

Abstract
Mass spectrometry-based bottom-up proteomics is rapidly evolving and routinely applied in large-scale biomedical studies. Proteases are a central component of every bottom-up proteomics experiment, digesting proteins into peptides. Trypsin has been the most widely applied protease in proteomics due to its characteristics. With ever-larger cohort sizes and possible future clinical application of mass spectrometry-based proteomics, the technical impact of trypsin becomes increasingly relevant. To assess possible biases introduced by trypsin digestion, we evaluated the impact of eight commercially available trypsins in a variety of bottom-up proteomics experiments and across a range of protease concentrations and storage times. To investigate the universal impact of these technical attributes, we included bulk HeLa cell lysate, human plasma, and single HEK293 cells, which were analyzed over a range of selected reaction monitoring (SRM), data-independent acquisition (DIA), and data-dependent acquisition (DDA) instrument methods on three LC-MS instruments. The quantification methods employed encompassed both label-free approaches and absolute quantification utilizing spike-in heavy-labeled recombinant protein fragment standards. Based on this extensive data set, we report variations between commercial trypsins, their source, and their concentration. Furthermore, we provide suggestions on the handling of trypsin in large-scale studies.
Introduction
The field of mass spectrometry (MS)-driven proteomics has had a major impact on biomedical research. Its application in precision medicine efforts has become more prominent in the past years, ranging from plasma proteome analysis of liquid biopsies to single-cell proteomics by MS (scp-MS).1,2 Increasingly larger cohorts are being analyzed and new techniques developed and standardized, while in unison moving the field closer toward the realms of clinical application.3−9 Especially high reproducibility and robustness are key to drive MS forward into clinical practice and impactful biological discoveries.
Due to their systemic representation and ease of access, biofluid-based biopsies such as serum and plasma are attractive samples for predictive biomarker panels that can be translated into clinical tests. However, for global MS-driven analysis, the complexity of the plasma proteome with its estimated 1010 dynamic range provides great challenges.10
Another technology of increasing interest within the realm of MS is scp-MS. It has emerged as a promising novel technique, which is moving away from being predominantly a technical exercise to having real-life biomedical implications.11,2,12 Here, the extremely limited input material and low abundance of proteins in an individual cell present major technical challenges, as summarized in refs (2, 11, 13−15). These challenges somewhat oppose the hurdles that the technology is facing in the plasma proteome field.
Both plasma and scp-MS have seen tremendous advances with the application of different MS acquisition techniques, labeling techniques, and sample preparation.8,16 When it comes to sample preparation, the field of plasma proteomics has seen major advancements, putting large efforts into optimizing and standardizing preparation techniques.7,17−19 The field of single-cell proteomics published its first joint recommendations in early 2023.20
However, irrespective of sample type, the vast majority of global, data-driven LC-MS proteomics relies on a bottom-up approach, where proteases are the driving force to turn denatured proteins into peptides. Trypsin has been used predominantly for this task, even though other proteases have been explored to increase proteome depth and quantification.21−24 Trypsin is a serine protease that is produced in the pancreas of most vertebrates. It has a high specificity for the cleavage C-terminal of lysine and arginine.25 Trypsin generates peptides that are favorable for electrospray ionization (ESI) MS approaches, combined with online C18 reverse-phase liquid chromatography.26 Furthermore, tryptic peptides benefit from the widely used CID and HCD fragmentation.27,28 However, as an enzyme, trypsin is susceptible to digestion and reconstitution conditions, which can impact enzymatic activity and digestion efficiency.17,29−31 Moreover, the choice of trypsin can impact the identified number of peptides and the number of missed cleavages.32,33 There have been several publications dating back a decade comparing trypsin performance of different vendors. In general, a variation between trypsins of different vendors has been reported.34,35 Others also looked into the differences between bovine and porcine trypsins.36 In the field of scp-MS, a significant impact of the protease concentration has been shown for two trypsins in a first publication.37
Today, there is a large variety of trypsins by different vendors on the market that are mainly recombinantly expressed or extracted from porcine pancreatic extracts. With an ever-increasing number of biological samples included in clinical cohorts, the relevance of exploring to what extent trypsin might act as a technical variable, and as a consequence, introducing batch effects, becomes more important. Here, the effect of trypsin vendors or storage time on batch effects must be addressed. This becomes even more relevant once the field transitions into real-time clinical testing, differing vastly from a well-planned analysis of retrospective clinical cohorts. Finally, another relevant aspect is the impact of trypsin concentration, especially in terms of peptide quantification and when comparing multiple data sets.
To address the impact of trypsin regarding future clinical applications of bottom-up proteomics, we hereby investigate to what extent we can detect experimental variations due to trypsin source, vendor, and storage time (Figure 1). Eight trypsins from four vendors were assessed in HeLa bulk cell lysate, human plasma, and single HEK293 cells. To assess the impact on different experimental setups, we evaluated data-dependent acquisition (DDA), data-independent acquisition (DIA), and selected reaction monitoring (SRM) to represent all commonly used data acquisition approaches. Furthermore, LFQ and absolute quantification were explored by incorporating heavy-labeled standards for the absolute quantification of 122 plasma proteins with SRM. Combined, we aim to provide an overview of the influence of trypsin on reproducibility and quantification when transitioning further into the clinical application of MS-driven proteomics.
Figure 1.
