

Abstract
Aim: Some observational studies suggested that atherosclerosis increased the risk of venous thromboembolism (VTE), and vice versa. However, the results were conflicting, and the causal relationship is yet to be established. Therefore, we applied Mendelian randomization (MR) analyses to assess the bidirectional causality between coronary heart disease (CHD) and VTE, deep venous thrombosis (DVT), and pulmonary embolism (PE).
Methods: A total of 184,305 individuals with CHD were included from the CARDIoGRAMplusC4D Consortium. Information on VTE, DVT, and PE were obtained from the FinnGen biobank. Genetic instruments for CHD and VTE were constructed using 37 and 12 single-nucleotide polymorphisms, respectively. Inverse-variance weighted meta-analysis under a random-effect model was used as the preliminary estimate. Five complementary MR methods were also used, including weighted median, MR-Egger, multivariable MR (adjusted for the body mass index), simple mode, and weighted mode methods.
Results: The genetically instrumented VTE (odds ratio [OR]: 1.05; 95% confidence interval [CI]: 1.00–1.11;P=0.06), DVT (OR: 1.03; 95% CI: 0.99–1.08;P=0.19), or PE (OR: 1.07; 95% CI: 0.98–1.16;P=0.11) showed no causal relationships with CHD. There was also no clear evidence showing the causal effects of CHD on VTE (OR: 1.00; 95% CI: 0.82–1.22;P=0.98), DVT (OR: 1.00; 95% CI: 0.79–1.27;P=0.97), or PE (OR: 0.98; 95% CI: 0.82–1.18;P=0.87). No pleiotropic bias was found in the MR analyses. As heterogeneity was significant, a random model was used to minimize the effect of heterogeneity.
Conclusions: No causal associations existed between CHD and VTE. Arterial and venous thromboses may represent separate entities.
Keywords: Coronary heart disease, Venous thromboembolism, Deep venous thrombosis, Pulmonary embolism, Bidirectional causality, Mendelian randomization
1. Introduction
It is generally believed that arterial and venous thrombotic disorders are separate entities, because of the noticeable anatomical differences, distinct pathophysiology (vascular wall lesions and high-shear stress versus stasis and hypercoagulability), and different treatment modalities (antiplatelet drugs versus anticoagulants). However, the concept has been challenged in recent studies 1) , as patients with venous thromboembolism (VTE) were found to have a higher prevalence of atherosclerosis 2) . In addition, atherosclerosis and VTE may share common risk factors 3) , which strengthen the connection between atherosclerosis and VTE. From a laboratory perspective, an association between atherosclerosis and VTE events is plausible as they share common features, such as platelet activation and coagulation 4) . Therefore, it is suggested that atherosclerosis and VTE are actually different presentations of the same disease. In some patients at high risk of atherosclerosis, VTE may occur as the first symptomatic cardiovascular event 5) . Likewise, atherosclerosis not only induces platelet activation and blood coagulation but also increases fibrin turnover, which may result in thrombotic complications. Activated platelets and coagulation factors have been found in the slow-flowing venous system 4) . Several studies have shown that fibrinogen, von Willebrand factor antigen, tissue plasminogen activator antigen, and D-dimer were all elevated in individuals with a history of ischemic coronary artery disease, creating a prothrombotic state in favor of VTE 4 , 6 , 7) . Hence, the link between VTE and atherosclerosis appeared to be reciprocal. However, other large-scale cohort studies revealed conflicting results that atherosclerosis did not increase the risk of VTE 8 , 9) .
It is noteworthy that these observational studies were limited in making causal inferences because of potential biases introduced by confounders and reverse causality. Currently, Mendelian randomization (MR) analysis is increasingly used to estimate causal inferences between exposures and outcomes. MR analysis resembles the random assignment of participants to treatment and control groups in a randomized controlled trial because the genetic variants are randomly assorted during gamete formation, which can minimize the effect of confounders and reverse causality. In this study, we performed bidirectional MR analyses to infer a causal association between coronary heart disease (CHD) and VTE. The evidence would provide crucial information on the causal relationship between CHD and VET, and whether one modification may lead to a decreased risk of the other.
