

Abstract
Calcium-dependent heart muscle contraction is regulated by the cardiac troponin protein complex (cTn), and specifically by the N-terminal domain of its calcium binding subunit (cNTnC). cNTnC contains one calcium binding site (site II), and altered calcium binding in this site has been studied for decades. It has been previously shown that cNTnC mutants which increase calcium sensitization may have therapeutic benefits, such as restoring cardiac muscle contractility and functionality post myocardial infarction events. Here we computationally characterized eight mutations for their potential effects on calcium binding affinity in site II of cNTnC. We utilized two distinct methods to estimate calcium binding: adaptive steered molecular dynamics (ASMD) and thermodynamic integration (TI). We observed a sensitizing trend for all mutations based on the employed ASMD methodology. The TI results showed excellent agreement with experimentally known calcium binding affinities in wildtype cNTnC. Based on the TI results, five mutants were predicted to increase calcium sensitivity in site II. This study presents an interesting comparison of the two computational methods, which have both been shown to be valuable tools in characterizing the impacts of calcium sensitivity in mutant cNTnC systems.
Graphical Abstract
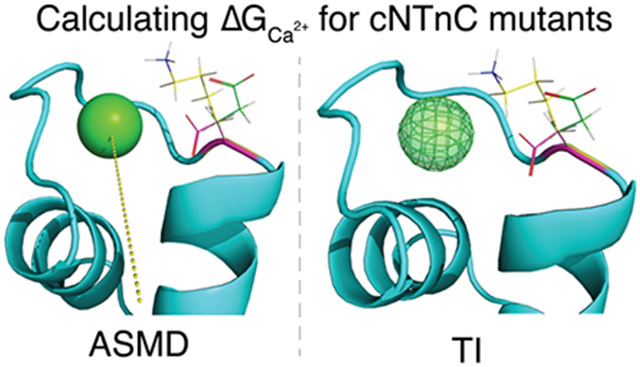
Introduction
Cardiac troponin (cTn) is a regulatory protein of calcium-dependent muscle contraction and relaxation. The cTn complex is comprised of three subunits: troponin T (cTnT), troponin I (cTnI), and troponin C (cTnC). The N-terminal region of cTnT interacts with tropomyosin (Tm) and anchors the protein complex to the thin filament, while its C-terminus forms interactions with both cTnI and cTnC 1, 2. cTnI is an extremely flexible protein that acts as an inhibitory subunit for actomyosin interaction 3. cTnC is a dumbbell-shaped protein with globular terminal domains linked by a long central helix 4, 5. The terminal domains consist of two helix-loop-helix (EF-hand) motifs, respectively. The C-terminal domain (cCTnC) contains two high-affinity binding sites (site III and site IV) for Ca2+ and Mg2+ ions; under physiological conditions these sites are constantly occupied by either ion. cCTnC, the structural domain of the cTnC subunit, facilitates key interactions related to anchoring the protein to the remaining cTn complex and the thin filament 6. In contrast, the N-terminal domain of cTnC (cNTnC) contains only one active binding site, site II 7. cNTnC is frequently referred to as the regulatory domain, due to the conformational change the complex undergoes once a calcium ion binds to site II. The conformational change results in the exposure of the cNTnC hydrophobic patch (cNTnC residues 20, 23, 24, 26, 27, 36, 41, 44, 48, 57, 60, 77, 80, and 81) which promotes binding of the cTnI switch peptide, thereby facilitating disassociation of cTnI from actin. As a result, Tm moves along the thin filament and exposes the myosin binding site. Myosin is then able to bind to actin forming a cross-bridge, a key step in muscle contraction.
Cardiac muscle contractility and function has been shown to be affected by mutations in cNTnC that alter the calcium sensitivity of site II. The mutational effect (sensitizing or desensitizing) depends on the identity and location of the substituted amino acid 7. There are several examples of previous attempts to rationally design and characterize calcium sensitivity altering mutants within the hydrophobic patch of the cNTnC. For instance, Tikunova and Davis designed five glutamine calcium sensitizing mutations (F20Q, V44Q, M45Q, L48Q, and M81Q) 7. Others have deigned calcium desensitizing mutations, such as I61Q 8 and D73N 9. The notable success of L48Q to restore cardiac muscle contractility and functionality post myocardial infarction (MI) event makes mutated cNTnC structures a promising option for treating heart failure via gene therapy techniques10,11. In addition to mutations modulating the calcium affinity of cNTnC, small molecules bound to the hydrophobic patch also exhibit a similar effect 12,13,14,15. However, there is a lack of exploration of calcium sensitizing mutations in the loop regions of the two EF-hand motifs. In order to bridge this gap, we explore several presumed calcium sensitizing mutations and characterize their effects via the computational methods adaptive steered molecular dynamics and thermodynamic integration.
Adaptive steered molecular dynamics (ASMD), developed by Hernandez and co-workers 16, functions similarly to traditional steered molecular dynamics (SMD). In both techniques, a pseudo particle applies a steering force in order to move along the predetermined reaction coordinate at a particular velocity. The nonequilibrium work conducted on the system during the simulation can be related to the difference in free energy between the initial and final states 17. However, the benefit of the ASMD methodology, is that the reaction coordinate can be separated into N smaller stages. For each stage, the Jarzynski average (JA)18 is calculated over the specified number of individual trajectories. The individual nonequilibrium trajectory’s work that most closely matches the JA serves as the initial position for the next sequential stage of the reaction coordinate. ASMD has been demonstrated to significantly reduce the number of nonequilibrium trajectories required to converge the potential mean force (PMF) compared to traditional SMD 19,20. Thermodynamic integration (TI) is an alchemical method utilized to determine the relative binding affinity of ligand to receptor 21,22,23,24. TI relates two states (initial and final) via a coupling parameter, , through a series of intermediate stages. corresponds to the initial state and corresponds to the final state. At each window a molecular dynamics simulation is performed to generate an ensemble of structures. The total energy of the system is taken to be its potential energy, and the derivative of the energy with respect to is utilized to calculate the free energy difference.