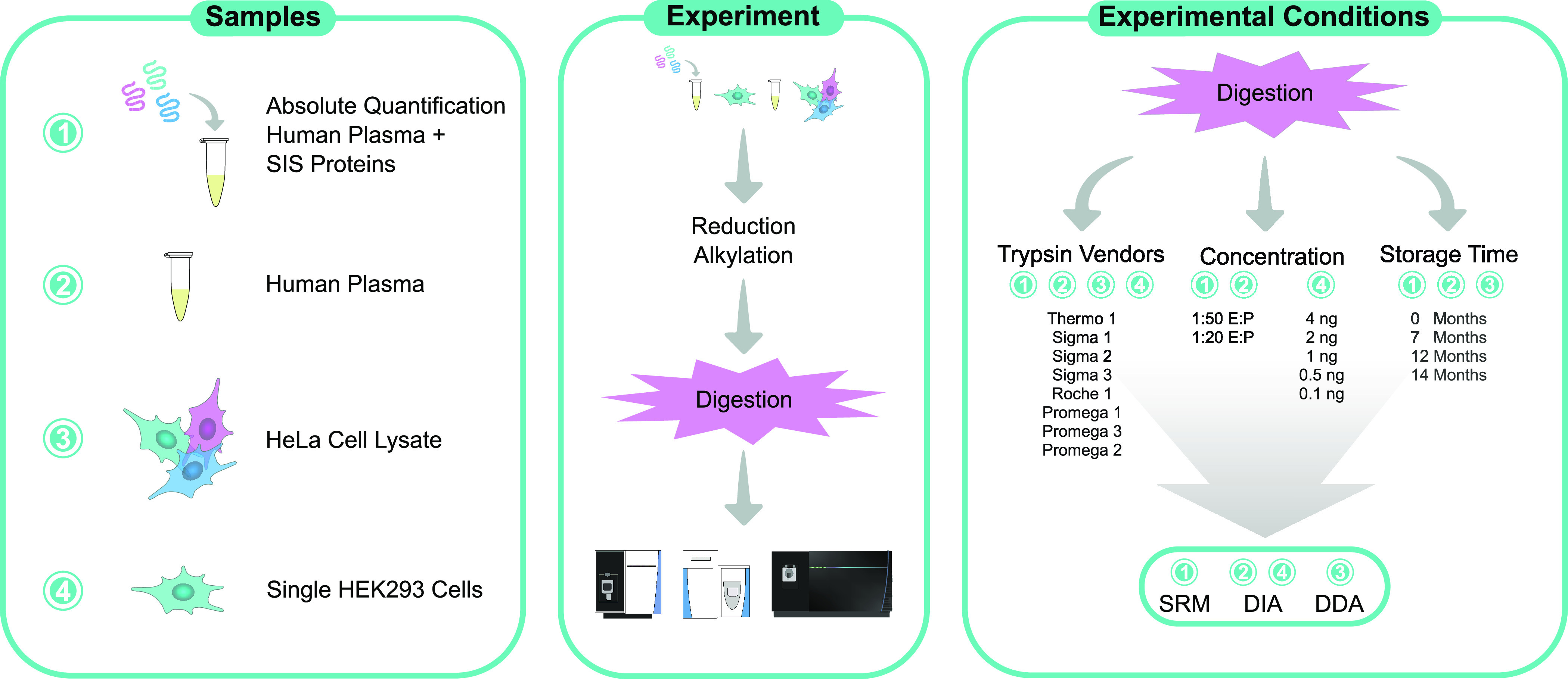
Overview of the experimental setup showing all sample types, their respective LC-MS analysis methods, the specific trypsins used, trypsin concentrations, and storage times that were investigated.
Experimental Design
This section summarizes the experimental design of this study. A detailed ‘Materials and Methods’ section can be found in the Supporting Information. Eight commercially available trypsins that were regularly described in the literature were compared within this work (Table 1). All trypsins corresponded to the porcine trypsin sequence. Three products were extracted from the porcine pancreas and five products were recombinantly expressed. Trypsins were stored and reconstituted according to vendors’ instructions, and samples were digested two weeks after the trypsins were received. To assess storage time-related impacts of trypsin, “Thermo 1“ was stored lyophilized for 0, 7, 12, and 14 months prior to sample preparation at the recommended temperature without interruption. Promega 1–3, “Sigma 3”, and “Thermo 1” were compared for single-cell experiments and stored at the recommended temperature for up to ∼4 weeks before use.
Table 1. Summary of Compared Trypsins.
| ID | vendor | product no. | source |
|---|---|---|---|
| Promega 1 | Promega | V5111 | porcine pancreatic extracts |
| Promega 2 | Promega | V5280 | porcine pancreatic extracts |
| Promega 3 | Promega | VA9000 | expressed in Pichia pastoris |
| Roche 1 | Roche | 3708985001 | expressed in Pichia pastoris |
| Sigma 1 | Sigma–Aldrich | EMS0006 | expressed in Pichia pastoris |
| Sigma 2 | Sigma–Aldrich | EMS0007 | expressed in Pichia pastoris |
| Sigma 3 | Sigma–Aldrich | EMS0004 | expressed in Pichia pastoris |
| Thermo 1 | Thermo Fisher Scientific | 90058 | porcine pancreatic extracts |
Sample Preparation
Trypsin from four different vendors was used to digest four replicates of human plasma and human plasma spiked with 201 stable isotope-labeled protein epitope signature tags (SIS-PrESTs, 13C and 15N labeled) at 1:50 and 1:20 enzyme to protein (E:P) ratios. SIS-PrESTs were spiked into human plasma at approximately 1:1 level to the corresponding endogenous protein in 1 μL of human plasma for absolute quantification of human plasma proteins (Supporting Table 1). SIS-PrESTs are recombinantly expressed protein standards that are up to 149 amino acids (AA) long.38 HeLa cell lysate was digested in a 1:50 E:P concentration. Each replicate contained 50 μg of protein derived from either HeLa cell lysate, 1 μL of human plasma, or 1 μL of human plasma spiked with SIS-PrESTs.
For the single-cell analysis, 187 single HEK293 cells were subjected to FACS sorting, and the cell lysate was digested with Promega 1–3, “Thermo 1”, and “Sigma 3” with 2 ng of trypsin per cell. For the comparison of trypsin concentrations, “Promega 2” was prepared in 0.1, 0.5, 1, 2, and 4 ng per cell.
Liquid Chromatography and Mass Spectrometers
Human plasma spiked with SIS-PrESTs was analyzed with an SRM assay that included 122 proteins with 253 peptides. SRM assays were run on an Ultimate 3000 nano-LC (Thermo Fisher Scientific) with a TSQ Altis (Thermo Fisher Scientific) MS. An Acclaim PepMap 100 trap column (75 μm × 2 cm, C18, 3 μm, 100 Å, Thermo Scientific) was used together with an analytical PepMap RSLC C18 column (150 μm × 15 cm, 2 μm, 100 Å, Thermo Fisher Scientific) on an EASY-Spray ion source.
HeLa cell digest was analyzed with DDA and plasma digest with a DIA method. Both methods were run on a Q-Exactive HF (Thermo Fisher Scientific) with an Ultimate 3000 nano-LC. 2 μg of the sample was injected into an Acclaim PepMap 100 trap column (75 μm × 150 mm, C18, 3 μm, 100 Å, Thermo Scientific) and separated on a 40 min linear gradient by an EASY-Spray HPLC column (75 μm × 250 mm, 2 μm, C18, 100 Å, Thermo Fisher Scientific).
The peptides derived from single cells were separated with the uPAC Neo Low Load analytical column, which was connected to the Ultimate 3000 RSLCnano system in a single-column setup. MS spectra were obtained with our recently developed wide window HRMS1 (WISH)–DIA method that is described in detail here.39,40 The FAIMS Pro interface was operated at a compensation voltage of −45 V connected to an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific).
Data Processing
LC-SRM/MS raw data was analyzed in Skyline.41 LC-DIA/MS raw files from plasma and single cells were analyzed using DirectDIA within the Spectronaut 17 environment. LC-DDA/MS raw files were analyzed using MaxQuant.42 Output tables were further analyzed in R.
