

Abstract
A ligninolytic peroxidase called versatile peroxidase, VP, (EC 1.11.1.16) is an iron-containing metalloenzyme. The most distinctive feature of this enzyme is its composite molecular framework, which combines lignin peroxidase’s capacity to oxidize compounds with high-redox potential with manganese peroxidase’s capacity to oxidize Mn2+ to Mn3+. In this study, we have extracted amino acid sequences from the Citrus sinensis source and subjected them to various computation tools to visualize the insight secondary and 3D structure, physicochemical properties, and validation of the structure which have not been studied so far to further investigate the catalytic efficiency and effectiveness of VP. The binding energies of HEME and HEME C (HEC) ligands with produced PDB (6rqf.1. A) have been also assessed, analyzed, and confirmed utilizing AutoDock. Binding energies were calculated using the AutoDock and validated by MD simulation using SCHRODINGER DESMOND. Most stable confirmation was achieved through a protein–ligand interaction study. Bio-technological use of VP in the biotransformation of β-naphthol has also been studied. The findings in the current study will have a substantial impact on proteomics, biochemistry, biotechnology, and possible uses of versatile peroxidase in the bio-remediation of different toxic organic compounds.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-023-03758-x.
Keywords: Metalloenzyme, Versatile peroxidase, In silico, Physicochemical, 3D structure, Protein–protein interaction
Introduction
Versatile peroxidase (VP) (EC 1.11.1.16) is a ligninolytic peroxidase with a hybrid molecular structure combining multiple oxidation-active sites. Mn2+ can be oxidized by VP enzymes in the same way that lignin peroxidase (LiP) and high-redox potential non-phenolic compounds can be oxidized by manganese peroxidase (MnP) and vice versa (Perez-Boada et al. 2005). The catalytic cycle of VP combines the cycles of the other fungal peroxidases, LiP and MnP(Ruiz‐Dueñas and Martínez 2009).VP’s catalytic versatility allows it to be used in Mn3+-mediated or Mn-independent reactions on low- and high-redox potential aromatic substrates. Although VP from P. eryngii catalyzes the oxidation of Mn2+ to Mn3+ by H2O2, it differs from classical MnP; in that it is manganese-independent, allowing it to oxidize substituted phenols and synthetic dyes, as well as the LiP substrate, veratryl alcohol (VA). In this gene family, Mn2+ plays two roles. It is both an inducer of MnP genes and a repressor of VPs transcription, and it is the best-known substrate for both MnP and VP enzymes (Camarero et al. 1999; Cohen et al. 2002; Salame et al. 2010). From a physical standpoint, versatile peroxidase (EC 1.11.1.16), a reactive 5 hydrogen peroxide oxidoreductase, is a HEME-containing protein that catalyzes the oxidative destruction of aromatic heterogeneous compounds utilizing hydrogen peroxide as an electron acceptor. The hybrid molecular architecture of this enzyme, which combines lignin peroxidase’s ability to oxidize high-redox potential chemicals with manganese peroxidase’s ability to oxidize Mn2+ to Mn3+, is its most distinguishing characteristic. An iron oxide complex is created by starting the catalytic process with the addition of hydrogen peroxide in the central HEME pocket (Scheme 1) (Ruiz-Duenas et al. 2008; Hofrichter 2002). The resting enzyme in the ferric state (Fe3+) reacts with H2O2 or any organic chemical on the distal side of HEME to create substance I, a Fe4+ oxo-porphyrin radical complex with activated HEME and a two-electron lacking complex. The next step involves the oxidation of compound I by two successive single-electron reactions with reducing substrates, yielding water and extremely reactive radical intermediates. Another intermediate enzyme compound II is produced in the first reduction step before being reduced back to the primary enzyme, ferric peroxidase. The very unstable radical intermediates undergo a series of non-enzymatic reactions that decompose ether bonds, de-methoxylated aromatic rings, cleave them internally, deprotonate them, and hydroxylate them to produce straightforward breakdown products. In cyclic redox events involving aryl alcohol oxidases and dehydrogenases, white rot fungus produces hydrogen peroxide, which is needed as an electron acceptor for these peroxidases to degrade lignin (Busse et al. 2013; Guillén and Evans 1994). The fact that VPs have three distinct active sites for substrate oxidation a site for the oxidation of Mn2+ to Mn3+, which functions as a diffusible mediator; a low-redox potential HEME-dependent binding pocket; and a high-redox potential surface-reactive tryptophan radical which connects to the HEME through a long-range electron-transfer pathway is one explanation for their functional promiscuity. They also contain two structural calcium ions, numerous glycosylation, and numerous disulfide linkages, which makes it more difficult for them to express themselves in heterologous hosts (Barber-Zucker et al. 2022). From the above-mentioned points, it is inferred that VP is a very important biotechnologically useful enzyme, and therefore, our research group has isolated and purified versatile peroxidase from Citrus sinensis which is a novel plant source known so far. Purification and characterization of the VP enzyme from Citrus sinensis have been published by our research group (Rai et al. 2020). To explore the catalytic efficiency and efficacy of VP further, we have retrieved amino acid sequences from the source Citrus sinensis and subjected them to various computation tools to visualize the insight physicochemical properties, secondary and 3D structure, and validation of the structure which have not been studied so far. In this study, the binding energy of HEME and HEME C (HEC) ligands with generated PDB (6rqf.1. A) has been also evaluated and studied using AutoDock and validated the results with molecular dynamics simulation study. The biotechnological application of VP in the biotransformation of β-naphthol was studied and found that the VP reacted efficiently and was eco-friendly.
Scheme 1.
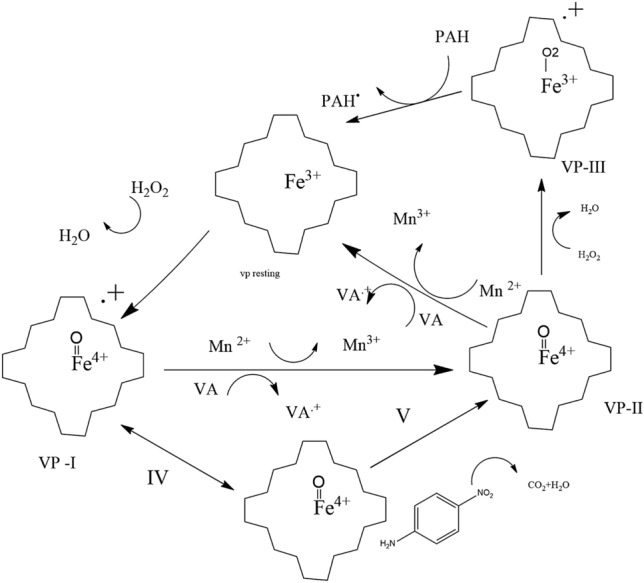
Variable peroxidase’s mechanism of substrate oxidation. Hydroperoxides oxidize the resting enzyme to a two-electron deficient compound I, which is then reduced by one electron to compound II and the natural enzyme
Methods
Protein sequence
FASTA formats of the different amino acid sequences were recovered from NCBI (National Center for Biotechnology Information). The homogeneity of the enzyme purified from the source Citrus sinensis was examined with SDS-PAGE by using the procedure of Weber and Osborn (Weber and Osborn 1969), and amino acid sequences were retrieved for the protein band which has been already done and reported by our research group (Rai et al. 2020).
