

Abstract
Aims
Calmodulinopathy due to mutations in any of the three CALM genes (CALM1–3) causes life-threatening arrhythmia syndromes, especially in young individuals. The International Calmodulinopathy Registry (ICalmR) aims to define and link the increasing complexity of the clinical presentation to the underlying molecular mechanisms.
Methods and results
The ICalmR is an international, collaborative, observational study, assembling and analysing clinical and genetic data on CALM-positive patients. The ICalmR has enrolled 140 subjects (median age 10.8 years [interquartile range 5–19]), 97 index cases and 43 family members. CALM-LQTS and CALM-CPVT are the prevalent phenotypes. Primary neurological manifestations, unrelated to post-anoxic sequelae, manifested in 20 patients. Calmodulinopathy remains associated with a high arrhythmic event rate (symptomatic patients, n = 103, 74%). However, compared with the original 2019 cohort, there was a reduced frequency and severity of all cardiac events (61% vs. 85%; P = .001) and sudden death (9% vs. 27%; P = .008). Data on therapy do not allow definitive recommendations. Cardiac structural abnormalities, either cardiomyopathy or congenital heart defects, are present in 30% of patients, mainly CALM-LQTS, and lethal cases of heart failure have occurred. The number of familial cases and of families with strikingly different phenotypes is increasing.
Conclusion
Calmodulinopathy has pleiotropic presentations, from channelopathy to syndromic forms. Clinical severity ranges from the early onset of life-threatening arrhythmias to the absence of symptoms, and the percentage of milder and familial forms is increasing. There are no hard data to guide therapy, and current management includes pharmacological and surgical antiadrenergic interventions with sodium channel blockers often accompanied by an implantable cardioverter–defibrillator.
Keywords: Calmodulin, Long QT syndrome, Catecholaminergic polymorphic ventricular tachycardia, Idiopathic ventricular fibrillation, Sudden death, Cardiomyopathies, Neurological disorders
Structured Graphical Abstract
Structured Graphical Abstract.

Number and percentages of patients with CALM variants and LQTS, CPVT, LQTS/CPVT overlap, IVF, UD, and SCD are reported in the upper left part of the graphical abstract together with two examples of LQTS and CPVT electrocardiograms. In the upper right part, it is shown a graphical representation of the reduction of all cardiac events and SCD between the original cohort (up to 2019) and the cases enrolled between 2019 and 2023. In the lower part, it is reported the number or percentage of CALM patients with cardiomyopathies, congenital heart defects, and neurological features. CALM, calmodulin; LQTS, long QT syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; ICD, implantable cardioverter–defibrillator; IVF, idiopathic ventricular fibrillation; UD, uncertain diagnosis; SCD, sudden cardiac death.
See the editorial comment for this article ‘Advancing personalized medicine: the power of collaborative registries of patients with inherited cardiac arrhythmia syndromes’, by A. Barsheshet and I. Goldenberg, https://doi.org/10.1093/eurheartj/ehad441.
Introduction
Within 10 years after the initial reports,1–3 calmodulin (CALM) mutations were catapulted to the forefront of clinical studies and research of arrhythmias of genetic origin for two main reasons: (i) the dramatic presentation of malignant, and often lethal, arrhythmias in infants and children and (ii) the novel insights into the clinical consequences of mutations affecting intracellular calcium homeostasis.
Calmodulin is a ubiquitous and essential calcium-binding protein (CaM) critically involved in countless intracellular signalling processes. Its uniqueness, and importance for life, is highlighted not only by its amino acid sequence being conserved perfectly among vertebrates but especially by the fact that in humans, there are three genes that encode a completely identical calmodulin protein. On this basis, one must expect that CALM mutations would be poorly compatible with life and, when present, would be associated with very serious clinical phenotypes.
The initial reports1,2 pointed indeed to life-threatening arrhythmias as the main features, with phenotypes close to catecholaminergic polymorphic ventricular tachycardia1 (CPVT) and long QT syndrome2 (LQTS). However, attention was called to the presence of neurological and neurodevelopmental deficits among some of the small number of initial patients.2 Brain injury secondary to cardiac arrest during early life was evident in some cases, but given that CALM is also highly expressed in the brain, a direct neurological involvement remained a possibility. Finally, a potential association with coexisting structural abnormalities and cardiomyopathies was noted but without conclusive data.
Our initial reports1,2 opened the gates to scattered reports mostly of anecdotal nature but all pointing to the fact, well known in the field of rare diseases, that when people begin to look in the right direction (i.e. screening for CALM mutations in genotype-negative young survivors of cardiac arrest), new cases are found. The rarity of the condition and the dismal responses to therapy highlighted the need for more data on which to base clinical management. Following the success of a similar operation initiated 45 years ago for LQTS,4,5 we established the International Calmodulinopathy Registry (ICalmR), and in 2019 we reported the initial data on 74 patients.6
Progress has clearly taken place. The CALM1–3 genes have been recognized for a definite association with LQTS7 and CPVT8 and now represent genes that should be screened in all LQTS and CPVT patients.9 However, new questions are emerging and not everything looks like it was initially described.
Since 2019, thanks to international collaboration, the ICalmR has almost doubled the number of enrolled patients and now offers novel information, which suggests that we might be observing a clinically relevant changing pattern. Here, we report the new data and the new implications.
Methods
The ICalmR was established in 2015 as an international, collaborative, observational registry with the main objectives of defining the clinical presentation, genetic background, natural history, and response to therapy in patients with CALM genetic variants.6 The modalities of data collection have been described.6 The addition of a significant number of new cases (n = 66) has allowed a comparison between the initial pattern of presentation and the current one, which reflects the expanded genetic screening for CALM variants recently implemented. In several cases, updated information from longer follow-up has resulted in a revised assessment of phenotypes or in upgrading the subject’s clinical features. Our definitions of clinical status—asymptomatic and symptomatic with syncope or major arrhythmic events (MAE), including aborted cardiac arrest (ACA), sudden cardiac death (SCD), and appropriate implantable cardioverter–defibrillator (ICD) shocks—remain unchanged.6
With the objective of approaching a genotype–phenotype correlation oriented towards the underlying mechanisms, we classified the patients, based on phenotype and clinical judgement, in the following groups: (i) LQTS, when an electrocardiogram (ECG) of the patient/victim/relative shows QT interval prolongation; (ii) CPVT, when the phenotype of the surviving patient or of a family member (FM), with the same genetic variant, fits with CPVT; this phenotype also includes those relatives in CPVT families who died suddenly; (iii) idiopathic ventricular fibrillation (IVF), when the surviving patient has a negative ECG and negative clinical investigations, and this includes FMs with the same genetic variant or who have died suddenly under age 30; (iv) LQTS/CPVT overlap, which refers to the presence in the same subject of both LQTS- and CPVT-like traits observed simultaneously or, more often, at distinct times; and (v) uncertain diagnosis (UD) with either (a) sudden death without an ECG and—after exclusion of a de novo mutation—the family examination is either unavailable or not informative or (b) an adequate and conclusive diagnostic investigation is unavailable for the survivor of a cardiac arrest and for the family. Within this group, we include victims in whom we have no, or insufficient, family information and who might not be de novo cases. Thus, this approach implies that the patients of the fourth group might as well belong to one of the first three groups. As in the LQTS International Registry,5 subjects with unexplained SCD at age < 40 years without genetic testing were considered genotype-positive for the same CALM variant found in first-degree relatives. Finally, the phenotype was classified as ‘atypical’ in very few cases, in which cardiac symptoms were lacking, or were not diagnostic, with predominant features of neurological–neurodevelopmental abnormalities.
The Ethics Committees of the co-ordinating (Istituto Auxologico Italiano IRCCS, Milan, Italy; Mayo Clinic, Rochester, MN, USA) and of enrolling centres approved the study.