Data Availability
DIA, SRM, and DDA raw files together with SRM Skyline documents, spectral libraries, and standard curves were uploaded to Panorama Public.43 Data is available through https://panoramaweb.org/Trypsin_Comparison.url with the Proteome-Xchange ID PXD042450.
Results and Discussion
DDA HeLa Cell Digest
HeLa cell lysate was digested in quadruplicate with eight commercially available trypsins at a 1:50 (E:P) ratio. No significant differences between the mean protein or peptide levels of the eight trypsins were identified (Figure 2A). On average, 14.4% (min 11.6% and max 20.6%) missed cleavages were detected for all peptides with clear statistical differences between the different trypsin manufacturers (Figure 2B, Supporting Table 2). While the Promega trypsins displayed a consistent number of missed cleavages, we could observe substantial variation between the Sigma trypsins. The lowest number of missed cleavages was identified in “Roche 1” (11.6%), and the highest number of missed cleavages resulted from digestion with “Sigma 2” (20.6%). Therefore, the identified number of peptides seems to be consistent between the eight different trypsins. However, the nature of identified peptides seems to vary between products, which could impact the quantification when comparing across different trypsins.
Figure 2.
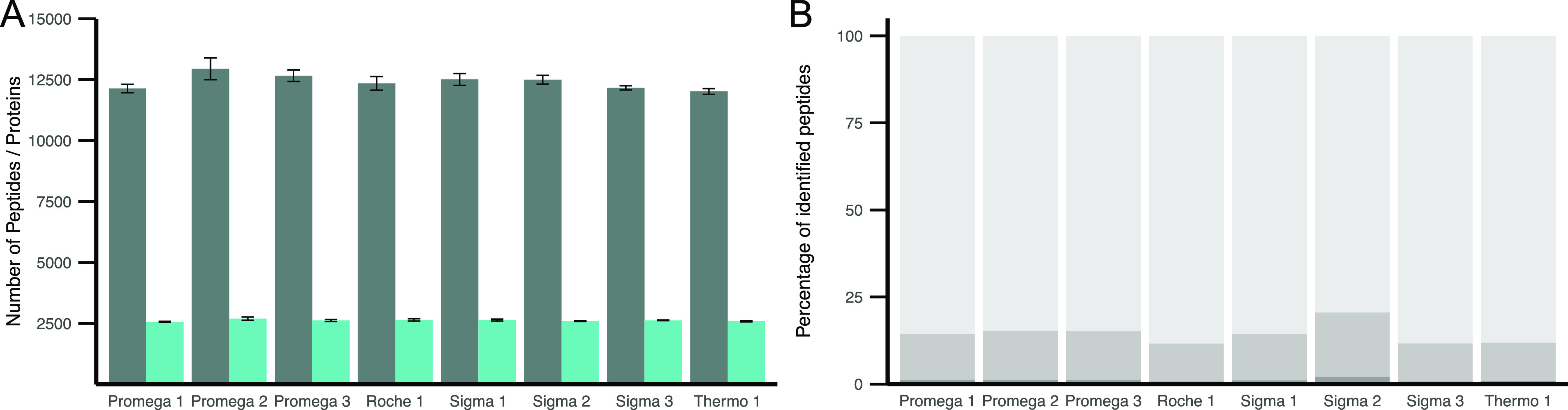
HeLa cell lysate digested with eight commercially available trypsins in a 1:50 E:P ratio. (A) Mean identified number of peptides (gray) and proteins (turquoise) out of four digestion replicates. Error bars indicate standard error (SE). No significant differences (<0.05) based on multiple t-test corrected for multiple testing by Bonferroni. (B) Percentage of missed cleavages in peptides identified in ≥3 replicates. 0 (light gray), 1 (medium gray), and 2 (dark gray) missed cleavages for each trypsin shown.
DIA Label-Free Plasma Analysis
To further compare the eight commercially available trypsins, we assessed their performance in a 1:20 and 1:50 E:P ratio in human plasma in triplicates acquired in DIA. Significant differences between the E:P ratio could be identified for the detected number of peptides but not proteins (Figure 3A, assessed by t-test corrected by Bonferroni). Before applying quality control measures as described in the methods section, we quantified 402 ± 15.4 proteins, which were filtered to 317 ± 13.5 to ensure reliable quantitative comparison (Figure 3B). Surprisingly, significantly more peptides were identified by all trypsins when using a higher E:P ratio (p = 0.0104, Supporting Table 3). This difference could also be observed at the protein level for six out of eight trypsins. The different trypsins delivered, for most parts, a homogeneous set of identified peptides (Figure 3C). The number of unique peptides identified by only one protease was below 30 except for “Sigma 2”, which displayed a surprisingly high number of unique peptides. This could potentially be related to the highest number of missed cleavages identified in the DDA and DIA data sets (Figure 3D). All trypsins extracted from the porcine pancreas displayed a lower number of unique peptides in lower E:P ratio. In contrast, four out of five recombinantly expressed trypsins displayed the opposite behavior. This relationship differed from the missed cleavages, which were substantially higher in the 1:50 than the 1:20 E:P ratio. This comparison of eight trypsins suggested that the peptide and protein numbers between the different proteases show only slight variation. However, an overall trend suggested higher proteome coverage at a 1:50 E:P ratio, which went hand in hand with an increasing number of missed cleaved peptides due to protease concentration (Supporting Figure 1).
Figure 3.