Analysis of primary sequence by using Expasy ProtParam tool
Expert protein analysis system (Expasy) was the first web service. A diverse catalog of bioinformatics resources developed by SIB Groups. A variety of life science and clinical research fields, including genomics, proteomics, and structural biology, as well as evolution and phylogeny, systems biology, and medical chemistry, are supported by this expandable and integrated gateway, which gives users access to over 160 databases and software tools. These tools are freely available and user-friendly. By using the amino acid sequence retrieved from the protein band of purified VP from Citrus sinensis, the physicochemical features of the protein sequences mainly molecular weight, theoretical isoelectric point (pI), amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) were characterized with the help of ProtParam tool (https://web.expasy.org/protparam) which is freely available tool. The acid dissociation constant (Ka) of a solution is pKa (the negative base-10 logarithm) values of the amino acids which are used to calculate the protein pI. The pKa value of amino acids is influenced by their side chain. It is crucial for determining the pH-dependent properties of a protein because every enzyme needs a particular pH to show its catalytic activity. The stability of the protein in a test tube can be determined using the instability index. A protein is stable if its instability index is below 40; if it is greater than 40, it may be unstable. GRAVY index measures a protein’s solubility; a protein’s hydrophobic nature is indicated by a positive GRAVY value, whereas a protein’s surface accessibility to water interactions is indicated by a negative GRAVY value. The amino acids’ hydropathy values are added together and divided by the number of residues in the sequence to yield the GRAVY value for a peptide or protein. The limitation of the ProtParam tool is protein cannot be post-translationally modified, and ProtParam will not be able to determine if your mature protein forms dimers or multimers. And all computations by ProtParam are based on either compositional data or the N-terminal amino acid.
Percentage of amino acid sequence in secondary structure
The secondary structure of proteins can be predicted using a variety of techniques based on their amino acid sequences. To increase the accuracy of protein secondary structure predictions, the self-optimized prediction method (SOPM) has been described. In a database comprising 126 chains of non-homologous (less than 25% identical) proteins, the SOPMA approach correctly predicts 69.5% of amino acids for a three-state description of the secondary structure (-helix, -sheet, and coil). Applying the tool ProtParam, the proportion of the versatile peroxidase’s amino acid sequence was assessed, and the secondary structure was predicted using a self-optimized prediction approach with alignment (SOPMA) secondary structure prediction (NPS@: SOPMA secondary structure prediction—IBCP) freely available tool. SOPMA is used to determine the secondary structural characteristics of the chosen protein sequences that are taken into consideration for the investigation.
Peptide structure
A tool called Pep-Draw (www.pepdraw.com) was developed to make it simpler to investigate peptides’ properties and chemical makeup. It allows users to visualize the fundamental peptide structure of an amino acid sequence and predict specific chemical properties like mass, charge, and hydrophobicity.
Mass: The total monoisotopic masses of each amino acid residue in the peptide make up the mass, also known as the formula weight. To calculate this, multiply the mass of all the fundamental atoms by the mass of the water, then added the atomic masses of all the side chain atoms.
Net charge: At a pH of 7, the residues’ net charge is the sum of their positive (basic) and negative (acidic) charges.
Hydrophobicity (Wimley–White scale): In this context, “hydrophobicity” refers to the free energy required to move a peptide from an aqueous environment to a hydrophobic one, such as octanol. The Wimley–White scale, which was established through experimentation, is the scale that is being utilized, and the hydrophobicity of a peptide is found by adding its Wimley–White hydrophobicities. The measurements are made in Kcal per mol. This assumes a pH of neutral.
Prediction of tertiary structure
The tertiary structure was characterized by SWISS-MODEL created on automated comparative modeling of protein. By offering the protein modeling service on the Internet, SWISS-MODEL established the field of automated modeling. It gives users access to a fully integrated platform for the sequence-to-structure analysis and modeling that combines the visualization tool Swiss-PdbViewer, the web-based Workspace, and the SWISS-MODEL Repository. To hide the computational complexity of structural bioinformatics and encourage bench scientists to utilize the continuously expanding structural knowledge available, this computational environment is made freely available to the scientific community. In fact, due to the favorable nature of comparative protein modeling and experimental structure determination, the availability of structural information has greatly grown for many organisms over the past decade. A bioinformatics technique called I-TASSER (Iterative Threading ASSEmbly Refinement) (http://zhanggroup.org/I-TASSER) uses amino acid sequences to forecast the three-dimensional structure model of protein molecules. By structurally matching structural models of the target protein to the recognized proteins in protein function databases, I-TASSER has been expanded to offer annotations on ligand sites, gene ontology, and enzyme commission.
Validation of protein structure
The structure evaluation was done to analyze the significance of the homology model, and PROCHECK, ERRAT, and VARIFY 3D tools were used to validate it from server SAVES. It has been in use since 2004 and is presently running version 6. It is an interactive validation server for five programs that are frequently used to validate protein structures. Some of these tools were created in the early 1990s using FORTRAN, and ProSA, while others have since been updated to use C + + . Every day, the server processes between 1000 and 2000 requests from users all over the world. However, starting a job over again is never difficult. You can choose any program to run independently using the same dashboard and are not required to run them all. The Ramachandran plot was created by SWISS-MODEL, and the PROCHECK was used to analyze the stereochemical quality of protein structures residue-by-residue. In biochemistry, the Ramachandran plot is also known as the [φ, ψ] plot. According to theory the angle of φ, ψ can have any value between + 180° and − 180°. Because of the structural hindrance of non-bonded atoms, a combination of all possible values in 3D space is not possible. The possible combination value of φ and ψ was first determined by G.N. Ramachandran, known as the Ramachandran plot. The permissible value of φ and ψ can be visualized in the two-dimensional plot of Ramachandran, where φ is plotted on the x-axis and ψ is plotted on the axis y (Apweiler et al. 2004; Weber and Osborn 1969; Yadav et al. 2017b; Pradeep et al. 2012). The program ERRAT is used to confirm protein crystallographically determined structures. In comparison with a database of reliable high-resolution structures, the error function is based on statistics of non-bonded atom–atom interactions in the reported structure. ProSA is also an interactive web application for predicting and modeling structures as well as refining and validating protein structures. VERIFY 3D assigns a structural class by the structure’s position and environment (alpha, beta, polar, nonpolar, etc.) and compares the results to good structures to ascertain whether an atomic model (3D) and its amino acid sequence (1D) are compatible. For good structure in the 3D/1D profile, at least 80% of the amino acids have received scores > = 0.2.
Prediction of protein function
The target amino acid sequence was submitted to the I-TASSER server, and COFACTOR and COACH predicted the biological annotations of the VP based on the I-TASSER structure prediction server (Yang et al. 2015). COFACTOR uses protein–protein networks and structural comparison to infer protein functions (ligand-binding sites, EC, and GO). The COFACTOR, TM-SITE, and S-SITE programs all produce results for function annotation on ligand-binding sites, and COACH is a meta-server technique that aggregates these data (Yang et al. 2015).