In a previous work, we developed an ASMD protocol to characterize the calcium binding affinity of site II in wildtype cNTnC, several known sensitizing mutations (F20Q, V44Q, M45Q, L48Q, and M81Q), and two desensitizing mutations (I61Q and D73N) 25. We successfully predicted the correct calcium binding affinity trends of those mutations compared to wildtype. Much work has also been done to study the dynamics and energetics of the hydrophobic patch opening and the potential effect of mutations in this region 26,27,28,29,30,31,32,33. Additionally, the dynamics and effects of mutations in other parts of the cTn complex, particularly cTnI, have been explored 34,35,36,37,38,39,40,41. Finally, thermodynamic integration has been previously applied to study calcium and magnesium binding to site II 42, 43. In this work, we rationally explore several presumed Ca2+ sensitizing mutations based on protein stability point mutation predictions and characterize their effects with ASMD and TI. We hope that this publication will be able to serve as a guide for potential future protein design studies. Furthermore, our work serves as an interesting comparison between ASMD and TI methods for predicting calcium binding affinities.
Methods
Identification of Explored Mutations based on Predicted Protein Stability
In order to predict protein stability, we used the Cologne University Protein Stability Analysis Tool (CUPSTAT) 44,45,46. This program makes use of structural environment-specific atom potentials and torsion angles to predict the difference in free energy of unfolding () between wild-type and mutant proteins. CUPSTAT was utilized to exhaustively predict all possible single point mutations in the wildtype cNTnC subunit. In order to identify mutations that could possibly cause increased calcium sensitivity, we sought mutants that destabilized the closed conformation, while concurrently stabilizing the open conformation of the cNTnC hydrophobic patch. In order to model the different states of calcium-bound cNTnC (closed and open) we used NMR models of the protein obtained from the Protein Data Bank (PDB). The most representative models of PDB IDs 1AP4 47 and 2KFX 48 were used to model the closed and open patch conformations respectively. 2KFX was selected to model the open conformation of Ca2+-bound cNTnC since this structure was obtained in absence of the cTnI switch peptide, making this the best model for comparison to 1AP4. However, the initial structure of 2KFX was determined with the known inhibitor W7 bound in the hydrophobic patch, therefore the ligand was removed prior to any computational analysis. We focused on mutations in the loop I and loop II regions and immediately adjacent residues. Eight mutations of interest were identified: D33H, D33M, L41W, V72D, V72N, F74E, F74R, and F77I.
Adaptive Steered Molecular Dynamics (ASMD)
The representative protein conformation of PDB 1AP4 served as the model for wildtype cNTnC and the base model from which all mutations were constructed using the protein mutagenesis tool in PyMOL 49. The N-terminal domain of cTnC has served as the blueprint for many impactful studies evaluating the role and influence of wildtype and mutant cardiac troponin in muscle contraction 8, 26, 27, 28, 29, 39, 50, 51, 52. Although the cNTnC model is rather simplistic to capture the function of the entire troponin complex in context of the muscle tissue, many experts in the field have utilized cNTnC to study the dynamics and calcium binding properties of site II. The initial protein structures for molecular dynamics of the wildtype and mutants models were solvated, minimized, and equilibrated as described in our previous work 25. For the point mutations located in loop I (D33H, D33M, and L41W), the ASMD pulls were calculated based on the distance between the center of mass of the carbons in the residues Ser69, Gly70, and Thr71 and the initial position of the Ca2+ ion. Hereafter, we will refer to this center of mass point as COM 1. The initial distance between COM 1 and the calcium ion was determined for all systems using the CPPTRAJ 53 distance function. The ASMD simulations were performed for a total distance of 20 Å, using a force constant of 7.2 kcal mol−1 Å−2 and a pulling speed of 0.5 Å/ns. All ASMD simulations were split into ten stages, and each stage was simulated for 4 ns (nstlim=2,000,000) with 50 trajectories per stage. For mutations located in loop II and adjacent (V72D, V72N, F74E, F74R, and F77I), we observed increased flexibility in the loop region resulting in the calcium ion being driven into the adjacent helices (helix C and D) while conducting ASMD pulls based on COM 1. Therefore, to compensate for this effect we employed a new center of mass point (COM 2) based on the carbons in the residues Asp67, Gly68, Ser69, Gly70, and Thr71. Similar to the mutations located in loop I, the distance between the starting position of the Ca2+ ion and COM 2 served as the initial distance for the ASMD simulations. Additionally, simulations based on this center of mass point were performed for a total distance of 20Å utilizing a force constant of 7.2 kcal mol−1 Å−2, pull speed of 0.5 Å/ns, and 50 trajectories per stage The initial distance for these systems was also obtained using the distance function of CPPTRAJ. The wildtype cNTnC system PMF was determined based on both center of mass points. Additionally, the PMF of the wildtype system was obtained utilizing a pulling speed of 0.1 Å/ns and the reaction coordinate partitioned into 20 stages. Each stage was simulated for 10 ns (nstlim=5,000,000) with 50 individual trajectories per stage. Upon completion of the SMD pulls for any stage (N) of the simulation, the JA was calculated via the “ASMD.py” script provided by Hernandez and colleagues in the AMBER Advanced Tutorial 26. The final coordinates of the trajectory whose work most closely matched the JA were used to initialize the subsequent stage (N+1) of the simulation. All ASMD simulations were performed with the GPU implementation of PMEMD54 from the AMBER1855 package.
