

Abstract
Despite complex pathways of drug disposition, clinical pharmacogenetic predictors currently rely on only a few high effect variants. Quantification of the polygenic contribution to variability in drug disposition is necessary to prioritize target drugs for pharmacogenomic approaches and guide analytic methods. Dexmedetomidine and fentanyl, often used in postoperative care of pediatric patients, have high rates of inter‐individual variability in dosing requirements. Analyzing previously generated population pharmacokinetic parameters, we used Bayesian hierarchical mixed modeling to measure narrow‐sense (additive) heritability () of dexmedetomidine and fentanyl clearance in children and identify relative contributions of small, moderate, and large effect‐size variants to . We used genome‐wide association studies (GWAS) to identify variants contributing to variation in dexmedetomidine and fentanyl clearance, followed by functional analyses to identify associated pathways. For dexmedetomidine, median clearance was 33.0 L/h (interquartile range [IQR] 23.8–47.9 L/h) and was estimated to be 0.35 (90% credible interval 0.00–0.90), with 45% of attributed to large‐, 32% to moderate‐, and 23% to small‐effect variants. The fentanyl cohort had median clearance of 8.2 L/h (IQR 4.7–16.7 L/h), with estimated of 0.30 (90% credible interval 0.00–0.84). Large‐effect variants accounted for 30% of , whereas moderate‐ and small‐effect variants accounted for 37% and 33%, respectively. As expected, given small sample sizes, no individual variants or pathways were significantly associated with dexmedetomidine or fentanyl clearance by GWAS. We conclude that clearance of both drugs is highly polygenic, motivating the future use of polygenic risk scores to guide appropriate dosing of dexmedetomidine and fentanyl.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
There is wide inter‐individual variation in pharmacokinetics of dexmedetomidine and fentanyl. Genomic contribution to this inter‐individual pharmacokinetic variation for both drugs remains poorly understood, as candidate gene analyses have failed to identify specific genes explaining a significant portion of the variability.
WHAT QUESTION DID THIS STUDY ADDRESS?
How much of the inter‐individual variability in dexmedetomidine and fentanyl clearance can be attributed to common single nucleotide variation in the genome? In addition, within this heritable component, what proportion can be attributed to variants with individually small, medium, or large effects?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Dexmedetomidine and fentanyl clearance are moderately heritable and highly polygenic traits, with small‐ and moderate‐effect variants accounting for the majority of estimated heritability.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Our findings of polygenic contribution to dexmedetomidine and fentanyl clearance highlight the importance of using genome‐wide approaches for the investigation of pharmacologic phenotypes. Our study motivates the application of heritability analyses to additional pharmacokinetic datasets. Our findings demonstrate the need to assemble much larger cohorts that will enable the development, validation, and eventual implementation of polygenic risk scores for genetically‐informed drug dosing in clinical practice.
INTRODUCTION
Precision medicine, and specifically the individualization of treatment decisions based on genetic variation (pharmacogenomics), is a rapidly evolving area of clinical care. Clinical pharmacogenomics is generally limited to the use of well‐studied genetic variants with high impact on drug outcomes. For example, the enzyme thiopurine S‐methyltransferase plays a vital role in metabolism of thiopurine drugs, and up to 10% of individuals in different populations are characterized as poor or intermediate metabolizers. In many clinical settings, genetic testing is performed to identify those with decreased enzyme activity prior to initiating therapy with a thiopurine agent, as alternate drugs or greatly reduced doses may be warranted to avoid severe toxicity. 1 , 2 , 3 More recently, a genome‐wide association study (GWAS) identified variants in a second gene, NUDT15, contributing to thiopurine toxicity, leading to the addition of NUDT15 variants to pharmacogenomic tests. 1 , 4 However, variants in these two genes are not fully predictive of thiopurine toxicity. 3 To more fully predict outcomes for drugs with residual variability or where no single pharmacogene has been identified, there is increasing focus on developing polygenic predictors of drug disposition and response. 5 , 6
Dexmedetomidine, an α2‐agonist, and fentanyl, an opioid, are frequently used post‐operatively in pediatric intensive care units (ICUs) to achieve sedation and analgesia. 7 , 8 , 9 , 10 Both are commonly dosed via continuous intravenous infusions using fixed weight‐based rates, which is suitable for some patients but might require titration due to over‐ or undersedation for others. Importantly, both over‐ and undersedation can be harmful to pediatric ICU patients. Oversedation has been associated with bradycardia, respiratory depression, increased need for mechanical ventilation, and prolonged ICU stays, whereas undersedation increases the risk for agitation, patient self‐injury, and anxiety. Both over‐ and undersedation have been reported to lead to development of delirium, withdrawal syndromes, neuromuscular weakness and atrophy, and post‐traumatic stress disorder. 11
The need for dosing adjustments of dexmedetomidine and fentanyl to maintain appropriate sedation is due in part to well‐described inter‐individual variation in pharmacokinetics. For both drugs, previous studies have shown wide variation among individuals in drug absorption, bioavailability, and clearance. 8 , 12 , 13 , 14 , 15 Specifically, inter‐individual variations in drug clearance rates in pediatric ICU patients have been reported as high as three‐fold for dexmedetomidine 16 and 10‐fold for fentanyl. 17 Pharmacogenomics research on dexmedetomidine and fentanyl pharmacokinetics has focused primarily on genes encoding enzymes known to play important roles in drug metabolism; these include cytochrome P450 (CYP) 2A6 and glucuronidation enzymes UGT1A4 and UGT2B10 for dexmedetomidine and CYP3A4 and CYP3A5 for fentanyl. To date, genes that have been identified explain little of the inter‐individual variability observed for clearance of both drugs. 18 , 19 , 20 The broader polygenic contribution to dexmedetomidine and fentanyl clearance has not been explored.
