

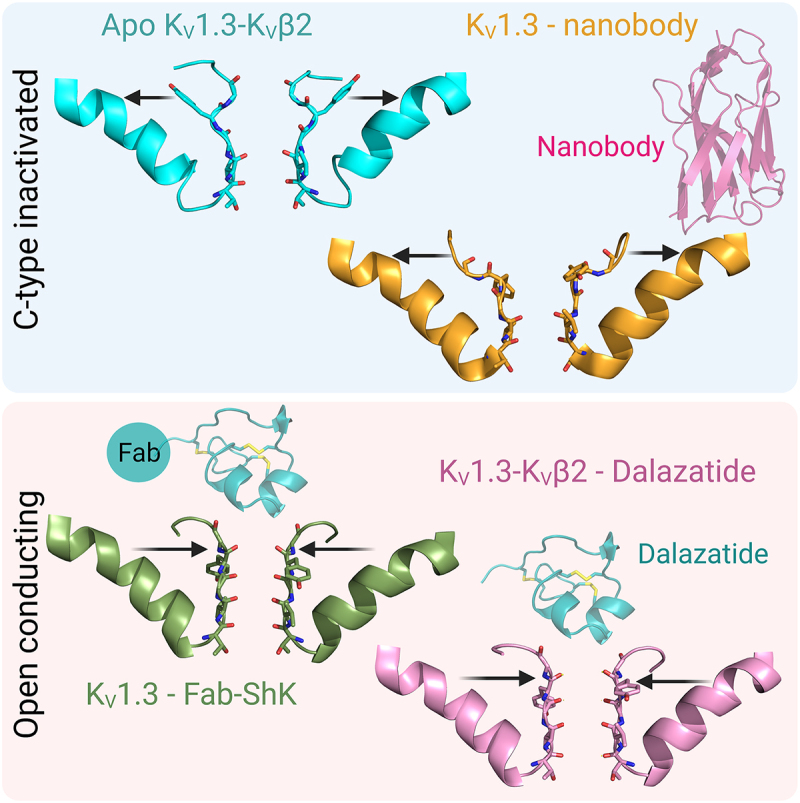
ABSTRACT
The voltage-gated potassium channel KV1.3 is an important therapeutic target for the treatment of autoimmune and neuroinflammatory diseases. The recent structures of KV1.3, Shaker-IR (wild-type and inactivating W434F mutant) and an inactivating mutant of rat KV1.2-KV2.1 paddle chimera (KVChim-W362F+S367T+V377T) reveal that the transition of voltage-gated potassium channels from the open-conducting conformation into the non-conducting inactivated conformation involves the rupture of a key intra-subunit hydrogen bond that tethers the selectivity filter to the pore helix. Breakage of this bond allows the side chains of residues at the external end of the selectivity filter (Tyr447 and Asp449 in KV1.3) to rotate outwards, dilating the outer pore and disrupting ion permeation. Binding of the peptide dalazatide (ShK-186) and an antibody-ShK fusion to the external vestibule of KV1.3 narrows and stabilizes the selectivity filter in the open-conducting conformation, although K+ efflux is blocked by the peptide occluding the pore through the interaction of ShK-Lys22 with the backbone carbonyl of KV1.3-Tyr447 in the selectivity filter. Electrophysiological studies on ShK and the closely-related peptide HmK show that ShK blocks KV1.3 with significantly higher potency, even though molecular dynamics simulations show that ShK is more flexible than HmK. Binding of the anti-KV1.3 nanobody A0194009G09 to the turret and residues in the external loops of the voltage-sensing domain enhances the dilation of the outer selectivity filter in an exaggerated inactivated conformation. These studies lay the foundation to further define the mechanism of slow inactivation in KV channels and can help guide the development of future KV1.3-targeted immuno-therapeutics.
KEYWORDS: Voltage-gated potassium channel, cryogenic electron microscopy, peptide toxin, molecular modeling and docking, T cells, autoimmune diseases
GRAPHICAL ABSTRACT
Introduction
The study of ion channels in immune cells began in 1984 with the discovery of voltage-gated potassium (K+) currents in human T lymphocytes [1–3]. Compounds known to inhibit neuronal K+ currents blocked the lymphocyte K+ current in a concentration-dependent manner and suppressed mitogen-triggered T cell proliferation with parallel potencies, suggesting a functional role for the K+ current in T cells [1,3–5]. The KV1.3/KCNA3 gene encoding this K+ current was identified in 1990 [6,7]. In the intervening decades, much has been learned about the function and structure of the voltage-gated KV1.3 channel. In this review, we summarize the functional role of KV1.3 in immune cells, its importance as a target for immunomodulatory therapeutics for autoimmune and neuroinflammatory diseases, and discuss recent cryogenic electron microscopy and molecular dynamics simulation studies that reveal the structure of the channel in different conformational states, as well as in complex with two classes of therapeutic leads.
Functional network of ion channels and ion transporters in immune cells
KV1.3 is a critical component of a network of ion channels and ion transporters that regulate calcium signaling, activation, proliferation, cytokine secretion and cellular homeostasis of immune cells [8,9]. Figure 1 shows the channel-transporter network in human T lymphocytes. Calcium entry occurs primarily through the calcium release-activated (CRAC) channel, which is composed of the proteins Orai1 and Orai2 in the plasma membrane, and Stim1 and Stim2 in the ER membrane [9,11–17]. KV1.3 and the calcium-activated KCa3.1 channel are the main conduits for K+ efflux [8,9,18,19]. Efflux of K+ through the two K+ channels hyperpolarizes the membrane potential, which in turn promotes further calcium entry through CRAC in a positive feedback loop. The network also contains channels and transporters for sodium, magnesium, zinc and chloride [9], but no functional voltage-gated calcium (CaV) channels are expressed in T cells [10] (Figure 1a). While K+ channels promote calcium signaling, efflux of chloride or influx of sodium dampens calcium entry and the calcium signal via membrane depolarization [9]. Similar KV1.3-containing channel-transporter networks are present in B lymphocytes, monocyte-macrophages, microglia and neutrophils [9,20–23] (Figure 2).
Figure 1.
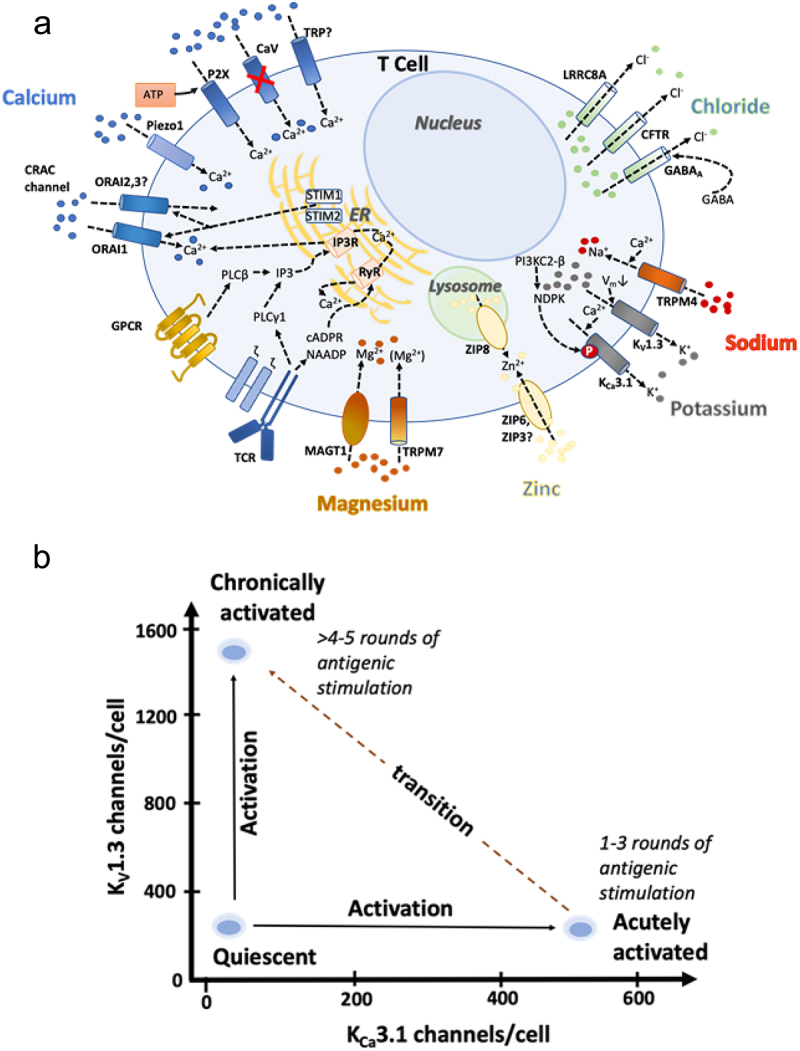
a) Network of ion channels and transporters in human T cells. KV1.3 and KCa3.1 are expressed in both the plasma membrane and inner mitochondrial membrane. Although CaV3.2, CaV3.3, CaV2.1 transcripts are present in T cells, they lack many 5’ exons and consequently no functional CaV channels are expressed in T cells [10]. This figure is modified and updated from a figure published in [9]. b) The number of KV1.3 and KCa3.1 channels per T cell in quiescent, acutely activated (1–3 rounds of antigenic stimulation) and chronically activated (>4–5 rounds of antigenic stimulation) cells.
Figure 2.
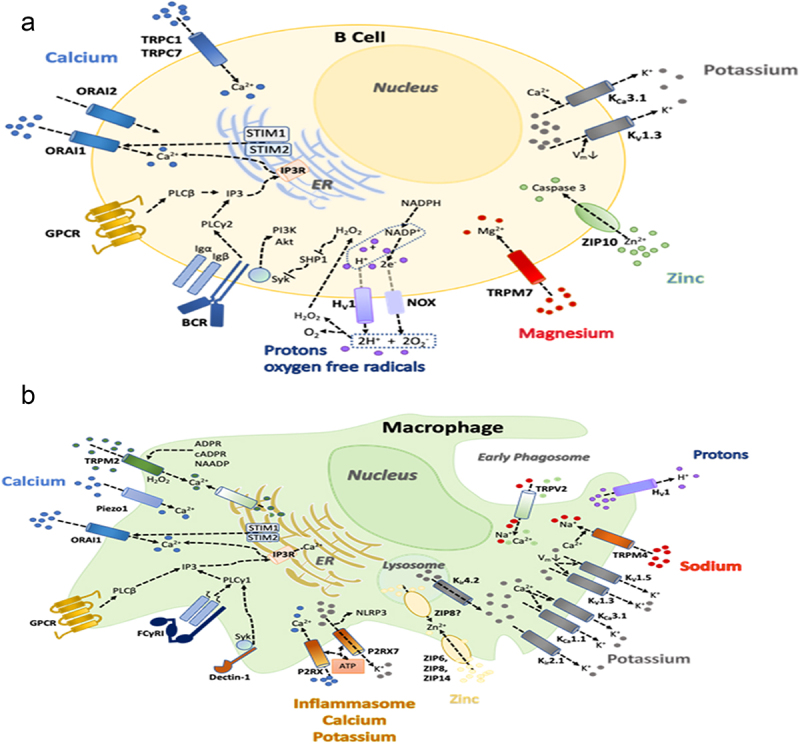
Network of ion channels and transporters in human B cells (a) and monocyte-macrophages (b). This figure is modified and updated from figures published in [9].