Thermodynamic Integration (TI)
Similar to the protocol for the ASMD simulations, the representative model of PDB 1AP4 served as the model for wildtype cNTnC, and the base from which all mutations where created in PyMOL using the protein mutagenesis tool. The wildtype and all mutant systems were prepared in the following manner. The individual models were solvated in a 12Å TIP3P 56 water box and neutralized with Na+ ions via the tLeap module of AMBER18. The protein was described with the forcefield ff14SB57. In order to complete the thermodynamic cycle, a system containing the Ca2+ ion was prepared using the tLeap module and solvated in a 12Å TIP3P water box. Simulations were conducted under NPT conditions using the Berendsen barostat 58 and periodic boundary conditions. The system was minimized for 2000 cycles and heated to 300 K using the Langevin thermostat 59 over 500 ps prior to the 5 ns production with a time step of 2 fs. The SHAKE algorithm was employed to constrain all bonds involving hydrogen atoms, and the Particle Mesh Ewald method60 was utilized to calculate electrostatic interactions of long distances with a cutoff of 10 Å.
The alchemical thermodynamic cycle used for ligand binding has been detailed previously by Leelananda and Lindert 61. In this implementation of TI, the method consisted of three steps for ligand (Ca2+) in protein: introduction of harmonic distance restraints, removal of electrostatic interactions, and removal of van der Waals forces. However, for the ligand in water system only two steps were necessary: removal of electrostatic interactions and removal of van der Waals forces. The specific distance restraints for all systems were calculated with the CPPTRAJ distance function. Restraints were created based on the distance between the Ca2+ ion and the following atoms: ASP67 CG, SER69 OG, and THR71 OG1. The coupling parameter increased incrementally by 0.1 from 0.0 – 1.0 for each transitional step of the thermodynamic cycle. During each simulation, values were collected every 2 ps resulting in 5000 data points per transitional step of for further analysis. The Multistate Bennett Acceptance Ratio (MBAR)62 was used to calculate the relative free energies of the simulations across all values of . Finally, analytical free energy corrections were made to account for the introduction of the distance restraints as described previously42. For each system, five independent TI runs were performed and the results averaged.
Results and Discussion
Prediction of Potential Calcium Sensitizing Mutations.
The Cologne University Protein Stability Analysis Tool (CUPSTAT) was employed to exhaustively predict the protein stability resulting from all possible single point mutations in the wildtype cNTnC subunit. We were particularly interested in mutations in the loop regions that were predicted to destabilize the closed conformation (model based on PDB 1AP4) while simultaneously predicted to stabilize the open conformation (model based on PDB 2KFX). We hypothesized that the stabilization of the open conformation will correlate with a lower free energy of hydrophobic patch opening and/or a slower Ca2+ disassociation rate , thereby leading to an increase in calcium sensitivity. Based on this line of thinking, we filtered the CUPSTAT predictions focusing on cases where torsion angles of the mutated residue were favorable in both open and closed conformations. The CUPSTAT predictions of all mutants that met these criteria are available in Table S1, where the bolded rows denote the mutations that were selected for simulations. We further narrowed the results for in silico characterization based on a predicted less than −0.7 kcal/mol, thus focusing our studies on the following mutations: D33H, D33M, L41W, V72N, F74E, F74R, and F77I. Although the mutation V72D did not meet our cutoff criteria ( kcal/mol), it was rationally selected for further analysis as we speculated that the inclusion of an additional negatively charge residue in the coordination sphere of site II might cause a divalent cation (i.e. Ca2+) to bind more tightly. Therefore, we ultimately selected eight potential calcium sensitizing mutations (D33H, D33M, L41W, V72D, V72N, F74E, F74R, and F77I) for further characterization via adaptive steered molecular dynamics and thermodynamic integration methods.
Characterization and Comparison of Potential Ca2+ Sensitizing Mutations with Adaptive Steered Molecular Dynamics and Thermodynamic Integration.
We simulated Ca2+ binding in cNTnC wildtype and mutant structures based on the CUPSTAT predictions with ASMD and TI. It has been well established that the accuracy of ASMD can be improved via two ways: using an increasingly slow pull speed and increasing the number of trajectories per stage 19,20, 25. In our previous work, we examined two different pull speeds and three different numbers of trajectories per stage. Ultimately, we settled on a pull speed of 0.5 Å/ns and 50 trajectories per stage (tps). Here, we explored the impact of an even slower velocity for wildtype cNTnC with the reaction coordinate separated into 20 stages. Figure S1 depicts two different JA PMFs of the wildtype cNTnC that differ by their pulling velocities and the number of stages that the reaction coordinate was segmented into (0.5 Å/ns with 10 stages and 0.1 Å/ns with 20 stages). We found the JA PMFs of these two simulations to be rather similar to one another (difference of only ~ 4kcal/mol). We considered this difference to be insignificant as the two PMFs overlapped in several of the individual work traces. These results supported our choice of pull speed (0.5 Å/ns) and number of stages (10) for ASMD of the mutations, as these parameters were less computationally expensive in comparison. Additionally, based on these results we hypothesize that when comparing PMFs at significantly slow pull velocities, the number of trajectories per stage has a more substantial impact on the accuracy of ASMD compared to the speed of the pull.
Figure 1A depicts two different JA PMFs of wildtype cNTnC that differed by the center of mass points for pulling calcium away from its initial bound position in site II. The JA PMF and the individual work traces for cNTnC based on COM 1 (identical to that published in our previous work) are depicted in light blue and light gray, respectively. For mutations located in and adjacent to loop II (V72D, V72N, F74E, F74R, and F77I), we observed increased flexibility in the loop region due to the substitution of larger residues. Upon visualizing the trajectories of the loop II mutant systems based on COM 1, we found that the calcium ion was being driven into the adjacent helices (helix C and D) leading to significantly higher PMFs. To remedy this biologically irrelevant occurrence, we created a new center of mass point, COM 2 (as described in the Methods section). The cNTnC system based off of COM 2 was simulated with a pull speed of 0.5 Å/ns, the reaction coordinate split into 10 stages, and 50 tps. The JA PMF and individual work traces for the ASMD pull based off of COM2 are depicted in Figure 1A as violet and dark gray, respectively. While the PMF for this system was slightly higher than that of the cNTnC system based on COM 1 for multiple calcium distances, there was overlap between the PMFs and both converged to a similar free energy. Figure 1B depicts the JA PMFs of wildtype and loop I mutant cNTnC structures based on COM 1. Figure 1C displays the JA PMFs of wildtype and loop II mutant cNTnC structures based on COM 2.