Genome‐wide approaches, including GWAS and Bayesian nonlinear modeling, take a broader approach by looking at the contribution of variants across the genome to a phenotype of interest. GWAS analyses individually examine millions of common genetic variants to identify those associated with a given phenotype. 21 Bayesian nonlinear models can quantify the collective contributions of large numbers of single nucleotide polymorphisms (SNPs) and estimate narrow‐sense heritability (), the proportion of phenotypic variation that can be attributed to additive influences of SNPs. 22 In contrast to GWAS‐based estimations of , Bayesian modeling can account for linkage disequilibrium by assuming that not all variants will have a non‐zero effect on the phenotype. The estimated can also be subdivided into relative contributions from genes with individually small, moderate, or large effects on the phenotype of interest. These genome‐wide approaches are complementary and frequently used in assessment of nonpharmacologic phenotypes, but their application within pharmacogenomics has been limited to date. We previously demonstrated validity of using one Bayesian method to explore pharmacologic outcomes, even when applied to small sample sizes, 23 and GWAS approaches have successfully detected pharmacogenomic signals despite small cohorts in a few examples where effect size of the variant(s) is very large, 24 , 25 including the NUDT15 association to thiopurine toxicity, as described above. 1 , 4
In this study, we used the results of previously performed population pharmacokinetic analyses 16 , 17 and deployed polygenic approaches to assess genomic contribution to dexmedetomidine and fentanyl clearance in children after cardiac surgery. We used BayesR, 26 , 27 , 28 an established Bayesian hierarchical analysis method, to estimate of dexmedetomidine and fentanyl clearance and assess the relative contribution of small‐, moderate‐, and large‐effect variants. We hypothesized that common genomic variation contributes substantially to inter‐individual variation in these phenotypes and that many variants with small effects on the phenotypes would cumulatively account for a greater proportion of heritability than few variants with large effects. We additionally assessed for specific variants that may be associated with dexmedetomidine or fentanyl clearance in our datasets using GWAS.
METHODS
Study design and data collection
The parent study supporting participant recruitment, specimen collection, and data collection was approved by the Vanderbilt University Medical Center Institutional Review Board and has been previously described in detail. 29 Pediatric patients undergoing surgery for congenital heart disease were voluntarily enrolled in this study via written parental informed consent and, when appropriate, informed assent from the patient. Enrollment of participants into the parent study began in 2007 and is ongoing; the genomic analyses presented here used data collected from April 2013 to October 2017. All participants were admitted to the pediatric cardiac ICU following surgery and underwent routine clinical care as determined by the primary clinical team, including selection and dosing of all medications. Study participants provided a blood or saliva sample for DNA extraction, and drug concentrations were measured from remnant blood samples collected during the course of clinical care. Participants were excluded from analysis if their surgery was canceled, if they had no genotype data available, if they did not survive to hospital discharge, or if they required extracorporeal membrane oxygenation postoperatively.
Study data were collected and stored using REDCap, a secure web application hosted at Vanderbilt University. 30 Medical history and demographic data were documented by the study team upon study enrollment. Surgical and clinical data were extracted from the electronic health record (EHR) by the study team during the hospital stay. Drug data, including dosing and times of administration, were extracted from the EHR and the Vanderbilt Enterprise Data Warehouse and standardized using the EHR2PKPD system. 31
This study was reviewed by the Institutional Review Board at Vanderbilt University Medical Center and determined to constitute non‐human subject research.
Drug concentration measurement and clearance calculations
The primary outcomes used for analysis were the clearances estimated per individual for each of the two target drugs, as previously described. 16 , 17 Briefly, dexmedetomidine and fentanyl dosing data were extracted from the EHR and drug concentrations were measured in remnant plasma specimens collected for research purposes using high‐throughput tandem mass spectrometry. Measured drug concentrations, drug dosing data, and covariate data collected for the parent study were used to generate a population pharmacokinetic model to estimate clearance of each drug, using Monolix 2021R. A two‐compartment model was selected as the base model for both drugs, and covariate modeling was performed using prespecified covariates.
For dexmedetomidine, the following covariates were considered in the population pharmacokinetic modeling based on previous research and biologic plausibility: UGT1A4, UGT2B10, and CYP2A6 variants, body weight, postnatal age, postmenstrual age, sex, Society of Thoracic Surgeons‐European Association for Cardio‐Thoracic Surgery (STAT) Congenital Heart Surgery Mortality score, cardiac bypass time, length of ICU stay, and serum creatinine. Of these, only body weight and postmenstrual age significantly improved model fit and remained in the final covariate model. 16
For fentanyl, CYP3A metabolizer status (based on CYP3A45 genotype), body weight, postnatal age, postmenstrual age, sex, patient‐reported race, serum creatinine, STAT score, cardiac bypass time, concomitant CYP3A inducers and inhibitors, and the first five principal components of ancestry were considered in the pharmacokinetic model. Body weight, postnatal age, STAT score, and CYP3A metabolizer status were included in the final covariate model. 17
For the final model for each drug, patient covariates, estimated model parameters, and the conditional mode of the individual random effect on clearance were used to calculate the estimated clearances used in this analysis.