KV1.3 as a therapeutic target for autoimmune and neuroinflammatory diseases
The expression of KV1.3 and KCa3.1 varies during activation and differentiation of T cells (Figure 1b). Upon activation by antigen, T cells up-regulate KCa3.1, and, when repeatedly stimulated by antigen, switch to up-regulating KV1.3 [24–27] (Figure 1b). This dichotomy in K+ channel expression between acutely-activated and chronically-activated T cells underlies the importance of KV1.3 as a therapeutic target in autoimmune diseases [8,9,18,19]. In patients with diverse autoimmune diseases (multiple sclerosis, type-1 diabetes mellitus, rheumatoid arthritis, psoriasis, autoimmune vasculitis), pathogenic auto-reactive T cells that have undergone repeated stimulation by the relevant autoantigen during the course of disease exhibit a KV1.3high pattern [8,9,18,19,25,28–33]. Studies with KV1.3 gene-knockout mice and pharmacological experiments with KV1.3-specific inhibitors demonstrate that KV1.3 is functionally important in pathogenic auto-reactive T cells [8,9,18,25,28–36]. Mice with the KV1.3 gene deleted are resistant to experimental autoimmune encephalomyelitis, a model for multiple sclerosis induced by immunization with myelin autoantigens [34]. In these KV1.3-knockout mice, myelin autoantigen-reactive T cells produce lower levels of inflammatory cytokines (IFN-γ, IL-17), secrete suppressive cytokine IL-10, and behave as FoxP3-independent suppressor T cells [34,35]. In other studies, KV1.3-specific inhibitors have been shown to suppress cytokine production and proliferation of pathogenic autoreactive T cells from patients with multiple sclerosis, type-1 diabetes mellitus, rheumatoid arthritis and autoimmune vasculitis; these inhibitors have also been shown to ameliorate disease in rodent models of multiple sclerosis, type-1 diabetes mellitus, rheumatoid arthritis, psoriasis and inflammatory bowel disease [8,9,18,19,25,28–33,36]. KV1.3 is also recognized as a therapeutic target to modulate the function of autoreactive B cells in granulomatosis with polyangiitis [32], disease-associated microglia in Alzheimer’s disease, Parkinson’s disease and ischemic stroke [37–45], macrophages [46] and neutrophils [22] in autoimmune and neuroinflammatory diseases [23]. In addition, mitochondrial KV1.3 is recognized as a target for the treatment of diverse tumors [47,48]. Since KV1.3-knockout mice and rats do not exhibit a deleterious phenotype, selective pharmacological targeting of KV1.3 should be safe [27,49–51]. Three KV1.3 inhibitors (dalazatide [31], DES-7144, an analog of DES-1 [36], and sI-544 [52] have progressed to human clinical trials, and other KV1.3 inhibitors (scorpion peptides, small molecules, nanobodies, knotbodies, and peptide-antibody fusions) are in pre-clinical development. The ideal KV1.3-targeting therapeutic would be potent, KV1.3-specific, and stable (not metabolized to a less selective inhibitor). Recent determinations of the structure of KV1.3 have shed light on the inactivated conformation of KV1.3 and on how peptide inhibitors interact with the channel.
Structure of KV1.3
Structure of KV1.3-KVβ2 in non-conducting, C-type inactivated conformation
The KV channel in immune cells is formed by four KV1.3 subunits assembled in the membrane, which bind to four accessory proteins designated KVβ2 (Figure 3a) [8,9,19]. The structure of human KV1.3 in complex with KVβ2 was determined recently by cryogenic electron microscopy (cryo-EM) [53]. In an initial 3D map at 3.1Å resolution, the cytosolic regions including KVβ2 and the intracellular tetramerization domain of Kv1.3 were well resolved, but the map displayed a lower resolution in the transmembrane region. Resolution was improved by subtracting density outside the transmembrane region using a soft mask around the detergent micelle, followed by 2D and 3D classification. From the analysis of signal-subtracted particles, a 3D map resolved to 3.4 Å with C4 symmetry applied was achieved for the transmembrane region. Models built from the maps of the transmembrane region and the cytosolic regions were combined [53] (PDB 7WF3).
Figure 3.
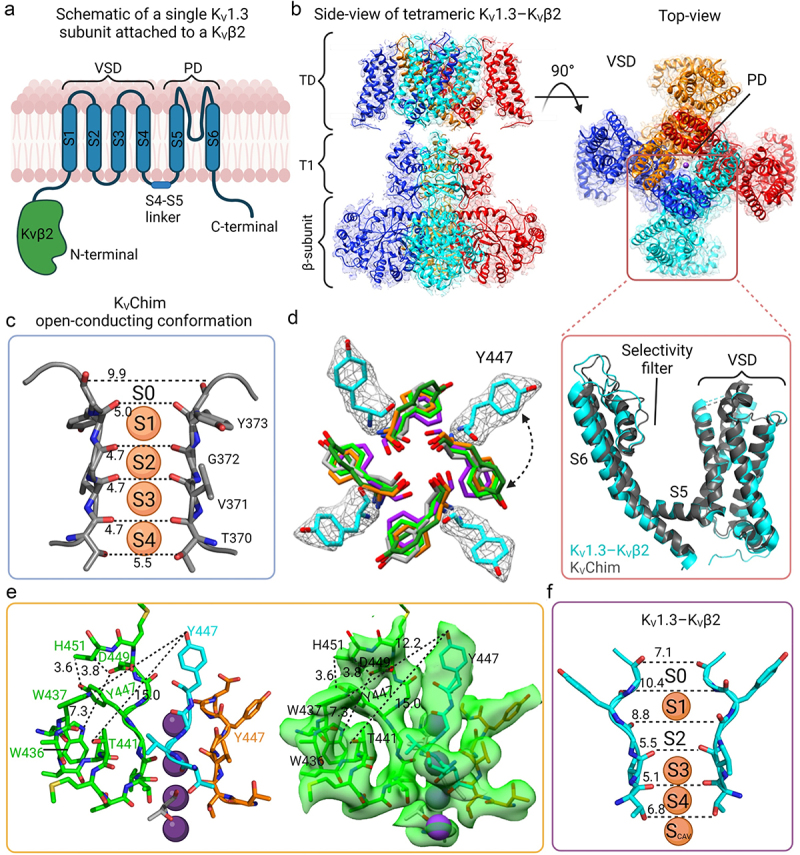
Structure of KV1.3-KVβ2 in non-conducting, C-type inactivated conformation. a) Schematic view of a single subunit of KV1.3 with the transmembrane region in blue and the N-terminal intracellular β-subunit in green. b) The cryo-EM density map of KV1.3-KVβ2 side-view (left) and top view (right). The inset shows a superimposition of the VSDs and selectivity filters of KV1.3-KVβ2 (cyan) (PDB 7WF3) and KVChim (gray) (PDB 2R9R). c) Selectivity filter of KVChim (PDB 2R9R) showing the distances between the carbonyl O atoms of two subunits and the four K+ in the selectivity filter at S1, S2, S3, S4 positions. d) Overlay of the KV1.3-KVβ2 Tyr447 (cyan) and equivalent aromatic residues in the selectivity filter of KVChim Tyr373 (gray), hERG F627 (orange) (PDB 5VA1), Eag-1 Phe439 (purple) (PDB 5K7L) and KcsA Tyr78 (neon green) (PDB 1K4C). The cryo-EM density of KV1.3-KVβ2 Tyr447 in white mesh is overlapped with the model represented as stick cyan. e) the four subunits of KV1.3-KVβ2 (green, cyan, orange and gray) with the new position of Tyr447 and D449 being stabilized by intra-subunit hydrogen bond interactions with His451, a residue at the external entrance to the KV1.3 pore. The residues of each subunit are represented as sticks. On the right, the cryo-EM density is represented as a transparent green surface. f) Selectivity filter of KV1.3-KVβ2 showing the difference in the position of the aromatic Tyr447 residue in the selectivity filter compared to KVChim in (c). The widened outer selectivity filter of KV1.3-KVβ2 shows K+ at ion-binding sites S1, S3, S4 but S2 is empty.
Figure 3b shows the cryo-EM density map of KV1.3-KVβ2 with fitted model viewed from the membrane plane (center) and the extracellular side (right). The pore domain (PD) is located at the center, with four voltage-sensing domains (VSDs) at the periphery [53]. The PD is formed by the association of S5 and S6 helices and the intervening P-loop from each of the four KV1.3 subunits. Each VSD is formed by the S1-S4 transmembrane segments from one KV1.3 subunit. The VSD of one subunit interacts with the S5 helix of a neighboring subunit in a domain-swapped configuration. The position of charged residues in the VSD indicates that the VSD is fully activated. In the PD, the S6 helical inner gate is in the open conformation [53]. The depolarized VSD and open S6 helical inner gate indicate that KV1.3 is in the voltage-activated state [53].
Voltage-activation causes KV channels to transition from the closed to an open-conducting conformation, and then into a non-conducting C-type (slow) inactivated conformation in a time-dependent manner [54–57]. The best structural example of a KV channel in the open-conducting conformation is the rat KV1.2-KV2.1 paddle chimera (KvChim; pore region is KV1.2) (PDB 2R9R) (Figure 3c). Hydrated K+ flows through the open inner gate into a central cavity, where it loses its hydration water and then passes in single file through a narrow selectivity filter into an outer vestibule (Figure 3c) [58,59]. Four K+ are present in the selectivity filter at ion-binding sites S1, S2, S3, S4 (Figure 3c) [58,59].
