

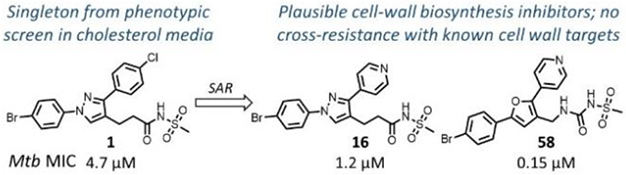
Abstract
Phenotypic whole cell high-throughput screening of a ~150,000 diverse set of compounds against Mycobacterium tuberculosis (Mtb) in cholesterol-containing media identified 1,3-diarylpyrazolyl-acylsulfonamide 1 as a moderately active hit. Structure-activity relationship (SAR) studies demonstrated a clear scope to improve whole cell potency to MIC values of <0.5 μM and a plausible pharmacophore model was developed to describe the chemical space of active compounds. Compounds are bactericidal in vitro against replicating Mtb and retained activity against multidrug resistant clinical isolates. Initial biology triage assays indicated cell wall biosynthesis as a plausible mode-of-action for the series. However, no cross-resistance with known cell wall targets such as MmpL3, DprE1, InhA and EthA was detected suggesting a potentially novel mode-of-action or inhibition. The in vitro and in vivo drug metabolism and pharmacokinetics profiles of several active compounds from the series were established leading to the identification of a compound for in vivo efficacy proof-of-concept studies.
Keywords: Tuberculosis; Mycobacterium tuberculosis; Phenotypic screen; TB Drug Discovery; 1,3-Diarylpyrazolyl acylsulfonamides
Graphical Abstract
INTRODUCTION
Tuberculosis (TB) continues to be a global epidemic most severely affecting the underprivileged sectors of society throughout the world. According to the World Health Organization (WHO) 2019 global TB report, 10 million people fell sick with TB in 2018 and 1.5 million people lost their lives.1 The most common drug regimen currently used for the treatment of drug-sensitive TB is 40-years old and consists of four drugs - rifampicin (RIF), isoniazid (INH), pyrazinamide (PZA) and ethambutol (EMB) - taken over a minimum period of six months. The long treatment duration along with unpleasant side-effects often leads to patient non-compliance, resulting in the development of drug resistant TB, viz. multidrug resistant (MDR) TB that is refractory to two frontline drugs RIF and INH and the extensively drug resistant (XDR) TB that develops additional resistance to any fluoroquinolone and at least one of the three injectable second-line drugs (amikacin, kanamycin, or capreomycin). Treatments for drug-resistant TB are fraught with limited choices of poorly efficacious medicines with severe side-effects and lengthy treatment durations of 18–24 months. Hence, there has been a strong global drive towards identifying safe new drugs with novel mode-of-actions (MoAs), which would be effective for treatment of drug-resistant TB and be included in combination regimens aimed at treatment shortening for drug-sensitive TB. Gratifyingly, a few recently approved drugs with novel MoAs such as bedaquiline,2 pretomanid,3 and delamanid4 have been added to the arsenal of the available TB drugs. Several follow-up drug candidates such as the DprE1 inhibitors Macozinone (MCZ, PBTZ-169)5 and TBA-7371,6 MmpL3 inhibitor SQ-109,7 QcrB inhibitor Telacebec (Q203)8 and the Leucyl-tRNA synthetase inhibitor GSK 30366569 are in the clinical development pipeline (Fig. 1). Other known drugs such as linezolid have also shown great promise in the treatment of MDR-TB and have encouraged pre-clinical efforts to discover safer analogues.10 Considering the significant attrition rates in clinical development and empirical nature of effective combination therapy toward shorter treatments, there is a continuous need to replenish the global TB drug pipeline with new bactericidal agents that avoid pre-existing clinical drug resistance, either through novel MoA or novel mode-of-inhibition (MoI) of clinically validated drug targets.
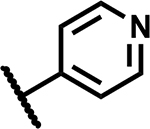
Figure 1.

New approved drugs and clinical candidate for TB
Mycobacterium tuberculosis (Mtb), the causative agent of TB, is known to adapt metabolically to the various nutrients available during its cycle of infection, persistence, and reactivation.11, 12 Fatty acids and cholesterol are important carbon sources for in vivo survival and pathogenesis of Mtb13, 14 even though this organism can utilize carbohydrates and carbon dioxide as carbon sources and needs glucose for in vivo persistence.15 There have been efforts to screen compound libraries against Mtb surviving on fatty acid and cholesterol media to identify compounds efficacious against various pathological forms of Mtb.16 Herein we report structure-activity relationship (SAR) studies and preliminary target identification studies of 1,3-diarylpyrazolyl-acylsulfonamides identified from the phenotypic screening of a compound library supplied by DuPont in cholesterol-containing media.
Results and Discussion.
Phenotypic hit with a novel mode-of-action.
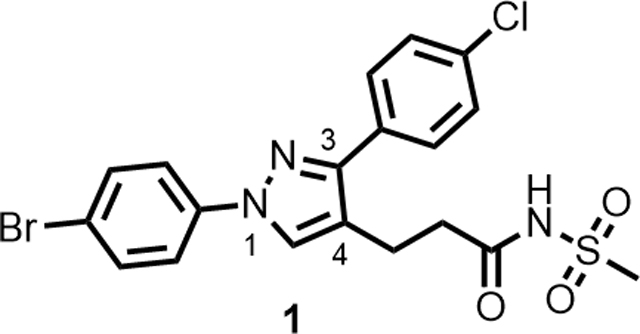
A high-throughput phenotypic whole cell screen of a ~150,000 diverse set of compounds from a DuPont Agrichemicals compound library against Mtb was conducted at the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIAID/NIH, U.S.) using Mtb in two different media, Middlebrook 7H9/DPPC/cholesterol/BSA/Tyloxapol (DPPC/chol) and Middlebrook 7H9/butyrate pH 6.0 /nitrite/BSA/Tyloxapol (butyrate pH6/nitrite). This screen identified ~2000 primary hits. The activity of these hits was confirmed by determining the minimum inhibitory concentrations (MICs) that lead to no visible growth from solid samples against Mtb under three conditions - DPPC/chol, and butyrate pH6/nitrite as well as 7H9/DPPC/casitone/Tyloxapol (DPPC/cas). Of the three media used, activity in DPPC/chol medium may closely simulate in vivo nutritional ambience for replicating Mtb whereas DPPC/cas is a protein-free minimal media used to identify weaker inhibitors that would be missed in standard serum containing media due to the effect of protein-binding. The low pH butyrate medium containing nitrite reflects some of the environmental stresses encountered by Mtb residing in macrophages in vivo, namely – acidic pH of the intraphagosomal environment of the macrophage and reactive nitrite intermediates (RNI) released from nitrite.14, 17 Among the few hits that confirmed from the screening campaign, compound 1 was identified as a singleton with moderate activity under the various media conditions; notably it did not show any previous literature reports of antitubercular activity (Table 1). Though 1 has a 1,3-diarylpyrazole core found in some known anti-Mtb actives,18, 19 it has a unique N-(methylsulfonyl)propanamide substituent at the pyrazole C4 position. It had been reported as a potassium channel modulator for the treatment of a variety of human disorders.20 Screening against various tool strains of Mtb indicated a novel MoA as described in the biology triage section of this manuscript. The compound was non-cytotoxic against HepG2 cell-line under both glucose and galactose containing media conditions at the highest concentration tested, minimizing the possibility of mitochondrial cytotoxicity.21 All these observations sparked further interest in exploring the potential of 1 to deliver a novel drug candidate for TB treatment. Detailed SAR and target identification studies were initiated as detailed in the following sections.
Table 1.
Hit triage of compound 1
| |
|---|---|
| MIC 7H9/ADC/Tw (µM) | 4.7 |
| MIC 7H9/glucose/BSA/Tx (µM) | 4.7 |
| MIC 7H9/DPPC/cholesterol/BSA/Tx (µM) | 4.7 |
| MIC 7H9/cholesterol/BSA/Tx (µM) | 4.7 |
| IC90 in 7H9/2.5mM butyrate/pH6/0.1mM nitrite (µM) | 6.25 |
| HepG2 IC50 glucose (µM) | >50 |
| HepG2 IC50 galactose (µM) | >50 |
| Solubility (µM) | 170 |
MIC - minimum inhibitory concentration against Mtb H37Rv; 7H9 - Middlebrook 7H9; ADC - Albumin-Dextrose-Catalase supplement; Tw – Tween 80; BSA – Bovine serum albumin; Tx – Tyloxapol; DPCC - Dipalmitoylphosphatidylcholine and cholesterol; IC50 - 50% inhibitory concentration; Solubility – Aqueous Solubility at pH 7.4
Synthesis.
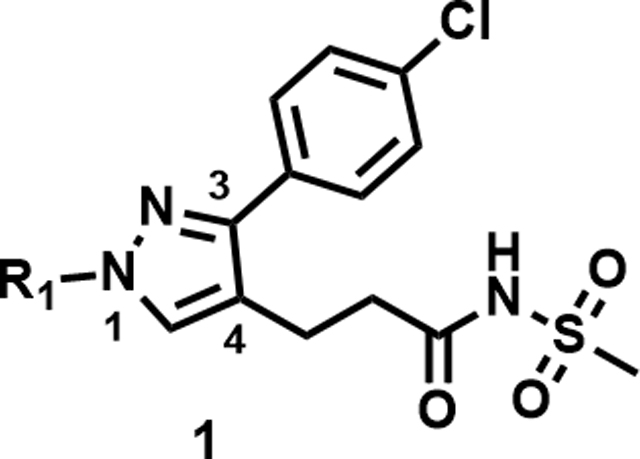
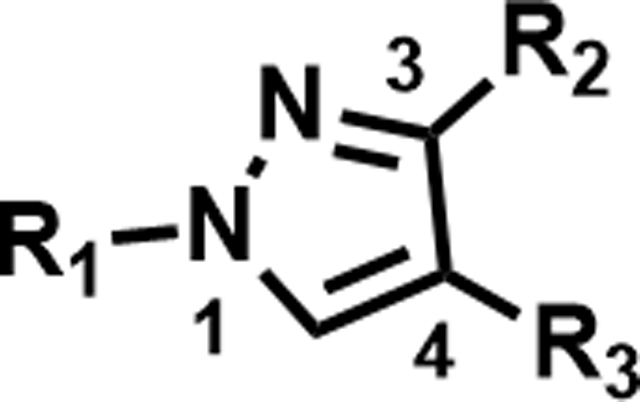
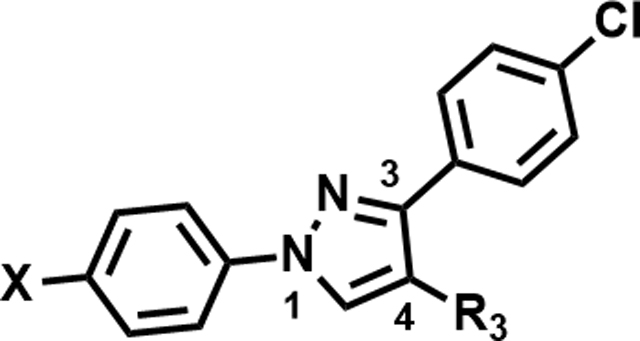
As the hit compound 1 was identified as a singleton from the screen, detailed SAR studies were needed to identify key pharmacophoric features and to expand the SAR scope for increasing anti-Mtb activity. To this end a systematic exploration of each substituent around the central pyrazole ring and variations of the pyrazole core were undertaken. Compounds were specifically synthesized to explore the SAR around substituents around the N1 (compounds 1–12, Table 2), C3 (compounds 13–25, Table 3), and C4 positions (compounds 26–45, Table 4). Furthermore, additional compounds were synthesized to explore replacements for the N-(methylsulfonyl)propanamide substituent at the C4-position (compounds 46–50, Table 5) and to vary the central pyrazole core (compounds 51–58, Table 6).
Table 2.
SAR at the N1-substituent
| |||
|---|---|---|---|
| Compound Number | R1 | MIC (µM) | Solubility (µM) |
| 1 |
|
4.7 | 170 |
| 2 |
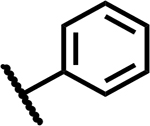
|
>50 | 200 |
| 3 |
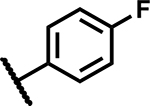
|
37 | 25 |
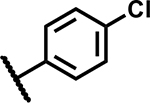
| 4 |
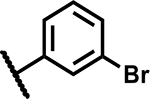
|
>50 | <5 |
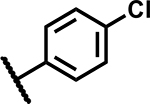
| 5 |
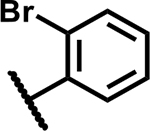
|
>50 | 165 |
| 6 |
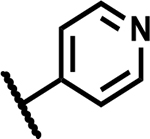
|
>50 | 190 |
| 7 |
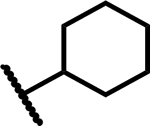
|
>50 | 200 |
| 8 |
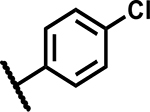
|
4.7 | 100 |
| 9 |
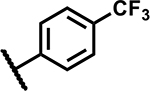
|
3.1 | 50 |
| 10 |
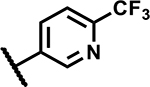
|
4.7 | 90 |
| 11 |
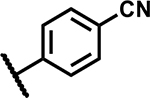
|
25 | 75 |
| 12 |
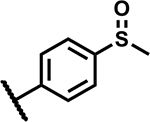
|
≥50 | 200 |
MICs were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media; Solubility was determined in aqueous pH 7.4 phosphate buffer simulating thermodynamic conditions.
Table 3.
SAR at C3-position
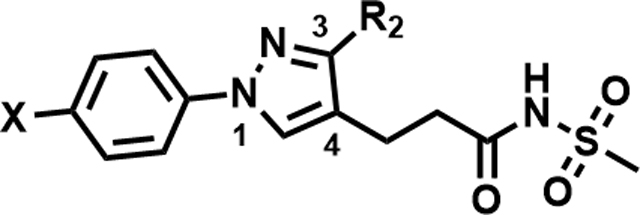
| ||||
|---|---|---|---|---|
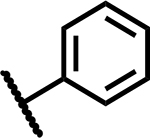
| Compound Number | X | R2 | MIC (µM) | Solubility (µM) |
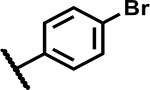
| 13 | Br |
|
2.3 | 200 |
| 14 | Br |
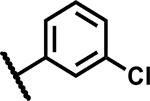
|
2.3 | 165 |
| 15 | Cl |
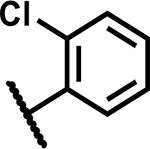
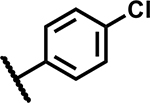
|
>50 | 90 |
| 16 | Br |
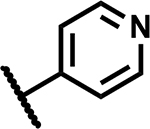
|
1.2 | 185 |
| 17 | Br |
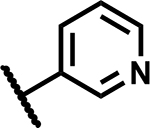
|
6.25 | 200 |
| 18 | Br |
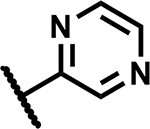
|
20 | 196 |
| 19 | Br |
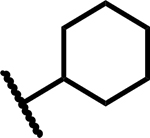
|
9.4 | 60 |
| 20 | Br |
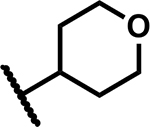
|
9.4 | 200 |
| 21 | Br |
|
>50 | 196 |
| 22 | Br |
|
50 | 200 |
| 23 | Br |
|
12.5 | <5 |
| 24 | Br |
|
25 | 160 |
| 25 | Br |
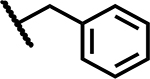
|
>50 | 200 |
MIC – were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media; Solubility – aqueous solubility at pH 7.
Table 4.
