

Abstract
Children of consanguineous unions carry long runs of homozygosity (ROH) in their genomes, due to their parents’ recent shared ancestry. This increases the burden of recessive disease in populations with high levels of consanguinity and has been heavily studied in some groups. However, there has been little investigation of the broader effect of consanguinity on patterns of genetic variation on a global scale. This study, which collected published genetic data and information about marriage practice from 395 worldwide populations, shows that reported preference for cousin marriage has a detectable association with the distribution of long ROH in this sample, increasing the expected number of ROH longer than 10 cM by a factor of 2.2. Variation in marriage practice and consequent rates of consanguinity are therefore an important aspect of demographic history for the purposes of modeling human genetic variation. However, reported marriage practices explain a relatively small proportion of the variation in ROH distribution, and consequently, population genetic data are only partially informative about cultural preferences.
Keywords: CONSANGUINITY, RUNS OF HOMOZYGOSITY, ROH, COUSIN MARRIAGE
Marriage practices have consequences for human genetic variation. One extensively debated and regulated practice is consanguinity, the union of closely related individuals. An estimated 10.4% of the world’s population comprises couples who are second cousins or closer and their offspring (Bittles and Black 2010; for additional estimates of worldwide consanguinity, see Romeo and Bittles 2014; see also Consang.net, https://www.consang.net/). The children of consanguineous marriages have longer runs of homozygosity (ROH) (Ceballos et al. 2018) and therefore carry a greater recessive disease burden (Szpiech et al. 2013), equivalent to an increase in child mortality or severe disease of approximately 3–4% (Bittles and Neel 1994; Sheridan et al. 2013; Mobarak et al. 2019). However, it is unclear to what extent ethnographic assessments of marriage practices (via either surveys or direct observation) at the population level translate to the genetic signature associated with cousin marriage.
Although actual levels of consanguinity must be correlated with patterns of genetic variation, there are three reasons that ethnographic assessments of cousin marriage prevalence might not be correlated. First, different cultures’ understanding of “cousin” marriage could cover different concepts. For example, some might have a clear distinction between first and second cousin marriage, while others consider all of these types the same. Second, cultural preferences for marriage may change rapidly. Ethnographic data may be outdated, or preferences may not persist long enough to have a detectable effect on patterns of genetic variation diagnostic of ongoing consanguinity. Finally, genomic signatures of consanguinity may be obscured by other demographic factors such as endogamy.
For these reasons, populations with a preference for cousin marriage may in fact demonstrate relatively few of the negative genetic consequences associated with increased homozygosity. For example, many marriages might occur between “cousins” who are actually quite distantly related. Alternatively, in many regions of the world, cousin marriage co-occurs with endogamy (marriage within defined subgroups such as castes) (Wall et al. 2020). Since endogamy can itself lead to excess recessive disease burden (Nakatsuka et al. 2017), the additional effect of consanguinity might be smaller. Finally, even though both endogamy and consanguinity, by increasing ROH, increase the risk of recessive disease in the short term, the same process exposes deleterious recessive alleles to selection, purging them more effectively from the population in the long term.
Most previous work on the relationship between consanguinity and genetic variation has focused on a limited number of populations. McQuillan et al. (2008) correlated pedigree-based measures of marriage practices to ROH in the Orcadian population. Kang et al. (2016) found a correlation between long ROH in present-day individuals and inbreeding coefficients for nine Jewish populations, with rates of consanguinity based on survey data. Arciero et al. (2021) found a strong correlation between survey-measured consanguinity and long ROH in a British Pakistani cohort. In the study most similar to ours, Pemberton and Rosenberg (2014) found a correlation of 0.349 between inbreeding coefficients estimated from genetic data and survey-based consanguinity data. However, their analysis was based on only 26 populations and did not correct for correlations among populations due to shared demographic history.
We therefore set out to test whether a cultural preference for cousin marriage is detectable in genetic data from a worldwide sample of 3,849 individuals from 395 populations. We categorized these populations into those prohibiting (43%), allowing (12%), and preferring (45%) cousin marriage using publicly available ethnographic sources. We found that ethnographic measures of preferences for cousin marriage are detectably related to the distribution of long ROH, increasing the expected number of ROH (NROH) longer than 10 Mb by 2.2× in populations preferring cousin marriages over those that prohibit it (P = 1.25 × 10−3), after controlling for 10 principal components (PCs) of genetic variation (gPCs), shorter ROH, and heterozygosity (collectively serving as proxies for demographic events such as bottlenecks, admixture, and endogamy). We show that this effect corresponds to an increase of 0.19 in absolute proportion of first cousin unions in populations where ethnographic data indicated a preference for the practice. Further, we found that populations preferring consanguinity show more long ROH than do geographically and genetically “close” consanguinity-prohibiting populations in a matched-pair design.
Methods
Simulations
First, we carried out simulations to investigate the effect of degree (proportion of cousin unions) and duration (number of generations) of consanguinity on the distribution of ROH. We used cousin-sim (Finke et al. 2021) to generate pedigrees, each of which was initialized with 1,000 unrelated founders and 1,000 individuals in subsequent generations for either 10 or 50 generations. In each generation, of all unions were between cousins, of which 62% were first-, 23% were second-, and 15% were third-degree cousins (NIPS and ICF 2019). The simulation does not enforce a mating pair to be cousins through a single path (e.g., a pair of first cousins may also be third cousins through a different path in the pedigree). The degree to which this occurs is a function of the population size, which in our case is 1,000. We used the pedigree as input to the pedigree simulator ped-sim (Caballero et al. 2019) with a Poisson crossover model and a refined genetic map of the human genome from Bherer et al. (2017) to simulate chromosomal segments and retained only those that were identical by descent within an individual. We refer to these segments as runs of homozygosity (ROH) to distinguish them from segments that are identical by descent between individuals.
We also calculated the expected number and total length of long ROH analytically using the model described in Ringbauer et al. (2021). Briefly, meioses separate the two chromosomes of an offspring of nth cousins. For such an offspring, the number of segments of length in the ith chromosome of length (in morgans) has the following density (Equation S8 of Ringbauer et al. 2021):
| (1) |
The integrals
yield the expected number and total length, respectively, of long (>10 cM or 0.1 M) ROH on the ith chromosome. This genetic length corresponds to approximately 10 Mb physical distance, on average, across the genome. We then computed the expected autosome-wide number and total length as
respectively, using the sex-averaged genetic map from Bherer et al. (2017) for chromosomal lengths (in morgans).
Genetic Data Collection and Processing
We collected genotype data generated with the Affymetrix Human Origins array from six reports (Pickrell et al. 2012; Lazaridis et al. 2014, 2016; Nakatsuka et al. 2017; Lipson et al. 2018; Jeong et al. 2019) and merged this with whole-genome sequence data from three further reports (Mallick et al. 2016; Pagani et al. 2016; Fan et al. 2019). We merged all data sets and removed (a) apparent duplicate samples (identified using PLINK --genome), (b) samples labeled as “questionable” or “ignore” in the original publications, and (c) samples where the population label was too generic to be useful (e.g., “scheduled_caste” in South Asian data). We retained the population labels as specified in the original reports except where there was an obvious typo or synonymous label. We merged populations that had the same labels or that differed only in capitalization or minor spelling differences. Our genetic data set contained 4,544 individuals from 488 populations (median number of individuals per population = 7; range = 1–71). We restricted to autosomes, leaving 469,421 SNPs.
