

Significance
Our study identified a dysregulation and hyperactivity of the critical metabolic process of the central carbon and energy metabolism, and metabolic flux related to the amino acid metabolism during the acute progressive phases of infection can promote the exhausted phenotype upon recovery leading to postviral fatigue syndrome in CCHFV (Crimean-Congo hemorrhagic fever virus) infection.
Keywords: Crimean-Congo hemorrhagic fever virus, genome-scale metabolic models, post viral fatigue
Abstract
Crimean-Congo hemorrhagic fever (CCHF) caused by CCHF virus (CCHFV) is one of the epidemic-prone diseases prioritized by the World Health Organisation as public health emergency with an urgent need for accelerated research. The trajectory of host response against CCHFV is multifarious and remains unknown. Here, we reported the temporal spectrum of pathogenesis following the CCHFV infection using genome-wide blood transcriptomics analysis followed by advanced systems biology analysis, temporal immune-pathogenic alterations, and context-specific progressive and postinfection genome-scale metabolic models (GSMM) on samples collected during the acute (T0), early convalescent (T1), and convalescent-phase (T2). The interplay between the retinoic acid-inducible gene-I-like/nucleotide-binding oligomerization domain-like receptor and tumor necrosis factor signaling governed the trajectory of antiviral immune responses. The rearrangement of intracellular metabolic fluxes toward the amino acid metabolism and metabolic shift toward oxidative phosphorylation and fatty acid oxidation during acute CCHFV infection determine the pathogenicity. The upregulation of the tricarboxylic acid cycle during CCHFV infection, compared to the noninfected healthy control and between the severity groups, indicated an increased energy demand and cellular stress. The upregulation of glycolysis and pyruvate metabolism potentiated energy generation through alternative pathways associated with the severity of the infection. The downregulation of metabolic processes at the convalescent phase identified by blood cell transcriptomics and single-cell type proteomics of five immune cells (CD4+ and CD8+ T cells, CD14+ monocytes, B cells, and NK cells) potentially leads to metabolic rewiring through the recovery due to hyperactivity during the acute phase leading to post-viral fatigue syndrome.
Tick-borne Crimean-Congo hemorrhagic fever virus (CCHFV) is a global threat for its pandemic potential. The virus is presently endemic in 49 countries in Africa, the Middle East, and Asia, and sporadic reports in European countries illustrate its increasing risk in the context of climate change. Even since January 2022, an unprecedented increase in Crimean-Congo Hemorrhagic Fever (CCHF) has been observed more than that in previous years (1, 2). Due to the biosafety level (BSL) 4 requirement for handling the CCHFV-infected materials and the high case fatality ratio (CFR) (10 to 50%) (3), little is known about the pathogenesis and host immune response dynamics during progressive CCHFV infection. Previously, we reported on the cross-sectional system-level transcriptomics profile in CCHFV-infected patients during the acute phase of infection (4). While this unique study identified the systemic alteration of host response, characterization of the early immune response against the CCHFV infection was not possible due to the nonavailability of the longitudinal samples. Though cross-sectional studies provide a snapshot of the viral pathogenesis and host immune response, it fails to provide an understanding of the dynamics of viral replication, immune response, and disease progression. Identifying the system-level host response using the peripheral blood that contains critical immune system components following infection provides a global view of how the host immune system mounts a response to the virus, attempting to clear it from the body.
Among the CCHFV-endemic countries, the CFR in Turkey is relatively low [5%] (5), with a robust public health system, expert clinicians, and good access to patient care providing a unique opportunity to study early immune response against the CCHFV. We hypothesized that the dynamic nature of the alteration of the transcriptome during the early phase of the infection to the recovery captures the entire temporal spectrum of virus-mediated disease dynamics and pathogenesis and responsible for postviral fatigue (PVF) upon recovery.
Here, we performed temporal genome-wide transcriptomics analysis using whole blood from a CCHF-cohort collected at Sivas Cumhuriyet University Hospital (SCUH), Turkey, from confirmed CCHF patients, on the hospitalization day (T0, acute phase), at discharge (T1, early convalescent phase), and median 30 d since the onset of the symptoms (T2, convalescent phase). Utilizing the advanced network-based systems biology analysis, digital cell quantification (DCQ), and temporal pathogenic alterations by time series clustering, we report on fundamental immune responses and metabolic rewiring as a consequence of infection that leads to the pathology of CCHF. We further developed the system-level context-specific genome-scale metabolic model (GSMM) for CCHFV infection, as recently described by us in COVID-19 (6), to identify the impact of viral infection in the metabolic process and rearrangement of intracellular metabolic fluxes during the progressive infection, which can be the target of the clinical intervention for CCHF.
Results
Characteristics of the Cohorts.
We recruited 30 hospitalized patients with CCHFV infection with a median time of 4 d (range 1 to 6 d) post symptoms onset (T0, n = 30) and followed them for 30 d. By using severity grading scores (SGS), 43% (13/30) patients were grouped into severity group 1 (SG-1, low risk of death), 30% (9/30) patients into severity group 2 (SG-2, intermediate risk of death), and 27% (8/30) patients into severity group 3 (SG-3, high risk of death). In the SG-3 group, five patients succumbed to death during hospitalization. Subsequent samples were collected from the survivors on discharge day with a median 10 (IQR: 9 to 11) days post symptoms onset (T1, n = 25) and a median duration of 30 (IQR: 28 to 33) days post symptoms onset (T2, n = 24). The patient severity grade and the sample collection are shown in Fig. 1A. Additionally, we have collected gender and age-matched CCHFV-seronegative samples healthy-control (HC, n = 30). The HCs were matched with the CCHFV patients regarding age (mean ± SD, years: 51.7 ± 17.4 vs. 50.4 ± 17.6) and gender (female: 17%). At recruitment, all patients (100%) had a history of fever and presented with nonspecific symptoms such as headache (97%), muscle ache (93%), weakness (90%), and nausea and/or vomiting (70%). Splenomegaly (77%), high-grade fever (70%), and liver enlargement (57%) were the most frequent signs among the patients (SI Appendix, Fig. S1). Bleeding was observed in half of the patients, including bleeding gum, epistaxis, hemoptysis, hematemesis, melena, hematuria, and vaginal bleeding (for females), which were specific for CCHFV infection. The cohort characteristics and clinical presentation at recruitment are provided in Dataset S1.
Fig. 1.

Temporal CCHFV pathogenesis and immune profiling: (A) Swimmer plot showing patient sampling information, including clinical features SGS, SG, and sampling time points. x axes represent days in the hospital, day 0 denotes the day of hospitalization, and y axes represent patient IDs ordered according to SG. (B) Patient-to-patient Spearman's correlation matrix clustering. The heatmap shows correlation coefficients computed using scaled TPM values of the top 5,000 varying genes based on median absolute deviation [HC (n = 22), T0 (n = 22), T1 (n = 23), T2 (n = 24)]. All correlations were highly significant (adjusted P < 0.0001) due to the high number of features used. Column and row annotations represent the patient cohort and disease severity group. (C) PCA plot visualizes the sample distribution. PCA was created using the scaled TPM values of top 5,000 varying genes based on the median absolute deviation. Ellipses represent 90% confidence space for each cohort, and point shapes correspond to the severity group. Centroid points of each cohort are computed and connected to each sample in the respective cohort with lines. (D) Bubble graph representing DCQ results. Bubble size and color gradient are relative to the median percentage of cell proportion estimated in each cohort. Asterisks represent a significant change in individual cell population from T0 to T2 by Friedman's test (adjusted P < 0.05). (E) Blood immune cell proportion using DCQ. Boxplot of DCQ results showing comparison among the four cohorts. Asterisks represent a significant change (adjusted P < 0.05) in the Mann–Whitney U test. (F) Network visualization of significant Spearman association (adjusted P < 0.001) between marker genes of cell types estimated in Fig. 1D in cohort HC, T0, T1, and T2, respectively. Bubble size corresponds to the mean TPM value. Nodes and edges represent marker genes of corresponding cell type and Spearman association, respectively.
Immune Cell Dynamics during CCHFV-Pathogenesis.
