

Abstract
CD4+ T helper 17 (TH17) cells protect barrier tissues but also trigger autoimmunity. The mechanisms behind these opposing processes remain unclear. Here, we found that the transcription factor EGR2 controlled the transcriptional program of pathogenic TH17 cells in the central nervous system (CNS), but not that of protective TH17 cells at barrier sites. EGR2 was significantly elevated in myelin-reactive CD4+ T cells from multiple sclerosis patients and mice with autoimmune neuroinflammation. The EGR2 transcriptional program was intricately woven within the TH17 cell transcriptional regulatory network and showed high interconnectivity with core TH17 cell-specific transcription factors. Mechanistically, EGR2 enhanced TH17 cell differentiation and myeloid cell recruitment to the CNS by upregulating pathogenesis-associated genes and myelomonocytic chemokines. T cell-specific deletion of Egr2 attenuated neuroinflammation without compromising the host’s ability to control infections. Our study shows that EGR2 regulates tissue- and disease-specific functions in pathogenic TH17 cells in the CNS.
The differentiation of TH17 cells is triggered by T cell receptor activation in the presence of interleukin (IL)-6 and transforming growth factor (TGF)-β11–3. While homeostatic TH17 cells can bolster the body’s defenses by promoting host-commensal homeostasis, tissue repair and epithelial barrier fortification, they can also transition into pathogenic TH17 cells in the presence of the pro-inflammatory cytokines IL-1β and IL-234–8. Although TH17 cells are implicated in various human inflammatory diseases9–12, their indiscriminate targeting may lead to increased susceptibility to infections, highlighting the need for selective targeting strategies aimed at pathogenic TH17 cells. Supporting this idea, Crohn’s disease patients who received the IL-17A blocking antibody secukinumab showed exacerbation of symptoms13, while in mice IL-17A was reported to have a beneficial role in maintaining the intestinal barrier integrity during inflammation14, 15. As such, identification of factors or pathways that selectively target pathogenic TH17 cells has fundamental clinical implications.
Regulatory network analyses have indicated the intricate interplay of the transcription factors that govern lineage commitment in TH17 cells. Pioneering transcription factors such as IRF4, BATF and STAT3, in addition to RORγt, exert control over the TH17 lineage-specific gene expression16–18. However, the dysregulated function of other transcriptional regulators may perturb the differentiation and function of TH17 cells, underscoring the need to further elucidate how the combinatorial expression of transcription factors shapes the TH17 response and drives protective or pathological outcomes. Nanowire-based siRNA perturbation studies have identified members of the EGR family of transcription factors, specifically EGR1 and EGR2, as potential regulators of the TH17 cell differentiation program18. Dysregulation of EGR-controlled genes has been observed in peripheral blood mononuclear cells (PBMCs) of multiple sclerosis (MS) patients and Egr2 polymorphisms have been associated with increased susceptibility to autoimmune diseases19–21, suggesting a potential link between EGR family members and TH17 cell-mediated immunopathology.
Here, we showed that the EGR family of transcription factors was selectively upregulated in myelin-reactive CD4+ T cells from MS patients. Specifically, we found that EGR2, but not EGR1, acted as a positive regulator of the TH17 cell program and was required for the development of pathogenic TH17 cells in experimental autoimmune encephalomyelitis (EAE). In contrast, EGR2 was dispensable for TH17 cell responses associated with tissue homeostasis or infection. This contextual requirement for EGR2 in TH17 cells was influenced by tissue-specific clues and the affinity and strength of TCR engagement. Collectively, our findings underscore the importance of the EGR2-specific gene regulatory network in effectively shaping pathogenic and non-pathogenic functional states of TH17 cells.
Results
EGR2 reinforces the TH17 transcriptional program
To investigate the pathogenic CD4+ T cell responses in MS, we analyzed the transcriptional profiles of myelin-reactive (Tet+) and myelin-nonreactive (Tet−) CCR6+CD4+ memory T cells from MS patients, using the publicly available RNA-seq dataset GSE6676322. Significant upregulation of EGR family of transcription factors was observed in Tet+ compared to Tet− CD4+ memory T cells from the same MS patient (Fig. 1a). Moreover, clustering analysis demonstrated that Tet+ CD4+ memory T cells from MS patients separated from healthy controls and Tet− CD4+ memory T cells from MS patients when pathogenicity-associated genes were clustered with EGR family transcription factors (Fig. 1b). These findings suggested that EGR transcription factors may be associated with disease-promoting functions of autoreactive CD4+ T cells from MS patients.
Fig. 1 |. EGR2 reinforces the TH17 transcriptional program.
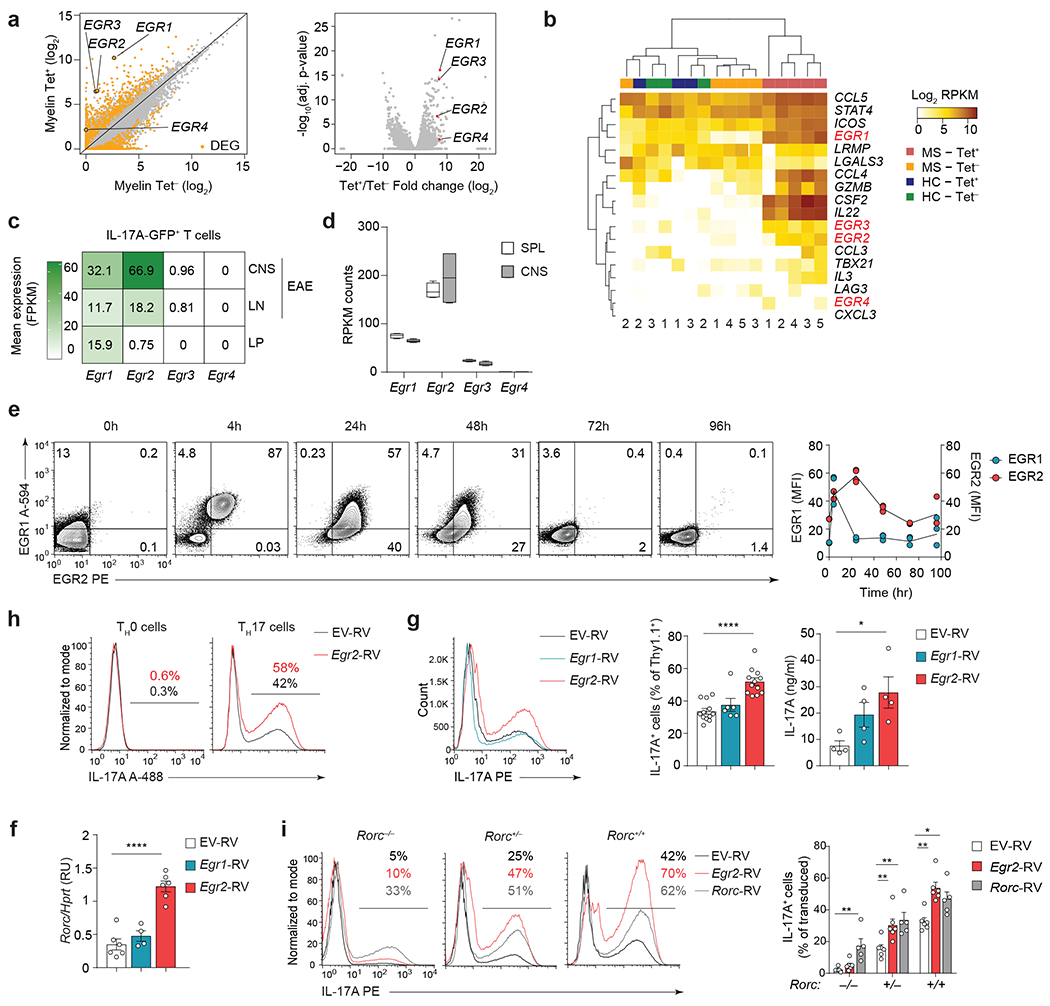
a, Scatter plot (left) and volcano plot (right) showing expression of EGR transcription factors in CCR6+ CD4+ T memory cells from HLA-DR4+ MS patients that bind to MOG97-109-loaded DR4 tetramers (Tet+) compared to CCR6+ memory CD4+ T cells from the same patients that do not bind to MOG97-109-loaded DR4 tetramers (Tet−) (GSE66763)22. b, Clustering of EGR transcription factors with previously defined TH17 cell pathogenicity-associated genes6 in MOG97-109-Tet+ or Tet− CCR6+ CD4+ T memory cells from HLA-DR4+ MS patients (MS Tet+ vs MS Tet−) and MOG97-109-Tet+ or Tet− CCR6+ CD4+ T memory cells from HLA-DR4+ healthy controls (HC Tet+ vs HC Tet−) (GSE66763)22. c, Expression levels of Egr1, Egr2, Egr3 and Egr4 transcripts in sorted IL-17A-GFP+ CD4+ T cells isolated from the CNS and LNs of MOG35-55-immunized Il17a-GFP reporter mice at the peak of EAE disease (day 15 post- immunization), and IL-17A-GFP+ CD4+ T cells isolated from the lamina propria of healthy Il17a-GFP reporter mice (GSE75105, GSE75106)16. Data are shown as fragments per kilobase of exon per million mapped fragments (FPKM). d, Expression levels of Egr1, Egr2, Egr3 and Egr4 transcripts in pathogenic MOG35-55 TCR transgenic (2D2) WT CD4+ T cells from the CNS of Tcrb−/− mice that received 2D2 WT TH17(β,6,23) cells (20 days post-transfer). Box plot depicts median (line), lower and upper quartiles, and whiskers depict 1 and 99 percentile values; n = 4 independent experiments (GSE168288). Data are shown as reads per kilobase per million (RPKM). e, Intranuclear staining of EGR1 and EGR2 proteins in wild-type naive CD62LhiCD25− 2D2 CD4+ T cells and 2D2 CD4+ T cells activated with plate-bound CD3+CD28 antibodies in the presence of TH17 cell-polarizing cytokines (IL-6+TGF-β1); n = 2 TH17 cultures examined in 2 independent experiments. f, Quantitative RT-PCR analysis of Rorc mRNA in TH17 cells transduced with empty virus (EV-RV), or retroviruses expressing Egr1 (Egr1-RV) or Egr2 (Egr2-RV). Combined data from n = 6 (EV-RV), n = 4 (Egr1-RV) and n = 6 (Egr2-RV) independent experiments. ****P < 0.0001; One-way ANOVA, followed by two-tailed unpaired Student’s t-test. g, Frequency of IL-17A+ cells (left) and production of IL-17A as measured by ELISA (right) in TH17 cells (IL-6+TGF-β1) transduced with EV-RV, Egr1-RV or Egr2-RV following 4h PMA+Iono stimulation. Combined data from n = 11 (EV-RV), n = 6 (Egr1-RV) and n = 11 (Egr2-RV) independent experiments and from n = 4 independent experiments (ELISA). ****P < 0.0001, *P < 0.05; One-way ANOVA, followed by two-tailed unpaired Student’s t-test. h, Intracellular staining for IL-17A in CD4+ T cells transduced with EV-RV, Egr1-RV or Egr2-RV under TH0 (IL-2) or TH17 cell-polarizing conditions (IL-6+TGF-β1). Histograms represent the frequency of IL-17A-producing cells within retrovirally-transduced CD4+ T cells. Data are representative for n = 3 independent experiments. i, IL-17A production in Rorc−/−, Rorc+/− and Rorc+/+ CD4+ T cells (on Bcl2l1Tg background) transduced with EV-RV, Egr2-RV or Rorc-RV under TH17 cell-polarizing conditions (IL-6+TGF-β1). The histograms and bar graphs represent the frequency of IL-17A-producing cells within retrovirally-transduced CD4+ T cells; n = 6 independent experiments. **P < 0.01, *P < 0.05; One-way ANOVA, followed by two-tailed unpaired Student’s t-test. Data are presented as mean ± s.e.m. in f,g, and i.
Transcriptional profile comparison of inflammatory IL-17A-GFP+ CD4+ T cells from MOG35-55-immunized Il17a-GFP reporter mice (EAE mice) and homeostatic IL-17A-GFP+ CD4+ T cells from the lamina propria (LP) of healthy Il17a-GFP reporter mice (GSE75105, GSE75106)16 revealed a specific upregulation of Egr2 in CNS-infiltrating TH17 cells (Fig. 1c). Additionally, RNA-seq and RT-PCR analyses indicated that among the EGR family members, Egr2 exhibited the highest expression in 2D2 (MOG35-55-specific TCR transgenic) TH17 cells isolated from the spleen and CNS of symptomatic wild-type (C57BL6) recipient mice in a passive transfer EAE model (Fig. 1d and Extended Data Fig. 1a,b). In MOG35-55-immunized Il17a-Cre R26ReYFP mice23, in which IL-17A-producing cells are permanently marked with yellow fluorescent protein (YFP) expression, specific upregulation of Egr2 was observed in IL-17A-YFP+ CD4+ T cells within the CNS, while Egr1 expression remained comparable between IL-17A-YFP+ and IL-17A-YFP− CD4+ T cells (Extended Data Fig. 1c).
Next, we conducted a time-course analysis of EGR1 and EGR2 protein expression in TH17 cells stimulated with IL-6 and TGF-β1. Both EGR1 and EGR2 were induced ‘early’, at 4h post-activation (Fig. 1e). However, EGR1 levels declined rapidly, while EGR2 expression peaked at 24h post-activation (Fig. 1e), indicating distinct kinetics of expression during the intermediate stage of TH17 cell differentiation. Retroviral expression of Egr2, but not Egr1, strongly enhanced the expression of Rorc (which encodes RORγt) in TH17 cells stimulated with IL-6 and TGF-β1 (Fig. 1f). Furthermore, retroviral expression of Egr2 significantly increased the frequency and amount of IL-17A in CD4+ T cells on a population basis (Fig. 1g), suggesting that EGR2, but not EGR1, promoted TH17 cell differentiation.
Transduction of IL-17A-producing (IL-17A-YFP+) CD4+ T cells with an Egr2-expressing retroviral vector (Egr2-RV) in the presence of TH17 cell-polarizing cytokines IL-6 and TGF-β1 resulted in a significant upregulation of Rorc and key RORγt-target genes (Il17a, Il17f, Il21 and Il22), compared to TH17 cells transduced with an empty retrovirus (EV-RV) (Extended Data Fig. 1d), suggesting that EGR2 may promote the TH17 cell-specific transcriptional program by enhancing RORγt expression. Retroviral expression of EGR2 in IL-2-cultured TH0 cells, which lack endogenous RORγt expression, did not induce IL-17A production (Fig. 1h), indicating that EGR2 alone was insufficient to drive TH0 cells towards the TH17 lineage in the absence of RORγt. Additionally, transduction of Rorc−/−CD4+ T cells with Egr2-RV did not promote TH17 lineage commitment under TH17 cell-polarizing conditions (IL-6+TGF-β1). However, transduction of Egr2-RV fully restored TH17 lineage commitment in Rorc+/− CD4+ T cells, similar to the effect observed with a Rorc-encoding retrovirus (Rorc-RV), which was used as a positive control (Fig. 1i), suggesting that at least one functional copy of Rorc was necessary for EGR2 to promote TH17 cell differentiation. Taken together, these findings showed that EGR2 promoted the differentiation of TH17 cells in a RORγt-dependent manner.
EGR2 is not required for TH17 lineage commitment
Studies using thymic CD2-Cre-mediated Egr2 deletion have reported that EGR2 inhibits TH17 cell development and function24. Deletion of Egr2 in thymocytes results in impaired positive selection, a reduced number of TCRhiCD4+ SP and CD8+ SP thymocytes25–27, and a lupus-like autoimmunity28, suggesting a potential contribution of T cell development defects to immune dysregulation. To circumvent the role of EGR2 in T cell development, we crossed Egr2f/f mice27, 29 to CD2-Cre mice30 to generate Egr2ΔT mice, in which Egr2 is specifically deleted in mature peripheral T cells (Extended Data Fig. 2a–c). To investigate the roles of EGR1 and EGR2 in TH17 lineage commitment, we cultured naïve-sorted CD4+ T cells from wild-type, Egr1−/−, Egr2ΔT and Egr1−/−Egr2ΔT mice with IL-6+TGF-β1 (TH17 cell-polarizing conditions). Egr1 and Egr2, but not Egr3, were expressed in wild-type TH17 cells (Fig. 2a), while compensatory expression of Egr1, Egr2 and Egr3 mRNA was observed in Egr2ΔT, Egr1−/− and Egr1−/−Egr2ΔT TH17 cells, respectively (Fig. 2a), suggesting cross-regulation and potential redundancy among EGR family members during TH17 cell differentiation. While deletion of one or two Egr factors had minimal impact, the frequency of IL-17A-producing Egr1−/−Egr2ΔTEgr3−/− CD4+ T cells was markedly reduced compared to wild-type CD4+ T cells (Fig. 2b,c and Extended Data Fig. 2d). EGR factors are induced in response to TCR signaling31, 32. To assess if they were essential for CD4+ T cell activation rather than TH17 lineage commitment, we examined the differentiation potential of Egr1−/−Egr2ΔTEgr3−/− CD4+ T cells into other TH lineages. We detected normal frequency of Egr1−/−Egr2ΔTEgr3−/− IFN-γ+ TH1 effector cells and slightly reduced frequency of Egr1−/−Egr2ΔTEgr3−/− IL-4+ TH2 cells compared to wild-type counterparts (Fig. 2d). These findings indicated the redundant contributions of EGR1, EGR2 and EGR3 to TH17 lineage commitment and their minimal impact on TH1 and TH2 differentiation.
