injected into tumor bearing mice at dose of 2×106/mouse at days 7 and 13. Tumor tissues were digested at day 19 for immune profiling analysis. For the survival analysis, mice were euthanized when tumor volume reached ~1000 mm3.
For CAR T therapy of acute lymphoblastic leukemia model, GFP+NALM6 cancer cells were inoculated into NSG mice (i.v, 1×106) at day 0. Human anti-CD19 CAR or anti-CD19-Ch25h CAR T cells were injected (1×106) into tumor bearing mice at day 10, and PBS injection was used as the control group. Peripheral blood was collected via retro-orbital bleeding at days 17, 24 and 31 and lymphocytes were used for flow cytometric analysis. For the survival analysis, mice were euthanized when became moribund.
Trogocytosis and viability assay in vivo
MC38 or MC38OVA cells were inoculated into the right flank of Rag1−/− mice (1×106, s.c). WT OT-I and Ch25h−/− OT-I splenocytes were stimulated in vitro with OVA peptide (0.5μg/ml for 48 hr). After stimulation, Ch25h−/− CTLs were stained with CFSE (1nM,) at 37°C for 10 min followed by wash with PBS for 3 times. Following wash, WT CTLs (1×106, CFSE negative) were mixed with CFSE-labeled Ch25h−/− CTLs at a ratio of 1:1 and re-suspended in 50μl PBS. This cell mixture was injected into MC38 or MC38OVA tumors. Twenty-four hr after injection, tumor tissues were digested into single cell suspension; and percentage of CD8+CFSE+, CD8+CFSE−, OVA−MHCI+CFSE+ and OVA−MHC+CFSE− cells were analyzed using flow cytometry.
For analysis of effects of TAK981 on trogocytosis, 1×106 MC38-OVA cells were injected (s.c) into right flank of Rag1−/− mice. 13 days after tumor inoculation, tumor bearing mice received OT1 CD8+ T cells injection (2×107/mouse, intratumorally) and TAK981 injection (15mg/kg, i.v) simultaneously. 24 hr after injection, tumor tissues were harvested and togocytosis and T cells exhaustion were analyzed with flow cytometry.
RNA sequencing analysis
WT OT-I or Ch25h−/− OT-I splenocytes were stimulated with OVA peptide (0.5 μg/ml for 48 hr) followed by treatment with FCM or TCM for 8 hr in vitro. Total RNA was extracted with RNeasy Plus Mini Kit (QIAGEN). These samples were then used for RNA sequencing carried out as previously described (Amorim et al., 2019).The adaptor-trimmed reads were aligned to the mouse genome mm10 by STAR v. 2.7.2d with default parameters. Expression levels for each gene were counted by VERSE v. 0.1.4. Normalization and differential expression (DE) analysis was conducted by DESeq2 v. 1.28.1 using Vehicle-treated group as the control. Significantly DE genes were defined as those with Bonferroni adjusted P-value < 0.01 and log2 (fold change) > 0.58 or < −0.58. Pathway enrichment analysis involves the statistical identification of particular biological function/process categories that are overrepresented in the specified gene collection. Total DE or down-/ up-regulated genes were submitted to the Kyoto Encyclopedia of Genes and Genomes, respectively. Significantly enriched pathways (P-value < 0.05) were determined through gene set enrichment analysis integrated in the R/Bioconductor package cluster Profiler v. 3.14.3. Seven KEGG pathways (‘00100’, ‘00120’, ‘00900’, ‘04975’, ‘04976’, ‘04979’, ‘05417’) were defined as the cholesterol related collection, of which P-values and fold changes were used for downstream heatmap plotting. Data have been submitted to Gene Expression Omnibus (GSE190702).