Statistical analysis
For continuous variables, normality was assessed by the Shapiro–Wilk test. Both QTc and follow-up times showed a skewed distribution. QTc data were summarized with mean ± SD in order to allow for comparisons with the results from other studies, including the 2019 original publication; age at onset and follow-up duration were presented as median and interquartile range (IQR, 25th–75th percentile); when clinically meaningful, the minimum–maximum range was also provided. Categorical variables were presented as absolute (n) and relative frequencies (%). The Mann–Whitney U test for continuous variables and the χ2 or Fisher exact tests for categorical variables were used to compare the clinical characteristics among genotype or phenotype groups. Binomial exact 95% confidence intervals (CI) were computed for the estimated proportions of cases. When comparing the occurrence of cardiac events between the two subsequent cohorts of enrolled cases, family membership was considered. The use of the generalized estimating equation (GEE) technique with a robust sandwich variance estimator or the inclusion of a random effect for family relatedness in a binary logistic regression model did not change the significance of the association between cohort membership and cardiac events. Thus, P-values from the standard Fisher exact test were reported. Event-free survival was described by Kaplan–Meier cumulative estimates. Time to first event was considered both for any cardiac event and for MAE. Two-sided P-values < .05 were considered statistically significant. IBM SPSS Statistics version 28.0 was used for computation.
Results
Registry population
To date, the ICalmR has enrolled 140 patients with 62 genetic variants (notably, all missense variants) in the CALM1, CALM2, and CALM3 genes (see Supplementary data online, Table S1).
Among all patients [72 males, 51%; median age at last follow-up 10.8 years (IQR 5–19)], 97 (69%) were index cases and the remaining 43 were FMs with the same variant. Fourteen FMs were part of one LQTS family,10 and 13 were part of one CPVT family.1
The main features of the study population are summarized in Table 1. According to the clinical classification, the breakdown of CALM variant–positive subjects into cardiac phenotypes is as follows: LQTS: 74 (53%); CPVT: 36 (26%); IVF: 7 (5%); LQTS/CPVT overlap: 10 (7%); UD: 11 (8%); and atypical: 2 (1%). Therefore, consistent with the original observation, CALM-LQTS and CALM-CPVT are the two most prevalent phenotypes and, together with CALM-LQTS/CPVT overlap, are contributing to 86% of the entire population and to 84% of the 103 symptomatic patients (Table 2).
Table 1.
Study population from the ICalmR
| Demographic and clinical characteristics | |
|---|---|
| Patients | 140 |
| Index cases | 97 (69) |
| Male sex | 72 (51) |
| Age at last FU, years, median (IQR) | 10.8 (5–19)a |
| Phenotype | |
| LQTS | 74 (53) |
| CPVT | 36 (26) |
| IVF | 7 (5) |
| LQTS/CPVT overlap | 10 (7) |
| UD | 11 (8) |
| Atypical | 2 (1) |
| Symptomatic | |
| Any cardiac eventb | 103 (74) |
| MAEc | 77 (55) |
| SCD | 26 (19) |
| Age at first event, years, median (IQR) | 4 (1.6–8) |
| Perinatal cardiac presentationd | 36 (26) |
| Cardiac structural comorbidities (n = 135) | 40 (30) |
| CHD | 20 (15) |
| Cardiomyopathy | 28 (21) |
| Neurological features (n = 111) | 35 (31.5) |
| Post-anoxic sequelae | 15 (13.5) |
| ACA-independent | 20 (18) |
LQTS, long QT syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; IVF, idiopathic ventricular fibrillation; UD, uncertain diagnosis; MAE, major arrhythmic events; ACA, aborted cardiac arrest; FU, follow-up.
Data are presented as n (%) unless otherwise indicated.
Extended follow-up of the original cohort also is included.
Arrhythmic syncope, ACA, SCD, or appropriate ICD shocks.
ACA, SCD, and appropriate ICD shocks.
Occurrence of symptoms (sinus bradycardia, marked QT prolongation, 2:1 atrioventricular block, T-wave alternans, and/or ventricular arrhythmias) from approximately the 28th week of gestation to the 28th day after birth.
Table 2.
Calmodulinopathy phenotypes and symptoms
| CALM-phenotype | All carriers | Any cardiac events | MAE |
|---|---|---|---|
| (n = 140)a | (n = 103)b,c | (n = 77)c,d | |
| LQTS | 74 (53) | 47 (64) | 36 (49) |
| CPVT | 36 (26) | 33 (92) | 20 (56) |
| IVF | 7 (5) | 5 (71) | 5 (71) |
| LQTS/CPVT overlap | 10 (7) | 7 (70) | 6 (60) |
| UD | 11 (8) | 11 (100) | 10 (91) |
| Atypical | 2 (1) | - | - |
Data are numbers and (%) of subjects.
LQTS, long QT syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; IVF, idiopathic ventricular fibrillation; MAE, major arrhythmic events; UD, uncertain diagnosis.
Percentage out of all 140 CALM patients.
Arrhythmic syncope, ACA, SCD, and appropriate ICD shocks.
Percentage in the corresponding phenotype group.
ACA, SCD, and appropriate ICD shocks.
Most patients (n = 103, 74%) were symptomatic, and by age 5.8 years (95% CI 4.2–7.5), 50% had already experienced an arrhythmic event (Figure 1A), with no difference by sex. Major arrhythmic events occurred in 77 (55%) subjects, with a median survival time of 10 years (95% CI 6.7–13.3) (Figure 1B). All 26 (19%) subjects with SCD died before age 16, at a mean age of 5.5 years (Figure 2). Even though calmodulinopathy remains associated with a high cardiac event rate (Figure 3A), when the first cohort6 (n = 74) was compared with the second one (n = 66), we observed a significant reduction in any cardiac event, MAE, appropriate ICD shocks, and SCD over a comparable length of follow-up from birth to last contact (Figure 3B). Specifically, the percentage with any cardiac event decreased from 85% (95% CI 75%–92%) to 61% (95% CI 48%–72%) (P = .001), while MAE decreased from 69% (95% CI 57%–79%) to 39% (95% CI 28%–52%) (P < .001), ICD interventions from 23% (95% CI 14%–34%) to 6% (95% CI 2%–15%) (P = .008), and SCD from 27% (95% CI 17%–39%) to 9% (95% CI 3%–19%) (P = .008) (Figure 3B). Probands showed a very similar pattern (Figure 3C). Adrenergic stimulation was the major trigger for cardiac events in the entire population, irrespective of phenotype (78%) and in the main subtypes (100% in CALM-CPVT cases, 68% in the CALM-LQTS group). However, especially in young children, MAE may also occur in the absence of any known arousal. Specifically, of the 23 subjects with ACA/SCD as a first cardiac event below age 2.5 years, 6 (26%) had their events while at rest or asleep. Furthermore, five young children had their cardiac events, fatal in three, during illnesses/infections, known or likely to have been febrile. Thirty-five CALM patients are alive and still asymptomatic at a median age of 12 years (IQR 3–35), and importantly, five have a normal cardiac phenotype and did not receive antiadrenergic therapy.
Figure 1.

Kaplan–Meier estimates of cumulative event-free survival of a first (A) cardiac event and (B) major arrhythmic event in the entire CALM population.
Figure 2.
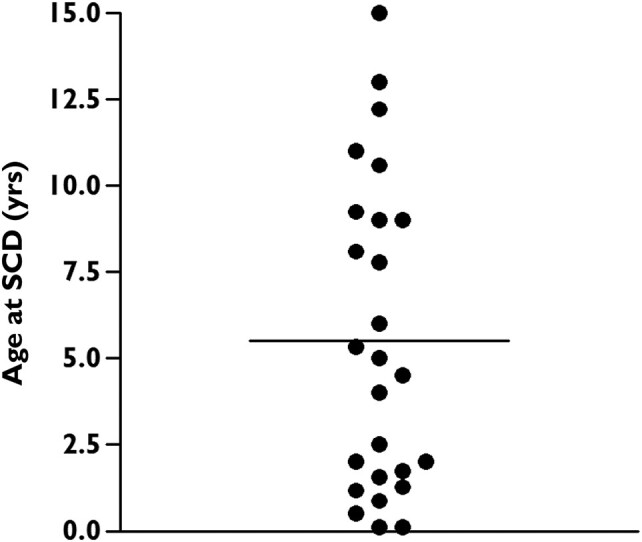
Age distribution of the 26 CALM patients who died suddenly among all 140 cases enrolled in the International Calmodulinopathy Registry. The horizontal bar represents the mean of values.