Fig. 2 |. EGR2 is not required for TH17 lineage commitment.
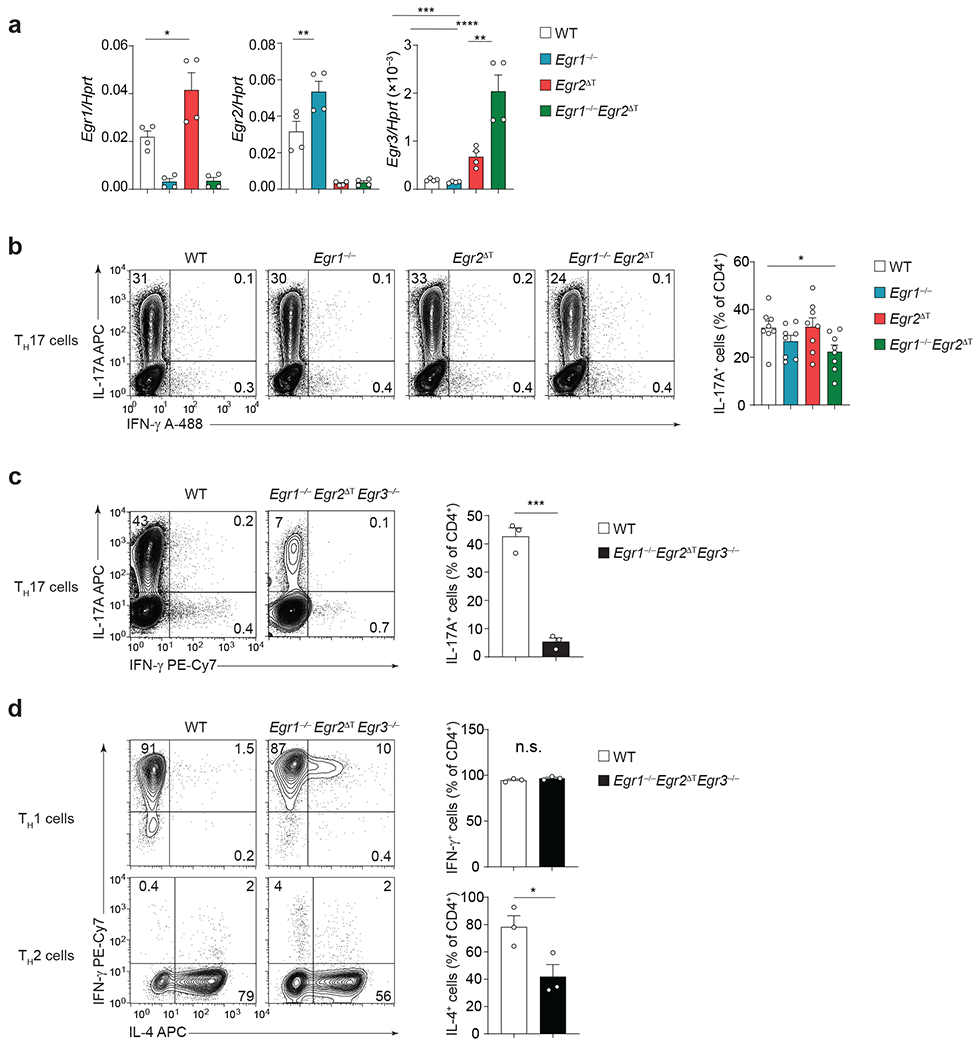
a, Quantitative RT-PCR analysis of Egr1, Egr2 and Egr3 mRNA expression in wild-type (WT), Egr1−/−, Egr2ΔT and Egr1−/−Egr2ΔT CD4+ T cells cultured with IL-6+TGF-β1 (TH17 cell-polarizing conditions) for 5 days, following ex vivo 4h stimulation with PMA+Iono; n = 4 from 2 independent experiments. *P < 0.05, **P = 0.0011, ***P = 0.0001, ****P < 0.0001; One-way ANOVA, followed by two-tailed unpaired Student’s t-test. b, IL-17A and IFN-γ production by WT, Egr1−/−, Egr2ΔT and Egr1−/−Egr2ΔT CD4+ T cells cultured with IL-6+TGF-β1 as in a was measured by flow cytometry following 4h stimulation with PMA+Iono. n = 7 independent experiments. *P < 0.05; One-way ANOVA, followed by two-tailed unpaired Student’s t-test. c-d, Frequency of cytokine-producing WT and Egr1−/−Egr2ΔTEgr3−/− CD4+ T cells cultured with IL-6+TGF-β1 as in a (c) and TH1- (IL-2+IL-12) or TH2 (IL-2+IL-4) polarizing conditions (d) following ex vivo 4h stimulation with PMA+Iono. n = 3 independent experiments. ***P < 0.001, n.s. = not significant; One-way ANOVA, followed by two-tailed unpaired Student’s t-test. Data are presented as mean ± s.e.m. in a-d.
EGR2-specific transcriptional regulatory network in TH17 cells
To examine the impact of EGR2 on the pathogenicity of TH17 cells, we focused on two distinct TH17 cell subsets: TH17 cells polarized with IL-6 and TGF-β1 (hereafter referred to as TH17(β,6)), which lack pathogenic potential7, 33, and highly pathogenic TH17 cells polarized in the presence of IL-23 (TH17(β,6,23)) or a combination of IL-1β, IL-6 and IL-23 (TH17(1,6,23))4, 6, 8, 34. We performed RNA-seq analysis to investigate the transcriptional changes in TH17(β,6) cells transduced with an Egr2-expressing retrovirus (TH17(β,6/Egr2) compared to TH17(β,6) cells transduced with an empty virus (TH17(β,6/EV)) or EV-transduced pathogenic TH17(β,6,23/EV) and TH17(1,6,23/EV) cells. Principal component analysis indicated that EGR2 had a profound impact on the transcriptional profile, as TH17(β,6/Egr2) cells clustered separately on PC1 from the other TH17 subsets (Fig. 3a and Supplementary Table 1). A comparable number of differentially expressed genes (DEGs) were identified in all pair-wise comparisons (Fig. 3b), suggesting that PC1 captured the major transcriptional program affected by EGR2. Pathway analysis of the DEGs indicated that EGR2-upregulated genes were linked to rheumatoid arthritis (RA), inflammatory bowel disease (IBD), regulation of actin cytoskeleton and cell adhesion molecules, while EGR2-downregulated genes were associated with focal adhesion, cell-matrix interactions and calcium signaling (Fig. 3c and Supplementary Tables 1 and 2). Notably, EGR2 exhibited a prominent effect on genes involved in cytokine-cytokine receptor interactions and chemokine signaling (Fig. 3c).
Fig. 3 |. EGR2-specific transcriptional regulatory network in TH17 cells.
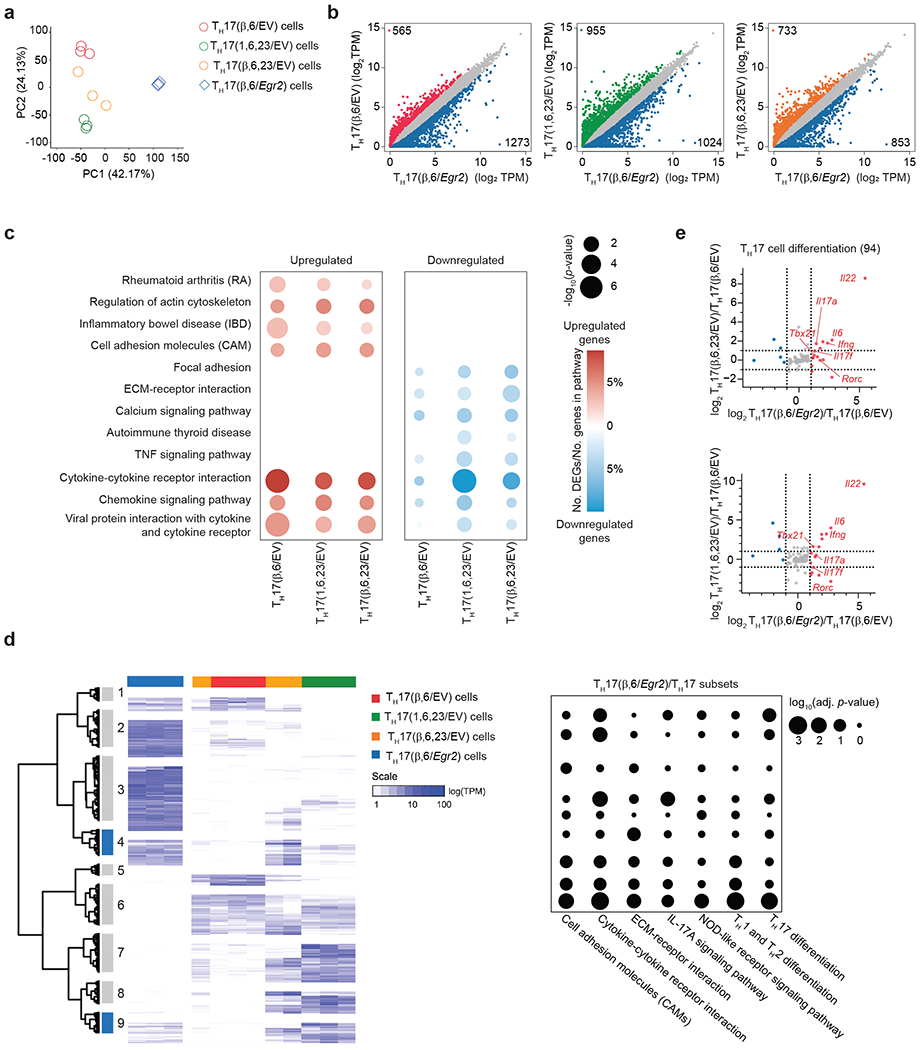
a, Scatter plot showing principal component analysis (PCA) of complete transcriptomes of TH17(β,6/EV), TH17(β,6/Egr2), TH17(β,6,23/EV) and TH17(1,6,23/EV) subsets. b, Scatter plot of differentially-expressed genes (DEGs) between TH17(β,6/Egr2) versus TH17(β,6/EV), TH17(β,6,23/EV) and TH17(1,6,23/EV) subsets. c, Dot-plot of selected Gene Ontology (GO) pathways that were up-regulated or down-regulated in TH17(β,6/Egr2) versus non-pathogenic TH17(β,6/EV) or pathogenic TH17(β,6,23/EV) and TH17(1,6,23/EV) subsets. Color indicates the percentage of DEGs in the selected GO pathways and the size indicates the −log10(p-value). d, Hierarchical clustering of DEGs in TH17(β,6/Egr2), non-pathogenic TH17(β,6/EV) or pathogenic TH17(β,6,23/EV) and TH17(1,6,23/EV) subsets. Clusters marked in blue contain genes induced in TH17(β,6/Egr2) cells and are highly expressed in TH17(β,6,23/EV) and TH17(1,6,23/EV) subsets, but not TH17(β,6) cells. Heat map shows log10-normalized transcript per million (TPM) expression level. Dot-plot shows adjusted p-value for selected KEGG pathway enrichments across all gene clusters. e, Ratio-ratio plots of log2 fold-change between TH17(β,6/Egr2) and TH17(β,6/EV) cells on x-axis versus fold-change between TH17(β,6,23/EV) (top) or TH17(1,6,23/EV) (bottom) and TH17(β,6/EV) on y-axis for all genes from the KEGG TH17 differentiation pathway (mmu04659). a-e Data represent biologically independent replicates per condition from n = 3 independent experiments.
Hierarchical clustering of the DEGs identified two gene clusters (clusters 4 and 9) highly expressed in (TH17(β,6/Egr2)), TH17((β,6,23/EV) and TH17(1,6,23/EV) cells, but not in TH17(β,6/EV) cells (Fig. 3d and Supplementary Table 1), suggesting these EGR2-induced genes may be linked to pathogenicity. KEGG pathway analysis showed that cluster 9 was enriched for pathways such as TH1-TH2 differentiation, TH17 differentiation, cell adhesion molecules, extracellular matrix receptor interactions and NOD-like receptors (Fig. 3d and Supplementary Tables 1 and 2). When focusing on TH17 differentiation (KEGG mmu04659), TH17(β,6/Egr2) cells exhibited higher expression of key TH17 signature genes, such as Rorc, Il17a, and Il22, as well as the TH1-signature genes, Tbx21 and Ifng, compared to TH17(β,6/EV) cells (Fig. 3e). These findings suggested that EGR2 might positively regulate the expression of Tbx21 and Ifng in TH17 cells, contributing to T cell-driven pathogenesis6, 35–37.
EGR2 is not essential for homeostatic TH17 cells
Commensal bacteria, such as segmented filamentous bacteria (SFB), induce homeostatic intestinal TH17 cells38 that support intestinal barrier integrity without eliciting tissue inflammation39, while infection-induced TH17 cells, which are responsible for pathogen clearance, can transiently damage tissues40. To investigate the role of EGR2 in the development of homeostatic TH17 cells, we analyzed the expression of EGR2 in CD4+Foxp3− T cells isolated from the LP of the small intestine and colon in SFB-colonized mice. Under steady-state conditions, only a small fraction of LP CD4+Foxp3− T cells expressed EGR2 protein (Fig. 4a) and both wild-type and Egr2Δ SFB-colonized mice showed a similar frequency of IL-17A-producing CD4+Foxp3− T cells (Fig. 4b). During C. rodentium infection, the frequency of EGR2-expressing CD4+Foxp3− T cells in the colon remained unchanged (Fig. 4c). Egr2ΔT CD4+ T cells produced higher amounts of IL-17A and IL-22 than wild-type CD4+ T cells and effectively controlled C. rodentium infection (Fig. 4d,e), indicating that EGR2 was not essential for the development and function of homeostatic and pathogen-induced TH17 cells in the intestine.
Fig. 4 |. EGR2 is not essential for homeostatic TH17 cells.
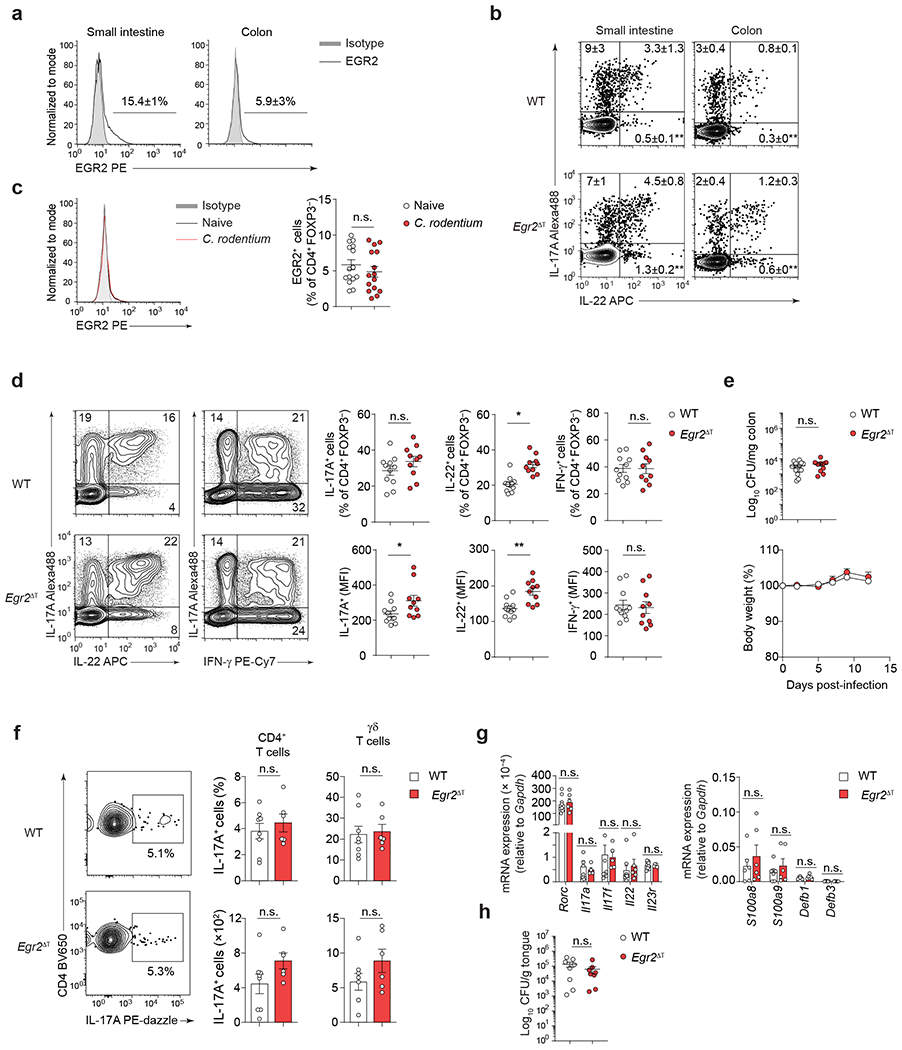
a, Percentage of EGR2-expressing CD4+FOXP3− T cells in the small intestine (n = 5) and colon (n = 15) of SFB-colonized mice at steady-state; 3 independent experiments. b, Percentage of IL-17A- and IL-22-producing CD4+FOXP3− T cells in the small intestine and colon of SFB-colonized WT (n = 5, small intestine; n = 8, colon) and Egr2ΔT (n = 5, small intestine; n = 7, colon) mice was determined by flow cytometry following ex vivo PMA+Iono stimulation; 2 (small intestine) and 3 (colon) independent experiments. **P < 0.01; two-tailed Student’s t-test. c, Percentage of EGR2-expressing CD4+FOXP3− T cells in the colon of naïve (n = 15) and Citrobacter rodentium-infected (n = 15) mice (12 days p.i.); 3 independent experiments. n.s. = not significant; two-tailed Student’s t-test. d, Frequency and MFI of IL-17A, IL-22 and IFN-γ production by colonic CD4+FOXP3− T cells from C.rodentium-infected WT (n = 11) and Egr2ΔT (n = 10) mice (day 12 p.i.) was determined by flow cytometry following ex vivo PMA+Iono stimulation; 3 independent experiments. **P < 0.01, *P < 0.05, n.s. = not significant; two-tailed Student’s t-test. e, Bacterial burden in the colon (12 days p.i.) and body weight (2, 5, 7, 9, 12 days p.i.) of Citrobacter rodentium-infected WT (n = 15) and Egr2ΔT (n = 10) mice, calculated as the percent difference between the original body weight (day 0) and the body weight on any given day; 3 independent experiments. n.s., not significant; two-tailed Student’s t-test. f, Frequency and absolute number of IL-17A-producing CD4+ T cells and γδ T cells in the gingiva of Candida albicans-infected WT (n = 8) and Egr2ΔT (n = 6) mice; 2 independent experiments. n.s., not significant; two-tailed Student’s t-test. g, Quantitative RT-PCR analysis of TH17 cell signature genes and IL-17-dependent antimicrobial peptide-encoding genes from tongue homogenates of WT (n = 9) and Egr2ΔT (n = 6) mice infected with Candida albicans; 2 independent experiments. n.s., not significant; Mann-Whitney U test. h, Candida albicans colony forming units (CFU) per gram of tongue tissue at day 5 post-infection; WT (n = 8) and Egr2ΔT (n = 6) mice; 2 independent experiments. n.s., not significant; two-tailed Student’s t-test. Data are presented as mean ± s.e.m. in a-h.