Metabolomics studies
WT OT-I+ or Ch25h−/− splenocytes were stimulated with OVA peptide (0.5μg/ml) for 48 hr followed by treatment with vehicle or TCM for 8 hr in vitro. These cells then were span down at 250g for 5 min followed by wash with 1ml PBS twice. After this, cell pellet was suspended in 1 mL of ice-cold (−48 °C) 80% (v/v) methanol: water followed by centrifuge the solution at −9°C, 11,500 g for 10 min. Finally, cells pellets were frozen in liquid nitrogen for 30s and stored until analysis. Samples were extracted using a biphasic extraction (Tambellini et al., 2013) and then subjected to lipidomics analysis by spotting on a 24 well PTFE slide and analysis by desorption electrospray ionization (Takats et al., 2004), on a Waters Xevo G2-XS qtof instrument. Pixels from technical replicates were averaged and 8564 features were detected. A targeted analysis was performed for 25HC m/z 385.3465, representing the [M+H-H20]+ ion of hydroxysterols (DeBarber et al., 2008).
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiment described here are the representative of at least three independent experiments (n≥5 mice for each group unless specifically indicated). For in vitro experiments, cells or tissues from each of these animals were processed (at least) in biological triplicates. All data here were shown as average ± S.E.M. Statistical analysis between two groups was conducted with 2-tailed Student t test and multiple comparisons was performed by using One-way ANOVA or two-way ANOVA analysis with Tukey’s multiple-comparison. Tumor growth curve analysis was conducted with Repeated-measure two-way ANOVA (mixed-model) with Tukey’s multiple-comparison. The Kaplan-Meier curves were used to analyze the survival data, and Cox regression was used to compute hazard ratio. No methods were used to determine whether the data met assumptions of the statistical approach. All statistical analysis was performed using GraphPad Prism 9 and the results of statistical analyses for each experiment are clearly indicated in the respective figures and in the Figure Legends. P values < 0.05 were considered significant.
Human databases analysis
We performed Kendall Correlation analysis between CD8A expression, which is used for measuring CD8+ T cell presence in the tumor microenvironment, and CH25H expression in human cancer tissues from GENT2. Weak positive association between CD8A and CH25H was generated using the ggpubr (ggpubr: ‘ggplot2’ Based Publication Ready Plots. R package version 0.4.0.) and ggplot2 (ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York, 2016) packages in the graphical and statistical software R (R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria). Timer 2.0 (Li et al., 2020) was used to analyze the correlation between immune cell infiltration and Ch25h expression in human melanoma. The expression of Ch25h in T cells was analyzed while controlling for age, gender, and disease stage. Those tumors that showed a survival benefit associated with increased Ch25h expression in T cells were then further analyzed for tumor purity and Ch25h expression.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the by the NIH/NCI R01 grants no. CA247803 and R01 CA240814 (to S.Y.F.), and P01 grant P01 CA165997 (to J.A.D., C.K., A.W. and S.Y.F.). We are grateful for an additional support from T32 CA115299 (to N.M.).
We thank the Human Immunology Core at the University of Pennsylvania for providing purified human leukocyte subsets for our research. We thank Clara Malekshahi for technical assistance, Dr. Bang Wang (Karolinska Institute) for help with analysis of RNA sequencing data, Dr. Evgeniy B. Eruslanov (University of Pennsylvania) for valuable advice, and Drs. Michael Milone, Malay Haldar, Ben Stanger, Andy Minn and Edmund K. Moon (University of Pennsylvania), Tsonwin Hai (Ohio State University) and Susan Ostrand-Rosenberg (University of Maryland) for the reagents. We are also grateful to Drs. Andrei Thomas-Tikhonenko (University of Pennsylvania) and Tsonwin Hai (Ohio State University) for critical reading of the manuscript and to Dr. A. Gamero (Temple University) and the members of the Koumenis, Minn, Chow and Fuchs laboratories (University of Pennsylvania) for suggestions and advice.
Footnotes
Declaration of interest:
A.B. and G.S. were employees of and stockholders in Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited producing TAK981, while engaged in the research project. Z.L., N.M. and S.Y.F. are listed as inventors on The University of Pennsylvania’s patent application related to the matter described in this manuscript. Other authors declare no conflict of interest.
