

Abstract
Mycobacterium tuberculosis, the bacterium responsible for tuberculosis, is a global health concern, affecting millions worldwide. This bacterium has earned a reputation as a formidable adversary due to its multidrug-resistant nature, allowing it to withstand many antibiotics. The development of this drug resistance in Mycobacterium tuberculosis is attributed to innate and acquired mechanisms. In the past, rifampin was considered a potent medication for treating tuberculosis infections. However, the rapid development of resistance to this drug by the bacterium underscores the pressing need for new therapeutic agents. Fortunately, several other medications previously overlooked for tuberculosis treatment are already available in the market. Moreover, several innovative drugs are under clinical investigation, offering hope for more effective treatments. To enhance the effectiveness of these drugs, it is recommended that researchers concentrate on identifying unique target sites within the bacterium during the drug development process. This strategy could potentially circumvent the issues presented by Mycobacterium drug resistance. This review primarily focuses on the characteristics of novel drug resistance mechanisms in Mycobacterium tuberculosis. It also discusses potential medications being repositioned or sourced from novel origins. The ultimate objective of this review is to discover efficacious treatments for tuberculosis that can successfully tackle the hurdles posed by Mycobacterium drug resistance.
1. Introduction
An evident increase in antimicrobial resistance over the past few decades has significantly impacted the administration of healthcare treatment. The early 1930s to 1960s were frequently regarded as the golden age of medicine due to the rapid discovery of new antibiotics. There have been no significant new drug discoveries since then, and this is primarily due to funding and ethical obstacles. According to the World Health Organization (WHO), antimicrobial resistance to prevalent antibiotics has been associated with lower respiratory and diarrheal diseases, HIV/AIDS, and malaria, which have a higher incidence of morbidity and mortality.1 Several factors can cause drug resistance, but excessive or improper medication use is the most common reason. The extensive use of antibiotics for humans and animals, such as ruminants and domestic pets, results in the release of residual or nonmetabolized antibiotic substances into the environment and causes environmental pollution.2,3
Pathogens are subjected to high environmental and genetic selection pressure, resulting in multidrug resistance strains over time. Usually, the resistance developed by microbes is intrinsic, meaning that a specific trait in microbes is expressed before exposure to a drug or antibiotic. Some microbes can acquire resistance through horizontal gene transfer (HGT), which includes conjugation, transformation, and transduction. Conjugation enables resistance transfer via a resistant plasmid.3,4 The transformation acquires resistance due to the transfer of purified DNA, which carries genes for resistance from microbes exposed to antibiotics. This is a common mechanism as the rate of DNA transfer is independent of the genetic relationship between the microbes. Lastly, the third gene transfer method, transduction, enables acquisition of phage-induced resistance. Escherichia coli, Bacteroides, Listeria monocytogenes, Serratia marcescens, and Pseudomonas aeruginosa are some examples that have intrinsically acquired resistance to antibiotics (Table 1).
Table 1. Different Kinds of Antibiotic-Resistant Microbes Associated with Threat Classes.
| classes
of threat |
|||
|---|---|---|---|
| critical threats | monentus threats | distress threats | watch list |
| Carbapenem-resistant Acinetobacter | Campylobacter | Erythromycin-resistant Group A Streptococcus | Azole-resistant |
| Candida auris | Candida ESBL-producing Enterobacteriaceae | Clindamycin-resistant Group A Streptococcus | Aspergillus fumigatus |
| Clostridioides difficile | Vancomycin-resistant Enterococci | Mycoplasma genitalium | |
| Carbapenem-resistant Enterobacteriacea | Pseudomonas aeruginosa | Bordetella pertussis | |
| Drug-resistant Neisseria gonorrheae | nontyphoidal Salmonella | ||
| Salmonella typhi | |||
| Shigella | |||
| Methicillin-resistant Staphylococcus aureus | |||
| Streptococcus pneumonia | |||
| Tuberculosis | |||
Unlike other ESKAPE pathogens, Mycobacterium tuberculosis (Mtb) sets itself apart with its distinct features. Instead of relying on extensive horizontal gene transfer (HGT) as its counterparts, Mtb develops resistance primarily through point mutations. It also exhibits limited genetic diversity and low recombination rates, which curtail the exchange of genetic material among the different strains. These attributes impede the success of treatments and contribute to the persistence of drug-resistant strains. Understanding these unique characteristics of Mtb is crucial for effective tuberculosis management and curbing of drug resistance.
Nonetheless, some microbes can acquire resistance via mutation during gene transfer.2 During gene transfer, however, the mutant gene is transferred to a microbe of the wild type, mutating its genes. This results in the development of antibiotic resistance. Therefore, this could explain why the transferred mutant gene is more fit and stable than the revertant before exposure to the selection force, namely antibiotic exposure.5,6
Similarly, the TB-causing agent Mycobacterium tuberculosis has a particular shape and set of traits that cause AMR. It is a rod-shaped bacterium generally between two and four micrometers long. One of its distinguishing features is the cell wall, which is thick and waxy and is mostly made of mycolic acids. Its resistance to numerous antimicrobial treatments results from the peculiar cell wall structure, making it challenging to eradicate. M. tuberculosis develops slowly, with a generation time of about 15–20 h. It is an obligate aerobe that needs oxygen for growth. It is also an acid-fast bacterium, which means that even after being exposed to acid, it retains the stain, making it possible to identify it using these methods. Additionally, M. tuberculosis is an intracellular pathogen with the capacity to live and grow inside the macrophages of the host, allowing it to escape the immune system and create long-lasting infections. These physical and distinguishing characteristics help M. tuberculosis to be harmful and resilient in spreading the disease and producing antibiotic resistance. Mycobacterium tuberculosis has demonstrated antibiotic resistance to essential antituberculosis drugs, i.e., isoniazid and rifampin, through its peculiar intrinsic-mutational properties. Infection with tuberculosis remains a global threat, with a high mortality rate. Due to the chromosomal mutation of bacteria, resistance to such drugs is observed.7 This article reviews the multidrug resistance of Mycobacterium tuberculosis and all plausible mechanisms by which it acquires or employs its innate resistance mechanisms. In addition to M. tuberculosis resistance mechanisms, this review will discuss the strategies used to produce novel drugs or reposition old ones to achieve potent bacteriocidal or bacteriostatic effects against M. tuberculosis infection.
Tuberculosis is a communicable disease spread from person to person by transmitting infectious droplets from the respiratory system. Active tuberculosis patients release these droplets into the air when they cough, sneeze, or breathe, which can infect those around them.4 Several factors include the number of bacteria in the infected person’s droplets, the length of time an individual is exposed to the germs, and the closeness of the individual to the source of the droplets, affecting the transmission risk. When a person is exposed to the germs that cause tuberculosis, their immune system responds by forming tubercles or granulomas around the bacteria. However, despite the immune system’s efforts to encase the bacteria, about 5% of individuals infected with TB will develop active tuberculosis within two years of the initial exposure.4,5
Additionally, another 10% of people with latent TB infection experience reactivation of their dormant tubercles at some point in their life. This makes it challenging to eliminate TB, as an estimated one-third of the global population has latent TB infection.6 The innate and adaptive-immune response, which includes alveolar-macrophages and dendritic cells, plays a significant role in determining an individual’s risk of developing tuberculosis. These immune cells work together to initiate a chain reaction of signaling pathways that activate additional immune-molecules, like inflammatory cytokines, heat shock proteins, and toll-like receptors.7 However, Mycobacterium tuberculosis has been shown to evade elimination by macrophages. To combat TB infection, immune cells like dendritic cells, neutrophils, monocytes, and lymphocytes are mobilized to the infection-site and organized into a granuloma around the infected macrophages (Figure 1). Chemokines and cytokines released in response to the local inflammatory environment facilitate this process. Researchers have found that increasing tumor necrosis factor (TNF) alpha levels can impede the spread of Mtb and the formation of granulomas. The granuloma is a hallmark TB pathology that traps the mycobacterium inside its walls, preventing it from reproducing.8 This structure is characterized by the presence of foam cells, which are formed from permanently active macrophages. In addition, the granuloma may develop a necrotic zone in its center, which makes it easier for the bacteria to disperse into the surrounding lung tissue via aerosols. Individuals with impaired immune systems, such as HIV infection, are at a higher risk of developing active TB.6,8 Therefore, it is critical to maintain a healthy immune system and take appropriate precautions to prevent the spread of TB, such as wearing a mask, practicing good hygiene, and seeking medical treatment if anyone experiences symptoms of tuberculosis.
Figure 1.

Development of tuberculosis (IL-1: inter-leukin1, IL-6: inter-leukin6, IL-12: inter-leukin12, IL-18: inter-leukin18, IFN-γ: γ-interferon, TNF-alpha: tumor-necrosis-factor α).8 Multiple immunological factors interact intricately to cause TB to develop. Interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-12 (IL-12), interleukin-18 (IL-18), -interferon (IFN-), and tumor necrosis-factor (TNF-alpha) are among those that have important functions. IL-1 functions as a proinflammatory cytokine that encourages the migration of immune cells to the site of infection and their activation there. The acute-phase response and inflammation are both influenced by IL-6. Natural killer (NK) cell activation and IFN production, both of which are crucial for the fight against tuberculosis by stimulating the immune system and activating macrophages, depend on IL-12. IFN- production is increased by the IL-18 and IL-12 working together. IFN- is a crucial cytokine in regulating mycobacterial development and is produced by T and NK cells. TNF-alpha, which is produced by activated macrophages, is essential for the development of granulomas and the control of infection.