Docking of protein and binding energy calculation using AutoDock
The software used is AutoDock tools 1.5.7 and AutoDock4.2.6 from the Scripps Research Institute, Inc. The hardware utilized is an ASUS-VivoBook14 with an 11th generation Intel(R) Core (TM) i5-1135G7 @ 2.40 GHz 2.42 GHz (USA). The ligands are downloaded from the protein data bank (http://www.rcsb.org) in SDF format, while the receptor protein’s structure is created from SWISS-MODEL Expasy in the PDB format. Unused molecules, like water molecules, were eliminated, and polar hydrogen and Kollman charges were added and saved as PDBQT format. Then, the ligand is processed which involves choosing the root and saving the file in PDBQT format after setting torsion. After that, the grid box’s size and the coordinates were adjusted. The binding sites of VP were defined with a rectangular grid box of 72 × 72 × 72 covering the entire grid points separated by 0.503 Å and the center of the grid box is 150.194, 148.116, and 175.849 Å. Lamarckian genetic algorithms have been used for docking searches, and the number of runs ranged from 10 to 100. Population sizes are 150, and the maximum number of runs for energy evaluation is 2,500,000. The maximum number of generations is 27,000, and the energy evaluation has typically been adjusted from medium to long for run options. The chosen rate of mutation probability and rate of the crossover are 0.02 and 0.8, respectively. The genetic algorithm (GA) crossover operator is a two-point crossover for all the runs. The primary parameter measured to evaluate the validity of the docking outcomes is the root-mean-square deviation (RMSD) value, where the linear relationship is observed between the number of runs. Observations of additional parameters include the free energy of binding (G), the inhibition constant (Ki), the number of hydrogen bonds, and the number of amino acid residues. By examining the re-rank score of the G and Ki values, postures are chosen for each run, with the conformation having the most negative G and the lowest Ki value, serving as a sample for the conformation of the total number of runs. These parameters are monitored to validate the variations in docking outcomes brought on by variations in the number of runs. The interaction of docked complexes was visualized by Biovia discovery studio and Molegro Molecular Viewer(MMV) (Forli et al. 2016; Pratama and Siswandono 2020).
Molecular dynamic (MD) simulation
Protein–ligand docking results only provide a static, image of the ligands’ binding orientation in the active site of the receptor, though being extensively used and have practical uses. So, after integrating the Newton equations of motion, MD simulation must be used to mimic the dynamic character of the system’s atoms as a function of time. In the previous investigation, it was shown that the majority of the versatile peroxidase’s active site was described as HEME. The docking result showed that Citrus sinensis versatile peroxidase has binding energies of − 16.99 and − 11.16 kcal/mol with HEC and HEM ligands, respectively. These two best-docked complexes have thus been simulated for 100 ns duration molecular dynamics starting from the optimal docking pose. Versatile peroxidase mainly contains HEME as a ligand. The MD simulation was carried out under a Linux environment on an HP Z2 workstation using DESMOND software. The complexes were immersed in a water bath of size (10 × 10 × 10 Ǻ) and 0.15 M salt concentration using the TIP4P solvent model. The whole system was neutralized using a suitable number of chloride and sodium ions. The complete systems were minimized separately for a maximum of 2000 iterations with a convergence threshold of 1.0 kcal/mol/Ǻ. In the last, the production run of MD simulation has been performed for 100 ns duration at 300 K temperature and 1 atm pressure with 2.0 fs time step using OPLS2005 force field as implemented in the academic DESMOND Maestro-Schrödinger suite 2022–1 interface program. Using the receptor atoms’ root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) of residues, a trajectory analysis was conducted to determine the stability and volatility of the protein complexes (Lee et al. 2021; Yadava et al. 2017).
Catalytic activity at HEC active site
The catalytic activity of the enzyme with β-naphthol as the substrate has been studied. The UV-spectra of β-naphthol substrate (control) in 50 mM phosphate buffer has a peak at 280 nm. One ml reaction mixture contained 5 mM substrate, 50 mM buffer, 0.5 mM H2O2, and 1.1 mg of VP enzyme. It is clear from the absorption spectra that the absorption depth at 280 nm decreased β-naphthol is being consumed by the enzyme as the reaction proceeds with time. Enzymes have a catalytic effect by binding efficiently to their substrates which confirms an ideal binding arrangement between the enzyme and the substrate and even more efficiently the transition state of the catalyzed reaction. This binding maximizes the enzyme’s ability to catalyze its reaction. We have also investigated the catalytic activity of enzymes with different substrates. HPLC for β-naphthol was analyzed.
Results and discussion
Amino acid sequences and program chart
Amino acid sequence:
The Expasy service offers a variety of tools in several fields of the life sciences, such as proteomics, genomics, phylogeny, systems biology, population genetics, and transcriptomics. It offers access to numerous scientific databases and software tools. Table 1 gives the physicochemical study of purified VP from Citrus sinensis by using Expasy–ProtParam to evaluate a protein sequence (Rai et al. 2020). ProtParam, which can be accessed via the URL (http://www.expasy.org/tools/protparam), is one of the protein analysis tools on the Expasy server. It is used to measure different physicochemical parameters for a provided protein. Only the protein sequence is used as an input when determining these parameters. The protein can be identified by an amino acid sequence, an accession number or ID from UniProtKB/Swiss-Prot, UniProtKB/TrEMBL, or both. The protein pI is derived using the pKa values of the amino acids. The side chain of amino acids has an impact on their pKa value. It is essential for figuring out a protein’s pH-dependent characteristics. The instability index can be used to gauge a protein’s stability in a test tube. If a protein’s instability index is below 40, it is thought to be stable; if it is more than 40, it may be unstable. The protein’s aliphatic index is the percentage of its volume made up of amino acids with aliphatic side chains, such as alanine, valine, isoleucine, and leucine to get the aliphatic index of a protein, and the aliphatic index is determined as follows:
N(Ala), N(Val), N(Ile), and N(Leu) stand for the mole percent (100 X mole fraction) of alanine, valine, isoleucine, and leucine, respectively. The side chains of valine (a = 2.9) and leu/isoleu (b = 3.9) around the side chain of alanine are represented by the variables “a” and “b.” The hydropathy assessments of each amino acid residue are aggregated and divided by the length or quantity of the sequence to produce the GRAVY score for a protein or peptide. An increasing positive score indicates greater hydrophobicity (Gasteiger et al. 2005; Artimo et al. 2012; Kyte and Doolittle 1982).
Table 1.
Physicochemical properties of the citrus sinensis amino acid sequence by using ProtParam
| Sl.no | Accession number | Source organism | No. of amino acid | Molecular weight | Theoretical PI | Total no. of negatively charged residue (asp + glu) | Total no. of positively charged (agr + lys) |
Instability index | Alphabetic index |
Grand average of hydrophobicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Citrus sinensis | 215 | 24,178.57 | 8.89 | 12 | 15 | 34.40 | 106.88 | 0.581 |
Amino acid composition and secondary structure prediction
Figure 1 displays the breakdown of several amino acids based on their percentage content. Leucine and valine were found to have the greatest compositions of all the amino acids. From the patterns of amino acid profiles and protein families, domains and active sites were able to deduce from the secondary structure of a protein. Significantly, the alpha helix predominates among the states shown in Fig. 2.
Fig. 1.
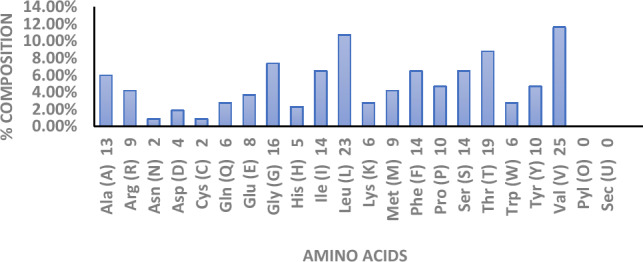
Amino acid composition with different percentages
Fig. 2.

Percentage basis composition of the different helices by using the SOMPA tool
Peptide structure prediction
Pep-Draw was developed to be a reliable and straightforward peptide analysis tool. It is highly useful for learners of the structure and properties of amino acids, as seen in Fig. 3. The peptide’s mass and length were determined to be 60 and 6969.4287, respectively (White and Wimley 1998).
Fig. 3.