REFERENCES
- Ahmed KA, Munegowda MA, Xie Y, and Xiang J (2008). Intercellular trogocytosis plays an important role in modulation of immune responses. Cell Mol Immunol 5, 261–269. 10.1038/cmi.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorim CF, Novais FO, Nguyen BT, Misic AM, Carvalho LP, Carvalho EM, Beiting DP, and Scott P (2019). Variable gene expression and parasite load predict treatment outcome in cutaneous leishmaniasis. Sci Transl Med 11. 10.1126/scitranslmed.aax4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggakusuma Romero-Brey I, Berger C, Colpitts CC, Boldanova T, Engelmann M, Todt D, Perin PM, Behrendt P, Vondran FW, et al. (2015). Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 62, 702–714. 10.1002/hep.27913. [DOI] [PubMed] [Google Scholar]
- Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, Pillai RV, McDowell HB, Thompson JW, Dubois LG, et al. (2017). The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun 8, 864. 10.1038/s41467-017-00910-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barysch SV, Stankovic-Valentin N, Miedema T, Karaca S, Doppel J, Nait Achour T, Vasudeva A, Wolf L, Sticht C, Urlaub H, and Melchior F (2021). Transient deSUMOylation of IRF2BP proteins controls early transcription in EGFR signaling. EMBO Rep 22, e49651. 10.15252/embr.201949651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JC, Andersen MH, Schrama D, and Thor Straten P (2013). Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother 62, 1137–1148. 10.1007/s00262-013-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri K, Llodra J, Roth EW, Tsai J, Gordo S, Wucherpfennig KW, Kam LC, Stokes DL, and Dustin ML (2014). Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 507, 118–123. 10.1038/nature12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, Dang EV, Reboldi A, and Yi T (2014). 25-Hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol 14, 731–743. 10.1038/nri3755. [DOI] [PubMed] [Google Scholar]
- Dance A (2019). Core Concept: Cells nibble one another via the under-appreciated process of trogocytosis. Proc Natl Acad Sci U S A 116, 17608–17610. 10.1073/pnas.1912252116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBarber AE, Lutjohann D, Merkens L, and Steiner RD (2008). Liquid chromatography-tandem mass spectrometry determination of plasma 24S-hydroxycholesterol with chromatographic separation of 25-hydroxycholesterol. Anal Biochem 381, 151–153. 10.1016/j.ab.2008.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decque A, Joffre O, Magalhaes JG, Cossec JC, Blecher-Gonen R, Lapaquette P, Silvin A, Manel N, Joubert PE, Seeler JS, et al. (2016). Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat Immunol 17, 140–149. 10.1038/ni.3342. [DOI] [PubMed] [Google Scholar]
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, and Harlin H (2006). Immune resistance orchestrated by the tumor microenvironment. Immunol Rev 213, 131–145. 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Geiss-Friedlander R, and Melchior F (2007). Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 8, 947–956. 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- Gold ES, Ramsey SA, Sartain MJ, Selinummi J, Podolsky I, Rodriguez DJ, Moritz RL, and Aderem A (2012). ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J Exp Med 209, 807–817. 10.1084/jem.20111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui J, Zahedi F, Ortiz A, Cho C, Katlinski KV, Alicea-Torres K, Li J, Todd L, Zhang H, Beiting DP, et al. (2020). Activation of p38alpha stress-activated protein kinase drives the formation of the pre-metastatic niche in the lungs. Nat Cancer 1, 603–619. 10.1038/s43018-020-0064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Wolford CC, and Chang YS (2010). ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expr 15, 1–11. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, Eyquem J, Zhao Z, Whitlock BM, Miele MM, et al. (2019). CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116. 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner H, Frank C, Dechant C, Repp R, Glennie M, Herrmann M, and Stockmeyer B (2007). Intimate cell conjugate formation and exchange of membrane lipids precede apoptosis induction in target cells during antibody-dependent, granulocyte-mediated cytotoxicity. J Immunol 179, 337–345. 10.4049/jimmunol.179.1.337. [DOI] [PubMed] [Google Scholar]