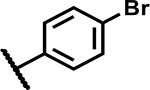
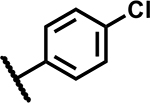
SAR at C4 N-sulfonylpropanamide
| |||||
|---|---|---|---|---|---|
| Compound Number | R1 | R2 | R3 | MIC (µM) | Solubility (µM) |
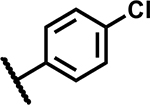
| 26 |
|
|
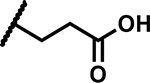
|
>50 | 155 |
| 27 |
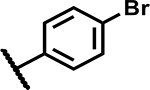
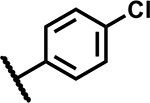
|
|
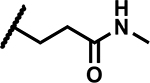
|
>50 | <5 |
| 28 |
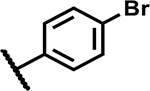
|
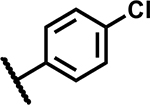
|
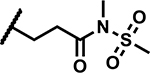
|
>50 | <5 |
| 29 |
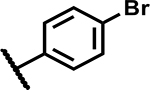
|
|
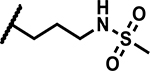
|
>50 | <5 |
| 30 |
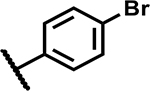
|
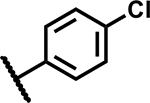
|
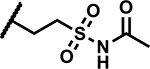
|
>50 | 165 |
| 31 |
|
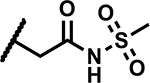
|
|
>50 | 200 |
| 32 |
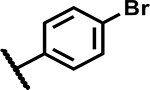
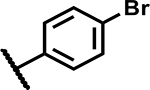
|
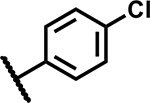
|
|
37 | 135 |
| 33 |
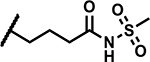
|
|
|
12.5 | 10 |
| 34 |
|
|
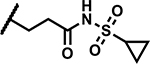
|
>50 | 5 |
| 35 |
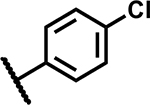
|
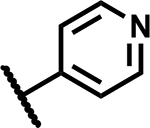
|
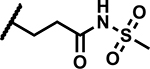
|
6.25 | ND |
| 36 |
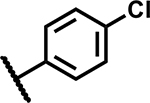
|
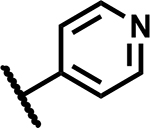
|
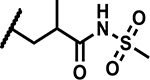
|
>50 | 190 |
| 37 |
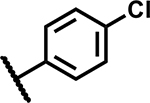
|
|
|
50 | 170 |
| 38 |
|
|
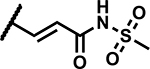
|
4.7 | 90 |
| 39 |
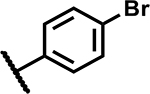
|
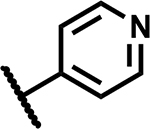
|
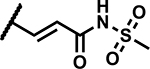
|
1.2 | 185 |
| 40 |
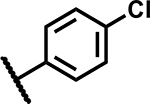
|
|
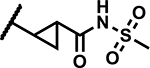
|
19 | 200 |
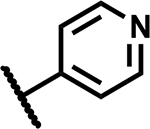
| 41 |
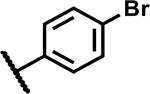
|
|
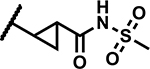
|
12.5 | 155 |
| 42 |
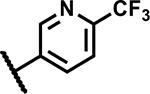
|
|
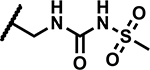
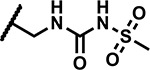
|
1.2 | 195 |
| 43 |
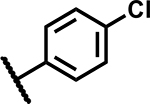
|
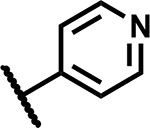
|
|
1.56 | 150 |
| 44 |
|
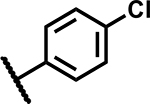
|
|
3.13 | <5 |
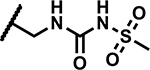
| 45 |
|
|
|
12.5 | ND |
MIC – were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media; Solubility – aqueous solubility at pH 7.
Table 5.
Heterocycles at C4
| ||||
|---|---|---|---|---|
| Compound Number | X | R3 | MIC (µM) | Solubility (µM) |
| 46 | Br |
|
>50 | 150 |
| 47 | Br |
|
50 | <5 |
| 48 | Cl |
|
>50 | 200 |
| 49 | Br |
|
19 | <5 |
| 50 | Cl |
|
25 | 175 |
MIC –were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media at day 14; Solubility – aqueous solubility at pH 7.
Table 6.
SAR at pyrazole core
| Compound Number | Structure | MIC (µM) | Solubility (µM) |
|---|---|---|---|
| 51 |
|
6.25 | 200 |
| 52 |
|
5.5 | 195 |
| 53 |
|
37 | 135 |
| 54 |
|
>50 | 200 |
| 55 |
|
>50 | 170 |
| 56 |
|
37 | 200 |
| 57 |
|
19 | 90 |
| 58 |
|
0.15 | 25 |
MIC – were measured in Middlebrook 7H9/Glu/BSA/Tyloxapol media; Solubility – aqueous solubility at pH 7.
Compound 1-25 and 35 were synthesized by modification of the reaction sequence described previously as depicted in Scheme 1.20 In short, the key intermediates, 4-formylpyrazoles (A) were synthesized by treating the appropriately substituted hydrazones with POCl3 in a Vilsmeier-Haack formylation reaction, which was followed by a regioselective Knövenagel condensation reaction with malonic acid to obtain the corresponding trans-acrylic acids B. Selective reduction of the acrylic acids in order to avoid concomitant halogen hydrogenolysis gave the corresponding propanoic acid intermediates C which were coupled with methanesulfonamide in the presence of CDI and DBU or, in a few cases, in the presence of DCC and DMAP to yield target compounds. The detailed synthetic schemes are described in Supplementary Information (Schemes S1 and S2). Synthesis of compounds 26-41 with modified alkyl linkers at C4 are described in Schemes S3–S6. Urea and carbamate derivatives 42-45 could be synthesized from common intermediate A via alcohols D and amines E as described in Scheme S7. Synthesis of compounds 46-50 with replacement of acylsulfonamide functionality are described in Schemes S8 and S9. Synthesis of compounds 51–58 with variations to the central pyrazole core are described in Schemes S10–S14.
Scheme 1. General synthetic scheme.

Reagents and conditions: (i) KOAc, EtOH, 80 °C; (ii) POCl3, DMF, 80 °C; (iii) Malonic acid, piperidine, pyridine, 90 °C; (iv) method 1: NH2NH2.H2O, MeOH, 80 °C; method 2: NBSH, Et3N, THF, 45 °C; (v) method 1: CH3SO2NH2, CDI, DBU, DMF, 90 °C, method 2: CH3SO2NH2, DCC, DMAP, 0 – 45°C; (vi) NaBH4, MeOH, 25 °C; (vii) PPh3, DIAD, NaN3, H2SO4, DCM, 0 °C to 25 °C; (viii) PPh3, THF/H2O, 60 °C; (ix) CDI, methanesulfonamide, DIPEA, DCM, DCE or DMF depending on the solubility of the substrate, 25 °C – 90 °C.
SAR at the N1-substituent.
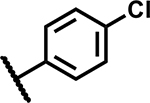
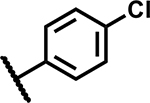
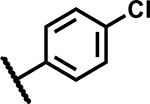
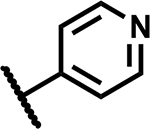
Mtb activity measured as MIC in Middlebrook 7H9/Glu/BSA/Tx media and aqueous solubility were monitored as first tier criteria towards selection of compounds for further characterization (Table 2). Compound 2 with an unsubstituted phenyl ring showed a much higher MIC (>50 μM) relative to 1. Replacement of p-bromo on the phenyl ring with p-fluoro, as in 3, or moving bromine to the -meta or -ortho positions as in 4 and 5, respectively, led to deterioration of activity (MICs ≥50 μM). Similarly, 6 with the phenyl ring replaced with 4-pyridyl, and 7 with a cyclohexyl ring showed much weaker MICs (≥50 μM). Compounds 11 and 12 with polar CN and methylsulfoxide groups respectively at the para position of the phenyl ring showed 5–10-fold weaker activity indicating the essentiality of hydrophobic substituents at this position. Only compounds 8 and 9 with p-chlorophenyl and p-CF3-phenyl, respectively, retained MICs similar to 1, albeit with lower aqueous solubility (100 and 50 μM, respectively). Compound 10 with a 4-CF3-3-pyridyl ring at N1 showed a comparable MIC to 1 with somewhat improved solubility (90 μM) compared to 9. Overall, variations at the N1-phenyl ring (R1) demonstrated the essentiality of an aryl ring with larger hydrophobic para-substituents for Mtb activity.
SAR at the C3-position.
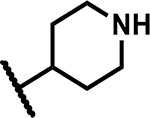
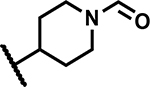
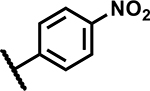
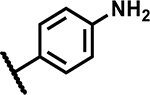
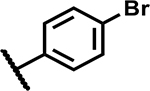
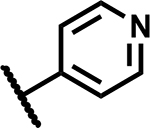
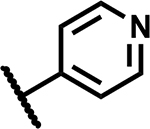
Compared to the N1 position, substituents at the C3-position (R2) of the pyrazole showed a wider scope for structural variation leading to slightly lower MICs (Table 3). Compound 13 with a hydrogen atom in place of Cl on the C3 phenyl group retained a similar MIC to 1. Compound 14 with m-Cl-phenyl also retained a similar MIC, but compound 15 with o-Cl-phenyl showed a >10-fold higher MIC. Compound 16 with a 4-pyridyl ring at C3 in place of Cl-phenyl improved the MIC ~4-fold, thus providing a more potent tool compound for MoA studies (see below). Compound 17 with a 3-pyridyl ring showed a MIC similar to compound 1, and 18 with a pyrazine ring showed a 5-fold higher MIC. Compounds 19 and 20 with saturated cyclohexyl and tetrahydropyran rings, respectively, at the C3-position showed MICs similar to 1, but 21 with a basic piperidine at C3 and its N-formyl derivative 22 showed much weaker activity (MICs ≥50 μM) indicating an intolerance towards polar functionality at this position for non-aromatic R2 substituents. Similarly, higher (3–5-fold) MICs were observed with 23 having a p-NO2-phenyl at the C3-position and the corresponding aniline derivative 24. Compound 25 with a C3-benzyl group also showed much weaker MIC, thus limiting the SAR scope to explore directly attached aryl, heteroaryl and saturated rings.
SAR at C4 N-(methylsulfonyl)propanamide.
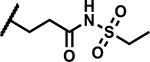
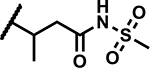
A few analogues were synthesized to determine the essentiality of the N-sulfonylpropanamide functionality linked to the pyrazole C4 position (R3, Table 4). The corresponding carboxylic acid 26 and N-methyl amide derivative 27 showed much weaker activity (MICs >50 μM) establishing the N-sulfonylpropanamide as a critical functionality for potency. Methylation of the sulfonamide NH led to 28 that showed a significant decrease in activity (MIC >50 μM) along with low aqueous solubility (<5 μM). Replacement of the carbonyl group with methylene (compound 29) led to a high MIC (>50 μM). The data points to the need for an acidic functionality (the sulfonylpropanamide) that has specific binding interaction with the target not seen with the carboxylic acid of 26. The specificity of the sulfonylpropanamide was further supported by swapping positions of the sulfonyl and carbonyl groups (compound 30) leading to high MICs (>50 μM). The length of alkyl chain linking the N-(methylsulfonyl)carbamoyl group to the pyrazole core was also critical to maintain lower MICs as demonstrated with 31 (MIC >50 μM) and 32 (MIC 37 μM) having shorter and longer linking alkyl chains to the pyrazole C4 position. The ethyl sulfonyl analogue 33 showed a 4-fold higher MIC, while the larger cyclopropylsulfonyl group was even less active (MIC >50 μM) indicating a limited scope around the sulfonyl alkyl group. Hence, the N-(methylsulfonyl)propanamide represents an optimal pharmacophore predicted to engage in key interactions with the target as delineated later.
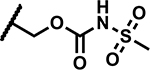
Modifications on the propanamide chain were then explored to better understand SAR and continue compound optimization, particularly to improve metabolic stability of the acylsulfonamide while maintaining the potency of the compounds (refer DMPK section). Addition of a methyl group either on α- or β-carbons of the propanamide chain led to compounds 36 and 37 with much weaker activity (MICs ≥50 μM) compared to the unsubstituted analogue 35 (MICs 6.25 μM) indicating a likely clash at the target level or perturbation of the preferred side chain binding conformation. Comparatively better MICs were observed by constraining the methyl group into a cyclopropane ring as in 40 and 41 (MICs 12.5–19 μM). The trans-acrylamide derivatives 38 and 39 showed more potent MICs (1.2–4.7 μM). Compounds 42, 43 and 44 with a sulfonylurea tethered to C4 and a net replacement of CH2 with NH showed lower MICs compared to the corresponding N-(methylsulfonyl)propanamide derivatives indicating a possible H-bond donor interaction from the urea NH with the target. Compound 45 with a sulfonylcarbamate functionality showed an 8-fold higher MIC relative to 43, supporting the hypothesis that there is a contribution to binding by the urea NH.
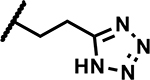
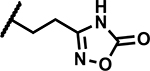
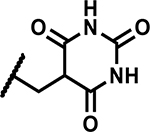
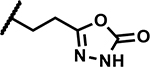
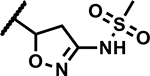
Further, we explored replacing the N-acylsulfonamide group with other functionality envisioned to similarly position an acidic group. (Table 5). Compound 46 and 47 with tetrazole and 1,2,4-oxadiazolone moieties attached to C4 via an ethyl linker and compound 48 with a pyrimidinetrione attached via a methylene linker showed much higher MICs (≥50 μM). Only 49 with an 1,3,4-oxadiazolone attached via an ethyl linker at the C4-position showed a lower MIC, about 4-fold higher than 1, albeit the low solubility (<5 μM) rendered the compound less interesting. Overall, there is a limited scope for replacement of the N-acylsulfonamide functionality. Compound 50 wherein the propanamide was cyclized into a dihydroisoxazole ring also retained an MIC within 4-fold of compound 1. Further scope for optimization of MICs using C4-functionalities as in compounds 49 and 50 by varying other substituents in the molecule remains to be explored.
SAR of the pyrazole core.
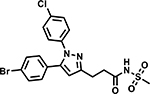
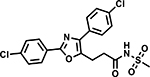
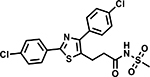
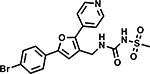
Table 6 shows the analogues synthesized to understand the importance of the relative positions of substituents on the pyrazole ring and the influence of alternative isosteric heterocycles. Compounds 51 and 52 retained the same relative position of all three substituents and the pyrazole nitrogen lone pair of 1 and 3 and, not surprisingly showed similar MICs. Analogues 53 and 54 with 1,4-diarylpyrazole and 3,5-diarylpyrazole cores also display the three substituents in the same relative orientation as 1 but shift the position of the pyrazole nitrogen lone pair resulting in much higher MICs (≥50 μM) and indicating an important target interaction for the pyrazole N2 of 1. 1,5-Diarylpyrazole 55 had a much higher MIC (≥50 μM), further confirming the importance of relative positions of aryl rings in maintaining potency. Replacement of the pyrazole with other 5-membered heterocycles as with 56 and 57 having oxazole and thiazole cores, respectively, showed 5–8-fold weaker MICs. Compound 58 with a furan core showed much more potent activity with MIC below 0.5 μM. The above observations indicate a smaller angle between the R1 and R2 substituents on the smaller pyrazole and furan heterocycles (150.3 ° and 125.5 ° respectively) are favored over what would be seen with the larger thiazole core (154.6 °) in 57. Loss of potency for oxazole 56 is consistent with the observation that polar atoms are not well tolerated at other position of 5-membered ring as seen for 53 and 54. The furan oxygen is favorable as is the 2-position N of the pyrazole of 1 and of 51. Improved activity of furan analogue 58 could be attributed to O being more electronegative than N, possibly making a stronger hydrogen bonding interaction with the target.