For each individual, we calculated the number of ROH (NROH) greater than 1, 2, 5, and 10 Mb (using the --homozyg command in PLINK). We then calculated the NROH greater than 1, 2, 5, and 10 cM by estimating the genetic length of each ROH using the combined recombination map from HapMap2 (International HapMap Consortium 2007). Note that, even when using genetic length, we considered only segments longer than 1 Mb. We then calculated the NROH as follows: NROH1 represents the NROH longer than 1 cM but shorter than 2 cM; NROH5, longer than 5 cM but shorter than 10 cM; and so on. Similarly, we calculated the total length of ROH (LROH) in each individual such that LROH1 represents the total LROH longer than 1 cM but less than 2 cM, and so on. We estimated the proportion of genotyped SNPs that are heterozygous by using the --het command in PLINK, excluding ROH called in each individual. Finally, we computed PCs of the merged data set, and also separately of individuals belonging to the 237 South Asian populations. We used PLINK v1.90 (Chang et al. 2015) and smartpca v16000 (Patterson et al. 2006) for all analyses.
Ethnographic Data Collection and Processing
We collected data on marriage practices from the following sources:
The electronic eHRAF (Human Relations Area Files) World Cultures database (https://ehrafworldcultures.yale.edu/ehrafe/, accessed March–June 2020), which catalogs a large set of ethnographic writing describing cultural and social aspects of different groups.
The Ethnographic Atlas (Murdock 1967), which describes the cultural practices of 1,291 societies.
Focused online searches for marital practices in specific groups. Sources include Google Scholar, Google Books, and other papers and books. To search for specific populations, we searched for terms such as “marriage”, “cousin marriage”, “endogamy”, “exogamy”, “consanguinity”, and “consanguineous” along with the name of the population.
We encoded the level of consanguinity in a population categorically as prohibited (coded 0), permitted (1), or preferred (2). When quantitative measures (e.g., prevalence of cousin unions in the population) were available, we designated populations to the above three categories based on the percentage of unions between first cousins and first cousins once removed. In such cases, a prevalence of 0–2.5% was encoded as prohibited, 2.5–20% as permitted, and 20% and above as preferred. When quantitative measures were not available, we analyzed ethnographic records for terms indicating the preference for cousin unions: groups that stated cousin marriage was “common,” “practiced,” “encouraged,” or equivalent were classified as preferred; “allowed,” “occasional,” or “present but uncommon,” as permitted; and “forbidden” or “barred” or marriage was “exogamous,” as prohibited. We classified groups where information was unclear or unavailable as “missing.” We gathered marriage practice information for 522 populations, which was reduced to 486 (and 3,849 individuals) after merging with the genetic data (see Table 1 and Supplementary Data File S1). This was further reduced to a final set of 395 after we removed populations with missing consanguinity information.
Table 1.
Consanguinity Preferences by Region
| Region | Consanguinity Preferencea | ||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | Missing | Total | |
| Americas | 8 | 0 | 8 | 2 | 18 |
| Central Asia and Siberia | 12 | 5 | 14 | 8 | 39 |
| East Asia | 11 | 5 | 11 | 1 | 28 |
| Middle East and North Africa | 3 | 5 | 22 | 1 | 31 |
| Oceania | 2 | 0 | 1 | 0 | 3 |
| South Asia | 64 | 25 | 93 | 54 | 236 |
| Sub-Saharan Africa | 33 | 3 | 16 | 11 | 63 |
| Western Eurasia | 37 | 6 | 11 | 14 | 68 |
0 = prohibited, 1 = accepted, 2 = preferred, missing = consanguinity could not be assigned.
To validate our consanguinity scoring, we tested whether our assignments correlated with individual survey data on marriage practices from the 2011 India Health and Development Survey (IHDS) (Desai and Vanneman 2018). We matched populations of individuals from the IHDS survey to population names in our genetics data using caste names provided in the IHDS data ( for the matched sample of Indian populations, for which a consanguinity score could be assigned). Our consanguinity assignments strongly correlated with the proportion of IHDS respondents answering yes to the following prompt: “Now, I would like to ask you some questions about marriage customs in your community (jati) for a family like yours. Do people marry a daughter to her cousin?” (Figure 1).
FIGURE 1.
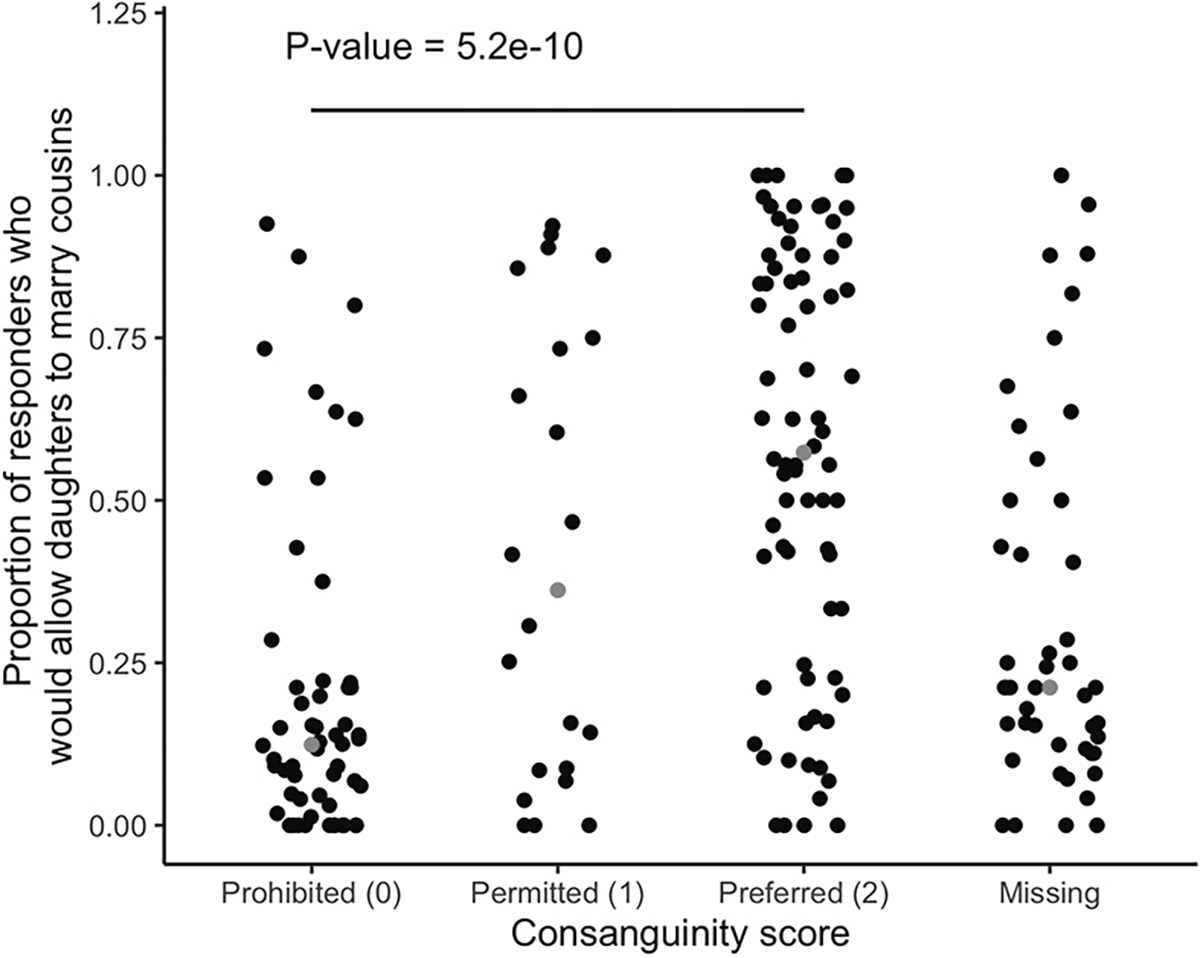
Our consanguinity scores correspond well with survey data on whether cousin marriage is permitted in an individual’s community. The x-axis shows the consanguinity score assigned to each population, and the y-axis shows the proportion of survey participants in the 2011 Indian Health and Development survey (Desai and Vanneman 2018) who responded that people in their community would marry a daughter to her cousin. Each point is a population (matched using the caste name variable from the survey; total after excluding samples for which a consanguinity score could not be determined); gray points represent the median for that category. The proportion of responders who would allow daughters to marry their cousins is significantly different across the three classes: prohibited, permitted, and preferred (ANOVA p-value = 5.2 × 10−10).