Genotyping, quality control, and processing
Genome‐wide SNPs were derived from either the Affymetrix Axiom Precision Medicine Research Array or Precision Medicine Diversity Array (Thermo Fisher Scientific) using Genome Reference Consortium Human Build 37 (GRCh37/hg19). The BayesR method is sensitive to population substructure, meaning that inclusion of individuals across different ancestries could confound analyses and result in spurious findings. Therefore, analyses were restricted to individuals of European ancestry as this population was the most prominent in our cohorts and the only population for which sample size was sufficient for BayesR. To do this, within each cohort (dexmedetomidine and fentanyl), genotype data from participants was combined with that of Hapmap European (Utah residents with Northern and Western European ancestry from the CEPH collection), Asian (Han Chinese in Beijing, China and Japanese in Tokyo, Japan), and African (African ancestry in Southwest USA; Luhya in Webuye, Kenya; Maasai in Kinyawa, Kenya; and Yoruba in Ibadan, Nigeria) populations to assess ancestry. Principal component (PC) analyses were performed, and, because individuals with self‐reported white race overlapped heavily with the HapMap European population and segregated from Asian and African HapMap populations, final analyses were restricted to participants within three standard deviations of the mean of PC1 and PC2 of self‐reported white participants (Figure S1). This approach effectively restricted our analyses to participants of European ancestry.
Genotype data were imputed to the TOPMed reference panel R2 using the TOPMed Imputation Server. Imputed gene dosages were converted to hard calls using PLINK2 and filtered by info scores greater than 0.8. SNPs were included if they met the following quality control (QC) parameters: minor allele frequency greater than 1% within the cohort, SNP genotyping rate greater than 98%, and Hardy–Weinberg equilibrium greater than 10−6. Sex chromosomes were excluded from analysis. Individuals were removed if they had a genotype call rate less than 98%, discrepancy between the genetically‐estimated sex and the sex assigned in the database, or a high degree of genetic relatedness to another sample using identity‐by‐descent analysis (Figure S2).
For all individuals who passed QC, any clearance values greater than the median + 3 × interquartile range [IQR] or less than the median − 3 × IQR were removed as outliers from the data set for analysis.
BayesR analysis
Following QC of data sets as described above, SNPs in high linkage disequilibrium (r 2 > 0.9) were removed. Residuals were then calculated from clearance values incorporating the first 20 PCs of ancestry. Residuals were used in the final Bayesian analysis because the BayesR software is not able to include covariates (Figure S2).
For each cohort, BayesR (version 1), a hierarchical Bayesian mixture model using Markov Chain Monte Carlo estimation, was used to fit all SNPs simultaneously to a model predicting drug clearance in order to give unbiased estimates of SNP effect sizes. 26 , 27 BayesR uses k different normal distributions to model the prior distribution of SNP effects, where the sum of the mixture proportions is constrained to unity. As in previous analyses, we set k = 4, 27 where each component was modeled as a normal distribution with a mean of 0 and a variance of 0, 0.01%, 0.1%, and 1% of the additive genetic variance, respectively, as shown: . In this model, β signifies a mixture of four zero‐mean normal distributions of SNP effects with a fixed relative variance for each mixture component, π represents mixture proportions constrained to sum to unity, and is additive genetic variance explained by SNPs. The mean and variance of the first component is set to zero to account for sparseness in the model. The components k1, k2, k3, and k4 are referred to as no‐effect, small‐effect (each SNP explaining 0.01% of the variance), moderate‐effect (each SNP explaining 0.1% of the variance), and large‐effect SNPs (each SNP explaining 1% of the variance), respectively. To estimate , the algorithm uses a Gibbs scheme to sample values from each unknown parameter's posterior distribution. 26 , 27
Narrow‐sense heritability () was calculated as where and (residual variance) were estimated by BayesR. The number of iterations was increased from the default 20,000 to 60,000 in our analyses to allow for convergence given the small sample sizes in our data sets. Default settings were used for all other prior distribution parameters. The 90% highest density credible intervals were calculated. Results were processed using custom R scripts.
Genomewide association studies
Dexmedetomidine and fentanyl cohorts used for GWAS were identical to those input into BayesR, and the same quality control steps were performed (Figure S2). SNPs in high linkage disequilibrium (r 2 > 0.7) were removed to reduce computational time, leaving 986,408 and 1,080,019 SNPs for final analysis for dexmedetomidine and fentanyl, respectively. PLINK software (versions 1.9 and 2.0) using linear regression was utilized to test each SNP for association with the continuous drug clearance phenotype. PLINK software was also used to extract the first 10 PCs, which were used as covariates. 32 Results were processed using custom R scripts, and Manhattan and quantile‐quantile (Q‐Q) plots were generated using the qqman package (version 0.1.8).
We performed post hoc functional analyses of variants with association p values less than or equal to 1 × 10−4 by GWAS using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) functional annotation tool. 33 Gene lists for each data set (dexmedetomidine and fentanyl) included all genes within 100,000 base pairs of those GWAS SNPs with association p values less than or equal to 1 × 10−4. Functional analyses using DAVID were restricted to gene ontology (molecular function, biological process, and cellular component), pathways (KEGG pathway, BioCarta, and biological biochemical image database), and diseases (OMIM and UniProt).
RESULTS
Study population characteristics
Characteristics of the dexmedetomidine and fentanyl cohorts are shown in Table 1 and summaries of surgical procedures incurred by participants in each cohort are shown in Table S1. The dexmedetomidine cohort included 354 participants, as described previously. 16 Due to sensitivity of the BayesR method to population substructure, 70 individuals were removed due to genotypes consistent with non‐European ancestry, classified as individuals with PCs outside three standard deviations of the mean PC1 or PC2 of self‐reported white participants (Figure S1) and one individual was removed due to high relatedness. Three additional individuals were excluded from analysis due to identification of log‐transformed clearance values as outliers; therefore, a total of 280 subjects were included for final analysis of dexmedetomidine clearance. The median postnatal age at the time of surgery was 20.7 months (IQR 5.2–72.8) and the median body weight was 9.6 kg (IQR 6.1–19.6). Median length of ICU stay was 3 days (IQR 2–7), and median cardiac bypass time was 1.6 h (1.1–2.4). Median individual dexmedetomidine clearance, as estimated by population pharmacokinetic modeling, 16 was 33.4 L/h (IQR 23.5–47.8).