2. Materials and Methods
2.1 Study Design
The single-nucleotide polymorphisms (SNPs) identified as genetic variants had to meet the following three assumptions: (1) SNPs were strongly associated with exposures; (2) SNPs were not related to any confounders of the exposure–outcome associations; and (3) SNPs only affected outcomes via exposures ( Fig.1 ) 10) . Ethics approval was not applicable to these analyses because all included genome-wide association studies (GWAS) data were publicly available and had been approved by the corresponding ethical review board.
Fig.1. Mendelian randomization model.
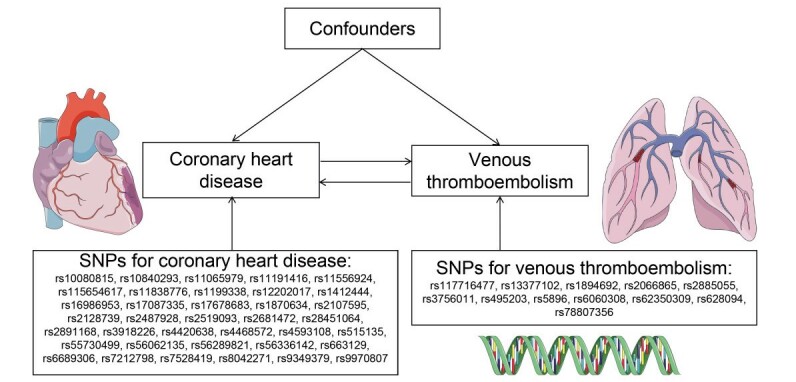
SNP: single-nucleotide polymorphism
2.2 Data Sources
These MR analyses used summary-level data from public GWAS on predominantly European individuals. GWAS summary statistics for CHD were obtained from the CARDIoGRAMplusC4D Consortium, which included 60,801 cases and 123,504 controls 11) . GWAS summary statistics for VTE (9176 cases and 209,616 controls), deep venous thrombosis (DVT; 4576 cases and 190,028 controls), and pulmonary embolism (PE; 4185 cases and 214,607 controls) were obtained from the FinnGen biobank.
2.3 Selection and Validation of SNPs
First, we selected SNPs associated with a genome-wide significance threshold exposure (P<5×10−8). Second, the independence of the selected SNPs was evaluated using the pairwise-linkage disequilibrium 12) , excluding the SNPs in linkage disequilibrium (r2>0.001 and clumping window <10,000 kb). Third, the F statistic was calculated to verify the strength of the SNP, deleting SNPs with an F statistic less than 10. The data were harmonized to ensure that SNP effects on exposure and outcome corresponded to the same allele.
2.4 MR Analyses
The inverse-variance weighted (IVW) meta-analysis under a random-effect model was utilized as the principal analysis. The following five methods, including weighted median, MR-Egger, multivariable MR, simple mode, and weighted mode, were also performed to ensure the robustness of the analyses. The weighted median method can provide valid estimates even if up to 50% of information comes from invalid genetic variants 13) . The MR-Egger method can assess and adjust the effect of horizontal pleiotropy of selected genetic variants 14) . Funnel plots can also detect horizontal pleiotropy if asymmetry exists. Multivariable MR analyses were performed by considering the body mass index (BMI) as a potential confounder or intermediator. Furthermore, a leave-one-out sensitivity analysis can analyze the influence of an individual SNP on the overall estimates. Cochrane’s Q value can assess heterogeneity among selected genetic variants. All statistical analyses were performed by utilizing the “TwoSampleMR” package in R software (version 4.0.3, R Foundation for Statistical Computing, Vienna, Austria).
3. Results
3.1 Effect of VTE on CHD
A total of 12 SNPs were included as genetic variants for VTE. According to the IVW method, the genetically instrumented VTE (odds ratio [OR]: 1.05; 95% confidence interval [CI]: 1.00–1.11; P=0.06), DVT (OR: 1.03; 95% CI: 0.99–1.08; P=0.19), or PE (OR: 1.07; 95% CI: 0.98–1.16; P=0.11) showed no relationships with CHD ( Fig.2 ) . Sensitivity analyses using weighted median and MR-Egger methods also revealed no significant associations between VTE or its subtypes with CHD, except for VTE in the multivariable MR method, where VTE increased the risk of CHD (OR: 1.05; 95% CI: 1.00–1.10; P=0.03) ( Table 1 ) . Sensitivity analyses using simple mode and weighted mode methods also yielded no significant associations ( Supplementary Table 1 ) . The MR-Egger method revealed no evidence of horizontal pleiotropy. However, the heterogeneity was significant in all analyses ( Table 2 ) . Therefore, IVW under a random model was applied to minimize the effect of heterogeneity. The scatter plot and forest plot based on all SNPs are shown in Supplementary Fig.1 and Supplementary Fig.2 , respectively. The leave-one-out sensitivity analysis in Supplementary Fig.3 indicates that the overall estimate was not influenced by an individual SNP. The funnel plots were symmetric in the IVW method, indicating no horizontal pleiotropy and confirming the results from the MR-Egger method ( Supplementary Fig.4 ) .