2. Structural Characteristics of the Mycobacterium tuberculosis Cell Wall
Mycobacterium tuberculosis is a bacterium that is responsible for tuberculosis in mammals.8 It is also the primary agent responsible for higher mortality rates associated with respiratory disease. The impermeability nature of the TB bacterial cell wall prevents Gram-staining reagents from penetrating the bacterium cell wall and makes it difficult to distinguish TB bacterium as Gram-positive or Gram-negative bacteria. An acid-fast staining is recommended for identifying bacteria. It has been well documented that the resistance to numerous effective drugs results from a unique cell wall composition (cell walls that have mycolic acid). The cell wall is the most critical factor in determining the shape of the bacterial cells. The Gram-stain distinguishes bacteria based on cell wall composition. The characteristics of peptidoglycan (PG) highlight the nature of Gram-staining. However, in Mycobacterial strains, the peptidoglycan (PG) is also associated with an unusual acid (mycolic acid: MA) and sugar moiety (arabinogalactan: AG) that render the cell impermeable to Gram-stain.9 In addition to PG, the above composition in the cell wall confers antimicrobial resistance to the bacteria. In addition, various drug efflux proteins play a crucial role in developing resistance.
In Figure 2, components above the peptidoglycan (PG) layer are considered to be the outer membrane. Apart from the components in the figure, various drug efflux pumps play an essential role in efluxing out the drug for treatment.10,76−78
Figure 2.

Cell wall composition of Mycobacterium tuberculosis that provides a resistance ability. A complex cell wall structure made up of mycolic acids, a peptidoglycan layer, particular lipids (lipomannan), proteins, and polysaccharides makes up the cell wall of Mycobacterium tuberculosis (TB). It gives the cells a waxy appearance, increases the cell wall’s impermeability, and aids M. tuberculosis in evading the immune system. The cell wall significantly influences the persistence and virulence of tuberculosis (TB) since it is essential for the survival, structural stability, and pathogenicity.
3. Adaptation Mechanisms of Drug-Resistant Mycobacterium tuberculosis Strains to the Host Lung Environment
Mycobacterium tuberculosis descended from an ancestral mycobacterium that could successfully infect humans and remain dormant in their bodies. Aerosolization may have played a significant role in the spread of the mycobacterial species due to the species’ superior hydrophobicity compared to its predecessors. Upon inhalation and infection, Mycobacterium tuberculosis encounters a variety of lung microenvironments, both extracellular (becoming reactivated in cavities or after escaping necrotic cells) and intracellular (within alveolar phagocytes like alveolar macrophages), including those encountered during the primary infection (Figure 3). The tuberculosis bacterium, Mycobacterium tuberculosis, is highly adaptable and can use the host’s energy supply and metabolic processes to evade the immune system of the host, flourish, and establish either an active or passive infection. These adaptations allowed the bacteria to thrive in the various habitats in which they have been found. The cell envelope of Mycobacterium tuberculosis is responsible for providing structural support and protecting the organism from osmotic changes. Four primary layers make up the envelope of Mycobacterium TB. These include a periplasmic-space and inner plasma membrane, a cell-envelope core of mycolic acids (MAs) and arabinogalactan (AG) covalently linked to peptidoglycan (PG), a peripheral lipid layer, and the capsule. Proteins are essential constituents of the Mycobacterium TB envelope and are actively being studied as prospective treatment targets, despite lipids and carbohydrates making up nearly 80% of the cell wall. Several proteins have been discovered that govern the permeability of the cell membrane of Mycobacterium TB. This regulation has significant implications for the development of treatment resistance.
Figure 3.

Interactions between drug-resistant Mycobacterium tuberculosis and the host at various stages of infection in the pulmonary microenvironment. Copyright [2021] [Allué-Guardia, García and Torrelles).11 (A) Inhaling drug-resistant Mycobacterium tuberculosis-containing droplets from a TB patient can cause infection. Drug-resistant Mycobacterium tuberculosis bacilli have altered cell envelope lipids. Drug-resistant Mycobacterium tuberculosis will reach the alveoli after bypassing upper respiratory tract barriers. The alveolar space has resident phagocytes called alveolar macrophages (AMs), while the interstitial space around the alveoli has IMs, DCs, neutrophils (N), and T cells. (B) Hydrolases (represented as scissors) in the alveolar lining fluid (ALF) can cleave and modify the Mycobacterium tuberculosis cell envelope, releasing cell envelope fragments into the alveolar space. This interaction occurs when drug-resistant Mycobacterium tuberculosis bacteria enter the alveolar space. (C) ALF-modified Drug-resistant Mycobacterium TB bacilli then engage with AMs, ATs, and other host’s innate immune cells (e.g., neutrophils, DCs). Drug-resistant Mycobacterium TB pieces are immunogenic and may draw neutrophils to the infection site, which may help resident resting AMs eliminate the infection. (D) These initial interactions will result in ALF-exposed drug-resistant Mycobacterium tuberculosis clearance, a successful infection driving primary active TB disease, or a latent infection defined by persisters in granulomas. This niche protects against anti-TB drugs, increasing the phenotype. Surrounding mesenchymal stem cells (MSCs) can decrease immune responses and offer a protective intracellular environment for M. tuberculosis B persistence. (E) Failure of granulomas to retain drug-resistant Mycobacterium tuberculosis can lead to reactivation and progression to active TB disease, with the development of drug-resistant Mycobacterium tuberculosis occurring extracellularly and causing lung tissue degradation and cavity formation. In this setting, an extracellular matrix (EM) composed of free fatty acids may be secreted by drug-resistant Mycobacterium tuberculosis, providing additional protection from TB medications.
The permeability of the cell wall of Mycobacterium tuberculosis is regulated by the export and synthesis of several proteins, including the mycobacterial membrane protein decaprenylphosphoryl-d-ribose 2′-epimerase (DprE), the Rv3143/Rv1524 axis, and Large 3 (MmpL3). Other proteins found in the cell wall of Mycobacterium tuberculosis function directly, rendering the antibiotic ineffective. Isoniazid (INH) is degraded into acetyl hydrazine and isonicotinic acid via the putative acetyltransferase Rv2170. Through this mechanism, some Mycobacterium tuberculosis strains can avoid the toxic effects of INH. Mycobacterium TB infection hinges on the cell envelope’s interaction with the host immune system. The infectious process of Mycobacterium tuberculosis and the development of treatment resistance require an understanding of the roles played by distinct exterior components of the cell envelope at different stages of infection. The mycobacterial cell envelope is essential not only for structural support but also for modifying the bacterium-host contact. Mycobacterium tuberculosis has a dynamic cell envelope because it is constantly regulated and reconstructed in response to the host microenvironment.
As a result of the infection, several different types of bacteria show differences in their cell envelopes. These factors contribute to the interaction between Mycobacterium tuberculosis and the host immune system and can also affect drug susceptibility. However, little is known about how temporal changes in the cell-wall of Mycobacterium tuberculosis are influenced by local environmental signals, which might lead to variable illness outcomes, especially in drug-resistant strains. Most of the data from in vitro investigations or particular disease periods failed to capture the entire temporal dynamics of this intricate and varied structure. Mycobacterium tuberculosis’s metabolic and physiological conditions before or during infection can also affect how it interacts with the host and the course of the illness.
4. Mechanism of Antimicrobial Resistance in Mycobacterium tuberculosis
Ethambutol, Pyrazinamide, Isoniazid, and Rifampicin are the first-line drugs used to treat tuberculosis caused by Mycobacterium tuberculosis.11 Ethambutol inhibits cell wall synthesis by targeting arabinosyl transferase. Pyrazinamide disrupts the energy metabolism and creates an acidic environment. Isoniazid inhibits mycolic acid synthesis by targeting InhA. Rifampicin binds to the RNA polymerase, inhibiting transcription. Using such combined therapies is curative and reduces the risk of relapse but increases the likelihood of drug-resistant strains.12,13 As the health system’s quality varies from person to person, the prevalence of drug-resistant strains of Mycobacterium tuberculosis increases globally, thereby increasing the difficulty of treating TB (WHO, 2016).14 There are several different mechanisms that Mycobacterium tuberculosis utilizes to develop drug resistance such as cell-wall impermeability, efflux-pumps, drug-degradation and modification, target-alteration, and target-mimicry (Figure 4). These mechanisms can be divided into two categories, which are as follows: innate (intrinsic) resistance mechanism and acquired (molecular) resistance mechanism.
Figure 4.

Resistance toward drugs shown by Mycobacterium tuberculosis. Mycobacterium tuberculosis (TB) bacteria have established different medication resistance mechanisms. When bacteria create enzymes that alter or destroy drugs, they lose effectiveness. Drug modification entails chemical changes to the drug’s molecule to prevent it from interacting with its target. Target modification describes genetic alterations that change the medicine’s target location, rendering it less vulnerable to the effects of the treatment. When the bacterium alters the drug’s target metabolic pathways or activities, it is said to have “targeted alteration.″ Proteins known as drug efflux pumps actively pump medicines out of bacterial cells to lower their intracellular concentration. The impenetrable cell wall of TB bacteria also increases medication resistance by preventing pharmaceuticals from entering the cell.
4.1. Intrinsic or Innate Resistance mechanism
The intrinsic drug resistance mechanism is responsible for the ability of Mycobacterium tuberculosis to counteract the cytotoxic effects of antibiotics. The inherent or innate drug resistance mechanisms contributing to Mycobacterium tuberculosis resistance are described below.