TABLE 1.
Dexmedetomidine and fentanyl cohort characteristics.
| Dexmedetomidine (n = 280) | Fentanyl (n = 346) | |
|---|---|---|
| Age (months) | 20.7 (5.2–72.8) | 8.5 (3.7–60.1) |
| Weight (kg) | 9.6 (6.1–19.6) | 7.7 (5.2–17.1) |
| Male sex | 145 (51.7%) | 184 (53.2%) |
| Self‐reported white race | 277 (98.9%) | 340 (98.3%) |
| Length of ICU stay (days) | 3 (2–7) | 4 (2–7) |
| Cardiac bypass time (h) | 1.6 (1.1–2.4) | 1.7 (1.1–2.5) |
| STAT score | 2 (1–3) | 2 (1–3) |
| Clearance (L/h) | 33.4 (23.5–47.8) | 8.1 (4.6–16.6) |
Note: Data are median (interquartile range) for continuous variables and NN (%) for categorical variables.
Abbreviations: ICU, intensive care unit; STAT, The Society of Thoracic Surgeons‐European Association for Cardio‐Thoracic Surgery.
The previously described fentanyl cohort included 434 participants 17 ; 87 subjects were removed due to being outside of three standard deviations of the first two PCs of self‐reported white participants (Figure S1) and one individual was removed due to high relatedness, leaving a total of 346 individuals for final analysis. The median postnatal age at the time of surgery was 8.5 months (IQR 3.7–60.1) and the median body weight was 7.7 kg (IQR 5.2–17.1). Median length of ICU stay was 4 days (IQR 2–7), and median cardiac bypass time was 1.7 h (1.1–2.5). Median fentanyl clearance, as estimated by population pharmacokinetic modeling, 17 was 8.1 L/h (IQR 4.6–16.6).
Heritability estimates of dexmedetomidine and fentanyl clearance
Dexmedetomidine clearance values from 280 individuals were log‐transformed and adjusted for the first 20 PCs (Figure S3). Covariates that were considered for inclusion in the population pharmacokinetic models used to calculate drug clearance values were not considered for inclusion in the BayesR model to estimate heritability. Residual values and 1,121,432 genetic variants were input into the BayesR model. The estimated additive genetic variance () was 0.13 with a residual variance () of 0.24, resulting in a calculated of 0.35 with a 90% highest density credible interval of 0.00–0.90 (Figure 1). As described above, the estimated was further subdivided into relative contributions from small‐, moderate‐, and large‐effect variants, which account for 0.01%, 0.1%, and 1% of , respectively. For dexmedetomidine, 36% of the estimated was accounted for by 2997 small‐effect variants, 26% was accounted for by 264 moderate‐effect variants, and 38% was accounted for by 57 large‐effect variants. Therefore, over half of the estimated of dexmedetomidine clearance was attributed to small and moderate effect‐size variants (Figure 1).
FIGURE 1.
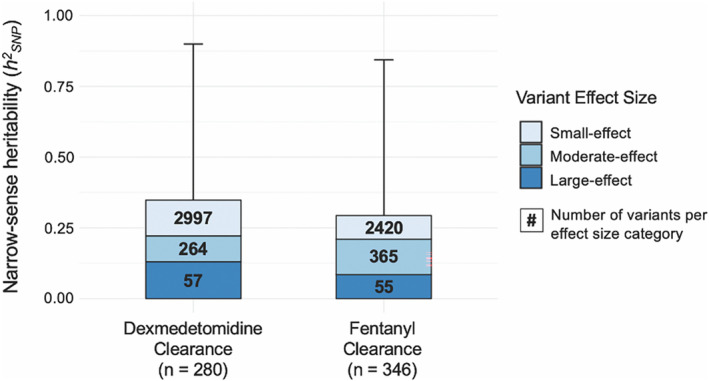
Narrow‐sense heritability () estimates of dexmedetomidine and fentanyl clearance. Total is further subdivided into relative contributions from large‐effect (dark blue), moderate‐effect (medium blue), and small‐effect (light blue) variants. Numerical values within each effect size box represent the number of variants that comprise each effect size category. Error bars represent 90% high‐density credible intervals for .
For fentanyl, clearance values from 346 individuals were adjusted using the first 20 PCs (Figure S3) and analyzed with 1,222,265 genetic variants using BayesR. The estimated was 0.25, was 0.61, and calculated was 0.29 with a 90% highest density credible interval of 0.00–0.84 (Figure 1). Relative contributions from small‐, moderate‐, and large‐effect variants were as follows: 29% of estimated was accounted for by 2420 small‐effect variants, 42% was accounted for by 365 moderate‐effect variants, and 38% was accounted for by 55 large‐effect variants. As with dexmedetomidine, more than half of the estimated of fentanyl clearance was attributed to small and moderate effect‐size variants (Figure 1).
Genome‐wide association analysis of dexmedetomidine and fentanyl clearance
Given the substantial heritability of clearance for both drugs and small number of large‐effect variants for each drug, we performed GWAS to identify any variants significantly associated with the outcomes. To our knowledge, these are the first GWAS of dexmedetomidine and fentanyl clearance performed to date. Q‐Q plots demonstrated mild inflation of smaller p values observed for dexmedetomidine and a relatively uniform distribution for fentanyl (Figure S4). For both dexmedetomidine and fentanyl clearance, GWAS failed to identify any variants meeting the genome‐wide statistical significance threshold of 5 × 10−8 (Figure 2).
FIGURE 2.