Peptide structure of the amino acid sequence by using the Pep-Draw tool
Tertiary modeling of amino acid sequence
The device SWISS-MODEL (http://swissmodel.expasy.org) and I-TASSER (zhanggroup.org) are used for automated comparative 3D modeling of protein structure from a phylogenetically related protein structure that is represented as a template, which was used to construct a three-dimensional (3D) structure modeling from the predicted amino acid sequence (Camacho et al. 2009). The best template for the homology model was selected based on the sequence identity percentage, QMEAN Z-scores, and global model quality estimate (GMQE). The pdb of 6rqf.1A from Spinacia oleracea has the highest resemblance to the amino acid sequence of the lemon leaf, with a sequence identity of 98.14% and a QMEAN Z-score of − 3.57, (Z-scores around 0.0 representing a “native-like” structure, while a “QMEAN” Z-score lower than − 4.0 generally denotes a model with low quality), and GMQE value of 0.80. In such an arrangement, the GMQE value ranging from zero to one is seen as having more reliability. In silico study VP of other sources have not been studied in this regard w.r.t all the parameters. Figure 4a illustrates the structure. Figure 4b shows the predicted ligand-binding site using I-TASSER. The molecular basis of proteins, their function, disease-related mutations, inhibitor designing based on structure, and pharmaceutical creation are all made feasible by analyzing the 3D structure of a protein (Benkert et al. 2011; Takio et al. 2022).
Fig. 4.
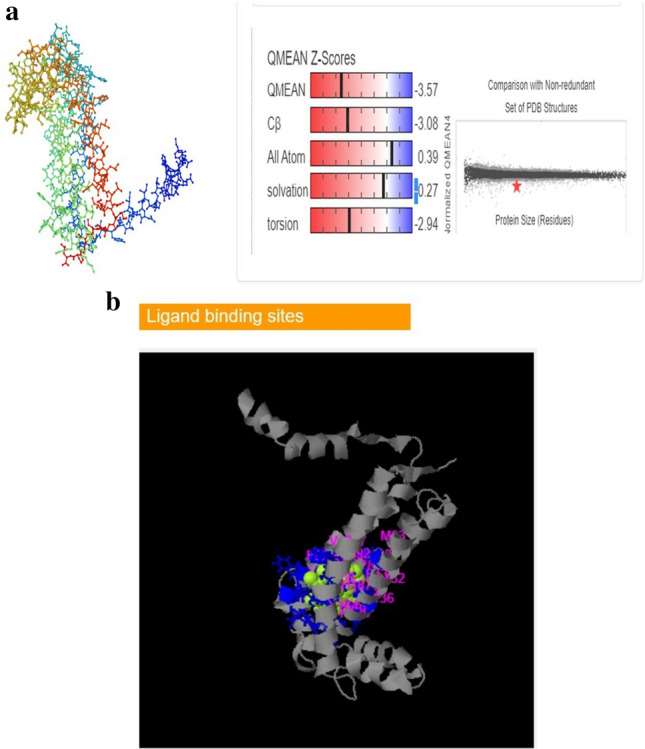
(a, b) Three-dimensional structure of predicted amino acid sequence with 215 residues by using (a) SWISS-MODEL, (b) ligand-binding site prediction by using I-TASSER
Validation of protein structure
By employing SWISS-MODEL Expasy (https://swissmodel.expasy.org) and POCHECK, Fig. 5a, b displays the Ramachandran plot of a predicted amino acid sequence of pure VP including 215 residues. In Fig. 5a, the PROCHECK validation showed a warning sign with 93.0% of the amino acid residues in most favored regions, 5.3% in additional allowed, 1.1% generously allowed, and 0.5% in disallowed regions. A good quality model has over 90% in the most favored region and less than 1% in the disfavored region. Figure 5b demonstrates 96.26% of favored Ramachandran. Since all amino acids except glycine have shorter non-bonding interatomic distances than their van der Waals radii, the white portions in this diagram depict sterically prohibited regions for those amino acids. Glycine is an exception because it lacks a side chain or is smaller in size. Because there are fewer obstacles, parallel beta (β), also known as dark green space, resembles the region that conformations find most desirable. Maximum values of φ and ψ of polypeptide bond breakdown are found in the alpha-helical or beta-sheet section of the Ramachandran plot. An amino acid-containing cyclic chain named proline limits its angle scales of φ value around − 60 for creating maximum conformationally controlled amino acid residue. The light green space corresponds that the structure of the polypeptide chain having an outer boundary distance of Van der Waals, which means that atoms are favored in this region to come a little nearer together. Due to different orientations of side chains D and L forms of amino acid according to their co-group; hence, they have favored different angle values of φ and ψ. From the ERRAT program, the current structure received an overall quality factor of 96.13%. For a high-quality model, the typical acceptability range is greater than 50%. From ProSA, the Z-score of the predicted model was − 1.5. A good protein model is one with a Z-score that is close to or greater than 0. The Z-score is defined as the energy difference in units of the ensemble’s standard deviation between the native fold and the average of a group of misfolds (Ramachandran and Sasisekharan 1968; Chen et al. 2010).
Fig. 5.

(a, b) Ramachandran plot of templet 6rqf.1.A by SWISS-MODEL Expasy
Result of function prediction
Gene ontology (GO) is a framework and collection of ideas for describing how genes work in all types of organisms. One of the important sources for bioinformatics is GO, which provides a comprehensive definition of protein function. There are three different ontologies: biological processes, cellular components, and molecular functions. To aid in the computational representation of biological systems, it was developed. A GO annotation establishes a connection between a particular gene product and a GO concept to describe the function of the gene. Depending on the I-TASSER structure prediction, COFACTOR and COACH predicted the function of VP. The initial I-TASSER model is matched to every structure in the PDB library using the TM-align structural alignment program after the structure assembly simulation. COFACTOR used structural comparison and protein–protein networks to infer protein functions (ligand-binding sites, EC, and GO). The COFACTOR, TM-SITE, and S-SITE programs all produce results for function annotation on ligand-binding sites. COACH is a meta-server technique that aggregates these data. In comparison with function annotations derived just from global structure comparison, COACH has undergone substantial training to identify biological functions from several sources of sequence and structural information. The top 10 homologous GO templates in the PDB that were produced using the I-TASSER server and had the highest TM-score (i.e., structural similarity) to the anticipated I-TASSER model are shown in Table 2. Among the top-scoring templates, a consensus GO word is displayed in Table 3. The average weight of the GO term, which is assigned based on the CscoreGO of the template, is what is meant by the GO-Score associated with each prediction. These proteins frequently perform similarly to the target due to their structural similarities. The GO keywords GO:0016491, GO:0045158, and GO:0020037 are linked to molecular functions in Table 3. A reversible chemical reaction in which the oxidation state of an atom or atoms inside a molecule is transformed is referred to as oxidoreductive catalysis, according to the definition given in the GO:0016491. One substrate act as an electron or hydrogen donor and is oxidized, whereas the other substrate acts as an electron or hydrogen acceptor and is reduced. Iron is a component of HEME, a substance specified by the GO:0020037 as attaching to a porphyrin (tetrapyrrole) ring. The GO:0022904 GO:0006810 and GO:0015979 are linked to the biological activity of the enzyme, which includes respiration electron transport, transport of substances (such as macromolecules, small molecules, ions), or movement of cellular components (such as complexes and organelles) into, out of, or within a cell, or between cells, or within a multicellular organism, using a transporter or transporter complex, a pore, or a motor protein, and photosynthesis. The GO:0070469 and GO:0016021 GO:0005743 The GO terms GO:0009535 and GO:0005886 are related to cellular elements. It specified the area outside of a cell’s outermost structure. It referred to the area outside the plasma membrane where cells that lack external encasing or protective structures are located. Fig. 6a–c contains the ancestor charts for GO: 0016491, GO: 0022904, and GO 0016021 (Basumatary et al. 2023).