Superimposition of KV1.3-KVβ2 with KVChim shows good alignment in the VSD, but structural differences are clearly visible in the selectivity filter (Figure 3b, right-bottom). A major difference is observed in the position of the aromatic residue in the selectivity filter [53]. This aromatic residue is in nearly identical positions in KVChim (Tyr373) and other K+ channels, both in the open-conducting (human ERG/KV11.1; PDB 5VA1; Phe367) or closed (rat Eag-1/KV10, PDB 5K7L, Phe439; bacterial KcsA, PDB 1K4C, Tyr78) conformations [53] (Figure 3d). However, in KV1.3-KVβ2, a rotation of the sidechain of Tyr447 by ~ 90 degrees leads to the hydroxyl oxygen of this residue adopting a position shifted 11 Å outwards from the position seen in these other K+ channels (Figure 3d) [53,57]. Another residue close to Tyr447 in the selectivity filter, Asp449, also swings outwards (Figure 3e) [53]. The outward positions of Tyr447 and Asp449 are stabilized by intra-subunit hydrogen bond interactions with His451, a residue at the external entrance to the KV1.3 pore (Figure 3e) [53]. Owing to the changed position of Tyr447 and Asp449, the outer selectivity filter of KV1.3 is significantly wider than in KVChim, while the inner selectivity filter is unchanged (Figure 3f) [53]. In the widened outer selectivity filter of KV1.3, K+ can be seen at ion-binding sites S1, S3 and S4, but S2 is empty (Figure 3f) [53]. Loss of the K+ at site S2 occurs because the carbonyl groups of residues Gly446 and Tyr447 are oriented away from the selectivity filter [53]. All-atom MD simulations over 1 μs following a voltage pulse show stochastic conduction of K+ through an unstable outer pore of KV1.3-KVβ2 [53]. This novel conformation with a dilated outer selectivity filter and reduced K+ occupancy represents the non-conducting C-type inactivated state of KV1.3-KVβ2 [53].
Structures of Shaker-IR, KVChim and KV1.3 in C-type inactivated conformation
Following the report described above in February 2022 [53], three new structures corroborated the finding that the dilated conformation of the selectivity filter represents the non-conducting slow inactivated state. Figure 4a shows the amino acid sequences of the pore regions of these three channels. In March 2022, Swartz and colleagues published cryo-EM structures of the fly Shaker-IR channel (PDB 7SIP) and its rapidly inactivating W434F mutant (PDB 7SJ1) [60]. The selectivity filter in Shaker-IR is in an open-conducting conformation resembling KVChim, while in Shaker-W434F it is in an inactivated conformation resembling KV1.3-KVβ2 (Figure 4b) [60]. In Shaker-W434F, Tyr445 and Asp447 in the selectivity filter are reoriented outwards and positioned near Thr449 (corresponding to Tyr447, Asp449 and His451 in KV1.3) (Figure 4a,b, right). Because of this rearrangement, the outer selectivity filter of Shaker-W434F is dilated and K+ occupancy in the outer filter is reduced (Figure 4b, right) [60]. In April 2022, Valiyaveetil and colleagues reported the structure of the rapidly-inactivating triple mutant (W362F+S367T+V377T) of KVChim (PDB 7SIT) (Figure 4a,c) [61]. In this structure too, outward reorientation of Tyr373 and Asp375 toward Thr377 (corresponding to Tyr447, Asp449 and His451 in KV1.3) (Figure 4a) resulted in widening of the outer selectivity filter and loss of K+ at the S1 and S2 sites (Figure 4c) [61]. In July 2022, Meyerson and collaborators published the structure of KV1.3 without KVβ2 [62]. The new KV1.3 structure showed a dilated outer selectivity filter with reduced K+ occupancy (Figure 4d) [62]. Interestingly, two dilated conformations were identified, designated D1 (PDB 7SSX) and D2 (PDB 7SSY) (Figure 4d). In D1 (Figure 4d, left), only Asp449 is oriented outwards, while in D2 (Figure 4d, right), both Tyr447 and Asp449 are rotated outwards, and K+ occupancy is reduced in the dilated outer filter [62]. The D2 conformation is identical to the C-type inactivated conformation in the structures of KV1.3-KVβ2 (PDB 7WF3), Shaker-IR W434F (PDB 7SJ1) and KVChim (W362F+S367T+V377T) (PDB 7SIT), while the D1 conformation appears to be an intermediate. KVβ2 does not appear to affect the inactivated conformation of the selectivity filter because structures containing this subunit (KV1.3-KVβ2, PDB 7WF3) and those lacking this subunit (Shaker-IR W434F, PDB 7SJ1; KVChim W362F+S367T+V377T, PDB 7SIT; KV1.3, PDB 7SSY) all exhibit the D2 inactivated conformation of the filter. In summary, comparison of the KV1.3, Shaker-IR and KVChim structures suggests that the three channels use similar mechanisms for C-type inactivation.
Figure 4.
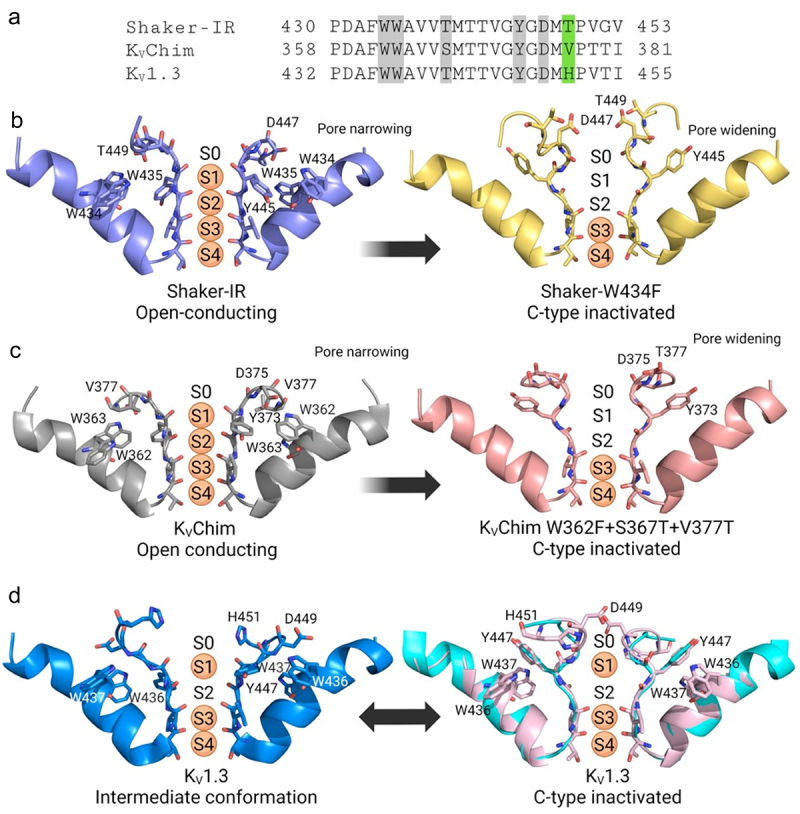
Transition from the open-conducting to the non-conducting C-type inactivated state of Shaker-IR, KVChim and KV1.3. a) amino acid sequence alignment of fly Shaker, rat KVChim and human KV1.3. Residues highlighted in gray are involved in the formation of hydrogen bonds that stabilize the open-conducting conformation. b) Shaker-IR (PDB 7SIP) selectivity filter is in an open-conducting conformation (left), while Shaker-W434F (PDB 7SJ1) selectivity filter is in a dilated C-type inactivated conformation (right). c) KVChim (PDB 2R9R) is in the open-conducting conformation (left) and the rapidly inactivating triple mutant (W362F+S367T+V377T) (PDB 7SIT) is in a dilated C-type inactivated conformation (right). d) KV1.3 with two dilated conformations designated D1 (PDB 7SSX) (left) and D2 (PDB 7SSY) (right). The D2 conformation is identical to the C-type inactivated conformation in the KV1.3-KVβ2 structure (cyan).
Another structure of KV1.3-KVβ2 (PDB 7EJI) with an overall resolution of 3.4 Å was published in 2021 [63]. However, the transmembrane region of this structure had low resolution, resulting in missing or weak side-chain densities in the EM density maps for critical residues in the selectivity filter (particularly Tyr447 and Asp449), pore domain and VSD (Arg364, Arg367, Arg373, Lys376 and Arg379 in the S4 helix; Phe306, Glu309 and Asp332 in the charge-transfer center). As this structure had limited resolution in the pore region and VSD, it has not been included in our structural comparisons.
High external K+ concentration is known to slow C-type inactivation of KV1.3 [64,65] and Shaker-IR [66]. Despite this dampening effect on C-type inactivation, the structure of KV1.3 determined in high K+ concentration and at 0 mV showed the inactivated dilated conformation of the selectivity filter, and a narrower conducting-conformation was only seen when peptide pore-blockers stabilized that conformation (as discussed below) [53,62]. In contrast, the structures of Shaker-IR and KVChim determined in elevated K+ concentrations and at 0 mV were in the open conducting conformation, and the dilated inactivated conformation was only seen in structures of these channels containing inactivation-promoting mutations [60,61]. These results suggest that KV1.3 is more prone to transition from the open-conducting conformation to the dilated inactivated conformation than either Shaker-IR or KVChim. Electrophysiological studies on Shaker-IR and KV1.2 mutants suggest that Shaker-IR is more susceptible to inactivation than KV1.2, and a key residue underlying this difference is Thr449 in Shaker-IR, corresponding to Val381 in KV1.2 (Val377 in KVChim) [67]. The corresponding residue in KV1.3, His451, stabilizes the dilated conformation of the inactivated channel by forming hydrogen-bonds with externally rotated Tyr447 and Asp449 (Figure 3e) [53]. Replacement of this His in KV1.3 with Val (corresponding residue in KV1.2 and KVChim) or Thr (corresponding residue in Shaker-IR) slows C-type inactivation, while replacement with Asn or Ser accelerates C-type inactivation, highlighting the importance of this residue for inactivation [68–70]. It would be interesting to determine if substitution of His for Thr449 in Shaker-IR or Val377 in KVChim (Val381 in KV1.2) rendered these channels as sensitive to C-type inactivation as KV1.3.