4.1.1. Impermeable Cell Wall
The cell wall of Mycobacterium tuberculosis has an unusual lipid composition and structure. It comprises three major components: mycolic acid, wax D, and cord factor.14 The outer mycomembrane of the cell wall contains arabinogalactan, and the inner one contains peptidoglycan. The membrane comprises two leaflets: lipids like phospholipid, trehalose mycolate, glycopeptidolipids, and lipoglycans are present in the outer leaflet, while long-chain mycolic acids are present in the inner leaflet.15 The cell wall is thicker and highly hydrophobic due to a large amount of lipids that prevent the diffusion of some antibiotics like rifampicin, macrolides, fluoroquinolones, and tetracycline.16 The arabinogalactan forms a hydrophilic barrier and ensures the impermeability of the hydrophilic compounds. This impermeability results in the accumulation of antibiotics in the surrounding the cell. The different cellular components, such as enzymes like β lactamase, which degrade β-lactam antibiotics, are released from the cell and detoxify or degrade the antibiotics.17
Due to impermeable cell walls, drug resistance can be overcome if enzyme and protein defects are present. Mycobacterium tuberculosis exhibits intrinsic resistance to Fosfomycin, an inhibitor of murA,18 a critical biosynthetic enzyme of peptidoglycan that covalently binds with a cysteine-residue in the active-site and changes it into aspartic acid. Trehalose dimycolate (TDM) plays a vital role in cell wall impermeability in Mycobacterium tuberculosis and is synthesized by proteins produced from the Antigen 85 (ag85 gene). So, the inactivation of the ag85 gene makes Mycobacterium tuberculosis susceptible to initial-line drugs and other major broad-spectrum antibiotics, indicating TDM is essential for the intrinsic resistance mechanism.19
The outer cell wall layer contains porins, i.e., pore-forming proteins,20 which help hydrophilic compounds and antibiotics enter the cell. So, if the number of porins is lower, along with the lipid nature of the cell wall, the entry of such hydrophilic compounds is prevented.19 The bacteria are susceptible to antibacterial agents through a channel protein called CpnT, which is present on the outer membrane. According to Danilchanka et al.,18 the cpnT mutant strain of Mycobacterium tuberculosis is more resistant.
4.1.2. Slow Metabolism
Some genes permit Mycobacterium tuberculosis to grow under microoxygen conditions; consequently, most synthesize triglyceride directly from acetyl CoA. Acetyl CoA is a necessary component of the tricitric acid (TCA) cycle, which slows the metabolic process. Mycobacterium tuberculosis has a slow generation rate, which contributes to the prevalence of the disease. The strain’s resistance to most antibiotics is due to its slow metabolism and lengthy generation time.21,22 Baek et al.23 reported that unstable antibiotics, such as Carbapenems, lose their activity faster than the Mycobacterium tuberculosis growth rate, so they cannot affect Mycobacterium tuberculosis.
4.1.3. Drug Efflux Pump
The efflux pump contains membrane-spacing proteins essential for bacterial metabolism, physiology, nutrient transport, toxin transport, and water or signaling molecule transport through the cell envelope.23,24 Nasiruddin et al.24 demonstrated the significance of efflux pumps in Mycobacterium tuberculosis’ intracellular growth within macrophages. Mycobacterial efflux pumps in Mycobacterium tuberculosis are closely associated with the expulsion of all antituberculous drugs, including ethambutol, fluoroquinolones, bedaquiline, clofazimine, isoniazid, rifampicin, and streptomycin.25 Upon macrophage infection, mycobacterial efflux pumps are activated; these pumps continue to function even after the infected cell has been released from the macrophage and cause the cell to resist drugs.26,27 Literature review reveals that most drug-resistant Mycobacteria strains have chromosomal mutations, resulting in the efflux pump’s overexpression and the consequent expulsion of the drugs. According to Milano et al.,28 a mutation in mmpR results in efflux pump overexpression, making the strain resistant to isoniazid.
Wang et al.29 identified five superfamilies of drug efflux pumps in Mycobacterium tuberculosis. The drug efflux pumps are members of (i) the multidrug and toxic-compound extrusion (MATE) superfamily, (ii) the resistance nodulation cell-division (RND) superfamily, (iii) the small-multidrug resistance (SMR) superfamily, (iv) the major-facilitator superfamily (MFS), and (v) the ATP-binding-cassette (ABC) superfamily. ABC pumps are primary transporter pumps that require ATP, whereas the other pumps are secondary transporter pumps that require proton motive forces. Antibiotic stress induces the expression of efflux pumps; for instance, isoniazid and rifampicin stress induce the overexpression of jefA, drrA, drrB, efpA, mmr, and Rv1217-Rv1218. According to Wang et al.,29 the overexpression of jefA confers isoniazid, streptomycin, and ethambutol resistance. Table 2 provides examples of antibiotic-resistant efflux pumps found in Mycobacterium tuberculosis. These findings demonstrate that efflux pumps are a significant factor in developing drug resistance in Mycobacterium tuberculosis.29
Table 2. List of Efflux Pumps in Mycobacterium tuberculosis for Antibiotics Resistance.
4.1.4. Degradation and Modification of Antitubercular Drugs
Mycobacterium tuberculosis can produce several enzymes that modify or degrade various antibiotic classes. Such enzyme examples include aminoglycosides β-lactams and macrolides. Antibiotics known as beta-lactams specifically target the bacterial transpeptidase, which disrupts the cell wall and ultimately causes cell death. Mycobacterium tuberculosis produces the Ambler class A β-lactamase (encoded by gene blaC) that confers β-lactam resistance.15
Antibiotics modified by methylation and acetylation cannot bind to their specific target proteins and are inactive. Thus, adding such chemical groups to specific sites of antibiotics renders them inactive.23 Aminoglycoside-modifying enzymes, such as acetyltransferase, are responsible for the acetylation of all 2′ amino groups in aminoglycosides. Antibiotics containing aminoglycosides include neomycin, ribostamycin, kanamycin, gentamycin, and tobramycin.31eis gene product is an intracellular protein that aids in the acetylation of drugs with the chemical structure of aminoglycosides or cyclic peptides. It is regarded as the most compelling example of drug inactivation by Mycobacterium tuberculosis. This process is known as an enzymatic modification. eis has been implicated in the acetylation-mediated inactivation of second line aminoglycoside antibiotics (such as Kanamycin A) and Cyclic peptide antibiotics (such as Capreomycin). eis also protects Mycobacterium tuberculosis from the human immune system by promoting macrophage survival. Accordingly, eis is essential for virulence and antibiotic resistance in Mycobacterium tuberculosis.16
4.1.5. Target Modification
The drugs are selective in their binding ability. Thus, target modification prevents the drug from binding to a specific target site and confers resistance to that drug. Target modification prevents the binding of lincosamides, macrolides, streptomycin, and rifampin to Mycobacterium tuberculosis. In addition to preventing the translocation of peptidyl-t RNA and inhibiting protein synthesis, these drugs inhibit Mycobacterial growth by reversibly binding to a specific site on the 50S subunit of rRNA. Table 3 provides examples of Mycobacterium tuberculosis genes responsible for modifying antibiotic-specific targets and conferring antibiotic resistance.32
Table 3. Genes of Mycobacterium tuberculosis Showing Antibiotic Resistance by Target Alteration.
| gene name | mechanism of action | antibiotic resistance |
|---|---|---|
| erm37 (Erythromycin resistance Methylase) | Methylation at the specific site of 23S rRNA and altering its structure | Macrolides |
| mfpA: encodes for pentapeptide repeat proteins | DNA gyrase binding protein | Quinolone |
| tlyA: encodes rRNA methyl transferase | Unmethylated ribosome | Capreomycin, Viomycin |
| gidB | Methylates 16S rRNA | Streptomycin |
| rbpA: RNA polymerase binding protein | Prevents binding of rifampicin to RNA Polymerase | Rifampicin |
4.1.6. Target Mimicry
Mycobacterium tuberculosis possesses many distinct mechanisms for resisting antibiotic treatment, including molecular mimicry. The binding of fluoroquinolones to DNA gyrase or topoisomerase inhibits DNA replication, transcription, repair, and degradation. Ultimately, it leads to cell death. MfpA, a Mycobacterial fluoroquinolone resistance protein A, is present in Mycobacterium tuberculosis and mimics DNA B-form in size shape, allowing it to bind with DNA gyrase and prevent fluoroquinolone from binding to DNA gyrase.32
4.2. Acquired Mechanism
The acquired drug resistance mechanism in Mycobacterium tuberculosis is primarily the result of chromosomal mutations. This mutation confers resistance in Mycobacterium tuberculosis via diverse mechanisms, including target alteration, elimination of the prodrug activation necessity, and overexpression of drug-specific targets. Table 4 summarizes some of the targeted genes of antibiotic-associated drug resistance according to the resistance mechanism.