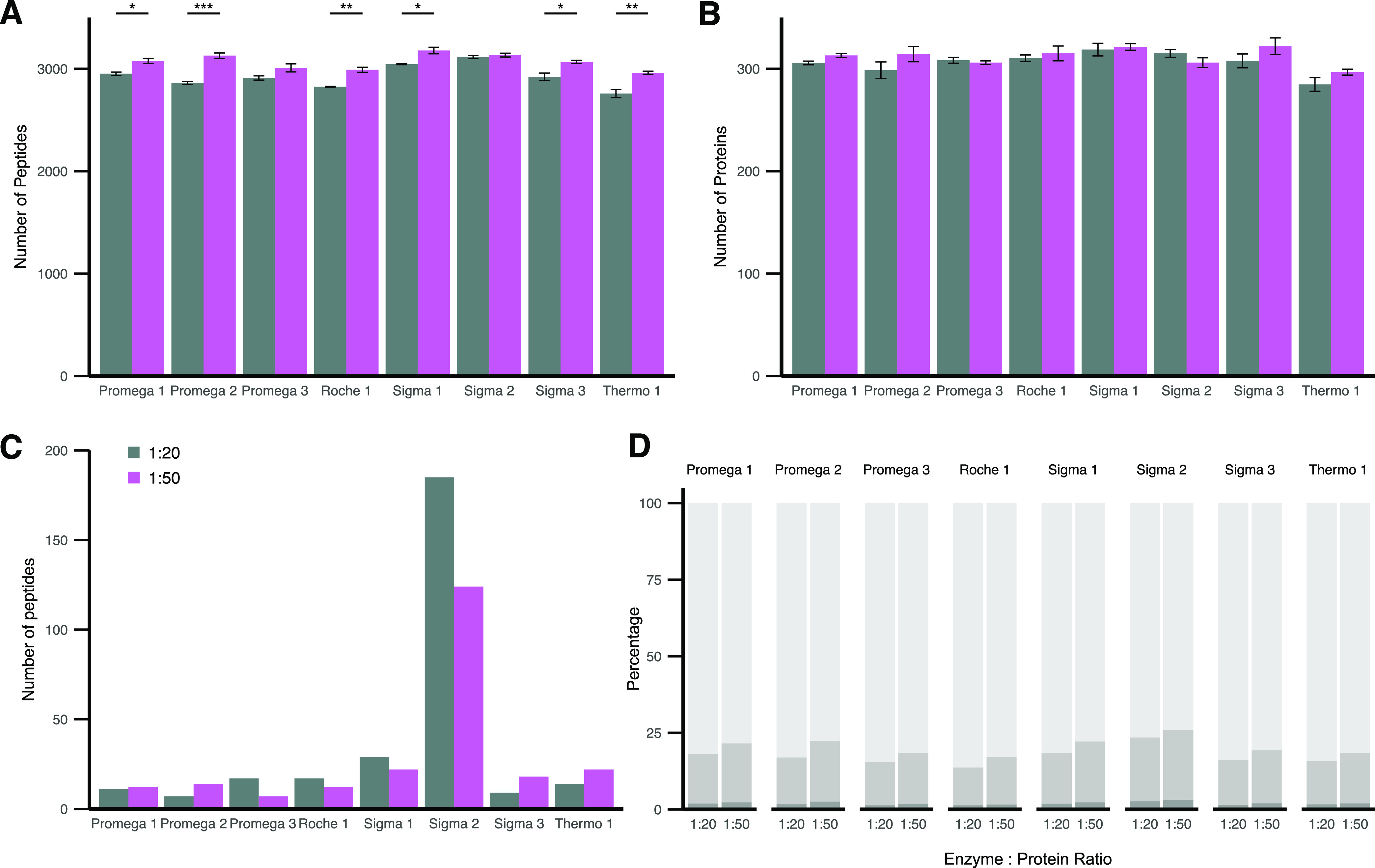
Human plasma digested with eight commercially available trypsins in a 1:20 (gray) and 1:50 (purple) E:P ratio. (A) Mean identified number of peptides out of three digestion replicates. (B) Mean identified number of proteins out of three digestion replicates. A–B Error bars indicate SE. Significant differences based on t-test corrected for multiple testing by Bonferroni are shown (*p < 0.05, **p < 0.01, and ***p < 0.0001). (C) Peptides only identified by the respective trypsin (≥50% of replicates) and not present in other trypsins. (D) Percentage of missed cleavages in peptides identified in ≥50% of replicates. 0 (light gray), 1 (medium gray), and 2 (dark gray) missed cleavages for each trypsin shown.
To assess the quantitative impact of trypsin, we first investigated the proteome depth that could be covered by each protease and the percentage of the total identified plasma proteome (361 proteins) covered by all trypsins (Figure 4A). While trypsin at a 1:20 E:P ratio could identify a mean of 84.1 ± 3.6% of the overall identified proteome (361 proteins), a 1:50 E:P ratio displayed a trend toward higher proteome coverage in six out of eight trypsins with a mean coverage of 85.7 ± 2.42%. “Thermo 1” displayed the overall lowest proteome coverage of 76.7% identified proteome in a 1:20 E:P ratio. This was in line with the previous results that also displayed the lowest number of peptides and proteins for “Thermo 1”.
Figure 4.
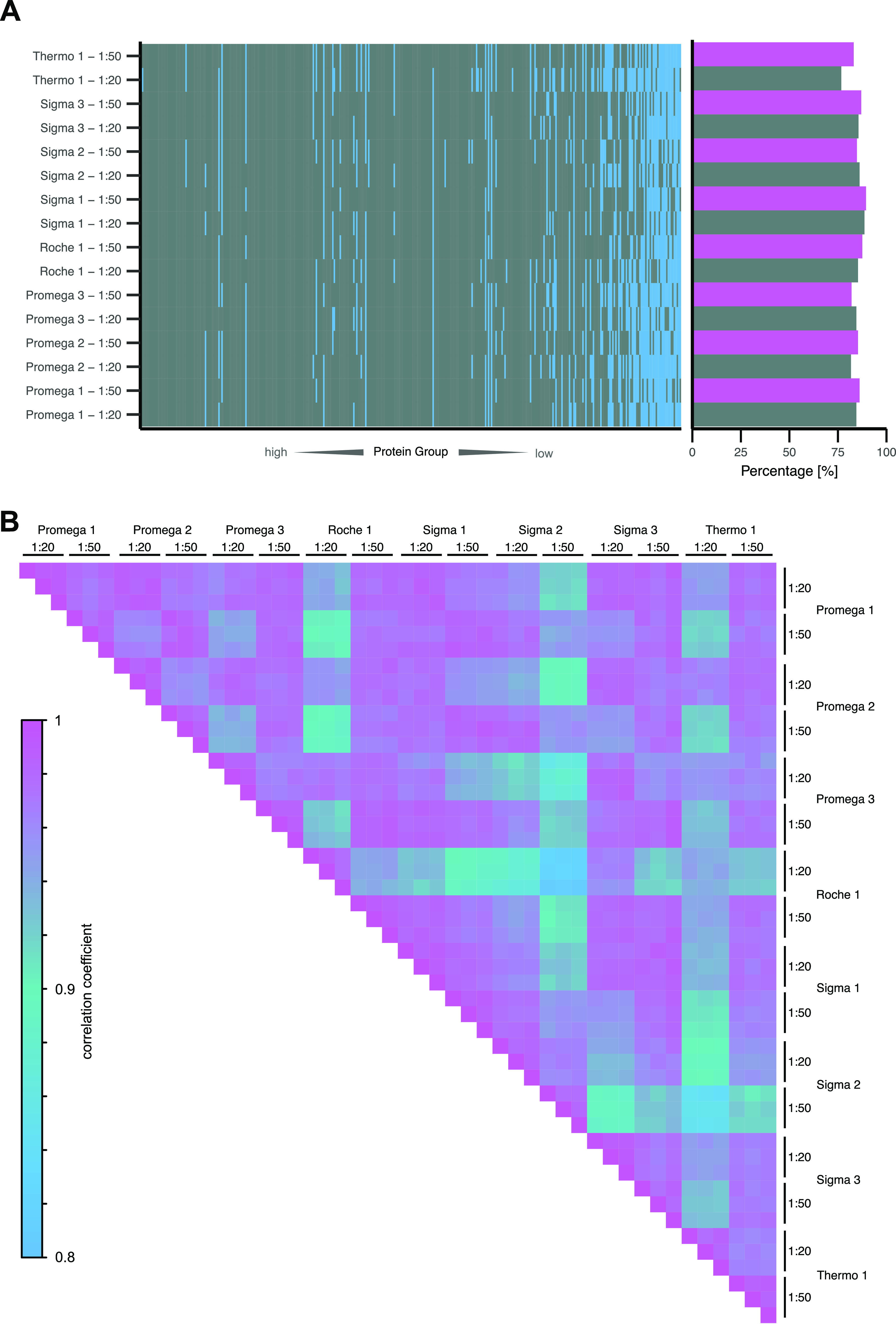
Proteome depth and quantitative correlation of plasma samples acquired in DIA and digested with eight commercially available trypsins in a 1:20 (gray) and 1:50 (purple) E:P ratio. (A) All protein groups identified in the total data set order by median quantity from highest (left) to lowest (right). Protein groups identified in ≥2 replicates are colored gray, and protein groups not identified are colored blue. Percentage of identified protein groups of the total number of identified protein groups in the data set shown to the right. (B) Pearson correlation between 1928 quantified peptides in three digestion replicates of eight trypsins in two different digestion concentrations. Peptide quantities are log2-scaled.