Figure 3.

Prevalence of symptomatic patients, also according to the type of arrhythmic events, in the entire CALM population (A), by time of enrolment in the International Calmodulinopathy Registry of all 140 cases (B), and in probands only (C).
CALM-LQTS
CALM-LQTS is the most frequent phenotype, diagnosed in the majority (n = 74, 53%) of the 140 patients and in 55 (57%) of the 97 probands. The QTc showed extreme prolongation (560 ± 79 ms) and was markedly longer in probands than in FMs (579 ± 78 vs. 488 ± 32 ms, P < .001). Forty-seven of the 74 CALM-LQTS patients (64%) were symptomatic, and the first cardiac event occurred very early, at a median age of 2 years (IQR 0.4–5.0). Thirty-six (49%) suffered MAE, and 11 (15%) died suddenly at a median age of 1.7 years (IQR 0.5–6.0). Of note, among those dying suddenly, five were not on therapy, three were on β-blocker therapy only (one at a low dose), one was on β-blockers and had a pacemaker (PM), and only two had a combination of PM, β-blockers, and mexiletine. Therefore, none of those dying suddenly had the triple therapy of full-dose β-blockers, sodium channel blockers, and left cardiac sympathetic denervation (LCSD), and clearly, none of them had an ICD. A perinatal presentation involved 34 patients (46%) who had a more prolonged QTc (612 ± 77 vs. 516 ± 49 ms, P < .001) and an earlier median age at onset of symptoms (0.63 vs. 3 years) compared with the remaining 40. However, they had a similar incidence of cardiac events [20/34, 59% (95% CI 41%–75%) vs. 27/40, 68% (95% CI 51%–81%), P = .48] and event severity [MAE, 16/34, 47% (95% CI 30%–65%) vs. 20/40, 50% (95% CI 34%–66%), P = .82], including a similar occurrence of SCD [5/34, 15% (95% CI 5%–31%) and 6/40, 15% (95% CI 6%–30%), respectively].
Among the 13 patients who survived a first cardiac event during early infancy, 3 had a dramatic presentation with multiple cardiac arrests (>10) in the subsequent 1–2 years. They are now 12–15 years old, and in two the arrhythmic burden has decreased with the combination of full/high doses of β-blockers (propranolol 2.5–10 mg/kg/day), mexiletine, or flecainide and with a dual-chamber ICD, plus right and LCSD in one. Indeed, from age 2–3 years onwards, one patient had no further cardiac events and the other had one ICD shock after skipping few medication doses. The third patient, with severe neurological consequences after the first arrhythmic episodes, suffered a stroke while on extracorporeal membrane oxygenation (ECMO).
Sodium channel blockers, mostly mexiletine or flecainide, were started in 26 CALM-LQTS patients, always in combination with β-blockers. Treatment was interrupted due to intolerance or inefficacy (no change in QT intervals and/or recurrences of arrhythmic episodes) in 10; among the remaining 16 on continued therapy, QTc shortened in 6.
CALM-CPVT
In 36 patients (20 index cases, 16 FMs; 53% males; mean QTc 421 ± 33 ms), clinical and ECG findings suggestive of CPVT were reported. Exertional arrhythmias observed at stress tests or at Holter monitoring were isolated premature ventricular contractions (PVCs), bigeminy, couplets, and ventricular tachycardias (VTs), more often polymorphic, rarely with a clear bidirectional feature (six cases). Almost all CALM-CPVT patients (33/36, 92%) were symptomatic with a first cardiac event at a median age of 5 years; 20 (56%) suffered MAE (4 SCD) at an age between 3 and 17 years. All cardiac events occurred in association with adrenergic stimuli, mostly (77%) during exercise. Of the 29 patients with known data on therapy and outcomes, all were on β-blockers, 8 had an ICD, 8 were on a sodium channel blocker (flecainide in 7), and only 3 had LCSD. Nine subjects experienced a breakthrough event during treatment; in five, this was likely associated with suboptimal medication compliance; one SCD occurred after several years of treatment with a selective β-blocker. None of them were on what is viewed presently as optimal antiarrhythmic therapy for CALM-CPVT (β-blockers, flecainide, and LCSD).
CALM-IVF
Two pathogenic variants (CALM1-p.F90L and CALM2-p.N98S) were associated with an IVF phenotype in seven patients (six from one family). According to our classification criteria, ventricular fibrillation (VF) episodes were documented in three subjects, SCD occurred in two siblings before age 11, and two other FMs were genotype-positive asymptomatic adults. A QTc within the normal range (mean 430 ± 25 ms), unremarkable exercise stress tests, and no significant arrhythmias during 24 h Holter monitoring, in association with normal findings at cardiac imaging, supported the CALM-IVF diagnosis. Aborted cardiac arrest and sudden death occurred at a median age of 10 years (range 4.6–16) with adrenergic stimulation. All three survivors and one subject implanted as primary prevention received an ICD, in which three of four delivered appropriate shocks during follow-up.
CALM-LQTS/CPVT overlap
In 10 subjects, a hybrid phenotype was determined by the overlap of LQTS- and CPVT-like features. In most, the initial presentation included QT interval prolongation, often a marked one (mean 495 ± 53 ms), which sometimes normalized on therapy during follow-up. The concomitant or subsequent appearance of exertional polymorphic ventricular ectopies and bidirectional VTs, along with a history of adrenergically induced cardiac events, accounted for the diagnosis of overlap, even when the CPVT phenotype appeared predominant over time. Also, among the seven symptomatic patients (five with ACA, one sudden death), the median age at onset of 6 years (IQR 4–9) was more similar to that of ‘typical CPVT’ cases (5, IQR 3–9).
Uncertain diagnosis
This group with major diagnostic uncertainty included 11 genotype-positive subjects from 9 families. Eight died suddenly in the absence of a diagnostic ECG. Death occurred at a median age of 4 years (range 10 months–9 years), in association with known adrenergic stimuli in five subjects and during sleep in three. A history of seizure-like episodes was reported in two children, and in two the autopsy showed dilated cardiac chambers. Of the two still alive, one child survived multiple VF episodes at age 3 associated with intercurrent illness and during sleep; the other, the mother of two of the eight victims of sudden death, had syncope while swimming. In both cases, the cardiac work-up was normal or inconclusive.
Atypical cases
The phenotype was classified as ‘atypical’ in two cases, in which cardiac symptoms were lacking or were not predominant. Specifically, a CALM1-p.E105K boy with a cardio-neurological phenotype showed a normal QTc on resting ECG but a prolonged QTc and abnormal T-waves on standing ECG, in association with a benign focal seizure susceptibility syndrome. A CALM2-p.I64M girl had a malformation/neurodevelopmental disorder with jejunal membranous atresia at birth, generalized joint laxity, some dysmorphic features, but otherwise normal ECG and echocardiographic features.
Cardiac structural abnormalities
In total, 40/135 (30%) patients with available data presented with at least one structural cardiac abnormality, either a cardiomyopathy and/or a congenital heart disease (CHD). Most (31, 78%) were CALM-LQTS patients, 7 (17.5%) CALM-CPVT or mixed LQTS/CPVT phenotype, and 2 (5%) UD.
Left ventricular non-compaction (Figure 4) was reported in 12 patients (9%), associated with left ventricular dysfunction and/or severe heart failure in 4, fatal in 2 below age 1.5. Hypertrophic cardiomyopathy (HCM) was reported in four subjects. Furthermore, in an additional 12 patients, other features of structural heart abnormalities were reported, including dilated cardiomyopathy with reduced ejection fraction (n = 2) and mild ventricular dilatation and hypertrophy (n = 5).
Figure 4.