The association between patient samples (Fig. 1B) and principal component analysis (Fig. 1C) showed distinct clustering of T0 and T1. The T2 samples were clustered near HC, suggesting recovery from the infection. To avoid the bias of the cell proportions due to the use of whole blood RNAseq (6), we performed the DCQ to estimate the cell type proportions from bulk blood RNAseq data computationally and adjusted for that. Drastic changes were noticed in several immune cell trajectories but normalized at the T2 time point (Fig. 1D). The myeloid cells, neutrophils, Natural Killer (NK) cells, and to some extent, T-cells were most affected during the infection (Fig. 1E and SI Appendix, Fig. S2). The classical monocytes (CM) were expanded during the T1 and T2 timepoint, while intermediate monocytes (IM) were expanded during the T0 time point compared to the HC. On the contrary plasmacytoid dendritic cells (pDCs), neutrophils, and NK cells were expanded during the T0 time point but came down to the HC level when the infection resolved. The pairwise coexpression landscape of marker genes of specific cell types (Fig. 1F) showed an altered correlation between the signature genes in the immune cells during the acute and early convalescent phase (T0 and T1) while normalized to the level of HC (Fig. 1F) at the convalescent phase (T2). These data indicate the altered immune cell dynamics and an impaired interaction between these cell types during CCHFV acute infection but repaired back when the infection was cleared.
System-Level Temporal CCHFV-Pathogenesis Signature.
We used time series clustering using TMixClust (7) to identify the trajectories of the gene expression profile alterations. Using the likelihood ratio test, we identified 2,504 genes significantly expressed across the three time points with a high degree of confidence (adjusted P < 0.001) (Fig. 2A). To understand their temporal expression dynamics, the time series clustering of the 2,504 genes identified four expression trajectory modules (M1 to M4) (Fig. 2B and Dataset S2). The expression of the genes (n = 879) in M1 initially decreased slowly between T0 and T1 time points and then decreased rapidly (Fig. 2B). For genes (n = 531) in M4, the expressions decreased drastically at the beginning and then slowly decreased until point T2. In contrast, the genes in M2 (n = 634) gradually increased over time, while in M3 (n = 460), the gene expression was first increased at the T1 while decreasing at the T2 (Fig. 2B). To avoid bias due to the cell type alterations, we performed differential gene expression (DGE) between the T0 and T2 after adjusting for cell types differing between the time points. We observed that 99% (2,495/2,504) genes significantly differed (adjusted P < 0.05) between the time points (Fig. 2C and Dataset S3). The gene set enrichment analysis (GSEA) of the four modules identified a distinct pattern of innate and adaptive immunity and metabolic processes (Fig. 2D). The significantly enriched pathways (adjusted P < 0.1) in M1 were mainly metabolic pathways, e.g., one carbon pool by folate and pyruvate metabolism. In the M3, metabolic pathways like oxidative phosphorylation (OXPHOS), glucagon signaling pathways, and adaptive immune response pathways like Th1 and Th2 receptor signaling processes indicate a dynamic nature of the metabolic process and adaptive immune response in CCHFV-pathogenesis (Dataset S4). The gene expression changes in M4 showed that the gene expression profile for the genes associated with the innate antiviral response pathways like RIG-I-like receptor, Nucleotide oligomerization domain (NOD)-like receptor, TNF signaling, and chemokine signaling NF-kB signaling was highly exertive at the acute phase of the disease (T0). Their demand decreased rapidly as the adaptive immunity came into play (T1), and after that, a slow decrease to the normal level. No significantly enriched pathways were found for M2 genes. We also checked the acute phase (T0) virus titer and its association with the module genes; a positive correlation was observed with M2 genes, while a negative association was observed with M1 and M3 genes related to the metabolic and adaptive immune response (Fig. 2E). Interestingly no correlation was observed between the virus titer and the genes related to the antiviral response and interferon (IFN) stimulating genes (ISGs), indicating either low-level virus titer can efficiently induce effective antiviral response and that is universal across the patients or ineffective suppression of virus production by the expressed ISGs. Though there were only three patients in SG-3, individual severity-specific trajectory analyses identified lower expression of module 1 and module 3 genes compared to the SG-1 and SG-2 (Fig. 2F), and there were statistical significance differences in T2 time point (SI Appendix, Fig. S3). No apparent change in the rate of viral decline was observed at the hospital discharge despite high viral titer in the SG-1 group (SI Appendix, Fig. S4). These data indicate a robust metabolic modulation toward the central carbon metabolisms (CCMs) (e.g., pyruvate metabolism, OXPHOS) and its association with the innate antiviral response. A metabolic exhausted phenotype was observed that was associated with severity despite a few samples.
Fig. 2.
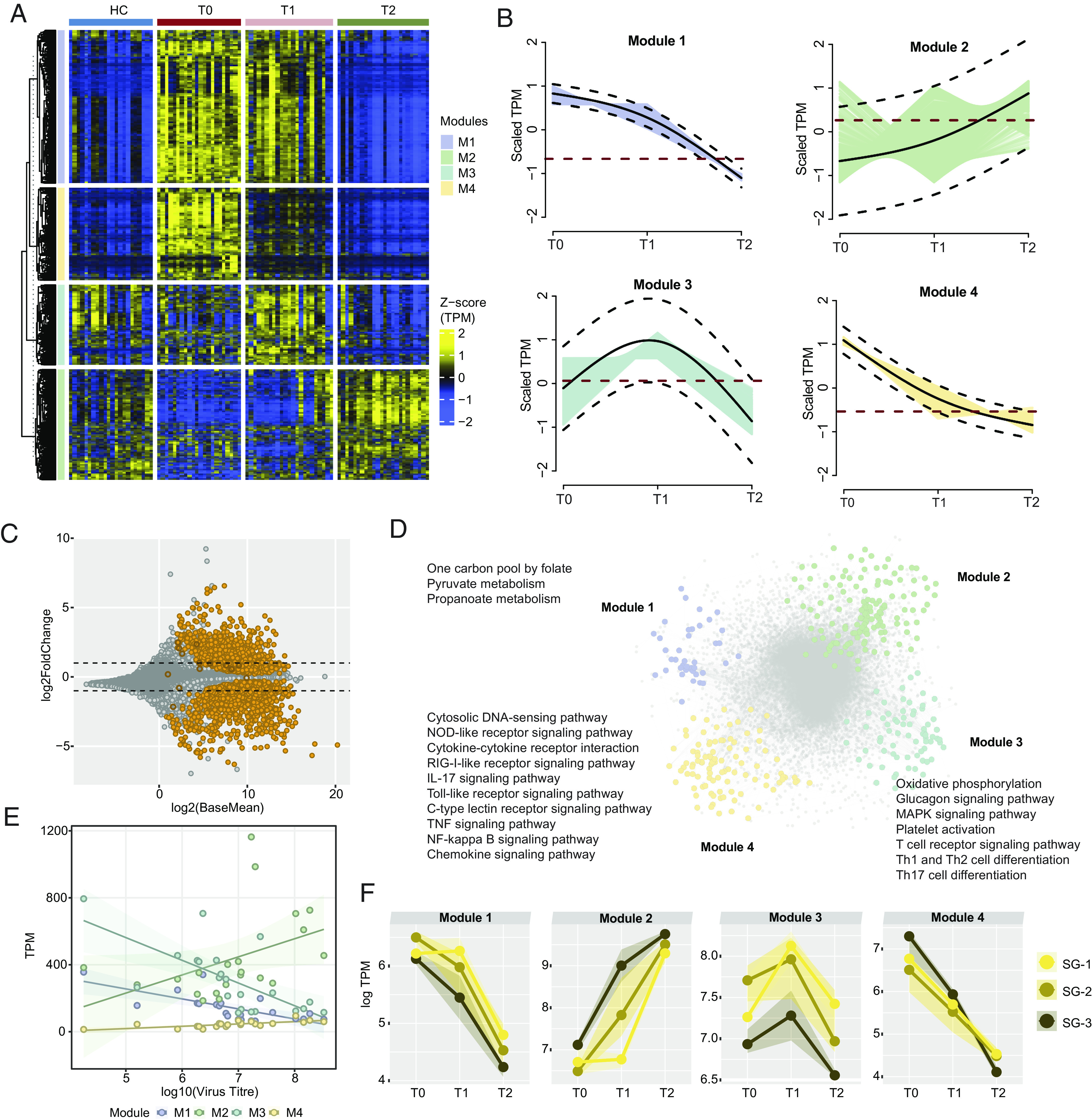
Immuno-metabolic trajectories of CCHFV-pathogenesis. (A) Heatmap of genes identified as significantly changing (adjusted P < 0.001) among the three times of infection [T0 (n = 21), T1 (n = 21), T2 (n = 21)] using likelihood ratio test. The heatmap shows TPM transformed and z-scaled expression values of the genes. Column annotation represents the cohorts, and row annotation represents four gene expression trajectory pattern modules computed using the TMixClust algorithm. (B) Modules identified using the TMixClust algorithm. Each module represents a specific trajectory of gene expression. The red dotted line denotes the mean scaled expression value of genes belonging to the corresponding module in a healthy state. (C) MA plot of DGE analysis between T0 and T2. Orange-colored circles denote genes visualized in Fig. 2A. Empty circles denote nonsignificant expression (adjusted P > 0.05). (D) Network of significant Spearman correlation (adjusted P < 0.001) at T0 between and among genes of each module shown in Fig. 2A and their first neighbors. Gray-colored nodes denote the first neighbor genes. Significantly enriched pathways (adjusted P < 0.05) are labeled beside corresponding modules. (E) Spearman association between virus titer and average expression of genes of each trajectory module shown in Fig. 2A. (F) Severity group-specific expression dynamics of each module genes over the three time points of infection. Average expression (log TPM) values of each module for each severity group are plotted on the y axis. The shaded region represents the second and third quartiles of expression.