By Pearson correlation of 1928 peptides quantified in each replicate of the DIA data set, we assessed the LFQ quantitative performance of each trypsin (Figure 4B). Intrareplicate correlations were very high, suggesting that the quantitative impact of sample preparation was neglectable. Besides that, all replicates showed a correlation above 0.8. However, “Sigma 2” displayed lower correlation coefficients at the 1:50 E:P ratio toward other proteases, which can be compensated for by a 1:20 E:P ratio. “Roche 1” and “Thermo 1” displayed a lower correlation coefficient at a 1:20 E:P ratio, which differed from the 1:50 E:P ratio. Therefore, the protease concentration seemed to impact the quantitative correlation more strongly than the choice of protease. However, unique proteases such as “Sigma 2” displayed an overall lower agreement with other proteases in terms of quantitative performance.
SRM SIS Protein-Based Plasma Protein Quantification
The trypsin concentration seemed to strongly impact the quantitative compatibility of plasma samples analyzed in LFQ DIA data. We further validated these impacts by spiking plasma with heavy-labeled protein standards (SIS-PrESTs) and quantified 223 peptides of 108 plasma proteins with absolute precision. The peptide concentration was determined in ratio to standard. We could observe a median CV of 4.4% between quadruplicates, suggesting high technical reproducibility. Overall, a digestion with a 1:20 E:P ratio led to significantly lower CVs (p = 0.0191) compared to a 1:50 E:P ratio (Supporting Table 4, Figure 5A). Similar to the DIA data set, we assessed the quantitative agreement of the different trypsins by Pearson correlation (Figure 5B). Overall, very high agreement between the quantified peptides could be observed. Interestingly, the impact of trypsin concentration was reduced in comparison to the LFQ DIA quantification. The targeted absolute quantification assays did not seem to be as susceptible to the impact of different trypsins and concentrations of trypsin as LFQ DIA sample sets.
Figure 5.

Quantitative variation of absolute quantified peptides based on ratio to standard to SIS-PrESTs related to commercially available trypsins and a 1:20 (gray) and 1:50 (purple) E:P ratio. (A) CV of 223 peptides quantified in quadruplicates. (B) Pearson correlation of log2 ratio to standard between 0.1 and 10 of 223 peptides identified in eight trypsins and two trypsin concentrations.
Digestion of Single Cells
To assess whether trypsin is impacting scp-MS in the same manner as high-load samples, 187 single HEK293 cells were digested with Promega 1–3, “Sigma 3”, and “Thermo 1”. While a high degree of technical consistency could be observed for all trypsins, we did observe differences on the quantitative performance of the five different trypsins, with “Promega 1”, “Promega 2”, and “Sigma 3” showing the highest similarity (Figure 6A). The overall variation between single-cell peptide quantities of different trypsins showed no clear differences, with an average CV between 20.2 and 28.0% (Figure 6B). Interestingly, the number of peptides identified in single cells varied mildly, but significantly, between two groups of trypsins. “Promega 1” displayed the lowest number of peptides and “Thermo 1” displayed the highest number (Figure 6C). This trend did not correspond to the above-described variation observed in high-load samples. As we observed a strong impact of the trypsin concentration on quantitative comparisons, we assessed five trypsin concentrations to evaluate this effect at the single-cell level. To this end, single cells were digested with 0.1, 0.5, 1, 2, and 4 ng using only “Promega 2”. “Promega 2” was chosen based on its widespread use in previous literature. Increasing the protease concentration from 0.1 to 0.5 ng boosted the peptide numbers moderately, yet significantly (p = 1.68 × 10–16) (Supporting Figure 2). However, for higher concentrations, no clear trend toward further increased peptide numbers could be observed. However, similar to the observations in high-load samples, the number of missed cleavages was reduced with increased trypsin concentration (Figure 6D). We could observe a mean decrease of missed cleavages of 11.3% between the highest and lowest concentrations. In summary, the choice of trypsin seemed to impact scp-MS experiments differently than high-load samples. However, we could report that both scp-MS and high-load samples displayed lower numbers of missed cleavages with higher trypsin concentrations.
Figure 6.