Fig.2. Mendelian randomization association of CHD with VTE.
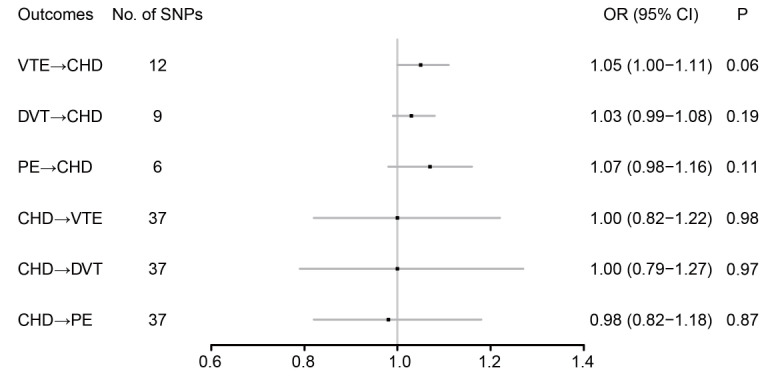
CHD: coronary heart disease; CI: confidence interval; DVT: deep venous thrombosis; OR: odd ratio; PE: pulmonary embolism; VTE: venous thromboembolism
Table 1. Sensitivity analyses using weighted median, MR-Egger, and MVMR methods.
| Outcomes | Weighted median | MR-Egger | MVMR | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |
| VTE→ CHD | 1.03 (0.98-1.08) | 0.21 | 1.01 (0.92-1.11) | 0.83 | 1.05 (1.00-1.10) | 0.03 |
| DVT→ CHD | 1.02 (0.98-1.05) | 0.36 | 1.02 (0.94-1.09) | 0.69 | 1.03 (1.00-1.07) | 0.07 |
| PE→ CHD | 1.03 (0.98-1.08) | 0.23 | 0.89 (0.75-1.06) | 0.27 | 1.02 (0.98-1.05) | 0.38 |
| CHD→ VTE | 0.97 (0.86-1.08) | 0.57 | 0.89 (0.54-1.45) | 0.63 | 0.98 (0.91-1.06) | 0.64 |
| CHD→ DVT | 1.02 (0.88-1.18) | 0.81 | 0.78 (0.44-1.40) | 0.41 | 0.94 (0.85-1.06) | 0.32 |
| CHD→ PE | 1.02 (0.88-1.17) | 0.83 | 0.92 (0.59-1.43) | 0.72 | 1.00 (0.89-1.11) | 0.93 |
CHD: coronary heart disease; CI: confidence interval; DVT: deep venous thrombosis; MR: Mendelian randomization; MVMR: multivariable Mendelian randomization; OR: odd ratio; PE: pulmonary embolism; VTE: venous thromboembolism
Supplementary Table 1. Sensitivity analyses using simple mode and weighted mode methods.
| Outcomes | Simple mode | Weighted mode | ||
|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |
| VTE→ CHD | 1.06 (0.96-1.17) | 0.28 | 1.02 (0.97-1.08) | 0.46 |
| DVT→ CHD | 1.02 (0.95-1.10) | 0.57 | 1.01 (0.97-1.05) | 0.61 |
| PE→ CHD | 1.01 (0.95-1.08) | 0.71 | 1.02 (0.97-1.08) | 0.47 |
| CHD→ VTE | 1.01 (0.80-1.27) | 0.94 | 1.00 (0.85-1.17) | 0.99 |
| CHD→ DVT | 1.02 (0.76-1.35) | 0.91 | 1.00 (0.84-1.20) | 0.97 |
| CHD→ PE | 0.99 (0.80-1.24) | 0.94 | 1.01 (0.86-1.19) | 0.87 |
CHD: coronary heart disease; CI: confidence interval; DVT: deep venous thrombosis; OR: odd ratio; PE: pulmonary embolism; VTE: venous thromboembolism
Table 2. Assessment of pleiotropy and heterogeneity.