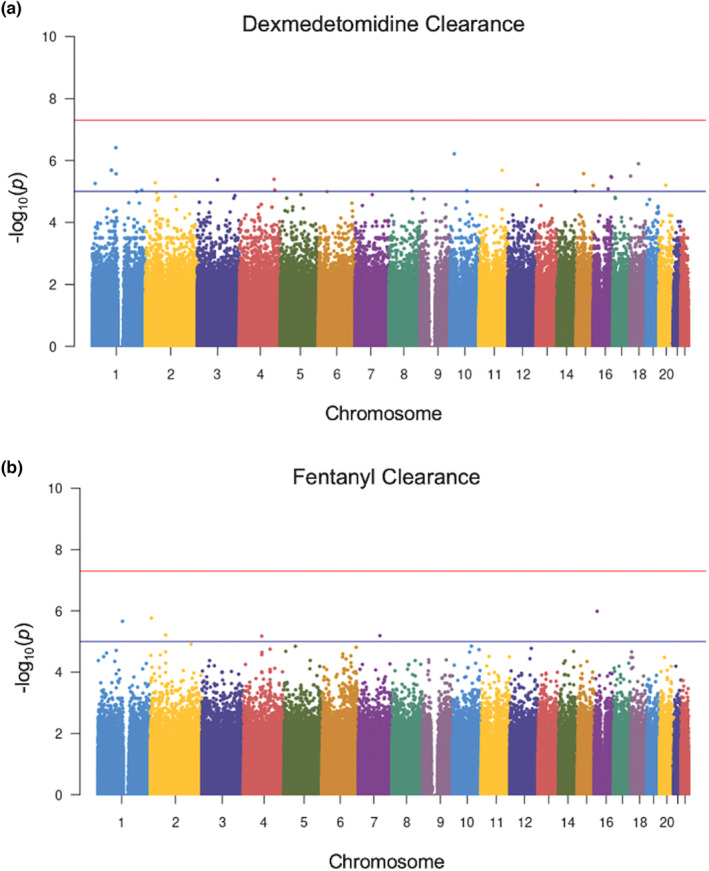
Manhattan plots showing genome‐wide association study results for dexmedetomidine (a) and fentanyl (b) clearance. For both plots, all genetic variants are shown as individual data points ordered by chromosome across the x‐axis. The y‐axis represents the negative log of the associated p value for each variant. The red line represents the standard genome‐wide significance threshold of 5 × 10−8. The blue line at 1 × 10−5 is suggestive of association. No variants in either the dexmedetomidine or fentanyl clearance analysis crossed this threshold to reach statistical significance.
Results of the GWAS analysis are reported in Tables S2 and S3. For dexmedetomidine clearance, the variants within 100,000 base pairs of coding regions with the strongest statistical associations (smallest p values) were: CACNB2, a voltage‐gated calcium channel; LRRC8C, which facilitates cAMP‐cGMP import across plasma membranes; and RAP1A, a member of the RAS oncogene family (Table S2). For fentanyl clearance, the variants within coding regions with the smallest p values were: CD58, involved in adhesion and activation of T cells; DYSF, which contributes to calcium‐mediated muscle contraction; and ORAI2, which plays a role in store‐operated calcium channel activity (Table S3).
We subsequently used the DAVID functional annotation tool to perform post hoc functional analyses on the variants with GWAS p values less than or equal to 1 × 10−4 within 100,000 bp of known genes (Tables S4 and S5). These analyses did not identify any statistically significant pathways or functions enriched for clearance of either drug. For dexmedetomidine, the top three gene ontology categories were anchoring junction, specific granule membrane, and integral component of plasma membrane (Table S6). Exogenous drug catabolism was among the top 10 pathways identified, which was associated with a total of four genes: CYP2A13, CYP2A7, CYP2F1, and NOS1. CYP2A13, CYP2A7, and CYP2F1 were all located within 100,000 base pairs of a single SNP on chromosome 19. For the fentanyl clearance data set, membrane, protein destabilization, and positive regulation of protein serine/threonine kinase activity were the top three gene ontology categories that emerged. No pathways related to drug metabolism were enriched for the fentanyl data set (Table S7).
DISCUSSION
In this study, we sought to explore the genomic contribution to variation among pediatric patients in clearance of two drugs commonly used after cardiac surgery: dexmedetomidine and fentanyl. Our data verify the clinical observation of large inter‐individual variability in drug clearance, as our pharmacokinetic analyses demonstrated an approximately two‐fold difference between the first and third quartile of estimated dexmedetomidine clearance and an almost four‐fold difference for fentanyl clearance, consistent with previous estimates. 16 , 17 We used the clearance data, coupled with genome‐wide genotype data, to employ a Bayesian hierarchical modeling methodology. Given that both estimations were greater than 0.25, inter‐individual variation in clearance of dexmedetomidine and fentanyl in this population is moderately heritable. Further, the observation that less than 40% of is attributable to large‐effect variants suggests that these traits are highly polygenic. The credible intervals for our observations are wide, which may be improved with larger sample sizes. These results suggest that, for both drugs, many genes with individually smaller effect sizes combine to comprise a substantial genomic contribution to drug clearance. Therefore, polygenic approaches combined with large sample sizes are necessary to gain a comprehensive understanding of genetic contribution to the individual pharmacokinetics of these drugs, which may ultimately be utilized, for example in the form of polygenic risk scores, in clinical practice to individualize therapy.