The above SAR demonstrates the specific structural requirements of compound 1 and analogues required to maintain anti-Mtb activity alluding to specific target engagement by the scaffold.
Representative Pharmacophore.
We have accordingly developed a pharmacophore hypothesis to summarize key structural features of the scaffold that may aid in efforts to further optimize this series. To describe the chemical space of the active acyl sulfonamide pyrazole compounds, a series of pharmacophore hypotheses were generated from the set of compounds using the Phase module from the Schrodinger molecular modelling suite.22 The activity cutoff was defined as MIC <25 μM in the 7H9/glucose/BSA/Tx assay among the set of 58 compounds in this series. An AARRR (A- acceptor, R – ring) pharmacophore hypothesis (Fig. 2) consisting of three aromatic rings (R, R1 and R2), and two hydrogen bond acceptors was found to best describe the chemical space of active compounds. A central aromatic ring feature (R) corresponds to the central pyrazole core with two more aromatic ring features corresponding to aromatic substitutions at the 1 and 3 positions (R1 and R2 respectively). The first acceptor feature (A1) can be found approximately 5.65 Å from the C4-position of the pyrazole core fitting the negatively charged N of the acyl sulfonamide moiety. At physiological pH, the acyl sulfonamide NH is deprotonated, creating an anion that could serve as a hydrogen bond acceptor. The second acceptor (A2) lies another 2.43 Å away forming a 158 ° angle between A1 and the central ring feature (Fig. 2A). A1 moves out of the plane formed by the three ring features forming a 95 ° dihedral angle around a line connecting the R feature and the A1 feature (Fig. 2B). In this set of compounds, R1 often seems, at a minimum, to require negatively charged acid groups as only compounds with a pKa below physiological pH have shown activity.
Figure 2:

Representations of the AARRR pharmacophore model. A) A 2D representation of the pharmacophore model with two of the ring features (R1 and R2) both shown to be about 4 Å from the central R feature forming and angle of 140° with each other. The first acceptor feature (A1) lies 5.65 Å on from R with the acceptor feature A2 lying an additional 2.59 Å on from A1 forming an angle of 158.4 ° with A1 and R. B) A 3D representation of the pharmacophore model showing the A1 and A2 features to be out of the plane formed by R, R1 and R2 with a −95.0 ° dihedral angle around a line from R to A1.
A representative active compound 43 fits all five of these features very closely (Fig. 3A). The pharmacophore fit of each ligand was determined via Phase docking with the fit evaluated using Phase fitness score.22 All active compounds featured in this manuscript fit the five features (Fig. 3B) very closely.
Figure 3.

A) Compound 43 fitted to the 5-feature AARRR pharmacophore hypothesis very closely. The three aromatic ring features are occupied by the chlorophenyl, pyrazole and pyridyl groups while the deprotonated sulfonamide fits the negative charge feature, and a sulfone O fits the acceptor feature; B) An overlay of all the active compounds in this set closely fitting all the five pharmacophore features. C) Compound 34 is an example of an inactive compound that is an excellent fit of the pharmacophore. This loss in activity is brought about by the bulky cyclopropyl group extending into a region outside of the pharmacophore. This region could be defined as an excluded volume. D) The inactive compounds from this set either fit the pharmacophore hypothesis fully or partially with many occupying spaces outside the definition of the pharmacophore suggesting potential for the definition of excluded volumes past the A2 feature and between the A1 and R1 features.
A bioisostere replacement of the acyl sulfonamide with the 1,3,4-oxadiazolone of 49 introduces an acid center with a predicted23 pKa of 7.87 +/− 0.4, in contrast to the predicted pKa of the sulfonamide of 43 of 3.72 +/− 0.23 thus lowering the propensity for an acceptor/negative charge to form at physiological pH. Since the oxadiazolone has a pKa close to physiological pH, both the protonation and deprotonated states need to be considered. Only the deprotonated form fits all five features of the pharmacophore hypothesis. This poorer fit explains the weaker MIC (19 μM) of 49 compared to 43 (1.56 μM) (Fig. S1c).
Compounds 28–31 were all inactive with poor pharmacophore fits for at least one reason as explained in supplementary section with Figure S1. Compounds 33 and 34 (Fig. 3C) with their respective ethyl and cyclopropyl attachments to the sulfone and 36 and 37 with methyl groups in positions α and β to the pyrazole core all showed near perfect fits for the pharmacophore model despite a complete loss of activity. These compounds contain additional bulk occupying regions not defined by the pharmacophore suggesting potential excluded volumes. Excluded volumes beyond the A2 feature and adjacent to the alkyl linker at the sites of potential branching would penalize these compounds. These excluded volumes would need more examples to be properly defined. An overlay of all the inactive compounds presented in the study (Fig. 3D) demonstrates many of them to occupy regions that could be considered for excluded volumes.
Compound 58 with a furan core fulfills all pharmacophore features (Fig. S1d) and showed potent activity whereas compounds 53, 54 and 56 with changes in the core ring (R) are examples of weak actives with good fits. Comparing the matched pair of inactive 53 and active 51 would suggest that a heteroaromatic acceptor N contributes to activity. However, small changes to the π-characteristics of the core caused by these rearrangements may also contribute to the lower activity. More data from analogues containing rearrangements of this core would be needed to confirm the contribution of this acceptor and warrant expanding the definition of this pharmacophore feature to include an acceptor corresponding to the 2-position of the pyrazole core.
Biology triage.
Compounds 1, 8, 43 and 58 were screened against various tool strains of Mtb to deconvolute the MoA for anti-Mtb activity. None of the compounds showed modulation in MICs against a cytochrome bd oxidase knockout mutant strain (Δcyd)24, 25 nor against a QcrB mutant (QcrBA317T), thereby eliminating the two targets of the respiratory pathway as a likely target (Table 7). Neither did the compounds elicit a positive response in the PrecA-LUX bioluminescence reporter assay,26 which is designed to detect modulation in recA expression, an indicator of DNA damage, hence ruling it out as a MoA for the compounds (Fig. S2 in supplementary information). However, the compounds showed sustained signals in the PiniB-LUX bioluminescence reporter assay that detects modulation in iniBAC operon expression,26 an indicator of cell wall biosynthesis being disrupted (Fig. S3). The prediction that the compounds inhibited cell wall biosynthesis was further confirmed by transcriptional profiling studies that showed upregulation in the genes involved in cell wall biosynthesis including many of the genes upregulated by isoniazid (INH) including acpM, fabD, kasA, kasB, accD6, rv2248, efpA, iniB, iniA and iniC (Table S3 and S4 in supplementary information). Despite these similarities, some of the genes that are upregulated during isoniazid treatment (ppsA, ppsB, ppsC, ppsD, ppsE and fbpC2) were downregulated during exposure to compounds 8 and 16 indicating differences in transcriptional responses. The compounds were screened against the mutant strains of MmpL3 and DprE1 which are frequently encountered cell wall targets (Table 7). Two strains carrying a mutation in MmpL3 (G253E, or G758A) were not resistant, arguing against MmpL3 as the target. Similarly, three strains carrying mutations in DprE1 (Y314C, P116S, or T314H), which confer resistance to other DprE1 inhibitors, were not resistant, suggesting DprE1 is not the target. In addition, other isogenic single-drug resistant strains of known cell wall targeting drugs such as INH and ethionamide (ETH) were also not cross-resistant to the compounds (Table 7). Taken together, these data suggest a MoA that most likely involves interference with Mtb cell wall biogenesis in a manner that does not lead to cross resistance to other cell wall inhibitors. Studies to determine specific MoA of this series are underway.
Table 7.
MICs against isogenic single-drug resistant mutant strains of Mtb
| MIC (µM) | |||||||
|---|---|---|---|---|---|---|---|
| Mtb Strain | 1 | 8 | 43 | 58 | RIF | INH | ETH |
| H37RvMA | 4 | 4 | 0.5 | 0.24 | 0.002 | 7.8 | 15.6 |
| ∆ cyd | 4 | 4 | 0.5 | 0.12 | 0.005 | ||
| QcrBA317T | 4 | 4 | 0.5 | 0.12 | 0.005 | ||
| MmpL3G253E | 4 | 4 | 1 | 0.24 | 0.005 | ||
| MmpL3G758A | 2 | 2 | 1 | 0.5 | 0.004 | ||
| DprE1Y314C | 3 | 4 | 2 | 0.24 | 0.005 | ||
| DprE1P116S | 0.5 | 0.5 | 0.12 | 0.12 | 0.002 | ||
| DprE1T314H | 0.5 | 0.5 | 0.12 | 0.12 | 0.002 | ||
| InhA-OE | 0.4 | 0.24 | <0.12 | <0.13 | 0.001 | 31.2 | |
| KatGT198A | 15.6 | 15.6 | 4 | 1 | 0.003 | >62.5 | >125 |
| EthAC253R | 8 | 8 | 2 | 0.5 | 0.002 | 7.8 | >125 |
InhA-OE: InhA overexpressor (H37Rv-LP:fabG1/inhA-c-15t); RIF: Rifampicin; INH: Isoniazid; ETH: Ethionamide
Bactericidality and activity against clinical isolates.
The bactericidal activity of representative compounds 1, 8 and 16 against replicating Mtb was evaluated through the exposure of Mtb cultures to various concentrations of compounds for 8 days and allowing the treated cultures to regrow on a fresh media after washing out the excess free drug (Fig. S4 in supplementary information). All the tested compounds showed 2–2.5 log CFU reduction at 1–4X MIC concentrations within 8 days indicating the bactericidal nature of the compounds. But the compounds were not active against non-replicating Mtb under nutrient starvation conditions. Compound 16 retained activity against 3 drug-sensitive, two mono-drug resistant and three MDR/XDR clinical isolates of Mtb indicating potential utility of the compounds from the series to treat drug-sensitive as well as drug-resistant TB (Table 8). The MIGIT (Mycobacteria Growth Indicator Tube) assay uses higher inoculum in liquid broth designed for faster growth of mycobacteria and involves longer incubation times relative to the Alamar Blue MIC microtiter plate assay used for the SAR investigation herein.27 Hence, MIC values between the two assays will not be comparable. Nonetheless, it is gratifying that the MIC shifts between multidrug resistant and susceptible Mtb strains in the MIGIT assays were relatively small (less than 4-fold) thereby suggesting broader clinical utility.
Table 8.
MICs of compound 16 against clinical Mtb isolates
| Mtb Isolate | Resistant Profiles | MIC of Compound 16 (µM)a |
|---|---|---|
| H37RvMa | Susceptible | ≤15 |
| S2371 | Susceptible | 30 |
| S1125 | Susceptible | 15–30 |
| MD55 | MDR: INH; RMP; EMB; PZA; SM | 60b |
| MD96 | XDR: INH; RIF; AM; KM; CAP; OFLX; EMB; ETH; PZA; SM | ≤30b |
| R88 | INH Mono-Resistant | 30b |
| R296 | MDR: INH; RIF; EMB; ETH | 60b |
| R3027 | RIF Mono-Resistant | 60b |
MIC were determined by MIGIT method
Values within 4-fold of MICs against drug-susceptible isolate are considered sensitive to the test compound; INH: Isoniazid; RIF: Rifampicin; AM: Amikacin; KM: Kanamycin; CAP: Capreomycin; OFLX: Ofloxacin; EMB: Ethambutol; ETH: Ethionamide; PZA: Pyrazinamide; SM: Streptomycin.
In vitro and in vivo DMPK profiles.
In general, the compounds in the series showed moderate to good metabolic stability in mouse, rat and human microsomes as presented in Table 9. The high plasma protein binding (PPB) observed for the compounds may be attributed to high albumin binding associated with the acidic acylsulfonamide functionality along with the high degree of lipophilicity. Compound 20 showed lower protein binding presumably due to lower lipophilicity imparted by tetrahydropyran ring attached to the C3 position. The compounds in Table 9 were also profiled for in vivo pharmacokinetics following intravenous dosing in mice.
Table 9.
In vitro and in vivo DMPK parametersa
| Compound | TPSA | LogD | Microsomal clearance H/R/M % remaining |
Human PPB fu |
t½ terminal (h) | Vd (L/kg) | CLb (mL/min/kg) | CLu (mL/min/kg) | AUC0 – t (min.µmol/L) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 81.1 | 2.4 | 90/74/71 | 0.004 | 2.0 | 0.2 | 1.18 | 295 | 3527 |
| 8 | 81.1 | 2.0 | 98/>99/87 | 0.023 | 3.6 | 1.2 | 2.56 | 111 | 4883 |
| 10 | 93.9 | 1.87 | 94/98/94 | 0.036 | 3.0 | 8.7 | 67.4 | 1872 | 134 |
| 16 | 93.9 | 0.93 | 85/94/79 | 0.009 | 1.3 | nd | > 200 | >20000 | 15 |
| 17 | 93.9 | 0.84 | 37/95/94 | 0.022 | 1.7 | 28.1 | 178 | 8091 | 31 |
| 20 | 90.3 | 0.55 | 86/83/77 | 0.07 | 2.3 | 29.3 | 145 | 2071 | 31 |
| 38 | 93.9 | 0.98 | 88/82/85 | 0.015 | 1.1 | nd | > 200 | >20000 | 8.7 |
| 40 | 93.9 | 1.06 | >99/92/96 | 0.01 | 0.9 | nd | > 200 | >20000 | 21 |
| 43 | 106 | 0.81 | 99/98/91 | 0.041 | 2.0 | nd | > 200 | >4878 | 29 |
| 45 | 103.2 | 2.73 | 97/97/98 | 0.03 | 5.8 | nd | > 200 | >20000 | 22 |
| 51 | 81.1 | 2.1 | >99/87/91 | 0.01 | 3.6 | 1.0 | 3.13 | 313 | 1359 |
| 58 | 101.6 | 0.36 | 98/87/85 | 0.007 | nd | nd | > 200 | >20000 | 20 |
in vivo mouse PK parameters calculated from non-compartmental analysis of intravenous dosing at 2 mg/kg; TPSA: total polar surface area; H/R/M: Human/Rat/Mouse; PPB: Plasma protein binding; fu: Fraction unbound; Vd: Volume of distribution; CLb: Total body clearance determined from whole blood; CLu: Unbound blood clearance determined from CLb and plasma protein binding (assuming blood to plasma ratio of 1). AUC: Area under the curve; nd: not determined.