Table 4. List of the Targeted Gene of Antibiotic Conferring Drug Resistance According to the Resistance Mechanism.
| resistance mechanism | antibiotic | target gene |
|---|---|---|
| Drug target alteration | Rifampicin | rpoB(35) |
| Isoniazid | inhA(36) | |
| Ethambutol | embB(37) | |
| Ethionamide | inhA(36) | |
| Fluoroquinolone | gyrA/B(38) | |
| Streptomycin | rrs(39) | |
| rpsL(40) | ||
| Amikacin | rrs(39) | |
| Kanamycin | rrs(33) | |
| Capreomycin | Rrs(39) | |
| Cycloserine | alr(34) | |
| Bedaquiline | atpe(41) | |
| Linezolid | rplc(42) | |
| rrl(43) | ||
| Abolition of prodrug activation | Isoniazd | katG(44) |
| Pyrazinamide | pncA(45) | |
| Ethionamide | ethA(46) | |
| p-amino salicylic acid | folC(47) | |
| Delamanide/Pretomanid | ddn(48) | |
| fgd1 | ||
| fibA/B/C | ||
| Abolition of drug target methylation | Capremycin | tlyA(49) |
| Overexpression of drug target | Cycloserine | alr promotor34 |
| Isoniazid | inhA promotor36 | |
| Ethionamide | ||
| Drug responsible for inactivating enzyme overexpression | Kanamycin | eis promotor50 |
| Mmmp15 efflux pump overexpression | Bedaquiline | Promotor/ mmpR51 |
| Clofazimine | ||
| Bypassing of drugs | p-amino salicylic acid | thyA(47) |
| Drug target substrate overabundance | Cycloserine | ald(34) |
4.2.1. Target Alteration
Target modification is the most prevalent mechanism of acquired resistance in Mycobacterium tuberculosis. In such instances, the drug can bind to a specific target site. Therefore, any change in target prevents the drug–drug target interaction and confers drug resistance. Mycobacterium tuberculosis frequently exhibits resistance to rifampicin, isoniazid, fluoroquinolone, aminoglycosides, cyclic peptides, para-aminosalicylic acid, and oxazolidinone due to mutations in drug target encoding genes or nucleotide substitutions in the operon that encodes the rRNA.33
4.2.2. Abolition of Prodrug Activation
Mycobacteria exhibit drug resistance when the antimycobacterial medication is in prodrug form and has not been activated. Examples of such drugs are isoniazid, pyrazinamide, ethionamide, para-aminosalicylic acid, delamanid, and pretomanid. Numerous point mutations and insertions/deletions in the chromosome may be responsible for prodrug inactivation.34
4.2.3. Overexpression of Drug Target
A mutation in the transcriptional repressor or promoter of the drug’s target site may result in overexpression of the target. Due to the inhibition of the drug’s effect, an excess of the target may result in the development of antibiotic resistance. Desjardins et al.35 cited isoniazid, ethambutol, and cycloserine as examples of antibiotics against which mycobacterial strains develop resistance due to drug target overexpression.
From all these studies, antibiotic resistance in Mycobacterium tuberculosis is due to a single cause and frequent gene mutation that encodes antibiotic resistance. Furthermore, chromosomal mutation, efflux pumps, slow metabolism, cell impermeability, antibiotic degradation or modification, and target modification are also crucial for making the strain antibiotic-resistant.
4.2.4. Hotspot Mutation in Genes for Drug Resistance
A mutation frequently found in a specific gene is known as a hotspot mutation. In the case of TB infection, a more significant occurrence of mutations in the gene is likely to elevate the likelihood of developing multidrug-resistant TB. Around 95% of mutations are associated with rifampicin drugs. The mutation is commonly found within 81 bp of the RIF resistance-determining region (RRDR) of the rpoB gene. This RRDR is a mutation hotspot region. The underlying mechanism is a nonsynonymous mutation that causes a change of one amino acid out of 12 amino acids with a compact side chain to an amino acid having a large side chain. It causes the inactivation of the RNA polymerase active site to bind and confer RIF resistance. The hotspot rpoB gene mutation confirmation using multiplex allele-specific PCR (MAS-PCR) proves that the mutation frequency was higher at codons 531, 526, and 516.52,53
4.3. Metabolic Bypass
In tuberculosis (TB), the term “metabolic bypass” refers to the ability of Mycobacterium tuberculosis, the bacterium that causes TB, to avoid or bypass metabolic pathways that are specifically targeted by antimicrobial medicines. These can be intrinsic and acquired processes that are essential for the bacterium’s development and survival. However, drug-resistant strains of M. tuberculosis may have mutations or changes in particular enzymes or metabolic pathways, which enable the bacteria to get around the inhibition produced by antimicrobial medications. Alternative routes or enzymes that make up for blocked or hindered metabolic processes can be included in metabolic bypass systems. By using these escape routes, the bacteria can continue carrying out crucial metabolic processes, including energy creation, biosynthesis, or uptake of nutrients, even while antimicrobial medications are present. Due to the drug-resistant strains’ metabolic adaptability, treatments often fail, and the illness persists despite efforts to eradicate it.
5. Emerging Antimicrobial Agents against Mycobacterium tuberculosis Infection
Mycobacterium tuberculosis infection has become more prevalent due to increased resistance mechanisms of previously discovered antimicrobial agents. This requires the development of novel antimicrobial agents to combat Mycobacterium tuberculosis infection. Several novel antimicrobial agents for tuberculosis infection are “Under Development.” The antituberculosis pharmaceutical market has been primarily associated with insufficient net profit opportunities and the financial return required to motivate pharmaceutical companies to develop new drugs.54,55 In addition, new compounds with antitubercular activity against Mycobacterium tuberculosis,56 clinical trials of the drug, and limited animal models to predict accurate required treatment duration are significant obstacles in new drug discovery.57,58 The treatment process must be made more effective in two ways: by modifying existing drugs and by identifying new drugs.
5.1. Enhancing the Application of Existing Agent
In the past, conventional antimicrobial agents for tuberculosis were effective, but resistance to these drugs and lengthy treatment duration are now their significant drawbacks. To effectively treat Mycobacterium tuberculosis infection, this results in the modification and revised dosage of old and existing medications. In clinical trials, higher doses of Rifamycin are administered, and the effective doses of each pre-existing agent are recorded,59 as mentioned in Table 5. It has been seen that increasing antitubercular medicine dosage is essential for adequately treating TB infections; however, it is not the case with all drugs. Increasing the dosage can lead to toxicity.75 Healthcare professionals seek to reach ideal therapeutic doses of antitubercular medications in the body to ensure maximal effectiveness against the bacterium. This method aids in overcoming potential drug resistance and ensures that the concentration of the medication is sufficient to stop bacterial development and eliminate the infection. Increasing the dosage can also ensure consistent treatment results by compensating for individual differences in medication metabolism and absorption, and careful monitoring is still required to analyze any toxic buildup in the body. Hence, to successfully treat TB, careful monitoring and dosage adjustments are essential to lowering the risk of treatment failure and the emergence of bacterial strains resistant to several drugs.
Table 5. Antimicrobial Agents (Rifampin): Description and Standard Doses Used for Treatment of Mycobacterium Strains.
| no. | antimicrobial agent | description | standard dose |
|---|---|---|---|
| 1. | Rifampin60−62 | Rifampin act by inhibiting the β subunit of RNA polymerase. Mutations in the rpoB gene cause resistance as the gene encoding for a subunit is necessary for the RNA polymerase enzyme. | 10 mg/kg |
| 2. | Rifapentine63−65 | Cyclopentyl rifamycin drug resistance is due to an increased dose (15 mg/kg). | 100 mg/kg (combination with moxifloxacin) |
| 3. | Rifabutin65−68 | The drug has been used for another Mycobacterial strain, i.e., Mycobacterium avium, and advanced HIV infection treatment. |
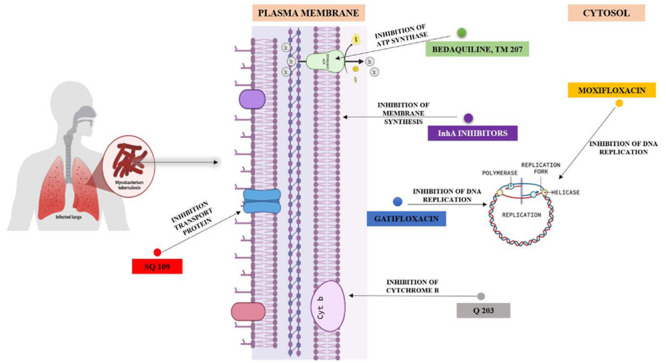
5.2. New Antimicrobial Agents for Tuberculosis
Searching for novel drugs requires that they have specific characteristics, such as the following: (i) should have a novel mechanism to neutralize cross-resistance; (ii) should have optimized properties of pharmacokinetic (PK)/pharmacodynamic (PD);59 (iii) should be a potent bactericidal to cut down the therapy duration; (iv) should not interfere with another drug that would allow combination. Attaining these characteristics as well as low production cost, drug stability, narrow spectrum, high tolerability, and, most importantly, a decreased rate of resistance re-emergence is the objective.69Table 6 lists novel antimicrobial agents and “Under Development”/”Under Clinical Trial” drugs or groups of antimicrobial agents for tuberculosis.