A 15-year-old male with CALM-LQTS and a pathogenic variant in CALM2 (Asn138Lys) displaying features of left ventricular non-compaction (A) and left ventricular hypertrophy with thickened papillary muscles (B).
Among the 28 CALM patients with coexisting signs of cardiomyopathies, left ventricular dysfunction was reported in 7. Mortality was high in this subgroup with three deaths and one with appropriate ICD shocks.
In 20 subjects, congenital atrial and/or ventricular septal defects, single or multiple, small or large, or requiring surgical closure or not, were present. Cardiomyopathies existed in eight of them.
Neurological–neurodevelopmental features
Of 111 evaluable patients, 35 (31.5%) had a neurological disorder, 15 reported post-anoxic sequelae after resuscitated ACA, and 20 [16 males (80%)] were thought to have primary neurological/neurodevelopmental features. Among the latter, attention-deficit/hyperactivity disorder (ADHD) (n = 5), seizures/epilepsy (n = 8), and autism spectrum disorder (n = 8) were the most frequently reported abnormalities, sometimes in association with cognitive deficits, ranging from mild to severe intellectual disability. Furthermore, among patients with primary neurological–neurodevelopmental features, the CALM1 gene was implicated more frequently, followed by CALM2 and CALM3 (Table 3).
Table 3.
CALM cases with likely primary neurodevelopmental/neurological features and concomitant structural abnormalities and/or congenital heart diseases
| Gene | Variant | Domain | Cardiological phenotype | Neurodevelopmental/neurological features | Structural abnormalities | Congenital heart diseases |
|---|---|---|---|---|---|---|
| CALM1 | p.N98S (p.Asn98Ser) | EF-hand III, Ca2+-chelation loop | LQTS/CPVT | Autism | - | - |
| CALM1 | p.N98S (p.Asn98Ser) | EF-hand III, Ca2+-chelation loop | CPVT | Mild intellectual disability, ADHD, epilepsy | - | - |
| CALM1 | p.E105A (p.Glu105Ala) | EF-hand III, Ca2+-chelation loop | LQTS/CPVT | Developmental disorder with hyperactivity | - | - |
| CALM1 | p.E105K (p.Glu105Lys) | EF-hand III, Ca2+-chelation loop | Atypical | Developmental delay, recurrent seizures (benign focal seizure susceptibility syndrome) | - | - |
| CALM1 | p.D132V (p.Asp132Val) | EF-hand IV, Ca2+-chelating | LQTS | Autism, ADHD, language disorder, amblyopia | - | - |
| CALM1 | p.E141V (p.Glu141Val) | EF-hand IV, Ca2+-chelation loop | LQTS | ADHD, dyslexia | Mildly impaired LV diastolic function, mild LA dilatation | - |
| CALM1 | p.F142L (p.Phe142Leu) | EF-hand IV | LQTS | Autism, intellectual disability | Non-compaction cardiomyopathy | - |
| CALM1 | p.F142L (p.Phe142Leu) | EF-hand IV | LQTS | Epilepsy, infantile spasms, EEG dysrhythmia | - | PFO, small aortopulmonary collateral from the distal arch |
| CALM1 | p.F142L (p.Phe142Leu) | EF-hand IV | LQTS | Autism, intellectual disability, developmental delay, epilepsy, arachnoid cyst fenestration | - | ASD, ostium secundum |
| CALM2 | p.T35I (p.Thr35Ile) | EF hand I | CPVT | Social communication disorder, gender dysphoria | - | - |
| CALM2 | p.E46K (p.Glu46Lys) | EF hand II | CPVT | Autism, epilepsy with abnormal EEG | - | PDA |
| CALM2 | p.E46K (p.Glu46Lys) | EF hand II | CPVT | Autism, severe intellectual disability | - | PDA |
| CALM2 | p.T63R (p.Thr63Arg) | EF-hand II, Ca2+-chelation loop | UD | Seizures | Dilated cardiac cavities at autopsy | - |
| CALM2 | p.I64M (p.Ile64Met) | EF-hand II, Ca2+-chelation loop | Absent | Malformation-neurodevelopmental disorder syndrome: jejunal membranous atresia, neurodevelopmental disorder, generalized joint laxity, some dysmorphic features | - | - |
| CALM2 | p.N98I | EF-hand III, Ca2+-chelation loop | LQTS | ADHD, dyslexia, dyspraxia | Concentric LV hypertrophy | |
| CALM2 | p.N138K (p.Asn138Lys) | EF-hand IV, Ca2+-chelation loop | LQTS | Mild intellectual disability, ADHD, Rolando focus at EEG | HCM | - |
| CALM3 | p.D96H (p.Asp96His) | EF-hand III, Ca2+-chelation loop | LQTS | Autism, developmental delay | - | PDA |
| CALM3 | p.N138K (p.Asn138Lys) | EF-hand IV, Ca2+-chelation loop | LQTS | Psychiatric disorders | - | - |
| CALM3 | p.E141G (p.Glu141Gly) | EF-hand IV, Ca2+-chelating | LQTS | Seizures | - | ASD |
| CALM3 | p.F142L (p.Phe142Leu) | EF-hand IV | LQTS | Autism, developmental delay | RV hypertrophy & dilatation | VSD, aortic overriding |
Cases with associated cardiac structural abnormalities are highlighted in grey.
ADHD, Attention deficit hyperactivity disorder; EEG, electroencephalogram; LV, left ventricle: LA, Left atrium; PFO, patent foramen ovale; ASD, atrial septal defect; PDA, patent ductus arteriosus; VSD, ventricular septal defect; RV, right ventricle; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; UD, uncertain diagnosis; CHD, congenital heart disease.
We identified 12 patients, mostly LQTS (n = 9), with coexistence of both primary neurological features and cardiac structural abnormalities.
Genetic features
In the 140 CALM-positive patients, a total of 62 single nucleotide substitutions were identified (18 CALM1, 30 CALM2, and 14 CALM3), leading to 59 distinct amino acid substitutions in the three CALM genes (see Supplementary data online, Table S1 for details). All but two variants were completely absent from public exome/genome databases (including gnomAD v2, with exome/genome data on >140 000 individuals). Manual curation and classification according to the American College of Medical Genetics and Genomics (ACMG) guidelines11 adjudicated 58/62 genetic variants as pathogenic or likely pathogenic (P/LP), while the remaining 4 were classified as variants of uncertain significance (VUS). More than 40% of these variants (23 P/LP and 4 VUS) stem from newly identified cases of this updated cohort, absent from the 2019 cohort.6
Most P/LP variants (49/58, 84%) resided in exons 5 and 6 of the CALM genes, mapping to the EF-hand motifs III and IV of CaM’s C-terminal lobe. These variants predominantly affected the Asp/D, Asn/N, and Glu/E amino acid residues in the EF-hand Ca2+-chelating loops responsible for Ca2+ binding (35/49, 71%), i.e. residues D94,96,130,132,134, N98 and E105,141. The prevalent exclusive phenotype associated with variants affecting these residues was LQTS (28/35 variants, 80%). At variance with the prevalent location of P/LP variants in EF-hands III and IV, three of the four variants classified as VUS stemmed from EF-hand I and the inter-EF-hand II/III linker.
Cascade parental screening, performed in 78 of the 97 index cases, showed that in 64 (82%), the culprit variant had not been inherited but was de novo; however, possible germline mosaicism was evaluated only in a minority of parents and was identified in 4 (5%). Somatic mosaicism of CALM2-p.D96G, in the absence of parental data, was identified in one CALM-LQTS case, which may also contribute to the unusually mild phenotype of the proband. In 13 of the 78 evaluable cases (11 P/LP, 2 VUS; 17%), at least one FM was found to carry the CALM variant identified in the index case, leading to a slight increase in the frequency of familial cases in the current cohort, compared with the 2019 cohort [7/34, 21% (95% CI 9%–38%) vs. 6/44, 14% (95% CI 5%–27%), P = .54].