Figure 1.

ASMD PMFs of Ca2+–bound wildtype and mutant cNTnC systems. All PMFs were obtained at constant NPT conditions. (A) The PMFs of wildtype cNTnC obtained using two different center of mass points. The PMF for the system (pull speed of 0.5 Å/ns and 10 stages) based on COM 1 is shown as the light blue line and the work traces for the 50 individual trajectories in each stage are shown in light grey lines. The PMF for the system (pull speed of 0.5 Å/ns and 10 stages) based on COM 2 is shown as the violet line and the work traces for the 50 individual trajectories in each stage are shown in dark grey lines. (B) JA PMFs of Ca2+ binding to wildtype cNTnC and loop I mutations based on COM 1 and a pull speed of 0.5 Å/ns, the reaction coordinate separated into 10 stages, and 50 individual trajectories per stage. (C) JA PMFs of Ca2+ binding to wildtype cNTnC and loop II mutations based on COM 2 and a pull speed of 0.5 Å/ns, the reaction coordinate separated into 10 stages, and 50 individual trajectories per stage.
The values calculated from the ASMD simulations () for all of the predicted sensitizing mutations followed the anticipated trend. Mutant V72D showed the greatest increase in predicted binding affinity with a of 5.9 kcal/mol. Mutations D33H, D33M, L41W, and F77I all showed a significant predicted increase in calcium binding with values of 4.9 kcal/mol, 3.5 kcal/mol, 4.5 kcal/mol, and 3.2 kcal/mol respectively. Mutant V72N showed a moderate increase in predicted calcium binding with an of 1.2 kcal/mol. Finally, the mutants F74E and F74R showed only a slight increase of calcium binding with values of 0.21 kcal/mol and 0.62 kcal/mol, respectively. The values between wildtype cNTnC and all single mutant cNTnC systems are summarized in Figure 2. was calculated using the following equation, indicating that all mutants were predicted to bind calcium more strongly than wildtype (positive values):
Figure 2.

Relative values between mutant and wildtype cNTnC structures as predicted by ASMD. values of Ca2+ binding were obtained from the respective PMFs at a distance of 15 Å from the calcium ion’s initial bound position in site II.
In our previous work, we have shown that the ASMD method significantly overestimates the relative free energy of calcium binding. Therefore, we additionally characterized the wildtype and mutant systems by means of thermodynamic integration.
Thermodynamic integration (TI) was performed in quintuplicate and averaged for wildtype and all proposed mutant cNTnC structures. Figure 3 illustrates a schematic version of the thermodynamic cycle implemented in our TI protocol. For all systems of ligand-bound protein and ligand in solvent, an ensemble of structures at each step was generated via a 5ns molecular dynamics simulation. In our application of the TI protocol the ligand of interest was the calcium ion. In Figures S2-S10, we provide plots of the values of the thermodynamic cycle for each simulated cNTnC system. The kinetics of calcium binding to the wildtype form of cNTnC have been previously described by Hazard and colleagues 63, via IAANS fluorescence titrations. The authors report the of Ca2+ for wildtype cTnC site II as 3 ± 1 μm ( kcal/mol). We found our relative free energy of calcium binding based on TI to be in good agreement with the experimental value for the wildtype system ( kcal/mol). The and values for wildtype and mutant cNTnC structures are provided in Table 1. All simulated mutant systems had either a negative effect (indicating increased predicted calcium binding affinity) or no effect. Mutant L41W was predicted to have the greatest effect on calcium binding with a kcal/mol and kcal/mol. Mutants F77I and F74R had the next largest impacts on predicted calcium binding affinity with values of −8.6 ± 1.0 kcal/mol ( kcal/mol) and −8.5 ± 0.5 kcal/mol ( kcal/mol), respectively. Mutants F74E and V72D showed moderate increases in predicted calcium binding affinity with values of −8.1 ± 1.5 kcal/mol ( kcal/mol) and −8.0 ± 1.7 kcal/mol ( kcal/mol), respectively. Mutations D33H and D33M showed relatively small differences in compared to wildtype with values of −7.6 ± 0.9 kcal/mol ( kcal/mol) and −7.3 ± 1.6 kcal/mol ( kcal/mol), respectively. Therefore, we believe that these mutations would have an insignificant impact on the calcium sensitivity of site II. The TI results for mutant V72N suggested that this mutant had no effect on calcium binding affinity, with kcal/mol and kcal/mol.
Figure 3.

Schematic thermodynamic cycle for thermodynamic integration calcium binding free energy calculation. cNTnC site II is shown in cyan with side chains of residues ASP67, SER69, and THR71 explicitly shown. When the ligand (Ca2+) is depicted as a green sphere, coulombic and vdW interactions with the chemical environment are present. However when the ligand is depicted as an orange sphere these nonbonded interactions are removed. The first step was to add harmonic distance restraints (depicted as dashed yellow lines) between the ligand and protein. These restraints prevent the ligand from leaving site II during the course of the simulation once the coulombic and vdW forces were removed. Next the coulombic and vdW interactions of the ligand were removed, and the ligand was subsequently removed from the protein system and placed into a water box. The coulombic and vdW interactions of the ligand were then restored to the ligand in the water box. Summing the free energy changes along the thermodynamic cycle resulted in the protein–ligand binding free energy. All systems were simulated in explicit solvent.