Effect of Consanguinity on Long ROH
We used Poisson and quasi-Poisson regression to model the effect of consanguinity on the number of long (>10 cM) ROH (NROH10). An expected LROH of >10 cM corresponds roughly to shared ancestry within the past five generations (since 10 cM corresponds to a recombination every 10 meioses), a plausible range for the ethnographic data. The consanguinity score of the population was treated as a nominal variable with three levels: prohibited, 0; permitted, 1; and preferred, 2. In all models, we included genome-wide heterozygosity, 10 genetic principal components (gPCs 1–10), and the number of shorter (1–2 cM and 2–5 cM) ROH (NROH1 and NROH2, respectively) as covariates to correct for other demographic events such as population bottlenecks.
We fitted models where each population was an observational unit, as well as models where each individual was treated as an observational unit. When each population was treated as an observational unit, we used the mean value of the variable (e.g., NROHs and gPCs) in the population and fitted both weighted (weights proportional to the number of individuals from each population) and unweighted models. When each individual was treated as the observational unit, we used individuals’ values of NROH and gPC and population-wide consanguinity scores. Finally, because our densest sampling was from South Asia, we also fitted models restricted to populations from this region.
The Poisson model assumes that the variance of the residuals is equal to the mean. We evaluated this assumption by calculating the dispersion parameter
where and are the observed and fitted values of NROH10, respectively, and is the residual degrees of freedom. Under a Poisson model, we expect . We formally evaluated this by testing whether followed a distribution with degrees of freedom. We fitted quasi-Poisson models when residuals were overdispersed and provide the dispersion parameter and the p-value in table captions when necessary.
To model the effect of consanguinity on the total length of ROH (LROH), we fitted a simple linear regression model with the logarithm of LROH as the response. We added 1 to LROH to avoid taking the logarithm of 0 for individuals without any detectable ROH. For clarity, we provide only results from the analysis of NROH in the main text and include results from LROH, which are qualitatively similar, in the supplementary material.
Paired Analysis
To further ensure that the relationship between consanguinity and NROH10 was not confounded by long-term demographic history, we identified population pairs that were matched genetically and geographically but differed in that one population preferred cousin unions and the other did not. To maximize genetic similarity and geographic proximity in selecting population pairs, we calculated genetic (Euclidean) distance using PC1–10 and geographic distances (using longitude and latitude) between populations that prefer consanguinity (score = 2) and those that either prohibit it or permit it (score ∈ {0, 1}). Then, we divided each distance matrix by its median (to account for differences in scale) and averaged the two to calculate a single distance matrix. We selected population pairs in ascending order of the distance between them such that each population was part of only one pair. We used a maximum distance cutoff between populations (determined visually) to ensure that remaining populations did not pair with populations that were genetically and geographically distant. We used a Wilcoxon signed-rank test to test for a difference in NROH10 between matched pairs.
Predictive Model
To predict consanguinity scores from genetic data, we created a binary variable that encoded populations prohibiting or permitting cousin marriage as 0 and populations preferring cousin marriage as 1. We fitted a logistic regression model where this binary variable was treated as the response and NROH10, NROH5, NROH2, NROH1, and heterozygosity were used as predictor variables. We also separately fitted a model with LROH10, LROH5, LROH2, LROH1, and heterozygosity as predictors. The performance of predictive models was evaluated using the receiver operating characteristic curve with a leave-one-out approach.
Results
Expected Effect of Consanguinity on ROH
Our simulations confirm that both the average total LROH and the NROH (of all lengths) increase as a function of increasing degree of consanguinity. As the practice continues for more generations, the effective population size decreases as a result, also leading to an increase in ROH. The simulation results also match the theoretical expectation derived for a single generation of cousin mating derived using Equation 1 (Ringbauer 2021) (Figure 2C,D, black points). In fact, the number of generations (10 or 50) has little impact on the average number or total length of long ROH (Figure 2C,D). This is because long ROH arise primarily due to cousin unions in the recent past and do not survive for many generations because of recombination. As a result, consanguinity occurring 50 generations ago will contribute little to the distribution of ROH10 in the present day. In contrast, the number of generations has a much greater impact on the number and length of all ROH (Figure 2A,B). Most ROH are short, and their distribution is sensitive to long-term demographic history (Ceballos et al. 2018; Arciero et al. 2021; for a more complete model that incorporates temporal changes in population size, see Severson et al. 2019, 2021). Thus, the increase in all ROH with number of generations largely reflects the small census population size of the simulation (1,000 individuals), with some small contribution from the continuation of cousin unions, which also reduces the effective population size (Severson et al. 2019, 2021).
FIGURE 2.