Although less than 40% of the estimated heritability for both dexmedetomidine and fentanyl clearance was attributed to variants with large effect sizes, their estimated contribution to heritability of both drugs was substantial. We therefore utilized GWAS in an attempt to identify any SNPs that were significantly associated with dexmedetomidine or fentanyl clearance. These analyses failed to identify any Bonferroni‐corrected statistically significant SNPs for clearance of either drug. Secondary functional analyses of genes with p values smaller than 1 × 10−4 using the DAVID functional annotation tool also did not pinpoint specific pathways or processes that were enriched within our data sets. For dexmedetomidine clearance, exogenous drug catabolism and the epoxygenase P450 pathway were among the top functional hits. However, neither of these pathways reached statistical significance and each involved only a few genes, all of which were located near a single variant identified by GWAS. Furthermore, these enzymes have not been previously reported to play a role in dexmedetomidine metabolism. For fentanyl clearance, no pathways or processes related to drug metabolism were enriched. In agreement with the results of the Bayesian analysis, these findings also suggest a highly polygenic contribution of the genome to dexmedetomidine and fentanyl clearance and support continued efforts toward utilization of polygenic approaches to investigate the pharmacokinetics of both drugs. It is also important to consider that drug metabolism and distribution are dependent on pathways across many tissue types 34 , 35 and are highly variable across the lifespan. 36 , 37 Therefore, future efforts aimed at defining pathways and predictive models across different tissue types and different ages may aid in the identification of novel genetic associations with drug outcomes.
Taken together, our data motivate collection of larger and more ancestrally diverse cohorts of dexmedetomidine‐ and fentanyl‐exposed individuals to develop and validate polygenic predictive models for these drugs, incorporating effects of many genes with a spectrum of effect size. Such predictors are now available for nonpharmacologic phenotypes such as type 2 diabetes, 38 coronary artery disease, 39 , 40 neurodegenerative disease, 41 , 42 and some cancers. 43 , 44 , 45 Polygenic risk predictors have been demonstrated to be especially useful for those individuals at the tail ends of the distribution for a given phenotype. 46 Applied to dexmedetomidine and fentanyl clearance, polygenic risk scores may be most useful for identifying individuals at the highest risk for undersedation or oversedation with standard drug dosing regimens secondary to differences in drug clearance. These approaches may be useful across a wide range of drugs with complex pharmacokinetic and/or pharmacodynamic pathways. Estimations of for drug traits may help identify the best target drugs for genomic approaches (i.e., those with high heritability).
Our study was limited by the application of modeling methodologies traditionally used with large sample sizes, such as Bayesian modeling and GWAS, to our small cohorts. This limitation likely resulted in decreased precision and wide credible intervals in our Bayesian analyses; however, Bayesian modeling has been previously used for pharmacogenomic analyses with similarly sized data sets. 23 Additionally, it has been demonstrated that in polygenic phenotypes accounted for by primarily small‐effect variants, as is true for both of our phenotypes, large sample sizes are required to detect significant variants with GWAS. 47 Therefore, given the small sample sizes, our GWAS analyses may be underpowered to detect the statistical association of some genes with our phenotypes and may have missed potentially significantly associated variants. Furthermore, development of clinically useful polygenic risk scores requires data sets much larger than those commonly available for pharmacologic outcomes. To combat this persistent limitation of pharmacogenomic research, we advocate for continued efforts toward curation of large data sets. To do so would require building scalable, sharable data structures and collaborative efforts both within and across international borders. In today's era, with the ubiquity of EHRs and advent of high throughput analytic techniques, such collaboration is both achievable and worthwhile.
Another limitation inherent in heritability studies is the need to restrict analyses to individuals of a single ancestry. Studies conducted in the United States and Europe often restrict analyses to European ancestry, and, as a result, the majority of publications in genomics include only individuals of European descent. 48 , 49 Our study was limited in a similar manner as our cohort was relatively small and currently lacks sufficient numbers of individuals of non‐European ancestries to perform analyses in other ancestries. It is paramount that we prioritize diversity within pharmacogenomics research; doing so will ensure applicability of findings across populations and foster new discoveries. We are continuously enrolling subjects in this cohort and hope to soon build large enough sample sizes to repeat these analyses for individuals of other ancestries. Additionally, methodological developments in datasets of diverse and complex ancestries would enable more ancestrally inclusive studies that may reveal important differences in inter‐individual drug variability.
Finally, both a limitation of our current study and an opportunity for future investigation is the application of these same methodologies to diverse pharmacodynamic outcomes. For dexmedetomidine and fentanyl, such outcomes may include standardized sedation scores. These analyses may identify a unique set of genes that influence the response to dexmedetomidine and fentanyl in this population. In order to be successful, these strategies must account for differences in pharmacokinetics, either through the use of robust modeling or through the measurement of drug concentrations.
In summary, our study demonstrates that the genome contributes substantially to inter‐individual variation in dexmedetomidine and fentanyl clearance in the pediatric cardiac surgery population. Clearance of both drugs was polygenic, with small and moderate effect‐size variants comprising the majority of estimated heritability, whereas no individual variant had a significant association with clearance of either drug. Taken together these findings motivate the development of polygenic predictors of drug responses with the ultimate goal of implementing genomically informed drug dosing into clinical practice.
AUTHOR CONTRIBUTIONS
M.L.S., A.M., N.T.J., M.L.W., L.C., and S.L.V.D. wrote the manuscript. M.L.S., A.M., N.T.J., M.L.W., T.E., J.D.M., L.C., P.K., and S.L.V.D. designed the research. M.L.S., A.M., N.T.J., M.L.W., J.B., and S.L.V.D. performed the research. M.L.S. analyzed the data.
FUNDING INFORMATION
This work is supported by the National Institutes of Health (NIH) National Institute of General Medical Sciences (NIGMS) R01 GM132204 to S.L.V. This work is also supported in part by NIH/NIGMS R01 GM124109 to LC and R01 HD084461 from the NIH Eunice Kennedy Shriver National Institute of Child Health & Human Development to P.J.K. A.M. is supported by a grant from the American Heart Association (20PRE35180088) and from the Vanderbilt Medical Scientist Training Program (T32GM007347). S.L.V. and P.J.K. are supported by P50HD106446. This study used REDCap, supported by UL1 TR000445 from NIH National Center for Advancing Translational Sciences.