Among the 140 subjects, a different frequency of symptoms was observed according to the different CALM genes involved. Indeed, cardiac events decreased from CALM1 [46/52, 88% (95% CI 77%–96%)], to CALM2 [37/53, 70% (95% CI 56%–82%)], to CALM3 [20/35, 57% (95% CI 39%–74%)] (P = .004). This pattern was observed for any event as well as for MAE [34/52, 65% (95% CI 51%–78%); 30/53, 57% (95% CI 42%–70%); 13/35, 37% (95% CI 21%–55%), respectively, P = .03].
Discussion
Unexpectedly, on the basis of the initial reports, as the number of cases with CALM mutations increases, the clinical picture blurs. This update from the ICalmR provides substantial new information that helps define the main phenotypic features of calmodulinopathy and shows that the clinical presentations are much more varied than initially reported. The extreme clinical severity, which was perhaps the most noticeable aspect in the first CALM patients, seems to have decreased as additional subjects are identified. Between the two extremes of CALM-LQTS and CALM-CPVT, intermediate forms—already noted—are now surfacing and appear more complex. In a few truly dramatic cases with neonatal presentation and multiple VF episodes, we now observe an almost sudden interruption of the arrhythmic events despite minor refinements in therapy, thus suggesting an almost spontaneous increase in cardiac electrical stability. The proportion of patients with de novo genetic variants has decreased, leading to a notable increase in familial and less severe cases. It is becoming evident that a non-negligible number of patients have some type of cardiomyopathy in addition to the primary channelopathy-like phenotype. Furthermore, the association with primary neurological features is emerging (Structured Graphical Abstract). Overall, these updated ICalmR data provide evidence for an expansion of clinical presentations within calmodulinopathy.
The changing pattern of calmodulinopathy
The initial reports1–3,12 of calmodulin missense variant–positive subjects were painting a very grim picture, largely characterized by life-threatening arrhythmias appearing in infancy and responding poorly to therapies. Indeed, most of the initial cases were implanted with an ICD and continued to receive a number of appropriate shocks. The initial and tragic impression, when managing them, was that these infants ‘were not made to live’.2 Most of these cases were de novo, thus contributing to the view that mutations in the CALM genes were not, or minimally, compatible with life. The phenotype of these patients seemed to mimic LQTS or CPVT, or something between the two.
The first publication of the Registry in 20196 contained glimpses of a potentially less catastrophic clinical presentation, but overall, the extreme severity of the clinical manifestations was unquestionable. Unexpectedly, but only at first glance, we are now witnessing significant changes. The proportion of de novo variants has decreased, and the corresponding increase in the number of familial cases implies that CALM variant–positive subjects can reach adulthood and are able to procreate, a marker of reduced clinical severity. Indeed, we are now identifying an increasing number of asymptomatic genotype-positive individuals. There is mounting evidence for either a better response to therapy or a diminishing frequency of arrhythmic events as the patients grow older.
This ‘changing pattern’ does not come as a complete surprise to those who remember the early days of LQTS. As shown in 1975,13 the mortality among the initial LQTS patients was staggering. Indeed, among the first 100 patients, almost 50% died suddenly at a very young age, until the introduction of antiadrenergic therapies reversed the situation. This is what happens with rare diseases. At first, the picture is dominated by the most severe cases who are the ones initially diagnosed; only when diagnosis becomes more common, milder cases are identified. In the first few years after 20121 and 2013,2 only a few tertiary centres, with their own genetic research laboratory, would test for CALM genetic variants in the few patients with dramatic phenotypes who had previously tested negative for LQTS-causing variants. Thus, it is obvious that the initial cases are severe. After our initial report of the ICalmR,6 the CALM genes were introduced in the commercial genetic panels, and a recent consensus document9 on the state of genetic testing for cardiac diseases stated that CALM genes should be screened in all LQTS patients. This caused a striking change as CALM genetic variants are now found even in patients not suspected to have a calmodulinopathy. This is how and why milder phenotypes are now being identified.
By the same token, the ‘typical presentation’, based on the initial cases, is now changing. It should be remembered that for several years, it was thought that the arrhythmic episodes of LQTS would always occur during sympathetic activation, such as physical or emotional stress.13 It was only in the late 1980s that in some LQTS families, sudden death was reported during sleep, a fact difficult to be understood at that time.14 However, 20 years later, we realized that sudden deaths during sleep were related to pathogenic variants in the cardiac sodium channel gene SCN5A.15 Similarly, when in 1979 it was proposed that some patients might have had LQTS despite a normal QT interval, the medical community reacted as against heresy.16 It took 20 years to prove it,17 and now everyone gives for granted the existence of genotype-positive and phenotype-negative LQTS patients. Thus, it should by now be clear that when dealing with the initial cases of a rare disease, great caution is required before making unwarranted statements about what are ‘typical or atypical’ clinical presentations.
Clinical manifestations and genotypes
The varied phenotypes associated with genetic variants in the CALM genes are confirmed and even accentuated by data from this new, larger cohort. Indeed, the black-and-white leaves room for a lot of grey. At this time, based on the genotype, one cannot always predict the phenotype: e.g. asymptomatic CALM variant–positive subjects may have children with a severe phenotype. Important for the families of the patients, the number of cases with a milder phenotype and greater probability of survival is increasing, in parallel with the increase in familial cases. These considerations have important implications also for the interpretation and classification of CALM genetic variants in accordance with the ACMG guidelines.11 The calmodulinopathy disease phenotypic spectrum has expanded, including complex forms and phenotypes ranging from ‘less severe’ to asymptomatic. This calls for caution concerning how the ACMG criteria will be applied in the future. For example, a CALM variant in an asymptomatic or mildly symptomatic individual should no longer be immediately dismissed as having a disease association. We now know that in calmodulinopathy, penetrance may be incomplete, and expressivity may be variable, albeit to a lesser extent than other arrhythmogenic diseases of genetic origin. Within the ICalmR, the CALM-LQTS subgroup continues to predominate. Even with the increased identification of FMs with a less severe phenotype, this subgroup remains the most severe with a very early onset of symptoms.
Most variants associated with LQTS are mapping to the EF-hand motifs III and IV, especially to those amino acid residues responsible for calcium binding. This is in line with the prevalent mechanistic explanation for CALM mutation–associated LQTS: a mutation-induced reduction in calmodulin C-domain (EF-hands III and IV, encoded by exons 5 and 6) Ca2+ binding leads to a reduction of the calmodulin-mediated Cav1.2 Ca2+-dependent inactivation,18 causing a delayed membrane repolarization and therefore a prolongation of QT intervals.
We now identify an increase in the LQTS/CPVT overlap group where the apparent change over time from ‘more LQTS’ to ‘more CPVT’ is forcing a constant reassessment of the diagnosis and calls for caution before hastily pigeonholing these patients in one or the other category. This clinical observation is supported by the functional evidence that CALM-LQTS variants also lead to dysregulation of RyR2 function.19 We coined the term ‘uncertain diagnosis’ when we realized that in some cases, the probability of LQTS or CPVT or IVF was equal and diagnosis with certainty was not possible and that sudden death victims could belong to one or another group. This is exemplified by a familial case in which the cardiac arrest of one child was initially interpreted as IVF, given the unexplained sudden death of a sibling, but was modified when later the survivor presented findings typical of CPVT.
Clinical management
As to management, the current numbers do not allow drawing firm conclusions but there is some encouraging news. Two infants with very frequent cardiac arrests and appropriate ICD shocks did eventually stabilize despite only minor therapeutic adjustments, remained symptom-free for several years, and are now teenagers. This may suggest an increase in cardiac electrical stability over time.
The analysis of all SCD cases showed that none were on full-dose combination therapy of nadolol or propranolol, plus mexiletine or flecainide, and LCSD. Based on this observation and on our long-standing clinical experience with channelopathies, we consider it reasonable—despite the lack of clear evidence—to recommend the full-dose triple therapy for symptomatic patients with a predominantly cardiac calmodulinopathy. As to sodium channel blockers, which continue to be used in several patients, it is admittedly difficult to state whether they are indeed useful.