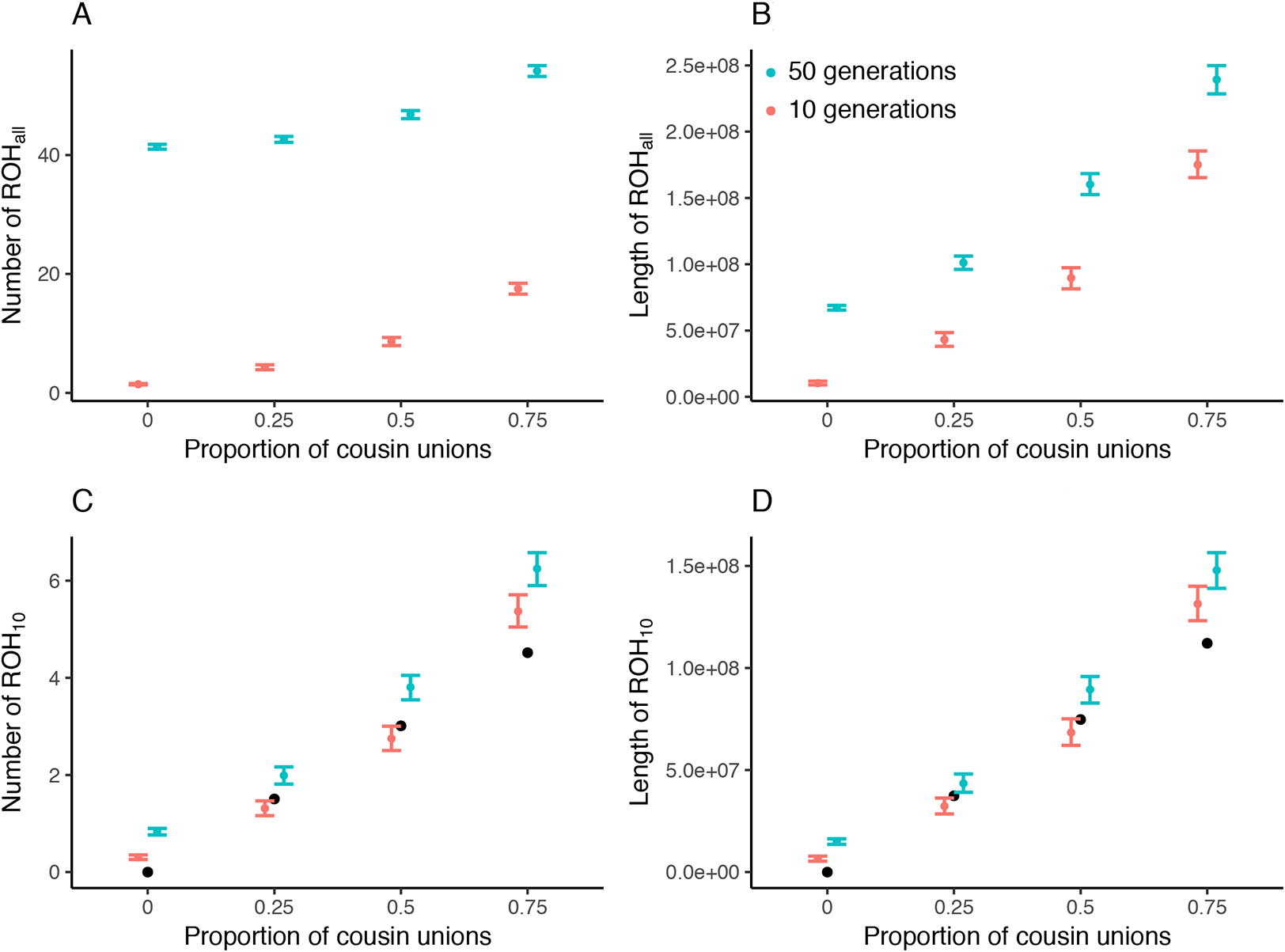
Distribution of the mean number (NROH; A and C) and mean length (LROH; B and D) of runs of homozygosity (ROH) observed in pedigrees simulated with varying degrees of consanguinity (proportion of unions between cousins) and numbers of generations for which the practice persisted: 10 (red) or 50 (blue). Colored points and whiskers represent the mean and 95% bootstrapped confidence interval, respectively, for all ROH (A and B) and for long (>10 cM) ROH (C and D). Black points represent the theoretical expectation from one generation of consanguinity using the model described in Ringbauer et al. (2021).
Geographic Variation in Cultural Preference of Consanguinity
We show the geographic variation in cultural preference for consanguinity in Figure 3 and Table 1. Preference varies significantly (ANOVA p-value = 1.22 × 10−6) across broad geographic regions (e.g., East Asia, northern Africa, and western Europe). As previously observed (Bittles and Black 2010), the Middle East and North Africa have a greater preference for consanguinity (mean consanguinity = 1.63 on our scale from 0 to 2). The lowest preference is in western Eurasia (mean consanguinity = 0.52). In South Asia, our region of densest sampling, preference for consanguinity is much higher in the south than in the north (Figure 3B) (Bittles 2002; Sharma et al. 2021).
FIGURE 3.
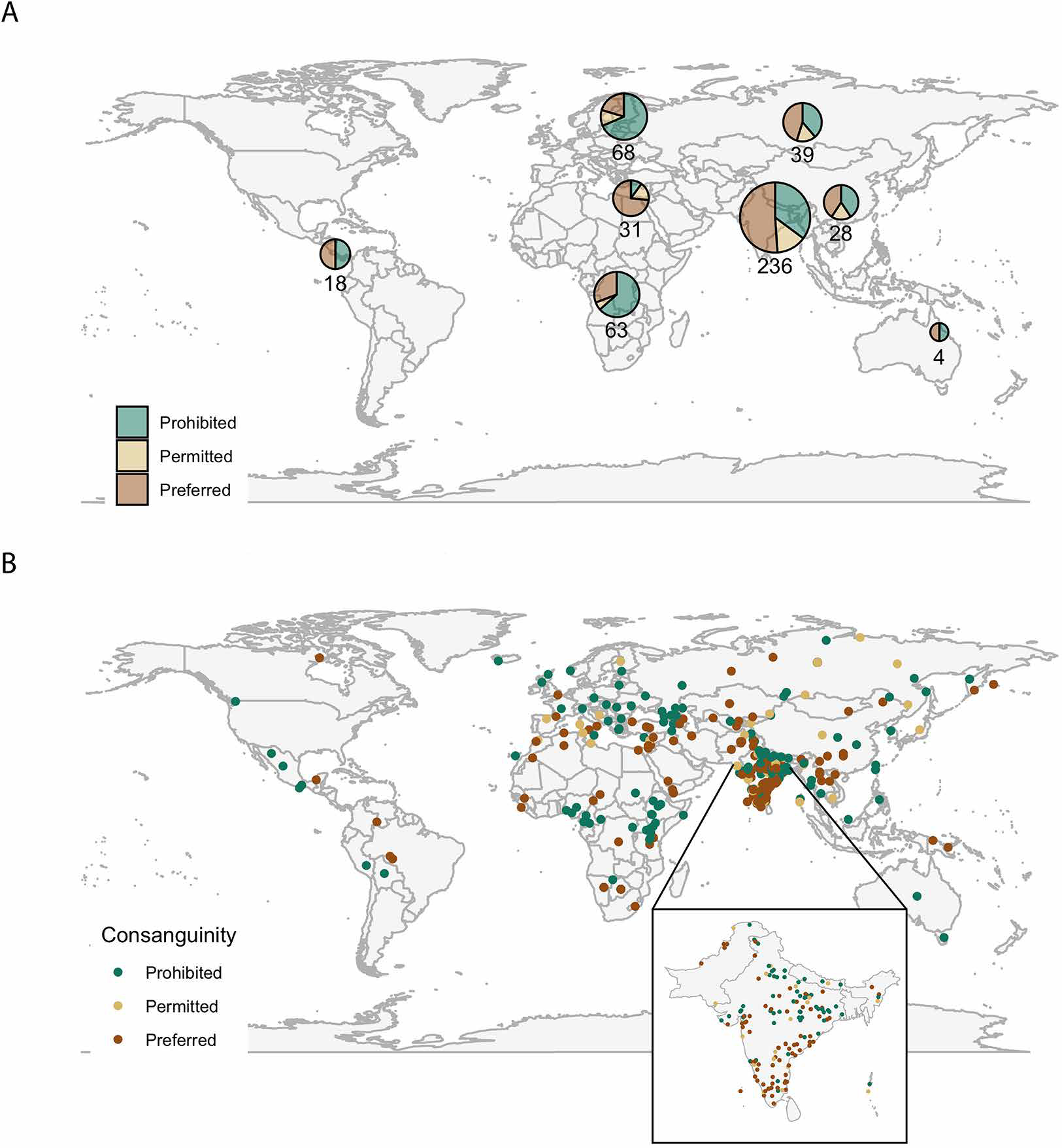
Consanguinity preferences across the world, by region (A) and by population (B). (A) The size of the pie charts and number below them correspond to the number of populations in the region across which information is aggregated.