Metabolic Perturbation Associated with Disease Severity.
Metabolic perturbation associated with disease severity. Acute viral illnesses can lead to metabolic alterations such as increased energy expenditure and dysregulation of various metabolic pathways. As we observed that the gene expression trajectory is linked with the metabolic process and innate antiviral response (Fig. 2), we aimed to identify the severity-specific alterations during the CCHFV infection. Due to the small sample size, we combined SG-2 and SG-3 (severe) and compared them with HC and SG-1 (mild). While comparing with HC, both the mild (Fig. 3A and Dataset S5) and severe groups (Fig. 3B and Dataset S6) showed upregulation of ISGs and chemokines. Interestingly, while comparing mild and severe, no specific pattern was observed, but there was upregulation of 98 genes and downregulation of 84 genes (Fig. 3C and Dataset S7). The directionally based GSEA identified upregulation of tricarboxylic acid cycle (TCA) in all the comparisons indicate an increased energy demand, altered metabolic requirements, and cellular stress (Fig. 3D). Interestingly, while comparing the mild with severe, there was upregulation of glycolysis and pyruvate metabolism that potentiated energy generation through alternative pathways due to impaired mitochondrial metabolism. Subsequent downregulation of the antiviral pathways indicates a negative correlation between the antiviral and central carbon metabolic process. The correlation between the IFN-stimulating gene with the genes involved in the TCA cycle, glycolysis, and pyruvate metabolism further supports this (Fig. 3E). Therefore, we can posit that the hyperactive metabolic process due to increased energy demands and the exhaustion of the antiviral response leads to severe disease in CCHFV patients during acute infection.
Fig. 3.
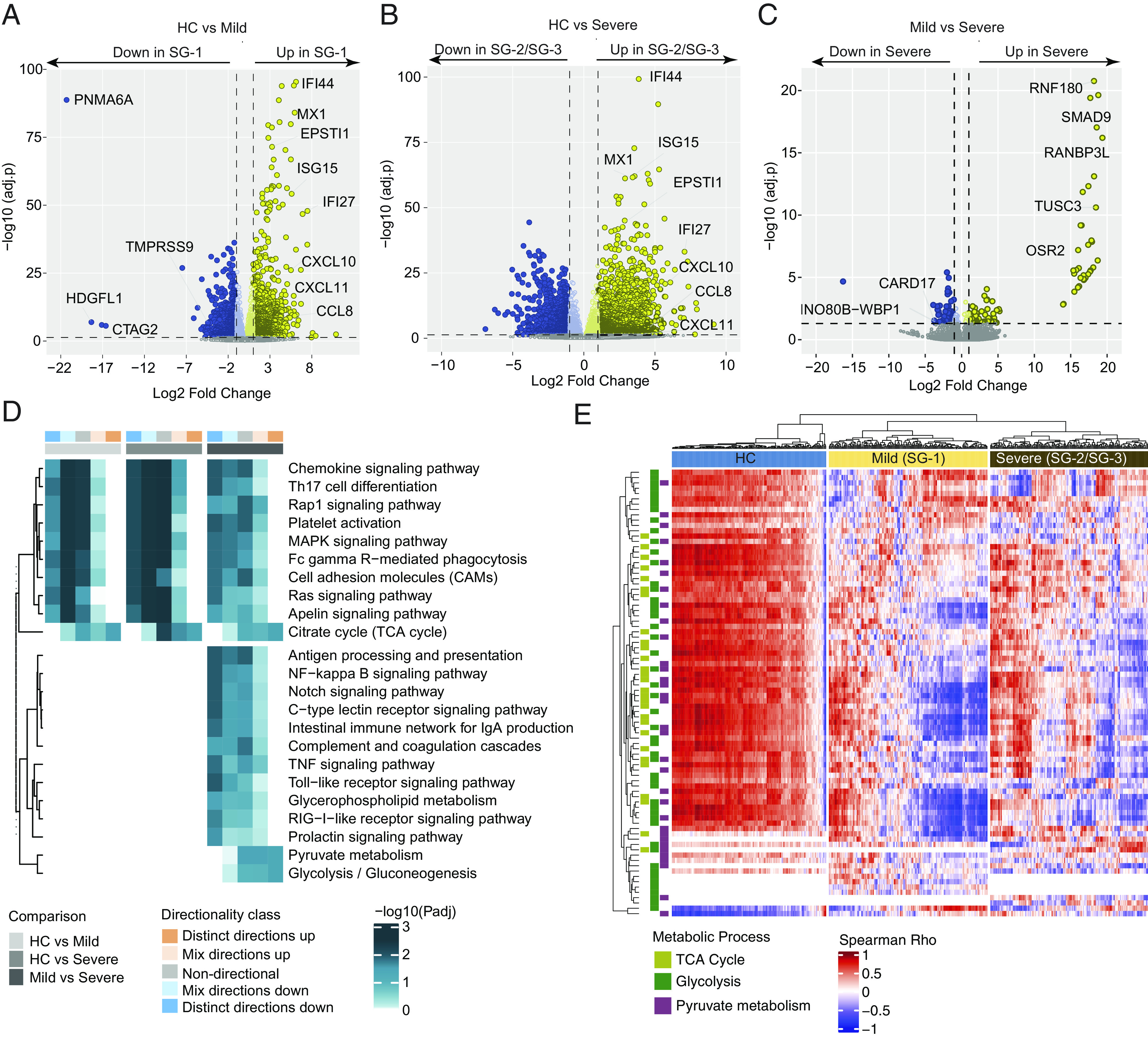
Severity-specific alterations during the CCHFV infection (A) Volcano plot showing expression changes in mild (SG-1) patients at T0 compared to HC. (B) Volcano plot showing expression changes in severe (SG-2/SG-3) patients at T0 compared to HC. (C) Volcano plot showing expression changes in severe (SG-2/SG-3) patients at T0 compared to mild (SG-1) patients at T0. Some of the essential genes related to antiviral responses are labeled. (D) Heatmap showing pathways significantly enriched (adjusted P < 0.05) in all the three pair-wise comparisons such as HC vs. mild (SG-1), HC vs. severe (SG-2/SG-3), and mild (SG-1) vs. severe (SG-2/SG-3) and uniquely enriched in mild (SG-1) vs. severe (SG-2/SG-3). The heatmap shows negative log scales adjusted P-values of different classes of directionality. Distinct directional classes of P-values calculated from gene statistics and expression direction were used to define up-regulated and down-regulated pathways. (E) Heatmap visualizing the association between IFN response genes (columns) and genes involved in the TCA cycle, glycolysis, and pyruvate metabolism pathways (rows) at T0. Column annotation represents patient groups and row annotation represents pathways. Spearman’s correlation coefficients were used to generate the heatmap.
Metabolic Models Identified Altered CCM during Acute CCHFV Infection.