Table 2.
CscoreGO is a combined measure for evaluating the global and local similarity between query and template protein
| Rank | CscoreGO | TM-score | RMSDa | IDENa | Cov | PDB Hit | Associated GO Terms |
|---|---|---|---|---|---|---|---|
| 1 | 0.90 | 0.9532 | 1.40 | 0.84 | 1.00 | 2e74A |
GO:0009579 GO:0016021 GO:0015979 GO:0046872 GO:0022904 GO:0016020 GO:0020037 GO:0022900 GO:0016491 GO:0009055 GO:0042651 GO:0006810 GO:0045158 |
| 2 | 0.73 | 0.9487 | 1.21 | 0.89 | 0.99 | 1q90B |
GO:0020037 GO:0016020 GO:0046872 GO:0009055 GO:0022900 GO:0022904 GO:0045158 GO:0009535 GO:0016491 GO:0009579 GO:0009536 GO:0016021 GO:0006810 GO:0015979 GO:0009507 |
| 3 | 0.67 | 0.9180 | 1.60 | 0.30 | 0.97 | 3cwbC |
GO:0070469 GO:0046872 GO:0005739 GO:0005743 GO:0022904 GO:0016491 GO:0016021 GO:0006810 GO:0009055 GO:0016020 GO:0022900 |
| 4 | 0.67 | 0.9162 | 1.59 | 0.31 | 0.97 | 1be3C |
GO:0070469 GO:0005743 GO:0005739 GO:0009055 GO:0016021 GO:0006810 GO:0016020 GO:0022900 GO:0016491 GO:0022904 GO:0046872 |
| 5 | 0.66 | 0.9602 | 1.16 | 0.33 | 0.99 | 2fynA |
GO:0005886 GO:0016020 GO:0016021 GO:0006810 GO:0022900 GO:0070469 GO:0046872 GO:0009055 GO:0016491 GO:0022904 |
| 6 | 0.62 | 0.9554 | 1.30 | 0.34 | 1.00 | 1zrtC |
GO:0005886 GO:0006810 GO:0009055 GO:0016020 GO:0016021 GO:0016491 GO:0022900 GO:0022904 GO:0046872 GO:0070469 |
| 7 | 0.61 | 0.9251 | 1.69 | 0.28 | 0.98 | 1ezvC |
GO:0022904 GO:0022900 GO:0016020 GO:0005743 GO:0009060 GO:0005739 GO:0016491 GO:0009055 GO:0005750 GO:0008121 GO:0006810 GO:0006122 GO:0070469 GO:0045153 GO:0046872 GO:0016021 |
| 8 | 0.42 | 0.5171 | 4.40 | 0.04 | 0.74 | 2izpA | GO:0005576 GO:0009405 |
| 9 | 0.29 | 0.5536 | 4.33 | 0.07 | 0.80 | 2p7nA | GO:0009405 |
| 10 | 0.28 | 0.5138 | 4.22 | 0.03 | 0.71 | 3pblA |
GO:0019835 GO:0016998 GO:0016798 GO:0016787 GO:0008152 GO:0003824 GO:0003796 GO:0009253 GO:0042742 GO:0007186 GO:0016021 |
Its range is [0–1], and higher values indicate more confident predictions. TM score measures global structural similarity between query and template protein. RMSDa is the RMSD between residues that are structurally aligned by TM align. IDENa is the percentage sequence identity in the structurally aligned region. Cov represents the coverage of global structural alignment and is equal to the number of structurally aligned residues divided by the length of the query protein
Table 3.
Consensus prediction of GO terms
| Molecular function | GO:0016491 | GO:0045158 | GO:0020037 | ||
| GO-score | 1.000 | .970 | 97 | ||
| Biological process | GO:0022904 | GO:0006810 | GO:0015979 | ||
| GO-score | 1.00 | 1.000 | .97 | ||
| Cellular component | GO:0016021 | GO:0070469 | GO:0005743 | GO:0009535 | GO:0005886 |
| GO-score | 1.000 | .960 | .890 | .730 | .66 |
Fig. 6.

(a, b, c) Predicted terms within the gene ontology hierarchy for molecular function, biological function, and cellular function. Confidently predicted terms are color-coded by CscoreGO
Docking of protein
The molecular interaction between a protein and a ligand was studied by AutoDock and BIOVIA discovery studio. The binding energy, ligand efficiency, dissolved energy of the hydrogen bond and van der Waal interaction, electrostatic energy, internal energy, torsional energy, free energy, inhibitory constant (Ki), and the cluster root-mean-square deviation (RMSD) values are all listed in Table 4. Ten conformations were achieved from the AutoDock tool; among them, the lowest binding energy was selected for visualization through BIOVIA discovery studio demonstrating that the protein was bounded to amino acids, namely ARG 87 and ARG 83, and were bounded on the surface of the HEC ligand as non-classical and classical hydrogen bonds, salt bridges, and electrostatic interactions, whereas PHE131, MET54, ALA90, ARG87, TYR58, TYR184, PHE44, HIS86, HIS187, LEU138, and PHE131 were bounded as hydrophobic alkyl, pi-alkyl, and pi-sigma. PRO192, VAL148, HIS86, PHE44, PHE131 hydrophobic alkyl and pi-alkyl contact, HEM ligand bound with amino acidTHR55, GLY51, HIS187 as hydrogen bond interaction, PRO192, VAL148, HIS86, PHE44, PHE131 hydrophobic alkyl and pi-alkyl interaction Hydrogen bonds, hydrophobic alkyl, and pi-alkyl interactions, all are summarized in Table 5. Using MMV software, an energy map of the docked HEC complex was created, where the colors green, sky blue, yellow, and blue space stand for steric favorable, hydrogen acceptor favorable, hydrogen donor favorable, and electrostatic favorable conditions as shown in Fig. 7 (Bathula et al. 2019; Kumar et al. 2022).
Table 4.
Properties of best-docked cluster obtained from AutoDock tool
| Name of the clusters | Binding energy (kcal/mol) | Ligand efficiency | Vdw-HB dissolved Energy kcal/mol | Electrostatic Energy kcal/mol | Internal energy kcal/mol | Torsional energy | cl RMS | Inhibition constant(ki) | Free energy kcal/mol |
|---|---|---|---|---|---|---|---|---|---|
| HEC | − 16.99 | − 0.4 | − 17.42 | − 1.96 | − 0.61 | 2.39 | < 2 | .353 pM | − 1377 |
| HEM | − 11.16 | − 0.26 | − 15.84 | 0.2 | 3.18 | 3.47 | < 2 | 6580 pM | − 1374.73 |
Table 5.
Interaction of amino acid residue of protein with ligand

Fig. 7.