- Huang JF, Yang Y, Sepulveda H, Shi W, Hwang I, Peterson PA, Jackson MR, Sprent J, and Cai Z (1999). TCR-Mediated internalization of peptide-MHC complexes acquired by T cells. Science 286, 952–954. 10.1126/science.286.5441.952. [DOI] [PubMed] [Google Scholar]
- Huangfu WC, Qian J, Liu C, Liu J, Lokshin AE, Baker DP, Rui H, and Fuchs SY (2012). Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 31, 161–172. 10.1038/onc.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudrisier D, Riond J, Garidou L, Duthoit C, and Joly E (2005). T cell activation correlates with an increased proportion of antigen among the materials acquired from target cells. Eur J Immunol 35, 2284–2294. 10.1002/eji.200526266. [DOI] [PubMed] [Google Scholar]
- Jhunjhunwala S, Hammer C, and Delamarre L (2021). Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 10.1038/s41568-021-00339-z. [DOI] [PubMed] [Google Scholar]
- Kandutsch AA, and Chen HW (1975). Regulation of sterol synthesis in cultured cells by oxygenated derivatives of cholesterol. J Cell Physiol 85, 415–424. 10.1002/jcp.1040850408. [DOI] [PubMed] [Google Scholar]
- Katlinskaya YV, Katlinski KV, Lasri A, Li N, Beiting DP, Durham AC, Yang T, Pikarsky E, Lengner CJ, Johnson FB, et al. (2016). Type I Interferons Control Proliferation and Function of the Intestinal Epithelium. Mol Cell Biol 36, 1124–1135. 10.1128/MCB.00988-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, et al. (2017). Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 31, 194–207. 10.1016/j.ccell.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labzin LI, Schmidt SV, Masters SL, Beyer M, Krebs W, Klee K, Stahl R, Lutjohann D, Schultze JL, Latz E, and De Nardo D (2015). ATF3 Is a Key Regulator of Macrophage IFN Responses. J Immunol 195, 4446–4455. 10.4049/jimmunol.1500204. [DOI] [PubMed] [Google Scholar]
- Li C, Deng YQ, Wang S, Ma F, Aliyari R, Huang XY, Zhang NN, Watanabe M, Dong HL, Liu P, et al. (2017). 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 46, 446–456. 10.1016/j.immuni.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, and Liu XS (2020). TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 48, W509–W514. 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightcap ES, Yu P, Grossman S, Song K, Khattar M, Xega K, He X, Gavin JM, Imaichi H, Garnsey JJ, et al. (2021). A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci Transl Med 13, eaba7791. 10.1126/scitranslmed.aba7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Bao X, Hu M, Chang H, Jiao M, Cheng J, Xie L, Huang Q, Li F, and Li CY (2020). Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 588, 693–698. 10.1038/s41586-020-2911-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Ortiz A, Verginadis II, Peck AR, Zahedi F, Cho C, Yu P, DeRita RM, Zhang H, Kubanoff R, et al. (2021). Regulation of intercellular biomolecule transfer-driven tumor angiogenesis and responses to anticancer therapies. J Clin Invest 131. 10.1172/JCI144225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, and Stancheva I (2007). A role for SUMO modification in transcriptional repression and activation. Biochem Soc Trans 35, 1389–1392. 10.1042/BST0351389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, Wang Q, Yang M, Kalady MF, Qian J, et al. (2019). Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 30, 143–156 e145. 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. (2009). Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17, 1453–1464. 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, and Karasuyama H (2021). The Role of Trogocytosis in the Modulation of Immune Cell Functions. Cells 10. 10.3390/cells10051255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Hori A, Toyoura S, and Yamaguchi SI (2021). Shaping of T Cell Functions by Trogocytosis. Cells 10. 10.3390/cells10051155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, Carbone CJ, Yu Q, Katlinski KV, Katlinskaya YV, et al. (2019). An Interferon-Driven Oxysterol-Based Defense against Tumor-Derived Extracellular Vesicles. Cancer Cell 35, 33–45 e36. 10.1016/j.ccell.