Mechanism of transition from open-conducting to non-conducting C-type inactivated conformation
Analysis of the structures described above suggests a mechanism for the transition from the open-conducting to the non-conducting C-type inactivated state. In the open-conducting conformation, a network of intra-subunit (Shaker-IR: Trp434-Asp447; KVChim: Trp362-Asp375; KV1.3: Trp436-Asp449) and inter-subunit (Shaker-IR: Trp435-Tyr445; Thr439-Tyr445; KVChim: Trp363-Tyr373; Ser367-Tyr373; KV1.3: Trp437-Tyr447, Thr441-Tyr447) hydrogen bonds stabilizes the outer pore (Figure 4), allowing optimal flow of K+ [53,57–62,71,72]. In Shaker-IR, rupture of the intra-subunit bond (Trp434-Asp447) speeds up C-type inactivation, while disruption of the two inter-subunit bonds renders the channel nonfunctional [71,72]. Similarly, rupture of the two inter-subunit bonds in KV1.2 results in nonfunctional channels [73]. In mouse KV1.3, the Asp402Asn mutation (Asp449 in human KV1.3) renders the channel nonfunctional, presumably by rupturing the intra-subunit bond, but concatenated dimers comprised of wild-type− and Asp402Asn−containing subunits exhibit rapid inactivation [74], like the Asp447Asn mutation in Shaker-IR [71]. Taken together, these results suggest that the intra-subunit bonds in KVChim, Shaker-IR and KV1.3 stabilize the open-conducting conformation and prevent C-type inactivation, while the two inter-subunit bonds are essential for structural integrity. The critical intra-subunit bond (Shaker-IR: Trp434-Asp447; KVChim: Trp362-Asp375; KV1.3: Trp436-Asp449) (Figure 4) ties the selectivity filter to the pore-helix. Its rupture causes the untethered selectivity filter to swing outwards (Figures 3d,e,f, 4b,c,d), allowing Tyr447 and Asp449 in KV1.3 to form intra-subunit hydrogen bonds with the external residue His451 (Figure 2e) (corresponding to KVChim: Val377; Shaker-IR: Thr449) (Figure 4). This outward positioning of Tyr447 and Asp449 widens the outer selectivity filter and perturbs ion coordination at sites S1 and S2, leading to the non-conducting C-type inactivated state.
In the next sections, we discuss KV1.3 inhibitors that operate by two distinct mechanisms, the first by narrowing the selectivity filter and occluding the pore, and the second by enhancing dilation of the selectivity filter in the non-conducting C-type inactivated conformation. We also discuss the importance of conformational dynamics in determining the interaction of pore-blocking peptides with KV1.3.
Structure of KV1.3 bound to pore-blocking peptides
Peptide inhibitors of KV1.3
Several classes of peptide toxins are potent blockers of KV1.3 [18,19,23]. ShK, a peptide produced by the Caribbean sea anemone Stichodactyla helianthus, blocks KV1.3, KV1.1 and KV1.6 with picomolar potency and KV1.2, KV3.2, KCa3.1 with nanomolar potency [75–77]. ShK contains 35 amino acid residues that are cross-linked by three disulfide bonds into a structure consisting of two short α-helices (residues 14–19 and 21–24), and an N-terminus with an extended conformation up to residue 8, followed by a pair of interlocking turns that resembles a 310-helix (Figure 5a, left) [78]. It contains no β-sheet and is quite distinct from the αβ fold found in scorpion toxins such as HsTX1 (PDB 1QUZ) (Figure 5a, right), which also block KV1.3 with picomolar affinity [79,80]. The structures of BgK, a peptide from the sea anemone Bunodosoma granuliferum, (PDB 1BGK) [81], and AcK1, a peptide from the human-infecting hookworm Ancylostoma caninum (PDB 2MD0) [82] are similar to that of ShK. Both BgK and AcK1 block KV1.3, albeit with significantly lower potency than ShK [82]. Over 20,000 peptides and protein domains with structures similar to ShK have now been identified, and are referred to as ShKT domains [83,84]. Proteins containing ShKT domains include metallopeptidases, prolyl-4-hydroxylases, tyrosinases, peroxidases, oxidoreductases. One such protein, human matrix metalloprotease 23 (MMP-23), uses a ShKT domain (PDB:2K72) to block KV1.3, while using other domains to trap the channel in intracellular compartments [85–87].
Figure 5.
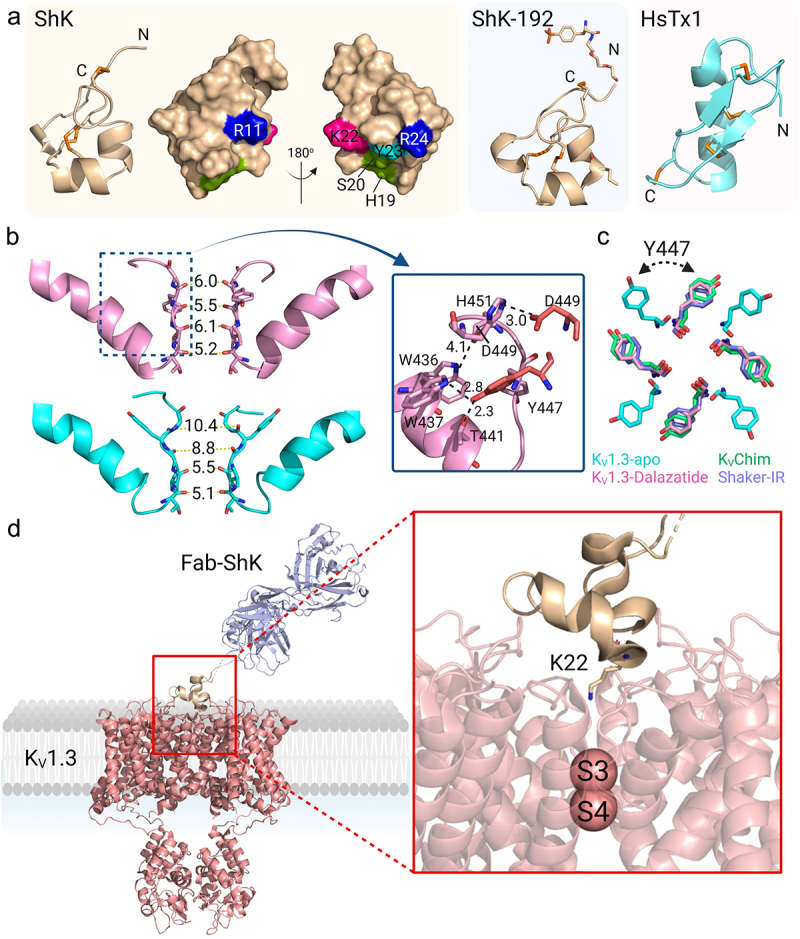
Structure of KV1.3 bound to pore-blocking peptides. a) KV1.3 inhibitory peptides. ShK represented as wheat-colored cartoon with the disulfide-bonds as orange sticks. The cluster of residues that interact with KV1.3 are represented as a surface: Arg11 and Arg24 in blue, His19 and Ser20 in green, Lys22 in pink, Tyr23 in cyan. ShK-192 (PDB 2K9E) is shown as a wheat-colored cartoon (middle) and HsTx1 (PDB 1QUZ) as a cyan-colored cartoon. b) Pore region of two subunits of the C-type inactivated apo KV1.3-KVβ2 (cyan) (PDB 7WF3) and dalazatide-KV1.3-KVβ2 (pink) (PDB 7WF4). The selectivity filter residues are shifted compared to the apo KV1.3-KVβ2, where the new position of Tyr447 formed inter-subunit hydrogen bonds Trp437-Tyr447 and Thr441-Tyr447. An inter-subunit Asp449-His451 hydrogen bond also stabilizes this conformation of the selectivity filter. c) Overlay of aromatic residues in the open-conducting conformations of KVChim (green), Shaker-IR (purple), apo KV1.3-KVβ2 (cyan) and dalazatide-KV1.3-KVβ2 (pink). d) KV1.3 bound to Fab-ShK (PDB 7SSV) with Lys22 inserted into the selectivity filter to occlude the channel pore.
The exquisite potency of ShK made it an attractive template for the development of drugs targeting KV1.3. However, its lack of selectivity necessitated development of analogs to improve selectivity for KV1.3 over other channels. As a first step, the surface of ShK involved in binding to KV1.3 was probed using alanine scanning, which identified a cluster of residues on one surface of ShK that interact with KV1.3 (Figure 5a) [88]. Complementary mutagenesis and double mutant cycle analysis then defined the ShK binding site in the external vestibule of KV1.3 [76,88,89], with a key interaction being occlusion of the outer selectivity filter by Lys22. Guided by this knowledge, nearly 400 ShK analogs were produced, including many with changes at the N-terminus. ShK-170, which incorporates a L-phosphotyrosine attached via an aminoethyloxyethyloxy-acetyl linker to the α-amino group of Arg1, blocked KV1.3 with an IC50 of 69 pM and > 100-fold selectivity for KV1.3 over other channels [90]. ShK-186, an analog of ShK-170 with the C-terminal carboxyl replaced with an amide to minimize digestion by carboxypeptidases, retained the selectivity of ShK-170 [91]. Another analog with the N-terminus extended by the residues EWSS ([EWSS]ShK), blocked KV1.3 with an IC50 of 34 pM and ~ 160-fold selectivity for KV1.3 over KV1.1 [92]. Analog ShK-192, with an N-terminal non-hydrolyzable para-phosphonophenylalanine (Ppa), norleucine at position 21, and C-terminal amidation, blocked KV1.3 with an IC50 of 140 pM and ~ 160-fold selectivity over KV1.1 (Figure 5a) [91]. Of all these analogs, only ShK-186, now renamed dalazatide, has progressed to human clinical trials. In a phase I clinical trial in patients with plaque psoriasis, dalazatide significantly suppressed inflammatory cytokines and chronically-activated T cells in the blood, and reduced the Psoriasis Area and Severity Index (PASI score) [31]. Dalazatide is now progressing to phase II clinical trials in patients with secondary progressive multiple sclerosis.
Interestingly, C-type inactivation reduces the affinity of dalazatide for KV1.3 [53], which was also the case for kaliotoxin, a scorpion peptide [93]. Kaliotoxin binding to a KV1.3-KcsA chimera grafted with the selectivity filter of KV1.3 has been shown by solid-state NMR and all-atom molecular dynamics simulation studies to narrow the outer selectivity filter, similar to changes that might occur as a channel recovers from C-type inactivation [94]. Since the KV1.3-KVβ2 structure is in the dilated C-type inactivated conformation, and because dalazatide’s potency is reduced by C-type inactivation, like kaliotoxin, it was of interest to determine if binding of dalazatide would narrow the outer selectivity filter of KV1.3, analogous to the effect of kaliotoxin on KV1.3-KcsA.