CALM complex phenotypes
The expansion of ICalmR has allowed the observation of a number of concomitant cardiac and extracardiac phenotypes. Specifically, coexisting cardiomyopathies and/or CHDs are not uncommon, as well as ACA-independent neurodevelopmental and/or neurological abnormalities (including autism, ADHD, intellectual disability, epilepsy, and seizures). There are patients who present with both abnormalities, and we observed one CALM-related neurological/neurodevelopmental phenotype in the absence of any cardiac features. Indeed, human arrhythmic CALM variants have now been demonstrated to both cause arrhythmic behaviour and affect neuronal function in Caenorhabditis elegans,20 supporting a fundamental evolutionary critical link between CALM integrity and optimal neurodevelopment.
Most cases belong to the CALM-LQTS subgroup in which impairment of calcium-dependent inactivation (CDI) of the cardiac Ca2+ channel Cav1.2 is the main underlying pathophysiological mechanism.18 These observations remind us of Timothy syndrome (TS), a syndromic, malignant arrhythmia condition caused by mutations in the CACNA1C-encoded Cav1.2 Ca2+ channel.21,22 Although initially described as a multisystem arrhythmia syndrome,21,22 the phenotypic spectrum of CACNA1C-TS has expanded to include forms with complex (arrhythmic and structural) cardiac-only phenotypes,23 arrhythmia-only phenotypes in the absence of TS features,24 and also primary neurodevelopmental/neurological phenotypes in the complete absence of a cardiac phenotype,25 as we are now observing in the ICalmR cohort.
The phenotypic similarities between calmodulinopathy and CACNA1C-associated disease are relevant because CaM and Cav1.2 are natural partners, with the latter being a major binding and modulation target of CaM. Equally important, both the Cav1.2 channel and CaM are also expressed in the brain,26,27 with the former regulating neuronal Ca2+ transients, while CaM’s binding to the C-terminal tail of Cav1.2 results in transcriptional regulation both of a subset of neuronal genes and of Cav1.2 itself in cardiomyocytes.26,28 In the latter, abnormal Ca2+ handling, also through impaired Cav1.2 function, has been described in various models of HCM.29 The proper regulation of Ca2+ and the correct functioning of Cav1.2 are probably crucial also for cardiac development, as demonstrated by a perturbation in Ca2+-handling pathways in cardiac tissues of children with different forms of CHDs.30
The above considerations and others31,32 imply that calmodulinopathy may be also syndromic with two important clinical implications for CALM patients. First, cardiac structural abnormalities should always be assessed because their presence could require a change in therapeutic strategies and could impact prognosis, as shown by two cases who died in the first year of life of heart failure. Second, detailed neurological evaluations should always be performed, and the possibility that neurological phenotypes may arise due to CALM mutations should be considered by neurologists in the differential diagnosis of patients coming to medical attention due to apparently isolated neurological/neurodevelopmental phenotypes.
Unexpected implications of ICalmR
The devastating psychological consequences of sudden infant death syndrome (SIDS) are often compounded by doubts about possible infanticide, at times with reason, at times without it.33,34 When dealing with rare conditions, significant progress has resulted from prospective registries.4,5 Sometimes, this progress has unforeseen consequences such as the one related to the fact that in 2003, an Australian woman was convicted by a jury of smothering and killing her four children over a 10-year period. Each child died suddenly and unexpectedly during sleep below age 2. In 2019, exome sequencing revealed that two of the children had a novel CALM variant (CALM2-p.G114R), inherited from the mother, and in 2021 we showed that this variant impairs CALM’s ability to bind calcium and to regulate Cav1.2 and RyR2.35 The deleterious effects of p.G114R are similar to those conferred by p.G114W and p.N98S, which are considered arrhythmogenic and to cause SCD in children. Thus, calmodulinopathy emerged as a possible explanation for a natural cause of their deaths.33 The subsequent events have been the object of wide international interest36,37 and are outside the scope of our present study (see Addendum).
The take-home message is that the progress in genetic discoveries, when integrated with carefully collected clinical data and presented as disease registries, independent of the more obvious implications for clinical management, has the potential of playing a major role also in the correct assessment of recurring familial sudden deaths of infants and children.
Limitations
This study has the unavoidable limitations inherent in any collaborative prospective registry. The multiplicity of data sources could favour non-uniformity of diagnostic and therapeutic approaches, potentially influencing data precision and outcomes. One limitation, though, is specific to the present registry and concerns the possibility of underestimating the frequency of pure or predominant neurological abnormalities. Indeed, so far, the interest for calmodulinopathy has been largely restricted to cardiology, and almost all the patients enrolled in the ICalmR were reported by cardiologists. Neurodevelopmental problems have infrequently been linked to CALM mutations, despite CALM being expressed in the brain, and a patient seen in a neurology centre is unlikely to be enrolled in ICalmR. The picture may change if and when neurologists will more frequently search CALM mutations in their patients and when there will be more crosstalk between neurology and cardiology.
Conclusions
The clinical phenotypes caused by CALM mutations are expanding, as more patients are being identified and carefully observed. Indeed, the number of intermediate forms including syndromic presentations is increasing. Despite the growing observation of mild cases and of patients responding to combination therapy, calmodulinopathy remains a most severe disease with an appalling frequency of very early sudden deaths, representing an intriguing challenge for the clinician and for the basic scientist. As pioneered 45 years ago for LQTS,4,5 our present report is impressively confirming that whenever a new and rare disease is identified, well-structured international registries, designed and handled by experienced investigators, provide the best resource of critical knowledge.
Addendum
On 6 June 2023, Judge Tom Bathurst, former Chief Justice of NSW, decided that ‘reasonable doubt’ existed and Mrs. Kathleen Folbigg was liberated after 20 years in jail, having been accused of having murdered her 4 infant children. Besides the depositions of several international expert witnesses, a major role in the reversal of the 2003 sentence to 40 years in prison was played by the evidence on the genotype-phenotype correlation provided by the present data of the ICamR reported to the Court.
Supplementary Material
Acknowledgements
We thank Pinuccia De Tomasi for expert editorial support and Alice Ghidoni for initial support in genetic variant classification. Special thanks are due to all the families represented in this registry. L.C., P.J.S., C.S., and F.D. are proud members of the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart (ERN GUARD-Heart). The open access charge was funded by BIBLIOSAN.
Contributor Information
Lia Crotti, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22, 20135 Milan, Italy; Department of Medicine and Surgery, University of Milano-Bicocca, Piazza dell'Ateneo Nuovo, 1, 20126 Milan, Italy.
Carla Spazzolini, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22, 20135 Milan, Italy.
Mette Nyegaard, Department of Health Science and Technology, Aalborg University, Aalborg, Denmark.
Michael T Overgaard, Department of Chemistry and Bioscience, Aalborg University, Aalborg, Denmark.
Maria-Christina Kotta, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22, 20135 Milan, Italy.
Federica Dagradi, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22, 20135 Milan, Italy.
Luca Sala, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22, 20135 Milan, Italy; Department of Biotechnology and Biosciences, University of Milano-Bicocca, Milan, Italy.
Takeshi Aiba, Division of Arrhythmia, National Cerebral and Cardiovascular Center, Suita, Japan.
Mark D Ayers, Department of Pediatrics, Division of Pediatric Cardiology, Indiana University School of Medicine, Indianapolis, IN, USA.
Anwar Baban, Member of the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart: ERN GUARD-Heart; Pediatric Cardiology and Arrhythmia/Syncope Units, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy.
Julien Barc, Université de Nantes, CHU Nantes, CNRS, INSERM, L’institut du Thorax, Nantes, France.
Cheyenne M Beach, Pediatric Cardiology, Yale School of Medicine, New Haven, CT, USA.
Elijah R Behr, Cardiology Section, Institute of Molecular and Clinical Sciences, St George’s University of London and Cardiovascular Clinical Academic Group, St George’s University Hospitals NHS Foundation Trust, UK.
J Martijn Bos, Departments of Cardiovascular Medicine, Pediatric and Adolescent Medicine, and Molecular Pharmacology & Experimental Therapeutics, Division of Heart Rhythm Services and Pediatric Cardiology, Windland Smith Rice Sudden Death Genomics Laboratory, Mayo Clinic, 200 First Street SW, Rochester, MN 55905, USA.