Table 1.
Thermodynamic integration results of calcium binding
| cNTnC Structure |
||
|---|---|---|
| WT | −7.2 ± 1.2 | - |
| D33H | −7.6 ± 0.9 | −0.4 ± 0.3 |
| D33M | −7.3 ± 1.6 | −0.1 ± 0.5 |
| L41W | −9.6 ± 0.9 | −2.4 ± 0.3 |
| V72D | −8.0 ± 1.7 | −0.8 ± 0.5 |
| V72N | −6.9 ± 0.7 | 0.4 ± 0.5 |
| F74E | −8.1 ± 1.5 | −0.9 ± 0.4 |
| F74R | −8.5 ± 0.5 | −1.3 ± 0.7 |
| F77I | −8.6 ± 1.0 | −1.4 ± 0.2 |
While ASMD has been shown previously to predict the correct trends of mutants altering calcium binding affinity, the method is prone to error. The loop region of site II is highly flexible and during the SMD trajectory the direction is likely to shift as it is dependent on the center of mass between alpha carbon atoms of multiple residues. If the loop region shifts dramatically, the calcium ion could be driven into the adjacent helices, thereby greatly increasing the energy of the PMF. Therefore, we believe this method to be suited for instances where the ASMD pull is dependent on more stable secondary structure (i.e., helix or sheet) which experiences less inherent dynamic motion. In regards to calculating calcium binding in site II of the cNTnC system, thermodynamic integration seems to be a more appropriate choice to correctly predict the relative free energy of binding. We found excellent agreement between our predicted and that of reported experimental for the wildtype cNTnC. Additionally, for all systems (wildtype and mutant) we observed a relatively low standard deviation between the five independent TI trials. Therefore, we have more confidence in the predicted of the proposed mutant systems. Based on the TI predictions, we suggest that mutants L41W, V72D, F74E, F74R, and F77I might exhibit a measurable calcium sensitizing effect. While we did not experimentally confirm the proposed mutations, we have previously simulated the correct trends in calcium binding affinity of sensitizing mutations (F20Q, V44Q, L48Q, M81Q) and desensitizing mutations (I61Q and D73N) utilizing the ASMD methodology 25. Additionally, we have used TI to accurately obtain the relative binding affinity for wildtype cNTnC and mutant D67A/D73A for calcium and magnesium ions 42, as well as the relative binding affinity of Mg2+ for several calcium sensitizing mutations such as L48Q and hypertrophic cardiomyopathy associated mutants Q50R and C84Y 43.
It is important to note that increased calcium sensitization of the myofilament and the cardiac troponin T subunit has been linked to cardiac arrhythmias 64,65,66,67. In cases of increased sensitization, the relaxation of cardiac muscle was impaired. However, the proposed sensitizing mutations in this work were intended to target the disease state (i.e., congestive heart failure) in which an individual exhibits a decreased sensitivity towards calcium binding. As such, they might be able to restore cardiac contractility and functionality post an initial loss of function.
Conclusion
We performed an exhaustive prediction of protein stability for all possible single point mutations in the cNTnC system, using the CUPSTAT webserver. Based on these results we studied eight mutations focused in cNTnC’s loop regions (loop I and loop II): D33H, D33M, L41W, V72D, V72N, F74E, F74R, F77I. We computationally characterized the effects of the mutations on altering calcium binding affinity via two methods: adaptive steered molecular dynamics and thermodynamic integration. With regards to the ASMD method, we showed that one reaches a convergence point, for this system, with increasingly slow pull speeds. Additionally, for instances where ASMD parameters are based on disordered regions, alternative methods may be more appropriate. Thermodynamic integration provided predictions to be in good agreement with experimentally determined calcium binding affinity for site II in wildtype cNTnC. Furthermore, the predicted of proposed mutations were in a much more realistic range with the greatest observed for mutant L41W (−2.372 ± 0.270 kcal/mol). Therefore, based on the TI results, we predict mutants L41W, V72D, F74E, F74R, and F77I to have a potential sensitizing effect on calcium binding affinity. We plan to experimentally validate these results and elucidate the impacts of the mutations on the KD of calcium binding in future works. In addition to experimental validation, in future work, we plan to simulate the proposed mutations in context of the whole troponin complex or even parts of thin filament. We believe this work will serve as a starting point for future design and characterization of novel mutations with therapeutic benefits.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Christopher Hadad for helpful discussions regarding the ASMD methodology. We would also like to thank the members of the Lindert lab for helpful discussions relating to this work. Finally, we would like to like to thank the Ohio Supercomputer Center68 for their valuable computational resources. This work was supported by the NIH (R01 HL137015 to S.L.).
Footnotes
Data and Software Availability
The wildtype cNTnC protein structures 1AP4 (closed conformation) and 2KFX (open conformation) were available in the Protein Data Bank (PDB) at https://www.rcsb.org/. All mutations were created using the wildtype cNTnC structure with the hydrophobic patch in the closed conformation as the base model within the PyMOL software using the protein mutagenesis tool. All ASMD and TI simulations were performed utilizing the AMBER18 software. Initial simulation file preparation scripts were obtained from the AMBER Advanced Tutorial 26 at http://ambermd.org/tutorials/advanced/tutorial26/. We have provided sample scripts for performing ASMD and TI and README files for each method in the supporting materials.
Supporting Information: Protocols, input file, and analysis scripts (ZIP); CUPSTAT calculations, additional ASMD and TI data (PDF).