Genetic Footprint of Cousin Marriages
The number of long (>10 cM) ROH (NROH10) was positively associated with the degree of consanguinity when each individual was treated as an observational unit ( individuals, , t-test p-value = 1.3 × 10−23; Table 2, Figure 4B). The estimated effect is consistent if we treat populations (as opposed to individuals) as observational units ( populations; Supplementary Table S1: unweighted , t-test p-value = 3.5 × 10−8; Supplementary Table S2: weighted , t-test p-value = 1.2 × 10−10; see Figure 4A). It is also consistent when we restrict the analysis to populations within South Asia, a region that is very diverse in marriage practices and where we have the largest density of observations (individual model with individuals, Supplementary Table S3: , t-test p-value = 4.5 × 10−12; population model with populations, Supplementary Table S4: unweighted , t-test p-value = 1.6 × 10−4; Supplementary Table S5: weighted , t-test p-value = 1.7 × 10−6). We observed similar results when we used LROH10 instead of NROH10 as response: Supplementary Table S6 gives results from a model where each individual is treated as an observational unit, and Supplementary Tables S7 and S8, from unweighted and weighted population models, respectively. For LROH10 models restricted to South Asia, see Supplementary Table S9 for the model for individuals and Supplementary Tables S10 and S11 for unweighted and weighted population models, respectively. That LROH10 and NROH10 give us similar results is expected, as both are strongly correlated in our data.
Table 2.
Quasi-Poisson Regression Fitted for Long (>10 cM) Runs of Homozygosity (NROH10; cM) for Individuals in the Global Sample
| Predictor Variable | Estimate | Std. Error | t-Value | Pr(>|t|) |
|---|---|---|---|---|
| Intercept | 0.95 | 1.34 | 0.71 | 4.80e-01 |
| NROH1 | −0.02 | 0.01 | −3.13 | 1.76e-03 |
| NROH2 | 0.06 | 0.00 | 17.22 | 4.62e-64 |
| Heterozygosity | −6.56 | 3.27 | −2.01 | 4.48e-02 |
| Consanguinity score = 1 | 0.30 | 0.10 | 3.01 | 2.67e-03 |
| Consanguinity score = 2 | 0.79 | 0.08 | 10.09 | 1.25e-23 |
| Central Asia and Siberia | 1.25 | 0.50 | 2.51 | 1.21e-02 |
| East Asia | 1.01 | 0.52 | 1.93 | 5.40e-02 |
| Middle East and North Africa | 0.70 | 0.53 | 1.30 | 1.93e-01 |
| Oceania | −3.30 | 2.05 | −1.61 | 1.08e-01 |
| South Asia | 1.10 | 0.53 | 2.09 | 3.70e-02 |
| Sub-Saharan Africa | 1.04 | 0.75 | 1.40 | 1.62e-01 |
| Western Eurasia | 0.39 | 0.53 | 0.74 | 4.57e-01 |
Abbreviations: NROH1, number of runs of homozygosity longer than 1 cM but shorter than 2 cM; NROH2, number of runs of homozygosity longer than 2 cM but shorter than 5 cM. Consanguinity scores of 1 and 2 represent populations that permit and prefer cousin unions, respectively. Regression (beta) coefficients for each region are expressed using NROH10 in the Americas as baseline. Ten genetic PCs (calculated for the full sample) were also included in the model, but their beta coefficients are not shown. Poisson dispersion parameter = 4.01, p-value = 0.
FIGURE 4.
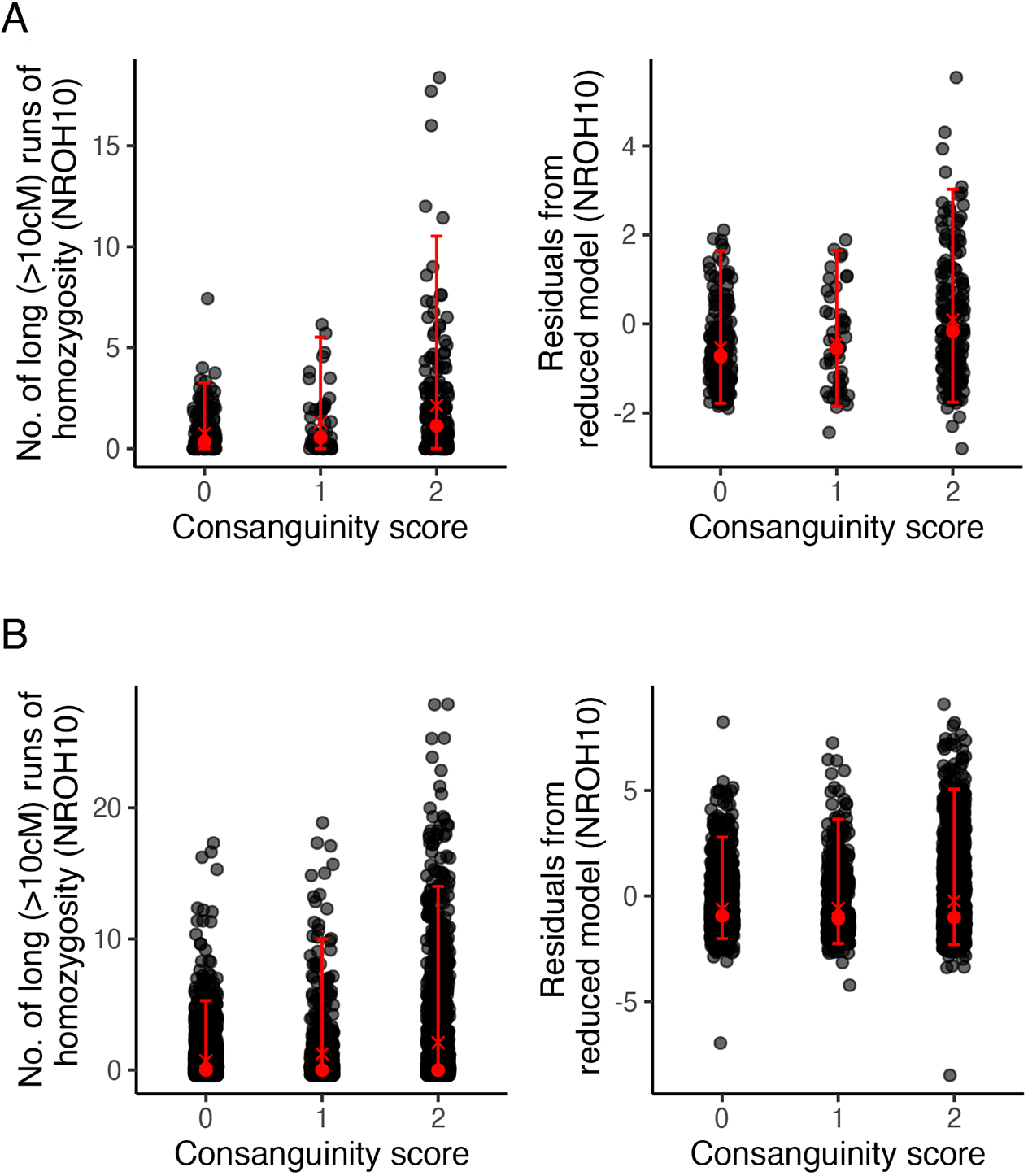
Relationship between consanguinity and number of long (>10 cM) runs of homozygosity (NROH10) by population (A) and by individual (B). Left panels show raw values of NROH10 on the y-axes; right panels show the residuals after all covariates (other than consanguinity score) have been regressed out. Dark gray points represent individual observations; red points represent median (circle) and mean (cross); and horizontal bars represent the 95% confidence intervals.