| Outcomes | Pleiotropy | Heterogeneity | ||
|---|---|---|---|---|
| Intercept | P | Q | P | |
| VTE→ CHD | 0.0123 | 0.29 | 34 | <0.01 |
| DVT→ CHD | 0.0068 | 0.60 | 26 | <0.01 |
| PE→ CHD | 0.0520 | 0.10 | 25 | <0.01 |
| CHD→ VTE | 0.0124 | 0.61 | 305 | <0.01 |
| CHD→ DVT | 0.0262 | 0.36 | 226 | <0.01 |
| CHD→ PE | 0.0069 | 0.75 | 119 | <0.01 |
CHD: coronary heart disease; DVT: deep venous thrombosis; PE: pulmonary embolism; VTE: venous thromboembolism
Supplementary Fig.1. Scatter plot of the association of CHD with VTE.
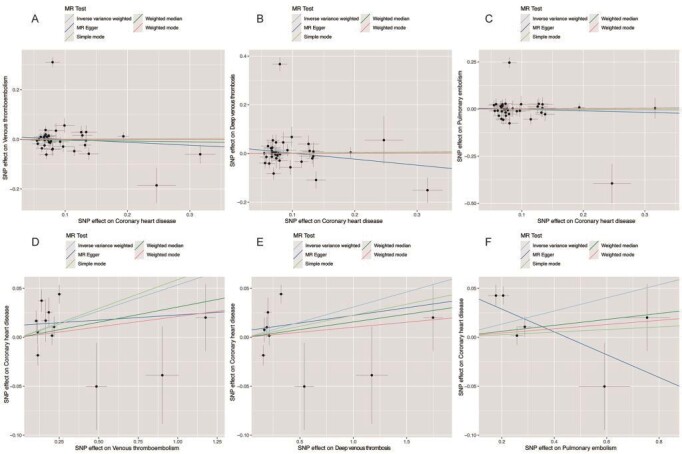
A: CHD→VTE; B: CHD→DVT; C: CHD→PE; D: VTE→CHD; E: DVT→CHD; F: PE→CHD
CHD: coronary heart disease; DVT: deep venous thrombosis; PE: pulmonary embolism; SNP: single-nucleotide polymorphism; VTE: venous thromboembolism
Supplementary Fig.2. Forest plot of the association of CHD with VTE.
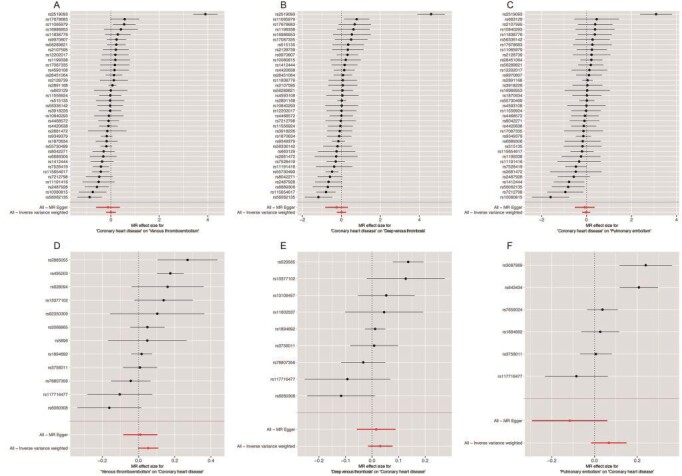
A: CHD→VTE; B: CHD→DVT; C: CHD→PE; D: VTE→CHD; E: DVT→CHD; F: PE→CHD
CHD: coronary heart disease; DVT: deep venous thrombosis; PE: pulmonary embolism; VTE: venous thromboembolism
Supplementary Fig.3. Leave-one-out sensitivity analysis of the association of CHD with VTE.
A: CHD→VTE; B: CHD→DVT; C: CHD→PE; D: VTE→CHD; E: DVT→CHD; F: PE→CHD
CHD: coronary heart disease; DVT: deep venous thrombosis; PE: pulmonary embolism; VTE: venous thromboembolism
Supplementary Fig.4. Funnel plot of the association of CHD with VTE.