Marina Cerrone, Inherited Arrhythmias Clinic, Leon H. Charney Division of Cardiology, NYU Grossmann School of Medicine, New York, NY, USA.
Peter Covi, Department of Pediatrics, University Hospital Salzburg, Paracelsus Medical University, Salzburg, Austria.
Bettina Cuneo, Department of Pediatrics, Section of Cardiology, University of Denver School of Medicine, Aurora, CO, USA.
Isabelle Denjoy, Centre de Référence Maladies Cardiaques Héréditaires Filière Cardiogen, Département de Rythmologie, Groupe Hospitalier Bichat-Claude Bernard, Paris, France.
Birgit Donner, Kardiologie, Universitäts-Kinderspital beider Basel (UKBB), Basel, Switzerland.
Adrienne Elbert, Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada.
Håkan Eliasson, Department of Women’s and Children’s Health, Karolinska Institutet, Stockholm, Sweden; Pediatric Cardiology C8:34, Karolinska University Hospital, Stockholm, Sweden.
Susan P Etheridge, Department of Pediatrics, Division of Pediatric Cardiology, University of Utah and Primary Children’s Hospital, Salt Lake City, UT, USA.
Megumi Fukuyama, Department of Cardiovascular Medicine, Shiga University of Medical Science, Shiga, Japan.
Francesca Girolami, Cardiology Unit, Meyer Children’s Hospital, Florence, Italy.
Robert Hamilton, Division of Cardiology, The Hospital for Sick Children (SickKids), Toronto, ON, Canada.
Minoru Horie, Department of Cardiovascular Medicine, Shiga University of Medical Science, Shiga, Japan.
Maria Iascone, Laboratorio di Genetica Medica, ASST Papa Giovanni XXIII, Bergamo, Italy.
Juan Jiménez Jaimez, Hospital Universitario Virgen de las Nieves, Instituto de Investigación Biosanitario IBS Granada, Spain.
Henrik Kjærulf Jensen, Department of Cardiology, Department of Clinical Medicine, Aarhus University Hospital, Aarhus University, K-8200 Aarhus N, Denmark.
Prince J Kannankeril, Department of Pediatrics, Vanderbilt University Medical Center, Nashville, TN, USA.
Juan P Kaski, Centre for Paediatric Inherited and Rare Cardiovascular Disease, Institute of Cardiovascular Science, University College London, Zayed Centre for Research into Rare Disease in Childhood, London, UK; Centre for Inherited Cardiovascular Diseases, Great Ormond Street Hospital, London, UK.
Naomasa Makita, National Cerebral and Cardiovascular Center, Suita, Japan; Sapporo Teishinkai Hospital, Sapporo, Japan.
Carmen Muñoz-Esparza, Member of the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart: ERN GUARD-Heart; Inherited Cardiac Disease Unit, Hospital Universitario Virgen Arrixaca, Murcia, Spain.
Hans H Odland, Department of Cardiology and Pediatric Cardiology, Section for Arrhythmias, Oslo University Hospital, Oslo, Norway.
Seiko Ohno, Department of Bioscience and Genetics, National Cerebral and Cardiovascular Center, Osaka, Japan.
John Papagiannis, Pediatric and Adult Congenital Heart Disease, Onassis Cardiac Surgery Center, Athens, Greece.
Alessandra Pia Porretta, Unité des Troubles du Rythme, Service de Cardiologie, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland.
Christopher Prandstetter, Medical Faculty, Johannes Kepler University Linz, Linz, Austria; Department of Pediatric Cardiology, Kepler University Hospital, Linz, Austria.
Vincent Probst, Service de Cardiologie, L’institut du Thorax, CHU Nantes, Nantes, France.
Tomas Robyns, Member of the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart: ERN GUARD-Heart; Department of Cardiovascular Diseases, University Hospitals Leuven, Leuven, Belgium.
Eric Rosenthal, Evelina London Children’s Hospital, St Thomas’ Hospital, London, UK.
Ferran Rosés-Noguer, Member of the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart: ERN GUARD-Heart; Lead Paediatric Cardiology Department, Vall d’Hebron University Hospital, Barcelona, Spain; Royal Brompton Hospital NHS Guy’s and St Thomas Foundation Trust, London, UK.
Nicole Sekarski, Unité de Cardiologie Pédiatrique, Département Médico-Chirurgical de Pédiatrie, CHUV | Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland.
Anoop Singh, Department of Pediatrics, Medical College of Wisconsin, Wauwatosa, WI, USA.
Georgia Spentzou, Bristol Royal Hospital for Children, Bristol, UK.
Fridrike Stute, Department of Pediatric Cardiology, University Heart & Vascular Center Hamburg, Hamburg, Germany.
Jacob Tfelt-Hansen, Member of the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart: ERN GUARD-Heart; Section of Genetics, Department of Forensic Medicine, Faculty of Medical Sciences, University of Copenhagen, Denmark; Department of Cardiology, The Heart Centre, Copenhagen University Hospital, Rigshospitalet, Copenhagen, Denmark.
Jan Till, Royal Brompton Hospital NHS Guy’s and St Thomas Foundation Trust, London, UK.
Kathryn E Tobert, Departments of Cardiovascular Medicine, Pediatric and Adolescent Medicine, and Molecular Pharmacology & Experimental Therapeutics, Division of Heart Rhythm Services and Pediatric Cardiology, Windland Smith Rice Sudden Death Genomics Laboratory, Mayo Clinic, 200 First Street SW, Rochester, MN 55905, USA.
Jeffrey M Vinocur, Pediatric Cardiology, Yale School of Medicine, New Haven, CT, USA.
Gregory Webster, Ann & Robert H. Lurie Children’s Hospital of Chicago, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Arthur A M Wilde, Member of the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart: ERN GUARD-Heart; Department of Cardiology, Amsterdam UMC Location University of Amsterdam, Amsterdam, The Netherlands; Amsterdam Cardiovascular Sciences, Heart Failure and Arrhythmias, Amsterdam, The Netherlands.
Cordula M Wolf, Center for Rare Congenital Heart Diseases, Department of Congenital Heart Defects and Pediatric Cardiology, German Heart Center Munich, Technical University Munich, School of Medicine & Health, Munich, Germany.
Michael J Ackerman, Departments of Cardiovascular Medicine, Pediatric and Adolescent Medicine, and Molecular Pharmacology & Experimental Therapeutics, Division of Heart Rhythm Services and Pediatric Cardiology, Windland Smith Rice Sudden Death Genomics Laboratory, Mayo Clinic, 200 First Street SW, Rochester, MN 55905, USA.
Peter J Schwartz, Istituto Auxologico Italiano IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Via Pier Lombardo 22, 20135 Milan, Italy.
Supplementary data
Supplementary data are available at European Heart Journal online.
Declarations
Disclosure of Interest
All authors declare no conflict of interest for this contribution.
Data Availability
Data are available upon reasonable request.
Funding
The ICalmR is one of the registries supported by ERN GUARD-Heart. This research was supported by the Italian Ministry of Health Ricerca Corrente ‘Registro internazionale delle calmodulinopatie’ to L.C., F.D., P.J.S., M.C.K., and C.S.; by the 2019-ATESP-0045 Fondo di Ateneo Quota Competitiva to L.C.; and partially by the Fondation Leducq grant 18CVD05 ‘Towards Precision Medicine with Human iPSCs for Cardiac Channelopathies’ to L.C., M.-C.K., L.S., and P.J.S. J.B., L.C., and P.J.S. were partially supported by the European Joint Programme on Rare Diseases: LQTS-NEXT grant. R.H. was supported by the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada, The Labatt Family Heart Centre, the Cartwright Family Fellowship, the Carter Heart Arrhythmia Trainee Fund and the Caitlin Elizabeth Morris fund. J.P.K. was supported by the Medical Research Council (MRC) Clinical Academic Research Partnership (CARP) Award (MR/T024062/1). G.R.W. was supported by an NIH K23HL130554 grant. A.A.M.W. was supported by the Netherlands Cardiovascular Research Initiative (CVON PREDICT-2). M.J.A. was supported in part by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program. M.T.O. was supported in part by the Danish Independent Research Council (Grant no. 2032–00333B) and the Lundbeck Foundation (Grant no. R324-2019-1933). H.K.J. was supported by the Novo Nordisk Foundation (Grant NNF 18OC0031258).