TH17 cells produce antimicrobial proteins, including β-defensins and S100 proteins, which play a role in fungal clearance at mucosal barrier sites 40. In an oral Candida infection model, both wild-type and Egr2 ΔT mice displayed similar induction of a type 17 response, as evidenced by the expression of the TH17-cell signature genes (Rorc, Il17a, Il17f, Il22, Il23r) and IL-17-dependent antimicrobial peptides/S100 proteins (Defb1, Defb3, S100a8 and S100a9), as well as control of fungal burden in the oral cavity (Fig. 4f–h). These findings indicated that EGR2 was not required for the generation and function of TH17 cells during homeostasis or during mucosal infections.
EGR2 is required for TH17 cell pathogenicity
In the MOG35-55-peptide immunization model of EAE, approximately 30% of the CNS-infiltrating CD4+ T cells expressed EGR2 protein at the peak of disease (Fig. 5a). Notably, EAE induced by MOG35-55 peptide immunization was attenuated in Egr2ΔT mice and in Egr2ΔIL17A mice, where Egr2 was specifically deleted in IL-17A-producing cells (Egr2f/f × Il17a-Cre+), compared to their wild-type (Egr2f/f) littermate controls (Fig. 5b,c). In a passive transfer model of EAE, the adoptive transfer of 2D2 Egr2ΔT TH17(β,6,23) cells resulted in significantly delayed and attenuated disease compared to the transfer of 2D2 wild-type TH17(β,6,23) cells, despite comparable frequency of IL-17A+ cells in both cultures before the transfer (Fig. 5d). In contrast, in the MOG35-55-peptide immunization model of EAE, Egr1−/− mice developed neuroinflammation as severe as wild-type mice (Fig. 5e), indicating that EGR2, but not EGR1, was required for TH17 cell-mediated neuropathology.
Fig. 5 |. EGR2 is required for TH17 cell pathogenicity.
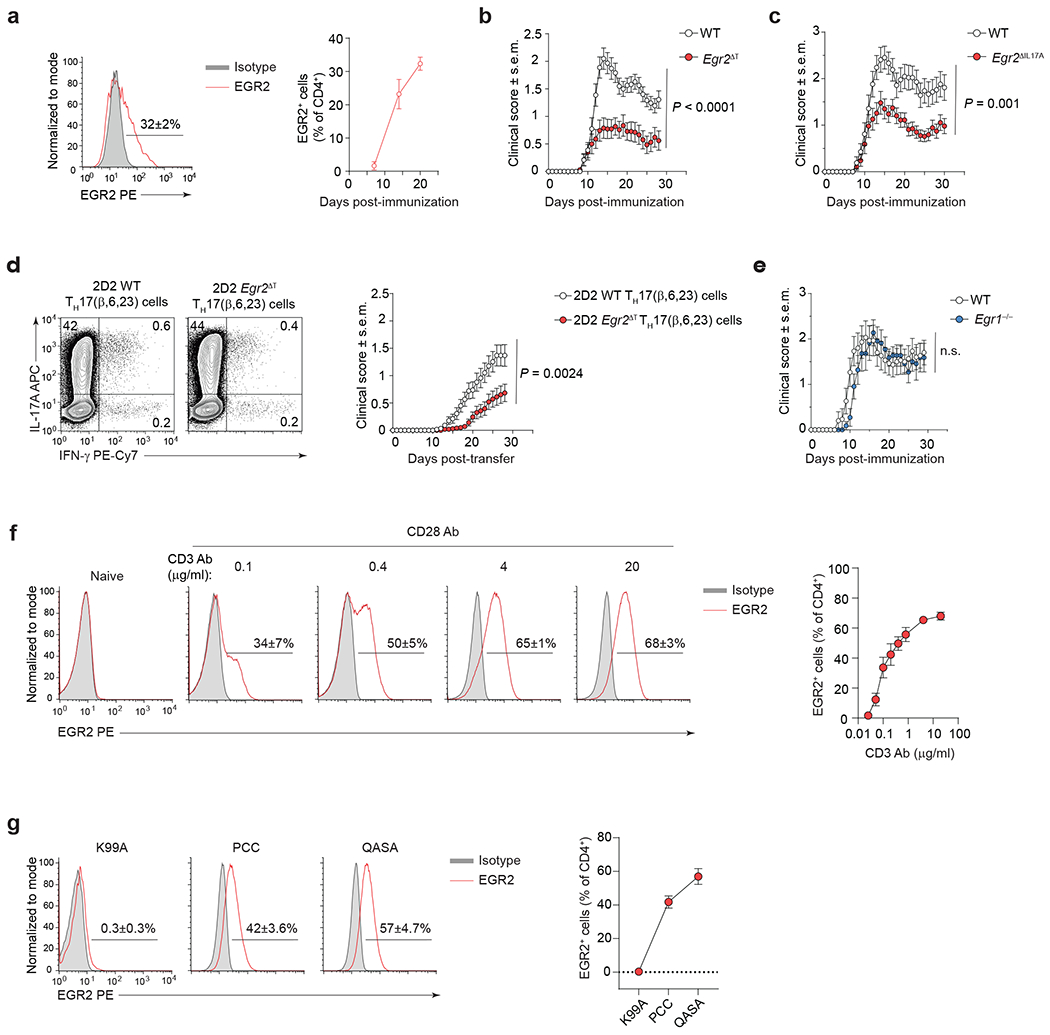
a, Intranuclear staining for EGR2 protein in CNS-infiltrating WT CD4+ T cells (red histogram) at the peak of EAE disease (20 days post-immunization with MOG35-55/CFA and Pertussis toxin). n = 10 mice per time point, 2 independent experiments. b, Mean clinical scores of WT (n = 31) and Egr2ΔT (n = 33) mice following MOG35-55-immunization as in a; 3 independent experiments. P < 0.0001; Two-way ANOVA. c, Mean clinical scores of WT (n = 21) and Egr2ΔIL17A (Egr2f/f × Il17a-Cre+) (n = 27) mice following MOG35-55-immunization as in a; 3 independent experiments. P = 0.001; Two-way ANOVA. d, Frequency of IL-17A- and IFN-γ-expressing 2D2 WT and 2D2 Egr2ΔT TH17(β,6,23) cells before the adoptive transfer following ex vivo PMA+Iono stimulation (left) and mean clinical scores of WT mice that received 7.5 x 106 2D2 WT (n = 59) or 2D2 Egr2ΔT (n = 49) TH17(β,6,23) cells intravenously (right); 5 independent experiments. P = 0.0024; Two-way ANOVA. e, Mean clinical scores of WT (n = 15) and Egr1−/− (n = 11) mice following MOG35-55-immunization as in a; 2 independent experiments. n.s. = not significant; Two-way ANOVA. f, Frequency of EGR2-expressing 2D2 (Vβ11+) TH17(β,6) cells (red histogram) 48h post-stimulation with increasing doses of plate-bound CD3 antibody and a fixed concentration of CD28 antibody (4 μg/ml) in the presence of TH17 cell-polarizing cytokines (IL-6+TGFβ1). g, Frequency of EGR2-expressing AND (Vβ3+) TH17(β,6) cells (red histogram) 48h post-stimulation with irradiated B10.BR splenocytes pulsed with a 6 μM concentration of PCC, K99A, or QASA peptide, in the presence of TH17 cell-polarizing cytokines (IL-6+TGFβ1). Combined data of 3 independent experiments (f,g). Data are presented as mean ± s.e.m. in a-g.
To gain insights into the context-dependent and/or tissue-specific roles of EGR2 in TH17 cells, we examined its impact in a colitis model. Naive CD4+ T cells obtained from wild-type and Egr2ΔT mice induced colitis of equal severity when adoptively transferred in Rag2−/− recipients intraperitoneally (Extended Data Fig. 3a). Notably, the expression of EGR2 protein was significantly diminished in CD4+Foxp3− T cells from the colon of colitis-induced mice at week 6 post-transfer, in comparison to CD4+Foxp3− T cells from the colon of healthy wild-type mice (naïve) (Extended Data Fig. 3a), suggesting that EGR2 did not regulate a pathogenic program in colonic CD4+ T cells during colitis.
In NKT cells, EGR2 expression is induced downstream of TCR-calcineurin-NFAT signaling41, which can be activated by PMA and ionomycin (Iono). Brief PMA+Iono stimulation markedly induced Egr2 mRNA in both non-pathogenic TH17(β,6) and pathogenic TH17(β,6,23) and TH17(1,6,23) cells, while the addition of the STAT3-activating cytokines, IL-6 and IL-23, had no effect on Egr2 expression in these cell subsets (Extended Data Fig. 3b). TCR signaling can impact the generation of self-reactive T cells42. To investigate whether the strength or changes in TCR affinity affected EGR2 expression, quantitative analysis of EGR2 protein expression in 2D2 (Vβ11+) CD4+ TH17(β,6) cells in the presence of increasing concentrations of CD3 antibody revealed that the frequency of EGR2+ TH17 cells was directly proportional to the strength of TCR signal (Fig. 5f), supporting the concept that the strength of TCR signaling may dictate clonal variations in EGR2 protein expression, thereby, influencing TH17 effector functions.
To assess the effect of changes in TCR affinity on EGR2 expression, we activated transgenic AND (Vβ3+) CD4+ TH17(β,6) cells with the native pigeon cytochrome C (PCC) peptide or altered peptide ligands that have equal affinity for the selecting I-Ek molecule but higher (QASA) or lower (K99A) affinity for TCR, respectively. While the native PCC peptide induced EGR2 protein expression in AND TH17(β,6) cells, the low affinity K99A peptide did not (Fig. 5g). The expression of EGR2 protein was higher in AND TH17(β,6) cells stimulated with the high affinity QASA peptide compared to the native PCC peptide (Fig. 5g). Thus, alterations in TCR affinity directly impacted EGR2 protein expression in TH17 cells, with autoreactive CD4+ T cells, which bear high-affinity TCRs, likely to express more EGR2 than microbe-specific CD4+ T cells, which bear low-affinity TCRs. These observations suggested that EGR2 expression in TH17 cells could be influenced by the strength and affinity of TCR engagement.
EGR2 drives regulatory network in pathogenic TH17 cells
We investigated the molecular mechanism of EGR2-driven TH17 cell pathogenicity through RNA-seq analysis on CD4+ T cells isolated from the spleen and CNS of T-cell-deficient (Tcrb−/−) mice that received either 2D2 wild-type or 2D2 Egr2ΔT TH17(β,6,23) cells during peak EAE (Extended Data Fig. 4a). Transcriptome analysis showed a similar gene expression profile between splenic 2D2 wild-type and 2D2 Egr2ΔT CD4+ T cells, with only 21 DEGs (Fig. 6a and Supplementary Table 1). However, CNS-infiltrating 2D2 Egr2ΔT CD4+ T cells exhibited significant dysregulation, with 129 down-regulated genes and 1623 up-regulated genes compared to 2D2 wild-type CD4+ T cells (Fig. 6a and Supplementary Table 1). ATAC-seq analysis showed that chromatin accessibility, as indicated by the number of accessible peaks, was not significantly different between 2D2 wild-type and 2D2 Egr2ΔT TH17(β,6) cells at the peak of EGR2 expression (40h post-activation with CD3+CD28 antibodies) (Extended Data Fig. 4b), suggesting that EGR2 did not regulate chromatin accessibility in TH17 cells.
Fig. 6 |. EGR2 drives regulatory network in pathogenic TH17 cells.
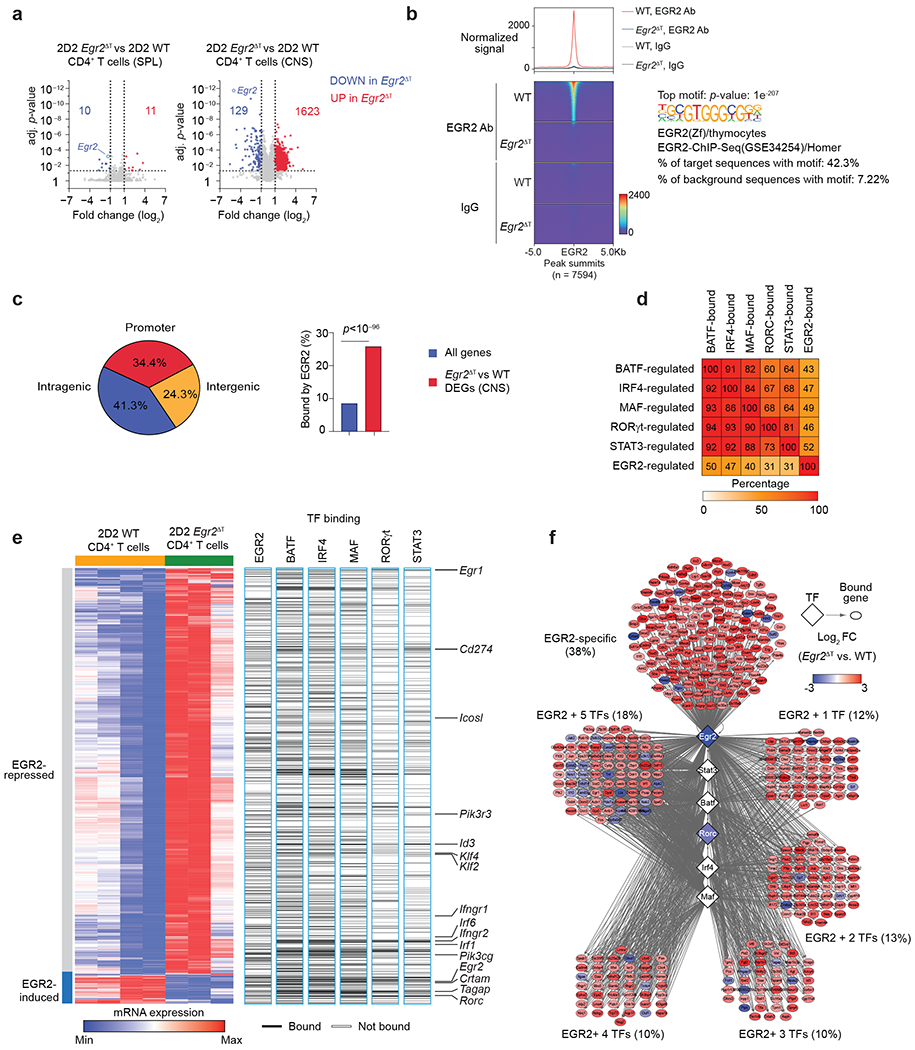
a, Volcano plots showing log2 fold-change on x-axis and adjusted p-value on y-axis for all measured transcripts. Red and blue points denote genes that were significantly up- or down-regulated in 2D2 Egr2ΔT CD4+ T cells compared to 2D2 WT CD4+ T cells from the spleen and CNS of T-cell-deficient (Tcrb−/−) mice that received either 2D2 WT or 2D2 Egr2ΔT TH17(β,6,23) cells (20 days post-transfer). Dotted lines indicate fold-change and p-value thresholds for DEGs. Data represent biologically independent replicates from n = 3 independent experiments. b, Histogram (top) and heatmap (bottom) of EGR2 signals, centered on peaks in 2D2 WT versus 2D2 Egr2ΔT TH17 (β,6) cells 40h post-activation with plate-bound CD3+CD28 antibodies (GSE226795). Both EGR2 antibody (top) and IgG controls (below) are shown. Right, the enrichment of the top motif underlying peaks in EGR2 Ab-fraction of 2D2 WT TH17 cells. c, Pie chart showing the distribution of EGR2-bound loci across the genome (promoter, intragenic, intergenic) (left) and bar graph of the proportion of DEGs bound by EGR2 versus all genes across the genome (right). d, Heatmap showing the percentage of BAT-, IRF4-, MAF-, RORγt-, STAT3 and EGR2-regulated genes that are also co-bound by each transcription factor. e, Heatmap showing DEGs between 2D2 WT and 2D2 Egr2ΔT CD4+ T cells from the CNS of Tcrb−/− mice that received either 2D2 WT or 2D2 Egr2ΔT TH17(β,6,23) cells (20 days post-transfer) as in a, and the binding of EGR2, BATF, IRF4, MAF, RORγt, and STAT317. f, Network diagram depicting EGR2-dependent genes bound by EGR2 alone or by EGR2 in combination with 1, 2, 3, 4, or all 5 core TH17-lineage specific transcription factors.