We used the analytical model of Ringbauer et al. (2021) to estimate the increase in consanguinity (measured as the proportion of unions among first cousins) between populations that were assigned a consanguinity score of 0 and those assigned a score of 2. The coefficient from the individual model (Table 2) translates to an increase of -fold in NROH10. In our data, populations with a score of 0 carried ~0.75 NROH10. Keeping all other covariates the same, this translates to an increase of NROH10 in populations with a score of 2. If we assume consanguinity is due only to first cousin unions and that children of first cousin unions carry, on average, ~8.7 NROH10, an increase in 1.66 NROH10 corresponds to an increase of 0.19 in absolute rate of first cousin unions in populations with a score of 2 relative to those with a score of 0.
Because the covariates in our regression models may not completely capture the effects and interactions of shared ancestry and culture, we confirmed this result with a matched-pair analysis. We identified pairs of populations matched for both genetic similarity and geographic proximity but dissimilar in cousin marriage practice and then tested for a difference in NROH within pairs (see Methods). We found that NROH10 significantly differed between such matched populations ( pairs; Figure 5), whereas other variables that are more sensitive to long-term demographic history such as NROH1, NROH2, NROH5, and heterozygosity are not (Table 3), consistent with results from the linear models. Removing populations with mean NROH10 > 5 did not change this result (Supplementary Table S12).
FIGURE 5.
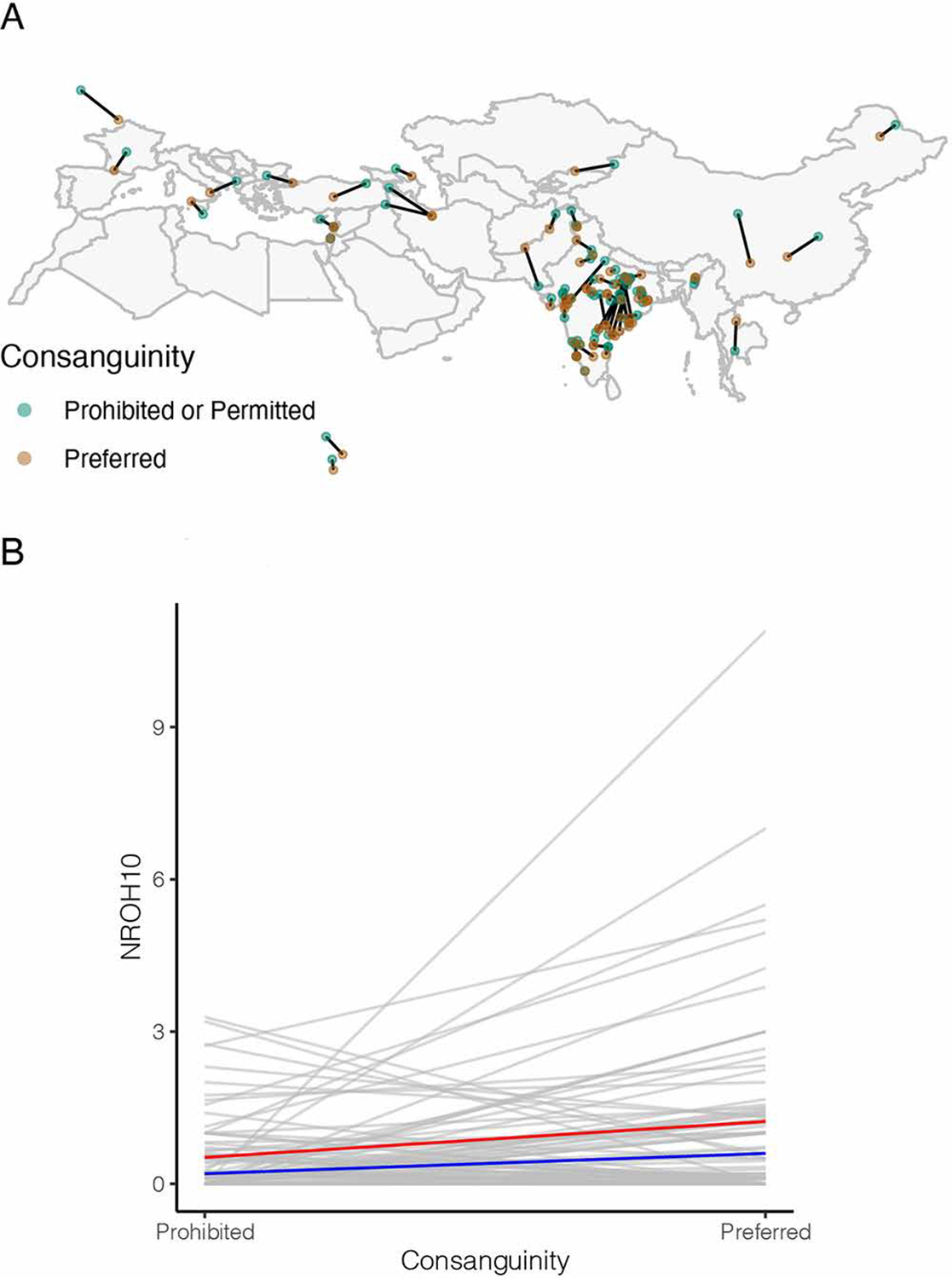
Paired comparison of NROH10 between populations where consanguinity is prevalent versus populations where it is not ( pairs). (A) Population pairs matched on genetic similarity and geographic similarity. One pair in Central America is not shown. (B) The mean number of long (>10 cM) runs of homozygosity (NROH10) is greater in populations that prefer cousin unions than in populations that prohibit them. The gray lines demonstrate comparisons between pairs of populations; the red and blue lines show the mean and median trend, respectively.
Table 3.
Wilcoxon Signed-Rank Test Results for Differences between 77 Pairs of Populations Preferring and Prohibiting Consanguinity
| Variable | Wilcoxon Signed-Rank Sum | P-Value |
|---|---|---|
| NROH10 | 1737.50 | 7.72e-04 |
| NROH5 | 1614.00 | 2.23e-01 |
| NROH2 | 1660.50 | 4.21e-01 |
| NROH1 | 1310.00 | 3.32e-01 |
| Heterozygosity | 1544.00 | 8.31e-01 |
Abbreviations: NROH10, number of runs of homozygosity longer than 10 cM; NROH5, number of runs of homozygosity longer than 5 cM but shorter than 10 cM; NROH2, number of runs of homozygosity longer than 2 cM but shorter than 5 cM; NROH1, number of runs of homozygosity longer than 1 cM but shorter than 2 cM.