Table 6. Description of Emerging Antimicrobial Agents for TB.
| antimicrobial agent | description | dose | drug status |
|---|---|---|---|
| Fluoroquinolones | |||
| A. Moxifloxacin59 | It is a broad-spectrum 8-methoxy fluoroquinolone drug. The drug has a bactericidal effect against both Gram-positive and Gram-negative microbes. The action mode is the inactivation or inhibition of prokaryotic bacterial DNA gyrase. | 400 mg per day | Approved |
| B. Gatifloxacin59,70 | Gatifloxacin also blocks DNA gyrase as moxifloxacin and prevents an essential step in chromosomal replication. | 400 mg with the combination of ethambutol, ethionamide, and gatifloxacin | Approved |
| Diarylquinolines and Respiratory Chain Inhibitors | |||
| A. Bedaquiline71,72,79 | Bedaquiline falls under the category of diarylquinolines: The drug comes under a new series of novel antitubercular drugs. The drug selectively and, more specifically, inhibits ATPase activity. This inhibition is seen in both replicating and dormant mycobacterial strains. The target of this antitubercular agent is the rotor ring, a part of F0F1 ATP synthase. The specific binding of the drug to the c subunit of the synthase occurs. However, efforts are still given for the development of second-generation diarylquinolines. The usage of drugs has been correlated with positive effects on cardiac liability. | Approved | |
| B. (Telacebec) Q 20369 | An imidazopyridine antitubercular compound that targets cytochrome b subunit (QcrB) of the cytochrome bc1 complex in the electron transport chain (ETC). The complex has a significant role in ATP synthesis also. The drug’s presence marks a rapid depletion of intracellular ATP and disrupts ATP homeostasis in dormant Mtb. The results are more promising than Bedaquiline. | Under clinical trial | |
| Inhibitors of MnpL (Mycobacterial membrane protein Large) 3 | Mycobacterial membrane protein Large (MmpL) is a family of transport proteins that transport metabolites, especially outside cells. | ||
| A. SQ 10959 | SQ 109 is highly potent toward bacteria and fungi lacking mycolic acids and latent cells with nonactive cell wall synthesis. In-depth knowledge of SQ 109 and its mechanism of action revealed that it inhibits cell respiration, menaquinone synthesis, and ATP synthesis. The wide application provides SQ 109 as an alternative and effective drug candidate for Multidrug resistance TB treatment. | Under clinical trial | |
| InhA inhibitors59 | Isoniazid is a cornerstone in tuberculosis treatment. Enoyl reductase InhA of mycobacteria is the molecular target of isoniazid. InhA is essential for mycolic acid synthesis, a prominent feature of the mycobacterial outer cell wall, and crucial for the organism’s growth and virulence. | Approved | |
However, this antimicrobial treatment may later be subjected to drug tolerance and the prevalence of TB. Regarding tuberculosis (TB) infection, drug tolerance and persistence are significant phenomena. The bacterium that causes TB, Mycobacterium tuberculosis, can develop a drug tolerance that makes it less sensitive to the actions of antibiotics. This tolerance enables the bacteria to endure drug exposure, which results in persistence of the infection. A subpopulation of bacteria called persister cells, which M. tuberculosis can produce, enter a latent or nonreplicating state and become highly resistant to antibiotics. The recurrence of TB and the failure of treatment are both believed to be significantly influenced by these persister cells. Henceforth, it is essential to understand the mechanisms behind drug tolerance and persistence in TB to develop new therapeutic approaches to target and eradicate these persistent bacterial populations, improve patient outcomes, and lessen the burden of drug-resistant TB.
5.3. Repositioning of Antitubercular Drugs
Using previously approved treatments to treat other medical conditions to treat tuberculosis (TB) is referred to as “repositioning of antitubercular drugs.” This approach aims to accelerate the discovery of new TB treatment alternatives by utilizing existing drugs’ well-established safety and efficacy properties.79 A crucial element of the repositioning method is finding drugs that have demonstrated antimycobacterial action or the ability to target specific M. tuberculosis pathways or mechanisms. By repurposing these pharmaceuticals, researchers and medical practitioners can circumvent some time-consuming and expensive phases often required in drug development such as preliminary clinical studies and safety testing.
Many commercially marketed drugs initially developed for other conditions have shown promise against TB. For instance, some antibiotics, antifungals, and even drugs used to treat other infectious diseases are proven to have antimycobacterial activity. Researchers may be able to create brand-new drug-resistant TB therapy regimens or discover other choices by evaluating how well they operate against M. tuberculosis.80 Numerous drug classes that exhibit antimycobacterial activity or may be utilized to treat tuberculosis have been found through scientific research. It has been proven that fluoroquinolones and macrolides, including clarithromycin and moxifloxacin, are effective against M. tuberculosis and are routinely used to treat various infectious diseases.81 These drugs work against mycobacteria by preventing vital bacterial functions, such as DNA replication and protein synthesis.
Additionally, M. tuberculosis has been successfully treated with antifungal drugs such as Posaconazole and itraconazole. These drugs combat mycobacteria and fungi’s crucial cellular processes.82 Research has shown that antifungal therapies can decrease the spread of TB strains’ resistant to therapy and improve the efficiency of other antitubercular drugs. Numerous already on-the-market drugs with other uses, such as antiretrovirals, have shown promise in treating TB. For instance, due to its antimycobacterial effects, the protease inhibitor lopinavir/ritonavir has been explored in conjunction with traditional anti-TB drugs.83M. tuberculosis has shown preclinical research resistance to M. statins, calcium channel blockers, and nonsteroidal anti-inflammatory drugs (NSAIDs).84
In addition to these specific drug classes, large-scale screening programs have been conducted to identify potential antitubercular activity in various chemical libraries. High-throughput screening and computational methods have been used to identify compounds specifically targeting specific M. tuberculosis pathways or processes. These techniques have led to identifying novel antimycobacterial medicines, some of which have reached the preclinical and clinical testing stages. Here are a few examples of drugs that have been repositioned for the treatment of tuberculosis (TB), together with relevant scientific data:
Bedaquiline: First developed as an antiarrhythmic drug and afterward changed into a TB drug. It stops the creation of energy and bacterial growth by focusing on the mycobacterial ATP synthase. Clinical trials have demonstrated its efficacy in treating multidrug-resistant tuberculosis (MDR-TB). According to a Phase IIb study, adding bedaquiline to a baseline regimen increased the percentage of patients with positive results compared to conventional treatment alone.85
Linezolid: Gram-positive infections are commonly treated with the antibiotic linezolid. Studies show that it also works against M. tuberculosis. In a clinical trial investigating it extensively for treating drug-resistant TB (XDR-TB), linezolid had a favorable therapeutic outcome in many patients. However, careful supervision is required because of the potential drawbacks associated with its use.86
Clofazimine, a drug once developed to treat leprosy, has shown antimycobacterial activity against M. tuberculosis. It has been altered for the treatment of drug-resistant TB. In preclinical and clinical trials, it has been shown that adding clofazimine to treatment regimens for MDR-TB and XDR-TB improves treatment outcomes and decreases the time to culture conversion.87
Nitazoxanide: An antiparasitic drug, nitazoxanide has shown encouraging antimycobacterial activity against M. tuberculosis. In vitro tests have demonstrated its ability to stop the growth of mycobacteria and cooperate with other anti-TB drugs. Preclinical studies in animal models have shown its efficacy in reducing the bacterial burden and increasing lung pathology in TB infection.88
Disulfiram: Originally used to treat alcoholism, disulfiram has been identified as a potential treatment for tuberculosis. Studies have shown that disulfiram inhibits the growth of mycobacteria by focusing on several bioenergetic and redox homeostasis-related pathways. Disulfiram was combined with traditional TB medication in the Phase II clinical trial, resulting in a considerably higher rate sputum culture conversion rate.89
The repositioning of drugs to treat tuberculosis offers the potential to speed up the development of new therapeutic options by leveraging current knowledge and resources. Drug-resistant tuberculosis can be treated, and efforts to identify and evaluate repurposed drugs show promise for improving patient outcomes.
6. Conclusion
Due to recent technological advancements and the overuse of antibiotics, microorganisms are developing resistance to various antimicrobial agents. We know that resistance continues to spread globally; however, because no effective treatment or prevention method is available, the mortality rate may significantly increase over the next few years. As a result, microbial disease prevention and treatment have become problematic. Therefore, it will be essential to discover novel chemotherapeutic agents to treat multidrug-resistant tuberculosis infections (MDR-TB). We have attempted to highlight some of the most promising classes of novel or repositioned antimicrobial agents for the infection caused by Mycobacterium tuberculosis. There is a need for additional research to improve drug development techniques, our understanding of mycobacterium biology and disease states, emerging technologies, and the clinical trials paradigm. Efforts were made to describe novel antimicrobial agents produced by diverse research groups. Emerging antimicrobial agents for Mycobacterium tuberculosis are categorized into two separate groups. One consists of the enhanced application of conventional medicines.
Investment in innovation and operational research is crucial for developing new antimicrobial medicines, vaccines, and diagnostic tools targeting critical microbes. Research funding gaps could be addressed through initiatives such as the Antimicrobial Resistance Multi-Partner Trust Fund (AMR MPTF), the Global Antibiotic Research & Development Partnership (GARDP), and the AMR Action Fund. While some governments, including Sweden, Germany, the USA, and the United Kingdom, are actively providing reimbursement, more initiatives are needed to find permanent solutions. In 2015, the Global Action Plan (GAP)73 on AMR was developed and endorsed by the World Health Assembly, the Food and Agriculture Organization of the United Nations (FAO), and the World Organization for Animal Health. The plan requires countries to develop and implement multisectorial national action plans to create sustainable progress. Before the GAP, global efforts to contain AMR were guided by the WHO global strategy for containment of Antimicrobial Resistance, developed in 2001 and provides a framework of interventions to slow the emergence and reduce the spread of AMR.72 Likewise, the Indian Ministry of Health formed a high-level committee in 2013 to discuss the use of newer TB drugs in India. WHO has also developed a Policy Implementation Package (PIP) to safely and effectively uptake new drugs or regimens. It provides a structure for introducing new TB drugs and/or regimens, complementing existing and new policy guidance on using new drugs for treating TB or MDR-TB (WHO, 2014).74 There are emerging antimicrobial agents for Mycobacterium tuberculosis agents, elaborated as two distinct groups. One includes enhanced application of conventional medicines, and the other includes new emerging antimicrobial agents, either in the clinical trial of the “Underdevelopment” pipeline or recently approved with defined doses.