The hit compound 1 had a low in vivo mouse blood clearance (Clb) and low volume of distribution (Vd) that translated to a high in vivo exposure. Replacing the N1 aryl group with a 4-CF3-3-pyridyl ring in 10, led to a dramatic increase in clearance and consequently lower exposure. Rapid Clb values were also observed for 16 and 17 with 4-pyridyl and 3-pyridyl groups, respectively, at the C3 position. The rapid in vivo clearance of these compounds was all the more problematic due to the relatively high PPB leading to exceedingly high unbound clearance (CLu) values calculated by correcting Clb for plasma free fraction (assuming blood to plasma ratio of 1).28 In vivo metabolite identification studies of 17 revealed that the high clearances were in part due to hydrolysis of the acylsulfonamide (data in supplementary information). Subsequent SAR exploration therefore focused on modifications that were designed to increase stability of the acylsulfonamide while maintaining the potency of the compounds. However, neither addition of a double bond to the acyl chain (38), nor cyclopropanation (40), replacement of the sulfonamide with a sulfonylurea (43), or sulfonylcarbamate (45) improved the clearances of the compounds. In addition, except for 10 which had a N1 CF3-3-pyridyl ring, all pyridyl compounds had total clearances that exceeded hepatic blood flow in mice (90 mL/min/kg), which suggests that extra-hepatic metabolism may be a significant mechanism of clearance. The high TPSA and low logD of these pyridyl derivatives (Table 7), suggests that the compounds have low lipoidal permeability and it is therefore likely that transporter-mediated clearance was also a key contributor to the observed total clearance.29, 30 This was further validated by performing in vivo metabolite identification studies on 43. Following intravenous dosing of the compound in mice, only unchanged compound was detected in blood, urine, and feces, suggesting that metabolism was not a major contributor to its clearance. Compounds, 1, 8 and 51 had high exposure on oral dosing (data not shown), with 8 in particular showing almost complete absorption (bioavailability >95%) even at high doses. This compound therefore had the best balance of in vitro potency and in vivo pharmacokinetics and was therefore selected for further in vivo proof-of-concept studies.
In vivo efficacy
In vivo efficacy testing was performed at an oral dose of 200 mg/kg in the acute Balb/c mouse model to evaluate the ability of 8 to inhibit replicating intracellular Mtb in lungs.31–34 Treatment was initiated 7 days after a low-dose aerosol infection with Mtb Erdman pFCA LuxAB strain32 and continued for 12 consecutive days. The compound was well tolerated during the treatment period but was not efficacious in reducing Mtb lung burdens in treated mice relative to the untreated mice as measured by quantification of relative light units (RLU) of the luciferase expressing Erdman strain from lung homogenate or by CFU counts obtained by plating serial dilutions of lung homogenate on agar plates. As a positive control, EMB at 100 mg/kg once a day led to a 2-log CFU reduction in this model. To explain the results, the free oral exposure of compound 8 in healthy mice at 200 mg/kg oral dose was compared with the MIC against the Mtb Erdman strain used for the efficacy studies (9.1 μM) and inhibitory concentration against intracellular Mtb (IC90 = 11 μM) (Fig. 4). In-study PK performed on the last 2 days of treatment matched closely with the healthy PK (data in SI). From the two analyses, it is evident that compound 8 at 200 mg/kg failed to achieve drug exposures necessary to exert an antimicrobial effect in vivo. While much higher or more frequent dosing may provide sufficient exposure to demonstrate efficacy, the underlying data suggests that further optimisation of antibacterial activity and/or PK is required to do so at lower doses that would feasibly predict human utility. Work towards achieving these improvements is in progress and will be reported in a future manuscript.
Figure 4.

Free exposures of 8 (calculated using human PPB) following a single oral dose of 200 mg/kg in mice.
Conclusion.
In summary, phenotypic screening of a DuPont compound library against Mtb in cholesterol-containing media identified 1,3-diarylpyrazolyl-acylsulfonamide 1 as a moderately active hit with a potentially novel MoA. The SAR studies described here demonstrate a clear scope to improve the MICs of compounds to <0.5 μM and a plausible pharmacophore model was developed to summarize SAR of the series and describe the chemical space of active compounds. Screening of the compounds against a panel of various tool strains of Mtb ruled out the involvement of known mechanisms and/or targets such as DNA damage and respiration but suggested involvement of cell wall damage in the mode-of-action. RNA microarray studies of Mtb cultures treated with the compounds confirmed the upregulation of genes that are involved in lipid metabolism/fatty acid biosynthesis. Isogenic single-drug resistant mutant strains of MmpL3, DprE1, InhA and EthA were not cross-resistant with the compounds indicating novel MoA or perhaps an alternative mode of inhibition of the targets. The compounds are bactericidal against replicating Mtb and retain activity against multidrug-resistant clinical isolates of Mtb. These features make the series an attractive chemical matter for further target identification and drug discovery efforts to potentially identify a novel clinical candidate for treatment of TB. Further drug optimization work is needed to improve upon both the Mtb activity of the series and the in vivo PK if compounds with efficacy in TB models of infection are to be realized.
Experimental
MIC testing and bioluminescence reporter assay.
MIC determinations were done as previously described.35 INH was included as a positive control. Briefly, Mtb H37Rv (ATCC 27294) was grown in Middlebrook 7H9/Glu/BSA/Tyloxapol consisting of Middlebrook 7H9 broth base (4.7 g/L)/ bovine serum albumin fraction V (5 g/L)/ dextrose (4 g/L)/ NaCl (0.81 g/L)/ 0.05% Tyloxapol to an OD650nm of 0.2 at which stage cells were diluted 1000-fold in the same medium. Compound was serially diluted in Middlebrook 7H9/Glu/BSA/Tyloxapol medium in round-bottom 96-well plates (Nunclon) at 50 μL/ well followed by addition of an equal volume of diluted cell suspension. Plates were incubated in sealed bags at 37 °C and growth recorded after 1- and 2-weeks of growth using an enlarging inverted mirror. The MIC was recorded as the compound concentration that completely inhibited all visible growth. Bioluminescence reporter assays were carried out as described by Naran et al.26
MICs against mutant strains.
Alamar Blue fluorescence-based broth microdilution assay was used to assess MICs of compounds against Mtb mutant strains, as described previously.36 MICs were determined after 7 days of growth in standard Middlebrook 7H9/Glycerol/ADC/Tween-80. For QcrB and cydA mutants, the growth measurement was monitored by OD600. MIC was defined as the concentration required to inhibit growth by 90%.
RNA extraction and Transcriptional Profiling.
RNA extraction from Mtb H37Rv (ATCC 27294) grown to an OD650 of 0.2 and subsequently treated for 6 h with 1X and 10X MIC of compound or vehicle control was performed using previously described methods.37 RNA was also extracted from control cultures treated for 6 h with isoniazid (2 μM) or solvent control. RNA (4 mg) yielding an RNA integrity number (RIN) of 8 or higher as determined by the Agilent 2100 Bioanalyzer was subsequently used for fluorescent-tagged cDNA synthesis with random hexamer (4.5 mg) (Invitrogen) in a final volume of 14.5 μL by sequential heat denaturation at 70 °C for 5 minutes, cooling on ice, addition of 5 μl of 5× First-Strand buffer, 1.25 μL 0.1 M DTT, 2.5 μL of dNTP mix (consisting of 5 mM each of dATP, dGTP, dTTP and 0.5 mM dCTP), 1 μL of 200 U/μL SuperScriptIII (Invitrogen), 1 μL of 40 U/μl RNAseOut, and 1 μL of Cy3 or Cy5-dCTP (GE) with incubation at 25 °C for 5 minutes and at 48 °C for 90 minutes. RNA was removed by adding 5 μL of 1M NaOH followed by incubation at 70 °C for 15 minutes. After neutralization with 5 μL of 1 M HCl, fluorescent cDNA was purified on Amicon Ultra-0.5 column (Millipor) according to the manufacturer’s recommendations. Fluorescent cDNA was analyzed with the Nanodrop ND-1000. Equal amounts (0.7 μg) of Cy3- and Cy5-labeled cDNA were hybridized to the Agilent SurePrint G3 4×44K custom oligonucleotide microarrays (design number 021966, 021362) in the TECAN HS Pro 4800 hybridization station. Hybridization was performed using Agilent 2x Gene expression hybridization HI-RPM buffer, and 10x Blocking Reagent at 65 °C for 17 h. Arrays were washed with Agilent Gene Expression Wash Buffer 1 at room temperature and Gene Expression Wash Buffer 2 at 37 °C. Slides were dried under nitrogen gas for 3 minutes at 30 °C and imaged using an Agilent high resolution DNA microarray scanner (model G2505C) at 5 μm resolution and 100/10% PMT dual scanning for XDR extended dynamic range. Agilent Feature Extraction software was used for image analysis.
Pharmacophore Hypothesis generation.
To generate the pharmacophore hypothesis, all compounds in this set were prepared using Maestro 12.438 to generate minimized 3D structures and determine the most likely protonation state at biological pH. A pharmacophore model was then developed using the Phase Develop Pharmacophore Model tool.22 The pharmacophore model was created from multiple ligands with the active ligands defined by an activity cutoff of MIC < 25 μM in the 7H9/glucose/BSA/Tx assay. The find best alignment method was chosen with the default hypothesis settings of 4 to 5 features and all the default pharmacophore features allowed. The pharmacophore hypotheses generated were then inspected and the hypothesis that best represented the dataset was selected and validated against a wider set of compounds from this series.
DMPK.
All protocols for in vitro DMPK studies and mouse PK studies are available in the supplementary document. Animal studies were conducted following guidelines and policies as stipulated in the UCT Research Ethics Code for Use of Animals in Research and Teaching, after review and approval of the experimental protocol by the UCT Senate Animal Ethics Committee (protocol FHS-AEC 013/032).
Chemistry.
All commercial reagents were purchased from Sigma-Aldrich, Combi-Blocks, Enamine, or Fluorochem and were used without further purification. Solvents were used as received unless otherwise stated. Analytical thin-layer chromatography (TLC) was performed on SiO2 plates on aluminium backing. Visualization was accomplished by UV irradiation at 254 and 220 nm. Flash column chromatography was performed using a Teledyne ISCO flash purification system with SiO2 60 (particle size 0.040–0.055 mm, 230–400 mesh). Purity of all final derivatives for biological testing was confirmed to be >95% as determined using an Agilent 1260 Infinity binary pump, Agilent 1260 Infinity diode array detector (DAD), Agilent 1290 Infinity column compartment, Agilent 1260 Infinity standard autosampler, and Agilent 6120 quadrupole (single) mass spectrometer, equipped with APCI and ESI multimode ionization source. Using a Kinetex Core C18 2.6 μm column (50 mm × 3 mm); mobile phase B of 0.4% acetic acid, 10 mM ammonium acetate in a 9:1 ratio of HPLC grade methanol and type 1 water, mobile phase A of 0.4% acetic acid in 10 mM ammonium acetate in HPLC grade (type 1) water, with flow rate of 0.9 mL/min, detector diode array (DAD). Or an Agilent UPLC–MS was used: Agilent Technologies 6150 quadrupole, ES ionization, coupled with an Agilent Technologies 1290 Infinity II series UPLC system Agilent 1290 series HPLC at two wavelengths 254 and 290 nm using the following conditions: Kinetex 1.7 μm Evo C18 100A, LC column 50 mm × 2.1 mm, solvent A of 0.1% (formic acid) water, solvent B of 0.1% (formic acid) acetonitrile. The structures of the intermediates and final products were confirmed by 1H NMR and mass spectrometry. 1H NMR spectra were recorded on a Bruker spectrometer at 300 or 400 MHz. Chemical shifts (δ) are given in ppm downfield from TMS as the internal standard. Coupling constants, J, are recorded in hertz (Hz). Synthesis details and data of intermediates are supplied in the Supporting Information (Section 1)
General sulfonamide coupling procedure for the synthesis of compounds 1–25, 27–29, 31–41, 51–57.
Method 1. CDI-mediated sulfonamide coupling.
To a solution of the appropriate acid (1 equiv) in DMF was added CDI (2 equiv) and the resulting reaction mixture was stirred for 10 min at 50 °C. Then the appropriate sulfonamide (1.2 equiv) and DBU (1.2 equiv) were added, and the resulting reaction mixture was heated to 90 °C for 16–48 h. The reaction mixture was then cooled to 25 °C, water was added (30 mL) and the aqueous layer was extracted with EtOAc (3 x). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude reaction mixture was purified by flash chromatography using ISCO Teledyne on 12 g RediSep Rf column and elution using a gradient of the appropriate solvent mixtures to yield the desired products.
Method 2: DCC-mediated sulfonamide coupling.
To the solution of the appropriate acid (1.00 equiv) and methanesulfonamide (1.20 equiv) in DMF were added DCC (2.50 equiv) and DMAP (1.50 equiv). The reaction mixture was stirred at 30 °- 40 °C for 16–48 h. The reaction mixture was quenched with water (5 mL) and pH was adjusted to 6 with 1N HCl. The aqueous phase was extracted with EtOAc (3 x). The organic phase was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica-gel column or preparative HPLC chromatography to yield the appropriate compound.
Method 3: Acid chloride coupling with sulfonamide.
To a solution of appropriate sulfonamide (5 equiv) in DCM was added Et3N (5 – 10 equiv). Then the appropriate acyl chloride (1 equiv) was added at 0 °C. The mixture was stirred at 15 °C to 25 °C for 16–24 h. The mixture was poured into 1N HCl and extracted with DCM. The combined organic phase was concentrated and purified by prep-HPLC or by silica gel column chromatography.
3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (1).
Method 3.
Yield: 19%. 1H NMR: (400MHz, DMSO-d6) δ 11.92–11.73 (m, 1H), 8.49–8.36 (m, 1H), 7.88–7.78 (m, 2H), 7.78–7.64 (m, 4H), 7.60–7.49 (m, 2H), 3.21 (s, 3H), 2.98–2.85 (m, 2H), 2.70–2.58 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.8, 149.5, 138.6, 132.7, 132.4 (2C), 131.8, 129.1 (2C), 128.7 (2C), 127.8, 120.1, 119.9 (2C), 118.4, 41.0, 35.7, 19.2. LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 482.0, 483.9; HPLC purity: >99%.
3-(3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (2).
Method 2.
Yield: 32%. 1H NMR: (400MHz, DMSO-d6) δ 12.09 – 11.57 (m, 1H), 8.45 – 8.34 (m, 1H), 7.89 – 7.81 (m, 2H), 7.81 – 7.71 (m, 2H), 7.62 –7.46 (m, 4H), 7.37 – 7.29 (m, 1H), 3.18 (s, 3H), 3.00 – 2.86 (m, 2H), 2.64 – 2.57 (m, 2H). LC/MS (APCI+): Calculated for C19H18ClN3O3S 403.08; Observed m/z [M+H]+ 404.1; HPLC purity: >99%.
3-(3-(4-chlorophenyl)-1-(4-fluorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (3).
Method 1.
Yield: 14%. 1H NMR (300 MHz, DMSO-d6) δ 11.72 (s, 1H), 8.36 (s, 1H), 7.91 – 7.84 (m, 2H), 7.78 – 7.71 (m, 2H), 7.59 – 7.52 (m, 2H), 7.37 (t, J = 8.8 Hz, 2H), 3.21 (s, 3H), 2.93 (t, J = 7.5 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H). LC/MS (ESI+): Calculated for C19H17ClFN3O3S 421.07; Observed m/z [M+H]+ 422.1; HPLC purity: 97%.
3-(1-(3-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (4).
Method 1.
Yield: 20%. 1H NMR (300 MHz, DMSO-d6) δ 11.72 (s, 1H), 8.48 (s, 1H), 8.09 (t, J = 1.9 Hz, 1H), 7.88 (dt, J = 7.2, 2.0 Hz, 1H), 7.81 – 7.73 (m, 2H), 7.60 – 7.43 (m, 4H), 3.21 (s, 3H), 2.93 (t, J = 7.5 Hz, 2H), 2.64 (t, J = 7.5 Hz, 2H). LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 481.9, 483.9; HPLC purity: 97%.
3-(1-(2-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (5).
Method 2.
Yield: 31%. 1H NMR (400 MHz, Chloroform-d) δ 7.74–7.65 (m, 5H), 7.60–7.58 (m, 3H), 7.45–7.43 (m, 1H), 3.27–3.25 (m, 3H), 3.13–3.07 (m, 2H), 2.64–2.58 (m, 2H). LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 482.0; 484.0; HPLC purity: >99%.
3-(3-(4-chlorophenyl)-1-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (6).
Method 2.