References
- (1).Jin JP; Chong SM Localization of the two tropomyosin-binding sites of troponin T. Arch Biochem Biophys 2010, 500, 144–150. DOI: 10.1016/j.abb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Heeley DH; Golosinska K; Smillie LB The effects of troponin T fragments T1 and T2 on the binding of nonpolymerizable tropomyosin to F-actin in the presence and absence of troponin I and troponin C. J Biol Chem 1987, 262, 9971–9978. [PubMed] [Google Scholar]
- (3).Ohtsuki I; Morimoto S Troponin. In Encyclopedia of Biological Chemistry (Second Edition), Lennarz WJ, Lane MD Eds.; Academic Press, 2013; pp 445–449. [Google Scholar]
- (4).Dvoretsky A; Abusamhadneh EM; Howarth JW; Rosevear PR Solution structure of calcium-saturated cardiac troponin C bound to cardiac troponin I. J Biol Chem 2002, 277, 38565–38570. DOI: 10.1074/jbc.M205306200. [DOI] [PubMed] [Google Scholar]
- (5).Herzberg O; Moult J; James MNG CONFORMATIONAL FLEXIBILITY OF TROPONIN C11Supported by the Medical Research Council of Canada, an Alberta Heritage Foundation of Medical Research Fellowship (to O.H.) and an Alberta Heritage Foundation for Medical Research Visiting Scientist Fellowship (to J.M.). In Calcium-Binding Proteins in Health and Disease, Norman AW, Vanaman TC, Means AR Eds.; Academic Press, 1987; pp 312–322. [Google Scholar]
- (6).Zot HG; Potter JD A structural role for the Ca2+-Mg2+ sites on troponin C in the regulation of muscle contraction. Preparation and properties of troponin C depleted myofibrils. J Biol Chem 1982, 257, 7678–7683. [PubMed] [Google Scholar]
- (7).Tikunova SB; Davis JP Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J Biol Chem 2004, 279, 35341–35352. DOI: 10.1074/jbc.M405413200. [DOI] [PubMed] [Google Scholar]
- (8).Wang D; McCully ME; Luo Z; McMichael J; Tu AY; Daggett V; Regnier M Structural and functional consequences of cardiac troponin C L57Q and I61Q Ca(2+)-desensitizing variants. Arch Biochem Biophys 2013, 535, 68–75. DOI: 10.1016/j.abb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).McConnell BK; Singh S; Fan Q; Hernandez A; Portillo JP; Reiser PJ; Tikunova SB Knock-in mice harboring a Ca(2+) desensitizing mutation in cardiac troponin C develop early onset dilated cardiomyopathy. Front Physiol 2015, 6, 242. DOI: 10.3389/fphys.2015.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shettigar V; Zhang B; Little SC; Salhi HE; Hansen BJ; Li N; Zhang J; Roof SR; Ho H-T; Brunello L; et al. Rationally engineered Troponin C modulates in vivo cardiac function and performance in health and disease. Nat. Commun 2016, 7, 10794. DOI: 10.1038/ncomms10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Feest ER; Steven Korte F; Tu AY; Dai J; Razumova MV; Murry CE; Regnier M Thin filament incorporation of an engineered cardiac troponin C variant (L48Q) enhances contractility in intact cardiomyocytes from healthy and infarcted hearts. J Mol Cell Cardiol 2014, 72, 219–227. DOI: 10.1016/j.yjmcc.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Aprahamian ML; Tikunova SB; Price MV; Cuesta AF; Davis JP; Lindert S Successful Identification of Cardiac Troponin Calcium Sensitizers Using a Combination of Virtual Screening and ROC Analysis of Known Troponin C Binders. J Chem Inf Model 2017, 57, 3056–3069. DOI: 10.1021/acs.jcim.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Coldren WH; Tikunova SB; Davis JP; Lindert S Discovery of Novel Small-Molecule Calcium Sensitizers for Cardiac Troponin C: A Combined Virtual and Experimental Screening Approach. J Chem Inf Model 2020, 60, 3648–3661. DOI: 10.1021/acs.jcim.0c00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li MX; Hwang PM Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. DOI: 10.1016/j.gene.2015.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cai F; Li MX; Pineda-Sanabria SE; Gelozia S; Lindert S; West F; Sykes BD; Hwang PM Structures reveal details of small molecule binding to cardiac troponin. J Mol Cell Cardiol 2016, 101, 134–144. DOI: 10.1016/j.yjmcc.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ozer G; Quirk S; Hernandez R Adaptive steered molecular dynamics: Validation of the selection criterion and benchmarking energetics in vacuum. J. Chem. Phys 2012, 136, 215104. DOI: 10.1063/1.4725183. [DOI] [PubMed] [Google Scholar]
- (17).Izrailev S; Stepaniants S; Isralewitz B; Kosztin D; Lu H; Molnar F; Wriggers W; Schulten K Steered Molecular Dynamics. In Computational Molecular Dynamics: Challenges, Methods, Ideas, Berlin, Heidelberg, 1999//, 1999; Deuflhard P, Hermans J; Leimkuhler B, Mark AE, Reich S, Skeel RD, Eds.; Springer Berlin Heidelberg: pp 39–65. [Google Scholar]
- (18).Jarzynski C. Nonequilibrium Equality for Free Energy Differences. Phys Rev Lett 1997, 78, 2690–2693. DOI: 10.1103/PhysRevLett.78.2690. [DOI] [Google Scholar]
- (19).Ozer G; Valeev EF; Quirk S; Hernandez R Adaptive Steered Molecular Dynamics of the Long-Distance Unfolding of Neuropeptide Y. J. Chem. Theory Comput 2010, 6, 3026–3038. DOI: 10.1021/ct100320g. [DOI] [PubMed] [Google Scholar]
- (20).Ozer G; Quirk S; Hernandez R Thermodynamics of Decaalanine Stretching in Water Obtained by Adaptive Steered Molecular Dynamics Simulations. J. Chem. Theory Comput 2012, 8, 4837–4844. DOI: 10.1021/ct300709u. [DOI] [PubMed] [Google Scholar]
- (21).Straatsma TP; Berendsen HJC; Postma JPM Free energy of hydrophobic hydration: A molecular dynamics study of noble gases in water. J. Chem. Phys 1986, 85, 6720–6727. DOI: 10.1063/1.451846. [DOI] [Google Scholar]