We observed a robust metabolic modulation during the CCHFV infection (Fig. 2), resembling our earlier study (4) and its association with the disease severity (Fig. 3). However, the trajectories of the metabolic process were yet to be discovered. We recently developed a systemic GSMM for Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (6) and HIV-1 (8) that can identify the alteration in the metabolic reaction and flux balance. Here, we developed context-specific (time-dependent postinfection) personalized and group-level GSMM and performed flux balance analysis (FBA) to identify unique metabolic reactions during the different courses of infection. Significant altered metabolic reactions (n = 546, Dataset S8) were identified during infections and in HC (Fig. 4A) linked with CCM and transport of the metabolites related to the CCM. During the acute T0 phase, the altered flux of essential (leucine and threonine) and nonessential amino acids (AA) (arginine, alanine, and glutamine) were observed compared to T1, T2, and HC, indicating high demand of the AA during the acute phase of infection (Fig. 4B). The transports of the key TCA cycle metabolites pyruvate, isocitrate, and alpha-ketoglutarate were also found to be altered (Fig. 4C). While looking at the metabolic reactions, dysregulated pentose phosphate pathway (PPP) and metabolic shift toward fatty acid oxidation were observed during the T0 phase (Fig. 4C). The mitochondrial TCA cycle was also altered, including the additional ATP production through OXPHOS (Fig. 4C), and the AA transports were also significantly associated (adjusted P < 0.05) with metabolic reactions (Fig. 4D). Our data thus indicate a high transportation of essential and nonessential AA, a shift toward fatty acid oxidation, and additional ATP generation through OXPHOS as the hallmark during the acute phase of infection that normalized to a certain extent during the recovery phase.
Fig. 4.
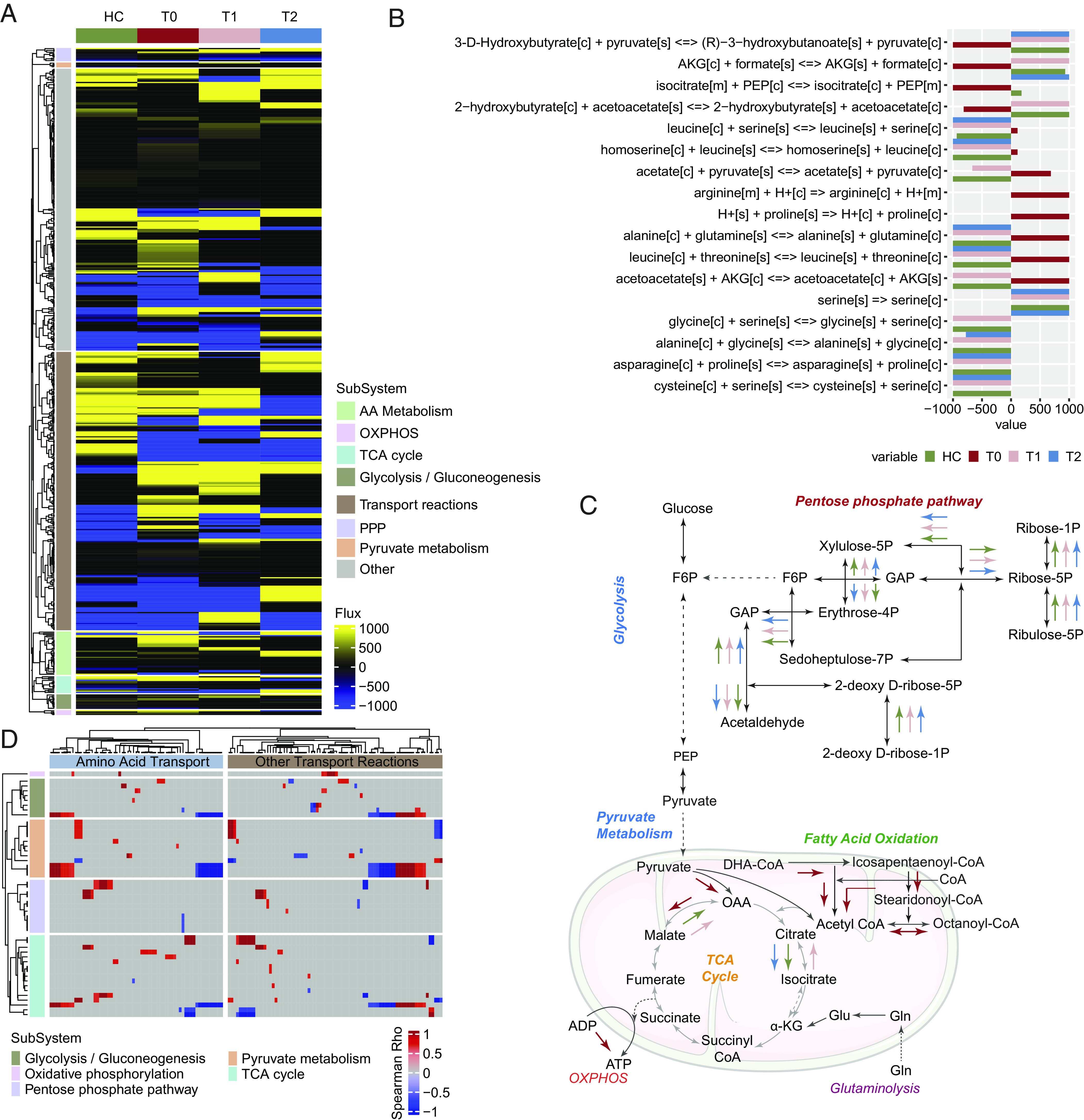
Context-specific postinfection genome-scale metabolic models in CCHFV infection (A) Heatmap of metabolic flux computed from FBA using cohort-specific genome-scale metabolic models. The heatmap shows that reactions were different among the four cohorts. The heatmap was generated using metabolic flux (mmol/h/gDCW) values calculated for each reaction. Column annotation shows the cohort, and row annotation shows metabolic subsystems corresponding to the reactions. (B) Transport reactions during the acute phase of infection differ from the other two time points and HC. Bars are colored corresponding to the cohort, and the reaction's metabolic flux value (mmol/h/gDCW) is plotted on the x axis. (C) A representative diagram of reactions that were part of various metabolic subsystems was found to be uniquely active or inactive during the acute phase. Colored arrows represent the estimated direction of metabolic turnover in the corresponding cohort. (D) Association between the metabolic flux of transport reactions and reactions part of CCM pathways at the acute stage of infection (T0). Metabolic flux values (mmol/h/gDCW) computed from individual sample-wise GSMM (personalized models) were used to compute Spearman's correlation coefficient. Column and row annotations represent transport reactions and CCM pathways, respectively.
Downregulation of Metabolic Pathways during Recovery.
We observed higher metabolic activity (Fig. 4) during the acute phase of infection that can potentially lead to metabolic insufficiency with altered signaling cascades at the convalescent phase (Fig. 4C). Therefore, to map the altered signaling cascades and metabolic rewiring following recovery, we performed DGE between the T2 and HC. The patient association matrix identified a clear separation of the HC and the T2 samples (Fig. 5A), indicating a leftover CCHFV imprint after recovery. The DGE, after adjustment for the cell type, identified upregulation of 1,853 genes and downregulation of 917 (adjusted P < 0.05). Among the genes, the persistence of high Epithelial Stromal Interaction 1 (ESPTI1), IFI27, and other inflammatory genes indicate residual inflammation (Fig. 5B and Dataset S9). The pathway enrichment analysis identified the downregulation of the metabolic processes like glycolysis, TCA cycle, OXPHOS, and related signaling cascades like PI3K-Akt/mTOR/HIF-1 signaling pathways indicative of energy maladjustment (Fig. 5C and Dataset S10). To further fingerprint which cells were metabolically impaired, we isolated CD4 T-cells, CD8 T-cells, monocytes, B-cells, and NK cells and performed LC-MS/MS-based sctProteomics (Single-Cell Type Proteomics) (Fig. 5D). Due to the low sample size and protein coverage, GSEA was performed using a consensus scoring approach (9) by incorporating the directionality of protein abundance. The GSEA identified that most metabolic pathways in CD8+ T cells, B cells, and NK cells were down-regulated at T2 (Fig. 5E and SI Appendix, Fig. S5), in line with the bulk RNAseq data, which indicated metabolic insufficiency at recovery. Interestingly, there was upregulation of the OXPHOS pathway in monocytes, and CD4+ T-cells can regulate the production of cytokines and enhanced respiratory burst with increased production of reactive oxygen species (ROS) (10). The self-reported health evaluation at the T2 time point further supported it, where 83% (20/24) of patients experienced fatigue that inhibited their daily activities after discharge. Other reported experiences, including musculoskeletal pain (75%), anorexia (50%), weight loss (50%), headache (38%), palpitation (38%), and sweating (38%), indicated PVF (Fig. 5F). Combining the blood transcriptomics and sctProteomics analysis, our proof-of-concept study suggested that metabolic rewiring during the recovery phase potentially leads to PVF in CCHF.