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Yoon BH, Kim SK, and Kim SY (2019). GENT2: an updated gene expression database for normal and tumor tissues. BMC Med Genomics 12, 101. 10.1186/s12920-019-0514-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Gabrilovich D, and Sotomayor EM (2007). Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25, 267–296. 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O, Goldstein I, and Kloog Y (2009). Intercellular exchange of proteins: the immune cell habit of sharing. FEBS Lett 583, 1792–1799. 10.1016/j.febslet.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Stasilojc G, Osterborg A, Blom AM, and Okroj M (2016). New perspectives on complement mediated immunotherapy. Cancer Treat Rev 45, 68–75. 10.1016/j.ctrv.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Takats Z, Wiseman JM, Gologan B, and Cooks RG (2004). Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306, 471–473. 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- Tambellini NP, Zaremberg V, Turner RJ, and Weljie AM (2013). Evaluation of extraction protocols for simultaneous polar and non-polar yeast metabolite analysis using multivariate projection methods. Metabolites 3, 592–605. 10.3390/metabo3030592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzana R, Eisenberg G, Merims S, Frankenburg S, Pato A, Yefenof E, Engelstein R, Peretz T, Machlenkin A, and Lotem M (2015). Human T cell crosstalk is induced by tumor membrane transfer. PLoS One 10, e0118244. 10.1371/journal.pone.0118244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmurugan R, Challa DK, Ram S, Ober RJ, and Ward ES (2016). Macrophage-Mediated Trogocytosis Leads to Death of Antibody-Opsonized Tumor Cells. Mol Cancer Ther 15, 1879–1889. 10.1158/1535-7163.MCT-15-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CM, Brennan VC, Gutierrez NM, Wang X, Wang L, and Yang WH (2013). SUMOylation of ATF3 alters its transcriptional activity on regulation of TP53 gene. J Cell Biochem 114, 589–598. 10.1002/jcb.24396. [DOI] [PubMed] [Google Scholar]
- Whiteside TL (2016). Exosomes and tumor-mediated immune suppression. J Clin Invest 126, 1216–1223. 10.1172/JCI81136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolford CC, McConoughey SJ, Jalgaonkar SP, Leon M, Merchant AS, Dominick JL, Yin X, Chang Y, Zmuda EJ, O’Toole SA, et al. (2013). Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J Clin Invest 123, 2893–2906. 10.1172/JCI64410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ST, Kreutzberger AJB, Lee J, Kiessling V, and Tamm LK (2016). The role of cholesterol in membrane fusion. Chem Phys Lipids 199, 136–143. 10.1016/j.chemphyslip.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang R, Case JB, Yutuc E, Ma X, Shen S, Gomez Castro MF, Liu Z, Zeng Q, Zhao H, Son J, et al. (2020). Cholesterol 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. Proc Natl Acad Sci U S A 117, 32105–32113. 10.1073/pnas.2012197117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yu P, Tomar VS, Chen X, Atherton MJ, Lu Z, Zhang HG, Li S, Ortiz A, Gui J, et al. (2022). Targeting PARP11 to avert immunosuppression and improve CAR T therapy in solid tumors. Nat Cancer. 10.1038/s43018-022-00383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, Li XL, Wang D, Huang XC, Mathis JM, Duan WM, Knight D, Shi R, Glass J, Zhang DQ, et al. (2008). Trogocytosis of MHC-I/peptide complexes derived from tumors and infected cells enhances dendritic cell cross-priming and promotes adaptive T cell responses. PLoS One 3, e3097. 10.1371/journal.pone.0003097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Asnani M, and Thomas-Tikhonenko A (2019). Escape From ALL-CARTaz: Leukemia Immunoediting in the Age of Chimeric Antigen Receptors. Cancer J 25, 217–222. 10.1097/PPO.0000000000000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-seq data have been deposited at GEO (accession number: GSE190702) and are publicly available as of the date of publication. Accession numbers are also listed in the key resources table. Original western blot images and all raw data used to create the graphs can be found in Data S1 (contains uncropped scans of Western blots and original data related to Figures 1–7 and S1–S4, S6 and S7)
No original code was generated for all data in this study.