- (22).Straatsma TP; Berendsen HJC Free energy of ionic hydration: Analysis of a thermodynamic integration technique to evaluate free energy differences by molecular dynamics simulations. J. Chem. Phys 1988, 89, 5876–5886. DOI: 10.1063/1.455539. [DOI] [Google Scholar]
- (23).Straatsma TP; McCammon JA Multiconfiguration thermodynamic integration. J. Chem. Phys 1991, 95, 1175–1188. DOI: 10.1063/1.461148. [DOI] [Google Scholar]
- (24).Straatsma TP; McCammon JA Computational Alchemy. Annu. Rev. Phys. Chem 1992, 43, 407–435. DOI: 10.1146/annurev.pc.43.100192.002203. [DOI] [Google Scholar]
- (25).Hantz ER; Lindert S Adaptative Steered Molecular Dynamics Study of Mutagenesis Effects on Calcium Affinity in the Regulatory Domain of Cardiac Troponin C. J Chem Inf Model 2021, 61, 3052–3057. DOI: 10.1021/acs.jcim.1c00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Stevens CM; Rayani K; Singh G; Lotfalisalmasi B; Tieleman DP; Tibbits GF Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations. J Biol Chem 2017, 292, 11915–11926. DOI: 10.1074/jbc.M116.770776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lim CC; Yang H; Yang M; Wang CK; Shi J; Berg EA; Pimentel DR; Gwathmey JK; Hajjar RJ; Helmes M; et al. A novel mutant cardiac troponin C disrupts molecular motions critical for calcium binding affinity and cardiomyocyte contractility. Biophys J 2008, 94, 3577–3589. DOI: 10.1529/biophysj.107.112896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lindert S; Kekenes-Huskey PM; McCammon JA Long-timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in troponin C. Biophys J 2012, 103, 1784–1789. DOI: 10.1016/j.bpj.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kekenes-Huskey PM; Lindert S; McCammon JA Molecular basis of calcium-sensitizing and desensitizing mutations of the human cardiac troponin C regulatory domain: a multi-scale simulation study. PLoS Comput Biol 2012, 8, e1002777. DOI: 10.1371/journal.pcbi.1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zamora JE; Papadaki M; Messer AE; Marston SB; Gould IR Troponin structure: its modulation by Ca2+ and phosphorylation studied by molecular dynamics simulations. Phys ChemChem Phys 2016, 18, 20691–20707, 10.1039/C6CP02610A. DOI: 10.1039/C6CP02610A. [DOI] [PubMed] [Google Scholar]
- (31).Bowman JD; Lindert S Molecular Dynamics and Umbrella Sampling Simulations Elucidate Differences in Troponin C Isoform and Mutant Hydrophobic Patch Exposure. J Phys Chem B 2018, 122, 7874–7883. DOI: 10.1021/acs.jpcb.8b05435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bowman JD; Coldren WH; Lindert S Mechanism of Cardiac Troponin C Calcium Sensitivity Modulation by Small Molecules Illuminated by Umbrella Sampling Simulations. J Chem Inf Model 2019, 59, 2964–2972. DOI: 10.1021/acs.jcim.9b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bowman JD; Lindert S Computational Studies of Cardiac and Skeletal Troponin. Front Mol Biosci 2019, 6, 68. DOI: 10.3389/fmolb.2019.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cheng Y; Lindert S; Kekenes-Huskey P; Rao VS; Solaro RJ; Rosevear PR; Amaro R; McCulloch AD; McCammon JA; Regnier M Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys J 2014, 107, 1675–1685. DOI: 10.1016/j.bpj.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cheng Y; Rao V; Tu AY; Lindert S; Wang D; Oxenford L; McCulloch AD; McCammon JA; Regnier M Troponin I Mutations R146G and R21C Alter Cardiac Troponin Function, Contractile Properties, and Modulation by Protein Kinase A (PKA)-mediated Phosphorylation. J Biol Chem 2015, 290, 27749–27766. DOI: 10.1074/jbc.M115.683045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dvornikov AV; Smolin N; Zhang M; Martin JL; Robia SL; de Tombe PP Restrictive Cardiomyopathy Troponin I R145W Mutation Does Not Perturb Myofilament Length-dependent Activation in Human Cardiac Sarcomeres. J Biol Chem 2016, 291, 21817–21828. DOI: 10.1074/jbc.M116.746172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lindert S; Cheng Y; Kekenes-Huskey P; Regnier M; McCammon JA Effects of HCM cTnI mutation R145G on troponin structure and modulation by PKA phosphorylation elucidated by molecular dynamics simulations. Biophys J 2015, 108, 395–407. DOI: 10.1016/j.bpj.2014.11.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cool AM; Lindert S Computational Methods Elucidate Consequences of Mutations and Post-translational Modifications on Troponin I Effective Concentration to Troponin C. J Phys Chem B 2021, 125, 7388–7396. DOI: 10.1021/acs.jpcb.1c03844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dewan S; McCabe KJ; Regnier M; McCulloch AD; Lindert S Molecular Effects of cTnC DCM Mutations on Calcium Sensitivity and Myofilament Activation-An Integrated Multiscale Modeling Study. J Phys Chem B 2016, 120, 8264–8275. DOI: 10.1021/acs.jpcb.6b01950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cheng Y; Lindert S; Oxenford L; Tu AY; McCulloch AD; Regnier M Effects of Cardiac Troponin I Mutation P83S on Contractile Properties and the Modulation by PKA-Mediated Phosphorylation. J Phys Chem B 2016, 120, 8238–8253. DOI: 10.1021/acs.jpcb.6b01859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cool A; Lindert S Umbrella Sampling Simulations Measure Switch Peptide Binding and Hydrophobic Patch Opening Free Energies in Cardiac Troponin. J Chem Inf Model 2022. DOI: 10.1021/acs.jcim.2c00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rayani K; Seffernick J; Li AY; Davis JP; Spuches AM; Van Petegem F; Solaro RJ; Lindert S; Tibbits GF Binding of calcium and magnesium to human