Yield: 23%. 1H NMR (400MHz, DMSO-d6) δ 8.67–8.62 (m, 2H), 8.62–8.57 (m, 1H), 8.22–8.14 (m, 1H), 7.89–7.85 (m, 2H), 7.81–7.76 (m, 2H), 7.60–7.55 (m, 2H), 3.00–2.97 (m, 3H), 2.90–2.86 (m, 2H); 1H NMR (400MHz, DMSO-d6 + D2O) δ 8.64–8.56 (m, 2H), 8.51–8.45 (m, 1H), 8.29–8.12 (m, 1H), 7.86–7.79 (m, 2H), 7.78–7.68 (m, 2H), 7.58–7.50 (m, 2H), 3.03 (s, 3H), 2.90–2.81 (m, 2H). LC/MS (APCI+): Calculated for C18H17ClN4O3S 404.07; Observed m/z [M+H]+ 405.0; HPLC purity: 98%.
3-(3-(4-chlorophenyl)-1-cyclohexyl-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (7).
Method 2.
Yield: 37%. 1H NMR (400 MHz, Chloroform-d) δ 7.74 (s, 1H), 7.57–7.55 (m, 2H), 7.42–7.40 (m, 2H), 7.35 (s, 1H), 4.14–4.10 (m, 1H), 3.26 (s, 3H), 3.04–3.00 (t, J = 7.2 Hz, 2H), 2.54–2.51 (t, J = 7.2 Hz, 2H), 2.20–2.17 (m, 2H), 1.94–1.91 (m, 2 H), 1.77–1.71 (m, 3H), 1.46–1.26 (m, 3H). LC/MS (APCI+): Calculated for C19H24ClN3O3S 409.12; Observed m/z [M+H]+ 410.0; HPLC purity: 96%.
3-(1,3-bis(4-chlorophenyl)-1H-pyrazol-5-yl)-N-(methylsulfonyl)propanamide (8).
Method 1.
Yield: 52%. 1H NMR (300 MHz, Methanol-d4) δ 8.14 (s, 1H), 7.84 – 7.75 (m, 2H), 7.76 – 7.66 (m, 2H), 7.54 – 7.46 (m, 4H), 3.21 (s, 3H), 3.10 –2.98 (m, 2H), 2.64 (t, J = 7.4 Hz, 2H). 13C NMR (151 MHz, Methanol-d4) δ 174.0, 151.9, 140.0, 135.1, 133.3, 132.9, 130.6 (2C), 130.5 (2C), 129.8 (2C), 129.0 (2C), 121.3 (2C), 41.4, 37.4, 20.4. LC/MS (ESI+): Calculated for C19H17Cl2N3O3S 437.04; Observed m/z [M+H]+ 438.1; HPLC purity: >99%.
3-(3-(4-chlorophenyl)-1-(4-(trifluoromethyl)phenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (9).
Method 3.
Yield: 11%. 1H NMR (400 MHz, Chloroform-d) 7.92 (s, 1H), 7.85–7.83 (d, J = 8.4 Hz, 2H), 7.70–7.63 (m, 4H), 7.44–7.42 (d, J = 8.8 Hz, 2H), 3.21 (s, 3H), 3.05–3.03 (m, 2H), 2.61–2.57 (m, 2H). LC/MS (ESI+): Calculated for C20H17ClF3N3O3S 471.06; Observed m/z [M+H]+ 472.1, 474.1; HPLC purity: 98%.
3-(3-(4-chlorophenyl)-1-(6-(trifluoromethyl)pyridin-3-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (10).
Method 1.
Yield: 22%. 1H NMR (400 MHz, Methanol-d4) δ 9.22 (d, J = 2.5 Hz, 1H), 8.42 (ddd, J = 8.6, 2.6, 0.8 Hz, 1H), 8.34 (d, J = 0.9 Hz, 1H), 7.92 (dd, J = 8.6, 0.7 Hz, 1H), 7.80 – 7.62 (m, 2H), 7.56 – 7.43 (m, 2H), 3.21 (s, 3H), 3.11 – 2.97 (m, 2H), 2.66 (t, J = 7.3 Hz, 2H). 13C NMR (151 MHz, Methanol-d4) δ 174.2, 153.4, 145.6 (q, J = 35.4 Hz), 141.1, 139.7, 135.5, 132.8, 130.6 (2C), 129.9, 129.4, 127.7, 125.7, 123.9, 122.7 (d, J = 10.1 Hz), 122.1, 41.4, 37.3, 20.5. LC/MS (APCI+): Calculated for C19H16ClF3N4O3S 472.06; Observed m/z [M+H]+ 473.0; HPLC purity: >99%.
3-(3-(4-chlorophenyl)-1-(4-cyanophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (11).
Method 3.
Yield: 33%. 1H NMR (400 MHz, Chloroform-d) δ 7.96 (s, 1H), 7.89–7.87 (m, 2H), 7.78–7.76 (m, 2H), 7.67–7.65 (m, 2H), 7.49–7.47 (m, 2H), 3.31 (s, 3H), 3.14–3.10 (m, 2H), 2.63–2.59 (m, 2H). LC/MS (APCI+): Calculated for C20H17ClN4O3S 428.07; Observed m/z [M+H]+ 429.1; HPLC purity: >99%.
3-(3-(4-chlorophenyl)-1-(4-(methylsulfinyl)phenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (12).
Step 1.
3-(3-(4-chlorophenyl)-1-(4-(methylthio)phenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide from acid 63l following sulfonamide coupling method 2 (Scheme S1). Yield: 40 mg (31%). 1H NMR (400 MHz, Chloroform-d) 7.83 (s, 1H), 7.67–7.65 (m, 4H), 7.47–7.46 (m, 3H), 7.45–7.34 (m, 2H), 3.28 (s, 3H), 3.12–3.08 (m, 2H), 2.62–2.58 (m, 2H), 2.54 (s, 3H). LC/MS (APCI+): Calculated for C20H20ClN3O3S2 449.06; Observed m/z [M+H]+ 450.0; HPLC purity: 95%.
Step 2.
To a solution of 3-(3-(4-chlorophenyl)-1-(4-(methylthio)phenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (35 mg, 77.78 μmol) in MeOH (200 μL) and THF (200 μL) was added NaIO4 (17 mg, 81.67 μmol, 5 μL). The mixture was stirred at 20 °C for 15 h. The reaction mixture was diluted with aqueous Na2SO3 (5 mL) and extracted with EtOAc (10 mL x 2). The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150×25×10um; mobile phase: [water (0.225%FA)-ACN]; B%: 35%-65%, 12min) to give 3-(3-(4-chlorophenyl)-1-(4-(methylsulfinyl)phenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (12). Yield: 21 mg (58%). 1H NMR (400 MHz, Chloroform-d) δ 7.90–7.88 (m, 3H), 7.73–7.66 (m, 4H), 7.48–7.46 (m, 2H), 3.30 (s, 3H), 3.13–3.09 (m, 2H), 2.78 (s, 3H), 2.67–2.64 (m, 2H). LC/MS (APCI+): Calculated for C20H20ClN3O4S2 465.06; Observed m/z [M+H]+ 466.0, 468.0; HPLC purity: >99%.
3-(1-(4-bromophenyl)-3-phenyl-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (13).
Method 2.
Yield: 29%. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (s, 1H), 7.62–7.60 (m, 2H), 7.57–7.38 (m, 7H), 3.18 (s, 3H), 3.06–3.02 (t, J = 7.2 Hz, 2H), 2.51–2.48 (t, J = 7.2 Hz, 2H). LC/MS (APCI+): Calculated for C19H18BrN3O3S 447.03; Observed m/z [M+H]+ 448.0; HPLC purity: 99%.
3-(1-(4-bromophenyl)-3-(3-chlorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (14).
Method 2.
Yield: 10%. 1H NMR (400 MHz, Chloroform-d) δ 7.83 (s, 1H), 7.70 (s, 1H), 7.64–7.53 (m, 5H), 7.43–7.34 (m, 2H), 3.26 (s, 3H), 3.07 (t, J =7.2 Hz, 2H), 2.58 (t, J =7.2 Hz, 2H). LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 482.0, 484.0; HPLC purity: >99%.
3-(3-(2-chlorophenyl)-1-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (15).
Method 1.
Yield: 9%. 1H NMR (300 MHz, Methanol-d4) δ 8.13 (d, J = 0.8 Hz, 1H), 7.73 (d, J = 8.9 Hz, 2H), 7.57 – 7.52 (m, 1H), 7.50 – 7.36 (m, 5H), 3.17 (s, 3H), 2.76 (t, J = 7.4 Hz, 2H), 2.58 – 2.43 (m, 2H). LC/MS (APCI+): Calculated for C19H17Cl2N3O3S 437.04; Observed m/z [M+H]+ 437.9, 439.9 (chlorine isotopic pattern); HPLC purity: >99%.
3-(1-(4-bromophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (16).
Method 3.
Yield: 19%. 1H NMR (400MHz, DMSO-d6) δ 8.74–8.63 (m, 2H), 8.51–8.47 (m, 1H), 7.88–7.83 (m, 2H), 7.74 (s, 4H), 3.10–3.07 (s, 3H), 2.99–2.93 (m, 2H), 2.59–2.55 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 172.3, 150.1 (2C), 147.9, 140.2, 138.5, 132.4 (2C), 128.4, 121.6 (2C), 121.2, 120.2 (2C), 118.8, 40.9, 36.1, 19.4. LC/MS (APCI+): Calculated for C18H17BrN4O3S 448.02; Observed m/z [M+H]+ 449.0; HPLC purity: 97%.
3-(1-(4-bromophenyl)-3-(pyridin-3-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (17).
Method 3.
Yield: 6%. 1H NMR (400 MHz, Chloroform-d) δ 8.79 (s, 1H), 8.45–8.44 (d, J = 4.4 Hz, 1H), 8.04–8.02 (d, J = 7.6 Hz, 1H), 7.83 (s, 1H), 7.56–7.50 (m, 4H), 7.31–7.30 (m, 1H), 3.19 (s, 3H), 3.01 (m, 2 H), 2.59–2.56 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 171.8, 147.7, 147.1, 146.6, 138.7, 137.0, 132.7, 132.6, 130.4, 128.1, 127.0, 124.6, 120.5, 120.2, 119.9, 41.7, 37.3, 19.8. LC/MS (APCI+): Calculated for C18H17BrN4O3S 448.02; Observed m/z [M+H] + 449.0, 451.0 (1:1) bromine isotopic pattern; HPLC purity: >99%.
3-(1-(4-bromophenyl)-3-(pyrazin-2-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (18).
Method 2.
Yield: 12%. 1H NMR (400 MHz, Chloroform-d) δ 9.44 (s, 1H), 8.67 (brs, 1H), 8.58 (s, 1H), 7.91 (s, 1H), 7.63 (q, J = 9.0 Hz, 4H), 3.28–3.19 (m, 5H), 2.79 (t, J = 7.1 Hz, 2H). LC/MS (APCI+): Calculated for C17H16BrN5O3S 449.02; Observed m/z [M+H]+ 449.8, 451.9 (1:1) bromine isotopic pattern; HPLC purity: 91%.
3-(1-(4-bromophenyl)-3-cyclohexyl-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (19).
Method 2.
Yield: 40%. 1H NMR (400 MHz, Chloroform-d) δ 8.17 (s, 1H), 7.65 (s, 1H), 7.52 (s, 4H), 3.30 (s, 3H), 2.93–2.82 (m, 2H), 2.60 (s, 3H), 1.88 (brs, 4H), 1.80–1.62 (m, 3H), 1.35 (s, 3H). LC/MS (APCI+): Calculated for C19H24BrN3O3S 453.07; Observed m/z [M+H]+ 454.0; HPLC purity: >99%.
3-(1-(4-bromophenyl)-3-(tetrahydro-2H-pyran-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (20).
Method 2.
Yield: 13%. 1H NMR (400MHz, DMSO-d6) δ 8.18 (s, 1H), 7.71–7.62 (m, 4H), 3.94–3.92 (m, 2H), 3.46–3.45 (m, 2H), 3.43–3.26 (m, 2H), 3.07 (s, 3H), 2.70–2.69 (m, 1H), 2.67–2.66 (m, 3H), 1.77–1.73 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 172.7, 155.6, 138.9, 132.2 (2C), 126.2, 119.7, 119.4 (2C), 117.4, 67.1 (2C), 40.9, 36.9, 32.6, 31.9 (2C), 18.4. LC/MS (APCI+): Calculated for C18H22BrN3O4S 455.05; Observed m/z [M+H] + 456.0; HPLC purity: 98%.
3-(1-(4-bromophenyl)-3-(piperidin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (21).
Step 1.
Tert-butyl 4-(1-(4-bromophenyl)-4-(3-(methylsulfonamido)-3-oxopropyl)-1H-pyrazol-3-yl)piperidine-1-carboxylate was obtained as a white solid. Yield: 60 mg (17%) from acid 67h by sulfonamide coupling method 2 (Scheme S2). LC/MS (APCI+): Calculated for C23H31BrN4O5S 554.12; Observed m/z [M+H-Boc]+ 455.0; HPLC purity: 83%.
Step 2.
The mixture of tert-butyl 4-(1-(4-bromophenyl)-4-(3-(methylsulfonamido)-3-oxopropyl)-1H-pyrazol-3-yl)piperidine-1-carboxylate (55 mg, 99.01 μmol) in TFA (100 μL) and DCM (1 mL) was stirred at 20 °C for 2 h. The reaction mixture was concentrated. The obtained residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150×25×10um; mobile phase: [water (0.225%FA)-ACN]; B%: 5%-35%, 12min) to give 3-(1-(4-bromophenyl)-3-(piperidin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (21) as a white solid. Yield: 27 mg (59%). 1H NMR (400MHz, DMSO-d6) δ 8.22 (s, 1H), 7.71–7.62 (m, 4H),3.36–3.33 (m, 3H), 3.06 (s, 3H), 2.77–2.63 (m, 2H), 2.49–2.27 (m, 2H), 2.01–1.91 (m, 4H). LC/MS (APCI+): Calculated for C18H23BrN4O3S 454.07; Observed m/z [M+H]+ 455.0; HPLC purity: 99%.
3-(1-(4-bromophenyl)-3-(1-formylpiperidin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (22).
Method 2.
Yield: 32%. 1H NMR (400MHz, DMSO-d6) δ 8.21 (s, 1H), 8.03 (s, 1H), 7.71–7.63 (m, 4H), 4.26–4.22 (m, 1H), 3.79–3.76 (m, 1H), 3.19 (s, 3H), 3.00–2.80 (m, 1H), 2.79–2.59 (m, 6H), 2.34–1.54 (m, 4H). LC/MS (APCI+): Calculated for C19H23BrN4O4S 482.06; Observed m/z [M+H]+ 483.1; HPLC purity: >99%.
3-(1-(4-bromophenyl)-3-(4-nitrophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (23).
Method 2.
Yield: 71%. 1H NMR (400 MHz, Methanol-d4) δ 8.38 – 8.30 (m, 2H), 8.22 (d, J = 6.3 Hz, 1H), 8.06 – 7.98 (m, 2H), 7.82 – 7.74 (m, 2H), 7.69 – 7.61 (m, 2H), 3.16 (s, 3H), 3.09 (t, J = 7.5 Hz, 2H), 2.65 (t, J = 7.5 Hz, 2H). LC/MS (ESI+): Calculated for C19H17BrN4O5S 492.01; Observed m/z [M+H]+ 493.0, 495.0 (1:1) bromine isotopic pattern; HPLC purity: 93%.
3-(3-(4-aminophenyl)-1-(4-bromophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (24).