KV1.3-KVβ2 bound to dalazatide
The structure of dalazatide bound to KV1.3 was determined at a resolution of 3.4 Å (PDB 7WF4) [53]. This structure showed large shifts in the positions of Tyr447 (11.8Å), Gly448 (2.0Å), Asp449 (3.7Å), Met450 (13.3 Å) and His451 (7.9 Å) in the outer pore region compared to C-type inactivated KV1.3-KVβ2 (Figure 5b) (PDB 7WF3) [53]. Most importantly, Tyr447 swung back toward the interior of the selectivity filter and superimposed well with the corresponding aromatic residues in the open-conducting conformations of KVChim and Shaker-IR (Figure 5b,c) [53]. In its new position, Tyr447 formed inter-subunit hydrogen bonds with Trp437 and Thr441 (Figure 5b), which correspond to the inter-subunit hydrogen bonds that stabilize the open-conducting conformation of KVChim (Trp363-Tyr373; Ser367-Tyr373) and Shaker-IR (Trp435-Tyr445; Thr439-Tyr445) [53,57,60] (Figure 4a). Inward movement of Asp449 was not sufficient to make the intra-subunit hydrogen bond Trp436-Asp449 that prevents C-type inactivation [53,71,72], but a compensatory inter-subunit hydrogen bond Asp449-His451 contributed to stabilizing the conformation (Figure 5b) [53]. The dalazatide-induced rearrangement caused the selectivity filter to narrow to the dimensions seen in the open-conducting conformation of KVChim and Shaker-IR (Figures 3c, 5b). Within the narrowed selectivity filter, K+ was seen at sites S2, S3 and S4, while the density observed at S1 was likely a pore-occluding dalazatide residue. Thus, binding of dalazatide stabilizes the KV1.3 selectivity filter in an open-conducting conformation, with K+ efflux prevented by the peptide occluding the outer entrance of the pore.
KV1.3 bound to Fab-ShK
Although EM density maps revealed a clear density for dalazatide in close proximity to residues in the outer pore of KV1.3 (His451, Met450, Asp449, Gly448), a model of dalazatide could not be built into the density because of the symmetry mismatch problem arising from an asymmetrical dalazatide molecule binding to a four-fold symmetrical KV1.3 [53]. Meyerson and colleagues solved this problem using an antibody-ShK fusion produced by Minotaur Therapeutics [62]. Since the antibody-ShK is divalent and could potentially crosslink Kv1.3 molecules, they generated a monovalent Fab-ShK for cryo-EM studies [62]. In electrophysiological studies, this Fab-ShK was found to completely block KV1.3 current at 10 nM [62]. They then solved the structure of KV1.3 bound to Fab-ShK at a resolution of 3.39 Å and successfully modeled ShK-Fab into the EM density. ShK bound to the outer vestibule of KV1.3, with Lys22 of ShK inserted into the pore and its ammonium group making contact with the backbone carbonyls of Tyr447 from all four KV1.3 subunits (Figure 5d) (PDB 7SSV) [62]. This configuration confirms our earlier results determined by electrophysiology, complementary mutagenesis and double mutant cycle analysis [76,88,89], and our recent molecular dynamics (MD) simulation studies described below [95]. Binding of Fab-ShK caused a narrowing of the outer selectivity filter and stabilization of the conductive conformation by inter-subunit hydrogen bonds Trp437-Tyr447 and Thr441-Tyr447, confirming our findings with dalazatide [53,62]. Thus, dalazatide and Fab-ShK stabilize the open-conducting conformation of the selectivity filter and block K+ flow by occluding the external pore.
Role of conformational dynamics in the interaction of ShK and the closely-related peptide HmK with KV1.3
In the course of defining the structure of dalazatide bound to KV1.3, it became apparent that conformational dynamics in the peptide had to be taken into account. Disulfide-rich peptides are generally thought to be relatively rigid, but NMR spectroscopy and MD simulations have demonstrated the presence of conformational dynamics and their potential importance for molecular recognition [96]. In the case of ShK, NMR has revealed significant flexibility in solution that also affects the dyad Lys22-Tyr23, which is important for KV1.3 block [97–99]. In the major conformation of ShK, Tyr23 is partially buried, but as a consequence of conformational dynamics, ShK samples at least one minor state where Tyr23 is more exposed. HmK, from the sea anemone Heteractis magnifica [100], shares 60% identity with ShK across their 35 amino acids and three disulfide bonds. Their C-terminal regions are largely identical but the N-terminal regions show different charge distributions (Figure 6a). They have similar secondary structure and both peptides display the key Lys-Tyr dyad [76,81,88,89]. Although the two peptides share high sequence identity and structural similarity, ShK blocks KV1.3 with much higher potency than HmK (ShK IC50 11 pM [76,101], HmK IC50 3 nM [102]).
Figure 6.
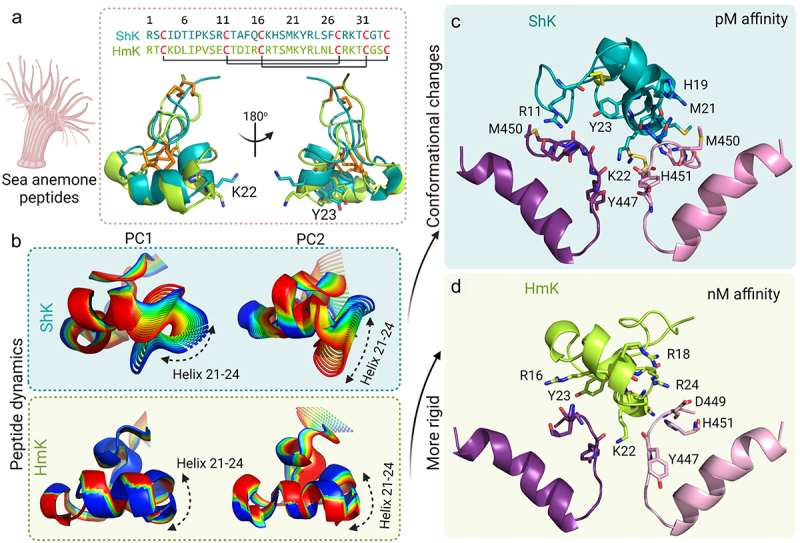
Role of conformational dynamics in the interaction of ShK and HmK with KV1.3. a) Structure and sequence of ShK (teal) and HmK (green). The disulfide bonds are represented as orange sticks and the Lys-Tyr dyad is shown as sticks. b) The 20 structures derived from PC1 and PC2 show that the dynamics of ShK are distributed throughout the peptide, with the helix 21–24 having a higher contribution, while HmK does not display significant dynamics, except at the N- and C-termini. The final frames of the 100 ns MD simulations show the interaction of KV1.3 (subunits A, and C colored pink and purple, respectively) with c) ShK (teal) and d) HmK (green). The residues in the selectivity filter and the outer vestibule of KV1.3 are the main residues interacting with the α-helix 21–24 of ShK, except for Arg11, which is located in the N-terminal region of the toxin. Both models show Lys22 occupying the pore.
ShK is more flexible than HmK
Using a combination of docking and long MD simulations (5 μs) we investigated the implications of conformational dynamics of ShK and HmK for binding to KV1.3 [95]. The ensemble of solution structures of ShK determined by NMR shows a range of configurations with disparate Lys22 Nη-Tyr23Oζ dyad heavy atom distances (PSB 1ROO) [78], but it was not known which configuration bound to KV1.3. In MD simulations of ShK in water the dyad distance varied between 3 and 12.5 Å, assuming three main configurations over the 5 μs trajectories [95]. In configuration 1, the dyad residues were close together (˜3 Å), associated with a partial unwinding of the helix residues 14–19 (adjacent to the helix 21–24 where the dyad is located), in configuration 2, an intermediate separation (˜5 Å) was observed, whereas in configuration 3, Lys22-Tyr23 were far apart (>7.5 Å). The same set of MD simulations were performed for HmK, but, in contrast with ShK, HmK has a considerably more rigid structure, with Lys22 Nζ and Tyr23 Oη remaining ˜3.25 Å apart over the 5 μs trajectory [95]. We used Principal Component Analysis (PCA) to investigate the fundamental motions in ShK and HmK by extracting the atomic positions of all atoms and building a covariance matrix, where the diagonalization gives the eigenvectors and eigenvalues that represents the principal motions [103]. Extracting the 20 structures from PC1 and PC2 for both peptides (Figure 6b), a clear difference in the dynamics is observed. While ShK displayed flexibility throughout its structure, with the helix 21–24 showing the greatest conformational changes, HmK was quite rigid except at N- and C-termini (Figure 6b). The C-terminal regions of ShK and HmK are largely identical, whereas several charge differences are present in the N-terminal region: Ile4 → Lys, Lys9 → Val, Arg11 → Glu, Ala14 → Asp, Gln16 → Arg, and His19 → Thr (ShK → HmK) (Figure 6a). These charge differences may strengthen intramolecular interactions in HmK compared to ShK, and thereby contribute to its greater rigidity.
Docking configurations of ShK and HmK in KV1.3 and comparison with cryo-EM structures
We further investigated whether the docking program HADDOCK [104] would be informative in comparing the three major configurations of ShK identified in MD simulations with the experimentally observed cryo-EM electron density map (PDB 7WF4). There are several literature reports suggesting that the binding of ShK to KV1.3 occurred by Lys22 occluding the channel pore [89,105] although alternative bound configurations have been proposed involving Lys22 lying between subunits of the KV1.3 channel instead of physically blocking the channel pore [106]. In contrast, electrophysiological assays of free- and membrane-tethered ShK were interpreted to indicate that Arg24 played a key role in channel blockade [102]. We therefore investigated the three major configurations of ShK using HADDOCK docking to KV1.3 [104]. The ShK configuration with best HADDOCK score and fewest violations compared to the cryo-EM density (PDB 7WF4) [53] had a dyad distance < 7.5 Å, which corresponds to configurations 1 or 2 in our MD simulations of ShK free in solution [95]. For HmK, where only one major state was identified, better HADDOCK scores were also found for configurations with Lys22 occupying the channel pore, suggesting that the difference in affinity is not due to alternative binding poses. We compared both ShK and HmK binding to KV1.3 through 100 ns MD simulations and monitored the peptide-channel interactions over the trajectories. This showed that in both ShK and HmK, the Lys22 side chain Nζ is stabilized in the pore through interactions with Tyr447 carbonyl oxygens of all four KV1.3 subunits (Figure 6c,d). The docked configuration of ShK is supported by the cryo-EM based structure of Fab-ShK bound to KV1.3 (Figure 5d) [62]. Intriguingly, the more flexible peptide ShK binds KV1.3 with higher affinity than the more rigid HmK, suggesting an interplay between peptide dynamics and channel binding that remains to be fully elucidated.