CONFLICT OF INTEREST STATEMENT
The authors declared no competing interests for this work.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
ACKNOWLEDGMENTS
The authors would like to thank the Research Immersion program at Vanderbilt University School of Medicine and Dr. Lea Davis for her advice and mentorship. The authors would also like to thank Dr. Sabrina Holley for her feedback and editing. This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University, Nashville, Tennessee.
Shannon ML, Muhammad A, James NT, et al. Variant‐based heritability assessment of dexmedetomidine and fentanyl clearance in pediatric patients. Clin Transl Sci. 2023;16:1628‐1638. doi: 10.1111/cts.13574
REFERENCES
- 1. Relling MV, Schwab M, Whirl‐Carrillo M, et al. Clinical pharmacogenetics implementation consortium guideline for thiopurine dosing based on tpmt and nudt 15 genotypes: 2018 update. Clin Pharmacol Ther. 2019;105(5):1095‐1105. doi: 10.1002/cpt.1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asadov C, Aliyeva G, Mustafayeva K. Thiopurine S‐methyltransferase as a pharmacogenetic biomarker: significance of testing and review of major methods. Cardiovasc Hematol Agents Med Chem. 2017;15(1):23‐30. doi: 10.2174/1871525715666170529091921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lennard L. Implementation of TPMT testing: TPMT testing. Br J Clin Pharmacol. 2014;77(4):704‐714. doi: 10.1111/bcp.12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang JJ, Landier W, Yang W, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33(11):1235‐1242. doi: 10.1200/JCO.2014.59.4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson D, Wilke MAP, Lyle SM, et al. A systematic review and analysis of the use of polygenic scores in pharmacogenomics. Clin Pharmacol Ther. 2022;111(4):919‐930. doi: 10.1002/cpt.2520 [DOI] [PubMed] [Google Scholar]
- 6. Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 2020;12(1):44. doi: 10.1186/s13073-020-00742-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plambech MZ, Afshari A. Dexmedetomidine in the pediatric population: a review. Minerva Anestesiol. 2015;81(3):320‐332. [PubMed] [Google Scholar]
- 8. Weerink MAS, Struys MMRF, Hannivoort LN, Barends CRM, Absalom AR, Colin P. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet. 2017;56(8):893‐913. doi: 10.1007/s40262-017-0507-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee S. Dexmedetomidine: present and future directions. Korean J Anesthesiol. 2019;72(4):323‐330. doi: 10.4097/kja.19259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ziesenitz VC, Vaughns JD, Koch G, Mikus G, van den Anker JN. Pharmacokinetics of fentanyl and its derivatives in children: a comprehensive review. Clin Pharmacokinet. 2018;57(2):125‐149. doi: 10.1007/s40262-017-0569-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Egbuta C, Mason KP. Current state of analgesia and sedation in the pediatric intensive care unit. J Clin Med. 2021;10(9):1847. doi: 10.3390/jcm10091847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oosten AW, Abrantes JA, Jönsson S, et al. Treatment with subcutaneous and transdermal fentanyl: results from a population pharmacokinetic study in cancer patients. Eur J Clin Pharmacol. 2016;72(4):459‐467. doi: 10.1007/s00228-015-2005-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iirola T, Aantaa R, Laitio R, et al. Pharmacokinetics of prolonged infusion of high‐dose dexmedetomidine in critically ill patients. Crit Care. 2011;15(5):R257. doi: 10.1186/cc10518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Välitalo PA, Ahtola‐Sätilä T, Wighton A, Sarapohja T, Pohjanjousi P, Garratt C. Population pharmacokinetics of dexmedetomidine in critically ill patients. Clin Drug Investig. 2013;33(8):579‐587. doi: 10.1007/s40261-013-0101-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuip EJM, Zandvliet ML, Koolen SLW, Mathijssen RHJ, van der Rijt CCD. A review of factors explaining variability in fentanyl pharmacokinetics; focus on implications for cancer patients: variability in fentanyl pharmacokinetics. Br J Clin Pharmacol. 2017;83(2):294‐313. doi: 10.1111/bcp.13129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. James NT, Breeyear JH, Caprioli R, et al. Population pharmacokinetic analysis of dexmedetomidine in children using real‐world data from electronic health records and remnant specimens. Br J Clin Pharmacol. 2022;88:2885‐2898. doi: 10.1111/bcp.15194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Williams ML, Kannankeril PJ, Breeyear JH, Edwards TL, Van Driest SL, Choi L. Effect of CYP3A5 and CYP3A4 genetic variants on fentanyl pharmacokinetics in a pediatric population. Clin Pharmacol Ther. 2022;111(4):896‐908. doi: 10.1002/cpt.2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou S, Skaar DJ, Jacobson PA, Huang RS. Pharmacogenomics of medications commonly used in the intensive care unit. Front Pharmacol. 2018;9:1436. doi: 10.3389/fphar.2018.01436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Magarbeh L, Gorbovskaya I, Le Foll B, Jhirad R, Müller DJ. Reviewing pharmacogenetics to advance precision medicine for opioids. Biomed Pharmacother. 2021;142:112060. doi: 10.1016/j.biopha.2021.112060 [DOI] [PubMed] [Google Scholar]
- 20. Kohli U, Pandharipande P, Muszkat M, et al. CYP2A6 genetic variation and dexmedetomidine disposition. Eur J Clin Pharmacol. 2012;68(6):937‐942. doi: 10.1007/s00228-011-1208-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uffelmann E, Huang QQ, Munung NS, et al. Genome‐wide association studies. Nat Rev Methods Primer. 2021;1(1):59. doi: 10.1038/s43586-021-00056-9 [DOI] [Google Scholar]