Fig. 5.
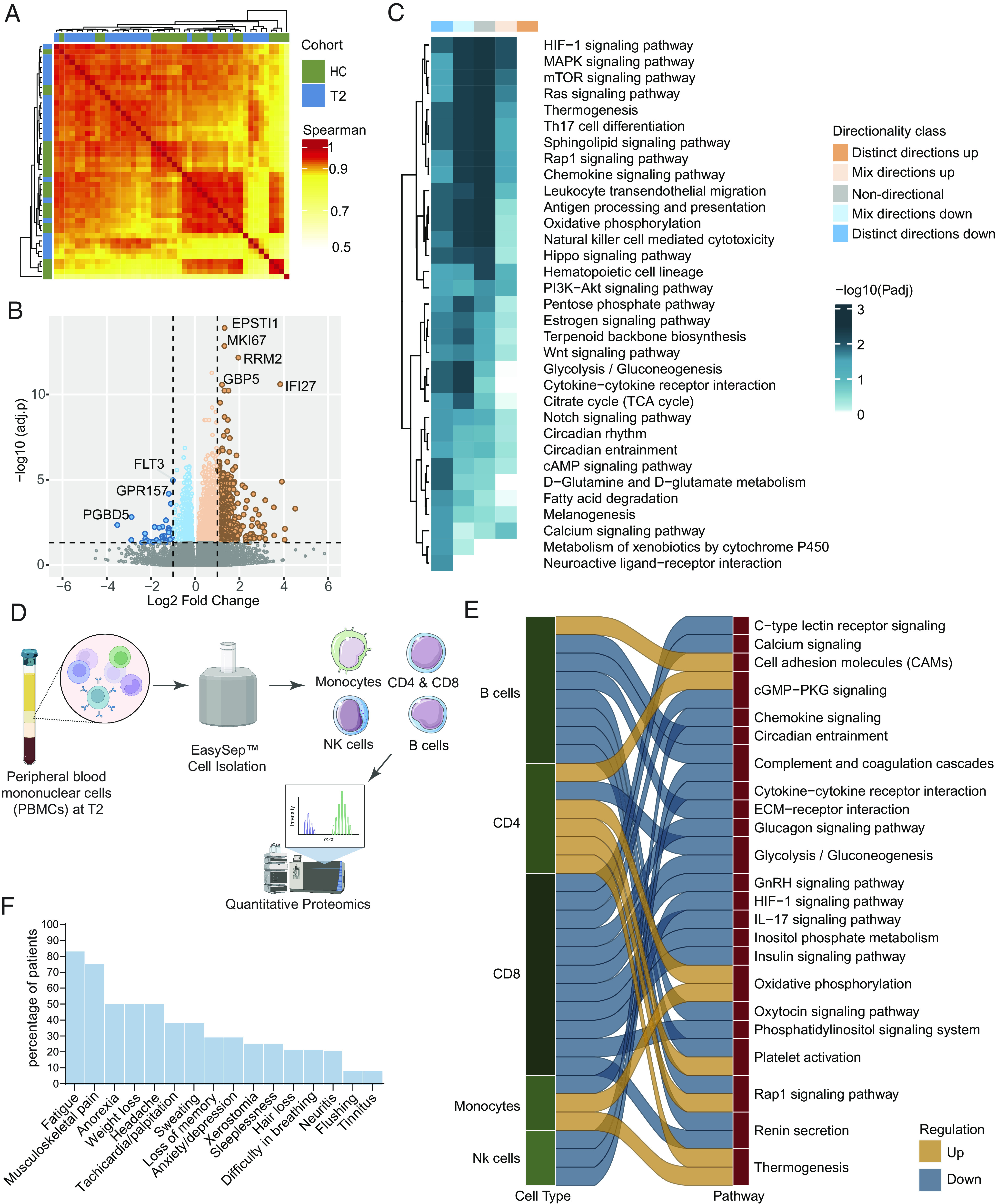
Postrecovery immune signature compared to the CCHFV-seronegative samples (A) Patient-to-patient correlation between patient samples using Spearman's correlation metric HC (n = 22) and T2 (n = 24). The heatmap shows correlation coefficients computed using scaled TPM values of the top 5,000 varying genes based on the median absolute deviation. All correlations were highly significant (adjusted P < 0.0001) due to the high number of features used. Column and row annotations represent patient cohorts. (B) Volcano plot of DGE analysis between cohorts. Negative log2 fold change values represent downregulation, and positive values represent upregulation at T2 compared to HC. The top significantly expressed genes are labeled. (C) Significantly enriched pathways at T2 compared to HC (adjusted P < 0.05). The heatmap shows negative log scales adjusted P-values of different classes of directionality. Distinct directional classes of P-values calculated from gene statistics and expression direction were used to define up-regulated and down-regulated pathways. (D) Experimental design of isolation of immune cells. Figure created with Biorender.com (E) Significantly up-regulated and down-regulated (adjusted P < 0.2) pathways in CCHFV convalescent patients compared to HC in each cell type computed from sctProteomics data analysis. (F) Proportion of the symptoms of the PVF syndrome in CCHFV-recovered patients.
Discussion
Our study discovered the fundamental understanding of the host immune response and the temporal spectrum of virus-mediated disease dynamics and pathogenesis following the CCHFV infection. We have identified the trajectory of antiviral immune responses (e.g., RIG-I-like receptor, NOD-like receptor signaling, and TNF signaling) and the impact of viral infection on host metabolism. Significantly, our data showed a correlation between the rearrangement of intracellular metabolic fluxes (e.g., toward the AA metabolism, dysregulated PPP, and metabolic shift toward OXPHOS and fatty acid oxidation at acute phase) and CCHF diseases. The upregulation of the TCA cycle during CCHFV infection, compared to the noninfected HC and between the severity groups (SG), indicated an increased energy demand and cellular stress. The upregulation of glycolysis and pyruvate metabolism potentiated energy generation through alternative pathways associated with the severity of the infection. Using the sctProteomics of five immune cells, we further reported that down-regulated metabolic processes potentially lead to metabolic insufficiency through the recovery due to hyperactivity during the acute phase leading to PVF. The upregulation of the OXPHOS pathway in monocytes can regulate the production of cytokines and enhance respiratory burst with increased production of ROS during the recovery phase (10).
Earlier studies have suggested that immune cells play a role in CCHFV pathogenesis through infection of specific cell types, e.g., mononuclear phagocytes, endothelial cells, and hepatocytes which are the main targets of infection (11). Though trajectory pathogenesis studies of the viral infection were not reported earlier in humans, DCQ-based immune cell identification was reported in the acute phase of Ebola (12), and SARS-CoV-2 (6) has been reported. For example, during the acute phase of Ebola virus disease (EVD), there was a reduction in the monocytes and CD4 T-cells and an increase in memory CD8 T-cells and NK cells (12). In contrast, in SARS-CoV-2, there was an increase in neutrophil and blood monocyte populations (CM and IM) (6). Interestingly, there was no change in CM in the early acute phase (T0) of CCHFV infection but an increase at the later T1 and T2 phases. Both the pDCs and mDCs were increased at the acute phase (T0) and hospital discharge (T1) but normalized at HC-level at the T2 time point, indicating a role of DCs during the early acute phase of CCHF infection. We, therefore, posit that CM plays the role of effective control and clearance of the CCHFV during the recovery phase.