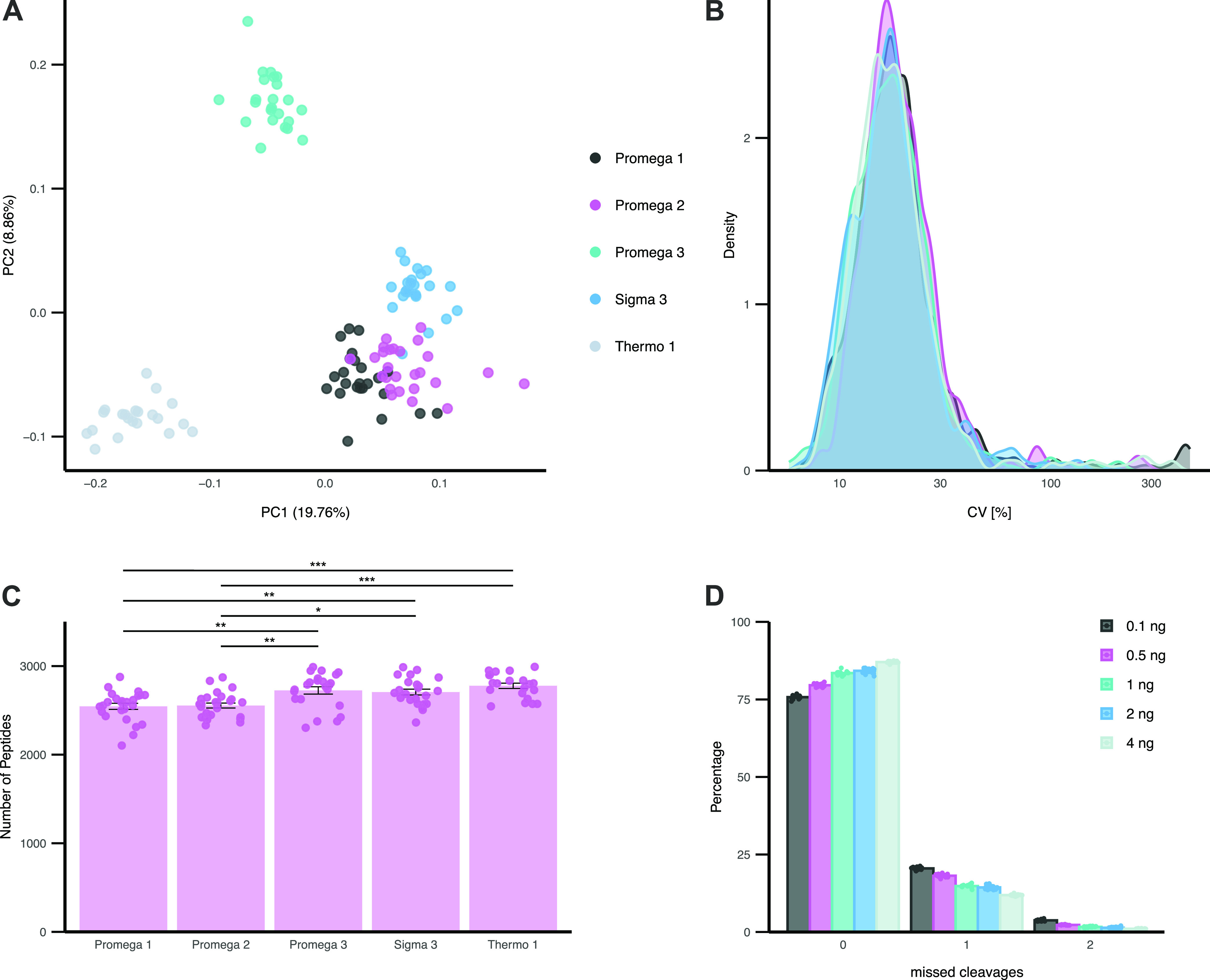
Single HEK293 cell proteomics profiling. (A) PCA of single HEK293 cells digested with five commercially available trypsins. PCA based on peptide level quantities of 610 peptides detected in all cells without imputation. (B) CV of 610 peptide quantities identified in all cells. (C) Mean identified number of peptides quantified for each commercial trypsin. Each cell is highlighted individually. Error bars indicate SE. Significant differences based on t-test corrected for multiple testing by Bonferroni are shown (*p < 0.05, **p < 0.01, and ***p < 0.0001). (D) Percentage of missed cleavages in single cells for 0.1, 0.5, 1, 2, and 4 ng of “Promega 2”. The mean of all single cells is shown as a bar, with replicate cells shown individually.
Trypsin Source Could Be a Significant Mediator of Trypsin Performance
In this work, we compared trypsins that were recombinantly expressed to those extracted from porcine pancreas as described in Table 1. To evaluate whether the source of purified trypsin had a significant effect on its performance, we searched the plasma samples acquired in DIA with the respective expression source proteome as background to the human proteome together with the AA sequence of trypsin. Overall, the number of peptides corresponding to the trypsin source made up 14–26 different proteins (Supporting Figure 3). In total, it contained 32 porcine and 35 P. pastoris proteins. The number of porcine peptides per trypsin was slightly higher than the number of P. pastoris peptides. Furthermore, we identified between 5 and 15 trypsin-derived specific peptides that were present in both trypsin concentrations of each vendor (Figure 7). These peptides were derived from trypsin itself and not the source of trypsin. The intensity of trypsin-derived peptides extracted from porcine pancreas was overall lower than the mean human peptide quantity of the given condition. In comparison, all trypsins expressed in P. pastoris except “Sigma 2” displayed higher trypsin-derived peptide intensities than the mean human peptide quantity. “Sigma 2” showed the overall lowest trypsin-derived peptide quantities in the whole data set. Trypsin-derived peptide quantities did not necessarily relate to the trypsin amount present during digestion as trypsins could differ in their autodegradation activity. While “Sigma 2” and “Thermo 1” displayed the overall lowest quantity of trypsin-derived peptides, they had the highest quantitative variations and lowest number of identified peptides and proteins in all high-load sample matrices and acquisition techniques. Overall, recombinantly expressed trypsins except “Sigma 2” displayed a higher quantity of trypsin-derived peptides with lower variation than the porcine counterpart. These findings could provide a possibility to control trypsin-induced biases in bottom-up proteomics. The trypsin-derived peptide quantities could be used as a measure of digestion efficiency and confirm protease concentration.
Figure 7.

Trypsin-related contaminations and autocleavage products identified in human plasma digested with trypsin of eight commercially available vendors and analyzed with DIA. Quantity of all tryptic peptides identified in both 1:20 (gray) and 1:50 (purple) trypsin and in ≥2 replicates. Median peptide intensity of ≥2 replicates of each peptide. The triangle displays the mean quantity of all human peptides present in ≥2 replicates.
Impact of Storage Time on Trypsin Performance
Finally, we evaluated the impact of storage time in the lyophilized form on “Thermo 1”, which was ordered and stored at four time points (0, 7, 12, and 14 months) prior to experiments. No significant differences between the identified protein or peptide numbers could be identified when analyzing HeLa cell digest acquired in DDA (Supporting Figure 4A). However, it is worth noting that the number of missed cleavages steadily increased slightly by mean 0.7% with an increased storage time (Supporting Figure 4B). This observed increase was, however, not as prominent as the observed variation between different trypsins, thereby suggesting merely a minor impact, which should be further validated. The quantitative performance in LFQ DIA data showed no significant variation from the mean (Supporting Figure 4C). By further looking into single peptide variation over time, we explored the ratio-to-standard variation of peptides in the SRM assay (Supporting Figure 4D). 68.6% of the peptides displayed a CV below 5% over digestion with “Thermo 1” stored over different periods of time (Supporting Figure 4E). Only 4.9% of the peptides displayed CVs over 10% in their ratio to standard. This indicates that the storage time of “Thermo 1” did not seem to impact the quantitative performance of the trypsin.