- 22. Mayhew AJ, Meyre D. Assessing the heritability of complex traits in humans: methodological challenges and opportunities. Curr Genomics. 2017;18(4):332‐340. doi: 10.2174/1389202918666170307161450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muhammad A, Aka IT, Birdwell KA, et al. Genome‐wide approach to measure variant‐based heritability of drug outcome phenotypes. Clin Pharmacol Ther. 2021;110(3):714‐722. doi: 10.1002/cpt.2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ozeki T, Mushiroda T, Yowang A, et al. Genome‐wide association study identifies HLA‐A*3101 allele as a genetic risk factor for carbamazepine‐induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20(5):1034‐1041. doi: 10.1093/hmg/ddq537 [DOI] [PubMed] [Google Scholar]
- 25. Ahlström S, Bergman P, Jokela R, et al. First genome‐wide association study on rocuronium dose requirements shows association with SLCO1A2. Br J Anaesth. 2021;126(5):949‐957. doi: 10.1016/j.bja.2021.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Erbe M, Hayes BJ, Matukumalli LK, et al. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high‐density single nucleotide polymorphism panels. J Dairy Sci. 2012;95(7):4114‐4129. doi: 10.3168/jds.2011-5019 [DOI] [PubMed] [Google Scholar]
- 27. Moser G, Lee SH, Hayes BJ, Goddard ME, Wray NR, Visscher PM. Simultaneous discovery, estimation and prediction analysis of complex traits using a Bayesian mixture model. Haley C, ed. PLOS Genet. 2015;11(4):e1004969. doi: 10.1371/journal.pgen.1004969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mollandin F, Rau A, Croiseau P. An evaluation of the predictive performance and mapping power of the BayesR model for genomic prediction. G3 (Bethesda). 2021;11(11):jkab225. doi: 10.1093/g3journal/jkab225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Driest SL, Marshall MD, Hachey B, et al. Pragmatic pharmacology: population pharmacokinetic analysis of fentanyl using remnant samples from children after cardiac surgery: population PK of fentanyl using remnant samples from children. Br J Clin Pharmacol. 2016;81(6):1165‐1174. doi: 10.1111/bcp.12903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377‐381. doi: 10.1016/j.jbi.2008.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi L, Beck C, McNeer E, et al. Development of a system for Postmarketing population pharmacokinetic and pharmacodynamic studies using real‐world data from electronic health records. Clin Pharmacol Ther. 2020;107(4):934‐943. doi: 10.1002/cpt.1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet. 2006;38(8):904‐909. doi: 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 33. Sherman BT, Hao M, Qiu J, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022;50(W1):W216‐W221. doi: 10.1093/nar/gkac194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piñero J, Gonzalez‐Perez A, Guney E, et al. Network, transcriptomic and genomic features differentiate genes relevant for drug response. Front Genet. 2018;9:412. doi: 10.3389/fgene.2018.00412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Drozdzik M, Busch D, Lapczuk J, et al. Protein abundance of clinically relevant drug‐metabolizing enzymes in the human liver and intestine: a comparative analysis in paired tissue specimens. Clin Pharmacol Ther. 2018;104(3):515‐524. doi: 10.1002/cpt.967 [DOI] [PubMed] [Google Scholar]
- 36. Mangoni AA, Jackson SHD. Age‐related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 2004;57(1):6‐14. doi: 10.1046/j.1365-2125.2003.02007.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Batchelor HK, Marriott JF. Paediatric pharmacokinetics: key considerations. Br J Clin Pharmacol. 2015;79(3):395‐404. doi: 10.1111/bcp.12267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Läll K, Mägi R, Morris A, Metspalu A, Fischer K. Personalized risk prediction for type 2 diabetes: the potential of genetic risk scores. Genet Med. 2017;19(3):322‐329. doi: 10.1038/gim.2016.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385(9984):2264‐2271. doi: 10.1016/S0140-6736(14)61730-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inouye M, Abraham G, Nelson CP, et al. Genomic risk prediction of coronary artery disease in 480,000 adults. J Am Coll Cardiol. 2018;72(16):1883‐1893. doi: 10.1016/j.jacc.2018.07.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Escott‐Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain. 2015;138(12):3673‐3684. doi: 10.1093/brain/awv268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Escott‐Price V, Nalls MA, Morris HR, et al. Polygenic risk of p arkinson disease is correlated with disease age at onset. Ann Neurol. 2015;77(4):582‐591. doi: 10.1002/ana.24335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. The Breast and Prostate Cancer Cohort Consortium (BPC3), The PRACTICAL (Prostate Cancer Association Group to Investigate Cancer‐Associated Alterations in the Genome) Consortium, The COGS (Collaborative Oncological Gene‐environment Study) Consortium et al. A meta‐analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet. 2014;46(10):1103‐1109. doi: 10.1038/ng.3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mavaddat N, Pharoah PDP, Michailidou K, et al. Prediction of breast cancer risk based on profiling with common genetic variants. J Natl Cancer Inst. 2015;107(5):djv036. doi: 10.1093/jnci/djv036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thomas M, Sakoda LC, Hoffmeister M, et al. Genome‐wide modeling of polygenic risk score in colorectal cancer risk. Am J Hum Genet. 2020;107(3):432‐444. doi: 10.1016/j.ajhg.2020.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khera AV, Chaffin M, Aragam KG, et al. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219‐1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nishino J, Ochi H, Kochi Y, Tsunoda T, Matsui S. Sample size for successful genome‐wide association study of major depressive disorder. Front Genet. 2018;9:227. doi: 10.3389/fgene.2018.00227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177(1):26‐31. doi: 10.1016/j.cell.2019.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538(7624):161‐164. doi: 10.1038/538161a [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7