

Abstract
Receptor‐interacting protein kinase 1 (RIPK1) mediates necroptosis and inflammation in various pathophysiologies, emerging as a pharmacological target for neurodegenerative and inflammatory indications. This phase I, first‐in‐human, placebo‐controlled study evaluated the safety, pharmacokinetics (PKs), and pharmacodynamics (PDs) of GFH312, an RIPK1 inhibitor, in healthy adults. Subjects received GFH312 as a single ascending dose up to 500 mg (part I) or once‐daily repeated doses up to 200 mg for 14 days (part II). PKs were assessed using plasma and cerebrospinal fluid (CSF); PDs were assessed by phospho‐RIPK1 levels. Seventy‐six subjects were enrolled between April 2021 and June 2022: 38 (part I) and 19 (part II) received GFH312; 14 and five received placebo, respectively. At least one treatment‐emergent adverse event (TEAE) occurred in 42.1% (part I) and 63.2% (part II) of subjects receiving GFH312, compared with 42.9% and 40.0% of subjects receiving placebo, respectively. The most common TEAE was headache (21.1%). Two treatment‐related TEAEs were reported in part I and four in part II. No serious TEAEs were reported. Systemic absorption was rapid; exposure (area under the concentration‐time curve from time zero to the last measurable concentration and maximum plasma concentration) increased with dose level. The GFH312 CSF concentration post 100 mg single dose was approximately fourfold higher than the half maximal inhibitory concentration of human monocyte‐derived macrophages necroptosis with expected central nervous system penetration. Subjects receiving GFH312 had decreased phospho‐RIPK1 levels in peripheral blood mononuclear cells postdose. In conclusion, GFH312 was well‐tolerated and demonstrated RIPK1 inhibition in healthy subjects. Ongoing studies will inform the use of GFH312 in potential indications.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Receptor‐interacting serine/threonine‐protein kinase 1 (RIPK1) is implicated in the pathophysiology of neurodegenerative and inflammatory conditions, and is a promising pharmacological target for treating neurological and inflammatory indications.
WHAT QUESTION DID THIS STUDY ADDRESS?
The study was a first‐in‐human assessment of the safety, pharmacokinetics, and pharmacodynamics of GFH312, an RIPK1 inhibitor, in healthy subjects after administrating a single dose or multiple doses in a dose‐escalating manner.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
A single dose of up to 500 mg and repeated doses once daily for 14 days of up to 200 mg was safe and showed promising RIPK1 inhibition in healthy subjects. The plasma concentration of GFH312 generally increased with increasing dosage.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The results demonstrated that GFH312 had a good safety profile, and will help inform future dose regimens in phase II studies for patients with neurological and inflammatory diseases.
INTRODUCTION
Necroptosis is a regulated necrotic cell death mechanism that can be activated under apoptosis‐deficient conditions. It is controlled by the kinase activity of receptor‐interacting protein kinase 1 (RIPK1) and its downstream mediators, RIPK3 and mixed lineage kinase domain‐like pseudokinase. 1 Necroptosis and RIPK1 activity have been implicated in many diseases, including amyotrophic lateral sclerosis (ALS), 2 multiple sclerosis, 3 Alzheimer's disease (AD), 4 , 5 stroke, 6 , 7 and others. 8
As the activation of tumor necrosis factor receptor (TNFR) 1 leads to RIPK1 activation, RIPK1 inhibitors were initially developed as a small‐molecule alternative to anti‐TNF antibody therapies for TNF‐driven autoimmune conditions. 8 , 9 Anti‐TNF therapeutics have failed in clinical trials for neurodegenerative diseases, 10 possibly due to the blockage of TNFR2 signaling, which appears essential for neuroprotection. 11 Given that RIPK1 inhibitors do not inhibit TNFR2, 8 , 9 they may offer advantages over anti‐TNF therapies in treating neurodegenerative diseases, such as AD, ALS, and Parkinson's disease.
RIPK1 has emerged as an important drug target as its kinase structure is highly amenable for developing specific pharmacological small‐molecule inhibitors. Studies in mice have shown different RIPK1 kinase dead knock‐in mutations do not display significant developmental defects 1 ; because necroptosis might be predominantly activated under pathological conditions, inhibiting this pathway is an attractive therapeutic option. 1 Studies of necrostatins, small‐molecule inhibitors of necroptosis, identified RIPK1 as a potential pharmacological target for inhibiting necroptosis 8 and mediates deleterious inflammatory cell death mechanisms involved in the pathogenesis of various diseases. 9 In vivo studies have shown beneficial effects of RIPK1 inhibitors in myocardial ischemia and reperfusion injury settings. 12 , 13 , 14 A few RIPK1 inhibitors have progressed to human clinical trials for the treatment of inflammatory and central nervous system (CNS) diseases. 15 , 16 , 17 , 18 , 19 , 20
Here, we report results from the phase I first‐in‐human study of GFH312 (ClinicalTrials.gov Identifier: NCT04676711), a highly selective small molecule targeting RIPK1 with potent in vitro inhibition of human RIPK1 kinase activity, with a 50% inhibitory concentration (IC50) of 40 nM, that has shown potent anti‐necroptosis effects in multiple cell line models, and protective effects in neurological and inflammatory disease models (F. Zhou, F. Xie, and Z. Huo, unpublished data).
METHODS
Study design
This was a phase I, first‐in‐human, randomized, double‐blinded, placebo‐controlled study, comprising a single ascending dose stage (part Ia) with an additional food effect cohort (part Ib), and a multiple ascending dose stage (part II). The full trial protocol can be found in Appendix S1. The study was conducted at a single site between April 14, 2021, and June 8, 2022, in Melbourne, Australia. Healthy male and female subjects aged 18–55 years, weighing greater than or equal to 50 kg with a body mass index within 18–32 kg/m2 were eligible. All subjects provided written informed consent. Full inclusion and exclusion criteria are listed in Appendix S2.