A nanobody stabilizes the C-type inactivated conformation of KV1.3
Van Hoorick and colleagues at Ablynx have developed and patented nanobodies targeting KV1.3 (patent WO2015193452A1) [107]. Meyerson and colleagues showed that one of these nanobodies, A0194009G09, significantly accelerated C-type inactivation of KV1.3, and determined the structure of KV1.3 bound to A0194009G09 to a resolution of 3.25 Å (PDB 7SSZ) [62]. Four nanobody molecules were attached to the KV1.3 tetramer, each bound to residues in the KV1.3 turret (Ala421, Pro424, Ser426, Gly427) and external loops of the VSD (Tyr265, Pro266) of one subunit (Figure 7a) [62]. Despite the lack of direct interaction with the selectivity filter, the nanobodies induced significant conformational changes in the outer selectivity filter, consistent with an exaggerated C-type inactivated conformation [62]. The selectivity filter residue Asp449 was oriented outwards, while Tyr447 adopted two positions, one similar to the D1 conformation of KV1.3 (PDB 7SSX), and the second with the hydroxyl group of Tyr447 oriented toward the extracellular space, a conformation designated D3 (PDB 8DFL), distinct from both D1 and D2 (PDB 7SSY) [62] (Figure 7b,c). The result was a widened outer selectivity filter with reduced K+ occupancy, a non-conducting C-type inactivated conformation. Although four nanobodies were seen bound in the structure, electrophysiological studies suggested that fewer nanobodies could bind to KV1.3, and higher occupancies sped up and promoted durable C-type inactivation [62].
Figure 7.
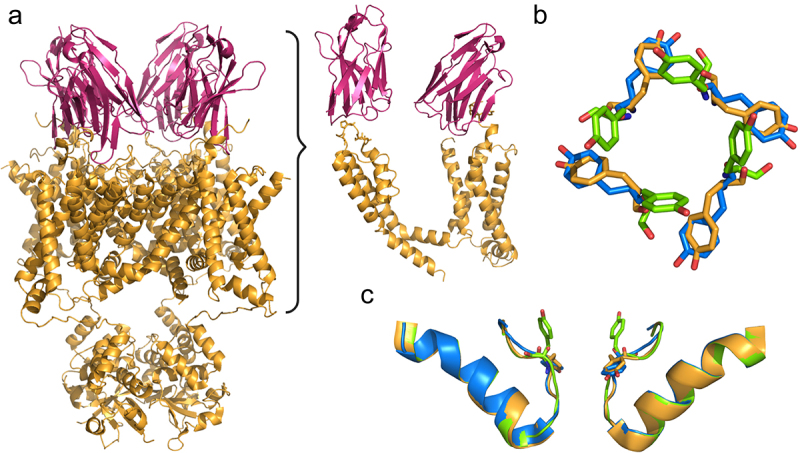
KV1.3 bound to the anti-KV1.3-nanobody A0194009G09. a) Four A0194009G09 nanobody molecules (pink) attached to the KV1.3 tetramer (yellow). A0194009G09 nanobody binds to residues in the KV1.3 turret and external loops of the VSD (A, right) (PDB 7SSZ). b) and c) Tyr447 adopted two conformations upon nanobody binding; the first conformation (yellow) is similar to D1 (PDB 7SSX, blue), while in the second conformation the hydroxyl group of Tyr447 is oriented toward the extracellular space in a conformation designated D3 (green) (PDB 8DFL), distinct from both D1 (PDB 7SSX) and D2 (PDB 7SSY).
Conclusion
Publication of the crystal structure of the bacterial potassium channel KcsA in 1998 [108] represented a major step forward in our understanding of the structural basis for K+ conduction. This structure enabled the development of more accurate homology models of the pore domains of voltage-gated potassium channels such as KV1.3, which in turn enhanced our understanding of how peptide ligands blocked the channel. Several of the current peptide inhibitors of KV1.3 were designed on the basis of these models. Subsequent structures of potassium channels further enhanced our under-standing of the open [59,109] and closed [110,111] conformations of KV channels. The recent determination by cryo-EM and X-ray crystallography structures of KV1.3, Shaker-IR (wild type and W434F mutant) and the inactivating KVChim mutant (W362F+S367T+V377T) reveal the mechanism whereby KV channels transition in a time-dependent manner from the open-conducting conformation into the non-conducting C-type inactivated state. This temporal change involves the breaking of a key intra-subunit hydrogen bond that tethers the selectivity filter to the pore-helix, allowing the selectivity filter to move outwards, resulting in dilation of the outer pore and the disruption of ion permeation. The recent structures of KV1.3 bound to various ligands enhance our understanding of KV1.3 pharmacology. Binding of the peptide blockers dalazatide and Fab-ShK to the KV1.3 outer vestibule was shown to narrow and stabilize the selectivity filter in the open-conducting conformation, with K+ efflux prevented by pore occlusion via the interaction of Lys22 of ShK with the backbone carbonyl of Tyr447 of KV1.3. Electrophysiological studies show that ShK blocks KV1.3 with considerably higher potency than its closely related analog HmK, despite being more flexible (as shown by MD simulations). In contrast, binding of anti-KV1.3 nanobody A0194009G09 to the turret and residues in the external loops of the VSD widens the outer selectivity filter in an exaggerated C-type inactivated conformation. These studies provide the framework for future studies to define the mechanism of slow inactivation and determine how small molecule and peptide inhibitors promote the slow inactivated conformation in KV channels, and will facilitate the design and development of next-generation KV1.3-targeted immuno-therapeutics.
Acknowledgments
We thank Ong Seow Theng and Stephanie Shee Min Goay for assistance with Figures 1 and 2. We thank Heike Wulff and Dorothy Wai for critiquing our manuscript. The publication costs for this paper were partially paid by the Washington Foundation for Molecular Pharmacology.
Funding Statement
This work was funded in part by the Australian Research Council Centre for Fragment-Based Design [IC180100021].
Disclosure statement
KGC is the co-inventor of a patent on dalazatide that has been licensed by the University of California Irvine to TEKv Therapeutics.
Data availability statement
All structures discussed in this review are available in the Protein Data Bank.
Author contributions
George Chandy and Ray Norton drafted and edited the manuscript. Karoline Sanches prepared the figures and figure legends and edited the manuscript.
References
- [1].DeCoursey TE, Chandy KG, Gupta S, et al. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307(5950):465–20. doi: 10.1038/307465a0 [DOI] [PubMed] [Google Scholar]
- [2].Matteson DR, Deutsch C.. K channels in T lymphocytes: a patch clamp study using monoclonal antibody adhesion. Nature. 1984;307(5950):468–471. doi: 10.1038/307468a0 [DOI] [PubMed] [Google Scholar]
- [3].Chandy KG, DeCoursey TE, Cahalan MD, et al. Voltage-gated potassium channels are required for human T lymphocyte activation. J Exp Med. 1984;160(2):369–385. doi: 10.1084/jem.160.2.369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cahalan MD, Chandy KG, DeCoursey TE, et al. A voltage-gated potassium channel in human T lymphocytes. J Physiol. 1985;358(1):197–237. doi: 10.1113/jphysiol.1985.sp015548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Price M, Lee SC, Deutsch C. Charybdotoxin inhibits proliferation and interleukin 2 production in human peripheral blood lymphocytes. Proc Natl Acad Sci U S A. 1989;86(24):10171–10175. doi: 10.1073/pnas.86.24.10171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Douglass J, Osborne PB, Cai YC, et al. Characterization and functional expression of a rat genomic DNA clone encoding a lymphocyte potassium channel. J Immunol. 1990;144(12):4841–4850. doi: 10.4049/jimmunol.144.12.4841 [DOI] [PubMed] [Google Scholar]
- [7].Grissmer S, Dethlefs B, Wasmuth JJ, et al. Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc Natl Acad Sci U S A. 1990;87(23):9411–9415. doi: 10.1073/pnas.87.23.9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev. 2009;231(1):59–87. doi: 10.1111/j.1600-065X.2009.00816.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. 2015;33(1):291–353. doi: 10.1146/annurev-immunol-032414-112212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Erdogmus S, Concepcion AR, Yamashita M, et al. Cavbeta1 regulates T cell expansion and apoptosis independently of voltage-gated Ca2+ channel function. Nat Commun. 2022;13(1):2033. doi: 10.1038/s41467-022-29725-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang SL, Yu Y, Roos J, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902–905. doi: 10.1038/nature04147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–185. doi: 10.1038/nature04702 [DOI] [PubMed] [Google Scholar]
- [13].Vig M, Peinelt C, Beck A, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312(5777):1220–1223. doi: 10.1126/science.1127883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang SL, Yeromin AV, Zhang XH, et al. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci U S A. 2006;103(24):9357–9362. doi: 10.1073/pnas.0603161103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Oh-Hora M, Yamashita M, Hogan PG, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9(4):432–443. doi: 10.1038/ni1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shaw PJ, Weidinger C, Vaeth M, et al. CD4+ and CD8+ T cell-dependent antiviral immunity requires STIM1 and STIM2. J Clin Invest. 2014;124(10):4549–4563. doi: 10.1172/JCI76602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vaeth M, Yang J, Yamashita M, et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat Commun. 2017;8(1):14714. doi: 10.1038/ncomms14714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chandy KG, Norton RS. Peptide blockers of KV1.3 channels in T cells as therapeutics for autoimmune disease. Curr Opin Chem Biol. 2017;38:97–107. doi: 10.1016/j.cbpa.2017.02.015 [DOI] [PubMed] [Google Scholar]
- [19].Wulff H, Christophersen P, Colussi P, et al. Antibodies and venom peptides: new modalities for ion channels. Nat Rev Drug Discov. 2019;18(5):339–357. doi: 10.1038/s41573-019-0013-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nguyen HM, Grossinger EM, Horiuchi M, et al. Differential KV1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia. 2017;65(1):106–121. doi: 10.1002/glia.23078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nguyen HM, Blomster LV, Christophersen P, et al. Potassium channel expression and function in microglia: plasticity and possible species variations. Channels (Austin). 2017;11(4):305–315. doi: 10.1080/19336950.2017.1300738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Immler R, Nadolni W, Bertsch A, et al. The voltage-gated potassium channel KV1.3 regulates neutrophil recruitment during inflammation. Cardiovasc Res. 2022;118(5):1289–1302. doi: 10.1093/cvr/cvab133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tajti G, Wai DCC, Panyi G, et al. The voltage-gated potassium channel KV1.3 as a therapeutic target for venom-derived peptides. Biochem Pharmacol. 2020;181:114146. doi: 10.1016/j.bcp.2020.114146 [DOI] [PubMed] [Google Scholar]