To a mixture of 3-(1-(4-bromophenyl)-3-(4-nitrophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (23) (100 mg, 0.203 mmol) and iron (53 mg, 0.953 mmol) in EtOH (2 mL) was added saturated solution of NH4Cl (1 mL) and heated to 80 °C for 18 h. The reaction mixture was cooled to 25 °C and separated the organic layer. The organic layer was dried over Na2SO4 and evaporated the solvent under reduced pressure. The residue was purified by flash silica gel column using DCM: MeOH (0–5% gradient) as eluents to afford 3-(3-(4-aminophenyl)-1-(4-bromophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (24) as a grey solid. Yield: 5 mg (5%). 1H NMR (400 MHz, Methanol-d4) δ 8.19 (s, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.5 Hz, 2H), 3.23 (s, 3H), 3.06 (t, J = 7.4 Hz, 2H), 2.68 (t, J = 7.4 Hz, 2H). LC/MS (APCI+): Calculated for C19H19BrN4O3S 462.04; Observed m/z [M+H]+ 463.1, 465.1 (1:1) bromine isotopic pattern; HPLC purity: 92%.
3-(3-benzyl-1-(4-bromophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (25).
Method 3.
Yield: 20%. 1H NMR (400 MHz, Chloroform-d) δ 7.71 (s, 1H), 7.58 (s, 4H), 7.36–7.32 (m, 5H), 4.09 (s, 2H), 3.21 (s, 3H), 2.74–2.71 (m, 2H), 2.06–2.02 (m, 2H). LC/MS (APCI+): Calculated for C20H20BrN3O3S 461.04; Observed m/z [M+H]+ 462.0; HPLC purity: >99%.
3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanoic acid (26).
To a solution of (E)-3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)acrylic acid (62a) (1.66 g, 4.11 mmol) in MeOH (150 mL) was added NH2NH2.H2O (16.47 g, 328.99 mmol, 16 mL) and the mixture was heated to 80 °C for 48 h (Scheme S1). The mixture was cooled to room temperature, diluted with ice-water (100 mL) and adjusted pH = 3 with 1 M HCl. The aqueous layer was extracted with DCM (50 mL x 3). The organic layer was washed with brine (50 mL), dried over anhydrous Na2SO4 and concentrated. To the residue was added petroleum ether (20 mL) slowly. The mixture was concentrated slowly to give some solid. The solid collected to give 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanoic acid (26) as a yellow solid. Yield: 1.80 g (99%). 1H NMR (400 MHz, Chloroform-d) δ7.85–7.75 (m, 1H), 7.59 (d, J =7.8 Hz, 6H), 7.48–7.38 (m, 2H), 3.02 (m, 2H), 2.67(m, 2H). LC/MS (APCI+): Calculated for C18H14BrClN2O 403.99; Observed m/z [M+H]+ 404.9, 407.0; HPLC purity: 92%.
3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-methylpropanamide (27).
Method 3.
To a solution of MeNH2/THF (2 M, 5 mL) was added the solution of 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanoyl chloride (68) (220 mg, 518.72 μmol) in DCM (2 mL) at −50 °C (Scheme S3). The mixture was stirred at −30 °C for ~3 h. The mixture was washed with water (5 mL) and concentrated. The residue was purified by prep-HPLC to give 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-methylpropanamide (27) as a light-yellow solid. Yield: 60 mg (27%). 1H NMR (300 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.85–7.81 (m, 2H), 7.77–7.75 (m, 2H), 7.72–7.70 (m, 2H), 7.56–7.54 (m, 2H), 2.89 (t, J = 7.6 Hz, 2H), 2.56 (d, J = 8.4 Hz, 3H), 2.43 (t, J = 7.6 Hz, 2H). LC/MS (APCI+): Calculated for C19H17BrClN3O 417.02; Observed m/z [M+H]+ 418.0, 420.0; HPLC purity: 98%.
3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-methyl-N-(methylsulfonyl)propanamide (28).
To a solution of N-methylmethanesulfonamide (116 mg, 1.06 mmol) in THF (5.00 mL) was added NaH 60% dispersion (45 mg, 1.13 mmol) at 0–10 °C. After stirred at 15 °C for 1 h, a solution of 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanoyl chloride (68) (300 mg, 707.35 μmol) in DCM (3.00 mL) was added at −10 °C via syringe. The mixture was stirred at 15 °C for another 2 h. The mixture was washed with brine (5 mL) and concentrated. The residue was purified by prep-HPLC to give 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-methyl-N-(methylsulfonyl)propanamide (28). Yield: 65 mg (17%). 1H NMR (300 MHz, Methanol-d4) δ 8.25–8.17 (m, 1H), 7.80–7.71 (m, 4H), 7.69–7.61 (m, 2H), 7.54–7.46 (m, 2H), 3.27 (s, 6H), 3.11–2.98 (m, 4H). LC/MS (APCI+): Calculated for C20H19BrClN3O3S 495.00; Observed m/z [M+H]+ 496.0, 498.0; HPLC purity: 98%.
N-(3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propyl)methanesulfonamide (29).
To a solution of 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propan-1-amine (72) (90 mg, 230.36 μmol) in DCM (3 mL) were added Et3N (93 mg, 921.44 μmol, 128 μL) and MsCl (36 μL) at 0–10 °C (Scheme S3). The mixture was stirred at 10 °C for 1 h. The mixture was quenched with water (2 mL) and extracted with DCM (5 mL x 2). The organic phase was concentrated and purified by reverse phase HPLC to give N-(3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propyl)methanesulfonamide (29) as a white solid. Yield: 68 mg (63%). 1H NMR (400MHz, DMSO-d6) δ 8.54 – 8.41 (m, 1H), 7.91 – 7.81 (m, 2H), 7.81 – 7.74 (m, 2H), 7.74 – 7.66 (m, 2H), 7.60 –7.48 (m, 2H), 7.12 – 7.00 (m, 1H), 3.10 – 2.96 (m, 2H), 2.89 (s, 3H), 2.76 – 2.69 (m, 2H), 1.87 – 1.73 (m, 2H). LC/MS (APCI+): Calculated for C19H19BrClN3O2S 467.01; Observed m/z [M+H]+ 468.0; HPLC purity: >99%.
N-((2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)sulfonyl)acetamide (30).
To the solution of 2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethane-1-sulfonamide (74) (150 mg, 340.34 μmol) and Et3N (69 mg, 680.68 μmol, 94 μL) in THF (4 mL) was added acetyl chloride (40 mg, 510.51 μmol, 36 μL) drop-wise in ice-bath (Scheme S4). The mixture was stirred at 40 °C for 16 h. The mixture was concentrated and separated between water (3 mL) and EtOAc (3 mL). The organic phase was dried over anhydrous Na2SO4 and concentrated. The residue was purified by prep-HPLC (column: Phenomenex Synergi C18150×25×10um; mobile phase: [water (0.225%FA)-ACN]; B%: 57%-87%, 10min) and then purified again by prep-HPLC (column: Phenomenex Gemini 150×25mmx10um; mobile phase: [water (0.05% ammonia hydroxide v/v)-ACN]; B%: 22%-52%, 10min) to afford N-((2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)sulfonyl)acetamide (30) as a white solid. Yield: 29 mg (17%). 1H NMR (400 MHz, DMSO-d6) δ 8.58 (s, 1H), 7.88–7.83 (m, 3H), 7.74–7.70 (m, 4H), 7.54–7.51 (m, 2H), 3.60–3.56 (m, 2H), 3.06–3.03 (m, 2H), 1.87 (s,3H). LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 482.0; HPLC purity: >99%.
2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)acetamide (31).
Method 2.
Yield: 11%.1H NMR (400 MHz, Chloroform-d) δ 8.05 (s, 1H), 7.88 (s, 1H), 7.67–7.60 (m, 6H), 7.49–7.47 (d, J = 8.4 Hz, 2H), 3.81 (s, 2H), 3.15 (s, 3H). LC/MS (APCI+): Calculated for C18H15BrClN3O3S 466.97; Observed m/z [M+H]+ 467.9, 470.0; HPLC purity: 99%.
4-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)butanamide (32).
Method 2.
Yield: 9%. 1H NMR (300 MHz, Methanol-d4) δ 8.20 (s, 1H), 7.78–7.72 (m, 4H), 7.66–7.64 (m, 2H), 7.50–7.48 (m, 2H), 3.17 (s, 3H), 2.79–2.75 (t, J = 7.6 Hz, 2H), 2.39–2.36 (t, J = 7.2 Hz, 2H), 1.99–1.95 (t, J = 7.6 Hz, 2H). LC/MS (APCI+): Calculated for C20H19BrClN3O3S 495.00; Observed m/z [M+H]+ 496.0; HPLC purity: 96%.
3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(ethylsulfonyl) propanamide (33).
Method 1.
Yield: 23%. 1H NMR (300 MHz, Methanol-d4) δ 8.14 (s, 1H), 7.73 (ddt, J = 7.5, 4.7, 2.4 Hz, 4H), 7.69 – 7.61 (m, 2H), 7.53 – 7.46 (m, 2H), 3.42 –3.35 (m, 2H), 3.05 (t, J = 7.3 Hz, 2H), 2.66 (t, J = 7.3 Hz, 2H), 1.25 (t, J = 7.4 Hz, 3H). LC/MS (ESI+): Calculated for C20H19BrClN3O3S 495.00; Observed m/z [M+H]+ 496.1; HPLC purity: 98%.
3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N-(cyclopropylsulfonyl) propanamide (34).
Method 1.
Yield: 25 mg (10%). 1H NMR (300 MHz, Methanol-d4) δ 8.15 (s, 1H), 7.80 – 7.67 (m, 4H), 7.68 – 7.62 (m, 2H), 7.54 – 7.46 (m, 2H), 3.05 (t, J = 7.3 Hz, 2H), 2.98 – 2.86 (m, 1H), 2.65 (t, J = 7.3 Hz, 2H), 1.26 – 1.13 (m, 2H), 1.08 – 0.98 (m, 2H). LC/MS (APCI+): Calculated for C21H19BrClN3O3S 507.00; Observed m/z [M+H]+ 508.1; HPLC purity: >99%.
3-(1-(4-chlorophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)propanamide (35).
Method 1.
Yield: 8%. 1H NMR (300 MHz, Methanol-d4) δ 8.68 – 8.58 (m, 2H), 8.21 (s, 1H), 7.89 – 7.80 (m, 4H), 7.57 – 7.48 (m, 2H), 3.23 (s, 3H), 3.14 (t, J = 7.4 Hz, 2H), 2.71 (t, J = 7.3 Hz, 2H). LC/MS (ESI+): Calculated for C18H17ClN4O3S 404.07; Observed m/z [M+H]+ 405.1; HPLC purity: >99%.
3-(1-(4-chlorophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-2-methyl-N-(methylsulfonyl)propanamide (36).
Method 1.
Yield: 18%. 1H NMR (300 MHz, Methanol-d4) δ 8.68 – 8.61 (m, 2H), 8.17 (s, 1H), 7.88 – 7.80 (m, 4H), 7.56 – 7.49 (m, 2H), 3.15 (s, 4H), 2.98 – 2.84 (m, 1H), 2.80 – 2.66 (m, 1H), 1.25 (d, J = 6.9 Hz, 3H). LC/MS (ESI+): Calculated for C19H19ClN4O3S 418.09; Observed m/z [M+H]+ 419.1; HPLC purity: >99%.
3-(1-(4-chlorophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)butanamide (37).
Method 1.
Yield: 21%. 1H NMR (300 MHz, Methanol-d4) δ 8.62 (d, J = 5.0 Hz, 2H), 8.28 (s, 1H), 7.94 – 7.76 (m, 4H), 7.51 (d, J = 8.9 Hz, 2H), 3.64 (q, J = 7.2 Hz, 1H), 3.10 (s, 3H), 2.75 – 2.54 (m, 2H), 1.35 (d, J = 7.2 Hz, 3H). LC/MS (ESI+): Calculated for C19H19ClN4O3S 418.09; Observed m/z [M+H]+ 419.1; HPLC purity: >99%.
(E)-3-(1-(4-chlorophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)acrylamide (38).
Method 1.
Isolated as hydrochloride salt. Yield: 21%. 1H NMR (300 MHz, DMSO-d6) δ 9.24 (s, 1H), 8.94 (d, J = 5.6 Hz, 2H), 8.10 (d, J = 5.7 Hz, 2H), 8.04 – 8.00 (m, 2H), 7.76 – 7.65 (m, 3H), 6.57 (d, J = 15.8 Hz, 1H), 3.32 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.4, 147.5, 145.5, 144.3 (2C), 137.4, 132.5, 132.1, 130.5, 129.7 (2C), 124.6 (2C), 120.9 (2C), 119.1, 118.6, 41.3. LC/MS (ESI+): Calculated for C18H15ClN4O3S 402.06; Observed m/z [M+H]+ 403.0, 404.9; HPLC purity: 98%.
(E)-3-(1-(4-bromophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)acrylamide (39).
Method 1.
Yield: 10%. 1H NMR (300 MHz, Methanol-d4) δ 9.02 – 8.90 (m, 3H), 8.49 – 8.43 (m, 2H), 7.97 – 7.87 (m, 3H), 7.81 – 7.71 (m, 2H), 6.61 (d, J = 15.5 Hz, 1H), 3.34 (s, 3H). LC/MS (ESI+): Calculated for C18H17BrN4O3S 448.02; Observed m/z [M+H]+ 448.8; HPLC purity: 99%.
2-(1-(4-chlorophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)cyclopropane-1-carboxamide (40).
Method 1.
Yield: 39%. 1H NMR (300 MHz, Methanol-d4) δ 8.91 – 8.83 (m, 2H), 8.60 – 8.52 (m, 2H), 8.41 (s, 1H), 7.96 – 7.89 (m, 2H), 7.62 – 7.55 (m, 2H), 3.35 (s, 3H), 2.78 – 2.68 (m, 1H), 1.96 (dt, J = 8.8, 4.7 Hz, 1H), 1.80 (dt, J = 9.3, 4.7 Hz, 1H), 1.58 (ddd, J = 8.1, 6.5, 4.3 Hz, 1H). 13C NMR (151 MHz, Methanol-d4) δ 172.1, 161.4, 161.1, 147.5, 147.1, 146.5, 144.1, 143.8, 138.5, 132.4, 128.7, 123.7, 123.3, 122.9, 120.4, 40.2, 24.3, 17.5, 15.6. LC/MS (ESI+): C19H17ClN4O3S 416.07; Observed m/z [M+H]+ 417.1; HPLC purity: >99%.
2-(1-(4-bromophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)-N-(methylsulfonyl)cyclopropane-1-carboxamide (41).
Method 1.
Yield: 22%. 1H NMR (600 MHz, Methanol-d4) δ 8.74 (d, J = 5.7 Hz, 2H), 8.33 – 8.26 (m, 3H), 7.83 – 7.75 (m, 2H), 7.70 – 7.62 (m, 2H), 3.29 (s, 4H), 2.63 (ddd, J = 8.9, 6.6, 4.7 Hz, 1H), 1.88 (dt, J = 8.2, 4.7 Hz, 1H), 1.73 (dt, J = 9.2, 4.7 Hz, 1H), 1.51 (ddd, J = 8.2, 6.6, 4.7 Hz, 1H). 13C NMR (151 MHz, Methanol-d4) δ 173.6, 163.1, 162.9, 162.6, 148.5, 148.1, 148.0, 145.9, 145.6, 139.9, 130.1, 125.0, 124.6, 121.8, 121.7, 41.6, 25.8, 19.0, 17.0. LC/MS (ESI+): calculated for C19H17BrN4O3S 460.02; Observed m/z [M+H]+ 460.9; HPLC purity: >98%.
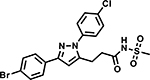
3-(3-(4-bromophenyl)-1-(4-chlorophenyl)-1H-pyrazol-5-yl)-N-(methylsulfonyl)propanamide (51).
Method 2.