In part Ia, subjects were randomized (3:1) to receive a single dose of either GFH312 or placebo. Seven ascending dose levels were sequentially tested: 5, 15, 45, 100, 200, 360, and 500 mg. Eight subjects were planned to be enrolled for each dose cohort; however, due to challenges in clinical operation during the coronavirus disease 2019 (COVID‐19) pandemic, seven, seven, and six subjects were enrolled in the 100, 360, and 500 mg cohorts, respectively. To escalate to the next dose cohort, both the study investigator and sponsor had to deem the preceding dose cohort to have an acceptable safety profile. In part Ib, the 100 mg dose cohort in part Ia was selected as a food effect cohort, based on attainment of adequate exposure and safety margin. After receipt of their dosage in part Ia, subjects in the food effect cohort underwent a washout period of greater than or equal to 7 days before receiving a single 100 mg dose of GFH312 30 min after a standard high‐fat meal. In part II, subjects were randomized (3:1) to receive either GFH312 or placebo once daily (q.d.) for 14 days at doses of 60, 120, or 200 mg. Safety evaluations up to 72 h (part I) and 24 h (part II) after the last dose for at least six of the subjects from each dose cohort were assessed before escalating to the next dose. GFH312 concentration in the cerebrospinal fluid (CSF) samples from the 100 mg dose cohort from part I and 120 mg q.d. cohort from part II was analyzed.
Both the study team and subjects were blinded to the treatment allocation. GFH312 and matching placebo tablets were prepared by GenFleet and administered by an unblinded pharmacist, based on a randomization list generated using block randomization by an independent unblinded statistician. The study protocol and any accompanying materials provided to the subject were reviewed and approved by the study's Human Research Ethics Committee. The study was conducted in accordance with the Declaration of Helsinki (Ethical Principles for Medical Research Involving Human Subjects); Integrated Addendum to International Conference on Harmonization (ICH) E6(R1): Guideline for Good Clinical Practice ICH E6(R2), annotated with comments by the Australian Therapeutic Goods Administration (TGA; 2018), and all other applicable regulatory requirements.
End points
The primary end point of the study was incidence of adverse events (AEs) and serious AEs (SAEs), change in laboratory values, electrocardiogram (ECG), vital signs, and physical examinations. Secondary end points included plasma concentration of GFH312 and pharmacokinetic (PK) parameters, CSF concentration, and CSF/plasma ratio of GFH312 following single and repeat doses in a single cohort. Exploratory end points included change in pharmacodynamic (PD) biomarker phospho‐RIPK1 (pRIPK1), S166 levels in peripheral blood mononuclear cells (PBMCs), and expression changes in a cytokine panel, including macrophage inflammatory protein 1α/β, interleukin (IL)‐35, IL‐23, IL‐6, IL‐2, IL‐33, and IL‐1b in the CSF samples.
Safety assessment
Safety assessments included AEs, SAEs, physical examination, vital signs, 12‐lead ECG and continuous telemetry, clinical laboratory measurements, weight, and pregnancy test. Part II also included suicide risk assessment using the Columbia‐Suicide Severity Rating Scale (C‐SSRS). The Study Monitoring Committee, which comprised the Principal Investigator and representatives from the sponsor and Contract Research Organization, reviewed the safety data before escalating to the next dose level.
All AEs were collected from the time the subject provided written informed consent, until 30 days after the last administration of the study drug. The National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0 was used to assess the severity of AEs.
Pharmacokinetic assessments
Whole blood, CSF, and urine samples were analyzed by a validated liquid chromatography–tandem mass spectrometry method, with a lower limit of quantification (LLOQ) of 2 ng/mL for plasma and urine, and 0.2 ng/mL for CSF. Timepoints for sample collection are detailed in Table S1. Blood samples were collected from all patients and centrifuged to extract plasma for PK determination. Urine samples were collected from subjects in part I.
The main PK parameters assessed included area under the concentration‐time curve (AUC), maximum concentration (C max), and time to maximum concentration (T max). The elimination rate constant (λ z), and its derived parameters (e.g., AUC from time zero to infinity [AUC0–inf], apparent clearance, apparent volume of distribution, and terminal half‐life [t 1/2]) were not reported if any of the following occurred: less than three data points, excluding C max, available at the terminal phase of the concentration‐time plot; adjusted coefficient of determination for the regression of λ z less than 0.75; or percentage of AUC extrapolation for the calculation of AUC0–inf greater than 20%.
Pharmacodynamic assessments
Whole blood and CSF specimens were collected predose and at different timepoints postdose, as indicated in Table S1. The pRIPK1 was assessed by isolating PBMCs from whole blood samples that were collected in 2 × 8 mL sodium citrate cell preparation tubes and shipped to 360 biolabs (Melbourne) at ambient temperature, stored at −80°C until analysis. The pRIPK1 concentration was measured by Meso Scale Discovery (MSD) enzyme‐linked immunosorbent assay. Analytes were bound to the capture antibody (biotinylated anti‐RIP Clone 38, BD Cat #610459) and detected with an anti‐pRIPK1 antibody (anti‐phospho‐RIP [Ser166] [DIL3S] CST Cat #65746). A SulfoTag‐conjugated secondary antibody was used before measuring luminescence (ECLu) on an MSD plate reader.
Statistical analysis
GFH312 plasma, urine, and CSF concentrations were summarized descriptively by nominal sampling times. PK parameters were calculated for each subject using a noncompartmental method in Phoenix WinNonlin version 8.3 (Certara), and summarized by dose level using descriptive statistics. For part I, dose proportionality was assessed using a power model with log‐transformed PK parameters of C max, AUC from time zero to the last measurable concentration (AUC0–t ) and AUC0–inf as the dependent variables, and log‐transformed dose level as the independent variable.
For PD data, due to their exploratory nature, no formal statistical analysis was conducted. For summaries and analyses, values below the LLOQ were imputed as ½*LLOQ.
Sample size
In parts I and II, eight subjects per dosing cohort were planned for enrollment, which is standard in first‐in‐human studies, and sufficient to meet the safety objectives. No formal sample size calculation was performed, and no power evaluation was provided. At a sample size of six subjects per cohort receiving GFH312, an AE of an underlying occurrence rate of greater than or equal to 30% had greater than or equal to 88% probability that at least one subject would report an AE.
RESULTS
Demographics
In part I, 52 subjects were enrolled, with 38 receiving GFH312 and 14 receiving placebo (Figure 1). Median age was 25.0 (range: 18–51) years, 53.8% of subjects were men, and 73.1% of the subjects were White. In part Ib, the washout period was 17 days for six subjects and 18 days for one subject in the fed cohort. In part II, 24 subjects were enrolled, with 19 receiving GFH312 and five receiving placebo. Median age was 31.0 (range: 20–49) years, 62.5% of subjects were men, and 75.0% of the subjects were White. Baseline characteristics for parts I and II are described in Table 1. In total, one subject withdrew early from part I, and three subjects withdrew early from part II. One subject from the 120 mg q.d. cohort in part II withdrew on day 12 due to personal reasons (patient ID: 103‐S255); a new subject (patient ID: 103‐S309) was enrolled as a replacement.