In contrast, the other consists of new antimicrobial agents undergoing clinical trials or recently approved with defined doses. In light of what has been said about the research gap, it should come as no surprise that the development of an antituberculosis medication is the top priority for making significant advances in the field of medicine in the years to come. Researchers must cultivate the field to wage an effective war against multidrug-resistant tuberculosis.
Acknowledgments
The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code 22UQU4350073DSR17.
The authors declare no competing financial interest.
References
- Anand U.; Nandy S.; Mundhra A.; Das N.; Pandey D. K.; Dey A. A review on anti-microbial botanicals, phytochemicals and natural resistance modifying agents from Apocynaceae family: Possible therapeutic approaches against multi-drug resistance in pathogenic microorganisms. Drug Resistance Updates 2020, 51, 100695 10.1016/j.drup.2020.100695. [DOI] [PubMed] [Google Scholar]
- Yan X. T.; Zhai Y. Q.; Cai Y. Y.; Guo Z.; Zhang Q. Q.; Ying G. G. Hypothetical scenarios estimating and simulating the fate of antibiotics: Implications for antibiotic environmental pollution caused by manure application. Sci. Total Environ. 2022, 822, 153177 10.1016/j.scitotenv.2022.153177. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Cai Q.; Zhang C.; Ouyang P.; Yu L.; Xu Y. Antibiotic resistance bacteria and antibiotic resistance genes survived from the extremely acidity posing a risk on intestinal bacteria in an in vitro digestion model by horizontal gene transfer. Ecotoxicol. Environ. Saf. 2022, 247, 114247 10.1016/j.ecoenv.2022.114247. [DOI] [PubMed] [Google Scholar]
- Goyal A. Horizontal gene transfer drives the evolution of dependencies in bacteria. Iscience. 2022, 25 (5), 104312 10.1016/j.isci.2022.104312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford N.; Ellington M. J. The emergence of antibiotic resistance by mutation. Clin. Microbiol. Infect. 2007, 13 (1), 5–18. 10.1111/j.1469-0691.2006.01492.x. [DOI] [PubMed] [Google Scholar]
- Reygaert W. C. An overview of the anti-microbial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4 (3), 482. 10.3934/microbiol.2018.3.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath M.; Gey van Pittius N. C.; Van Helden P. D.; Warren R. M.; Warner D. F. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J. Anti-microb. Chemother. 2014, 69 (2), 292–302. 10.1093/jac/dkt364. [DOI] [PubMed] [Google Scholar]
- Mehta K.; Sharma P.; Mujawar S.; Vyas A. Role of Anti-microbial Peptides in Treatment and Prevention of Mycobacterium tuberculosis: A Review. Int. J. Pept. Res. Ther. 2022, 28 (5), 132. 10.1007/s10989-022-10435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imran M.; Khan S. A.; Asdaq S. M.; Almehmadi M.; Abdulaziz O.; Kamal M.; Alshammari M. K.; Alsubaihi L. I.; Hussain K. H.; Alharbi A. S.; Alzahrani A. K. An insight into the discovery, clinical studies, compositions, and patents of macozinone: A drug targeting the DprE1 enzyme of Mycobacterium tuberculosis. J. Infect. Public Health 2022, 15 (10), 1097–107. 10.1016/j.jiph.2022.08.016. [DOI] [PubMed] [Google Scholar]
- Maitra A.; Munshi T.; Healy J.; Martin L. T.; Vollmer W.; Keep N. H.; Bhakta S. Cell wall peptidoglycan in Mycobacterium tuberculosis: An Achilles’ heel for the TB-causing pathogen. FEMS Microbiol. Rev. 2019, 43 (5), 548–75. 10.1093/femsre/fuz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allué-Guardia A.; García J. I.; Torrelles J. B. Evolution of Drug-Resistant Mycobacterium tuberculosis Strains and Their Adaptation to the Human Lung Environ. Front. Microbiol. 2021, 12, 612675 10.3389/fmicb.2021.612675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailo C. V.; Zami Z.; Lalremruata R.; Sanga Z.; Fela V.; Kharkongor F.; Chhakchhuak L.; Chhakchhuak Z.; Laldinmawii G.; Kumar D.; Kumar N. S. MGIT sensitivity testing and genotyping of drug resistant Mycobacterium tuberculosis isolates from Mizoram, Northeast India. Indian J. Med. Microbiol. 2022, 40 (3), 347–53. 10.1016/j.ijmmb.2022.06.003. [DOI] [PubMed] [Google Scholar]
- Eldholm V.; Norheim G.; von der Lippe B.; Kinander W.; Dahle U. R.; Caugant D. A.; Mannsåker T.; Mengshoel A. T.; Dyrhol-Riise A. M.; Balloux F. Evolution of extensively drug-resistant Mycobacterium tuberculosisfrom a susceptible ancestor in a single patient. Genome Biol. 2014, 15 (11), 1–1. 10.1186/s13059-014-0490-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization Global Tuberculosis Report. WHO/HTM/TB 2016.13; WHO, 2016.
- Hett E. C.; Rubin E. J. Bacterial growth and cell division: a mycobacterial perspective. Microbiol. Mol. Biol. Rev. 2008, 72 (1), 126–56. 10.1128/MMBR.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. Antibiotic resistance mechanisms in M. tuberculosis: an update. Arch. Toxicol. 2016, 90, 1585–604. 10.1007/s00204-016-1727-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygli S. M.; Borrell S.; Trauner A.; Gagneux S. Anti-microbial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol. Rev. 2017, 41 (3), 354–73. 10.1093/femsre/fux011. [DOI] [PubMed] [Google Scholar]
- Danilchanka O.; Pires D.; Anes E.; Niederweis M. The Mycobacterium tuberculosis outer membrane channel protein CpnT confers susceptibility to toxic molecules. Antimicrob. Agents Chemother. 2015, 59 (4), 2328–36. 10.1128/AAC.04222-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A.; Kumar M.; Kumar A.; Kanchan K. Comprehensive review on mechanism of action, resistance and evolution of anti-mycobacterial drugs. Life Sci. 2021, 274, 119301 10.1016/j.lfs.2021.119301. [DOI] [PubMed] [Google Scholar]
- Nasiri M. J.; Haeili M.; Ghazi M.; Goudarzi H.; Pormohammad A.; Imani Fooladi A. A.; Feizabadi M. M. New insights in to the intrinsic and acquired drug resistance mechanisms in mycobacteria. Front. Microbiol. 2017, 8, 681. 10.3389/fmicb.2017.00681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehtram A.; Shariq M.; Ali S.; Quadir N.; Sheikh J. A.; Ahmad F.; Sharma T.; Ehtesham N. Z.; Hasnain S. E. Teleological cooption of Mycobacterium tuberculosis PE/PPE proteins as porins: Role in molecular immigration and emigration. Int. J. Med. Microbiol. 2021, 311 (3), 151495 10.1016/j.ijmm.2021.151495. [DOI] [PubMed] [Google Scholar]
- Pandey A. K.; Sassetti C. M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Nat. Acad. Sci. 2008, 105 (11), 4376–80. 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S. H.; Li A. H.; Sassetti C. M. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 2011, 9 (5), e1001065 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasiruddin M.; Neyaz M. K.; Das S. Nanotechnology-Based Approach in Tuberculosis Treatment. Tuberc. Res. Treat. 2017, 2017, 4920209. 10.1155/2017/4920209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laws M.; Jin P.; Rahman K. M. Efflux pumps in Mycobacterium tuberculosis and their inhibition to tackle anti-microbial resistance. Trends Microbiol. 2022, 30 (1), 57–68. 10.1016/j.tim.2021.05.001. [DOI] [PubMed] [Google Scholar]
- Malinga L. A.; Stoltz A.; Van; der Walt M. Efflux pump mediated second-line tuberculosis drug resistance. Mycobact. Dis. 2016, 6 (03), 1–9. [Google Scholar]
- Park J. S.; Lee J. Y.; Lee Y. J.; Kim S. J.; Cho Y. J.; Yoon H. I.; Lee C. T.; Song J.; Lee J. H. Serum levels of anti-tuberculosis drugs and their effect on tuberculosis treatment outcome. Anti-microb. Agents Chemother. 2016, 60 (1), 92–8. 10.1128/AAC.00693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano A.; Pasca M. R.; Provvedi R.; Lucarelli A. P.; Manina G.; Ribeiro A. L.; Manganelli R.; Riccardi G. Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5–MmpL5 efflux system. Tuberculosis 2009, 89 (1), 84–90. 10.1016/j.tube.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Wang K.; Pei H.; Huang B.; Zhu X.; Zhang J.; Zhou B.; Zhu L.; Zhang Y.; Zhou F. F. The expression of ABC efflux pump, Rv1217c–Rv1218c, and its association with multi-drug resistance of Mycobacterium tuberculosis in China. Curr. Microbiol. 2013, 66, 222–6. 10.1007/s00284-012-0215-3. [DOI] [PubMed] [Google Scholar]