Yield: 25%. 1H NMR (400 MHz, Chloroform-d) δ 8.00 (s, 1H), 7.72–7.70 (m, 2H), 7.56–7.45 (m, 6H), 6.53 (s, 1H), 3.30 (s, 3H), 3.11–3.07 (t, J = 7.2 Hz, 2H), 2.69–2.65 (t, J = 7.2 Hz, 2H). LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 482.0; HPLC purity: 99%.
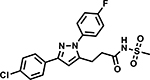
3-(3-(4-chlorophenyl)-1-(4-fluorophenyl)-1H-pyrazol-5-yl)-N (methylsulfonyl)propanamide (52).
Method 2.
Yield: 20%. 1H NMR (300 MHz, Methanol-d4) δ 7.85 – 7.76 (m, 2H), 7.64 – 7.53 (m, 2H), 7.46 – 7.39 (m, 2H), 7.39 – 7.28 (m, 2H), 6.71 (s, 1H), 3.21 (s, 3H), 3.00 (t, J = 7.3 Hz, 2H), 2.71 (t, J = 7.3 Hz, 2H). LC/MS (ESI+): Calculated for C19H17ClFN3O3S3 421.07; Observed m/z [M+H]+ 422.1; HPLC purity: 96%.
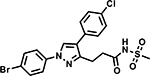
3-(1-(4-bromophenyl)-4-(4-chlorophenyl)-1H-pyrazol-3-yl)-N-(methylsulfonyl)propanamide (53).
Method 2.
Yield: 15%. 1H NMR (400 MHz, Chloroform-d) δ 10.70 (s, 1H), 8.00 (s, 1H), 7.64–7.45 (m, 4H), 7.43–7.41 (m, 2H), 7.34–7.32 (m, 2H), 3.32 (s, 3H), 3.19–3.16 (t, J = 6.4 Hz, 2H), 2.89–2.86 (t, J = 6.4 Hz, 2H). LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 482.0; HPLC purity: 95%.
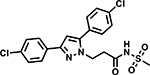
3-(3,5-bis(4-chlorophenyl)-1H-pyrazol-1-yl)-N-(methylsulfonyl)propanamide (54).
Method 1.
Yield: 11%. 1H NMR (300 MHz, Methanol-d4) δ 7.89 – 7.79 (m, 2H), 7.60 – 7.49 (m, 4H), 7.45 – 7.36 (m, 2H), 6.73 (s, 1H), 4.45 (t, J = 6.5 Hz, 2H), 3.14 (s, 3H), 2.97 (t, J = 6.5 Hz, 2H). LC/MS (ESI+): Calculated for C19H17Cl2N3O3S 437.04; Observed m/z [M+H]+ 438.0; HPLC purity: >99%.
3-(5-(4-bromophenyl)-1-(4-chlorophenyl)-1H-pyrazol-3-yl)-N-(methylsulfonyl)propanamide (55).
Method 2.
Yield: 34%. 1H NMR (400 MHz, Chloroform-d) δ 10.71 (s, 1H), 7.50–7.49 (m, 2H), 7.38–7.36 (m, 2H), 7.27–7.25 (m, 2H), 7.11–7.09 (m, 2H), 6.37 (s, 1H), 3.30 (s, 3H), 3.13–3.11 (t, J = 6.0 Hz, 2H), 2.88–2.84 (t, J = 6.0 Hz, 2H). LC/MS (APCI+): Calculated for C19H17BrClN3O3S 480.99; Observed m/z [M+H]+ 482.0; HPLC purity: >99%.
3-(2,4-bis(4-chlorophenyl)oxazol-5-yl)-N-(methylsulfonyl)propanamide (56).
Method 3.
Yield: 11%. 1H NMR (400 MHz, Methanol-d4) δ 8.06 (d, J = 6.80 Hz, 2H), 7.76 (d, J = 6.80 Hz, 2H), 7.54 (d, J = 6.80 Hz, 2H), 7.49 (d, J = 6.80 Hz, 2H), 3.32 (t, J = 5.60 Hz, 2H), 3.22 (s, 3H), 2.85 (t, J = 7.20 Hz, 2H). LC/MS (APCI+): Calculated for C19H16Cl2N2O4S 439.31; Observed m/z [M+H]+ 441.0. HPLC purity: 96%.
3-(2,4-bis(4-chlorophenyl)thiazol-5-yl)-N-(methylsulfonyl)propanamide (57).
Yield: 20%. 1H NMR (400 MHz, Methanol-d4) δ 7.98 – 7.90 (m, 2H), 7.73 – 7.62 (m, 2H), 7.55 – 7.45 (m, 4H), 3.29 (d, J = 7.2 Hz, 2H), 3.20 (s, 3H), 2.73 (t, J = 7.2 Hz, 2H). LC/MS (ESI+): Calculated for C19H16Cl2N2O3S 454.00; Observed m/z [M+H]+ 455.0. HPLC purity: >99%.
General procedure for the synthesis of carbamate 45 and ureas (42–44 and 58).
To a solution of methanesulfonamide (1.5–2.5 equiv) in DCM or DMF was added DIPEA (7.5 equiv) and CDI (0.5–2.5 equiv) at 0 °C. The resulting mixture was stirred at 25 °C for 16 h. Then the appropriate amine or alcohol (1 equiv) dissolved in DCM or DMF (depending on the solubility) was added and heated at 45 °C-90 °C for 24–48 h. Solvents were evaporated under reduced pressure. Crude product was purified by preparative HPLC or silica gel column chromatography.
N-(((3-(pyridin-4-yl)-1-(6-(trifluoromethyl)pyridin-3-yl)-1H-pyrazol-4-yl)methyl)carbamoyl)methanesulfonamide (42).
Yield: 31%. 1H NMR (400 MHz, Methanol-d4) δ 9.35 (d, J = 2.36 Hz, 1H), 8.85 (d, J = 5.96 Hz, 2H), 8.66 (s, 1H), 8.56 (dd, J = 2.36, 8.42 Hz, 1H), 8.37 (d, J = 6.48 Hz, 2H), 8.02 (d, J = 8.60 Hz, 1H), 4.67 (s, 2H), 3.25 (s, 3H). LC/MS (APCI+): Calculated for C17H15F3N6O3S 440.40; Observed m/z [M+H]+ 441.1.
N-(((1-(4-chlorophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)methyl)carbamoyl)methanesulfonamide (43).
Yield: 19%. 1H NMR (400 MHz, Methanol-d4) δ 8.88 (d, J = 5.44 Hz, 2H), 8.53 (d, J = 5.20 Hz, 2H), 8.49 (s, 1H), 7.93 (d, J = 7.72 Hz, 2H), 7.58 (d, J = 7.72 Hz, 2H), 4.68 (s, 2H), 3.26 (s, 3H). 13C NMR (151 MHz, Methanol-d4) δ 154.5, 149.8 (2C), 148.9, 143.7, 139.7, 133.7, 130.9, 130.7, 123.9, 123.7, 121.7, 121.5, 121.2, 120.6, 41.8, 35.5. LC/MS (APCI+): Calculated for C17H16ClN5O3S 405.86; Observed m/z [M+H]+ 406.1; HPLC purity: 97%.
N-(((1,3-bis(4-chlorophenyl)-1H-pyrazol-4-yl)methyl)carbamoyl)methanesulfonamide (44).
Yield: 15%. 1H NMR (300 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.51 (s, 1H), 7.94 – 7.88 (m, 2H), 7.80 – 7.74 (m, 2H), 7.63 – 7.53 (m, 4H), 6.85 (t, J = 5.4 Hz, 1H), 4.38 (d, J = 5.4 Hz, 2H), 3.22 (s, 3H). LC/MS (APCI+): Calculated for C18H16Cl2N4O3S 438.03; Observed m/z [M+H]+ 438.9, 440.9; HPLC purity: 96%.
(1-(4-chlorophenyl)-3-(pyridin-4-yl)-1H-pyrazol-4-yl)methyl (methylsulfonyl)carbamate (45).
Yield: 26%. 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 8.81 (s, 1H), 8.71 (s, 2H), 7.95–7.98 (m, 2H), 7.79–7.80 (m, 2H), 7.62–7.64 (m, 2H), 5.29 (s, 2H), 3.24 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 152.4, 150.7, 150.5, 149.5, 139.7, 138.3, 132.0, 131.7, 130.1, 130.0 (2C), 122.3, 120.8, 120.5, 116.8, 58.4, 41.2. LC/MS (APCI+): Calculated for C17H15ClN4O4S 406.84; Observed m/z [M+H]+ 407.1; HPLC purity: 98%.
N-(((5-(4-bromophenyl)-2-(pyridin-4-yl)furan-3-yl)methyl)carbamoyl)methanesulfonamide (58).
Yield: 29%. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.76 (d, J = 6.1 Hz, 2H), 8.00 (d, J = 6.1 Hz, 2H), 7.90 – 7.84 (m, 2H), 7.74 – 7.67 (m, 2H), 7.21 (s, 2H), 4.51 (d, J = 5.7 Hz, 2H), 3.24 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.2, 157.9, 153.5, 152.6, 146.0, 143.9, 140.1, 132.1 (2C), 129.9, 128.0, 126.3 (2C), 122.1, 119.9, 110.9, 41.4, 35.3. LC/MS (ESI+): Calculated C18H16BrN3O4S 449.00; Observed m/z [M+H]+ 450.0, 452.0 (1:1) bromine isotopic pattern; HPLC purity: 96%.
Synthesis of compounds with heterocyclic rings at C4 – 46–50.
5-(2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)-1H-tetrazole (46).
To a solution of 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanenitrile (93) (100 mg, 258.62 μmol) in isopropanol (1 mL) and water (500 μL) was added ZnBr2 (58 mg, 258.62 μmol, 13 μL) and NaN3 (50 mg, 775.86 μmol, 27 μL) (Scheme S8). The mixture was stirred at 120 °C for 15 h. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (10 mL x 2). The combined organic layers were dried over Na2SO4 and concentrated. The residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150×30mmx4um; mobile phase: [water (0.225%FA)-ACN]; B%: 60%-90%,12min) to afford 5-(2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)-1H-tetrazole (46) as a white solid. Yield: 21 mg (19%). 1H NMR (300 MHz, DMSO-d6) δ 8.52 (s, 1H), 7.85–7.83 (m, 2H), 7.77–7.70 (m, 4H), 7.56–7.54 (m, 2H), 3.22–3.18 (m, 2H), 3.12–3.10 (m, 2H). LC/MS (APCI+): Calculated for C18H14BrClN6 428.02; Observed m/z [M+H]+ 429.0; HPLC purity: >99%.
3-(2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)-1,2,4-oxadiazol-5(4H)-one (47).
Step 1.
To a mixture of 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanenitrile (93) (1.00 g, 2.59 mmol) and NH2OH.H2O (270 mg, 3.89 mmol) in water (5 mg, 259.00 μmol) and EtOH (10 mL) was added a solution of NaOMe (168 mg, 3.11 mmol) in MeOH (4 mL) (Scheme S8). The mixture was stirred at 90 °C for 6 h. The mixture was poured into water (10 mL) and stirred for 15 minutes. The solid was collected to afford (Z)-3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N’-hydroxypropanimidamide (94) as a light-yellow solid. Yield: 1 g (crude). LC/MS (APCI+): Calculated for C18H16BrClN4O 418.02; Observed m/z [M+H]+ 419.0; HPLC purity: 69%.
Step 2.
To a solution of (Z)-3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)-N’-hydroxypropanimidamide (94) (250 mg, 411.01 μmol) in DMF (800 μL) and EtOH (2 mL) was added NaOMe (4 M, 206 μL). The mixture was stirred at 15 °C for 0.5 h and diethylcarbonate (304 mg, 2.57 mmol, 310 μL) was added. The mixture was stirred at 80 °C for another 16 h. The mixture was concentrated and diluted with water (5 mL). The aqueous phase was adjusted to pH=7 with 1 N HCl and extracted with EtOAc (5 mL x 3). The organic phase was concentrated and purified by prep-HPLC (column: Phenomenex Synergi C18 150×25×10um; mobile phase: [water (0.225%FA)-ACN]; B%: 65%-95%, 11min) to afford 3-(2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)-1,2,4-oxadiazol-5(4H)-one (47) as a white solid. Yield: 58 mg (32%). 1H NMR (400 MHz, Chloroform-d) δ 9.41 (s, 1H), 7.86 (s, 1H), 7.66–7.54 (m, 6H), 7.47–7.40 (m, 2H), 3.17–3.02 (m, 2H), 2.88–2.77 (m, 2H). LC/MS (APCI+): Calculated for C19H14BrClN4O2 444.00; Observed m/z [M+H]+ 445.0; HPLC purity: >99%.
5-((1,3-bis(4-chlorophenyl)-1H-pyrazol-4-yl)methyl)pyrimidine-2,4,6(1H,3H,5H)-trione (48).
To a solution of 1,3-bis(4-chlorophenyl)-1H-pyrazole-4-carbaldehyde (61h) (0.150 g, 0.473 mmol) and pyrimidine-2,4,6(1H,3H,5H)-trione (0.064 g, 0.500 mmol) in a mixture of EtOH (10 mL) and DMF (10 mL) (Scheme S9). The reaction mixture was heated in a pressure tube to 80 °C for 16 h. The resulting suspension was cooled to 25 °C, NaBH4 (0.038 g, 1.004 mmol) was added and the resulting reaction was stirred for 1 h at 25 °C to yield a clear orange solution. The solvents were evaporated in vacuo to afford a residue. To the obtained residue were added water (80 mL) and some NaCl and hexane (70 mL) and EtOAc (10 mL) and the resulting suspension was acidified with HCl to pH = 2 and stirred for 1 h. The resulting precipitate was filtered off, yielding a white solid. The water layer was reextracted with EtOAc (80 mL x 2), and the combined organic phases were dried over Na2SO4, evaporated and purified by SiO2 flash chromatography [CH2Cl2/iPrOH 95/5 to 90/10], yielding 5-((1,3-bis(4-chlorophenyl)-1H-pyrazol-4-yl)methyl)-5-hydroxypyrimidine-2,4,6(1H,3H,5H)-trione (0.027 g, 0.059 mmol, 12%) as a slightly yellowish solid (by-product). The white precipitate showed broad signals in the NMR [due to B(OH)3 contamination] and was therefore dissolved in MeOH (80 mL) and HCl conc (1 mL) and the solvent was evaporated. This procedure was repeated, to give 5-((1,3-bis(4-chlorophenyl)-1H-pyrazol-4-yl)methyl)pyrimidine-2,4,6(1H,3H,5H)-trione (48) as a yellow solid. Yield: 162 mg (78%). 1H NMR (300 MHz, DMSO-d6) δ 11.77 (s, 2H), 8.18 (s, 1H), 7.86 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.4 Hz, 2H), 3.66 (s, 2H), 3.39 (s, 1H). LC/MS (ESI+): Calculated for C20H14Cl2N4O3 428.04; Observed m/z [M+H]+ 429.1, 431.0; HPLC purity: 98%.
5-(2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)-1,3,4-oxadiazol-2(3H)-one (49).
Step 1.
The reaction mixture of 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanoic acid (26) (500 mg, 1.23 mmol), Et3N (36 mg, 356.70 μmol, 49 μL) and NH2NH2.H2O (157 mg, 3.08 mmol, 153 μL) in toluene was stirred at 130 °C for 3 h (Scheme S8). The mixture was separated between water (3 mL) and EtOAc (3 mL). The aqueous phase was extracted with EtOAc (3 mL x 2). The combined organic phase was washed with brine (5 mL), dried with anhydrous Na2SO4, filtered and concentrated to afford crude 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanehydrazide (95) as a yellow solid. Yield: 250 mg (48%). LC/MS (APCI+): Calculated for C18H16BrClN4O 418.02; Observed m/z [M+H]+ 419.0; HPLC purity: 67%.