FIGURE 1.
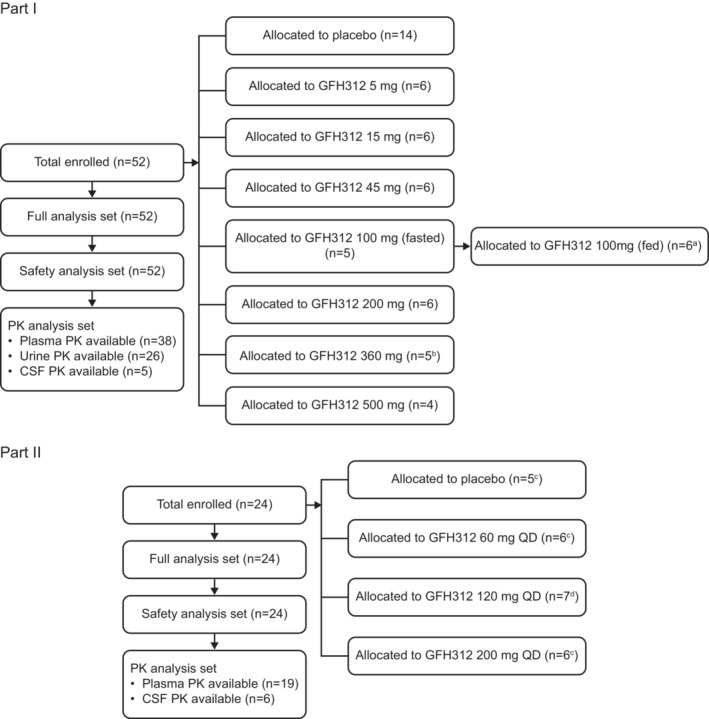
Study flow diagram. aOne subject mistakenly received placebo in Part Ib. bOne subject withdrew early due to travel restrictions during the coronavirus disease 2019 (COVID‐19) pandemic. cOne subject discontinued treatment early due to adverse events. dOne subject withdrew due to personal reasons. CSF, cerebrospinal fluid; PK, pharmacokinetics; QD, once daily.
TABLE 1.
Demographic and baseline characteristics in part I and part II.
| Part I | Part II | |||||
|---|---|---|---|---|---|---|
| GFH312 | Placebo | Total | GFH312 | Placebo | Total | |
| (N = 38) | (N = 14) | (N = 52) | (N = 19) | (N = 5) | (N = 24) | |
| Age, years, mean ± SD | 28.5 ± 9.1 | 24.7 ± 7.1 | 27.5 ± 8.7 | 32.2 ± 7.8 | 33.8 ± 6.3 | 32.5 ± 7.4 |
| Sex, n (%) | ||||||
| Female | 20 (52.6) | 4 (28.6) | 24 (46.2) | 5 (26.3) | 4 (80.0) | 9 (37.5) |
| Male | 18 (47.4) | 10 (71.4) | 28 (53.8) | 14 (73.7) | 1 (20.0) | 15 (62.5) |
| Race, n (%) | ||||||
| White | 27 (71.1) | 11 (78.6) | 38 (73.1) | 13 (68.4) | 5 (100.0) | 18 (75.0) |
| Asian | 10 (26.3) | 3 (21.4) | 13 (25.0) | 2 (10.5) | 0 | 2 (8.3) |
| American Indian or Alaskan native | 1 (2.6) | 0 | 1 (1.9) | 3 (15.8) | 0 | 3 (12.5) |
| Black or African American | 0 | 0 | 0 | 1 (5.3) | 0 | 1 (4.2) |
| Weight, kg, mean ± SD | 69.2 ± 12.5 | 73.2 ± 11.5 | 70.3 ± 12.2 | 74.5 ± 11.3 | 73.1 ± 7.5 | 74.2 ± 10.5 |
| Height, cm, mean ± SD | 171.7 ± 12.3 | 173.8 ± 7.5 | 172.2 ± 11.2 | 173.9 ± 7.5 | 171.8 ± 8.2 | 173.5 ± 7.5 |
| BMI, kg/m2, mean ± SD | 23.4 ± 2.6 | 24.2 ± 3.1 | 23.6 ± 2.8 | 24.6 ± 3.3 | 25.0 ± 4.0 | 24.7 ± 3.4 |
Abbreviations: BMI, body mass index; SD, standard deviation.
Safety
Part I
Across part I, all subjects had 100% compliance (total dose administered divided by total planned dose, multiplied by 100) for GFH312. At least one treatment‐emergent AE (TEAE) occurred in 16 of 38 (42.1%) subjects who received GFH312 and six of 14 (42.9%) who received placebo. All TEAEs were grade 1 (mild) in severity (Table 2). Comparing the fasted (n = 5) and fed (n = 6) GFH312 100 mg cohorts, three of five (60.0%) fasted subjects experienced at least one TEAE, whereas no TEAEs occurred for the fed subjects.
TABLE 2.
Summary of treatment‐emergent adverse events.