Predicting Marriage Practice from Genetic Data
Finally, we asked to what extent it is possible to predict reported marriage preferences based on the genomic distributions of ROH. To do so, we created a binary variable that encoded populations either prohibiting or preferring cousin marriage as 0 and populations preferring cousin marriage as 1. We predicted this binary variable using logistic regression with NROH1, 2, 5, and 10 and heterozygosity as predictors. Separately, we also predicted consanguinity with LROH1, 2, 5, and 10 and heterozygosity. We do not include region or gPCs in the regressions, which is conservative. Using leave-one-out cross-validation, we found that classification power is low (area under the curve = 0.63 with NROH and 0.64 with LROH) but generally well calibrated (i.e., probability of cousin marriage predicted by the model matches the observed probability) (Figure 6).
FIGURE 6.
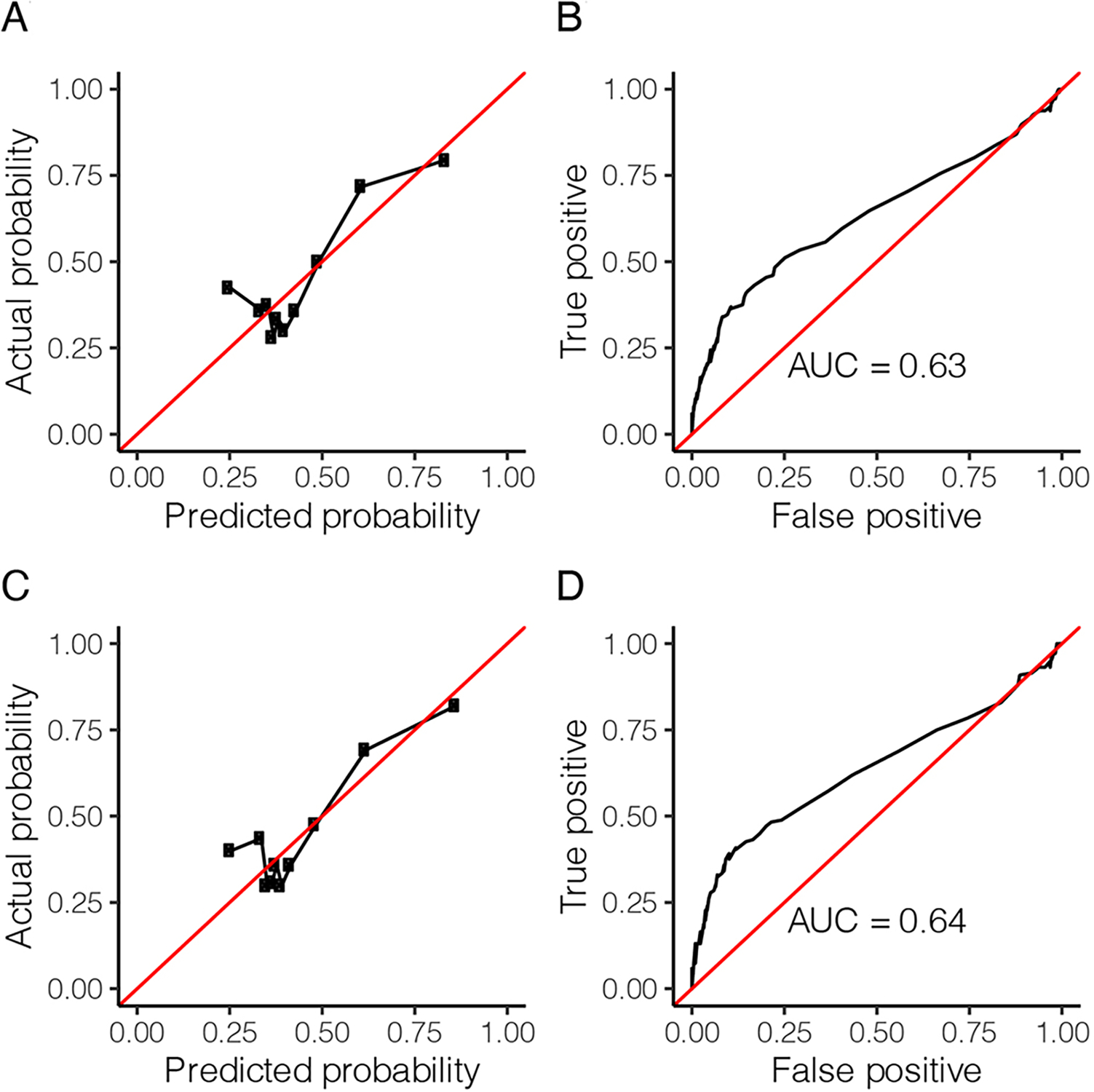
Results from predictive model using number of runs of homozygosity (NROH; A and B) or total length of runs of homozygosity (C and D). The model is well calibrated (A and C), and the area under the receiver operating characteristic (ROC) curve (AUC) as the prediction cutoff varies is ~0.64 (B and D).
Discussion
The negative genetic consequences of marriage between close relatives are well known, yet the practice continues among millions of households around the world. Consanguinity creates long ROH in the genome, increasing the risk of recessive disease in the offspring. We found a statistically significant relationship between ethnographic assessments of cousin marriage practice and long ROH in 3,849 individuals across 396 worldwide populations. Based on analytical expectations, this suggests that populations with a reported cultural preference for cousin marriage have, on average, actual first cousin marriage rates approximately 0.19 greater than those that prohibit it. For example, if 1% of unions in populations prohibiting consanguinity are between first cousins, we expect the rate of such unions in populations preferring consanguinity to be ~20%. This translates to an increase in child mortality or severe genetic disease of less than 1%—a cost that may in many cases be outweighed by the social and economic benefits of cousin marriage (Paul and Spencer 2008; Bittles and Black 2010).
Indeed, while geneticists have focused on the deleterious aspects of cousin marriage, social scientists have primarily focused on the benefits, such as keeping land within the family line (Anderson 1986; Stone 1977), creating greater assurance that a marrying daughter will receive better treatment in her new household (Do et al. 2013; Mobarak et al. 2019), marrying a daughter to her cousin as barter for obtaining a cousin-bride for a son (Edlund 2018), or parents having a strong preference for the social status of their in-laws (Edlund 1999). Less emphasis has been placed on integrating these benefits into a broader cost-benefit framework that ultimately determines the emergence, persistence, and potential decline of cousin marriage practice at the population level. This suggests that future work should incorporate genetic data, which might provide more accurate estimates of actual prevalence of consanguinity, as a cost metric for this practice.
Our study has a number of limitations—some unavoidable. Our classification of marriage practices is based on literature search, and many of our classifications may be incorrect or fail to reflect complexity or structure within named groups. Similarly, the genetic data we assembled typically have little information about sampling. Group labels may be misleading or incorrect, and we may have linked them incorrectly to the ethnographic data. In addition, since we are, to first order, measuring whether the individuals in our sample have consanguineous parents, our results could be biased if the sampled individuals were not representative of the wider populations. With more detailed data, many of these limitations can be avoided (e.g., Kang et al. 2016), although at the cost of limiting the geographic and cultural scope of the study. Despite these limitations, we did detect the expected associations, indicating that we captured an important part of the effect, although our estimates of magnitude are likely underestimates.