Conclusions
In this study, we investigated the impact of eight commercial trypsins on commonly used bottom-up proteomics workflows across a range of samples. Besides the trypsin itself, different protease concentrations, sample matrices, instrument acquisition techniques, and protease storage times were investigated. The resulting data set allowed us to unravel some of the technical implications of proteases in bottom-up proteomics while the field is transitioning further into large-scale clinical and biomedical research applications. In high-load samples, we observed a low variation between the trypsins regarding the pure number of proteins or peptides identified in each sample and in terms of proteome depth. Differences only became apparent when looking into the unique peptides that each protease generates and the number of missed cleavages. From a quantitative standpoint, the majority of the trypsins performed similarly in each given concentration. In scp-MS, however, the choice of trypsin seemed to display a stronger impact on the number of peptides and their quantitative performance. On the other hand, trypsins displayed similar technical variation between cells, and minor cell heterogeneity was apparent irrespective of which trypsin was used. Unexpectedly, the concentration of trypsin had the most drastic impact on the quantitative and qualitative performance of trypsin across all sample types. Higher trypsin concentrations resulted in more comparable protein quantitation and gave rise to lower numbers of missed cleavages. Surprisingly, however, the higher trypsin concentration did not result in higher numbers of identified proteins or peptides for plasma and HeLa bulk digest. In scp-MS, however, we did observe increased numbers of peptides, which plateaued in the higher concentrations.
Our results indicate that trypsins with higher quantitative variation also showed the lowest quantity of trypsin-derived peptides in the samples. As all trypsins display a rather low number of contaminating peptides identified in plasma samples, this does not need to be a specific criterion for trypsin selection. However, all recombinantly expressed trypsins except “Sigma 2” displayed higher concordance between their trypsin-derived peptide quantities. This could suggest that recombinantly expressed trypsins might be easier to interchange with one another than trypsins from different sources. However, further evidence is needed. Furthermore, we see indications that the monitoring of trypsin peptides in data sets could provide an opportunity to compensate for trypsin-related biases in bottom-up proteomics.
When investigating the effects of long-term storage of a single trypsin, we could not observe any strong storage time-related impacts on quantitative levels. This is a first indication that long-term trypsin storage might be a way to compensate for trypsin-related quantitative variations in bottom-up proteomics analysis of large cohorts. As storage time does not seem to impact the trypsin performance to a strong degree, it might be useful to reduce trypsin variability by preordering large batches of trypsin produced in one batch to compensate for trypsin concentration variation. However, to validate the relevance of these results for specific experimental setups, these variations should be assessed for each specific trypsin and digestion condition (including the trypsin concentration used). It must be noted that the digestion conditions have previously been described to have a major impact on the protease digestion performance and the presented findings here might not necessarily translate to other digestion protocols.17
In summary, we compared eight commercially available trypsins on three different biological samples that are frequently used in biomedical research. We observed minor differences in performance between trypsins, with the trypsin concentration having the largest effect on the quantitative performance of bottom-up experiments. Notably, the use of recombinant protein standard for absolute quantification provided a way to reduce the protease-related bias in comparison to LFQ. This could be a key aspect to consider while the field moves further into clinical applications. Therefore, we suggest paying special attention to the choice of trypsin when studying large cohorts with bottom-up proteomics and validate its reproducibility. Certain trypsins might be interchangeable with one another, which is an important aspect to consider when moving forward into clinical applications. However, further studies in this regard are needed.
Acknowledgments
We thank Christian Gnann and Sarah Line Skovbakke for providing cells for the conducted experiments. Furthermore, we thank the protein factory of the Human Protein Atlas program and the Science for Life Laboratory for their contributions. Ulrich auf dem Keller acknowledges the Novo Nordisk Foundation Young Investigator Award (NNF16OC0020670 to U.a.d.K). Work in the Cell Diversity Lab was funded by the following grants to E.M.S.: (1) reference number NNF21OC0071016 from the Novo Nordisk Foundation and (2) case no. 2067-00053B from the Independent Research Fund Denmark. V.P. is funded by a Leo Foundation grant (LF-OC-21-000832). P.A.-F. is funded by a Danish Cancer Society grant (R324-A17978).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.3c02543.
Materials and methods: sample preparation, LC-MS/MS acquisition, SRM assay development, and data processing; Figure 1: Missed cleavages in DIA plasma data set; Figure 2: Number of peptides quantified in single cells at five trypsin concentrations; Figure 3: Porcine, Pichia pastoris, and trypsin-specific peptides in DIA data set; Figure 4: Storage time validation of trypsin (PDF)
Table 1: SIS-PrEST concentration and sequence information; Table 2: Statistical test of missed cleavages in DDA data set; Table 3: Statistical test of trypsin grouped by concentration; Table 4: Statistical test of peptide CVs grouped by concentration (XLS)
Author Contributions
# F.D. and E.M.S. contributed equally to this work.
The authors declare the following competing financial interest(s): The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: FE, DK and AH are co-founders of ProteomEdge AB. The other authors declare no conflict of interest.