| Adverse events, n (%) a | Part I | Part II | ||||||
|---|---|---|---|---|---|---|---|---|
| GFH312 | Placebo | GFH312 | Placebo | |||||
| (N = 38) | (N = 14) | (N = 19) | (N = 5) | |||||
| Any grade | Grade 2 | Any grade | Grade 2 | Any grade | Grade 2 | Any grade | Grade 2 | |
| Any TEAEs | 16 (42.1%) | 0 | 6 (42.9%) | 0 | 12 (63.2%) | 3 (15.8%) | 2 (40.0%) | 0 |
| TEAEs which occurred in ≥2 subjects in GFH312 group or TEAEs with severity of grade 2 | ||||||||
| Headache | 3 (7.9%) | 0 | 1 (7.1%) | 0 | 4 (21.1%) | 0 | 1 (20.0%) | 0 |
| Procedural pain | 3 (7.9%) | 0 | 1 (7.1%) | 0 | 2 (10.5%) | 0 | 1 (20.0%) | 0 |
| Muscle twitching | 2 (5.3%) | 0 | 1 (7.1%) | 0 | 0 | 0 | 1 (20.0%) | 0 |
| Post‐procedural complication | 0 | 0 | 1 (7.1%) | 0 | 2 (10.5%) | 0 | 0 | 0 |
| Nausea | 0 | 0 | 0 | 0 | 2 (10.5%) | 0 | 0 | 0 |
| Back pain | 0 | 0 | 0 | 0 | 2 (10.5%) | 1 (5.3%) | 0 | 0 |
| Positional vertigo | 0 | 0 | 0 | 0 | 1 (5.3%) | 1 (5.3%) | 0 | 0 |
| Blood creatinine increased | 0 | 0 | 0 | 0 | 1 (5.3%) | 1 (5.3%) | 0 | 0 |
| Treatment‐related TEAEs | ||||||||
| Any TEAEs | 2 (5.3%) | 0 | 2 (14.3%) | 0 | 4 (21.1%) | 1 (5.3%) | 1 (20.0%) | 0 |
| Diarrhea | 0 | 0 | 1 (7.1%) | 0 | 0 | 0 | 0 | 0 |
| Muscle twitching | 1 (2.6%) | 0 | 1 (7.1%) | 0 | 0 | 0 | 1 (20.0%) | 0 |
| Headache | 1 (2.6%) | 0 | 0 | 0 | 3 (15.8%) | 0 | 0 | 0 |
| Blood creatinine increased | 0 | 0 | 0 | 0 | 1 (5.3%) | 1 (5.3%) | 0 | 0 |
Note: TEAEs are defined as adverse events that occurred following the first administration of study medication.
Abbreviation: TEAE, treatment‐emergent adverse event.
No ≥grade 3 adverse events were observed in the study.
The most common TEAEs were procedural pain and headache, each occurring in three of 38 (7.9%) subjects in the GFH312 group. Most headache events lasted for 1–2 days and resolved without medical intervention. Treatment‐related TEAEs occurred in two of 38 (5.3%) subjects in the GFH312 group (headache and muscle twitching in one subject each) and two of 14 (14.3%) subjects in the placebo group (diarrhea and muscle twitching in one subject each). All muscle twitching events were transient and resolved without medical intervention. Full information on TEAEs by dose cohort is presented in Tables S2 and S3; information on AEs for each patient is presented in Table S4. There were no clinically significant abnormalities reported for laboratory measures, vital signs, physical findings or ECG recordings, and no SAEs or TEAEs leading to treatment discontinuation.
Part II
Across part II, the mean compliance for GFH312 was 92.9%. At least one TEAE occurred in 12 of 19 (63.2%) subjects who received GFH312 and two of five (40.0%) subjects who received placebo (Table 2). Of the GFH312 group, the highest severity of TEAEs was grade 1 in 11 (57.9%) subjects, and grade 2 in three subjects (15.8%; one receiving 60 mg q.d. had positional vertigo, one receiving 200 mg q.d. had increased blood creatinine, and one receiving 120 mg q.d. had back pain); all TEAEs in the placebo group were grade 1.
The most common TEAE was headache, occurring in four of 19 (21.1%) subjects in the GFH312 group; two were from the GFH312 120 mg q.d. group who had undergone lumbar puncture for CSF sampling. All headache events resolved in 1 day without medication intervention. Cardiac TEAEs occurred in two of 19 (10.5%) subjects who received GFH312 and one of five (20.0%) subjects who received placebo, which were cases of transient arrhythmias that were assessed to be unrelated to the study drug by the investigator. In the GFH312 group, four of 19 (21.1%) subjects had treatment‐related TEAEs: one subject had elevated blood creatinine (Grade 2) and three subjects experienced headache (all Grade 1). In the placebo group, 1/5 (20.0%) subject had a treatment‐related TEAE of muscle twitching. There was a trend for elevations in serum creatinine in higher‐dose cohorts, both in absolute terms and in proportion to the baseline value. The proportionate increase appeared similar in both the GFH312 and placebo groups, although the highest absolute serum creatinine values were seen in subjects who received the highest dose of GFH312 (200 mg q.d.), including two subjects whose creatinine level was above the upper limit of normal. There were no trends observed for serum urea levels, no significant abnormalities of the urinalysis in any subject, and the serum creatinine levels decreased toward baseline in all subjects after cessation of study drug. No other clinically significant abnormalities in the safety parameters, including suicide risk assessment using C‐SSRS, were reported in part II. No SAEs were reported. Three subjects had TEAEs that led to discontinuation of the study drug, with two subjects in the GFH312 group experiencing TEAEs of elevated blood creatinine and ventricular tachycardia, respectively, and one subject in the placebo group experiencing muscle twitching, procedural pain, paresthesia, and burning sensation following a lumbar puncture.
Pharmacokinetic
Part I
A summary of the PK parameters of GFH312 for part I is shown in Table 3. Systemic absorption of GFH312 was rapid, with concentrations exceeding LLOQ occurring at the first PK timepoint of 30 min for all dose cohorts (Figure 2a). Median T max ranged from 2.1 to 6.0 h. Geometric mean AUC0–t and C max post‐single dose ranged from 1072.9 to 30,890.5 h ng/mL and 118.1 to 1294.9 ng/mL, respectively. The drug was not dose proportional across dose cohorts of 5–500 mg. The dose‐proportionality assessment for dose cohorts of 45–360 mg reported a slope of 0.91 (90% confidence interval [CI]: 0.71–1.12) for AUC0–t , 0.87 (90% CI: 0.64–1.09) for AUC0–inf, and 0.63 (90% CI: 0.47–0.79) for C max. The AUC increased in an approximately proportional manner, but C max increased slightly less than proportionally for dose cohorts of 45–360 mg. The 500 mg cohort had comparatively lower dose‐normalized exposure, possibly due to saturation in drug absorption. The intersubject variability was high, with the coefficient of variation ranging between 9.4 and 52.8% for C max, 16.8 and 62.3% for AUC0–t, and 16.9 and 62.4% for AUC0–inf.
TABLE 3.
