

SUMMARY
Ligand-to-metal charge transfer (LMCT) using stoichiometric copper salts has recently been shown to permit decarboxylative C–N bond formation via an LMCT/radical polar crossover (RPC) mechanism; however, this method is unable to function catalytically and cannot successfully engage unactivated alkyl carboxylic acids, presenting challenges to the general applicability of this approach. Leveraging the concepts of ligand-to-metal charge transfer (LMCT) and radical-ligand-transfer (RLT), we herein report the first photochemical, iron-catalyzed direct decarboxylative azidation. Simply irradiating an inexpensive iron nitrate catalyst in the presence of azidotrimethylsilane allows for a diverse array of carboxylic acids to be converted to corresponding organic azides directly with broad functional group tolerance and mild conditions. Intriguingly, no additional external oxidant is required for this reaction to proceed, simplifying the reaction protocol. Finally, mechanistic studies are consistent with a radical mechanism and suggest that the nitrate counteranion serves as an internal oxidant for turnover of the iron catalyst.
eTOC blurb
Decarboxylative azidation is a powerful approach to prepare carbon-nitrogen bonds in bioactive molecules. However, previous methods require strong oxidants, precious metals, complex reagents, and/or carboxylic acid pre-activation, limiting widespread adoption. We report a visible-light-induced, iron-catalyzed approach, leveraging ligand-to-metal charge tansfer (LMCT) and radical ligand transfer (RLT) for direct decarboxylative azidation both activated and unactived (alkyl) carboxylic acids Intriguingly, mechanistic results demonstrate the intrinsic oxidative ability of nitrate counterion allows the transformation to proceed catalytically without additional external oxidant.
Graphical Abstract
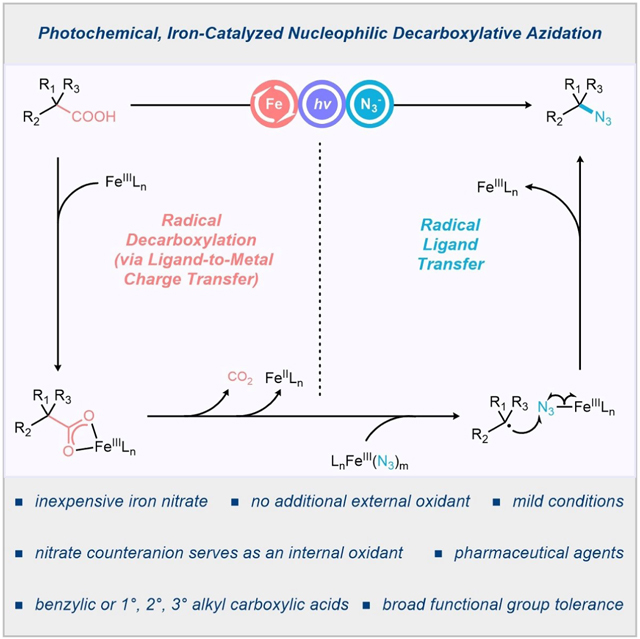
Carbon–Nitrogen bonds are among the most prevalent and important linkages in bioactive molecules; however, the direct use of common chemical feedstocks such as carboxylic acids to form C–N bonds remain challenging. Recent progress has shown ligand-to-metal charge transfer (LMCT) to be a powerful mechanism to activate carboxylic acids for subsequent reactions, including C–N bond formation; however, these methods are not catalytic, requiring stoichiometric metal to proceed, and unable to engage unactivated carboxylic acids. Here we show simple iron salts can photocatalyze decarboxylative C–N bond formation for a wide variety of activated and unactivated acids using nitrate as a terminal oxidant. Preliminary studies suggest this reaction performs radical ligand transfer (RLT) as the C–N bond forming step, revealing a new approach
INTRODUCTION
C–N bonds are some of the most ubiquitous linkages in pharmaceutical science, with the average small molecule pharmaceutical agent containing 2.3 nitrogen atoms1 and C–N bond-forming Buchwald-Hartwig couplings appearing in 8%2 of reported medicinal chemistry routes (Figure 1, Panel A). Thus, there is strong demand for new methods for C–N bond formation, particularly those arising from orthogonal precursors to the Csp2–X electrophiles required for cross coupling with nitrogenous fragments.
Figure 1. Importance and Approaches to Decarboxylative C–N Bond Formation.

Yoon has recently disclosed an approach to decarboxylative C–N bond formation of activated carboxylic acids using tandem ligand-to-metal charge transfer (LMCT)/radical polar crossover (RPC) using stoichiometric copper, whereas our approach to decarboxylative C–N bond formation via tandem LMCT/radical ligand transfer (RLT) using catalytic iron.
A recent and powerful approach to C–N bond formation was demonstrated by Yoon and coworkers, where carboxylic acids and N-containing functional groups such as sulfonamides, carbonates, and amides can be coupled in a light-driven decarboxylative coupling reaction using copper as a mediator (Figure 1, Panel B).3 This intriguing reaction relies on the ability of copper(II) carboxylates to undergo a ligand-to-metal charge transfer (LMCT) reaction upon photoexcitation, resulting in formation of a carboxylic radical intermediate and a reduced copper(I) complex.4–9 The carboxylic radical can then rapidly decarboxylate to generate a carbon-centered radical which, in the presence of excess copper(II) species, can undergo an oxidative radical-polar crossover (RPC) to furnish a carbocation intermediate which can be trapped by the nitrogen-containing nucleophilic fragment to form the final C–N coupled product.10 This tandem LMCT/RPC approach is able to form a wide variety of functionalized products under mild, light-driven conditions and is an invaluable addition to synthetic strategy. However, there remain several challenges for LMCT-mediated C–N bond formation.