A: CHD→VTE; B: CHD→DVT; C: CHD→PE; D: VTE→CHD; E: DVT→CHD; F: PE→CHD
CHD: coronary heart disease; DVT: deep venous thrombosis; PE: pulmonary embolism; VTE: venous thromboembolism
3.2 Effect of CHD on VTE
A total of 37 SNPs were included as genetic variants for CHD. There was also no clear evidence showing the causal effects of CHD on VTE (OR: 1.00; 95% CI: 0.82–1.22; P=0.98), DVT (OR: 1.00; 95% CI: 0.79–1.27; P=0.97), or PE (OR: 0.98; 95% CI: 0.82–1.18; P=0.87) ( Fig.2 ) . Sensitivity analyses using the weighted median, MR-Egger, and multivariable MR methods also revealed no significant associations between CHD with VTE or its subtypes. Sensitivity analyses using simple mode and weighted mode methods also yielded no significant associations between CHD and VTE ( Supplementary Table 1 ) . The MR-Egger method revealed no evidence of horizontal pleiotropy. However, the heterogeneity was significant in all analyses ( Table 2 ) . The scatter plot and forest plot based on all SNPs are shown in Supplementary Fig.1 and Supplementary Fig.2 , respectively. The leave-one-out sensitivity analysis indicated that the overall estimate was not influenced by an individual SNP ( Supplementary Fig.3 ) . The funnel plots also revealed no horizontal pleiotropy ( Supplementary Fig.4 ) .
4. Discussion
Using genetic variants associated with CHD and VTE/DVT/PE, our bidirectional MR analyses showed that CHD had no causal relationships with VTE/DVT/PE. Vice versa, VTE/DVT/PE had no causal relationships with CHD.
Although it is generally believed that arterial and venous thrombotic disorders are separate entities, several studies have suggested an association between arterial and venous thromboses 3 , 15 , 16) . If the relationship is confirmed, then the currently used agents for the prevention and treatment of arterial diseases, such as antiplatelet therapy and statins, could be considered for managing VTE. Vice versa, the agents for the prevention and treatment of VTE could be considered for the management of arterial events. Concordant with this assumption, a collaborative overview of 53 randomized trials (8400 patients) of antiplatelet therapy suggested that a few weeks of antiplatelet therapy produced a highly significant reduction in DVT 17) . Aspirin is effective not only in preventing arterial events but also in treating VTE 18) . Likewise, low-molecular-weight heparin, a pivotal drug for treating VTE, was found to be helpful in treating arterial events 19) . A reduced incidence of VTE was also found in patients treated with statins in a retrospective cohort study 20) . However, the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) trial did not find that 40 mg/d of pravastatin decreased the risk of VTE compared with placebo in individuals aged between 70 and 82 years (hazard ratio [HR]: 1.42; 95% CI: 0.80–2.52; P=0.23) 21) .
The potential association between VTE and atherosclerosis was assessed for the first time in 2003. Prandoni et al. found that the prevalence of carotid plaques was more common in patients with spontaneous thrombosis (47.1%) than in patients with secondary thrombosis (27.4%) or in controls without thrombosis (32.0%). Their results remained unchanged after adjustment for risk factors for atherosclerosis and became even more evident in elderly patients 3) . After that, numerous relevant studies have been published, providing conflicting results. In the prospective Atherosclerosis Risk in Communities study, 13,081 individuals were followed for a mean of 12.5 years to assess the association between subclinical atherosclerosis (carotid intima-media thickness or carotid plaque) and VTE, during which 225 first VTE events were identified. Although a higher risk of VTE was observed across quartiles of baseline carotid intima-media thickness, this association was nonsignificant after adjustments for age, sex, and ethnicity. Adjustments for BMI and diabetes further weakened the association. The presence of carotid plaques at baseline also had no association with VTE 8) . Similarly, in the Cardiovascular Health Study, 4108 individuals without baseline clinical cardiovascular diseases were included and followed for a median of 11.7 years. Subclinical atherosclerosis did not affect overall or idiopathic VTE, yet overall (relative risk [RR]: 0.60; 95% CI: 0.39–0.91) and idiopathic VTE (RR: 0.32; 95% CI: 0.18–0.59) decreased the risk of subclinical atherosclerosis 9) . Unlike the above null effects of atherosclerosis on VTE, the population-based, case–cohort Norwegian study found that subjects with a family history of CHD had a higher incidence of VTE 15) . Another retrospective case–control study also found that coronary artery calcium increased the risk of VTE (OR: 4.3; 95% CI: 1.9–10.1) 16) .