GFH312 PK parameters after a single dose in part I.
| PK parameter, geomean (geoCV%) | GFH312 | GFH312 | GFH312 | GFH312 | GFH312 | GFH312 | GFH312 | GFH312 |
|---|---|---|---|---|---|---|---|---|
| 5 mg | 15 mg | 45 mg | 100 mg | 100 mg (Fed) | 200 mg | 360 mg | 500 mg | |
| (N = 6) | (N = 6) | (N = 6) | (N = 5) | (N = 6) | (N = 6) | (N = 5) | (N = 4) | |
| C max (ng/mL) | 118.1 (18.1) | 359.9 (35.3) | 334.8 (52.8) | 549.5 (38.1) | 806.9 (9.4) | 793.5 (28.2) | 1294.9 (15.5) | 1165.2 (29.9) |
| T max (h) a | 3.0 (2.0–4.0) | 2.1 (2.0–4.0) | 3.0 (2.0–4.2) | 6.0 (2.0–6.0) | 4.0 (2.0–6.0) | 4.0 (2.0–6.0) | 4.0 (1.0–4.0) | 4.0 (2.0–4.5) |
| AUC0–t (h ng/mL) | 1072.9 (16.8) | 3405.4 (35.4) | 4754.7 (40.9) | 8713.9 (62.3) | 8943.3 (51.3) | 18,525.1 (34.5) | 30,890.5 (52.4) | 29,636.0 (30.8) |
| AUC0–inf (h ng/mL) | 1144.2 (16.9) | 3452.9 (34.6) | 4956.6 (39.1) | 8879.0 (62.4) | 10,531.5 (34.9) b | 17,992.7 (27.9) c | 29,583.1 (62.2) d | 31,533.0 (28.4) |
| t 1/2 (h) | 6.2 (27.6) | 7.2 (27.0) | 13.7 (34.4) | 11.0 (16.2) | 8.8 (28.6) b | 16.1 (43.7) c | 16.4 (52.5) d | 17.6 (44.8) |
| CL/F (L/h) | 4.4 (16.9) | 4.3 (34.6) | 9.1 (39.1) | 11.3 (62.4) | 9.5 (34.9) b | 11.1 (27.9) c | 12.2 (62.2) d | 15.9 (28.4) |
| V z/F (L) | 39.0 (18.2) | 45.1 (25.5) | 179.2 (60.1) | 178.4 (52.7) | 120.4 (40.7) b | 257.7 (57.4) c | 287.1 (10.6) d | 403.0 (65.4) |
Abbreviations: AUC0–inf, area under the drug concentration‐time curve from time zero to infinity; AUC0–t , area under the drug concentration‐time curve from time zero to the last quantifiable concentration; AUC_%Extrap, percentage of AUC extrapolation for the calculation of AUC0–inf; C max, maximum concentration; CL/F, apparent clearance; geoCV%, geometric coefficient of variation; geomean, geometric mean; PK, pharmacokinetic; R 2adj, adjusted coefficient of determination for the regression of the λ z; SD, standard deviation; T max, time to maximum concentration; t 1/2, terminal half‐life; V z/F, apparent volume of distribution; λ z, elimination rate constant.
T max is reported as median (range).
n=5, λ z cannot be calculated accurately in one subject due to R 2adj less than 0.75.
n=5, λ z cannot be calculated accurately in one subject as AUC_%Extrap exceeds 20%.
n=3, λ z cannot be calculated accurately in two subjects as AUC_%Extrap exceeds 20%.
FIGURE 2.
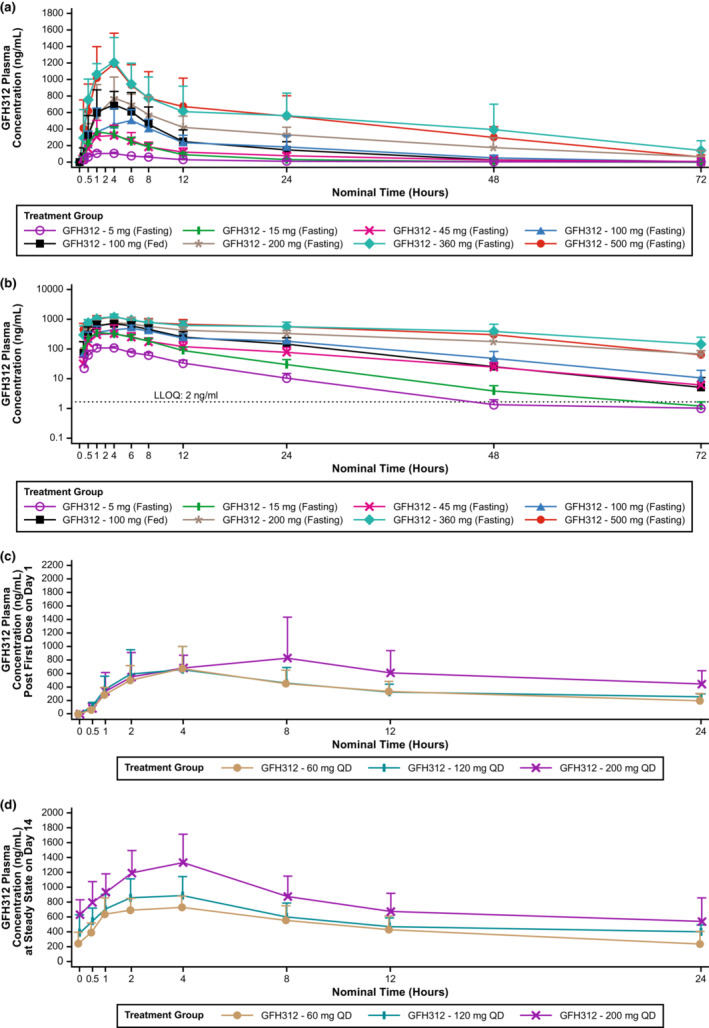
Plasma concentration over time for GFH312. (a) Plasma concentration after a single dose of GFH312 in part I, linear scale. (b) Plasma concentration after a single dose of GFH312 in part I, log scale. (c) Plasma concentration after the first dose of GFH312 in part II. (d) Plasma concentration of GFH312 at steady‐state on day 14 in part II. LLOQ, lower limit of quantification; QD, once daily.
Geometric mean t 1/2 were similar between the 5 mg (6.2 h) and 15 mg (7.2 h) dose groups. Over the dose levels of 45–360 mg, geometric mean t 1/2 was approximately consistent in fasted condition, ranging from 11.0 to 16.4 h; for the 500 mg dose cohort, geometric mean t 1/2 was 17.6 h.
Compared with the fasted 100 mg cohort, the fed cohort had more rapid systemic absorption (median T max 4.0 vs. 6.0 h) and a higher geometric least squares (LS) mean C max (816.2 vs. 549.5 mg/mL). Geometric LS mean AUC0–t was comparable between the fed and fasted cohorts (Table S5).