Ethical Approval
See Methods
Pre-registered Clinical Trial Number
None supplied.
References
- 1. Nyegaard M, Overgaard MT, Søndergaard MT, Vranas M, Behr ER, Hildebrandt LL, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet 2012;91:703–712. 10.1016/j.ajhg.2012.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013;127:1009–1017. 10.1161/CIRCULATIONAHA.112.001216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marsman RF, Barc J, Beekman L, Alders M, Dooijes D, Van Den Wijngaard A, et al. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol 2014;63:259–266. 10.1016/j.jacc.2013.07.091 [DOI] [PubMed] [Google Scholar]
- 4. Schwartz PJ. The idiopathic long QT syndrome: the need for a prospective registry. Eur Heart J 1983;4:529–531. 10.1093/oxfordjournals.eurheartj.a061517 [DOI] [PubMed] [Google Scholar]
- 5. Moss AJ, Schwartz PJ. 25th anniversary of the International Long-QT Syndrome Registry: an ongoing quest to uncover the secrets of long-QT syndrome. Circulation 2005;111:1199–1201. 10.1161/01.CIR.0000157069.91834 [DOI] [PubMed] [Google Scholar]
- 6. Crotti L, Spazzolini C, Tester DJ, Ghidoni A, Baruteau AE, Beckmann BM, et al. Calmodulin mutations and life-threatening cardiac arrhythmias: insights from the International Calmodulinopathy Registry. Eur Heart J 2019;40:2964–2975. 10.1093/eurheartj/ehz311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 2020;141:418–428. 10.1161/CIRCULATIONAHA.119.043132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walsh R, Adler A, Amin AS, Abiusi E, Care M, Bikker H, et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur Heart J 2022;43:1500–1510. 10.1093/eurheartj/ehab687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilde AAM, Semsarian C, Márquez MF, Shamloo AS, Ackerman MJ, Ashley EA, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) . Expert consensus statement on the state of genetic testing for cardiac diseases. Europace 2022;24:1307–1367. 10.1093/europace/euac030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kato K, Isbell HM, Fressart V, Denjoy I, Debbiche A, Itoh H, et al. Novel CALM3 variant causing calmodulinopathy with variable expressivity in a 4-generation family. Circ Arrhythm Electrophysiol 2022;15:e010572. 10.1161/CIRCEP.121.010572 [DOI] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Makita N, Yagihara N, Crotti L, Johnson CN, Beckmann B-M, Roh MS, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet 2014;7:466–474. 10.1161/CIRCGENETICS.113.000459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J 1975;89:378–390. 10.1016/0002-8703(75)90089-7 [DOI] [PubMed] [Google Scholar]
- 14. Schwartz PJ, Locati E, Priori SG, Zaza A. The long Q-T syndrome. In: Zipes DP and Jalife J (eds.), Cardiac Electrophysiology: From Cell to Bedside: Philadelphia: W.B. Saunders Co., 1990, 589–605. [Google Scholar]
- 15. Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001;103:89–95. 10.1161/01.cir.103.1.89 [DOI] [PubMed] [Google Scholar]
- 16. Schwartz PJ. The long QT syndrome. In: Kulbertus HE, Wellens HJJ (eds.), Sudden Death: The Hague: M Nijhoff Publishers bv; 1980, 358–378. [Google Scholar]
- 17. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long QT syndrome. Clinical impact. Circulation 1999;99:529–533. 10.1161/01.CIR.99.4.529 [DOI] [PubMed] [Google Scholar]
- 18. Limpitikul WB, Dick IE, Joshi-Mukherjee R, Overgaard MT, George ALJR, Yue DT. Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol 2014;74:115–124. 10.1016/j.yjmcc.2014.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Søndergaard MT, Liu Y, Brohus M, Guo W, Nani A, Carvajal C, et al. Diminished inhibition and facilitated activation of RyR2-mediated Ca2+ release is a common defect of arrhythmogenic calmodulin mutations. FEBS J 2019;286:4554–4578. 10.1111/febs.14969 [DOI] [PubMed] [Google Scholar]
- 20. Jensen HH, Frantzen MT, Wesseltoft JL, Busuioc AO, Møller KV, Brohus M, et al. Human calmodulin mutations cause arrhythmia and affect neuronal function in C. elegans. Hum Mol Genet 2023;32:2068–2083. 10.1093/hmg/ddad042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004;119:19–31. 10.1016/j.cell.2004.09.011 [DOI] [PubMed] [Google Scholar]
- 22. Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A 2005;102:8089–8096; discussion 8086–8088. 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boczek NJ, Ye D, Jin F, Tester DJ, Huseby A, Bos JM, et al. Identification and functional characterization of a novel CACNA1C-mediated cardiac disorder characterized by prolonged QT intervals with hypertrophic cardiomyopathy, congenital heart defects, and sudden cardiac death. Circ Arrhythm Electrophysiol 2015;8:1122–1132. 10.1161/CIRCEP.115.002745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boczek NJ, Best JM, Tester DJ, Giudicessi JR, Middha S, Evans JM, et al. Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ Cardiovasc Genet 2013;6:279–289. 10.1161/CIRCGENETICS.113.000138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodan LH, Spillmann RC, Kurata HT, Lamothe SM, Maghera J, Jamra RA, et al. Phenotypic expansion of CACNA1C-associated disorders to include isolated neurological manifestations. Genet Med 2021;23:1922–1932. 10.1038/s41436-021-01232-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 2011;3:a003947. 10.1101/cshperspect.a003947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. GTEx Consortium . The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell 2006;127:591–606. 10.1016/j.cell.2006.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han L, Li Y, Tchao J, Kaplan AD, Lin B, Li Y, et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res 2014;104:258–269. 10.1093/cvr/cvu205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu Y, Feng W, Zhang H, Li S, Wang D, Pan X, et al. Ca²+-regulatory proteins in cardiomyocytes from the right ventricle in children with congenital heart disease. J Transl Med 2012;10:67. 10.1186/1479-5876-10-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jensen HH, Brohus M, Nyegaard M, Overgaard MT. Human calmodulin mutations. Front Mol Neurosci 2018;11:396. 10.3389/fnmol.2018.00396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nyegaard M, Overgaard MT. The International Calmodulinopathy Registry: recording the diverse phenotypic spectrum of un-CALM hearts. Eur Heart J 2019;40:2976–2978. 10.1093/eurheartj/ehz463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. American Academy of Pediatrics, Hymel KP; Committee on Child Abuse and Neglect; National Association of Medical Examiners . Distinguishing sudden infant death syndrome from child abuse fatalities. Pediatrics 2006;118:421–427. 10.1542/peds.2006-1245 [DOI] [PubMed] [Google Scholar]
- 34. Schwartz L Z. The origin of maternal feelings of guilt in SIDS. Relationship with the normal psychological reactions of maternity. Ann N Y Acad Sci 1988;533:132–144. 10.1111/j.1749-6632.1988.tb37242.x [DOI] [PubMed] [Google Scholar]
- 35. Brohus M, Arsov T, Wallace DA, Jensen HH, Nyegaard M, Crotti L, et al. Infanticide vs inherited cardiac arrhythmias. Europace 2021;23:441–450. 10.1093/europace/euaa272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cave D. She was imprisoned for killing her 4 children. But was it their genes all along? New York Times March 8, 2021: https://www.nytimes.com/2021/03/08/world/australia/kathleen-folbigg-child-murder-genetics.html.
- 37. Phillips N. She was convicted of killing her four children. Could a gene mutation set her free? Nature 2022;611:218–223. 10.1038/d41586-022-03577-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon reasonable request.