- [24].Beeton C, Wulff H, Barbaria J, et al. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci U S A. 2001;98(24):13942–13947. doi: 10.1073/pnas.241497298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wulff H, Calabresi PA, Allie R, et al. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J Clin Invest. 2003;111(11):1703–1713. doi: 10.1172/JCI16921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fung-Leung WP, Edwards W, Liu Y, et al. T cell subset and stimulation strength-dependent modulation of T cell activation by Kv1.3 blockers. PLoS One. 2017;12(1):e0170102. doi: 10.1371/journal.pone.0170102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chiang EY, Li T, Jeet S, et al. Potassium channels KV1.3 and KCa3.1 cooperatively and compensatorily regulate antigen-specific memory T cell functions. Nat Commun. 2017;8(1):14644. doi: 10.1038/ncomms14644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rus H, Pardo CA, Hu L, et al. The voltage-gated potassium channel KV1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci U S A. 2005;102(31):11094–11099. doi: 10.1073/pnas.0501770102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Beeton C, Wulff H, Standifer NE, et al. KV1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A. 2006;103(46):17414–17419. doi: 10.1073/pnas.0605136103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kundu-Raychaudhuri S, Chen YJ, Wulff H, et al. KV1.3 in psoriatic disease: PAP-1, a small molecule inhibitor of Kv1.3 is effective in the SCID mouse psoriasis–xenograft model. J Autoimmun. 2014;55:63–72. doi: 10.1016/j.jaut.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tarcha EJ, Olsen CM, Probst P, et al. Safety and pharmacodynamics of dalazatide, a KV1.3 channel inhibitor, in the treatment of plaque psoriasis: a randomized phase 1b trial. PLoS One. 2017;12(7):e0180762. doi: 10.1371/journal.pone.0180762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Land J, Lintermans LL, Stegeman CA, et al. KV1.3 channel blockade modulates the effector function of B cells in Granulomatosis with polyangiitis. Front Immunol. 2017;8:1205. doi: 10.3389/fimmu.2017.01205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lintermans LL, Stegeman CA, Munoz-Elias EJ, et al. KV1.3 blockade by ShK186 modulates CD4+ effector memory T-cell activity of patients with Granulomatosis with polyangiitis. Rheumatology (Oxford). 2023. doi: 10.1093/rheumatology/kead192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gocke AR, Lebson LA, Grishkan IV, et al. Kv1.3 deletion biases T cells toward an immunoregulatory phenotype and renders mice resistant to autoimmune encephalomyelitis. J Immunol. 2012;188(12):5877–5886. doi: 10.4049/jimmunol.1103095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Grishkan IV, Tosi DM, Bowman MD, et al. Antigenic stimulation of KV1.3-deficient Th cells gives rise to a population of Foxp3-independent T cells with suppressive properties. J Immunol. 2015;195(4):1399–1407. doi: 10.4049/jimmunol.1403024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Unterweger AL, Jensen MO, Giordanetto F, et al. Suppressing KV1.3 ion channel activity with a novel small molecule inhibitor ameliorates inflammation in a humanised mouse model of ulcerative colitis. J Crohns Colitis. 2021;15(11):1943–1958. doi: 10.1093/ecco-jcc/jjab078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fordyce CB, Jagasia R, Zhu X, et al. Microglia KV1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25(31):7139–7149. doi: 10.1523/JNEUROSCI.1251-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sarkar S, Nguyen HM, Malovic E, et al. KV1.3 modulates neuroinflammation and neurodegeneration in Parkinson’s disease. J Clin Invest. 2020;130(8):4195–4212. doi: 10.1172/JCI136174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rangaraju S, Dammer EB, Raza SA, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13(1):24. doi: 10.1186/s13024-018-0254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Maezawa I, Nguyen HM, Di Lucente J, et al. KV1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: preclinical proof of concept. Brain. 2018;141(2):596–612. doi: 10.1093/brain/awx346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ramesha S, Rayaprolu S, Bowen CA, et al. Unique molecular characteristics and microglial origin of KV1.3 channel-positive brain myeloid cells in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2021;118(11):e2013545118. doi: 10.1073/pnas.2013545118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang X, Liang P, Zhang Y, et al. Blockade of KV1.3 potassium channel inhibits microglia-mediated neuroinflammation in epilepsy. Int J Mol Sci. 2022;23(23):14693. doi: 10.3390/ijms232314693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nicolazzo JA, Pan Y, Di Stefano I, et al. Blockade of Microglial KV1.3 potassium channels by the peptide HsTX1[R14A] attenuates lipopolysaccharide-mediated neuroinflammation. J Pharm Sci. 2022;111(3):638–647. doi: 10.1016/j.xphs.2021.11.003 [DOI] [PubMed] [Google Scholar]
- [44].Pan Y, Kagawa Y, Sun J, et al. Peripheral administration of the KV1.3-blocking peptide HsTX1[R14A] improves cognitive performance in senescence accelerated SAMP8 mice. Neurotherapeutics. 2023;20(4):1198–1214. doi: 10.1007/s13311-023-01387-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen YJ, Cui Y, Singh L, et al. The potassium channel KV1.3 as a therapeutic target for immunocytoprotection after reperfusion. Ann Clin Transl Neurol. 2021;8(10):2070–2082. doi: 10.1002/acn3.51456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mei Y, Fang C, Ding S, et al. PAP-1 ameliorates DSS-induced colitis with involvement of NLRP3 inflammasome pathway. Int Immunopharmacol. 2019;75:105776. doi: 10.1016/j.intimp.2019.105776 [DOI] [PubMed] [Google Scholar]
- [47].Leanza L, Romio M, Becker KA, et al. Direct pharmacological targeting of a mitochondrial ion channel selectively kills tumor cells in vivo. Cancer Cell. 2017;31(4):516–531 e10. doi: 10.1016/j.ccell.2017.03.003 [DOI] [PubMed] [Google Scholar]
- [48].Kadow S, Schumacher F, Kramer M, et al. Mitochondrial KV1.3 channels as target for treatment of multiple Myeloma. Cancers (Basel). 2022;14(8):1955. doi: 10.3390/cancers14081955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Koni PA, Khanna R, Chang MC, et al. Compensatory anion currents in KV1.3 channel-deficient thymocytes. J Biol Chem. 2003;278(41):39443–39451. doi: 10.1074/jbc.M304879200 [DOI] [PubMed] [Google Scholar]
- [50].Fadool DA, Tucker K, Perkins R, et al. KV1.3 channel gene-targeted deletion produces “Super-Smeller mice” with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron. 2004;41(3):389–404. doi: 10.1016/S0896-6273(03)00844-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xu J, Wang P, Li Y, et al. The voltage-gated potassium channel KV1.3 regulates peripheral insulin sensitivity. Proc Natl Acad Sci U S A. 2004;101(9):3112–3117. doi: 10.1073/pnas.0308450100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Clinicaltrials.gov . A first-in-human study to evaluate the safety, tolerability, and efficacy of Si-544 in adults with atopic dermatitis. https://classic.clinicaltrials.gov/ct2/show/NCT053833782022.
- [53].Tyagi A, Ahmed T, Jian S, et al. Rearrangement of a unique KV1.3 selectivity filter conformation upon binding of a drug. PNAS. 2022;119(5):e2113536119. doi: 10.1073/pnas.2113536119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Panyi G, Sheng Z, Deutsch C. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys J. 1995;69(3):896–903. doi: 10.1016/S0006-3495(95)79963-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Panyi G, Deutsch C. Cross talk between activation and slow inactivation gates of Shaker potassium channels. J Gen Physiol. 2006;128(5):547–559. doi: 10.1085/jgp.200609644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hoshi T, Armstrong CM. C-type inactivation of voltage-gated K+ channels: pore constriction or dilation? J Gen Physiol. 2013;141(2):151–160. doi: 10.1085/jgp.201210888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ong ST, Tyagi A, Chandy KG, et al. Mechanisms underlying C-type inactivation in KV channels: lessons from structures of human KV1.3 and fly Shaker-IR channels. Front Pharmacol. 2022;13:924289. doi: 10.3389/fphar.2022.924289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309(5736):897–903. doi: 10.1126/science.1116269 [DOI] [PubMed] [Google Scholar]
- [59].Long SB, Tao X, Campbell EB, et al. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450(7168):376–382. doi: 10.1038/nature06265 [DOI] [PubMed] [Google Scholar]
- [60].Tan XF, Bae C, Stix R, et al. Structure of the Shaker KV channel and mechanism of slow C-type inactivation. Sci Adv. 2022;8(11):eabm7814. doi: 10.1126/sciadv.abm7814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Reddi R, Matulef K, Riederer EA, et al. Structural basis for C-type inactivation in a Shaker family voltage-gated K+ channel. Sci Adv. 2022;8(16):eabm8804. doi: 10.1126/sciadv.abm8804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Selvakumar P, Fernandez-Marino AI, Khanra N, et al. Structures of the T cell potassium channel KV1.3 with immunoglobulin modulators. Nat Commun. 2022;13(1):3854. doi: 10.1038/s41467-022-31285-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu S, Zhao Y, Dong H, et al. Structures of wild-type and H451N mutant human lymphocyte potassium channel KV1.3. Cell Discov. 2021;7(1):39. doi: 10.1038/s41421-021-00269-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Grissmer S, Cahalan MD. Divalent ion trapping inside potassium channels of human T lymphocytes. J Gen Physiol. 1989;93(4):609–630. doi: 10.1085/jgp.93.4.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Levy DI, Deutsch C. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys J. 1996;70(2):798–805. doi: 10.1016/S0006-3495(96)79619-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lopez-Barneo J, Hoshi T, Heinemann SH, et al. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Recept Channels. 1993;1(1):61–71. [PubMed] [Google Scholar]