Mean (±SD) CSF concentration of GFH312 in the 100 mg dose group was 26.6 (±9.0) ng/mL at 4 h postdose, and 24.4 (±15.4) ng/mL at 8 h postdose. Mean CSF/plasma concentration ratio was 0.056 and 0.058 at 4 and 8 h postdose, respectively; the mean CSF/plasma free concentration ratio was 2.98 and 3.09 at 4 and 8 h postdose, respectively (Table S6). The urine PK parameters of GFH312 are presented in Table S7. Overall, mean urine concentration of GFH312 increased with increasing dose administered, and the cumulative urinary excretion of GFH312 was low across all dose levels.
Part II
Mean plasma concentrations of GFH312 over time on day 1 and day 14 are shown in Figure 2b,c. Similar to the single‐dose cohorts in part I, systemic absorption of GFH312 was rapid, with concentrations exceeding LLOQ occurring at the first PK timepoint of 30 min for all dose cohorts (Table 4). Median T max ranged from 4.0 to 4.1 h on day 1 and median steady‐state T max,(ss) ranged from 2.0 to 4.0 h on day 14. Geometric mean C max ranged from 587.7 to 946.1 ng/mL on day 1 and geometric mean C max,ss ranged from 738.0 to 1290.2 ng/mL on day 14; geometric mean C max,ss increased with increasing GFH312 doses. The accumulation ratio (RAC max and RAAUC) was less than two‐fold across all dose cohorts, indicating limited accumulation following multiple GFH312 doses.
TABLE 4.
GFH312 PK parameters postdose on day 1 and day 14 in part II.
| GFH312 | GFH312 | GFH312 | |
|---|---|---|---|
| 60 mg q.d. (N = 6) | 120 mg q.d. (N = 7) | 200 mg q.d. (N = 6) | |
| Day 1, geomean (geoCV%) | |||
| C max (ng/mL) | 587.7 (61.0) | 587.8 (63.1) | 946.1 (47.9) |
| T max (h) a | 4.0 (4.0–4.1) | 4.0 (2.0–23.7) | 4.1 (2.0–8.3) |
| AUC0–t (h ng/mL) | 7770.9 (50.4) | 8473.9 (40.9) | 13,031.9 (35.1) |
| Day 14, geomean (geoCV%) | |||
| C max,ss (ng/mL) | 738.0 (22.5) b | 921.7 (28.3) c | 1290.2 (29.0) e |
| C min,ss (ng/mL) | 249.3 (51.3) b | 362.8 (32.6) c | 543.6 (33.5) e |
| T max,ss (h) a | 4.0 (1.0–4.1) b | 2.0 (2.0–3.9) c | 4.0 (2.0–4.0) e |
| AUC0–tau (h ng/mL) | 10,656.5 (34.4) b | 12,973.5 (24.1) c | 18,963.7 (30.6) e |
| RAAUC | 1.3 (39.3) b | 1.3 (55.2) d | 1.6 (35.2) f |
| RACmax | 1.1 (47.4) b | 1.5 (62.4) c | 1.2 (45.8) e |
Abbreviations: AUC0–t , area under the drug concentration‐time curve from time zero to the last quantifiable concentration; AUC0–tau, area under the drug concentration‐time curve from time zero to end of dosing period; AUC0–24, area under the drug concentration‐time curve from time zero to 24 h; C max, maximum concentration; C max,ss, maximum concentration at steady‐state; C min,ss, minimum concentration at steady‐state; geoCV%, geometric coefficient of variation; geomean, geometric mean; PK, pharmacokinetic; QD, once daily; RAAUC, accumulation ratio calculated by AUC; RACmax, accumulation ratio calculated by C max; T max, time to maximum concentration; T max,ss, time to maximum concentration at steady‐state.
T max and T max,ss are reported as median (range).
n = 5, one subject discontinued treatment on day 7 due to adverse event.
n = 6, one subject discontinued treatment on day 12 due to personal reasons.
n = 5, one subject discontinued treatment on day 12 due to personal reasons, and for another subject, AUC0–24 on day 1 and RAAUC could not be calculated.
n = 5, one subject discontinued treatment on day 8 due to an adverse event.
n = 3, for three subjects, AUC0–24 on day 1 and RAAUC could not be calculated.
The mean (±SD) CSF concentration of GFH312 in the 120 mg q.d. dose group at day 14 was 58.2 (±15.1) ng/mL at 4 h post‐dose. Mean (±SD) CSF to plasma concentration ratio and mean (±SD) CSF to plasma free concentration ratio for GFH312 at day 14, 4 h postdose, was 0.07 (±0.02) and 3.59 (±0.89), respectively (Table S6).
PD analysis
In part I, subjects in the GFH312 group achieved up to 80% decrease from baseline in pRIPK1 level at 2 h, which was maintained for over 24 h. In part II, subjects in the GFH312 group achieved sustained decrease in pRIPK1 level during the 14‐day daily dosing period, and a partial recovery was observed 1 day post‐last dose (Figure 3). Cytokine levels were generally below the LLOQs both at baseline and after 14 days of dosing. The only exception was IL‐6, which was detected in three pre‐ or postdose samples from different subjects and was not considered meaningful. 21
FIGURE 3.
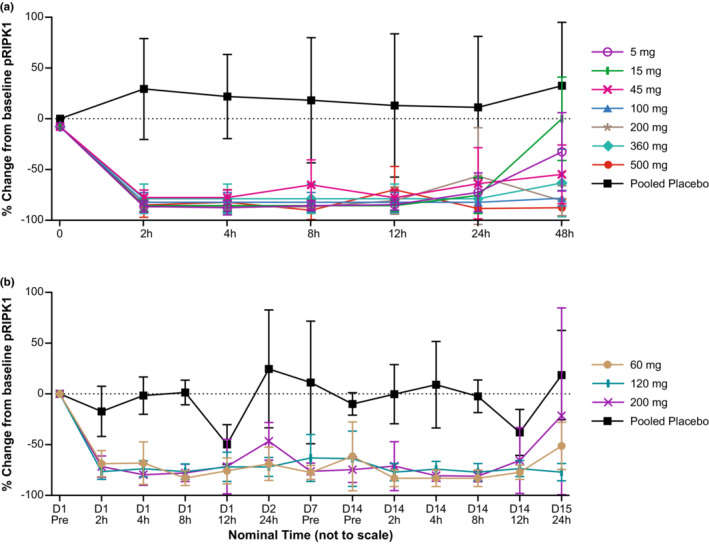
The pRIPK1 mean change (±SD) from baseline in (a) part I and (b) part II. D, day; pRIPK1, phospho‐receptor‐interacting serine/threonine‐protein kinase 1; pre, predose; SD, standard deviation.