Given the transient expression of EGR2 protein in TH17 cells (4-40h post-activation) (Fig. 1e) and the limited recovery of CD4+ T cells from the CNS during EAE (~1 × 105/mouse), we used in vitro polarized 2D2 wild-type and 2D2 Egr2ΔT TH17(β,6) cells at 40h post-activation, coinciding with the peak EGR2 expression, to conduct comprehensive genome-wide mapping of EGR2 occupancy (Extended Data Fig. 5a). CUT&Tag (cleavage under target and tagmentation) analysis revealed that EGR2 predominantly bound within promoters (34.4%) or intragenic regions (41.3%) of the genome (Fig. 6b,c and Supplementary Table 3). Integration of the EGR2-transcriptional program with BATF, IRF4, MAF, RORγt and STAT3 regulatory networks in TH17 cells17, showed significant overlap, with up to 50% of EGR2-dependent genes being bound by at least one other TH17 lineage-specific transcription factor, and vice versa (Fig. 6d). EGR2-bound and repressed genes in CNS-infiltrating 2D2 wild-type CD4+ T cells included known (Cd274, Id3) and novel target genes, such as Egr1, Ifngr1, Ifngr2, Icosl, Pik3r3, Irf1, Irf6 and the stemness-associated transcription factors Klf2 and Klf4 (Fig. 6e and Extended Data Fig. 5b; Supplementary Tables 1 and 3). EGR2 and at least one other TH17 lineage-specific transcription factor co-regulated genes including Egr2, Tagap and Rorc, while Crtam was specifically upregulated by EGR2 (Fig. 6e,f). Although multiple EGR2 binding sites were detected in the Il17a promoter (Extended Data Fig. 5c), CUT&Tag analysis did not reveal EGR2-specific peaks at the Il17a locus (Extended Data Fig. 5b). However, chromatin immunoprecipitation (ChIP)-PCR indicated comparable enrichment of EGR2 binding to the Il17a promoter as observed for the EGR2 target genes Rorc, Crtam and Lag3 (Extended Data Fig. 5d). In promoter-driven luciferase assays in HEK293 cells, EGR2 directly trans-activated Rorc and Il17a transcription in a dose-dependent manner (Extended Data Fig. 5e), indicating that EGR2 directly regulated the transcription of TH17 signature genes, including Rorc and Il17a.
An interactive network diagram revealed that EGR2 regulated a subset of genes independently (38%), while also co-regulating genes with one or more core TH17 lineage-specific transcription factors (63%) (Fig. 6f). Notably, EGR2 and all five core TH17 cell-specific transcription factors (BATF, IRF4, STAT3, RORγt, and MAF) bound to the Egr1 and Egr2 genes in TH17(β,6) cells (Fig. 6e,f). While Egr1 was repressed, Egr2 was induced by EGR2 (Fig. 6e,f), suggesting a mutually exclusive pattern of Egr1 and Egr2 expression and the self-reinforcing nature of the EGR2-specific transcriptional program. These findings highlight the intricate regulatory interactions of EGR2 with core TH17 lineage-specific transcription factors and its role in shaping the TH17 cell transcriptional landscape.
EGR2 controls pathogenesis-associated genes in TH17 cells
RNA-seq of CD4+ T cells from the CNS of Tcrb−/− mice that received either 2D2 wild-type or 2D2 Egr2ΔT TH17(β,6,23) cells showed that EGR2-upregulated genes in CNS-infiltrating CD4+ T cells included Tbx21, Runx1, and Rorc (Fig. 7a), known transcriptional regulators of pathogenic TH1-like TH17 cells35. Furthermore, in the MOG35-55-peptide immunization model of EAE, the numbers of IL-17A+ and IFN-γ+ CD4+ T cells were significantly reduced in Egr2ΔT mice compared to wild-type mice (Fig. 7b). Expression of 10 out of 16 pathogenicity-associated genes6 (Il22, Ccl3, Lgals3, Lrmp3, Ccl4, Casp1, Tbx21, Csf2, Gzmb, Lag3) was higher in TH17(β,6)/Egr2 cells than TH17(β,6)/EV cells (Fig. 7c). Conversely, 2D2 Egr2ΔT CD4+ T cells isolated from the CNS of Tcrb−/− recipients at the peak of disease had lower expression of the same pathogenicity-associated genes compared to 2D2 wild-type CD4+ T cells (Fig. 7c), indicating that EGR2 promoted the expression of pathogenicity-associated gene in TH17 cells. T-BET, a critical transcriptional regulator of pathogenic TH17 cells4, 6, 35, was strongly induced in TH17(β,6)/Egr2 cells compared to TH17(β,6)/EV cells (Fig. 7d), suggesting that EGR2 enhanced the pathogenic potential of TH17 cells by regulating the expression of T-BET. However, Tbx21 was not a direct genomic target of EGR2 in CUT&Tag analysis (Extended Data Fig. 5b), suggesting an indirect regulation through upregulation of Runx1 (Fig. 7e), known to induce Tbx2135.
Fig. 7 |. EGR2 controls pathogenesis-associated genes in TH17 cells.
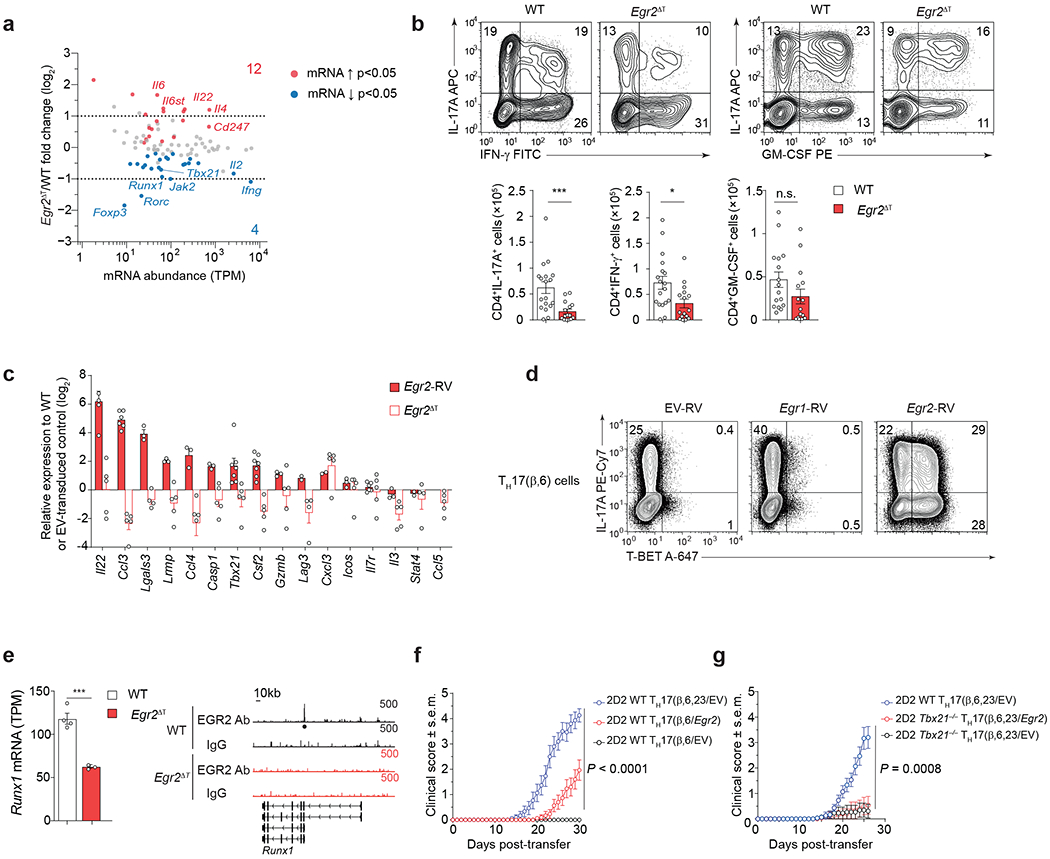
a, MA plot showing transcript abundance in TPM (x-axis) and log2 fold-change (y-axis) for all genes included in KEGG pathway TH17 cell differentiation (mmu04659). Red and blue dots show genes significantly (p-value < 0.05) up- and down-regulated in 2D2 Egr2ΔT CD4+ T cells compared to 2D2 WT CD4+ T cells from the CNS of Tcrb−/− mice that received either 2D2 WT or 2D2 Egr2ΔT TH17(β,6,23) cells (20 days post-transfer). Data represent biologically independent replicates from n = 3 independent experiments. TPM, transcript per million. b, Intracellular cytokine staining of CD4+ T cells isolated from the CNS of WT (n = 15) and Egr2ΔT (n = 15) mice at the peak of disease (14 days post-immunization with MOG35-55/CFA and pertussis toxin). Representative contour plots show the frequency of IL-17A-, IFN-γ- and GM-CSF-producing CD4+ T cells following ex vivo PMA+Iono stimulation. Bar graphs represent the numbers of cytokine producing CD4+ T cells from 3 independent experiments. ***P < 0.001, *P < 0.05; two-tailed unpaired Student’s t-test. c, Expression levels of ‘pathogenicity-associated’ genes in TH17(β,6/Egr2) cells was measured by RT-PCR and normalized to TH17(β,6/EV) cells (solid red bars). Data represent biologically independent replicates per condition from n = 3 independent experiments. Quantitative RT-PCR analysis of ‘pathogenicity-associated’ genes in 2D2 Egr2ΔT CD4+ T cells isolated from the CNS of Tcrb−/− recipients at the peak of disease (20 days post-transfer) and normalized to CNS-infiltrating 2D2 WT CD4+ T cells (white bars). Data represent biologically independent replicates per condition from n = 4 independent experiments. d, Frequency of IL-17A-producing and T-BET-expressing TH17(β,6) cells transduced with EV-RV, Egr1-RV and Egr2-RV was measured by flow cytometry following ex vivo PMA+Iono stimulation; n = 2 independent experiments. e, Abundance of Runx1 transcripts in 2D2 WT and 2D2 Egr2ΔT CD4+ T cells from the CNS of Tcrb−/− mice that received either 2D2 WT or 2D2 Egr2ΔT TH17(β,6,23) cells (20 days post-transfer); Genome browser tracks at the Runx1 locus, showing EGR2 binding by CUT&Tag. The black dot represents EGR2 peak in 2D2 WT TH17(β,6) 40h post-stimulation with plate-bound CD3+CD28 antibodies. Track heights are labeled at the top-right corner of each track. f, Mean clinical scores of Tcrb−/− mice that received 3.75 x 106 2D2 WT TH17(β,6/EV) cells, 2D2 WT TH17(β,6/Egr2) cells or 2D2 WT TH17(β,6,23/EV) cells; n = 14 mice per group; 2 independent experiments. P < 0.0001; Two-way ANOVA. g, Mean clinical scores of Tcrb−/− mice that received 3.75 x 106 2D2 WT TH17(β,6,23/EV) cells, 2D2 Tbx21−/− TH17(β,6,23/EV) cells or 2D2 Tbx21−/− TH17(β,6,23/Egr2) cells; n = 14 mice per group; 2 independent experiments. P = 0.0008; Two-way ANOVA.
2D2 TH17(β,6)/Egr2 cells induced EAE while 2D2 TH17(β,6)/EV cells did not (Fig. 7f). Notably, retroviral expression of Egr2 did not restore the pathogenicity of 2D2 Tbx21−/− TH17(β,6,23) cells (Fig. 7g), indicating the essential role of T-BET in EGR2-driven pathogenic TH17 cell development. Additionally, ectopic expression Egr1 did not upregulate pathogenicity-associated genes (Ccl3, Lgals3, Ccl4, Casp1 and Tbx21) in TH17(β,6) cells (Fig. 7d and Extended Data Fig. 5e). Together, these findings highlight EGR2 as the principal regulator of the pathogenicity-associated transcriptional program in TH17 cells during EAE, including the induction of the TH1-specific transcriptional regulator T-BET.
EGR2 controls CNS homing of encephalitogenic TH17 cells
We investigated the role of EGR2 in immune cell trafficking to the CNS. Transduction of Egr2 into non-pathogenic TH17(β,6) cells led to increased expression of genes associated with T cell migration, including chemokines and chemokine receptors, similar to pathogenic TH17(β,6,23/EV) and TH17(1,6,23/EV) cells (Fig. 8a). In the MOG35-55-peptide immunization model of EAE, Egr2ΔT mice showed a significant decrease in CD4+ T cell accumulation in the CNS relative to wild-type mice (Fig. 8b–c), despite comparable rates of T cell priming in the lymph nodes and reactivation of CD4+ T cells in the CNS post-MOG35-55 peptide immunization (Extended Data Fig. 6a). The expression of IL-1R, critical for the expansion of autoreactive TH17 cells during EAE43, was similar between Egr2ΔT and wild-type CD4+ T cells (Extended Data Fig. 6b). No differences were observed in cell-cycling (Ki67) and apoptosis (annexin V) and the differentiation of effector CD4+ T cells in the draining lymph nodes was comparable between Egr2ΔT and wild-type mice (Extended Data Fig. 6c–e). Collectively, these findings indicated that EGR2 was dispensable for CD4+ T cell activation, differentiation, proliferation, and survival, but controlled the tissue-specific homing and function of pathogenic TH17 cells in the CNS. In a Toxoplasma gondii infection model, which relies on the recruitment of TH1 cells to the CNS44, the migration TH1 cells to the CNS and their production of the effector cytokines IFN-γ and GM-CSF were similar in Egr2ΔT and wild-type mice (Extended Data Fig. 7a–b), underscoring the specific role of EGR2 in regulating TH17 cell recruitment and localized function within the CNS.
Fig. 8 |. EGR2 controls CNS homing of encephalitogenic TH17 cells.