- [67].Wu X, Gupta K, Swartz KJ. Mutations within the selectivity filter reveal that KV1 channels have distinct propensities to slow inactivate. J Gen Physiol. 2022;154(11). doi: 10.1085/jgp.202213222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Busch AE, Hurst RS, North RA, et al. Current inactivation involves a histidine residue in the pore of the rat lymphocyte potassium channel RGK5. Biochem Biophys Res Commun. 1991;179(3):1384–1390. doi: 10.1016/0006-291X(91)91726-S [DOI] [PubMed] [Google Scholar]
- [69].Nguyen A, Kath JC, Hanson DC, et al. Novel nonpeptide agents potently block the C-type inactivated conformation of KV1.3 and suppress T cell activation. Mol Pharmacol. 1996;50(6):1672–1679. [PubMed] [Google Scholar]
- [70].Somodi S, Varga Z, Hajdu P, et al. pH-dependent modulation of KV1.3 inactivation: role of His399. Am J Physiol Cell Physiol. 2004;287(4):C1067–76. doi: 10.1152/ajpcell.00438.2003 [DOI] [PubMed] [Google Scholar]
- [71].Pless SA, Galpin JD, Niciforovic AP, et al. Hydrogen bonds as molecular timers for slow inactivation in voltage-gated potassium channels. Elife. 2013;2:e01289. doi: 10.7554/eLife.01289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lueck JD, Mackey AL, Infield DT, et al. Atomic mutagenesis in ion channels with engineered stoichiometry. Elife. 2016;5. doi: 10.7554/eLife.18976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang Y, Zhang X, Liu C, et al. Regulation of K+ conductance by a hydrogen bond in KV2.1, KV2.2, and KV1.2 channels. Membranes (Basel). 2021 Mar 9;11(3):190. doi: 10.3390/membranes11030190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Aiyar J, Rizzi JP, Gutman GA, et al. The signature sequence of voltage-gated potassium channels projects into the external vestibule. J Biol Chem. 1996;271(49):31013–31016. doi: 10.1074/jbc.271.49.31013 [DOI] [PubMed] [Google Scholar]
- [75].Castañeda O, Sotolongo V, Amor AM, et al. Characterization of a potassium channel toxin from the caribbean sea anemone Stichodactyla helianthus. Toxicon. 1995;33(5):603–613. doi: 10.1016/0041-0101(95)00013-C [DOI] [PubMed] [Google Scholar]
- [76].Kalman K, Pennington MW, Lanigan MD, et al. ShK-Dap22, a potent KV1.3-specific immunosuppressive polypeptide. J Biol Chem. 1998;273(49):32697–32707. doi: 10.1074/jbc.273.49.32697 [DOI] [PubMed] [Google Scholar]
- [77].Chi V, Pennington MW, Norton RS, et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon. 2012;59(4):529–546. doi: 10.1016/j.toxicon.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Tudor JE, Pallaghy PK, Pennington MW, et al. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat Struct Mol Biol. 1996;3(4):317–320. doi: 10.1038/nsb0496-317 [DOI] [PubMed] [Google Scholar]
- [79].Rashid MH, Huq R, Tanner MR, et al. A potent and KV1.3-selective analog of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases. Sci Rep. 2014;4(1):4509. doi: 10.1038/srep04509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lebrun B, Romi-Lebrun R, Martin-Eauclaire MF, et al. A four-disulphide-bridged toxin, with high affinity towards voltage-gated K+ channels, isolated from Heterometrus spinnifer (Scorpionidae) venom. Biochem J. 1997;328(Pt 1):321–327. doi: 10.1042/bj3280321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Dauplais M, Lecoq A, Song J, et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J Biol Chem. 1997;272(7):4302–4309. doi: 10.1074/jbc.272.7.4302 [DOI] [PubMed] [Google Scholar]
- [82].Chhabra S, Chang SC, Nguyen HM, et al. KV1.3 channel-blocking immunomodulatory peptides from parasitic worms: implications for autoimmune diseases. FASEB J. 2014;28(9):3952–3964. doi: 10.1096/fj.14-251967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].UniProt C, Martin M-J, Orchard S. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 2023;51(D1):D523–D531. doi: 10.1093/nar/gkac1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shafee T, Mitchell ML, Norton RS. Mapping the chemical and sequence space of the ShKT superfamily. Toxicon. 2019;165:95–102. doi: 10.1016/j.toxicon.2019.04.008 [DOI] [PubMed] [Google Scholar]
- [85].Rangaraju S, Khoo KK, Feng ZP, et al. Potassium channel modulation by a toxin domain in matrix metalloprotease 23. J Biol Chem. 2010;285(12):9124–9136. doi: 10.1074/jbc.M109.071266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Galea CA, Nguyen HM, George Chandy K, et al. Domain structure and function of matrix metalloprotease 23 (MMP23): role in potassium channel trafficking. Cell Mol Life Sci. 2014;71(7):1191–1210. doi: 10.1007/s00018-013-1431-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Nguyen HM, Galea CA, Schmunk G, et al. Intracellular trafficking of the KV1.3 potassium channel is regulated by the prodomain of a matrix metalloprotease. J Biol Chem. 2013;288(9):6451–6464. doi: 10.1074/jbc.M112.421495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Rauer H, Pennington M, Cahalan M, et al. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J Biol Chem. 1999;274(31):21885–21892. doi: 10.1074/jbc.274.31.21885 [DOI] [PubMed] [Google Scholar]
- [89].Lanigan MD, Kalman K, Lefievre Y, et al. Mutating a critical lysine in ShK toxin alters its binding configuration in the pore-vestibule region of the voltage-gated potassium channel, KV1.3. Biochemistry. 2002;41(40):11963–11971. doi: 10.1021/bi026400b [DOI] [PubMed] [Google Scholar]
- [90].Beeton C, Pennington MW, Wulff H, et al. Targeting effector memory T cells with a selective peptide inhibitor of KV1.3 channels for therapy of autoimmune diseases. Mol Pharmacol. 2005;67(4):1369–1381. doi: 10.1124/mol.104.008193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Pennington MW, Beeton C, Galea CA, et al. Engineering a stable and selective peptide blocker of the KV1.3 channel in T lymphocytes. Mol Pharmacol. 2009;75(4):762–773. doi: 10.1124/mol.108.052704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Chang SC, Huq R, Chhabra S, et al. N-Terminally extended analogs of the K+ channel toxin from Stichodactyla helianthus as potent and selective blockers of the voltage-gated potassium channel KV1.3. FEBS J. 2015;282(12):2247–2259. doi: 10.1111/febs.13294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zachariae U, Schneider R, Velisetty P, et al. The molecular mechanism of toxin-induced conformational changes in a potassium channel: relation to C-type inactivation. Structure. 2008;16(5):747–754. doi: 10.1016/j.str.2008.01.018 [DOI] [PubMed] [Google Scholar]
- [94].Lange A, Giller K, Hornig S, et al. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440(7086):959–962. doi: 10.1038/nature04649 [DOI] [PubMed] [Google Scholar]
- [95].Sanches K, Prypoten V, Chandy KG, et al. Interaction of the inhibitory peptides ShK and HmK with the voltage-gated potassium channel KV1.3: role of conformational dynamics. J Chem Inf Model. 2023;63(10):3043–3053. doi: 10.1021/acs.jcim.2c01237 [DOI] [PubMed] [Google Scholar]
- [96].Sanches K, Wai DCC, Norton RS. Conformational dynamics in peptide toxins: implications for receptor interactions and molecular design. Toxicon. 2021;201(15):127–140. doi: 10.1016/j.toxicon.2021.08.020 [DOI] [PubMed] [Google Scholar]
- [97].Sher I, Chang SC, Li Y, et al. Conformational flexibility in the binding surface of the potassium channel blocker ShK. Chembiochem. 2014;15(16):2402–2410. doi: 10.1002/cbic.201402295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Meirovitch E, Tchaicheeyan O, Sher I, et al. Structural dynamics of the potassium channel blocker ShK: SRLS analysis of 15N relaxation. J Phys Chem B. 2015;119(49):15130–15137. doi: 10.1021/acs.jpcb.5b07875 [DOI] [PubMed] [Google Scholar]
- [99].Iwakawa N, Baxter NJ, Wai DCC, et al. Conformational exchange in the potassium channel blocker ShK. Sci Rep. 2019;9(1):19307. doi: 10.1038/s41598-019-55806-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gendeh GS, Young LC, de Medeiros CL, et al. A New potassium channel toxin from the sea anemone Heteractis magnifica: isolation, cDNA cloning, and functional expression. Biochemistry. 1997;36(38):11461–11471. doi: 10.1021/bi970253d [DOI] [PubMed] [Google Scholar]
- [101].Pennington MW, Chang SC, Chauhan S, et al. Development of highly selective KV1.3-blocking peptides based on the sea anemone peptide ShK. Mar Drugs. 2015;13(1):529–542. doi: 10.3390/md13010529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhao R, Dai H, Mendelman N, et al. Tethered peptide neurotoxins display two blocking mechanisms in the K+ channel pore as do their untethered analogs. Sci Adv. 2020;6(10):eaaz3439. doi: 10.1126/sciadv.aaz3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11(3):205–217. doi: 10.1002/prot.340110305 [DOI] [PubMed] [Google Scholar]
- [104].De Vries SJ, Van Dijk M, AMJJ B. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5(5):883–897. doi: 10.1038/nprot.2010.32 [DOI] [PubMed] [Google Scholar]
- [105].Rashid MH, Kuyucak S. Affinity and selectivity of ShK toxin for the KV1 potassium channels from free energy simulations. J Phys Chem B. 2012;116(16):4812–4822. doi: 10.1021/jp300639x [DOI] [PubMed] [Google Scholar]
- [106].Jin L, Wu Y. Molecular mechanism of the sea anemone toxin ShK recognizing the KV1.3 channel explored by docking and molecular dynamic simulations. J Chem Inf Model. 2007;47(5):1967–1972. doi: 10.1021/ci700178w [DOI] [PubMed] [Google Scholar]
- [107].Stortelers C, Pinto-Espinoza C, Van Hoorick D, et al. Modulating ion channel function with antibodies and nanobodies. Curr Opin Immunol. 2018;52:18–26. doi: 10.1016/j.coi.2018.02.003 [DOI] [PubMed] [Google Scholar]
- [108].Doyle DA, Morais Cabral J, Pfuetzner RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69 [DOI] [PubMed] [Google Scholar]
- [109].Wang W, MacKinnon R. Cryo-EM structure of the open human ether-a-go-go-related K+ channel hERG. Cell. 2017;169(3):422–430 e10. doi: 10.1016/j.cell.2017.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zhou Y, Morais-Cabral JH, Kaufman A, et al. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414(6859):43–48. doi: 10.1038/35102009 [DOI] [PubMed] [Google Scholar]
- [111].Whicher JR, MacKinnon R. Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science. 2016;353(6300):664–669. doi: 10.1126/science.aaf8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All structures discussed in this review are available in the Protein Data Bank.