Our earlier study reported system-level host's metabolic reprogramming toward CCM with distinct upregulation of OXPHOS during acute CCHFV infection (8). This corroborated with the transcriptomics or proteomics performed in vitro infection models in cancer cell lines (13) and in vivo infection studies in nonhuman primates (14) and mice (15), showing dysregulation of the metabolic process and upregulation in the IFN pathways. The temporal quantitative proteomics analysis by liquid chromatography–mass spectrometry (LC–MS) in the CCHFV-infected human hepatocellular carcinoma cell line, Huh7 model reported by us showed dynamic changes in the ISGs and metabolic process during the progressive replication (4). To our knowledge to date, no study has investigated a longitudinal immune response against CCHFV pathogenesis in patients. Cross-sectional studies also suggested that the regulation of the immune response was mediated by high levels of regulatory cytokine IL-10, which inhibits cell-mediated immunity by down-regulating IL-12 expression in CCHF patients (16). It can also inhibit the mediators of inflammation through the modulation of the antigen-presenting cells (17). The cumulative information obtained from both in vitro and in patients thus provides details on the viral pathogenesis and the cellular response during different phases of infection that can help to identify critical targets for therapeutic interventions and inform the development of effective treatments and vaccines.
Being a single-stranded RNA virus, CCHFV can be recognized by cellular RNA receptors such as RIG-I/MDA5, leading to IFN production, or sensed by NOD-like receptors that lead to inflammasome activation, critical events in inflammatory immune response processes (18, 19). CCHFV has been shown to induce ISG56, MxA, and IFN-β (19), and blocking of IFN signaling can cause attenuation of these ISGs (20) even in lethal CCHF infections in mice (21, 22). The virus has also evolved mechanisms to hijack these processes, and in virus infection models, IFN had no inhibitory effect on the virus replication postinfection (23, 24). We have observed that the ISGs were up-regulated during the acute phase of the disease, a probable initial response to the virus infection, and showed a downregulation when the adaptive immune responses set in at T2 following a conventional pattern observed during virus infections. However, it was difficult to ascertain whether the ISGs had any protective effect from the virus or whether they could overcome the antiviral responses since no correlation was observed between the virus titer and viral sensing and antiviral response pathways in module M4. As our study was restricted to the CCHFV patients who survived the infection, we can speculate a positive role of the ISGs in controlling CCHFV pathogenesis.
We also report a robust metabolic modulation toward the CCMs (e.g., pyruvate metabolism, OXPHOS) and its association with the antiviral response. The severely altered transports of AA and the key TCA cycle metabolites during the acute phase of infection indicate the hypermetabolic state during the infection. Interestingly, the dysregulated PPP and metabolic shift toward fatty acid oxidation and the additional ATP production through OXPHOS during the T0 phase indicate altered energy metabolism to provide more energy to the cells to counter the infection. This hypermetabolic state during the acute phase led to metabolic insufficiency with altered signaling cascades at the convalescent phase despite restoring the metabolic process. The observation supports that the essential metabolic processes (e.g., glycolysis, TCA cycle, and OXPHOS) and the regulatory signaling cascades (Akt/mTOR/HIF-1 signaling) were down-regulated at T2 than HC. This is further recapitulated in immune cell sctProteomics, where most metabolic pathways were down-regulated in the CCHFV-convalescent samples. Most of the metabolic processes, including glycolysis and its regulator, HIF-1 signaling, and IL-17 signaling, were down-regulated in CD8+ T-cells in CCHFV-convalescent samples. The IL-17 signaling pathways play an essential role in host defense, including the CD8+ T cell cytotoxicity stimulation, as observed in the West Nile Virus (WNV) infection (25). As metabolic reprogramming is critical for CD8+ T-cells activation, proliferation, virus clearance, and protection against secondary infections (26), reprogramming the impaired metabolic pathways to promote CD8+ T‐cell could tailor desirable CD8+ T‐cell immune responses early, as observed in the HCV infection (27).
Several viral infections, like Dengue fever (28), WNV (29), SARS-CoV-2 (30), EVDs (31), and Marburg viruses (32), impact the neuropsychiatric systems upon complications present PVF, which may linger for months after infection. The PVF presents similar to chronic fatigue syndrome, characterized by fatigue/weakness, headache, dizziness, musculoskeletal pain, and cognitive and sleep disorders, and could be linked to dysregulated CCM (33) and microbial dysbiosis impacting the gut–brain axis (34). It could be associated with virus-specific pathophysiologic changes, immunologic aberrations, and inflammatory damage during acute infection. Several clinical studies have reported the acute phase of symptoms in CCHF patients. However, the clinical manifestations of CCHF during the postacute phase have not been widely investigated. During the convalescent phase (10 to 20 d after symptoms onset), the survivors experience fatigue, tachycardia, hair loss, neuritis, hearing loss, loss of memory, and bradycardia for a long time (35). During the convalescent period, 83% of CCHF patients in our cohort reported having fatigue, evidenced by a relationship between the PVF and metabolic insufficiency upon recovery. Herein, we postulate that the high prevalence of fatigue among CCHF patients, both in the acute and convalescent phases, can be explained partly by dysregulated metabolic pathways during infection, as reported in COVID-19 (36, 37). A better understanding of the immuno-metabolic mechanism of the PVF in CCHF patients can identify therapeutic targets for better and faster recovery.
In conclusion, our study identified the temporal systemic antiviral response following the CCHFV infection and metabolic rearrangement. Dysregulation and hyperactivity of the critical metabolic process of the central carbon and energy metabolism and metabolic flux related to the AA metabolism during the acute progressive phases of infection can potentially promote the exhausted phenotype upon recovery. Reprogramming the impaired metabolic pathways to improve the antiviral mechanism by harnessing immunometabolic regulations may pave the way for early recovery and improve the quality of life.
Materials and Methods
Ethics Statement and Biosafety.
This study was approved by the Regional Ethics Committees, Stockholm (Dnr. 2017-/1712-31/2), and the Sivas Cumhuriyet University, Turkey (2020-07/03). All patients and/or their relatives were informed about the purpose of the study and signed a consent form before sample collection. The Institutional Biosafety Committee (IBC) of Turkey by Turkish Ministry of Health (Turkish MoH) has established clear protocols to prevent the transmission of CCHFV in both hospital and laboratory settings. These protocols involve the proper use of personal protective equipment and equipment, as well as safe transfer of samples. Regular training is provided to researchers, healthcare workers, and laboratory staff to effectively manage the biohazards associated with CCHFV, following approved local IBC guidelines of Turkish MoH. To handle the serial blood samples and estimate serum CCHFV levels, a BSL-2 laboratory situated on the same campus was utilized. The transfer of samples between SCUH and the BSL-2 laboratory adhered to established guidelines developed by the local IBC. The extracted RNA from the samples was subsequently transported to Sweden.
Cohort Characteristics.
We recruited 30 patients hospitalized with CCHFV infection between 15th April and 18th June 2021 with a median time of 4 d (range 1 to 6 d) after the onset of symptoms (T0). By using SGS, 43% (13/30) patients were grouped into severity group 1 (SG-1, low risk of death), 30% (9/30) patients into severity group 2 (SG-2, intermediate risk of death), and 27% (8/30) patients into severity group 3 (SG-3, high risk of death). Samples were collected from 25 patients on discharge day (T1). Third samples were collected from 24 patients (lost to follow-up:1) with a median duration of 30 d (IQR: 28 to 33 d) post symptoms onset (T2). The patient and healthy control characteristics in Dataset S1, admission symptoms, and signs for CCHF patients are presented in SI Appendix, Fig. S1. In the SG-3 group, 5 (63%) of 8 patients succumbed to death, and the CFR for the cohort was 17% (5 out of 30). The convalescent phase of 24 CCHF patients attended the outpatient clinic of Infectious Diseases and Clinical Microbiology at SCUH with a median duration of 30 d (range 19 to 35 d) post symptoms onset (T2, convalescent phase) to check their health status. At the time of the clinic visit, the patients were questioned for possible postviral symptoms. Whole blood was collected in Tempus™ Blood RNA Tube (Thermo Fisher, USA) and EDTA tube for peripheral blood mononuclear cells (PBMCs).
RNA Sequencing (RNASeq).
The RNA was extracted from the whole blood using Tempus™ Spin RNA Isolation Kit (ThermoFisher, USA), followed by library preparation using SMARTer® Stranded Total RNA Sample Prep Kit–Pico Input Mammalian (Takara Bio Europe AB, Sweden). Illumina NovaSeq6000 platform in paired-end mode was used for the RNASeq. After excluding the low RNA integrity number samples and failed library (n = 5), RNASeq was successful for 69 CCHFV-infected samples for all time points and 22 HC, of which all three-time points samples were available in 21 patients.