- Hameed H. M. A.; Islam M. M.; Chhotaray C.; Wang C.; Liu Y.; Tan Y.; Li X.; Tan S.; Delorme V.; Yew W. W.; Liu J.; Zhang T. Molecular targets related drug resistance mechanisms in MDR-, XDR-, and TDR-Mycobacterium tuberculosis strains. Front. Cell Infect. Microbiol. 2018, 8, 114. 10.3389/fcimb.2018.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Shi W.; Zhang S.; Hao X.; Maslov D. A.; Shur K. V.; Bekker O. B.; Danilenko V. N.; Zhang Y. Mutations in efflux pump Rv1258c (Tap) cause resistance to pyrazinamide, isoniazid, and streptomycin in M. tuberculosis. Front. Microbiol. 2019, 10, 216. 10.3389/fmicb.2019.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labby K. J.; Garneau-Tsodikova S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med. Chem. 2013, 5 (11), 1285–309. 10.4155/fmc.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munita J. M.; Arias C. A. Mechanisms of antibiotic resistance. Microbiol Spectr 2016, 4, 1–37. 10.1128/microbiolspec.VMBF-0016-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell E. A.; Korzheva N.; Mustaev A.; Murakami K.; Nair S.; Goldfarb A.; Darst S. A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001, 104 (6), 901–12. 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Desjardins C. A; Cohen K. A; Munsamy V.; Abeel T.; Maharaj K.; Walker B. J; Shea T. P; Almeida D. V; Manson A. L; Salazar A.; Padayatchi N.; O'Donnell M. R; Mlisana K. P; Wortman J.; Birren B. W; Grosset J.; Earl A. M; Pym A. S Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat. Genet. 2016, 48 (5), 544–551. 10.1038/ng.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damtie D.; Woldeyohannes D.; Mathewos B. Review on molecular mechanism of first line antibiotic resistance in Mycobacterium tuberculosis. Mycobact Dis. 2014, 4 (6), 174. 10.4172/2161-1068.1000174. [DOI] [Google Scholar]
- Hazbon M. H.; Brimacombe M.; Bobadilla del Valle M.; Cavatore M.; Guerrero M. I.; Varma-Basil M.; Billman-Jacobe H.; Lavender C.; Fyfe J.; Garcia-Garcia L.; Leon C. I.; Bose M.; Chaves F.; Murray M.; Eisenach K. D.; Sifuentes-Osornio J.; Cave M. D.; Ponce de Leon A.; Alland D. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2006, 50 (8), 2640–9. 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plinke C.; Cox H. S.; Zarkua N.; Karimovich H. A.; Braker K.; Diel R.; Rüsch-Gerdes S.; Feuerriegel S.; Niemann S. embCAB sequence variation among ethambutol-resistant Mycobacterium tuberculosis isolates without embB 306 mutation. J. Anti-microb Chemother. 2010, 65 (7), 1359–67. 10.1093/jac/dkq120. [DOI] [PubMed] [Google Scholar]
- Che Y.; Song Q.; Yang T.; Ping G.; Yu M. Fluoroquinolone resistance in multidrug-resistant Mycobacterium tuberculosis independent of fluoroquinolone use. Eur. Respir. J. 2017, 50 (6), 1701633 10.1183/13993003.01633-2017. [DOI] [PubMed] [Google Scholar]
- Maus C. E.; Plikaytis B. B.; Shinnick T. M. Molecular analysis of cross-resistance to capreomycin, kanamycin, amikacin, and viomycin in Mycobacterium tuberculosis. Anti-microb. Agents Chemother. 2005, 49 (8), 3192–7. 10.1128/AAC.49.8.3192-3197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier A.; Kirschner P.; Bange F. C.; Vogel U.; Böttger E. C. Genetic alterations in streptomycin-resistant Mycobacterium tuberculosis: mapping of mutations conferring resistance. Anti-microb. Agents Chemother. 1994, 38 (2), 228–33. 10.1128/AAC.38.2.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitric E.; Verhasselt P.; Koul A.; Andries K.; Hoffner S.; Andersson D. I. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Anti-microb. agents Chemother. 2010, 54 (3), 1022–8. 10.1128/AAC.01611-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckert P.; Hillemann D.; Kohl T. A.; Kalinowski J.; Richter E.; Niemann S.; Feuerriegel S. rplC T460C identified as a dominant mutation in linezolid-resistant Mycobacterium tuberculosis strains. Anti-microb. Agents Chemother. 2012, 56 (5), 2743–5. 10.1128/AAC.06227-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillemann D.; Rüsch-Gerdes S.; Richter E. In vitro-selected linezolid-resistant Mycobacterium tuberculosis mutants. Anti-microb. Agents Chemother. 2008, 52 (2), 800. 10.1128/AAC.01189-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollela V. R.; Namburete E. I.; Feliciano C. S.; Macheque D.; Harrison L. H.; Caminero J. A. Detection of katG and inhA mutations to guide isoniazid and ethionamide use for drug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2016, 20 (8), 1099–104. 10.5588/ijtld.15.0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. T.; Malik S. I.; Ali S.; Masood N.; Nadeem T.; Khan A. S.; Afzal M. T. Pyrazinamide resistance and mutations in pncA among isolates of Mycobacterium tuberculosis from Khyber Pakhtunkhwa, Pakistan. BMC Infect. Dis. 2019, 19, 1–7. 10.1186/s12879-019-3764-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlock G. P.; Metchock B.; Sikes D.; Crawford J. T.; Cooksey R. C. ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Anti-microb. Agents Chemother. 2003, 47 (12), 3799–805. 10.1128/AAC.47.12.3799-3805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y.; Thiede J. M.; Kordus S. L.; McKlveen E. J.; Turman B. J.; Baughn A. D. Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Anti-microb. Agents Chemother. 2015, 59 (9), 5097–106. 10.1128/AAC.00647-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haver H. L.; Chua A.; Ghode P.; Lakshminarayana S. B.; Singhal A.; Mathema B.; Wintjens R.; Bifani P. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Anti-microb. Agents Chemother. 2015, 59 (9), 5316–23. 10.1128/AAC.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monshupanee T.; Johansen S. K.; Dahlberg A. E.; Douthwaite S. Capreomycin susceptibility is increased by TlyA-directed 2′-O-methylation on both ribosomal subunits. Mol. Microbiol. 2012, 85 (6), 1194–203. 10.1111/j.1365-2958.2012.08168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambli P.; Ajbani K.; Nikam C.; Sadani M.; Shetty A.; Udwadia Z.; Georghiou S. B.; Rodwell T. C.; Catanzaro A.; Rodrigues C. Correlating rrs and eis promoter mutations in clinical isolates of Mycobacterium tuberculosis with phenotypic susceptibility levels to the second-line injectables. Int. J. Mycobacteriol. 2016, 5 (1), 1–6. 10.1016/j.ijmyco.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg G. V.; Keller P. M.; Stucki D.; Trauner A.; Borrell S.; Latshang T.; Coscolla M.; Rothe T.; Homke R.; Ritter C.; Feldmann J.; Schulthess B.; Gagneux S.; Bottger E. C. Acquired resistance to bedaquiline and delamanid in therapy for tuberculosis. New Eng. J. Med. 2015, 373 (20), 1986–1988. 10.1056/NEJMc1505196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A.; Ali H.; Yasmin A.; Baig M.; Ullah A.; Kazmi A.; Ahmed M. A.; Albadrani G. M.; El-Demerdash F. M.; Bibi M.; Abdel-Daim M. M.; Ali I.; Hussain S. Unveiling the Antibiotic Susceptibility and Antimicrobial Potential of Bacteria from Human Breast Milk of Pakistani Women: An Exploratory Study. BioMed. Research International 2023, 6399699 10.1155/2023/6399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaw M. T.; Emran N. A.; Lin Z. Mutations inside rifampicin-resistance determining region of rpoB gene associated with rifampicin-resistance in Mycobacterium tuberculosis. J. Infect. Public Health 2018, 11 (5), 605–10. 10.1016/j.jiph.2018.04.005. [DOI] [PubMed] [Google Scholar]
- Sufyan M.; Daraz U.; Hyder S.; Zulfiqar U.; Iqbal R.; Eldin S. M.; Rafiq F.; Mahmood N.; Shahzad K.; Uzair M.; Fiaz S.; Ali I. An overview of genome engineering in plants, including its scope, technologies, progress and grand challenges. Functional & Integrative Genomics 2023, 23 (2), 119. 10.1007/s10142-023-01036-w. [DOI] [PubMed] [Google Scholar]
- O’Brien R. J.; Nunn P. P. The need for new drugs against tuberculosis: obstacles, opportunities, and next steps. Am. J. Respir. Crit. Care Med. 2001, 163 (5), 1055–8. 10.1164/ajrccm.163.5.2007122. [DOI] [PubMed] [Google Scholar]
- Mitchison D. A. Shortening the treatment of tuberculosis. Nat. Biotechnol. 2005, 23 (2), 187–8. 10.1038/nbt0205-187. [DOI] [PubMed] [Google Scholar]
- Mohite P.; Nahar D.; Pawara R.; Alqahtani T.; Eldin S. M.; Mukerjee N.; Al-Tawaha A. M. S.; Iqbal R.; Bawazeer S.; Ali I. Triazolopyridine, a leitmotif of synthetic methods and pharmacological attributes: an extensive review. Arabian Journal of Chemistry 2023, 16. 10.1016/j.arabjc.2023.105181. [DOI] [Google Scholar]