(a, b) Energy map of template 6rqf.1 drawn by using MMV software was green, sky blue, yellow, and red space representing sterically favorable, hydrogen acceptor favorable, hydrogen donor, favorable, and electrostatic, respectively
Molecular dynamics simulation
The molecular dynamic simulation for better-docked poses of the HEC complex is conformation-1 and of the HEME complex is conformation-10 obtained from the docking file done by AutoDock. An all-atom 100 ns molecular dynamics simulations have been done by using Desmond. The changes in molecular conformation and intermolecular interaction potential as a function of simulation interval are predicted exclusively at the molecular level. RMSD, RMSF, and non-bonded interactions are essential aspects when using MD simulation to analyze the stability and flexibility of the versatile protein with the HEC and HEME complex. RMSD values for the protein’s (alpha) carbon atoms were determined concerning the MD simulation’s first posture and compared to each docked receptor ligand complex’s structural variations and quality. Protein-fit ligands were analyzed for RMSD and RMSF value during 100 ns interval of molecular dynamics simulation. Herein, the RMSD variation of Cα atoms of vp-HEC fit exhibited an increase in the beginning and reaches 6.5 Ǻ during 0 to 10 ns dynamics. After that, approximately constant RMS deviation is exhibited centered around 6.8 with small fluctuations of ± 0.5 Ǻ. The RMS deviation of ligand fitted within the active site of the protein, during entire dynamics, is about 2.0 Ǻ which is far below the RMS deviation of protein Cα atoms. In the case of the vp-HEM complex, the RMSD variation of Cα-atoms of the protein acquires the constant value of about 7.0 Ǻ within a very short duration of the dynamics. It remains constant up to the dynamics of 55 ns; thereafter, a bump is seen during 55 to 65 ns dynamics, which reduces to 6.8 ± 0.3 Ǻ at the end of 65 ns and maintains the constant value during the rest of dynamics. Protein-fit ligand RMS deviation demonstrates similar variations but with a lesser value of 4.5 Ǻ as compared to protein RMSD. The average root-mean-square fluctuations (RMSF) of each residue have also been calculated in the case of both complexes vp-HEC and vp-HEM. In both complexes, the end side residues demonstrate > 6.0 Ǻ fluctuations. The next larger fluctuations (> 4.0 Ǻ) are exhibited by the residues numbered 153–164, in both cases. Minimum fluctuations are exhibited by residues numbered 125–135 and 180–190 having RMSF values less than 1.2 Ǻ as shown in Fig. 8a–d. There are four types of protein–ligand interactions, hydrogen bonds, hydrophobic bonds, and ionic and water bridges. The stacked bar charts of the interactions with various residues of the binding site are normalized throughout the simulation. HIS86 shows hydrophobic, water bridge, and hydrogen bond, ARG83 and ARG87 show strong hydrogen interaction and water bridge interaction, HIS187 is hydrophobic, and the hydrogen bond and PHE131 show hydrophobic interaction with HEC ligand. In the case of the HEME ligand, the highest hydrophobic interaction was shown by PHE52, the highest H-bond interaction was shown by HIS187, and maximum water bridge was formed by GLY51. Very lowest interaction is shown by GLN47, ALA53, TYR58, ARG83, GLY135, and PHE189. Hydrogen bonds play an important role in drug specificity. The geometrical orientation of the hydrogen bond between the donor and acceptor atom should be 2.5 A and bond angle ≥ 120° between a donor and hydrogen atom, and the bond angle should be ≥ 90 between the acceptor and hydrogen atom. Hydrophobic interaction is divided into three sub-groups, namely π cation, π–π interaction, and some nonspecific interaction. The formation of π-cations, π–π interactions, and various nonspecific interactions are possible when aromatic and charged groups are brought together within 4.5 Ǻ; aromatic groups interact with one another face-to-face or edge-to-edge, or other nonspecific hydrophobic groups are brought together within 3.6 Ǻ. Within 3.7 Ǻ, two oppositely charged groups can interact ionically and by interactions with hydrogen-bonded protein ligands that are mediated by water molecules. All protein–ligand contact with 2d interaction is given in Fig. 9a–d. To assess conformational changes in HEC and HEM ligand–protein complexes further, we used gyration (Rg) analysis. The Rg is defined as the mass-weighted RMSD of a collection of atoms from their common center of mass. As a result, this research helps us understand the protein’s overall dimensions. In both the structures, the average radius of Rg decreases with simulation and finally attains the values 21 Å for the HEC complex and 22 Å for the HEM complex. The solvent-accessible surface area (SASA) is the center of the solvent molecule’s center as it rolls over a protein’s van der Waal surface. It is calculated using the “rolling ball” algorithm, which uses a sphere of solvent to probe the surface. The probe radius affects the observed surface area, with a 1.4 A probe determining the accessible molecular surface (Kumar et al. 2012). Understanding protein structure and function is crucial, and amino acid residues’ location influences protein folding and stability shown in Fig. 10a, b (Purohit et al. 2008). Additionally, interactions between additional residues in the binding pocket and conformational of the rotatable bond changes throughout the simulation are shown in Figure S1a–d, (Supplementary Information). As described in the literature (Bharadwaj et al. 2021), the free energy of binding of the protein–ligand complexes has been estimated using the MMGBSA method. The net binding free energy is calculated as the difference between the minimized complex’s free energies and the sum of ligand and receptor free energies. Even if more computationally challenging, the MMGBSA free energy estimations ordinarily provide a noteworthy correlation with that of experimentally observed activities (Lyne et al. 2006). The various energy terms in the MMGBSA calculations of both HEC and HEME ligands within the binding site in their best-docked poses are presented in Table 6.
Fig. 8.

RMSD of Cα atoms of protein and protein-fit ligands, and root-mean-square fluctuations (RMSF) of the protein residues of vp-HEC complex (a, b) and vp-HEME complex (c, d), respectively
Fig. 9.

Normalized protein–ligand interaction bar chart and ligand–protein docking after simulation of 100 ns where (a, b) protein–HEC ligand and (c, d) protein–HEME ligand
Fig. 10.

(a) Radius of gyration of HEC complex (in black color) and HEM complex (in red color). (b) Solvent-accessible surface area (SASA) of HEC-protein complex (in black color) and HEM protein complex structure complex (in red color)
Table 6.
Net free energy of binding (∆Gbind) and its various components, e.g., coulomb energy (∆Gbind_coulomb), lipophilic (∆Gbind_lipo), solvation (∆Gbind_solvant), and van der Waals (∆Gbind_vdw) energy terms as obtained through MMGBSA free energy calculations
| Ligand | ∆Gbind (kcal/mol) |
∆Gbind_coulomb (kcal/mol) |
∆Gbind_covalent (kcal/mol) |
∆Gbind_lipo (kcal/mol) |
∆Gbind_solvant (kcal/mol) |
∆Gbind_vdw (kcal/mol) |
|---|---|---|---|---|---|---|
| HEC | − 122.959 | − 68.253 | 5.661 | − 27.739 | 52.960 | − 69.575 |
| HEME | − 52.035 | − 21.118 | 14.484 | − 22.873 | 16.749 | − 37.338 |
Bioconversion reaction
To study the bioconversion degradation of β-naphthol, we preliminary studied the change in UV spectra in a 1 ml reaction mixture containing 5 mM substrate, 50 mM buffer, 0.5 mM H2O2, and 1.1 mg of VP enzyme at 0-, 30-, and 60-min time interval at room temperature. The spectrophotometric analysis of the reaction of β-naphthol treated with the VP enzyme in the presence of H2O2 revealed that this enzyme has the potential to break down them into distinct products. The intensity peak of 5 mM β-naphthol is observed at 2.6, and on adding the enzyme, it is reduced to 0.6. The peak of 1,2-naphthalene-dione at retention time 14.5–14.9 m/z value 158 and the peak of 2-hydroxycinnamic acid at retention time 18.5–18.8 m/z value 164 are displayed in Fig. 11a–c based on data from various literature surveys and the HPLC–MS library.
Fig. 11.