DCQ.
The cell type proportions in each sample are computed using a method designed from an algorithm adapted from Estimating the Proportions of Immune and Cancer Cells (38) and sample-wise normalized gene expression data [TPM (transcript per million)] as recently described by us (6).
Transcriptomics Analysis.
The raw fastq reads were processed using nf-core pipeline rnaseq v3.3 (39). DGE analysis with adjustment for confounding factors such as age, gender, cell type proportion, and other possible factors was performed using R/Bioconductor package DESeq2 v1.26.0 (40). The unwanted and hidden variation factor in the data was computed using the Bioconductor package RUVSeq v1.28.0 (41) and added to the DESeq2 design matrix. R/Bioconductor package PIANOv2.2.0 (42) was used for KEGG gene-set enrichment analysis (nperm = 1,000, geneset statistic=mean) for differentially expressed genes, and gseapy v0.10.5 (43, 44) was used for genes in trajectory modules. The likelihood ratio test implemented in the DESeq2 package was used to find genes expressed among the three infection time points. Expression trajectory modules were identified using a Gaussian mixed-effects model implemented in the R/Bioconductor package TMixClust v1.16.0 (7). The number of clusters was optimized based on the silhouette width calculated by the package.
Genome-Scale Metabolic Modeling (GSMM) and FBA.
Context-specific metabolic model reconstruction was performed by incorporating normalized expression data on human reference genome-scale metabolic models obtained from Metabolic Atlas (45). We have generated personalized (individual sample) and group-specific (using average gene expression) metabolic models. The model reconstruction was performed using Task-driven Integrative Network Inference for Tissues (tINIT) algorithm (46, 47). A TPM value of 1 was used as the expression threshold for the process. Reconstruction of the metabolic model was performed by Matlab implementation of the tINIT algorithm (https://github.com/SysBioChalmers/Human-GEM). RAVEN toolbox v2.4.0 (48) was used for FBA with ATP hydrolysis as the objective function.
Isolation of Cell Subsets from PBMCs.
The subset of PBMCs at the T2 time point (6 each CCHFV-positive and HC) was thawed at 37 °C and washed two times in FACS buffer (PBS, 2 mM EDTA, and 2% FBS) by 5 min centrifugation at 1,500 rpm. Before cell separation, the PBMCs were treated with DNase I solution (100 μg/mL) (Stemcell #07900) for 15 min. Cell populations were isolated in three batches. Batch 1: CD4+ T cells (EasySep Human CD4 positive selection kit II, Stemcell #17852) with flow through used for monocyte isolation (EasySep Human monocyte isolation kit, Stemcell #19359), batch 2: CD8+ T cells (EasySep Human CD8 Positive Selection Kit II, Stemcell #17853) with flow through used for B cell isolation (EasySep Human B cell isolation kit, Stemcell #17954), and batch 3: NK cell isolation (EasySep Human NK cell isolation kit, Stemcell #17955). All isolations were performed using EasySep isolation columns according to the manufacturer's protocols. After isolation, cells were washed twice with PBS, and the cell pellet snap was frozen. The purity of the isolation protocols was evaluated using flow cytometry for CD4+ T cells [anti-CD3 (clone OKT3, Biolegend #317306) and anti-CD4 (clone SK3, Biolegend #344654)], CD8+ T cells [anti-CD3 (clone OKT3, Biolegend #317306) and anti-CD8 (clone RPA-T8, Biolegend #301038)], monocytes [anti-CD3 (clone OKT3, Biolegend #317306), anti-CD14 (clone M5E2, BD Bioscience #555398), and anti-CD16 (clone 3G8, BD Bioscience #560195)], B cells [anti-CD3 (clone OKT3, Biolegend #317306) and anti-CD19 (clone HIB19, BD Bioscience #562294)], and NK cells [anti-CD3 (clone OKT3, Biolegend #317306) and anti-CD56 (clone B159, BD Bioscience 562289)]. Viability dye LIVE/DEAD fixable Near-IR dead cell stain (Invitrogen #L34975) was complemented in each stain and required on BD LSRFortessa (BD Bioscience). The purity of the samples is given in SI Appendix, Fig. S6.
sctProteomics.
Protein was extracted from cell pellets, and liquid chromatography with tandem mass spectrometry (LC-MS/MS) was performed at the Proteomics Biomedicum core facility, Karolinska Institutet using an analytical workflow previously described (8, 49) with some modifications. In brief, the cell pellets were solubilized with 10 µL of 8M urea and 10 µL of 0.2% ProteaseMAX (Promega) in 100 mM Tris-HCl, pH 8.5 with 1 µL of 100× protease inhibitor cocktail (Roche) following sonication in the water bath for 5 min. Samples were probed and sonicated with VibraCell probe (Sonics & Materials, Inc.) for 30 s, with pulse 2/2, at 20% amplitude after adding 69 µL Tris-HCl buffer and 10 µL acetonitrile (ACN). Protein concentration was measured by BCA assay (Thermo Scientific). An aliquot of 2 or 5 µg samples (CD8, NK, and CD4, monocyte, B cells, respectively) was taken for further preparation, including reduction with dithiothreitol (Sigma) at 37 °C for 45 min and alkylation with iodoacetamide for 30 min at room temperature (RT) in the dark. Proteolytic digestion was performed by adding 0.4 or 1 µg sequencing grade trypsin (Promega) in an enzyme-to-protein ratio of 1:5, incubating overnight at 37 °C. Following clean up on the C18 Hypersep plate with 5-µL bed volume (Thermo Fisher Scientific), the biological samples were labeled with TMTpro reagents in random order adding 150 µg TMT-reagent in 30 µL dry ACN and incubating at RT for 2 h. The reaction was quenched before combining and replicated into an analytical sample in a vial. The TMTpro-labeled samples were analyzed by LC-MS/MS as described previously (49, 50). Acquiring raw data files were used for protein identification and reporter ion abundance-based quantification with Proteome Discoverer v2.5 (Thermo Fisher Scientific) using the Mascot Server v5.1 search engine against the human protein database (SwissProt). The results were filtered with 5% FDR using the Percolator. The data were normalized using R package NormalyzerDE v1.12.0 (25), and differential expression analysis was performed using R package limma v3.50.0.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Acknowledgments
The study is funded by Swedish Research Council Grants 2018-05766 and 2017-03126 and EU-H2020 CCHFVaccine to A.T.M., and 2021-00993 to U.N., Sivas Cumhuriyet University Rectorate, Scientific Research Projects (CUBAP) Turkey 2020-RGD-040 to N.E. U.N. acknowledges the support received from the Swedish Research Council Grants 2018-06156, and 2021-01756. We would like to acknowledge the support from the National Genomics Infrastructure, Science for Life Laboratory for RNAseq and Proteomics Biomedicum; and Karolinska Institute, Solna, for LC-MS/MS analysis. The computations were performed using resources provided by Swedish National Infrastructure for Computing (SNIC) through the Uppsala Multidisciplinary Center for Advanced Computational Science under Project SNIC2017-550. Part of the work presented in the manuscript was included in Dr. A.T.A. Ph.D. dissertation entitled, “Multi-dimensional omics approaches to dissect natural immune control mechanisms associated with RNA virus infections”, completed at Karolinska Institute, April 2023 available at https://openarchive.ki.se/xmlui/handle/10616/48491.
Author contributions
U.N. designed research; A.T.A., N.E., S.S.-A., B.B., A.N.P., G.B., M.A., S.A., V.S., Á.V., and S.G. performed research; N.E., B.B., R.H., G.B., M.A., A. Mardinoglu, R.B., I.C., A. Mirazimi, and U.N. contributed new reagents/analytic tools; A.T.A., S.S.-A., B.B., A.N.P., S.A., V.S., Á.V., R.B., S.G., and I.C. analyzed data; A. Mardinoglu, A. Mirazimi, and U.N. supervision; and A.T.A., N.E., S.S.-A., R.H., A. Mardinoglu, V.S., Á.V., R.B., S.G., A. Mirazimi, and U.N. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. G.P. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
The data have been deposited with links to BioProject accession number PRJNA916211 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) (51). All the codes are available at GitHub: https://github.com/neogilab/CCHF_GSMM (52). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD038929 (53).