Finally, we also briefly explored the potential of using the present-day relationship between long ROH and population-level assessments of cousin marriage to predict the marriage preferences for populations with unavailable ethnographic information (e.g., ancient populations). In particular, our results suggest that it may be difficult to predict population-level cultural preferences for ancient populations using genetic data, for three reasons. First, the distribution of ROH can be affected by factors other than consanguinity that might be unknown for ancient populations. Second is the limitation of small sample sizes of ancient populations. If, in a sample of 10 individuals, we find zero individuals whose parents are cousins, the 95% binomial confidence interval for the population proportion is approximately 0–0.32. This proportion spans virtually the entire range of probabilities observed in present-day populations and is therefore not very informative about the actual practice of consanguinity in the population. Third, cultural preferences may not necessarily be concordant with actual practice in a particular sample—the lack of consanguinity in a particular sample may reflect contingency rather than a lack of cultural preference, and vice versa. Overall, there are limitations to how much genetic data can tell us about culture, and it must be integrated with other types of information to address questions around the existence, history, and the health and economic effects of consanguinity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Zachary Szpiech and Shai Carmi for helpful comments. This work was supported by National Institute of General Medical Sciences award R35GM133708. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
LITERATURE CITED
- Anderson NF 1986. Cousin marriage in Victorian England. J. Fam. Hist. 11:285–301. [Google Scholar]
- Arciero E, Dogra SA, Malawsky DS et al. 2021. Fine-scale population structure and demographic history of British Pakistanis. Nat. Commun. 12:7,189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bherer C, Campbell CL, and Auton A. 2017. Refined genetic maps reveal sexual dimorphism in human meiotic recombination at multiple scales. Nat. Commun. 8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittles AH 2002. Endogamy, consanguinity and community genetics. J. Genet. 81:91–98. [DOI] [PubMed] [Google Scholar]
- Bittles AH, and Black ML 2010. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proc. Natl. Acad. Sci. U. S. A. 107:1,779–1,786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittles AH, and Neel JV 1994. The costs of human inbreeding and their implications for variations at the DNA level. Nat. Genet. 8:117–121. [DOI] [PubMed] [Google Scholar]
- Caballero M, Seidman DN, Qiao Y et al. 2019. Crossover interference and sex-specific genetic maps shape identical by descent sharing in close relatives. PLoS Genet. 15:e1007979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos FC, Joshi PK, Clark DW et al. 2018. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 19:220–234. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC et al. 2015. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai S, and Vanneman R 2018. India Human Development Survey-II (IHDS-II), 2011–12. Ann Arbor, MI: Interuniversity Consortium for Political and Social Research. [Google Scholar]
- Do Q-T, Iyer S, and Joshi S 2013. The economics of consanguineous marriages. Rev. Econ. Stat. 95:904–918. [Google Scholar]
- Edlund L 1999. Son preference, sex ratios, and marriage patterns. J. Polit. Econ. 107:1,275–1,304. [Google Scholar]
- Edlund L 2018. Cousin marriage is not choice: Muslim marriage and underdevelopment. AEA Pap. Proc. 108:353–357. [Google Scholar]
- Fan S, Kelly DE, Beltrame MH et al. 2019. African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations. Genome Biol. 20:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke K, Kourakos M, Brown G et al. 2021. Ancestral haplotype reconstruction in endogamous populations using identity-by-descent. PLoS Comput. Biol. 17:e1008638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong C, Balanovsky O, Lukianova E et al. 2019. The genetic history of admixture across inner Eurasia. Nat. Ecol. Evol. 3:966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JT, Goldberg A, Edge MD et al. 2016. Consanguinity rates predict long runs of homozygosity in Jewish populations. Hum. Hered. 82:87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Nadel D, Rollefson G et al. 2016. Genomic insights into the origin of farming in the ancient Near East. Nature 536:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis I, Patterson N, Mittnik A et al. 2014. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson M, Cheronet O, Mallick S et al. 2018. Ancient genomes document multiple waves of migration in Southeast Asian prehistory. Science 361:92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick S, Li H, Lipson M et al. 2016. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillan R, Leutenegger A-L, Abdel-Rahman R et al. 2008. Runs of homozygosity in European populations. Am. J. Hum. Genet. 83:359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobarak AM, Chaudhry T, Brown J et al. 2019. Estimating the health and socioeconomic effects of cousin marriage in South Asia. J. Biosoc. Sci. 51:418–435. [DOI] [PubMed] [Google Scholar]
- Murdock GP 1967. Ethnographic atlas: A summary. Ethnology 6:109–236. [Google Scholar]
- Nakatsuka N, Moorjani P, Rai N et al. 2017. The promise of discovering population-specific disease-associated genes in South Asia. Nat. Genet. 49:1,403–1,407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIPS and ICF (National Institute of Population Studies [Pakistan] and ICF International Inc.). 2019. Pakistan Demographic and Health Survey 2017–2018. Islamabad, PK, and Rockville, MD: NIPS and ICF. https://dhsprogram.com/pubs/pdf/FR354/FR354.pdf. [Google Scholar]
- Pagani L, Lawson DJ, Jagoda E et al. 2016. Genomic analyses inform on migration events during the peopling of Eurasia. Nature 538:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, and Reich D 2006. Population structure and eigenanalysis. PLoS Genet. 2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul DB, and Spencer HG 2008. “It’s ok, we’re not cousins by blood”: The cousin marriage controversy in historical perspective. PLoS Biol. 23:2,627–2,630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton TJ, and Rosenberg NA 2014. Population-genetic influences on genomic estimates of the inbreeding coefficient: A global perspective. Hum. Hered. 77: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Patterson N, Barbieri C et al. 2012. The genetic prehistory of southern Africa. Nat. Commun. 3:1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringbauer H, Novembre J, and Steinrücken M 2021. Parental relatedness through time revealed runs of homozygosity in ancient DNA. Nat. Commun. 12:5,425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo G, and Bittles AH 2014. Consanguinity in the contemporary world. Hum. Hered. 77:6–9. [DOI] [PubMed] [Google Scholar]
- Severson AL, Carmi S, and Rosenberg NA 2019. The effect of consanguinity on between-individual-identity-by-descent sharing. Genetics 212:305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson AL, Carmi S, and Rosenberg NA 2021. Variance and limiting distribution of coalescence times in a diploid model of a consanguineous population. Theor. Popul. Biol. 139:50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Kalam MA, Ghosh S et al. 2021. Prevalence and determinants of consanguineous marriage and its types in India: Evidence from the National Family Health Survey, 2015–2016. J. Biosoc. Sci. 53:566–576. [DOI] [PubMed] [Google Scholar]
- Sheridan E, Wright J, Small N et al. 2013. Risk factors for congenital anomaly in a multiethnic birth cohort: An analysis of the Born in Bradford study. Lancet 382:1,350–1,359. [DOI] [PubMed] [Google Scholar]
- Stone L 1977. Family, Sex and Marriage in England 1500–1800. New York: Harper and Row. [Google Scholar]
- Szpiech ZA, Xu J, Pemberton TJ et al. 2013. Long runs of homozygosity are enriched for deleterious variation. Am. J. Hum. Genet. 93:90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Sathirapongsasuti JF, Gupta R et al. 2020. South Asian patient population genetics reveal strong founder effects and high rates of homozygosity—New resources for precision medicine. Preprint, 10.1101/2020.10.02.323238v1. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.