Any additional information required to analyze the data reported in this study is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti- mouse CD8-APC/Cy7 | Biolegend | Cat# 100714; RRID: AB_312753 |
| anti-mouse CD69-PE | Biolegend | Cat# 104507; RRID: AB_313110 |
| anti-mouse PD-1-BV605 | BioLegend | Cat# 135219; RRID: AB_11125371 |
| anti-mouse LAG3-PE/Cy7 | BioLegend | Cat# 125226; RRID: AB_2715764 |
| anti-mouse CD366-BV421 | BioLegend | Cat# 134019; RRID: AB_2814028 |
| anti-mouse Annexin V-FITC | BioLegend | Cat# 640906; RRID: AB_2561292 |
| anti-mouse CD45-APC/Cy7 | BioLegend | Cat# 157203; RRID: AB_2876533 |
| anti-mouse CD3-PE | BioLegend | Cat# 100206; RRID: AB_312663 |
| anti-mouse CD8-AF700 | BioLegend | Cat# 100730; RRID: AB_493703 |
| anti-mouse CD69-BV421 | BioLgend | Cat# 104545; RRID: AB_2686969 |
| anti-mouse PD-1-PE/Cy7 | BioLegend | Cat# 109110; RRID: AB_572017 |
| anti-mouse LAG3-BV650 | BioLegend | Cat# 125227; RRID: AB_2687209 |
| Anti-mouse Annexin V-APC | BioLegend | Cat# 640941; RRID: AB_2616657 |
| anti-hCD19-APC | Biolegend | Cat# 392504; RRID: AB_2728416 |
| anti-human CD19 CAR-PE | Acrobiosystems | Cat#FM3-HPY53 |
| anti-human CD8-PE/Cy7 | BioLegend | Cat# 344750; RRID: AB_2687201 |
| anti-mouse CD4-APC/Cy7 | BioLegend | Cat# 100414; RRID: AB_312699 |
| anti-mouse CD19-PE | BioLegend | Cat# 115507; RRID: AB_313642 |
| anti-mouse NK1.1-APC | BioLegend | Cat# 108709; RRID: AB_313396 |
| Biotin anti-mouse CD3 | BioLegend | Cat# 100243; RRID: AB_2563946 |
| Alexa Fluor-594 streptavidin | BioLegend | N/A |
| Alexa Fluor-488 anti-mouse CD8 | BioLegend | Cat# 100723; RRID: AB_389304 |
| Rabbit anti-mouse ATF3 antibody | Cell Signaling | Cat#18665S |
| Anti-mouse SUMO1 | Invitrogen | Cat# 33–2400; RRID: AB_2533109 |
| VeriBlot for IP Detection Reagent HRP | Abcam | N/A |
| Goat anti-mouse HRP-conjugated antibody | Cell Signaling | Cat# 7076S |
| Rat anti-mouse β-tubulin antibody | Cell Signaling | Cat# 2146 |
| PE-anti-mouse H-2Kb bounds to SIINFEKL antibody | BioLegend | Cat# 141603; RRID: AB_10897938 |
| Recombinant Anti-Mesothelin antibody | Abcam | Cat# ab196235 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Healthy human peripheral T cells | Human Immunology Core at the University of Pennsylvania | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Prostaglandin E2 | Sigma | Cat# P0409 |
| DMSO | Sigma | Cat# 20–139 |
| VEGF165 | Sigma | Cat# V5765 |
| 1,1’-dioctadecyl-3,3,3’,3’tetramethylindodicarbocyanine | Biotium | Cat# 60014 |
| Latrunculin A | Sigma | Cat# L5163 |
| 25-hydroxycholesterol | Sigma | Cat# H1015 |
| GW3965 | Sigma | Cat# G6295 |
| Golgi stop | BD | Cat# BDB554724 |
| Transwell Chambers | Millipore | Cat# CLS3422–48EA |
| DNase I | Roche | Cat# 10104159001 |
| Collagenase D | Roche | Cat# 11088882001 |
| Percoll | Sigma | Cat# 17–0891-01 |
| Lymphoprep™ | Stem Cell | Cat# 07851 |
| Recombinant human IL-2 | Sigma | Cat# I0523 |