DISCUSSION
This study was the first‐in‐human assessment of the safety, PKs, and PDs of GFH312, a small molecule targeting RIPK1. A single dose of up to 500 mg and repeated doses of up to 200 mg q.d. for 14 days were evaluated and found to be safe, with promising PD data in healthy subjects. There was high intersubject variability in PK parameters across all dose cohorts, but generally plasma concentrations increased with increasing doses of GFH312.
Safety
GFH312 was well‐tolerated for single doses up to 500 mg and repeat doses up to 200 mg q.d., with no reports of serious TEAEs or life‐threatening or fatal events with single or multiple doses (Table 2). In the single‐dose cohorts, all TEAEs were grade 1 in severity. There was no apparent trend of increasing frequency or severity of treatment‐related TEAEs with increasing dose, and the proportion of TEAEs was similar between treatment and placebo groups. In the multiple‐dose cohorts, there was a trend toward an increase in frequency of treatment‐related TEAEs with increasing dose; however, the study was not powered to conclude significant differences in treatment‐related TEAEs between doses due to the small sample size.
In part II, four subjects receiving GFH312 had TEAEs classified as “injury, poisoning, and procedural complications”: two (10.5%) were post‐procedural complication, two (10.5%) were procedural pain, and one (5.3%) was procedural headache (Table S3). All four subjects were from the GFH312 120 mg q.d. cohort who underwent lumbar puncture for CSF sampling, and the TEAEs were likely due to the procedure. In comparison, procedural pain occurred in one subject from the 120 mg placebo group. In part II, mean creatinine levels were elevated for all GFH312 dose levels and in the placebo group, but still within the normal range. Some possible explanations for the elevated levels were discussed by the Safety Review Committee, including strenuous exercise, meals rich in creatinine, GFH312 effect on renal blood flow or inhibition of creatinine excretion through renal transporters, and interference of GFH312 or the excipients with creatinine assay, but no definite conclusions can be made at this stage. There is currently no evidence that GFH312 affects the musculoskeletal system; based on the TEAEs classified as musculoskeletal and connective tissue disorders in the study, only three cases of muscle twitching were considered related to GFH312 treatment, in which one occurred after the lumbar puncture procedure. The subjects did not have any symptoms or laboratory abnormalities indicative of rhabdomyolysis. Moreover, another recent study in healthy Chinese subjects showed that subjects receiving GFH312 did not experience an increase in creatinine levels or musculoskeletal system AEs (unpublished data). To our knowledge, there is no clinical evidence demonstrating that RIPK1 inhibitors have an increased risk of rhabdomyolysis or other musculoskeletal AEs.
Similar to GFH312, other RIPK1 inhibitors, such as SAR443060 and GSK2982772, were found to be well‐tolerated in healthy subjects, with all TEAEs rated as mild or moderate, and no SAEs reported. 18 , 20
Pharmacokinetics
With a single dose of GFH312, the mean plasma concentration of GFH312 increased with dose level, except between the 15 and 45 mg dose levels (Figure 2a). Exposures (AUC0–t and C max) increased with increasing dose over the 5–360 mg range, but did not increase when dosage was increased from 360 to 500 mg (Table 3), which may be due to saturation in drug absorption at dose levels above 360 mg, or due to small sample size. Both 5 and 15 mg cohorts had substantially higher dose‐normalized exposure than other dose cohorts; in these cohorts, GFH312 was administered with 5 mg tablets. For the 45 mg dose cohort, one 5 mg tablet and two 20 mg tablets were used, and in cohorts above 45, 20 and 100 mg tablets were used. Hence, the higher dose‐normalized exposure in the 5 and 15 mg cohorts could be due to the use of 5 mg tablets, which may have a higher rate and extent of drug absorption than the 20 and 100 mg tablets. This is supported by the results of an in‐house in vitro analysis, which showed that GFH312 5 mg tablets underwent faster dissolution compared with the 20 and 100 mg tablets.
There was no food effect on AUC0–t and AUC0–inf (ratio of geometric least squares means 1.03; 90% CI: 0.91–1.16), but systemic absorption of GFH312 after dosing on day 1 for the fed cohort was more rapid than that for the fasted cohort (Table S5). Mean C max for the 100 mg cohort was higher for the fed cohort than the fasted cohort, which may be caused by a greater absorption rate of GFH312 under fed status.
In part II, at days 7, 13, and 14 predose, geometric mean concentrations were similar within each dose level, suggesting that the plasma concentration approached steady‐state ~7 days after administration. On day 1, mean C max and AUC0–t were similar between 60 mg q.d. and 120 mg q.d. cohorts, but higher for the 200 mg q.d. cohort (Table 4). There was an obvious increment in GFH312 exposure post‐first dose on day 1 and at steady‐state on day 14 when increasing dosage from 120 to 200 mg q.d.; however, the increase in exposure was less than proportional compared with the increase in dose. PK exposure on day 1 in the 60 mg q.d. cohort was comparable with the 100 mg single‐dose and 120 mg q.d. cohorts. Moreover, the C max,ss, C min,ss, and AUCo–tau on day 14 were similar between the 60 mg q.d. and 120 mg q.d. cohorts, and the C max/dose and AUC0–t /dose at steady‐state was much higher for the 60 mg q.d. cohort than the 120 and 200 mg q.d. cohorts. No potential factors have been found to explain the high exposure in the 60 mg q.d. cohort after analyzing demographics, data checking formulation profile and dosing information, and investigating the bioanalytical process of PK samples.
The GFH312 CSF concentration of subjects after a single 100 mg dose was approximately four‐fold higher than the IC50 of human monocyte‐derived macrophages necroptosis. Together with the CSF/plasma concentration ratio and CSF/plasma free concentration ratio in both parts I and II, these results confirmed that GFH312 has CNS penetration, signifying the potential of GFH312 as a treatment for neurodegenerative conditions. 2 , 4 , 5 Further studies investigating GFH312 in these disease states would be warranted to clarify the underlying mechanisms.