Therefore, it seems that positive associations between markers of subclinical atherosclerotic CHD and risk of VTE were mainly found in case–control studies 3 , 15 , 16) , whereas that was not the case in prospective cohort studies 8 , 9) . Although previous case–control studies found a link between carotid plaques and a higher VTE risk 3) , the recruited control group was not fully representative of the case-derived population and may lead to an overestimation of the true effect. This issue is more obvious when the sample size of the control group is small. In prospective cohort studies, however, atherosclerosis may change over time. It is argued that long-term follow-up between the baseline measurement and event may introduce regression dilution bias, resulting in underestimation of the true relationship 22) . To overcome this issue, the atherosclerosis status was repeatedly measured within the same individual during follow-up in the Tromsø Study. Plaque formation (HR: 1.00; 95% CI: 0.98–1.02) or progression of carotid plaque size (HR: 0.96; 95% CI: 0.84–1.11) had no effects on VTE risk. After multivariable adjustment, the results remained unchanged 23) . These results corroborated our finding that CHD had no causal effect on VTE/DVT/PE.
The effect of VTE on atherosclerosis was investigated for the first time in a prospective study that included 360 patients with the first episode of PE. Compared with patients with PE associated with transient risk factors, patients with idiopathic PE had a higher incidence of arterial cardiovascular events (3.2% vs. 0.4%; RR: 7.2; 95% CI: 1.71–30.45). After adjusting for age, the difference remained unchanged 24) . However, as individuals free from VTE were not included, the risk in the general VTE population was not validated. Later, Schulman et al. found higher mortality rates from acute myocardial infarction and stroke in patients with previous VTE 25) . Bova et al. also found that the rate of acute myocardial infarction, ischemic stroke, or peripheral arterial disease was more common in patients with unprovoked VTE than in control subjects. After adjusting for cardiovascular risk factors, the difference remained unchanged 2) . In a cohort study based on nationwide Danish medical databases, 25,199 patients with DVT events, 16,925 patients with PE events, and 163,566 population controls were analyzed. Patients with DVT had higher risks of myocardial infarction (RR: 1.60; 95% CI: 1.35–1.91) and stroke (RR: 2.19; 95% CI: 1.85–2.60) in the first-year follow-up. Patients with PE also had higher risks of myocardial infarction (RR: 2.60; 95% CI: 2.14–3.14) and stroke (RR: 2.93; 95% CI: 2.34–3.66) in that year. During the subsequent 20-year follow-up, the incidences of arterial events remained higher in patients with VTE than in matched controls 26) . Therefore, it seems that VTE may increase the risk of CHD. However, as in any observational study, unrecognized confounders may be present despite the extensive evaluation of patient characteristics.
As genetic variants are randomly allocated before birth, we can minimize the effect of confounders and reverse causation on outcomes by using genetic variants strongly associated with exposure as instrumental variables. We found no causal associations between CHD and VTE/DVT/PE, and vice versa. Concordant with our results, it is commonly believed that if an association between CHD and VTE exists, the mechanism is probably due to the shared common risk factors between the two diseases 27) . However, an individual participant data meta-analysis of nine prospective studies revealed that in the unadjusted models, nearly all cardiovascular risk factors (hypertension, hyperlipidemia, diabetes, and smoking) showed positive associations with VTE. After adjusting for age, sex, and BMI, these risk factors did not increase VTE risk any more, except for current smoking 28) . This finding corroborated that the pathogenesis of venous disease was different from that of atherosclerotic disease. Arterial and venous thromboses may represent separate entities. The previously reported significant associations between VTE and atherosclerosis may be explained by not fully accounting for confounders.
Limitations
Our study is the first to use the MR approach to investigate the directional link between CHD and VTE/DVT/PE. Several sensitivity analyses also confirmed the robustness of our results. Nonetheless, several limitations deserve our attention. First, the results of the current MR analyses may not be generalizable to non-European populations, given that most GWAS primarily enrolled European individuals. Besides, given the lack of detailed data, we could not make specific statements about the relative risk according to subgroups, such as males versus females.