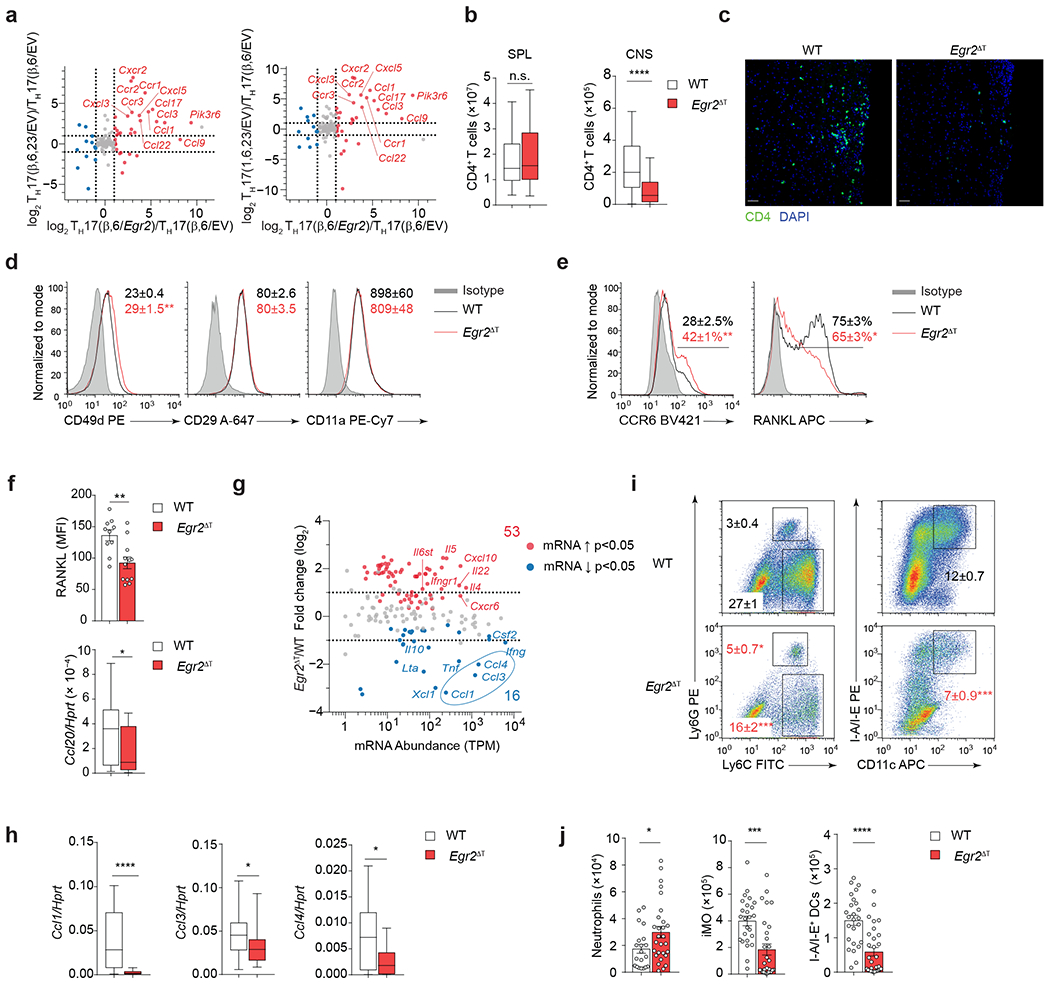
a, Ratio-ratio plot of log2 fold-change between TH17(β,6/Egr2) and TH17(β,6/EV) cells on x-axis versus fold-change between TH17(β,6,23/EV) cells (left) or TH17(1,6,23/EV) cells (right) and TH17(β,6/EV) on y-axis for all genes from the KEGG chemokine signaling pathway (mmu04062). b, Absolute numbers of CD4+ T cells isolated from spleens and CNS of MOG35-55 peptide immunized WT (n = 55) and Egr2ΔT (n = 51) mice 14 days post-immunization. Box plot depicts median (line), lower and upper quartiles, and whiskers depict 1 and 99 percentile values; combined data from 12 independent experiments. ****P < 0.0001, n.s. = not significant; two-tailed unpaired Student t-test. c, Longitudinal spinal cord sections of MOG35-55-peptide immunized WT (n = 4) and Egr2ΔT (n = 5) mice stained with CD4 antibody (green) and nuclear dye DAPI (blue) 14 days post-immunization. Line = 50 μm; representative images from 2 independent experiments. d, Mean fluorescence intensity ± s.e.m. of CD49d (integrin α4), CD29 (β1 integrin), CD11a (integrin αL) on CD4+ T cells from the CNS of WT (n = 18) and Egr2ΔT (n = 18) mice 14 days post-immunization as in c. e, Frequency of CCR6- and RANKL-expressing CD4+ T cells from the CNS of WT (n = 10) and Egr2ΔT (n = 10) mice 14 days post-immunization as in c. Representative staining from 4 independent experiments in d and e; **P < 0.01, *P < 0.05, two-tailed unpaired Student t-test. f, RANKL expression (MFI) on CNS-infiltrating CD4+ T cells from the CNS of WT (n = 10) and Egr2ΔT (n = 10) 14 days post-immunization as in c (top). **P < 0.01, *P < 0.05, two-tailed unpaired Student t-test. Quantitative RT-PCR of Ccl20 mRNA expression in spinal cord homogenates of MOG35-55-peptide immunized WT (n = 28) and Egr2ΔT (n = 18) mice 14 days post-immunization (bottom). Box plot depicts median (line), lower and upper quartiles, and whiskers depict 1 and 99 percentile values; combined data from 3 independent experiments. *P < 0.05; Mann-Whitney U test. g, MA plot showing transcript abundance in TPM (x-axis) and log2 fold-change (y-axis) for all genes included in the KEGG cytokine-cytokine receptor pathway (mmu04060). Red and blue dots show genes significantly (p-value < 0.05) up- and down-regulated in 2D2 Egr2ΔT CD4+ T cells compared to 2D2 WT CD4+ T cells from the CNS of Tcrb−/− mice that received either 2D2 WT or 2D2 Egr2ΔT TH17(β,6,23) cells (20 days post-transfer). h, Quantitative RT-PCR of Ccl1, Ccl3 and Ccl4 mRNA expression in spinal cord homogenates of MOG35-55-peptide immunized WT (n = 28) and Egr2ΔT (n = 18) mice 14 days post-immunization; combined data from 3 independent experiments. ****P < 0.0001, *P < 0.05, Mann-Whitney U test. i-j, Frequency (i) and numbers (j) of neutrophils (Ly6G+Ly6C+), inflammatory monocytes (Ly6G−Ly6C+) and MHC Class II-expressing dendritic cells (I-A/I-E+ CD11c+). Cells originated from the CNS of MOG35-55 peptide immunized WT (n = 20) and Egr2ΔT (n = 28) mice 14 days post-immunization; combined data from 4 independent experiments. ***P < 0.001, *P < 0.05, two-tailed Mann-Whitney U test. Mean values ± s.e.m. are reported in b, d-f, i-j.
Encephalitogenic T cells rely on expression of the integrins VLA-4 (α4β1) and LFA-1 (αLβ2) for their migration into the CNS45–47. CNS-infiltrating CD4+ T cells in MOG35-55 immunized Egr2ΔT mice had normal expression of α4 (CD49d), β1 (CD29) and αL (CD11a) integrins compared to wild-type mice (Fig. 8d), indicating that impaired cell accumulation in the CNS of Egr2ΔT mice was not due to reduced expression of essential adhesion molecules. Within the CNS, TH17 cell-derived RANKL (encoded by Tnfsf11) triggers astrocyte-derived CCL20 production, which is essential for the CCR6-dependent recruitment of TH17 cells to the CNS parenchyma48, 49. We observed a higher frequency of CCR6+ CD4+ T cells in the CNS of Egr2ΔT mice compared to wild-type mice (Fig. 8e); however, Egr2ΔT CD4+ T cells exhibited suboptimal upregulation of RANKL within the CNS (Fig. 8e,f). Consistently, the expression of Ccl20 mRNA was significantly lower in spinal cord homogenates from MOG35-55 immunized Egr2ΔT mice compared to wild-type mice (Fig. 8f), suggesting that EGR2 expression in TH17 cells might facilitate their infiltration into the CNS by regulating the RANKL-CCL20 axis. In addition, Egr2ΔT mice exhibited a significant reduction in the expression of the myelomonocytic chemokines Ccl1, Ccl3 and Ccl4 in spinal cord homogenates compared to wild-type mice (Fig. 8g,h and Supplementary Table 1). Correspondingly, the recruitment of inflammatory monocytes and MHC class II-expressing dendritic cells to the CNS was significantly diminished in MOG35-55 immunized Egr2ΔT mice compared to wild-type mice (Fig. 8i,j). These data indicated that Egr2ΔT CD4+ T cells had suboptimal activation of the RANKL-CCL20 pathway, diminished production of myelomonocytic chemokines and limited immune cell recruitment to the CNS (Extended Data Fig. 8), highlighting the critical involvement of EGR2 in the tissue-specific homing and function of TH17 cells and the pathogenesis of autoimmune disorders affecting the CNS.
Discussion
Here we showed the upregulation of EGR transcription factors in myelin-reactive memory CD4+ T cells from MS patients compared to healthy controls, highlighting their involvement in MS pathogenesis. EGR2 promoted TH17 cell differentiation program in a RORγt-dependent manner. The transcriptional program induced by EGR2 was intricately integrated within the TH17 transcriptional regulatory network established by the core TH17-specific transcription factors, particularly at the Egr1 and Egr2 loci, where we detected the binding of EGR2, BATF, IRF4, STAT3, RORγt and MAF. While Egr1 was repressed, Egr2 was induced by EGR2, establishing a feed-forward reinforcement of the EGR2 transcriptional program.
In the context of autoimmune neuroinflammation, EGR2 promoted the expression of genes associated with the pathogenicity and tissue-specific homing of TH17 cells to the CNS. Our observation that EGR2 specifically facilitated the accumulation of TH17 cells, but not TH1 cells, suggested that the migration of TH1 and TH17 cells to the CNS is controlled by different mechanisms. Indeed, TH1 and TH17 cells display differential utilization of T cell-associated integrins α4β1 and αLβ2 to invade CNS parenchyma47. EGR2 was not required for the expression of these integrins or the TH17 cell-specific chemokine receptor CCR6. Instead, EGR2 was required for optimal upregulation of RANKL on CNS-infiltrating CD4+ T cells and the expression of Ccl20. Additionally, EGR2 controlled the production of myelomonocytic chemokines and the recruitment of inflammatory monocytes and dendritic cells to the CNS.
Because Egr2 is induced in response to TCR signaling, it was possible that disruption of Egr2 would have a broad effect on CD4+ T cell responses. However, our findings indicated that this was not the case. CD4+ T cells could still differentiate into IFN-γ-producing TH1 cells in the absence of Egr1, Egr2 and Egr3. Moreover, T cell-restricted Egr2 deficiency did not compromise cytokine production in vivo following T. gondii, C. albicans and C. rodentium infections. Thus, inhibition of EGR2 transcriptional activity in CD4+ T cells will unlikely compromise the beneficial and protective functions of T helper cells.
While multiple factors are likely to determine the induction of EGR2 in TH17 cells, our data suggested that tissue-specific cues in the CNS had an important role. CD4+ T cells from the intestinal barrier tissue had limited expression of EGR2 under both steady-state and inflammatory conditions (infection and colitis), while ~30% of CD4+ T cells in the CNS expressed EGR2 during autoimmune neuroinflammation. Deficiency of Egr2 had the greatest impact on the gene expression program in the CNS compared to spleen. Another contributing factor to differential EGR2 expression in TH17 cells may be their activation state. Autoreactive TH17 cells could be more activated than infection-induced TH17 cells due to persistent antigen stimulation in the CNS compared to the more acute exposure to microbial antigens. Here we showed that the affinity and strength of TCR engagement determined clonal differences in EGR2 expression in TH17 cells, with the highest levels of EGR2 detected following strong or high-affinity TCR engagement.
Understanding the etiology of complex diseases necessitates a comprehensive exploration of the modular framework of gene regulation. While lineage-specific transcription factors establish the identity of specific cell lineages, it is the coordinated expression of genes within distinct transcriptional modules that determines the context-specific functions of helper T cells. In this regard, our study provides novel insights into the role of EGR2 as an accessory transcription factor that modulates the transcriptional program of pathogenic TH17 cells, particularly in the context of autoimmune neuroinflammation.
Methods
Mice
All animals were housed at the National Cancer Institute (NCI) animal facility under specific pathogen free (SPF) conditions and used between 8 – 10 weeks of age. All studies were performed under animal protocols approved by the NCI Animal Care and Use Committee. C57BL/6J (Stock No. 000664), 2D2 TCR transgenic mice (TCRMOG, Stock No. 00691251), Tcrb−/− (Stock No. 002118), hCD2-Cre (Stock No. 02740630), Il17a-Cre (Stock No. 01687923), B10.BR (Stock No. 004804), and R26R-EYFP (Stock No. 006148) mice were purchased from the Jackson laboratory. Egr2f/f mice were generated and previously characterized by W. J. Leonard (NHLBI)27. Triple Egr1−/−Egr2ΔTEgr3−/− mice were generated by crossing hCD2-Cre (Stock No. 027406) to the germline Egr1−/−52, Egr2f/f29, germline Egr3−/−53 mouse line, which was developed and kindly provided by D. L. Wiest (Fox Chase Cancer Center). Tbx21-ZsGreen reporter mice were generated and previously characterized by J. Zhu (NIAID)54. Rorc−/− mouse line55 on Bcl2l1Tg56 background was generated and maintained in-house. AND TCR transgenic mice were kindly provided by A. Singer (NCI).
Peptides
The native pigeon cytochrome C (PCC) peptide and its altered peptide ligands, K99A and QASA, were synthesized by GenScript Biotech (New Jersey). The peptides have similar binding to I-Ek 57, 58, and have a single amino acid substitution at residue 99 (K99A) or an insertion of 4 amino acids between residues 99 and 100 (QASA), which decreased and increased affinity for TCR, respectively. The amino acid sequences for each peptide are as follows: PCC (88-104): KAERADLIAYLKQATAK; K99A: KAERADLIAYLAQATAK; and QASA: KAERADLIAYLKQASAQ ATAK.
CD4+ T helper differentiation protocols
Naïve CD4+ T cells were purified by pre-enrichment of CD4+ T cells from pooled spleens and lymph nodes using CD4+ T negative selection kit (Stem Cell Technologies), followed by flow cytometric sorting of CD4+ CD62Lhi CD25− T cells (BD FACSAria™ Fusion Cell Sorter). Naïve CD4+ T cells were activated with soluble anti-CD3 antibody (2 μg/ml; 145-2C11; BioXCell) in the presence of irradiated splenocytes (2000 rad) at a 5:1 ratio and cultured for 5 days in the presence of polarizing cytokines. For TH0 cells: hIL-2 (200 U/ml; NCI Biological Resources Branch Preclinical Repository); TH1 cells: hIL-2 (200 U/ml), mIL-12 (10 ng/ml; Miltenyi Biotec) and anti-IL-4 antibody (10 μg/ml; 11B11; BioXCell); TH2 cells: hIL-2 (200 U/ml), mIL-4 (10 ng/ml; Miltenyi Biotec) and anti-IFN-γ antibody (10 μg/ml; XMG1.2; BioXCell); TH17(β,6) cells: mIL-6 (30 ng/ml; Miltenyi Biotec), hTGF-β1 (3 ng/ml; Miltenyi Biotec), anti-IL-4 antibody (10 μg/ml; 11B11; BioXCell) and anti-IFN-γ antibody (10 μg/ml; XMG1.2; BioXCell); TH17(β,6,IL-23) cells: mIL-6 (30 ng/ml; Miltenyi Biotec), hTGF-β1 (3 ng/ml; Miltenyi Biotec), anti-IL-4 antibody (10 μg/ml; 11B11; BioXCell) and anti-IFN-γ antibody (10 μg/ml; XMG1.2; BioXCell) for 36 hrs, followed by 3 days of culture in the presence of mIL-23 (10 ng/ml; R&D Systems); pathogenic TH17(1,6,IL-23) cells: mIL-1β (10 ng/ml; Miltenyi Biotec), mIL-6 (20 ng/ml; Miltenyi Biotec), mIL-23 (20 ng/ml; R&D Systems), anti-IL-4 antibody (10 μg/ml; 11B11; BioXCell) and anti-IFN-γ antibody (10 μg/ml; XMG1.2; BioXCell).
In vitro stimulation assays
Naïve 2D2 (Vβ11+) TCR transgenic CD4+ T cells were activated with increasing concentration of plate-bound anti-CD3 antibody (0.025, 0.05, 0.1, 0.2, 0.4, 0.8, 4, and 20 μg/ml; 145-2C11; BioXCell) and a fixed concentration of anti-CD28 antibody (4 μg/ml; 37.51; BioXCell) under TH17(β,6) cell-polarizing conditions. For peptide stimulation assays, naïve AND (Vβ3+) TCR transgenic CD4+ T cells were stimulated with irradiated (2000 rad) B10.BR splenocytes pulsed with the PCC, K99A, or QASA peptide (final 6 μM concentration) under TH17(β,6) cell-polarizing conditions. EGR2 expression was measured after 48 hours of activation by intranuclear staining using PE-conjugated anti-EGR2 antibody (erongr2; Thermo-Fisher Scientific eBiosciences).
Retrovirus packaging and transduction
To produce packaged retrovirus particles, 293HEK cells were transiently transfected with packaging plasmids (Gag/Pol, Eco-Env) and retroviral plasmids encoding Thy1.1, Egr1-Thy1.1, Egr2-Thy1.1 or Rorc-Thy1.1 using X-tremeGENE9 DNA Transfection Reagent (Sigma-Aldrich Roche). Supernatants were collected 48 hrs after transfection, and retroviruses were concentrated using PEG Virus Precipitation Kit (Abcam) according to manufacturer’s instructions. Retroviral stocks were stored at −80°C until use. For retroviral transduction, naïve sorted CD4+ T cells were spinfected with retroviruses within 24h of activation at 2000 rpm for 1 hour at RT in the presence of 8 μg/ml of hexadimethrine bromide (Sigma-Aldrich Roche). Cells were cultured in fresh medium in the presence of polarizing cytokines for additional 4 days, after which cells were harvested for flow cytometry or transcriptional analysis.
Active and passive induction of EAE and disease analysis
Eight-to-ten-week old mice were immunized subcutaneously with 100 μg of MOG35-55 peptide (MEVGWYRSPFSRVVHLYRNGK) emulsified in complete Freund’s adjuvant supplemented with Mycobacterium tuberculosis H37Ra extract (Difco), followed by Pertussis toxin (List Biological Laboratories) injections (150 ng/mouse) on days 0 and 2, as described previously50. For passive EAE, naïve 2D2 (MOG35-55 TCR transgenic) CD4+ T cells were cultured under non-pathogenic TH17(β,6) or pathogenic TH17(β,6,IL-23) conditions for 5 days (as described above). On the 5th day of culture, TH17 cells were reactivated on plates pre-coated with anti-CD3 (2 μg/ml; 145-2C11; BioXCell) and anti-CD28 (2 μg/ml; PV1; BioXCell) antibodies in the presence of polarizing cytokines for 48h before adoptive transfer into C57BL6 (7.5 × 106 cells/mouse) or Tcrb−/− (3.75 × 106 cells/mouse) recipients. Classical EAE symptoms were scored daily according to standard criteria: 0, asymptomatic; 1, flaccid tail; 2, hind-limb weakness and impaired righting ability; 3, hind-limb paralysis; 4, front- and hind-limb paralysis; 5, moribund or death. Method for isolation of spinal cord tissue for immunohistology and CNS-infiltrating cells for flow cytometry was followed as described previously50.