| Ovalbumin (257–264) chicken | Sigma | Cat# S7951 |
| Anti-PD-1 | Bio X cell | Cat# BE0146 Clone: RMP1–14 |
| CFSE | Biolegend | Cat# 79898 |
| Dynabeads™ Mouse T-Activator CD3/CD28 | Gibco | Cat# 11453D |
| Dynabeads™ Human T-Activator CD3/CD28 | Gibco | Cat# 11132D |
| CountBright™ Absolute Counting Beads | Invitrogen | Cat# C36950 |
| Fetal Bovine Serum | Hyclone | Cat# SH30071.03 |
| High-Capacity RNA-to-cDNA™ Kit | Applied Biosystems | Cat# 4387406 |
| Gibco™ L-Glutamine | Gibco | Cat# 25–030-081 |
| Intracellular Fix & Perm Buffer set | eBioscience | Cat# 88–8824 |
| Lipofectamin2000 | Invitrogen | Cat# 52887 |
| Retronectin | Takara | Cat# T100B |
| RBC lysis buffer | Biolegend | Cat# 420302 |
| Critical commercial assays | ||
| ONE-Glo™ Luciferase Assay System | Promega | Cat# E6120 |
| Power SYBR Green PCR Master Mix | Thermo Fisher | Cat# 4367659 |
| SimpleChIP Plus Enzymatic Chromatin IP Kit | Cell Signaling | Cat# 9005S |
| RNeasy Kits | Qiagen | Cat# 74004 |
| EasySep™ Mouse CD8+ T Cell Isolation Kit | Stem Cell | Cat# 19853 |
| EasySep™ Mouse T Cell Isolation Kit | Stem Cell | Cat# 19851 |
| EasySep™ Mouse CD4+ T Cell Isolation Kit | Stem Cell | Cat# 19852 |
| Deposited data | ||
| Bulk RNA-sequence data | This paper | GEO: GSE190702 |
| Raw data | This paper | Data S1 |
| Experimental models: Cell lines | ||
| Human: Phoenix-ECO | ATCC | Cat# CRL-3214™ |
| Mouse: MC38 | ATCC | N/A |
| Mouse: MC38 OVA | Cancer Cell, 2017, 31: 194–207 | N/A |
| Mouse: MC38-OVA-Luciferase | Nat Cancer 2022 May 30. doi: 10.1038/s43018–022- 00383–0 | N/A |
| Mouse: Hepa1–6 | ATCC | Cat# CRL-1830™ |
| Mouse: B16F10 | ATCC | Cat# CRL-6475™ |
| Mouse: MH6499c4 | Immunity 49, 178–193 | N/A |
| Human: NALM6 | ATCC | Cat# CRL-3273™ |
| Human: EM-Meso-GFP-Luciferase | Oncoimmunology, 2018, VOL. 7, NO. 3, e1395997 | N/A |
| Mouse: NIH 3T3 | ATCC | Cat# CRL-1658™ |
| Experimental models: Organisms/strains | ||
| Mouse: WT C57BL/6 | Jackson Laboratory | Cat# 000664 |
| Mouse: Ch25h−/− C57BL/6 | Jackson Laboratory | Cat# 016263 |
| Mouse: Ch25hΔCD8 C57BL/6 | This paper | N/A |
| Mouse: Atf3ΔCD8 C57BL/6 | This paper | N/A |
| Mouse: OT1 C57BL/6 | Jackson Laboratory | Cat# 003831 |
| Mouse: Rag1−/− C57BL/6J | Jackson Laboratory | Cat# 002216 |
| Mouse: NSG NOD/ShiLtJ | Jackson Laboratory | Cat# 005557 |
| Oligonucleotides | ||
| Primers for q-PCR, see table S1 | This paper | N/A |
| Recombinant DNA (plasmid) | ||
| Lenti-p-EF1α-Meso-scFV | 10.1073/pnas.0813101106. | N/A |
| Lenti-p-EF1α-Meso-scFV-T2A-Ch25h | This paper | N/A |
| Retro-p-CD19-scFV-BBZ-MSGV | 10.1016/j.cell.2021.08.004. | N/A |
| Retro-p-CD19-scFV-BBZ-T2A-Ch25h MSGV | This paper | N/A |
| Software and algorithms | ||
| Prism 9 | GraphPad Prism9 | https://www.graphpad.com/ |
| FlowJo 7.6 version | FlowJo | https://www.flowjo.com/ |
| Image J | Image J | https://imagej.nih.gov/ij/ |
| Other | ||