5. Conclusions
Our comprehensively bidirectional MR analyses suggested no causal associations between CHD and VTE/DVT/PE. This finding confirms that VTE and atherosclerosis represent separate entities. The previously reported significant associations may be explained by not fully accounting for confounders.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFC1700503), CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1-009), and the Twelfth Five-Year Planning Project of the Scientific and Technological Department of China (2011BAI11B02).
Conflict of Interest
All authors declared no conflicts of interest.
References
- 1).Prandoni P: Venous thromboembolism and atherosclerosis: is there a link? J Thromb Haemost, 2007; 5 Suppl 1: 270-275 [DOI] [PubMed] [Google Scholar]
- 2).Bova C, Marchiori A, Noto A, Rossi V, Daniele F, Santoro C, Ricchio R, De Lorenzo R, Umbaca R and Prandoni P: Incidence of arterial cardiovascular events in patients with idiopathic venous thromboembolism. A retrospective cohort study. Thromb Haemost, 2006; 96: 132-136 [PubMed] [Google Scholar]
- 3).Prandoni P, Bilora F, Marchiori A, Bernardi E, Petrobelli F, Lensing AW, Prins MH and Girolami A: An association between atherosclerosis and venous thrombosis. N Engl J Med, 2003; 348: 1435-1441 [DOI] [PubMed] [Google Scholar]
- 4).Koenig W, Rothenbacher D, Hoffmeister A, Griesshammer M and Brenner H: Plasma fibrin D-dimer levels and risk of stable coronary artery disease: results of a large case-control study. Arterioscler Thromb Vasc Biol, 2001; 21: 1701-1705 [DOI] [PubMed] [Google Scholar]
- 5).Ageno W, Becattini C, Brighton T, Selby R and Kamphuisen PW: Cardiovascular risk factors and venous thromboembolism: a meta-analysis. Circulation, 2008; 117: 93-102 [DOI] [PubMed] [Google Scholar]
- 6).Wilhelmsen L, Svärdsudd K, Korsan-Bengtsen K, Larsson B, Welin L and Tibblin G: Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med, 1984; 311: 501-505 [DOI] [PubMed] [Google Scholar]
- 7).Folsom AR, Wu KK, Rosamond WD, Sharrett AR and Chambless LE: Prospective study of hemostatic factors and incidence of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation, 1997; 96: 1102-1108 [DOI] [PubMed] [Google Scholar]
- 8).Reich LM, Folsom AR, Key NS, Boland LL, Heckbert SR, Rosamond WD and Cushman M: Prospective study of subclinical atherosclerosis as a risk factor for venous thromboembolism. J Thromb Haemost, 2006; 4: 1909-1913 [DOI] [PubMed] [Google Scholar]
- 9).van der Hagen PB, Folsom AR, Jenny NS, Heckbert SR, O’Meara ES, Reich LM, Rosendaal FR and Cushman M: Subclinical atherosclerosis and the risk of future venous thrombosis in the Cardiovascular Health Study. J Thromb Haemost, 2006; 4: 1903-1908 [DOI] [PubMed] [Google Scholar]
- 10).Emdin CA, Khera AV and Kathiresan S: Mendelian Randomization. Jama, 2017; 318: 1925-1926 [DOI] [PubMed] [Google Scholar]
- 11).Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikäinen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, König IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimäki T, Loos RJF, Melander O, Metspalu A, März W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O’Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ and Farrall M: A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet, 2015; 47: 1121-1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Machiela MJ and Chanock SJ: LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics, 2015; 31: 3555-3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Burgess S, Bowden J, Fall T, Ingelsson E and Thompson SG: Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology, 2017; 28: 30-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Bowden J, Davey Smith G and Burgess S: Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol, 2015; 44: 512-525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Quist-Paulsen P, Naess IA, Cannegieter SC, Romundstad PR, Christiansen SC, Rosendaal FR and Hammerstrøm J: Arterial cardiovascular risk factors and venous thrombosis: results from a population-based, prospective study (the HUNT 2). Haematologica, 2010; 95: 119-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Hong C, Zhu F, Du D, Pilgram TK, Sicard GA and Bae KT: Coronary artery calcification and risk factors for atherosclerosis in patients with venous thromboembolism. Atherosclerosis, 2005; 183: 169-174 [DOI] [PubMed] [Google Scholar]