Mouse infections
For Citrobacter rodentium infections, mice were inoculated orally by gavage with 108 colony-forming units (CFU) of C. rodentium strain DBS100 or ICC169 (nalidixic acid-resistant, a gift from G. Frankel, Imperial College London) (in PBS) cultured for 18 hours in Luria-Bertani (LB) broth at 37°C as previously described59. For quantification of bacterial burden after infection with C. rodentium ICC169, cecum and colon were collected, weighed, placed in tubes containing 2.8-mm stainless beads (Sigma-Aldrich), and homogenized using a Precellys 24 tissue homogenizer (Bertin Instruments), followed by serial dilution and plating on LB agar plates containing nalidixic acid. Colonies were then counted after 16 hours of incubation at 37°C, and CFUs per milligram of tissue were calculated. For Toxoplasma gondii infections, mice were inoculated orally by gavage with 5 cysts of T. gondii strain ME49 (clone C1) obtained from the brains of chronically infected mice as previously described60. For Candida albicans infections, yeast cells from the SC5314 strain were grown and suspended in HBSS at 107/ml. Mice were anesthetized with ketamine and xylazine and cotton swabs soaked in the C. albicans solution were inserted under the mouse tongue for 90 minutes as previously described61, 62. To determine the number of C. albicans CFUs, the mice were euthanized at day 5 post-infection, their tongues were weighed, homogenized with a tissue homogenizer (Omni International) in PBS, and the homogenate was plated onto yeast, peptone, dextrose plates, CFUs were counted after a 24 – 48-hour incubation at 37°C. In separate experiments, mouse tongues were harvested at day 1 post-infection, homogenized using a tissue homogenizer (Omni International) in TRIzol (Invitrogen). mRNA was extracted using the RNeasy kit (Qiagen) and converted to cDNA using the qScript cDNA Supermix kit (Quanta BioSciences). Quantitative RT-PCR was performed with TaqMan detection using PerfeCTa qPCR FastMix (Quanta BioSciences). Primers were purchased from ThermoFisher: S100a8 (Mm00496696_m1), S100a9 (Mm00656925_m1), Defb1 (Mm004328_ m1), Defb3 (Mm01614469_m1), and Gapdh (Mm99999915_g1). All quantitative RT-PCR reactions were performed using QuantStudio 3 Real-Time PCR System (Applied Biosystems). Reactions were performed in duplicate, and the average cycle threshold (CT) was used for subsequent calculations. For each specimen, relative expression was calculated by normalizing to Gapdh.
Antibodies and flow cytometry
Fluorochrome-labeled or biotinylated antibodies of the following specificities were purchased from BD Pharmingen, BioLegend, Thermo-Fisher Scientific eBiosciences or Cell Signaling Technology: CCR6 (29-2L17), CD11b (M1/70), CD11c (HL3), CD3ε (145-2C11), CD4 (RM4-5), CD8α (5H10), CD5 (53-7.3), CD69 (H1.2F3), IL-1R/CD121a (JAMA-147), RANKL (IK22/5), GM-CSF (MP1-22E9), IFN-γ (XMG1.2), IL-4 (11B11), IL-17A (TC11-18H10.1), IL-22 (IL22JOP), CD49d (R1-2), CD11a (M17/4), CD29 (HMβ1-1), Ki67 (B56), Ly-6C (HK1.4), Ly-6G (1A8), I-A/I-E (M5), Thy1.1 (OX-7), TCRβ (H57), Vβ11 (RR3-15), RORγt (AFKJS-9), EGR1 (44D5), EGR2 (erongr2), FOXP3 (FJK-16s), streptavidin. To evaluate cytokine expression, ex vivo isolated or in vitro differentiated CD4+ T cells were stimulated with phorbol 12-myristate 13-acetate (50 ng/ml, Sigma-Aldrich) and ionomycin (1 μM, Sigma-Aldrich) for 4 hours, including the addition of monensin (3 μM) to block cytokine secretion in the last 2 hours of stimulation. Cell surface staining, fixation/permeabilization and intracellular cytokine staining were performed as previously published50. Intracellular staining of EGR1, EGR2, Ki67, and IL-22 was performed after fixation/permeabilization using the Foxp3/Transcription Factor Staining kit (ThermoFisher Scientific eBiosciences). The Annexin V Apoptosis Detection Kit APC was used following manufacturer’s protocol (ThermoFisher Scientific). Data were acquired on a BD LSR Fortessa or LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Immunohistology
At the peak of disease, mice were transcardially perfused with PBS and spinal cord were harvested and fixed in 4% paraformaldehyde for 12 hours at 4°C. Tissues were transferred into 30% sucrose at 4°C for 24 hours before embedding in Tissue-Tek OCT (Sakura). Frozen spinal cords were cut into 10-μm slices using a cryostat (Leica). Slices were post-fixed with 1% PFA for 10 minutes, blocked in PBS 1% bovine serum albumin for 30 minutes at room temperature before staining overnight at 4°C using anti-CD4 antibody (RM4-5, Invitrogen) diluted in PBS 1% bovine serum albumin. After 3 washes, anti-rat antibody conjugated to Alexa647 (Abcam) was added to the slices and incubated 4 hours at 4°C. The slices were washed, counterstained with DAPI (Life Technologies), mounted using Vectashield Antifade Mounting Medium (Vector Laboratories) and sealed. Images were acquired using Yokogawa CSU-W1 Spinning Disk Confocal on Nikon T2i Eclipse Microscope and processed using ImageJ.
Plasmids
cDNAs encoding Egr1, Egr2 or Rorc were cloned into a retroviral plasmid expressing Thy1.1 marker (pMSCV-IRES-Thy1.1) or into pcDNA3.1(−) expression vector (Thermo-Fisher Scientific Invitrogen). Plasmids pGL3-Rorc, pGL4-Il17a, pGL4-Il22 contain 2kb genomic DNA sequence upstream of the start codon of Rorc, Il17a and Il22 genes, respectively; pGL3, pGL4 and pRL plasmids were purchased from Promega.
Dual luciferase reporter assay
The ability of EGR1 and EGR2 to transactivate −2kb Rorc-, Il17a- and Il22-promoters was determined using Promega’s Dual Luciferase Reporter Assay System (E1910) according to manufacturer’s instructions. For each sample, firefly luciferase readings were normalized to pRL-TK renilla luciferase activity and to empty vector (EV) control. For analysis of dose-dependent effects, HEK293 cells were transfected with Rorc-, Il17a- and Il22-promoter luciferase reporter plasmids, pRL-TK, and increasing concentrations of EGR1 or EGR2. All transfection reactions were normalized for the total amount of DNA.
Quantitative real-time PCR
Total RNA from CD4+ T cells or spinal cord homogenates was extracted using Direct-zol RNA MicroPrep Plus (R2060, ZymoResearch) following manufacturer’s instructions. Reverse transcription was performed using high-capacity cDNA reverse transcription kit (Applied Biosystems). For quantitative RT-PCR, we used the SyberGreen PCR system (Applied Biosystems) and analyzed on a QuantStudio 6 Flex instrument (Thermo Fisher). mRNA expression was calculated by the ΔCt method, relative to housekeeping genes Hprt or Gapdh. RT-PCR primer sequences (5’ to 3’ sequences, forward and reverse strand) listed in the Supplementary Table 4.
ChIP-qPCR
To test for EGR2 binding to the Rorc and Il17a promoters, we performed chromatin immunoprecipitation (ChIP)-qPCR in 48h activated TH17(β,6) cells. Naïve 2D2 CD4+ T cells were activated with plate-bound anti-CD3 antibody (20 μg/ml) and anti-CD28 antibody (4 μg/ml) for 48h in the presence of TH17(β,6) cell-polarizing conditions. TH17(β,6) cells were fixed in 1% formaldehyde in PBS (final concentration) for 10 minutes at RT with gentle shaking. ChIP assays were performed using SimpleChIP Enzymatic Chromatin IP Kit (Cat. No. 9005; Cell Signaling) using rabbit anti-mouse EGR2 antibody (Cat. No. 43020; Abcam) or rabbit Ig control (Cat. No. 2729S; Cell Signaling), following manufacturer’s instructions. Primers specific for test regions containing EGR binding sites and for control regions were used to amplify the ChIP-enriched and input DNA by SYBR Green real-time PCR (Applied Biosystems). Data are presented as percentage of input DNA. Primers are listed (5’ to 3’ sequence, forward and reverse strand) in Supplementary Table 5.
Bulk RNA-Seq
RNA was extracted from splenic and CNS-infiltrating CD4+ T cells at the peak of disease, and from TH17(β,6/EV), TH17(β,6/Egr2), TH17(β,6,23/EV) and TH17(1,6,23/EV) cells after 4hr PMA and ionomycin stimulation, using QIAshredder columns and RNase Plus Mini kit (Qiagen). RNA samples with an RNA integrity number (RIN) >8 (TapeStation Analysis, Agilent Technologies) were sequenced on HiSeq 4000 using Illumina TruSeq Stranded mRNA Library Prep and paired-end sequencing. All samples were sequenced at Biomedical Informatics and Data Science (BIDS) Directorate (Frederick National Laboratory for Cancer Research). The samples have 44 – 66 million pass filter reads with >90% bases above the quality score of Q30. Reads of the samples were trimmed for adapters and low-quality bases using Trimmomatic software before alignment with the reference genome (Mouse – mm10) and the annotated transcripts using STAR version 2.5.2b in 2-pass mode. Gencode mouse annotations M21 were used. The average mapping rate of all samples is 98%; unique alignment is above 88%. Mapping statistics are calculated using Picard software. RSEM version 1.3.0 was used to perform gene-level read counting. HTSfilter was used to purge non-expressed genes63. edgeR was used to normalize counts, calculate differentially expressed genes (DEG) and transcripts per million (TPM)64. Genes with a TPM value smaller than 2 in all genotypes/conditions were filtered out. Subsequently, an offset of 1 was added to the TPM values for each gene for downstream analysis. Prcomp was used for PCA, and Hclust was used for Euclidian clustering with complied DEG as input (Supplementary Table 1). Clusterprofiler was used for hypergeometric testing of DEG and cluster-derived gene sets against the KEGG gene ontology database (Supplementary Table 2)65. Heatmaps were rendered with pheatmap and all other plots with ggplot2 or DataGraph (Visual Data Tools Inc). Raw and processed sequencing data available from NCBI Gene Expression Omnibus under accession number GSE168288.
FastATAC-Seq
FastATAC-Seq was performed according to a published protocol66 with minor modifications. Twenty thousand cells were pelleted and washed with 50 μl 1 × PBS. After pelleting the nuclei by centrifuging at 500 × g for 10 min, the pellets were re-suspended in 50 μl transposase mixture (25 μl of 2x TD buffer, 2.5 μl of TDE1, 0.5 μl of 1% digitonin, 22 μl of nuclease-free water) (Cat. No. 20034198, Illumina; Cat. No. G9441, Promega). The reaction was incubated at 37°C with shaking at 300 rpm for 30 min. Fragmented DNA fractions were purified using a QIAGEN MinElute kit and amplified with 10 or 11 cycles of PCR based on the amplification curve. Once the libraries were purified using a QIAGEN PCR cleanup kit, they were further sequenced for 50 cycles (paired-end reads) on NextSeq 550 (Illumina). FastATAC-Seq reads from three biological replicates for each sample were mapped to the mouse genome (mm10 assembly) using Bowtie 1.1.167. In all cases, redundant reads were removed using FastUniq68. Only one mapped read to each unique region of the genome that was less than 175 bp was kept and used in peak calling. Regions of open chromatin were identified by MACS (version 1.4.2)69 using a p value threshold of 1 × 10−5. Only regions called in all three replicates were used in downstream analysis. Peak annotation and motif analysis were performed with the Hypergeometric Optimization of Motif EnRichment program (HOMER) version 4.11.170 using the “annotatePeaks.pl peak_file mm10 -size 4000 -hist 100 -ghist” and “findMotifsGenome.pl peak_file mm10 motif_folder -size given -preparsedDir tmp 2>out.” The heatmap were generated by “pheatmap” package in R 4.2.2 (R Development Core Team, 2018).
CUT&Tag
CUT&Tag was performed using the established bench top CUT&Tag V.3 protocol (https://www.protocols.io/view/bench-top-cut-amp-tag-kqdg34qdpl25/v3) with the following specifications. Briefly, isolated nuclei from in vitro polarized mouse 2D2 WT or 2D2 Egr2ΔT TH17(β,6) cells were prepared after cell sorting of live (propidium iodide−) cells. 6 × 105 ConA-bound nuclei per genotype were resuspended in antibody buffer and distributed equally into four conventional 1.5 ml tubes. Antibodies were diluted at 1:50 for anti-EGR2 (Clone: erongr2, CUST06000, Invitrogen, Custom Stock Dilution: 0.5 µg/ml) and 1:100 for Rat IgG (16-4321-82, Invitrogen, Custom Stock Dilution: 1 µg/mL) and incubated with nuclei for 1 hour at RT. Rabbit anti-rat (ab102248, Abcam) secondary antibody was used at 1:100 dilution. pA-Tn5 was sourced pre-loaded (C01070001, Diagenode). Prepared libraries were normalized and prepared for sequencing via NovaSeq SP (Illumina) at the NHLBI Genomics Core. Raw reads were processed using CutRunTools/2020062971 with the mm10 mouse genome as reference. EGR2 peaks were called by SEACR v1.3 (Sparse Enrichment Analysis for CUT&RUN)72 with options ‘0.01 non stringent’, all fragment sizes included. EGR2 and Rat IgG CUT&Tag bigwig files for WT and Egr2ΔT nuclei were generated using deepTools v3.5.1 bamCoverage with options ‘extendReads and normalizeUsing RPKM’. To further filter peaks, RPKM normalized signal +/− 200bp from peak summits was calculated by deepTools computeMatrix via sum. Bona fide EGR2 peaks were established as those with EGR2 signal at least 4-fold higher than IgG in WT nuclei and at least 4-fold higher than EGR2 signal in Egr2ΔT nuclei and excluded from the blacklist (mm10). RPKM normalized EGR2 and IgG CUT&Tag signals +/−5kb from filtered EGR2 peaks were visualized with deepTools. EGR2 peaks and signal files were visualized in IGV v2.4.14. Filtered peaks were ranked by SEACR calculated total signal within peaks. EGR2 known motif scoring was evaluated by HOMER v4.11.1 findMotifsGenome.pl with option -size 200 and EGR2 peaks were annotated to genes and features with annotatePeaks.pl (Supplementary Table 3). TH17 genes regulated by BATF, IRF4, MAF, RORγt, and STAT3 were defined using data from Ciofani et al.17, as genes differentially expressed between transcription factor wild-type vs transcription factor knock-out TH17 cells and bound by that transcription factor in wild-type TH17 cells. Heatmaps were drawn using Morpheus (https://software.broadinstitute.org/morpheus/) and networks visualized using Cytoscape 373.
Statistics
Data are presented as mean ± s.e.m. Statistical tests were selected based on data distribution using GraphPad Prism software. Two-tailed Student’s t-test and Mann-Whitney U test were applied to determine statistical significance of parametric and nonparametric datasets, respectively. One-way and two-way ANOVA were used when appropriate. ****P < 0.0001, ***P < 0.001, **P < 0.01 *P < 0.05.
Softwares
Graphics presented in Extended Data Figures were created with BioRender.com.
Data availability
The data that support the findings of the present study are available from the corresponding author upon request (vanja.lazarevic@nih.gov). There are no restrictions on data availability. Raw and processed data are deposited to GEO repository under the following accession numbers: RNA-Seq (GSE168288), FastATAC-Seq (GSE224960) and CUT&Tag (GSE226795).
Code availability
No custom-made code was used in the analysis. The pipelines for analysis can be obtained by e-mailing alejandro.villarino@miami.edu (RNA-Seq), hiroyuki.nagashima@nih.gov (FastATAC-seq) and daniel.chauss@nih.gov (CUT&Tag).
Extended Data
Extended Data Fig. 1. EGR2 reinforces TH17 differentiation program in a RORgt-dependent manner.
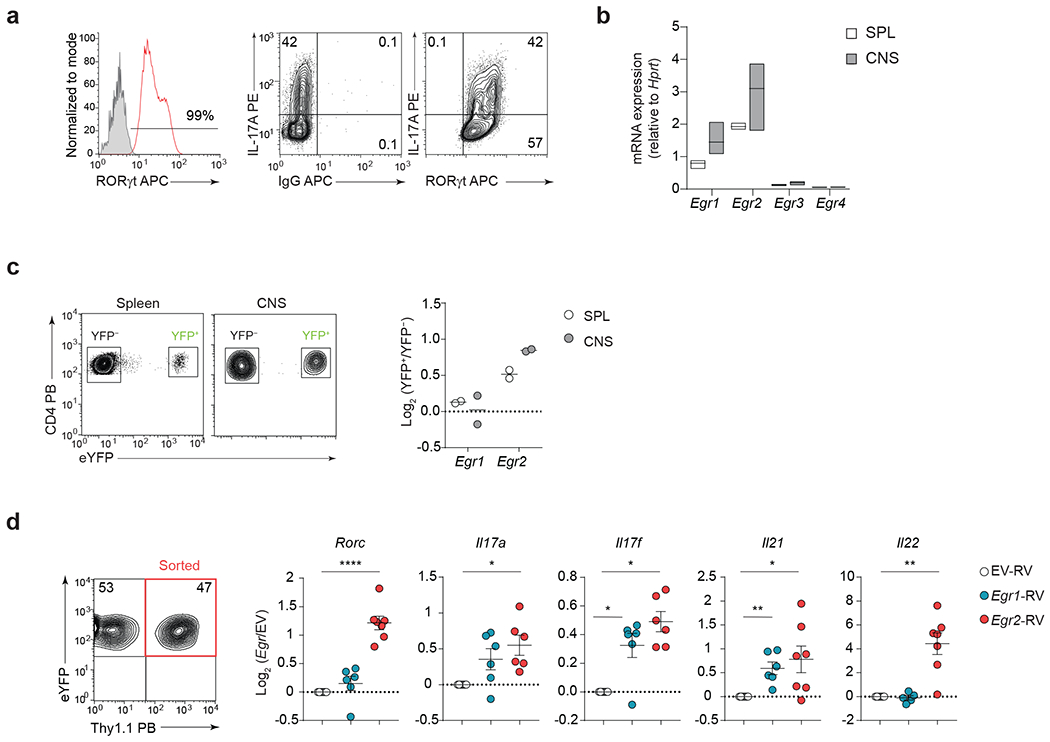
a, Representative flow plots showing the frequencies of RORγt- and IL-17A-expressing 2D2 TH17(β,6,23) cells before the adoptive transfer. Data are representative of n = 3 independent experiments. b, Quantitative RT-PCR analysis of Egr1, Egr2, Egr3, and Egr4 mRNA expression in pathogenic 2D2 WT CD4+ T cells from the CNS of Tcrb−/− mice that received 2D2 WT TH17(β,6,23) cells (20 days post-transfer). Box plot depicts median (line), lower and upper quartiles. Data represent biologically independent replicates from n = 3 independent experiments. c, Quantitative RT-PCR analysis of Egr1 and Egr2 mRNA expression in sorted YFP+ and YFP− CD4+ T cell populations isolated from the spleen and CNS of MOG35-55 immunized Il17a-Cre R26ReYFP fate-mapping mice. Data are presented as the log2 fold-change in the relative expression of Egr1 and Egr2 in YFP+ over YFP− CD4+ T cells. Data represent biologically independent replicates from n = 2 independent experiments. d, Quantitative RT-PCR analysis of Rorc, Il17a, Il17f, Il21 and Il22 mRNA in TH17 (β,6) cells transduced with empty virus (EV-RV), or retroviruses expressing Egr1 (Egr1-RV) or Egr2 (Egr2-RV). Mean values ± s.e.m. are reported. Data represent biologically independent replicates from n = 6 independent experiments. ****P < 0.0001, **P < 0.01, *P < 0.05; two-tailed Student’s t-test.