(a) Changes spectra of UV at 0-, 30-, and 60-min time intervals at room temperature (b) HPLC analysis of the degradation of β-naphthol of 5 mM (black line) and reaction mixture after 1 h (red line) (where, 1 ml reaction mixture contained 5 mM substrate, 50 mM buffer, 0.5 mM H2O2, and 1.1 mg of VP enzyme)
Discussion
Computational techniques have shown to be an excellent supplement to our knowledge of the protein universe and its characteristics. In silico study is considered one of the most advantageous methods for examining the structural and functional features of the protein and makes a significant contribution to computational biology. To assess the structural and functional features of plant versatile peroxidase enzymes, an investigation was carried out utilizing various computational methods, including ProtParam, SOPMA, pep-draw, SWISS-MODEL, I-TASSER, PROCHECK, ERRAT, and ProSA. The current work has comprised protein profiling, homology modeling, investigation of functionality, identification of functional domains, protein–ligand interaction, and bioconversion of toxic aromatic hydrocarbon. The additional information offered in this paper will advance proteomics research and the creation of new bioinformatics methods. The investigation of enzyme–ligand interactions will aid in the creation of an innovative approach to the active site of the VP enzyme. These findings will open the door to the use of oxidoreductase enzymes in genetic engineering, such as genetic mutation for the synthesis of synthetic proteins, biotechnological applications, and degradation study of toxic organic pollutants. Due to their superior efficiency in the bioconversion of inorganic and organic chemicals compared to conventional methods, plant-based oxidoreductase enzyme has drawn attention. These factors include their strong catalytic activity, high specificity, ease of storage, low cost, and ease of use. The current work defined the structural and functional elements and will confirm the amino acid sequence of VP extracted from citrus sinensis as a powerful biocatalyst for ecological remediation. The percentage of sequence identity between the target and templates has a significant impact on the accuracy that may be anticipated from homology modeling. A reliable structure is typically produced when there is more than 50% sequence identity. Every enzyme requires a specific pH to demonstrate its catalytic activity, and it is possible to determine a protein’s pH-dependent property and stability from its physicochemical properties. The secondary structure of a protein can be determined from patterns of amino acid profiles, protein families, and active sites. Finding out the peptide structure is very helpful for understanding the structure and characteristics of proteins and for identifying their functional characteristics. Based on the Ramachandran plot, ERRAT score, and Z-score from program ProSA, it can be concluded the 3D homology model of VP obtained was good. Gene ontology (GO) is a framework for understanding gene function in organisms, providing a comprehensive definition of protein function. It is used in bioinformatics to represent biological systems and establish connections between gene products and GO concepts (Basumatary et al. 2023; Takio et al. 2022). Without COFACTOR, some bioinformatics analysis tools can only predict monomers. However, there are several homology modeling limitations in the in silico investigation. Proteins cannot undergo post-translational modifications in the protParam tool; hence, ProtParam cannot tell whether your mature protein forms dimers or multimers. ProtParam bases every calculation on either compositional information or the N-terminal amino acid, and the SAVES server only maintains the results files for a job for one week due to the high volume of requests it receives from users around the world. Molecular dynamics simulations of vp-HEC and vp-HEME complexes reveal strong interactions with His187 residues, HEC ligands with ARG83, HIS86, and ARG87, and HEME ligands with MET54 and THR55 residues. Both compounds transit quickly to the binding site and remain stable, with HEC showing better binding capabilities. Between 0 and 5000 ps, major fluctuation has been noticed, and Rg represents the compactness of the complexes. A lesser value of Rg indicates a more stable complex. From the SASA data analysis, it is observed that with time the value of HEM complex SASA is greater than HEC complex. By observing SASA analysis, the enlargement of the HEC and HEM complex structure due to a change in flexibility was further confirmed (Singh et al. 2022; Kumar et al. 2012; Purohit et al. 2008). After observing binding energy, RMSD, RMSF, Rg, and SASA values, it was confirmed that HEC shows better binding capabilities with protein. In the field of environmentally friendly green chemistry, the VP enzyme isolated from citrus sinensis leaf extract is proven to be effective in the degradation of polyaromatic hydrocarbons. They have been employed to degrade thousands of resistant inorganic and organic contaminants, including phenols, amines, dyes, insecticides, and many more. Interaction with different organic and inorganic ligands with this VP protein is yet to be explored (Yadav et al. 2017a).
Conclusion
The fundamental facts of the similarity and preservation of VPs among the closely linked plant taxonomic groupings were discovered by the extremely detailed computer modeling of protein 3D structures. The interaction between the protein and the ligand focused on the functional residues that made up the (active sites) and a favorable docking pose with the least amount of binding energy was used. The various catalytic sites for potential modification for desirable features relevant to a variety of sectors could be disclosed by in silico predictions and validation of adaptable peroxidase three-dimensional structures. To increase productivity and efficiency, uncover new sources using a metagenomics technique, and try guided evolution to include desired functional traits, and the in silico study provides a strong platform for wet-lab operations. The analysis of new relevant proteins and complexes for more reliable and effective regulation of their functional activity can be done using high-resolution experimental data for protein–ligand interactions. The most stable conformation has been achieved through a protein–ligand interaction study, which can be explored for further study in catalytic processes. Molecular dynamics simulation of the two better-docked complexes VP-HEME C and VP-HEME have been carried out for 100 ns production runs. MD simulation shows that both the complexes VP-HEC and VP-HEME show interactions with His187 residue throughout the whole dynamics. The strong interactions are exhibited by the HEC ligand with the residues ARG83, HIS86, and ARG87. HEME ligands show prominent hydrogen bonding interactions with MET54 and THR55 residues. Based on RMSD, RMSF, and hydrogen bonding interactions during the entire dynamics and MMGBSA binding free energy estimations, it can be concluded that HEC and HEME both compounds transit quickly to the binding site and remain stable during the dynamics. However, compound HEC shows better binding capabilities as compared to HEME binding with the protein. As a result, it is possible to conclude that the only plant source versatile peroxidase isolated from citrus sinensis contains HEC (HEME C) ligand in its active site. In addition, the study’s findings could support future uses of these plant VP enzymes in the depolymerization of resistant lignin, and other degradation and wastewater treatment. It will be easier to extract from the plant than to handle microorganisms.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The Department of Chemistry and CRF of NERIST is well-acknowledged for providing the necessary facilities. I am very thankful for the GATE fellowship provided by NERIST
Abbreviations
- VP
Versatile peroxidase
- LiP
Lignin peroxidase
- MnP
Manganese peroxidase
- WRF
White rot fungus
- NCBI
National Center for Biotechnology Information
- BLAST
The Basic Local Alignment Search Tool
- FASTA
Fast alignment
- pI
Isoelectric point
- SOPMA
Self-optimized prediction method with alignment
- SAVESv6.0
Structure validation server
- PLIP
Protein–ligand interaction profiler
- STRING
Search tool for the retrieval of interacting genes/proteins
- Hh
Alpha helix
- Ee
Extended strand
- Tt
Beta-turn
- Cc
Random coil
- RMSD
Root-mean-square deviation
- RMSF
Root-mean-square fluctuation
- MMGBSA
Molecular mechanics with generalized born and surface area solvation
Authors’ contributions
The primary, secondary, and peptide structure analyses and modeling were completed and written manuscript by RAH. The data analysis and interpretation, responsibility, and accountability for the contents of the article were done by MY. MD Simulation and MMGBSA energy calculation has been done by UY and his student SN. NR has done the preliminary study of versatile peroxidase enzymes. HSY was credited for the methodology of the communicated work.
Funding
Not applicable.
Data availability
Data will be provided on request to corresponding author.
Declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Research involving human participants and/or animals
The research work does not involve any human or animal participants.