- 17).Collaborative overview of randomised trials of antiplatelet therapy--III: Reduction in venous thrombosis and pulmonary embolism by antiplatelet prophylaxis among surgical and medical patients. Antiplatelet Trialists’ Collaboration. Bmj, 1994; 308: 235-246 [PMC free article] [PubMed] [Google Scholar]
- 18).Karthikeyan G, Eikelboom JW, Turpie AG and Hirsh J: Does acetyl salicylic acid (ASA) have a role in the prevention of venous thromboembolism? Br J Haematol, 2009; 146: 142-149 [DOI] [PubMed] [Google Scholar]
- 19).Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE and Comerota AJ: Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest, 2008; 133: 454s-545s [DOI] [PubMed] [Google Scholar]
- 20).Ray JG, Mamdani M, Tsuyuki RT, Anderson DR, Yeo EL and Laupacis A: Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med, 2001; 161: 1405-1410 [DOI] [PubMed] [Google Scholar]
- 21).Freeman DJ, Robertson M, Brown EA, Rumley A, Tobias ES, Frölich M, Slagboom PE, Jukema JW, de Craen AJ, Sattar N, Ford I, Gaw A, Greer IA, Lowe GD and Stott DJ: Incident venous thromboembolic events in the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER). BMC Geriatr, 2011; 11: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Prandoni P: Links between arterial and venous disease. J Intern Med, 2007; 262: 341-350 [DOI] [PubMed] [Google Scholar]
- 23).Småbrekke B, Rinde LB, Hald EM, Njølstad I, Mathiesen EB, Johnsen SH, Hansen JB, Braekkan SK and Lijfering WM: Repeated measurements of carotid atherosclerosis and future risk of venous thromboembolism: the Tromsø Study. J Thromb Haemost, 2017; 15: 2344-2351 [DOI] [PubMed] [Google Scholar]
- 24).Becattini C, Agnelli G, Prandoni P, Silingardi M, Salvi R, Taliani MR, Poggio R, Imberti D, Ageno W, Pogliani E, Porro F and Casazza F: A prospective study on cardiovascular events after acute pulmonary embolism. Eur Heart J, 2005; 26: 77-83 [DOI] [PubMed] [Google Scholar]
- 25).Schulman S, Lindmarker P, Holmström M, Lärfars G, Carlsson A, Nicol P, Svensson E, Ljungberg B, Viering S, Nordlander S, Leijd B, Jahed K, Hjorth M, Linder O and Beckman M: Post-thrombotic syndrome, recurrence, and death 10 years after the first episode of venous thromboembolism treated with warfarin for 6 weeks or 6 months. J Thromb Haemost, 2006; 4: 734-742 [DOI] [PubMed] [Google Scholar]
- 26).Sørensen HT, Horvath-Puho E, Pedersen L, Baron JA and Prandoni P: Venous thromboembolism and subsequent hospitalisation due to acute arterial cardiovascular events: a 20-year cohort study. Lancet, 2007; 370: 1773-1779 [DOI] [PubMed] [Google Scholar]
- 27).Wattanakit K, Lutsey PL, Bell EJ, Gornik H, Cushman M, Heckbert SR, Rosamond WD and Folsom AR: Association between cardiovascular disease risk factors and occurrence of venous thromboembolism. A time-dependent analysis. Thromb Haemost, 2012; 108: 508-515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Mahmoodi BK, Cushman M, Anne Næss I, Allison MA, Bos WJ, Brækkan SK, Cannegieter SC, Gansevoort RT, Gona PN, Hammerstrøm J, Hansen JB, Heckbert S, Holst AG, Lakoski SG, Lutsey PL, Manson JE, Martin LW, Matsushita K, Meijer K, Overvad K, Prescott E, Puurunen M, Rossouw JE, Sang Y, Severinsen MT, Ten Berg J, Folsom AR and Zakai NA: Association of Traditional Cardiovascular Risk Factors With Venous Thromboembolism: An Individual Participant Data Meta-Analysis of Prospective Studies. Circulation, 2017; 135: 7-16 [DOI] [PMC free article] [PubMed] [Google Scholar]