Extended Data Fig. 2. EGRs function redundantly during TH17 cell differentiation.
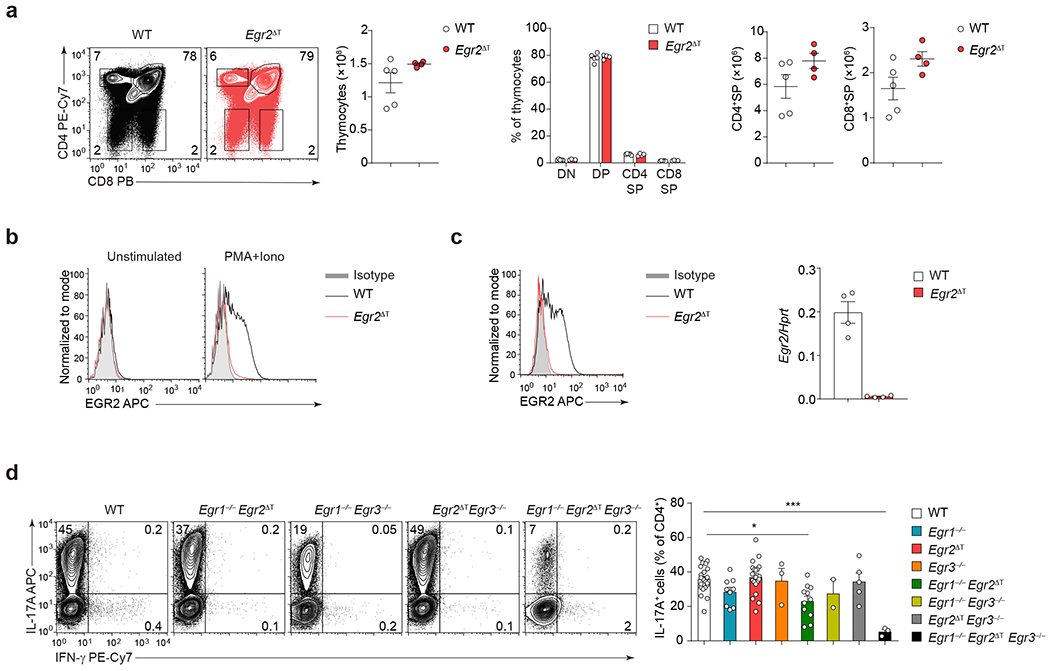
a, Frequency of DN (CD4−CD8−), DP (CD4+CD8+), CD4SP (CD4+CD8−) and CD8SP (CD4−CD8+) thymocytes, and absolute numbers of total thymocytes, CD4SP and CD8SP, in 8-wk old WT (n = 5) and Egr2ΔT (n = 4) mice from 2 independent experiments. b, Histograms showing ex vivo EGR2 protein expression in unstimulated and stimulated (PMA+Iono) splenic WT and Egr2ΔT CD4+ T cells; n = 2 independent experiments. c, EGR2 protein expression (left) and Egr2 mRNA abundance (right) in WT and Egr2ΔT TH17 cells (IL-6 + TGF-β1) following PMA+Iono stimulation. Data represent biologically independent replicates from (n = 4) independent experiments. d, Representative contour plots and bar graphs depict the frequency of IL-17A-producing WT (n = 22), Egr1−/− (n = 9), Egr2ΔT (n = 20), Egr3−/− (n = 3), Egr1−/−Egr2ΔT (n = 11), Egr1−/−Egr3−/− (n = 2), Egr2ΔTEgr3−/− (n = 5) and Egr1−/−Egr2ΔTEgr3−/− (n = 3) CD4+ T cells cultured under TH17-cell polarizing conditions as in c. **P < 0.01, *P < 0.05, two-tailed Student’s t-test. Mean values ± s.e.m. are shown in a, c, d.
Extended Data Fig. 3. EGR2 is not expressed in CD4+ T cells during colitis.
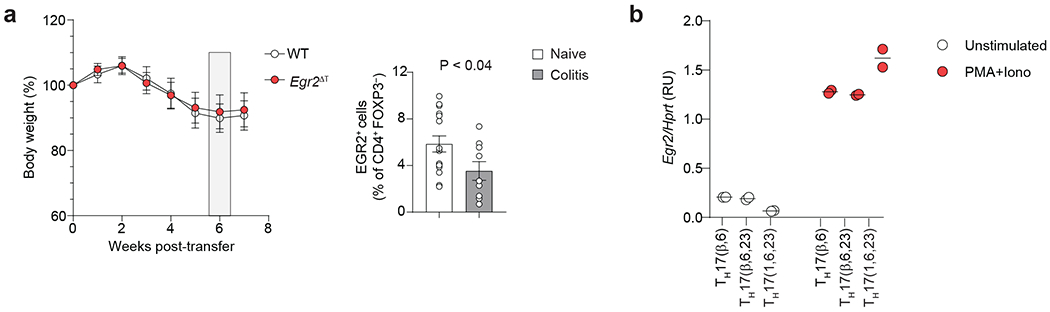
a, (Left) Combined weight loss curve of Rag2−/− recipients after intraperitoneal injection of naive CD45RBhiCD25− CD4+ T cells isolated from WT (n = 15) or Egr2ΔT (n = 15) mice. Data are presented as percent of original body weight (measured on day 0). Combined data from n = 5 independent experiments. (Right) Bar graph depicts the frequency of EGR2+ CD4+FOXP3− T cells isolated from the colon of healthy (naive) WT (n = 15) mice and Rag2−/− recipients of naïve WT CD4+ T cells at 6 weeks post-transfer (colitis) (n = 9); combined data from 5 (naïve) and 3 (colitis) independent experiments. b, RT-PCR analysis of Egr2 mRNA expression in unstimulated or stimulated (PMA+Iono) TH17(β,6), TH17(β,6,23) and TH17(1,6,23) cells. Egr2 mRNA was normalized to the house-keeping Hprt gene. Data represent biologically independent replicates per condition from n = 2 independent experiments. Mean values ± s.e.m. are reported in a-b.
Extended Data Fig. 4. EGR2 does not control chromatin accessibility in TH17 cells.
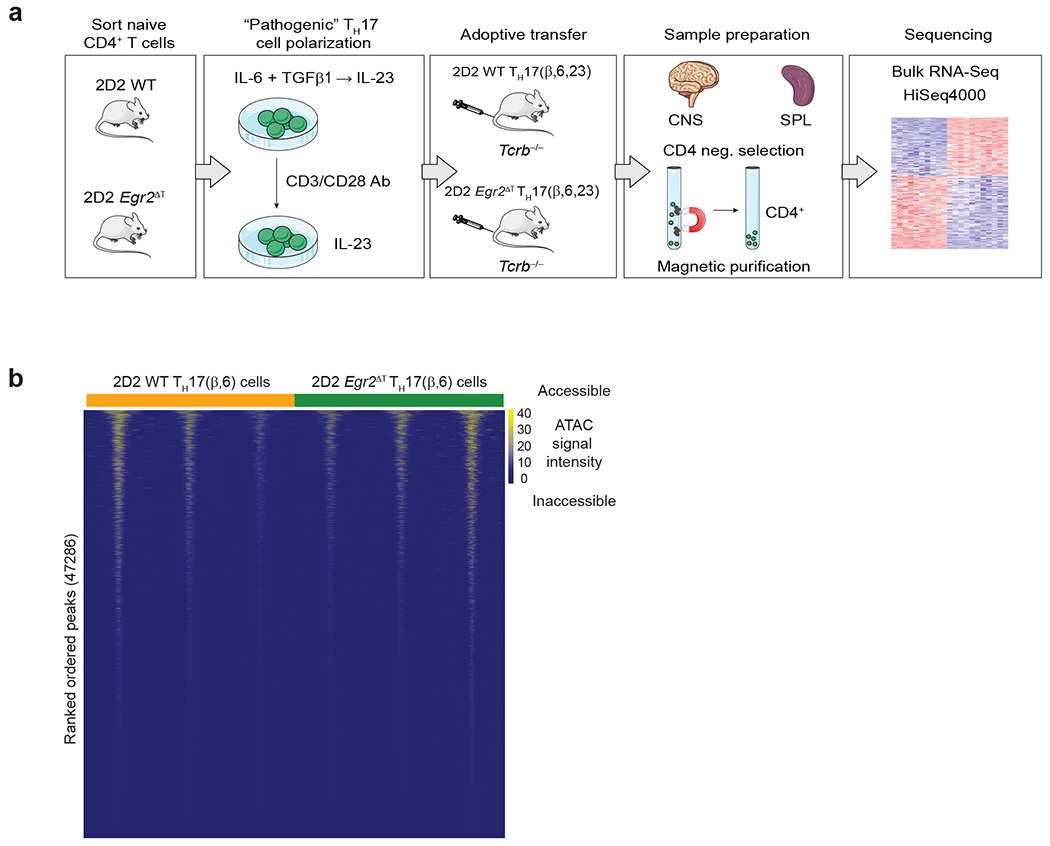
a, Bulk RNA-seq Workflow. Naïve 2D2 WT and 2D2 Egr2ΔT CD4+ T cells were activated and differentiated under pathogenic TH17 cell-polarizing conditions (TH17(β,6,23)) for 5 days. Differentiated TH17(β,6,23) cells were reactivated by plate-bound CD3+CD28 antibodies in the presence of IL-23 for 48h before adoptive transfer into Tcrb−/− recipients. At the peak of the disease (day 20 post-transfer), donor CD4+ T cells were purified from spleens and CNS of the Tcrb−/− recipient mice using CD4 negative selection for RNA profiling and library was sequenced on a HiSeq4000. Three independent experiments were performed. b, Heatmap illustrating dynamics of chromatin accessibility in 2D2 WT and 2D2 Egr2ΔT TH17(β,6) cells (GSE224960). Data represent n = 3 biologically independent replicates per condition.
Extended Data Fig. 5. EGR2 binds to and transactivates Rorc and Il17a promoters.
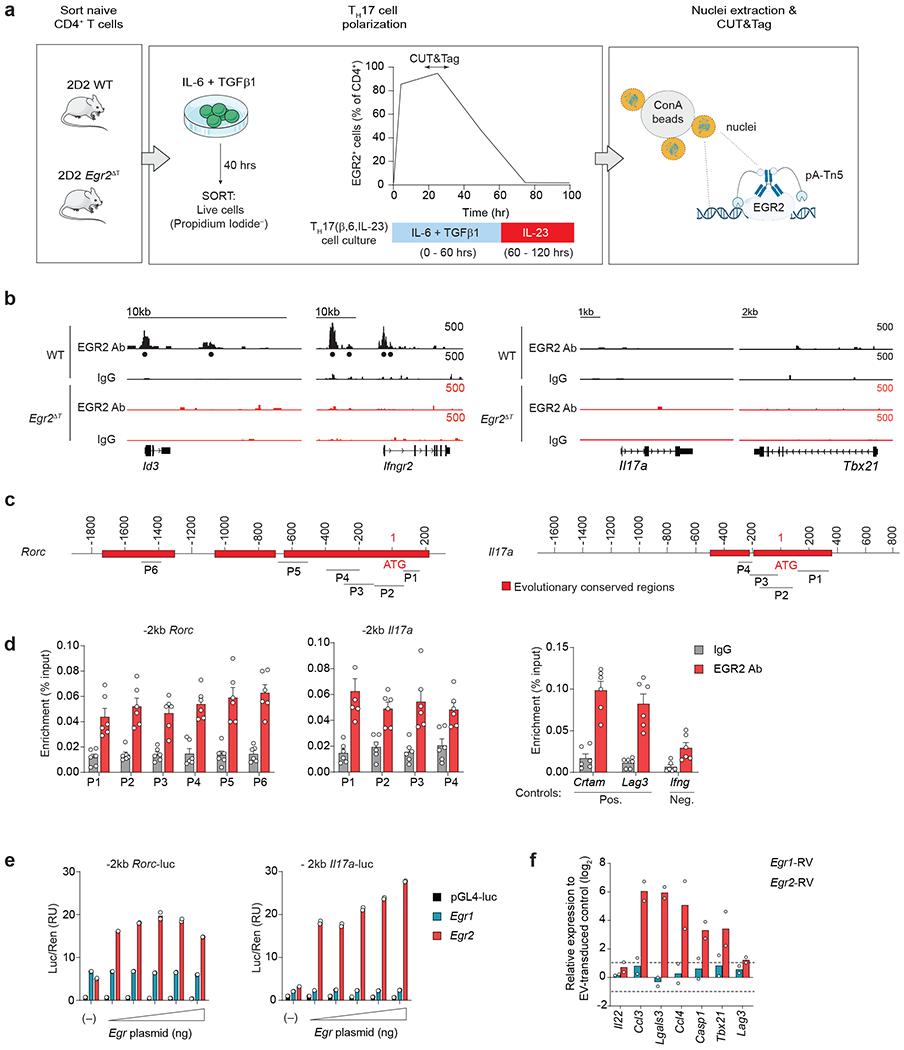
a, CUT&Tag Workflow. CUT&Tag sequencing was performed on nuclei isolated from live (propidium iodide− sorted) 2D2 WT and 2D2 Egr2ΔT TH17(β,6) cells at the peak of EGR2 expression (40h post-activation) using EGR2 antibody or IgG control. b, (Left) Genome browser tracks at Id3 and Ifngr2, showing EGR2 binding in 2D2 WT TH17(β,6) cells. (Right) Genome browser tracks at Il17a and Tbx21 showing no EGR2 binding in 2D2 WT TH17(β,6) cells. c, Evolutionary conserved regions (ECRs) and EGR binding sites within each ECR of Rorc and Il17a genes were analyzed using ECR browser (https://ecrbrowser.dcode.org/) and JASPAR (http://jaspar.genereg.net/). ChIP primers (P) to determine EGR2 binding to predicted EGR binding sites were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). d, EGR2 ChIP-PCR analysis of 2D2 TH17(β,6) cells 48h after activation with plate-bound CD3+CD28 antibodies, showing EGR2-specific binding to Rorc and Il17a promoters. Crtam intron and Lag3 core promoters were used as positive controls. Ifng promoter and IgG control antibody were used as negative controls. Results are presented as percent of input DNA. Data are shown as mean ± s.e.m. and represent biologically independent replicates from n = 3 independent experiments. e, Firefly luciferase activity (normalized to Renilla) driven by the 2kb genomic DNA sequences upstream of the start codon (ATG) of Rorc and Il17a was measured in the presence of increasing doses of Egr1- or Egr2-expressing plasmids in HEK293 cells. Data represent technical duplicates and are representative of n = 3 independent experiments. f, Quantitative RT-PCR analysis of ‘pathogenicity-associated’ genes in TH17(β,6) cells transduced with Egr1-RV or Egr2-RV and normalized to empty virus (EV-RV)-transduced control TH17(β,6) cells. RT-PCR analysis was performed on RNA isolated from sorted retrovirally-transduced cells. Data represent biologically independent replicates per condition from n = 2 independent experiments.
Extended Data Fig. 6. EGR2 is not required for CD4+ T cell activation.