Supplementary Material
References
- Geyer P. E.; Holdt L. M.; Teupser D.; Mann M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942 10.15252/msb.20156297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoof E. M.; Furtwängler B.; Üresin N.; et al. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 3341 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnielli C. M.; Macedo C. C. S.; De Rossi T.; et al. Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer. Nat. Commun. 2018, 9, 3598 10.1038/s41467-018-05696-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner C. B.; Demichev V.; Wendisch D.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cells Syst. 2020, 11, 11–24.e4. 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortin G. L.; Carr S. A.; Anderson N. L. Introduction: Advances in Protein Analysis for the Clinical Laboratory. Clin. Chem. 2010, 56, 149–151. 10.1373/clinchem.2009.132803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant R. P.; Hoofnagle A. N. From Lost in Translation to Paradise Found: Enabling Protein Biomarker Method Transfer by Mass Spectrometry. Clin. Chem. 2014, 60, 941–944. 10.1373/clinchem.2014.224840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Whiteaker J. R.; Carr S. A.; et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry-based assays. Clin. Chem. 2016, 62, 48–69. 10.1373/clinchem.2015.250563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin A.; Khan S.; Kislinger T. Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin. Proteomics 2020, 17, 17 10.1186/s12014-020-09283-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl J.; Kurth F.; Kappert K.; et al. Quantitative protein biomarker panels: a path to improved clinical practice through proteomics. EMBO Mol. Med. 2023, 15, e16061 10.15252/emmm.202216061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N. L.; Anderson N. G. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 2002, 1, 845–867. 10.1074/mcp.R200007-MCP200. [DOI] [PubMed] [Google Scholar]
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks J.; et al. Increasing the throughput of sensitive proteomics by plexDIA. Nat. Biotechnol. 2022, 2022, 50–59. 10.1038/s41587-022-01389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petelski A. A.; Emmott E.; Leduc A.; et al. Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc. 2021, 16, 5398–5425. 10.1038/s41596-021-00616-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Piehowski P. D.; Zhao R.; et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018, 9, 882 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtwängler B.; Üresin N.; Motamedchaboki K.; et al. Real-Time Search Assisted Acquisition on a Tribrid Mass Spectrometer Improves Coverage in Multiplexed Single-Cell Proteomics. Mol. Cell. Proteomics 2022, 21, 100219 10.1016/j.mcpro.2022.100219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosius V.; Schoof E. M. Recent advances in the field of single-cell proteomics. Transl. Oncol. 2023, 27, 101556 10.1016/j.tranon.2022.101556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proc J. L.; Kuzyk M. A.; Hardie D. B.; et al. A Quantitative Study of the Effects of Chaotropic Agents, Surfactants, and Solvents on the Digestion Efficiency of Human Plasma Proteins by Trypsin. J. Proteome Res. 2010, 9, 5422. 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignjatovic V.; Geyer P. E.; Palaniappan K. K.; et al. Mass Spectrometry-Based Plasma Proteomics: Considerations from Sample Collection to Achieving Translational Data. J. Proteome Res. 2019, 18, 4085–4097. 10.1021/acs.jproteome.9b00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume J. E.; Manning W. C.; Troiano G.; et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020, 11, 3662 10.1038/s41467-020-17033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto L.; Aebersold R.; Cox J.; et al. Initial recommendations for performing, benchmarking and reporting single-cell proteomics experiments. Nat. Methods 2023, 20, 375–386. 10.1038/s41592-023-01785-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney D. L.; Wenger C. D.; Coon J. J. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 2010, 9, 1323–1329. 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giansanti P.; Tsiatsiani L.; Low T. Y.; Heck A. J. R. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- Woessmann J.; Kotol D.; Hober A.; Uhlén M.; Edfors F. Addressing the Protease Bias in Quantitative Proteomics. J. Proteome Res. 2022, 21, 2526–2534. 10.1021/acs.jproteome.2c00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinitcyn P.; Richards A. L.; Weatheritt R. J.; et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat. Biotechnol. 2023, 1–11. 10.1038/s41587-023-01714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; Ong S.-E.; Mann M. Trypsin Cleaves Exclusively C-terminal to Arginine and Lysine Residues*. Mol. Cell. Proteomics 2004, 3, 608–614. 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- Fenn J. B.; Mann M.; Meng C. K.; Wong S. F.; Whitehouse C. M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Vandermarliere E.; Mueller M.; Martens L. Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom. Rev. 2013, 32, 453–465. 10.1002/mas.21376. [DOI] [PubMed] [Google Scholar]
- Tsiatsiani L.; Heck A. J. R. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- Lin Z.; Ren Y.; Shi Z.; et al. Evaluation and minimization of nonspecific tryptic cleavages in proteomic sample preparation. Rapid Commun. Mass Spectrom. 2020, 34, e8733 10.1002/rcm.8733. [DOI] [PubMed] [Google Scholar]
- Niu B.; Martinelli II M.; Jiao Y.; et al. Nonspecific cleavages arising from reconstitution of trypsin under mildly acidic conditions. PLoS ONE 2020, 15, e0236740 10.1371/journal.pone.0236740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. Z.; DeMarco M. L. Manipulating trypsin digestion conditions to accelerate proteolysis and simplify digestion workflows in development of protein mass spectrometric assays for the clinical laboratory. Clin. Mass Spectrom. 2017, 6, 1–12. 10.1016/j.clinms.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Broek I.; Smit N. P. M.; Romijn F. P.; et al. Evaluation of interspecimen trypsin digestion efficiency prior to multiple reaction monitoring-based absolute protein quantification with native protein calibrators. J. Proteome Res. 2013, 12, 5760–5774. 10.1021/pr400763d. [DOI] [PubMed] [Google Scholar]
- Loziuk P. L.; Wang J.; Li Q.; et al. Understanding the role of proteolytic digestion on discovery and targeted proteomic measurements using liquid chromatography tandem mass spectrometry and design of experiments. J. Proteome Res. 2013, 12, 5820–5829. 10.1021/pr4008442. [DOI] [PubMed] [Google Scholar]
- Burkhart J. M.; Schumbrutzki C.; Wortelkamp S.; Sickmann A.; Zahedi R. P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteomics 2012, 75, 1454–1462. 10.1016/j.jprot.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Bunkenborg J.; Espadas G.; Molina H. Cutting edge proteomics: Benchmarking of six commercial trypsins. J. Proteome Res. 2013, 12, 3631–3641. 10.1021/pr4001465. [DOI] [PubMed] [Google Scholar]
- Walmsley S. J.; Rudnick P. A.; Liang Y.; et al. Comprehensive analysis of protein digestion using six trypsins reveals the origin of trypsin as a significant source of variability in proteomics. J. Proteome Res. 2013, 12, 5666. 10.1021/pr400611h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger M.; Müller E.; Dürnberger G.; Pichler P.; Mechtler K. Robust and Easy-to-Use One-Pot Workflow for Label-Free Single-Cell Proteomics. Anal. Chem. 2023, 95, 4435–4445. 10.1021/acs.analchem.2c05022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiler M.; Straube W. L.; Lundberg E.; Uhlen M.; Mann M. A Protein Epitope Signature Tag (PrEST) Library Allows SILAC-based Absolute Quantification and Multiplexed Determination of Protein Copy Numbers in Cell Lines*. Mol. Cell. Proteomics 2012, 11, O111.009613 10.1074/mcp.O111.009613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosius V.; Aragon-Fernandez P.; Üresin N.; Kovacs G.; Phlairaharn T.; Furtwängler B.; op de Beeck J.; Skovbakke S. L.; Goletz S.; Thomsen S. F.; auf dem Keller U.; Nath Natarajan K.; Porse B. T.; Schoof E. M.. Exploration of cell state heterogeneity using single-cell proteomics through sensitivity-tailored data-independent acquisition Nat. Commun. 2022, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Y.; Bateman N. W.; Gallien S.; et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nat. Commun. 2020, 11, 5248 10.1038/s41467-020-18904-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; et al. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Sharma V.; Eckels J.; Taylor G. K.; et al. Panorama: A Targeted Proteomics Knowledge Base. J. Proteome Res. 2014, 13, 4205–4210. 10.1021/pr5006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DIA, SRM, and DDA raw files together with SRM Skyline documents, spectral libraries, and standard curves were uploaded to Panorama Public.43 Data is available through https://panoramaweb.org/Trypsin_Comparison.url with the Proteome-Xchange ID PXD042450.