Step 2.
To the solution of 3-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)propanehydrazide (95) (250 mg, 595.66 μmol) in THF (5 mL) was added CDI (145 mg, 893.49 μmol). The mixture was stirred at 60 °C for 4 h under nitrogen atmosphere. The mixture was poured into water (5 mL) and extracted with EtOAc (10 mL x 3). The organic phase was concentrated and purified by prep-HPLC (column: Phenomenex Synergi C18 150×25×10um; mobile phase: [water (0.225%FA)-ACN]; B%: 65%-95%, 10min) to afford 5-(2-(1-(4-bromophenyl)-3-(4-chlorophenyl)-1H-pyrazol-4-yl)ethyl)-1,3,4-oxadiazol-2(3H)-one (49) as a white solid. Yield: 93 mg (34%). 1H NMR (300 MHz, DMSO-d6) δ 8.57 (s, 1H), 7.86–7.82 (m, 2H), 7.76–7.69 (m, 4H), 7.59–7.51 (m, 2H), 3.05–2.96 (m, 2H), 2.94–2.87 (m, 2H). LC/MS (APCI+): Calculated for C19H14BrClN4O2 444.00; Observed m/z [M+H]+ 445.0; HPLC purity: 98%.
N-(5-(1,3-bis(4-chlorophenyl)-1H-pyrazol-4-yl)-4,5-dihydroisoxazol-3-yl)methanesulfonamide (50).
To a solution of methanesulfonamide (250 mg, 2.63 mmol) in DMF (2 mL) was added NaH (100 mg, 2.71 mmol) and the reaction mixture was stirred for 3 min under nitrogen atmosphere. Then a solution of 5-(1,3-bis(4-chlorophenyl)-1H-pyrazol-4-yl)-3-bromo-4,5-dihydroisoxazole (97) (0.100 g, 0.229 mmol) in DMF (2 mL) was added and the mixture was heated to 90 °C in a pressure tube for 16 h (Scheme S9). The cooled solution was diluted with NaHCO3 solution (50 mL) and extracted with EtOAc (2 × 50 mL). The organic phase was dried over Na2SO4, evaporated and the material was purified by flash chromatography (Et2O/HCOOH 99.5/0.5 to 99/1) yielding N-(5-(1,3-bis(4-chlorophenyl)-1H-pyrazol-4-yl)-4,5-dihydroisoxazol-3-yl)methanesulfonamide (50) after crystallization from CH2Cl2/diisopropylether. Yield: 28 mg (27%). 1H NMR (300 MHz, Methanol-d4) δ 8.50 (s, 1H), 7.86 (d, J = 9.0 Hz, 2H), 7.80 (d, J = 8.6 Hz, 2H), 7.55–7.47 (m, 4H), 5.71 (t, J = 10.0 Hz, 1H), 3.47 (dd, J = 16.7, 9.8 Hz, 1H), 3.31 – 3.24 (m, 1H), 3.21 (s, 3H). LC/MS (ESI+): Calculated for C19H16Cl2N4O3S 450.03; Observed m/z [M+H]+ 450.8, 452.8; HPLC purity: >99%.
Supplementary Material
ACKNOWLEDGMENT
DuPont Chemicals, USA for donating the compound library. Dr. Sridevi Bashayam, Dr. Jayanth Thiruvellore, and Mr. Rajkumar Dhinakaran, from Syngene, India and the chemistry team from WuXi AppTec, China for profound chemistry support. Tando Ntsabo and Nicole Cardoso from TB biology, H3D for performing MIC and biology triage assays. Technical staff at in vitro DMPK and parasitology divisions of H3D for excellent technical assistance in generating all in vitro DMPK and cytotoxicity data, Marianna de Kock and Claudia Spies, SAMRC and Stellenbosch University, South Africa for excellent technical assistance in generating MICs against clinical TB isolates.
Funding Sources
The project was funded through Global Health Grants (Number OPP1066878) received from the Bill and Melinda Gates Foundation, the Division of Intramural Research of the NIAID/ NIH, and the Strategic Health Innovation Partnerships (SHIP) unit of the South African Medical Research Council (SAMRC). The University of Cape Town, SAMRC, and South African Research Chairs Initiative of the Department of Science and Innovation, administered through the South African National Research Foundation are gratefully acknowledged for support (K.C.).
ABBREVIATIONS
- TB
tuberculosis
- MIC
minimum inhibitory concentration
- SAR
structure-activity relationship
- RT
room temperature
- MeOH
methanol
- EtOH
ethanol
- EtOAc
ethyl acetate
- DIPEA
diisopropylethylamine
- THF
tetrahydrofuran
- DMF
N,N-dimethylformamide
- BuOH
butanol
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- POCl3
Phosphorus(V) oxychloride
- SOCl2
thionyl chloride
- NaBH4
sodium borohydride
- NaCNBH3
Sodium cyanoborohydride
- LAH
lithium aluminum hydride
- mCPBA
meta-chloroperbenzoic acid
- AUC
area under the curve
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Synthetic schemes, Synthesis of intermediates, Analytical spectra for lead compounds, microbiology assays, in vitro DMPK assays, mouse pharmacokinetic studies, In vivo efficacy studies (PDF)
Data from RNA microarray experiment (XLS)
Molecular formula strings and some data (CSV)
REFERENCES
- 1.World Health Organization Global Tuberculosis Report 2019. https://www.who.int/tb/global-report-2019 (2020-August-13).
- 2.Pontali E; Sotgiu G; D’Ambrosio L; Centis R; Migliori GB Bedaquiline and multidrug-resistant tuberculosis: A systematic and critical analysis of the evidence. European Respiratory Journal 2016, 47, 394–402. [DOI] [PubMed] [Google Scholar]
- 3.Our Pipeline: Pretomanid. https://www.tballiance.org/portfolio/compound/pretomanid (April 19, 2021).
- 4.Xavier A; Lakshmanan M Delamanid: A new armor in combating drug-resistant tuberculosis. Journal of Pharmacology and Pharmacotherapeutics 2014, 5, 222–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarov V; Mikušová K Development of Macozinone for TB treatment: An update. Applied Sciences 2020, 10, 2269. [Google Scholar]
- 6.Early bactericidal activity of TBA-7371 in pulmonary tuberculosis. https://clinicaltrials.gov/ct2/show/NCT04176250 [Google Scholar]
- 7.Sacksteder KA; Protopopova M; Barry CE; Andries K; Nacy CA Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiology 2012, 7, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Jager VR; Dawson R; van Niekerk C; Hutchings J; Kim J; Vanker N; van der Merwe L; Choi J; Nam K; Diacon AH Telacebec (Q203), a new antituberculosis agent. New England Journal of Medicine 2020, 382, 1280–1281. [DOI] [PubMed] [Google Scholar]
- 9.Tenero D; Derimanov G; Carlton A; Tonkyn J; Davies M; Cozens S; Gresham S; Gaudion A; Puri A; Muliaditan M; Rullas-Trincado J; Mendoza-Losana A; Skingsley A; Barros-Aguirre D First-time-in-human study and prediction of early bactericidal activity for GSK3036656, a potent leucyl-tRNA synthetase inhibitor for tuberculosis treatment. Antimicrobial Agents and Chemotherapy 2019, 63, e00240–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schecter GF; Scott C; True L; Raftery A; Flood J; Mase S Linezolid in the treatment of multidrug-resistant tuberculosis. Clinical Infectious Diseases 2010, 50, 49–55. [DOI] [PubMed] [Google Scholar]
- 11.Niederweis M Nutrient acquisition by mycobacteria. Microbiology 2008, 154, 679–692. [DOI] [PubMed] [Google Scholar]
- 12.Warner DF Mycobacterium tuberculosis metabolism. Cold Spring Harbor Perspectives in Medicine 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilburn KM; Fieweger RA; VanderVen BC Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis. Pathogens and Disease 2018, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brzostek A; Pawelczyk J; Rumijowska-Galewicz A; Dziadek B; Dziadek J Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. Journal of Bacteriology 2009, 191, 6584–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marrero J; Trujillo C; Rhee KY; Ehrt S Glucose phosphorylation is required for Mycobacterium tuberculosis persistence in mice. PLOS Pathogens 2013, 9, e1003116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Grau MA; Valcarcel ICG; Early JV; Gessner RK; de Melo CS; de la Nava EMM; Korkegian A; Ovechkina Y; Flint L; Gravelle A; Cramer JW; Desai PV; Street LJ; Odingo J; Masquelin T; Chibale K; Parish T Synthesis and biological evaluation of aryl-oxadiazoles as inhibitors of Mycobacterium tuberculosis. Bioorganic & Medicinal Chemistry Letters 2018, 28, 1758–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold B; Pingle M; Brickner SJ; Shah N; Roberts J; Rundell M; Bracken WC; Warrier T; Somersan S; Venugopal A; Darby C; Jiang X; Warren JD; Fernandez J; Ouerfelli O; Nuermberger EL; Cunningham-Bussel A; Rath P; Chidawanyika T; Deng H; Realubit R; Glickman JF; Nathan CF Nonsteroidal anti-inflammatory drug sensitizes Mycobacterium tuberculosis to endogenous and exogenous antimicrobials. Proceedings of the National Academy of Sciences 2012, 109, 16004–16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salunke SB; Azad AK; Kapuriya NP; Balada-Llasat JM; Pancholi P; Schlesinger LS; Chen CS Design and synthesis of novel anti-tuberculosis agents from the celecoxib pharmacophore. Bioorg Med Chem 2015, 23, 1935–43. [DOI] [PubMed] [Google Scholar]
- 19.Yadlapalli RK; Chourasia OP; Vemuri K; Sritharan M; Perali RS Synthesis and in vitro anticancer and antitubercular activity of diarylpyrazole ligated dihydropyrimidines possessing lipophilic carbamoyl group. Bioorg Med Chem Lett 2012, 22, 2708–11. [DOI] [PubMed] [Google Scholar]
- 20.Nardi A.-n.; Christensen J., Kejser; Jones D, Spencer. Novel pyrazole derivatives useful as potassium channel modulators. WO 2009/003921 Al, 08.01.2009, 2008. [Google Scholar]
- 21.Swiss R; Will Y Assessment of mitochondrial toxicity in HepG2 cells cultured in high-glucose- or galactose-containing media. Current Protocols in Toxicology 2011, 49, 2.20.1–2.20.14. [DOI] [PubMed] [Google Scholar]
- 22.Dixon SL; Smondyrev AM; Knoll EH; Rao SN; Shaw DE; Friesner RA PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. Journal of Computer-Aided Molecular Design 2006, 20, 647–671. [DOI] [PubMed] [Google Scholar]
- 23.ACD/Labs, Release 2019.2.2, Advanced Chemistry Development, Inc. In Toronto, Ontario, Canada, 2020. [Google Scholar]
- 24.Arora K; Ochoa-Montaño B; Tsang PS; Blundell TL; Dawes SS; Mizrahi V; Bayliss T; Mackenzie CJ; Cleghorn LAT; Ray PC; Wyatt PG; Uh E; Lee J; Barry CE; Boshoff HI Respiratory flexibility in response to inhibition of cytochrome oxidase in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 2014, 58, 6962–6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moosa A; Lamprecht DA; Arora K; Barry CE; Boshoff HIM; Ioerger TR; Steyn AJC; Mizrahi V; Warner DF Susceptibility of Mycobacterium tuberculosis cytochrome oxidase mutants to compounds targeting the terminal respiratory oxidase, cytochrome c. Antimicrobial Agents and Chemotherapy 2017, 61, e01338–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naran K; Moosa A; Barry CE; Boshoff HIM; Mizrahi V; Warner DF Bioluminescent reporters for rapid mechanism of action assessment in tuberculosis drug discovery. Antimicrobial Agents and Chemotherapy 2016, 60, 6748–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ardito F; Posteraro B; Sanguinetti M; Zanetti S; Fadda G Evaluation of BACTEC Mycobacteria Growth Indicator Tube (MGIT 960) automated system for drug susceptibility testing of Mycobacterium tuberculosis. Journal of Clinical Microbiology 2001, 39, 4440–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rowland M Protein binding and drug clearance. Clinical Pharmacokinetics 1984, 9, 10–17. [DOI] [PubMed] [Google Scholar]
- 29.Varma MVS; Chang G; Lai Y; Feng B; El-Kattan AF; Litchfield J; Goosen TC Physicochemical property space of hepatobiliary transport and computational models for predicting rat biliary excretion. Drug Metabolism and Disposition 2012, 40, 1527–1537. [DOI] [PubMed] [Google Scholar]
- 30.Smith DA Evolution of ADME science: Where else can modeling and simulation contribute? Molecular Pharmaceutics 2013, 10, 1162–1170. [DOI] [PubMed] [Google Scholar]
- 31.Gao W; Kim J-Y; Anderson JR; Akopian T; Hong S; Jin Y-Y; Kandror O; Kim J-W; Lee I-A; Lee S-Y; McAlpine JB; Mulugeta S; Sunoqrot S; Wang Y; Yang S-H; Yoon T-M; Goldberg AL; Pauli GF; Suh J-W; Franzblau SG; Cho S The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrobial Agents and Chemotherapy 2015, 59, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho SH; Warit S; Wan B; Hwang CH; Pauli GF; Franzblau SG Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 2007, 51, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akester JN; Njaria P; Nchinda A; Le Manach C; Myrick A; Singh V; Lawrence N; Njoroge M; Taylor D; Moosa A; Smith AJ; Brooks EJ; Lenaerts AJ; Robertson GT; Ioerger TR; Mueller R; Chibale K Synthesis, structure–activity relationship, and mechanistic studies of aminoquinazolinones displaying antimycobacterial activity. ACS Infectious Diseases 2020, 6, 1951–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ray PC; Huggett M; Turner PA; Taylor M; Cleghorn LAT; Early J; Kumar A; Bonnett SA; Flint L; Joerss D; Johnson J; Korkegian A; Mullen S; Moure AL; Davis SH; Murugesan D; Mathieson M; Caldwell N; Engelhart CA; Schnappinger D; Epemolu O; Zuccotto F; Riley J; Scullion P; Stojanovski L; Massoudi L; Robertson GT; Lenaerts AJ; Freiberg G; Kempf DJ; Masquelin T; Hipskind PA; Odingo J; Read KD; Green SR; Wyatt PG; Parish T Spirocycle MmpL3 inhibitors with improved hERG and cytotoxicity profiles as inhibitors of Mycobacterium tuberculosis growth. ACS Omega 2021, 6, 2284–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh S; Park Y; Engelhart CA; Wallach JB; Schnappinger D; Arora K; Manikkam M; Gac B; Wang H; Murgolo N; Olsen DB; Goodwin M; Sutphin M; Weiner DM; Via LE; Boshoff HIM; Barry CE Discovery and structure–activity-relationship study of N-alkyl-5-hydroxypyrimidinone carboxamides as novel antitubercular agents targeting decaprenylphosphoryl-β-d-ribose 2′-oxidase. Journal of Medicinal Chemistry 2018, 61, 9952–9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh V; Donini S; Pacitto A; Sala C; Hartkoorn RC; Dhar N; Keri G; Ascher DB; Mondésert G; Vocat A; Lupien A; Sommer R; Vermet H; Lagrange S; Buechler J; Warner DF; McKinney JD; Pato J; Cole ST; Blundell TL; Rizzi M; Mizrahi V The inosine monophosphate dehydrogenase, Guab2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infectious Diseases 2017, 3, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boshoff HIM; Myers TG; Copp BR; McNeil MR; Wilson MA; Barry CE The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: Novel insights into drug mechanisms of action. Journal of Biological Chemistry 2004, 279, 40174–40184. [DOI] [PubMed] [Google Scholar]
- 38.Schrödinger Release 2020–2: Maestro, Schrodinger2020–2: New York, NY, 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.