Pharmacodynamics
In this study, all dose levels of GFH312 in both parts I and II resulted in a rapid decrease of pRIPK1 levels from baseline, whereas subjects in the placebo group maintained a consistently low or undetectable pRIPK1 level (Figure 3), which is expected, given that the subjects in the study were healthy volunteers. A single dose of 5 mg GFH312 caused the pRIPK1 level to fall below the LLOQ, which restricted further analysis of PK‐PD correlations. The decrease in pRIPK1 levels was sustained for 24 h postdose for part I, and from day 1 to day 14 for part II. The PD results indicate that GFH312 at a dose level as low as 5 mg is sufficient to produce an effective and sustained inhibition of pRIPK1; as pRIPK1 expression is a reliable indicator of RIPK1 kinase activity, 22 , 23 GFH312 could potentially be used in neurodegenerative and inflammatory diseases that involve activation of RIPK1 and necroptosis. 8 , 9 , 12 Low or undetectable cytokine levels were observed in most patients, which is expected in healthy subjects.
CONCLUSION
In conclusion, a single dose of up to 500 mg and repeated doses of up to 200 mg q.d. for 14 days of GFH312 were safe and showed promising RIPK1 inhibition in healthy subjects. There was high intersubject variability in PK parameters, but plasma levels generally increased with increasing doses of GFH312. The CSF data suggest that GFH312 has exposure in the CNS, and may potentially be useful for treating neurological indications. These data suggest that GFH312 is a promising agent that targets RIPK1 with a large therapeutic window, and will inform dose regimens in proposed future studies.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. Y.W. and H.S. designed the study. J.L performed the study. S.W., W.Z., C.Z., H.Z., and J.W. analyzed the data.
FUNDING INFORMATION
This study was sponsored by GenFleet Therapeutics (AUSTRALIA) PTY LTD.
CONFLICT OF INTEREST STATEMENT
S.W., W.Z., H.Z., J.W., C.Z., H.S., and Y.W. are employees of Genfleet Therapeutics, Shanghai, China. H.S., Y.W., and H.Z. hold shares in Genfleet Therapeutics, Shanghai, China. J.L. was the Principal Investigator of the study and Chief Medical Officer of Nucleus Network, Melbourne, Australia.
Supporting information
Appendix S1
Appendix S2
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
ACKNOWLEDGMENTS
The authors thank all persons who participated in this study and the investigators and research staff at the study site. Medical writing support was provided by Zhi Yang Loh of Nucleus Global Asia Pacific, funded by GenFleet Therapeutics, in accordance with Good Publication Practice (GPP 2022) guidelines.
Lickliter J, Wang S, Zhang W, et al. A phase I randomized, double‐blinded, placebo‐controlled study assessing the safety and pharmacokinetics of RIPK1 inhibitor GFH312 in healthy subjects. Clin Transl Sci. 2023;16:1691‐1703. doi: 10.1111/cts.13580
REFERENCES
- 1. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci USA. 2019;116:9714‐9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ito Y, Ofengeim D, Najafov A, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ofengeim D, Ito Y, Najafov A, et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10:1836‐1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caccamo A, Branca C, Piras IS, et al. Necroptosis activation in Alzheimer's disease. Nat Neurosci. 2017;20:1236‐1246. [DOI] [PubMed] [Google Scholar]
- 5. Ofengeim D, Mazzitelli S, Ito Y, et al. RIPK1 mediates a disease‐associated microglial response in Alzheimer's disease. Proc Natl Acad Sci USA. 2017;114:E8788‐E8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang S, Wang Y, Li D, Wu J, Si W, Wu Y. Necrostatin‐1 attenuates inflammatory response and improves cognitive function in chronic ischemic stroke mice. Medicines (Basel). 2016;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. King MD, Whitaker‐Lea WA, Campbell JM, Alleyne CH Jr, Dhandapani KM. Necrostatin‐1 reduces neurovascular injury after intracerebral hemorrhage. Int J Cell Biol. 2014;2014:495817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mifflin L, Ofengeim D, Yuan J. Receptor‐interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov. 2020;19:553‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1‐mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20:19‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. The lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group . TNF neutralization in MS: results of a randomized, placebo‐controlled multicenter study. Neurology. 1999;53:457‐465. [PubMed] [Google Scholar]
- 11. Dong Y, Fischer R, Naudé PJW, et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci USA. 2016;113:12304‐12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karunakaran D, Geoffrion M, Wei L, et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu YR, Xu HM. Protective effect of necrostatin‐1 on myocardial tissue in rats with acute myocardial infarction. Genet Mol Res. 2016;15:gmr.15027298. [DOI] [PubMed] [Google Scholar]
- 14. Oerlemans MIFJ, Liu J, Arslan F, et al. Inhibition of RIP1‐dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia–reperfusion in vivo. Basic Res Cardiol. 2012;107:270. [DOI] [PubMed] [Google Scholar]
- 15. Weisel K, Scott N, Berger S, et al. A randomised, placebo‐controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ Open Gastroenterol. 2021;8:e000680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grievink HW, Heuberger JAAC, Huang F, et al. DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin Pharmacol Ther. 2020;107:406‐414. [DOI] [PubMed] [Google Scholar]
- 17. Grievink HW, Heuberger JAAC, Huang F, et al. CORRIGENDUM: DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin Pharmacol Ther. 2021;109:1678‐1679. [DOI] [PubMed] [Google Scholar]
- 18. Weisel K, Berger S, Thorn K, et al. A randomized, placebo‐controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res Ther. 2021;23:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weisel K, Berger S, Papp K, et al. Response to inhibition of receptor‐interacting protein kinase 1 (RIPK1) in active plaque psoriasis: a randomized placebo‐controlled study. Clin Pharmacol Ther. 2020;108:808‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vissers M et al. Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: randomized, placebo‐controlled, double‐blind phase I/Ib studies in healthy subjects and patients. Clin Transl Sci. 2022;15:2010‐2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gruol DL. IL‐6 regulation of synaptic function in the CNS. Neuropharmacology. 2015;96:42‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laurien L, Nagata M, Schünke H, et al. Autophosphorylation at serine 166 regulates RIP kinase 1‐mediated cell death and inflammation. Nat Commun. 2020;11:1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geng J, Ito Y, Shi L, et al. Regulation of RIPK1 activation by TAK1‐mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017;8:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7