cardiac Troponin C. J Biol Chem 2021, 100350. DOI: 10.1016/j.jbc.2021.100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rayani K; Hantz ER; Haji-Ghassemi O; Li AY; Spuches AM; Van Petegem F; Solaro RJ; Lindert S; Tibbits GF The effect of Mg2+ on Ca2+ binding to cardiac troponin C in hypertrophic cardiomyopathy associated TNNC1 variants. The FEBS Journal 2022, n/a, 10.1111/febs.16578. DOI: 10.1111/febs.16578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Parthiban V; Gromiha MM; Schomburg D CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res 2006, 34 (Web Server issue), W239–242. DOI: 10.1093/nar/gkl190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Parthiban V; Gromiha MM; Hoppe C; Schomburg D Structural analysis and prediction of protein mutant stability using distance and torsion potentials: role of secondary structure and solvent accessibility. Proteins 2007, 66, 41–52. DOI: 10.1002/prot.21115. [DOI] [PubMed] [Google Scholar]
- (46).Parthiban V; Gromiha MM; Abhinandan M; Schomburg D Computational modeling of protein mutant stability: analysis and optimization of statistical potentials and structural features reveal insights into prediction model development. BMC Struct Biol 2007, 7, 54. DOI: 10.1186/1472-6807-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Spyracopoulos L; Li MX; Sia SK; Gagné SM; Chandra M; Solaro RJ; Sykes BD Calcium-Induced Structural Transition in the Regulatory Domain of Human Cardiac Troponin C. Biochemistry 1997, 36, 12138–12146. DOI: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- (48).Hoffman RM; Sykes BD Structure of the inhibitor W7 bound to the regulatory domain of cardiac troponin C. Biochemistry 2009, 48, 5541–5552. DOI: 10.1021/bi9001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).The PyMOL Molecular Graphics System; Schrödinger, LLC. [Google Scholar]
- (50).Skowronsky RA; Schroeter M; Baxley T; Li Y; Chalovich JM; Spuches AM Thermodynamics and molecular dynamics simulations of calcium binding to the regulatory site of human cardiac troponin C: evidence for communication with the structural calcium binding sites. J Biol Inorg Chem 2013, 18, 49–58. DOI: 10.1007/s00775-012-0948-2. [DOI] [PubMed] [Google Scholar]
- (51).Karplus M; McCammon JA Molecular dynamics simulations of biomolecules. Nat Struct Biol 2002, 9, 646–652. DOI: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- (52).Wang D; Robertson IM; Li MX; McCully ME; Crane ML; Luo Z; Tu AY; Daggett V; Sykes BD; Regnier M Structural and functional consequences of the cardiac troponin C L48Q Ca(2+)-sensitizing mutation. Biochemistry 2012, 51, 4473–4487. DOI: 10.1021/bi3003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Roe DR; Cheatham TE PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput 2013, 9, 3084–3095. DOI: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- (54).Götz AW; Williamson MJ; Xu D; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput 2012, 8, 1542–1555. DOI: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TEI; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; et al. AMBER 2018 2018. [Google Scholar]
- (56).Jorgensen WL; Madura JD Quantum and statistical mechanical studies of liquids. 25. Solvation and conformation of methanol in water. J Am Chem Soc 1983, 105, 1407–1413. DOI: 10.1021/ja00344a001. [DOI] [Google Scholar]
- (57).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 2015, 11, 3696–3713. DOI: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR Molecular dynamics with coupling to an external bath. J Chem Phys 1984, 81, 3684–3690. DOI: 10.1063/1.448118. [DOI] [Google Scholar]
- (59).Loncharich RJ; Brooks BR; Pastor RW Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide. Biopolymers 1992, 32, 523–535. DOI: 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- (60).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald method. J Chem Phys 1995, 103, 8577–8593. DOI: 10.1063/1.470117. [DOI] [Google Scholar]
- (61).Leelananda SP; Lindert S Computational methods in drug discovery. Beilstein J Org Chem 2016, 12, 2694–2718. DOI: 10.3762/bjoc.12.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Shirts MR; Chodera JD Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 2008, 129, 124105. DOI: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Hazard AL; Kohout SC; Stricker NL; Putkey JA; Falke JJ The kinetic cycle of cardiac troponin C: calcium binding and dissociation at site II trigger slow conformational rearrangements. Protein Sci 1998, 7, 2451–2459. DOI: 10.1002/pro.5560071123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Huke S; Knollmann BC Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol 2010, 48, 824–833. DOI: 10.1016/j.yjmcc.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Landstrom AP; Dobrev D; Wehrens XHT Calcium Signaling and Cardiac Arrhythmias. Circ Res 2017, 120, 1969–1993. DOI: 10.1161/CIRCRESAHA.117.310083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Fiset C; Giles WR Cardiac troponin T mutations promote life-threatening arrhythmias. J Clin Invest 2008, 118, 3845–3847. DOI: 10.1172/JCI37787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Sutanto H; Lyon A; Lumens J; Schotten U; Dobrev D; Heijman J Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog Biophys Mol Biol 2020, 157, 54–75. DOI: 10.1016/j.pbiomolbio.2020.02.008. [DOI] [PubMed] [Google Scholar]
- (68).Ohio Supercomputer Center. Ohio Technology Consortium of the Ohio Board of Regents, 1987.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.