Informed consent
NA.
References
- Apweiler R, Bairoch A, Wu CH, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artimo P, Jonnalagedda M, Arnold K, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40:W597–W603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber-Zucker S, Mindel V, Garcia-Ruiz E, et al. Stable and functionally diverse versatile peroxidases designed directly from sequences. J Am Chem Soc. 2022;144:3564–3571. doi: 10.1021/jacs.1c12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basumatary D, Saikia S, Yadav HS, Yadav M. In silico analysis of peroxidase from Luffa acutangula. 3 Biotech. 2023;13:25. doi: 10.1007/s13205-022-03432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathula R, Lanka G, Muddagoni N, et al. Identification of potential Aurora kinase-C protein inhibitors: an amalgamation of energy minimization, virtual screening, prime MMGBSA and AutoDock. J Biomol Struct Dyn. 2019;2:2. doi: 10.1080/07391102.2019.1630318. [DOI] [PubMed] [Google Scholar]
- Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj S, Rao AK, Dwivedi VD, et al. Structure-based screening and validation of bioactive compounds as Zika virus methyltransferase (MTase) inhibitors through first-principle density functional theory, classical molecular simulation and QM/MM affinity estimation. J Biomol Struct Dyn. 2021;39:2338–2351. doi: 10.1080/07391102.2020.1747545. [DOI] [PubMed] [Google Scholar]
- Busse N, Wagner D, Kraume M, Czermak P. Reaction kinetics of versatile peroxidase for the degradation of lignin compounds. Am J Biochem Biotechnol. 2013;9:365–394. doi: 10.3844/ajbbsp.2013.365.394. [DOI] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:1–9. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarero S, Sarkar S, Ruiz-Dueñas FJ, et al. Description of a versatile peroxidase involved in the natural degradation of lignin that has both manganese peroxidase and lignin peroxidase substrate interaction sites. J Biol Chem. 1999;274:10324–10330. doi: 10.1074/jbc.274.15.10324. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen R, Yarden O, Hadar Y. Lignocellulose affects Mn2+ regulation of peroxidase transcript levels in solid-state cultures of Pleurotus ostreatus. Appl Environ Microbiol. 2002;68:3156–3158. doi: 10.1128/AEM.68.6.3156-3158.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli S, Huey R, Pique ME, et al. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. Proteom Protoc Handb. 2005;2:571–607. doi: 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]
- Guillén F, Evans CS. Anisaldehyde and veratraldehyde acting as redox cycling agents for H2O2 production by Pleurotus eryngii. Appl Environ Microbiol. 1994;60:2811–2817. doi: 10.1128/aem.60.8.2811-2817.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofrichter M. lignin conversion by manganese peroxidase (MnP) Enzyme Microb Technol. 2002;30:454–466. doi: 10.1016/S0141-0229(01)00528-2. [DOI] [Google Scholar]
- Kumar A, Rajendran V, Sethumadhavan R, Purohit R. In silico prediction of a disease-associated STIL mutant and its affect on the recruitment of centromere protein J (CENPJ) FEBS Open Bio. 2012;2:285–293. doi: 10.1016/j.fob.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Singh VK, Kayastha AM. Molecular modeling, docking and dynamics studies of fenugreek (Trigonella foenum-graecum) α-amylase. J Biomol Struct Dyn. 2022;2:1–16. doi: 10.1080/07391102.2022.2144458. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lee KE, Bharadwaj S, Sahoo AK, et al. Determination of tyrosinase-cyanidin-3-O-glucoside and (−/+)-catechin binding modes reveal mechanistic differences in tyrosinase inhibition. Sci Rep. 2021;11:24494. doi: 10.1038/s41598-021-03569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyne PD, Lamb ML, Saeh JC. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J Med Chem. 2006;49:4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- Perez-Boada M, Ruiz-Duenas FJ, Pogni R, et al. Versatile peroxidase oxidation of high redox potential aromatic compounds: site-directed mutagenesis, spectroscopic and crystallographic investigation of three long-range electron transfer pathways. J Mol Biol. 2005;354:385–402. doi: 10.1016/j.jmb.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Pradeep NV, Anupama AK, Pooja J. Categorizing phenomenal features of α-amylase (Bacillus species) using bioinformatic tools. Adv Lif Sci Technol. 2012;4:27–31. [Google Scholar]
- Pratama MRF, Siswandono S. Number of runs variations on Autodock 4 do not have a significant effect on RMSD from docking results. Pharm Pharmacol. 2020;8:476–480. doi: 10.19163/2307-9266-2020-8-6-476-480. [DOI] [Google Scholar]
- Purohit R, Rajasekaran R, Sudandiradoss C, et al. Studies on flexibility and binding affinity of Asp25 of HIV-1 protease mutants. Int J Biol Macromol. 2008;42:386–391. doi: 10.1016/j.ijbiomac.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Rai N, Yadav M, Yadav HS. Purification and characterization of versatile peroxidase from citrus sinensis leaf extract and its application in green chemistry. Anal Chem Lett. 2020;10:524–536. doi: 10.1080/22297928.2020.1833751. [DOI] [Google Scholar]
- Ramachandran GNT, Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283–437. doi: 10.1016/S0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- Ruiz-Duenas FJ, Morales M, Mate MJ, et al. Site-directed mutagenesis of the catalytic tryptophan environment in Pleurotus eryngii versatile peroxidase. Biochemistry. 2008;47:1685–1695. doi: 10.1021/bi7020298. [DOI] [PubMed] [Google Scholar]
- Ruiz-Dueñas FJ, Martínez ÁT. Microbial degradation of lignin: how a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microb Biotechnol. 2009;2:164–177. doi: 10.1111/j.1751-7915.2008.00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salame TM, Yarden O, Hadar Y. Pleurotus ostreatus manganese-dependent peroxidase silencing impairs decolourization of Orange II. Microb Biotechnol. 2010;3:93–106. doi: 10.1111/j.1751-7915.2009.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Bhardwaj VK, Purohit R. Computational targeting of allosteric site of MEK1 by quinoline-based molecules. Cell Biochem Funct. 2022;40:481–490. doi: 10.1002/cbf.3709. [DOI] [PubMed] [Google Scholar]
- Takio N, Yadav M, Yadav HS. In silico studies on catalases from plant sources. Authorea Prepr. 2022;2:2. [Google Scholar]
- Weber K, Osborn M. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J Biol Chem. 1969;244:4406–4412. doi: 10.1016/S0021-9258(18)94333-4. [DOI] [PubMed] [Google Scholar]
- White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta Reviews Biomembr. 1998;1376:339–352. doi: 10.1016/S0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- Yadav M, Rai N, Yadav HS. The role of peroxidase in the enzymatic oxidation of phenolic compounds to quinones from Luffa aegyptiaca (gourd) fruit juice. Green Chem Lett Rev. 2017;10:154–161. doi: 10.1080/17518253.2017.1336575. [DOI] [Google Scholar]
- Yadav M, Yadav S, Yadav D, Yadav KDS. In-silico analysis of manganese peroxidases from different fungal sources. Curr Proteomics. 2017;14:201–213. doi: 10.2174/1570164614666170203165022. [DOI] [Google Scholar]
- Yadava U, Yadav VK, Yadav RK. Novel anti-tubulin agents from plant and marine origins: Insight from a molecular modeling and dynamics study. RSC Adv. 2017;7:15917–15925. doi: 10.1039/C7RA00370F. [DOI] [Google Scholar]
- Yang J, Yan R, Roy A, et al. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be provided on request to corresponding author.