- Karakousis P, Parry Z, Klinkenberg L, Pinn M, Nuermberger E, Grosset J. Towards establishing a high-burden guinea pig model for TB chemotherapy, abstr. 11. Abstr. 1st Int. In Workshop Clin. Pharmacol.; Tuberc. Drugs, Toronto, Canada, 2008. [Google Scholar]
- van den Boogaard J.; Kibiki G. S.; Kisanga E. R.; Boeree M. J.; Aarnoutse R. E. New drugs against tuberculosis: problems, progress, and evaluation of agents in clinical development. Anti-microb. Agents Chemother. 2009, 53 (3), 849–62. 10.1128/AAC.00749-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman W. J.; Gallicano K.; Peloquin C. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin. Pharmacokinet. 2001, 40, 327–41. 10.2165/00003088-200140050-00002. [DOI] [PubMed] [Google Scholar]
- Diacon A. H.; Patientia R. F.; Venter A.; Van Helden P. D.; Smith P. J.; McIlleron H.; Maritz J. S.; Donald P. R. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Anti-microb. Agents Chemother. 2007, 51 (8), 2994–6. 10.1128/AAC.01474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacon A. H.; Pym A.; Grobusch M.; Patientia R.; Rustomjee R.; Page-Shipp L.; Pistorius C.; Krause R.; Bogoshi M.; Churchyard G.; Venter A.; Allen J.; Palomino J. C.; De Marez T.; van Heeswijk R. P.G.; Lounis N.; Meyvisch P.; Verbeeck J.; Parys W.; de Beule K.; Andries K.; Neeley D. F. M. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. New Eng. J. Med. 2009, 360 (23), 2397–405. 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- Rothstein D. M. Rifamycins, alone and in combination. Cold Spring Harbor Perspect. Med. 2016, 6 (7), a027011 10.1101/cshperspect.a027011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis B.; Lamb H. M. Rifapentine. Drugs 1998, 56, 607–16. 10.2165/00003495-199856040-00008. [DOI] [PubMed] [Google Scholar]
- Brogden R. N.; Fitton A. Rifabutin: a review of its anti-microbial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 1994, 47, 983–1009. 10.2165/00003495-199447060-00008. [DOI] [PubMed] [Google Scholar]
- Dickinson J. M.; Mitchison D. A. In vitro activity of new rifamycins against rifampicin-resistant M. tuberculosis and MAIS-complex mycobacteria. Tuberc. 1987, 68 (3), 177–82. 10.1016/0041-3879(87)90053-5. [DOI] [PubMed] [Google Scholar]
- Heifets L. B.; Lindholm-Levy P. J.; Iseman M. D. Rifabutine: minimal inhibitory and bactericidal concentrations for Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 1988, 137 (3), 719–21. [DOI] [PubMed] [Google Scholar]
- Truffot-Pernot C.; Giroir A. M.; Maury L.; Grosset J. Study of the minimal inhibitory concentration of rifabutine (Ansamycin LM 427) for Mycobacterium tuberculosis, Mycobacterium xenopi and Mycobacterium avium-intracellulare. Rev. Mal. Respir. 1988, 5 (4), 401–406. [PubMed] [Google Scholar]
- Hoagland D. T.; Liu J.; Lee R. B.; Lee R. E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Delivery Rev. 2016, 102, 55–72. 10.1016/j.addr.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag P. P.; Grant S. S.; Lewis T. A.; Gordon K.; Kawate T.; Fitzgerald M.; Nietupski R.; Gomez J.; White E. L.; Maddox C.; Nebane N. M.. Identification of a Chemically Validated Target in Replicating and Non-Replicating Mycobacterium Tuberculosis with the Aid of a Small Molecule Probe; Probe Reports from the NIH Molecular Libraries Program, 2015. [PubMed]
- Andries K.; Verhasselt P.; Guillemont J.; Gohlmann H. W. H.; Neefs J.-M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Sci. 2005, 307 (5707), 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Mendelson M.; Matsoso M. P. The World Health Organization Global Action Plan for anti-microbial resistance: guest editorial. S. Afr. Med. J. 2015, 105 (5), 325. 10.7196/SAMJ.9644. [DOI] [PubMed] [Google Scholar]
- Policy Implementation Package for New TB Drug Introduction; World Health Organization, 2014. [Google Scholar]
- Prasad R.; Singh A.; Gupta N. Adverse drug reactions in tuberculosis and management. Indian J. Tuberc. 2019, 66 (4), 520–532. 10.1016/j.ijtb.2019.11.005. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan L. Mycobacterium tuberculosis pathogenicity viewed through the lens of molecular Koch’s postulates. Curr. Opin. Microbiol. 2020, 54, 103–11. 10.1016/j.mib.2020.01.011. [DOI] [PubMed] [Google Scholar]
- Berg S.; Kaur D.; Jackson M.; Brennan P. J. The glycosyltransferases of Mycobacterium tuberculosis—roles in the synthesis of arabinogalactan, lipoarabinomannan, and other glycoconjugates. Glycobiol. 2007, 17 (6), 35R–56R. 10.1093/glycob/cwm010. [DOI] [PubMed] [Google Scholar]
- Besra G. S.; Bolton R. C.; McNeil M. R.; Ridell M.; Simpson K. E.; Glushka J.; Van Halbeek H.; Brennan P. J.; Minnikin D. E. Structural elucidation of a novel family of acyltrehaloses from Mycobacterium tuberculosis. Biochemistry 1992, 31 (40), 9832–9837. 10.1021/bi00155a040. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Takkar P. Repositioning of Isatin hybrids as novel anti-tubercular agents overcoming pre-existing antibiotics resistance. Med. Chem. Res. 2021, 30, 847–876. 10.1007/s00044-021-02699-5. [DOI] [Google Scholar]
- Sharma A.; De Rosa M.; Singla N.; Singh G.; Barnwal R. P.; Pandey A. Tuberculosis: an overview of the immunogenic response, disease progression, and medicinal chemistry efforts in the last decade toward the development of potential drugs for extensively drug-resistant tuberculosis strains. J. Med. Chem. 2021, 64 (8), 4359–4395. 10.1021/acs.jmedchem.0c01833. [DOI] [PubMed] [Google Scholar]
- De Groote M. A.; Huitt G. Infections due to rapidly growing mycobacteria. Clin. Infect. Dis. 2006, 42 (12), 1756–1763. 10.1086/504381. [DOI] [PubMed] [Google Scholar]
- Serpi M.; Ferrari V.; Pertusati F. Nucleoside derived antibiotics to fight microbial drug resistance: new utilities for an established class of drugs?. J. Med. Chem. 2016, 59 (23), 10343–10382. 10.1021/acs.jmedchem.6b00325. [DOI] [PubMed] [Google Scholar]
- Van Heeswijk R. P. G.; Dannemann B.; Hoetelmans R. M. W. Bedaquiline: a review of human pharmacokinetics and drug–drug interactions. J. Anti-microb Chemother. 2014, 69 (9), 2310–2318. 10.1093/jac/dku171. [DOI] [PubMed] [Google Scholar]
- Kroesen V. M.; Gröschel M. I.; Martinson N.; Zumla A.; Maeurer M.; van der Werf T. S.; Vilaplana C. Non-steroidal anti-inflammatory drugs as host-directed therapy for tuberculosis: a systematic review. Front. Immunol. 2017, 8, 772. 10.3389/fimmu.2017.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley K. E; Rosenkranz S. L; Conradie F.; Moran L.; Hafner R.; von Groote-Bidlingmaier F.; Lama J. R.; Shenje J.; De Los Rios J.; Comins K.; Morganroth J.; Diacon A. H; Cramer Y. S; Donahue K.; Maartens G.; Alli O.; Gottesman J.; Guevara M.; Hikuam C.; Hovind L.; Karlsson M.; McClaren J.; McIlleron H.; Murtaugh W.; Rolls B.; Shahkolahi A.; Stone L.; Tegha G.; Tenai J.; Upton C.; Wimbish C. QT effects of bedaquiline, delamanid, or both in patients with rifampicin-resistant tuberculosis: a phase 2, open-label, randomised, controlled trial. Lancet Infect. Dis. 2021, 21 (7), 975–983. 10.1016/S1473-3099(20)30770-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashemian S. M. R.; Farhadi T.; Ganjparvar M. Linezolid: a review of its properties, function, and use in critical care. Drug Des., Devel. Ther. 2018, 12, 1759–1767. 10.2147/DDDT.S164515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler J. A.; Maartens G.; Meintjes G.; Wasserman S. Clofazimine for the treatment of tuberculosis. Front. Pharmacol. 2023, 14, 1100488 10.3389/fphar.2023.1100488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K.; Ahmed F.; Sharma T.; Grover A.; Agarwal M.; Grover S. Potential Repurposed Drug Candidates for Tuberculosis Treatment: Progress and Update of Drugs Identified in Over a Decade. ACS Omega 2023, 8, 17362. 10.1021/acsomega.2c05511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi N.; Basham C.A.; Johnston J.C.; Khan F.A.; Migliori G.B.; Centis R.; D'Ambrosio L.; Jassat W.; Davies M.A.; Schwartzman K.; Campbell J.R. CO105 The Association of Sars-Cov-2 Infection and Tuberculosis Disease with Unfavorable Treatment Outcomes: A Systematic Review. Value Health 2023, 26 (6), S34. 10.1016/j.jval.2023.03.178. [DOI] [PMC free article] [PubMed] [Google Scholar]