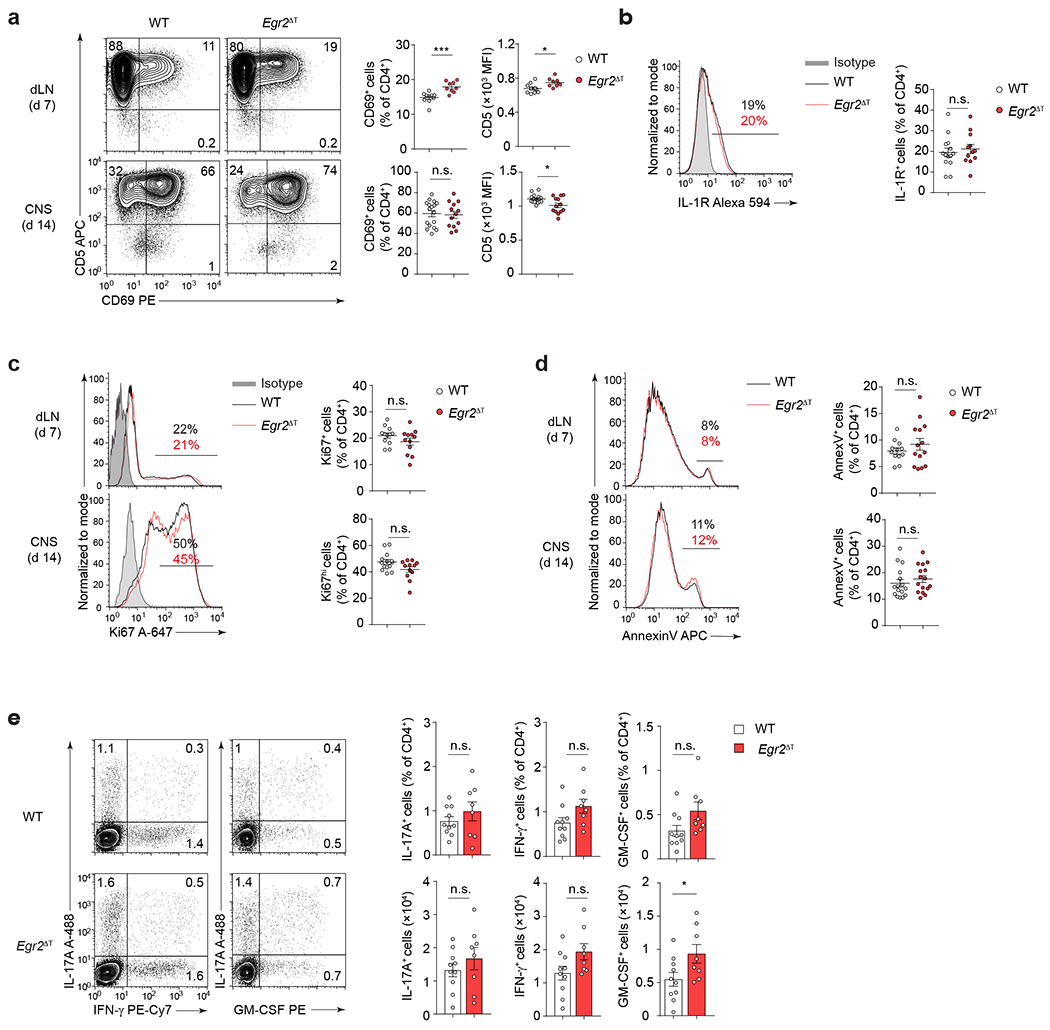
a, CD5 and CD69 protein expression (MFI, frequency) in CD4+ T cells isolated from draining lymph nodes (dLN) and CNS of WT (n = 17) and Egr2ΔT (n = 13) mice following immunization with MOG35-55/CFA and Pertussis toxin. ***P < 0.001, *P < 0.05, n.s. = not significant, two-tailed Mann-Whitney U test. b, IL-1R expression in CNS-infiltrating CD4+ T cells from WT (n = 14) and Egr2ΔT (n = 12) mice 14 days post-immunization as in a. n.s. = not significant, two-tailed Student’s t-test. c-d, Expression of Ki67 marker of proliferation (c) and Annexin V marker of apoptosis (d) in CD4+ T cells isolated from draining lymph nodes and CNS of WT (n = 14, Ki67; n = 16, AnnexinV) and Egr2ΔT (n = 12, Ki67; n = 16, AnnexinV) mice post-immunization as in a. n.s. = not significant, two-tailed Student’s t-test. e, Contour plots depict representative intracellular cytokine staining for IL-17A, IFN-γ and GM-CSF and bar graphs summarize the frequency and the absolute numbers of IL-17A-, IFN-γ- and GM-CSF-producing CD4+ T cells in the draining lymph nodes of WT (n = 10) and Egr2ΔT (n = 8) mice 7 days post-immunization as in a; Data are represented as mean± s.e.m. and are combined from 3 (a-d) and 2 (e) independent experiments.
Extended Data Fig. 7. EGR2 is not required for TH1 cell migration to CNS.
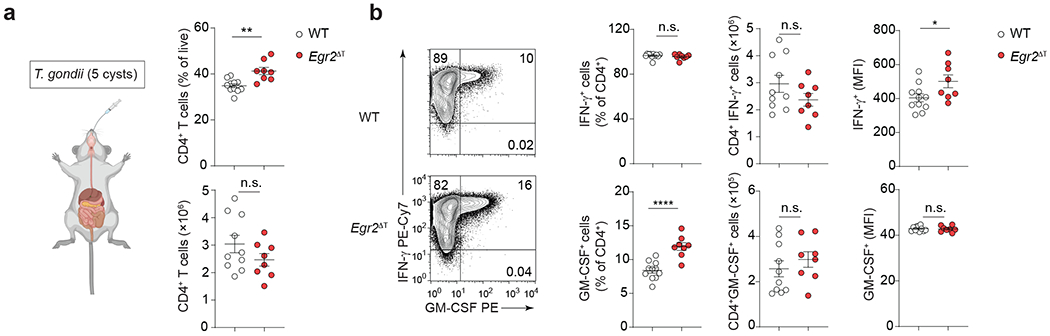
a, Percentage and number of CD4+ T cells in the CNS of Toxoplasma gondii infected WT (n = 10) and Egr2ΔT (n = 8) mice (14 days post-infection). **P < 0.01, n.s. = not significant, two-tailed unpaired Student t-test. b, Cytokine production by CD4+ T cells in spleen and CNS of WT (n = 10) and Egr2ΔT (n = 8) mice (14 days post-infection). Mean values ± s.e.m. are reported, combined data from 2 independent experiments (a-b). ****P < 0.0001, *P < 0.05, n.s. = not significant, two-tailed Student’s t-test.
Extended Data Fig. 8. EGR2 drives regulatory network in pathogenic TH17 cells.
EGR2-regulated module of the TH17 differentiation program controls TH17 cell migration, recruitment of myelomonocytic cells, and the expression of pathogenicity-associated genes.
Extended Data Fig. 9. Gating strategy.
Live cells were identified by their negative staining for the live/dead marker in comparison to FSC-H. Single cells were gated based on their FSC-W versus FSC-H parameters, while lymphocytes were distinguished by their size, using SSC-H versus FSC-H. CD4+ T cells were further characterized by their co-expression of CD4 and TCRβ, and in the intestine, they were further classified as FOXP3− Teff cells or FOXP3+ Treg cells. Monocytes, neutrophils, and dendritic cells were identified using the previously described gating strategy50.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program of the NIH National Cancer Institute, Center for Cancer Research (ZIA BC011765), National Institute of Allergy and Infectious Diseases (ZIA AI001175), National Institute of Diabetes and Digestive and Kidney Diseases (ZIA DK075149 to BA), National Heart, Lung, and Blood Institute, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health. The authors gratefully acknowledge R. Germain, R. Bosselut, A.Singer, and D. Hafler for scientific discussions and critical reading of the manuscript. We would like to thank all members of NCI (EIB) flow cytometry core facility, S. Sharrow, A. Crossman, L. Granger and T. Adams for their expert technical help with flow cytometry and cell sorting. We thank the members of the CCR Sequencing Facility at the Frederick National Laboratory for Cancer Research for their help during sample preparation, sequencing, and data processing. Special thanks to members of the NIAMS Sequencing Core Facility (S. Dell’Orso and F. Naz) and the NIAMS Bioinformatics lab (Biodata Mining and Discovery Section), H.-W. Sun, K. Jiang and A. Uhlman. This work used the computational resources of the NIH High-Performance Computing Biowulf Cluster. We are grateful to M. Lu for technical assistance and genotyping.
Footnotes
Competing Interest Statement
The authors declare no competing interests.
References
- 1.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL & Kuchroo VK Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR & Weaver CT Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM & Stockinger B TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W & O’Shea JJ Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467, 967–971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA & Cua DJ IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201, 233–240 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A & Kuchroo VK Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13, 991–999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T & Cua DJ TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 8, 1390–1397 (2007). [DOI] [PubMed] [Google Scholar]
- 8.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ & Cua DJ The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol 10, 314–324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleinschek MA, Boniface K, Sadekova S, Grein J, Murphy EE, Turner SP, Raskin L, Desai B, Faubion WA, de Waal Malefyt R, Pierce RH, McClanahan T & Kastelein RA Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J Exp Med 206, 525–534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, Evans JG, Cimaz R, Bajaj-Elliott M & Wedderburn LR Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A 107, 14751–14756 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM & Fugger L Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172, 146–155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J & Ouyang W Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445, 648–651 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, Karczewski J, Pezous N, Bek S, Bruin G, Mellgard B, Berger C, Londei M, Bertolino AP, Tougas G, Travis SP & Secukinumab in Crohn’s Disease Study, G. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693–1700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, Blumenschein WM, Judo M, Ayanoglu G, McClanahan TK, Li X & Cua DJ Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 43, 727–738 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Connor W Jr., Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK & Flavell RA A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol 10, 603–609 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, Kuchroo VK, Park H & Regev A Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 163, 1400–1412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R & Littman DR A validated regulatory network for Th17 cell specification. Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D, Park H, Kuchroo VK & Regev A Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myouzen K, Kochi Y, Shimane K, Fujio K, Okamura T, Okada Y, Suzuki A, Atsumi T, Ito S, Takada K, Mimori A, Ikegawa S, Yamada R, Nakamura Y & Yamamoto K Regulatory polymorphisms in EGR2 are associated with susceptibility to systemic lupus erythematosus. Hum Mol Genet 19, 2313–2320 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Riveros C, Mellor D, Gandhi KS, McKay FC, Cox MB, Berretta R, Vaezpour SY, Inostroza-Ponta M, Broadley SA, Heard RN, Vucic S, Stewart GJ, Williams DW, Scott RJ, Lechner-Scott J, Booth DR, Moscato P & Consortium ANMSG A transcription factor map as revealed by a genome-wide gene expression analysis of whole-blood mRNA transcriptome in multiple sclerosis. PLoS One 5, e14176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ & Brant SR Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39, 596–604 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC & Hafler DA Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med 7, 287ra274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ & Stockinger B Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol 12, 255–263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miao T, Raymond M, Bhullar P, Ghaffari E, Symonds AL, Meier UC, Giovannoni G, Li S & Wang P Early growth response gene-2 controls IL-17 expression and Th17 differentiation by negatively regulating Batf. J Immunol 190, 58–65 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Lauritsen JP, Kurella S, Lee SY, Lefebvre JM, Rhodes M, Alberola-Ila J & Wiest DL Egr2 is required for Bcl-2 induction during positive selection. J Immunol 181, 7778–7785 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawson VJ, Weston K & Maurice D Early growth response 2 regulates the survival of thymocytes during positive selection. Eur J Immunol 40, 232–241 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Du N, Kwon H, Li P, West EE, Oh J, Liao W, Yu Z, Ren M & Leonard WJ EGR2 is critical for peripheral naive T-cell differentiation and the T-cell response to influenza. Proc Natl Acad Sci U S A 111, 16484–16489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu B, Symonds AL, Martin JE, Kioussis D, Wraith DC, Li S & Wang P Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J Exp Med 205, 2295–2307 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taillebourg E, Buart S & Charnay P Conditional, floxed allele of the Krox20 gene. Genesis 32, 112–113 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Vacchio MS, Wang L, Bouladoux N, Carpenter AC, Xiong Y, Williams LC, Wohlfert E, Song KD, Belkaid Y, Love PE & Bosselut R A ThPOK-LRF transcriptional node maintains the integrity and effector potential of post-thymic CD4+ T cells. Nat Immunol 15, 947–956 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carleton M, Haks MC, Smeele SA, Jones A, Belkowski SM, Berger MA, Linsley P, Kruisbeek AM & Wiest DL Early growth response transcription factors are required for development of CD4(−)CD8(−) thymocytes to the CD4(+)CD8(+) stage. J Immunol 168, 1649–1658 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Shao H, Kono DH, Chen LY, Rubin EM & Kaye J Induction of the early growth response (Egr) family of transcription factors during thymic selection. J Exp Med 185, 731–744 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jager A, Dardalhon V, Sobel RA, Bettelli E & Kuchroo VK Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 183, 7169–7177 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q & Dong C Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30, 576–587 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, Lee YK, Weaver CT, Yagi R & Lazarevic V The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-gamma-producing T helper 17 cells. Immunity 40, 355–366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, Peng H, Cravens PD, Racke MK & Lovett-Racke AE T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med 206, 1549–1564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO & Weaver CT Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K & Littman DR Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, Li Y, Kelly G, Chakravarty P, Shoaie S, Gutierrez MG & Stockinger B The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 51, 77–89 e76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M & Gaffen SL Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206, 299–311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lazarevic V, Zullo AJ, Schweitzer MN, Staton TL, Gallo EM, Crabtree GR & Glimcher LH The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nat Immunol 10, 306–313 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka A, Maeda S, Nomura T, Llamas-Covarrubias MA, Tanaka S, Jin L, Lim EL, Morikawa H, Kitagawa Y, Akizuki S, Ito Y, Fujimori C, Hirota K, Murase T, Hashimoto M, Higo J, Zamoyska R, Ueda R, Standley DM, Sakaguchi N & Sakaguchi S Construction of a T cell receptor signaling range for spontaneous development of autoimmune disease. J Exp Med 220 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mufazalov IA, Schelmbauer C, Regen T, Kuschmann J, Wanke F, Gabriel LA, Hauptmann J, Muller W, Pinteaux E, Kurschus FC & Waisman A IL-1 signaling is critical for expansion but not generation of autoreactive GM-CSF+ Th17 cells. EMBO J 36, 102–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki Y, Orellana MA, Schreiber RD & Remington JS Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240, 516–518 (1988). [DOI] [PubMed] [Google Scholar]
- 45.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L & Karin N Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 356, 63–66 (1992). [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Kai H, Chang F, Shibata K, Tahara-Hanaoka S, Honda S, Shibuya A & Shibuya K A critical role of LFA-1 in the development of Th17 cells and induction of experimental autoimmune encephalomyelytis. Biochem Biophys Res Commun 353, 857–862 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Rothhammer V, Heink S, Petermann F, Srivastava R, Claussen MC, Hemmer B & Korn T Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J Exp Med 208, 2465–2476 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerrini MM, Okamoto K, Komatsu N, Sawa S, Danks L, Penninger JM, Nakashima T & Takayanagi H Inhibition of the TNF Family Cytokine RANKL Prevents Autoimmune Inflammation in the Central Nervous System. Immunity 43, 1174–1185 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B & Sallusto F C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 10, 514–523 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Kwong B, Rua R, Gao Y, Flickinger J Jr., Wang Y, Kruhlak MJ, Zhu J, Vivier E, McGavern DB & Lazarevic V T-bet-dependent NKp46(+) innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat Immunol 18, 1117–1127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA & Kuchroo VK Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197, 1073–1081 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Topilko P, Schneider-Maunoury S, Levi G, Trembleau A, Gourdji D, Driancourt MA, Rao CV & Charnay P Multiple pituitary and ovarian defects in Krox-24 (NGFI-A, Egr-1)-targeted mice. Mol Endocrinol 12, 107–122 (1998). [DOI] [PubMed] [Google Scholar]
- 53.Tourtellotte WG & Milbrandt J Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nat Genet 20, 87–91 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K & Paul WE The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37, 660–673 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y & Littman DR An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol 5, 64–73 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Chao DT, Linette GP, Boise LH, White LS, Thompson CB & Korsmeyer SJ Bcl-XL and Bcl-2 repress a common pathway of cell death. J Exp Med 182, 821–828 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rogers PR, Dubey C & Swain SL Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. J Immunol 164, 2338–2346 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Rogers PR, Grey HM & Croft M Modulation of naive CD4 T cell activation with altered peptide ligands: the nature of the peptide and presentation in the context of costimulation are critical for a sustained response. J Immunol 160, 3698–3704 (1998). [PubMed] [Google Scholar]
- 59.Bouladoux N, Harrison OJ & Belkaid Y The Mouse Model of Infection with Citrobacter rodentium. Curr Protoc Immunol 119, 19 15 11–19 15 25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O’Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME & Belkaid Y Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 31, 772–786 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Solis NV & Filler SG Mouse model of oropharyngeal candidiasis. Nat Protoc 7, 637–642 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, Chauss D, Harrison OJ, Alejo J, Williams DW, Pittaluga S, Lee CR, Bouladoux N, Swamydas M, Hoffman KW, Greenwell-Wild T, Bruno VM, Rosen LB, Lwin W, Renteria A, Pontejo SM, Shannon JP, Myles IA, Olbrich P, Ferre EMN, Schmitt M, Martin D, Genomics, Computational Biology, C., Barber DL, Solis NV, Notarangelo LD, Serreze DV, Matsumoto M, Hickman HD, Murphy PM, Anderson MS, Lim JK, Holland SM, Filler SG, Afzali B, Belkaid Y, Moutsopoulos NM & Lionakis MS Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 371 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rau A, Gallopin M, Celeux G & Jaffrezic F Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics 29, 2146–2152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W & Robinson MD Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8, 1765–1786 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, Majeti R & Chang HY Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu H, Luo X, Qian J, Pang X, Song J, Qian G, Chen J & Chen S FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7, e52249 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W & Liu XS Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H & Glass CK Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu Q, Liu N, Orkin SH & Yuan GC CUT&RUNTools: a flexible pipeline for CUT&RUN processing and footprint analysis. Genome Biol 20, 192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meers MP, Tenenbaum D & Henikoff S Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B & Ideker T Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of the present study are available from the corresponding author upon request (vanja.lazarevic@nih.gov). There are no restrictions on data availability. Raw and processed data are deposited to GEO repository under the following accession numbers: RNA-Seq (GSE168288), FastATAC-Seq (GSE224960) and CUT&Tag (GSE226795).