Supporting Information
References
- 1.World Health Organisation, WHO, Situation report Iraq, Week 31 (World Health Organization, WHO Regional office for the Eastern Mediterranean, 2022).
- 2.Mukherjee D., Roy S., Iturburu A., Jaiswal V., Crimean Congo hemorrhagic fever in COVID 19 times, an emerging concern in Iraq. Ann. Med. Surg. (Lond) 80, 104273 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mertens M., Schmidt K., Ozkul A., Groschup M. H., The impact of Crimean-Congo hemorrhagic fever virus on public health. Antiviral Res. 98, 248–260 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Neogi U., et al. , Multi-omics insights into host-viral response and pathogenesis in Crimean-Congo hemorrhagic fever viruses for novel therapeutic target. Elife 11, e76071 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yilmaz G. R., et al. , The epidemiology of Crimean-Congo hemorrhagic fever in Turkey, 2002–2007. Int. J. Infect. Dis. 13, 380–386 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Ambikan A. T., et al. , Multi-omics personalized network analyses highlight progressive disruption of central metabolism associated with COVID-19 severity. Cell Syst. 13, 665–681.e4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golumbeanu M., et al. , Proteo-transcriptomic dynamics of cellular response to HIV-1 infection. Sci. Rep. 9, 213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambikan A. T., et al. , Genome-scale metabolic models for natural and long-term drug-induced viral control in HIV infection. Life Sci. Alliance 5, e202201405 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Svensson Akusjärvi S., et al. , Peripheral blood CD4(+)CCR6(+) compartment differentiates HIV-1 infected or seropositive elite controllers from long-term successfully treated individuals. Commun. Biol. 5, 357 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iles K. E., Forman H. J., Macrophage signaling and respiratory burst. Immunol. Res. 26, 95–105 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Burt F. J., et al. , Immunohistochemical and in situ localization of Crimean-Congo hemorrhagic fever (CCHF) virus in human tissues and implications for CCHF pathogenesis. Arch. Pathol. Lab. Med. 121, 839–846 (1997). [PubMed] [Google Scholar]
- 12.Liu X., et al. , Transcriptomic signatures differentiate survival from fatal outcomes in humans infected with Ebola virus. Genome Biol. 18, 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mo Q., et al. , Transcriptome profiling highlights regulated biological processes and type III interferon antiviral responses upon Crimean-Congo hemorrhagic fever virus infection. Virol. Sin. 38, 34–46 (2023), 10.1016/j.virs.2022.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnold C. E., et al. , Host response transcriptomic analysis of Crimean-Congo hemorrhagic fever pathogenesis in the cynomolgus macaque model. Sci. Rep. 11, 19807 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golden J. W., et al. , The host inflammatory response contributes to disease severity in Crimean-Congo hemorrhagic fever virus infected mice. PLoS Pathog. 18, e1010485 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saksida A., et al. , Interacting roles of immune mechanisms and viral load in the pathogenesis of crimean-congo hemorrhagic fever. Clin. Vaccine Immunol. 17, 1086–1093 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peyrefitte C. N., et al. , Differential activation profiles of Crimean-Congo hemorrhagic fever virus- and Dugbe virus-infected antigen-presenting cells. J. Gen. Virol. 91, 189–198 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Lerolle S., Freitas N., Cosset F. L., Legros V., Host cell restriction factors of Bunyaviruses and viral countermeasures. Viruses 13, 784 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spengler J. R., et al. , RIG-I mediates an antiviral response to Crimean-Congo hemorrhagic fever virus. J. Virol. 89, 10219–10229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scholte F. E. M., et al. , Crimean-Congo hemorrhagic fever virus suppresses innate immune responses via a ubiquitin and ISG15 specific protease. Cell Rep. 20, 2396–2407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindquist M. E., et al. , Exploring Crimean-Congo hemorrhagic fever virus-induced hepatic injury using antibody-mediated type I interferon blockade in mice. J. Virol. 92, e01083-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zivcec M., et al. , Lethal Crimean-Congo hemorrhagic fever virus infection in interferon α/β receptor knockout mice is associated with high viral loads, proinflammatory responses, and coagulopathy. J. Infect. Dis. 207, 1909–1921 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber F., Mirazimi A., Interferon and cytokine responses to Crimean Congo hemorrhagic fever virus; an emerging and neglected viral zonoosis. Cytokine Growth Factor Rev. 19, 395–404 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson I., et al. , Crimean-Congo hemorrhagic fever virus delays activation of the innate immune response. J. Med. Virol. 80, 1397–1404 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Acharya D., et al. , Interleukin-17A promotes CD8+ T cell cytotoxicity to facilitate west nile virus clearance. J. Virol. 91, e01529-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta S. S., Wang J., Chen M., Metabolic reprogramming in CD8(+) T cells during acute viral infections. Front. Immunol. 11, 1013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolski D., et al. , Early transcriptional divergence marks virus-specific primary human CD8(+) T cells in chronic versus acute infection. Immunity 47, 648–663.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalimuddin S., et al. , Chronic sequelae complicate convalescence from both dengue and acute viral respiratory illness. PLoS Negl. Trop. Dis. 16, e0010724 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia M. N., et al. , Evaluation of prolonged fatigue post-West Nile virus infection and association of fatigue with elevated antiviral and proinflammatory cytokines. Viral. Immunol. 27, 327–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O' Mahony L., et al. , Impact of long COVID on health and quality of life. HRB Open Res. 5, 31 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott J. T., et al. , Post-Ebola syndrome, Sierra Leone. Emerg. Infect. Dis. 22, 641–646 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauer M. P., Timen A., Vossen A., van Dissel J. T., Marburg haemorrhagic fever in returning travellers: An overview aimed at clinicians. Clin. Microbiol. Infect. 21s, e28–e31 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Armstrong C. W., McGregor N. R., Butt H. L., Gooley P. R., Metabolism in chronic fatigue syndrome. Adv. Clin. Chem. 66, 121–172 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Proal A., Marshall T., Myalgic encephalomyelitis/chronic fatigue syndrome in the Era of the human microbiome: Persistent pathogens drive chronic symptoms by interfering with host metabolism, gene expression, and immunity. Front. Pediatr. 6, 373 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ergönül O., Crimean-Congo haemorrhagic fever. Lancet Infect. Dis. 6, 203–214 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sadlier C., et al. , Metabolic rewiring and serotonin depletion in patients with postacute sequelae of COVID-19. Allergy 77, 1623–1625 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scherer P. E., Kirwan J. P., Rosen C. J., Post-acute sequelae of COVID-19: A metabolic perspective. Elife 11, e78200 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Racle J., Gfeller D., EPIC: A tool to estimate the proportions of different cell types from bulk gene expression data. Methods Mol. Biol. 2120, 233–248 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Ewels P. A., et al. , The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Risso D., Ngai J., Speed T. P., Dudoit S., Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Väremo L., Nielsen J., Nookaew I., Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 41, 4378–4391 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen E. Y., et al. , Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subramanian A., et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson J. L., et al. , An atlas of human metabolism. Sci. Signal. 13, eaaz1482 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agren R., et al. , Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 8, e1002518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agren R., et al. , Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 10, 721 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H., et al. , RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 14, e1006541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Appelberg S., et al. , Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 9, 1748–1760 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Végvári Á., Rodriguez J. E., Zubarev R. A., Single-cell chemical proteomics (SCCP) interrogates the timing and heterogeneity of cancer cell commitment to death. Anal. Chem. 94, 9261–9269 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ambikan A., Transcriptome-based systems-level temporal immune-metabolic profile in CCHF virus infection. NCBI BioProject. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA916211. Deposited 27 December 2022.
- 52.Ambikan A., neogilab/CCHF_GSMM: CCHF R Codes (Version cchf v1) [Computer software]. 10.5281/zenodo.8268766. Deposited 21 August 2023. [DOI]
- 53.Perez-Riverol Y., et al. , The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543-D552 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Data Availability Statement
The data have been deposited with links to BioProject accession number PRJNA916211 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) (51). All the codes are available at GitHub: https://github.com/neogilab/CCHF_GSMM (52). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD038929 (53).