The first remaining challenge for C–N bond formation via LMCT is the absence of catalytic methods. The Yoon LMCT/RPC approach is overall oxidative, requiring two single electron oxidations by copper(II) species in order to generate product. As no suitable terminal oxidant has been found to regenerate the active copper(II) state, two equivalents of copper(II) salt are required for the reaction to proceed. A second outstanding challenge is engaging substrates unable to form stabilized carbocations via RPC oxidation. The LMCT/RPC decarboxylative C–N coupling is most successful with aryl acetic acids capable of forming benzylic carbocations stable enough to be engaged via intermolecular nucleophilic attack in preference to unimolecular elimination to form alkenes. Despite efforts to engage aliphatic, unactivated carboxylic acids in this reaction, alkenes were the sole observed products.11 1-Adamantane carboxylic acid was the one exception to this trend; however, the inability of the adamantyl 3° cation to undergo elimination via geometric constraints represents a special case that is not generalizable.
Contemplating these two challenges, we wondered if judicious redesign of the reaction approach might permit catalytic C–N bond formation via an LMCT manifold. First, we theorized that we might be able to regenerate the active LMCT complex by identifying a chemoselective oxidant which would not interfere with either the LMCT or C–N bond forming step. Further, it would be ideal if this oxidant were cheap, earth abundant, and simple to separate both it and its reduction products from the reaction mixture. Second, we hypothesized that non-adamantyl unactivated carboxylic acids might be able to react productively by avoiding the RPC elementary step, impeding alkene formation though avoiding explicit radical oxidation.10 Our recent work in radical alkene difunctionalization suggested that radical ligand transfer (RLT) might be a viable alternative to RPC, allowing for a nucleophilic nitrogen-containing ligand of an earth abundant transition metal complex to be directly transferred to the radical intermediate without direct radical oxidation.12 However, similar to LMCT and RPC, RLT results in single electron reduction of the transition metal complex, requiring a suitable oxidant to regenerate the active ligand transfer complex after the reaction.
Toward implementing these two changes and realizing a photocatalytic C–N bond formation, we envisioned an iron photocatalyzed decarboxylative azidation reaction. Iron has recently been demonstrated to perform LMCT chemistry with a variety of anionic ligands, including carboxylic acids.6,13–18 Intriguingly, iron is also known to undergo RLT chemistry readily, with the “radical rebound” step of cytochrome P450 being a hydroxyl RLT.19–22 Thus, we faced the tantalizing possibility of using iron for both LMCT and RLT steps, dramatically simplifying an otherwise complex cooperative catalytic reaction. However, RLT reactivity is most established and general for simple anionic ligands, including azide, leading us to target azide as the nitrogen-atom donor in this reaction.12, 23–27 Further, decarboxylative azidation has been demonstrated to be a valuable transformation in its own right; however, current methods require strong oxidants, precious metals, complex reagents, and/or carboxylic acid pre-activation, presenting significant barriers to their widespread adoption.28–32 Finally we wondered if nitrate, a weakly-coordinating anion that contains nitrogen in the +5 oxidation state, might be able to serve as a selective terminal oxidant in light of recent examples of its oxidation ability in transition metal catalysis.33,34 Using the nitrate counteranion would have the added benefit of permitting the use of iron(III) nitrate, a cheap, commodity chemical, as our as both our iron and oxidant source.
Putting this design into practice, we report the first photochemical, nucleophilic decarboxylative azidation using tandem iron-catalyzed LMCT/RLT (Figure 1, Panel C). This conceptually-novel design provides hitherto the mildest and simplest conditions to access diverse range of aliphatic azides selectively. Without complex ligand design, expensive radical trapping agents or redundant pre-activation of carboxylic acids, this photochemical, decarboxylative azidation proceeds smoothly with only iron salt and nucleophilic azide source! Importantly, the protocol doesn’t require external oxidant, possibly due to intrinsic oxidative ability of nitrate counterion on the catalyst,35 which allows the transformation to proceed with only catalytic amount of iron salt, providing a ‘redox-neutral’ transformation. Further, simple aliphatic carboxylic acids can be engaged in this chemistry; however, their reactivity is lower than that of activated carboxylic acids.
RESULTS
Optimization of reaction conditions
Initially, we investigated the feasibility of this nucleophilic decarboxylative azidation induced by iron-mediated ligand-to-metal charge transfer (LMCT) decarboxylation and radical ligand-transfer (RLT) by using 2-phenylpropanoic acid as a model substrate, nucleophilic trimethylsilyl azide (TMSN3) as the azide source, and stoichiometric iron nitrate under the irradiation of 390 nm purple light at room temperature. This preliminary test successfully delivered the corresponding azide product 1 in 80% yield, exhibiting the dual-role of the iron nitrate in this transformation (entry 1, Figure 2). After confirming the reactivity of iron nitrate, we attempted to evaluate the possibility of performing this reaction in catalytic conditions. To our delight, reducing the iron nitrate loading to 20 mol% did not significantly impact the yield of azide 1 (76%, entry 2, Figure 2), clearly demonstrating catalysis with respect to iron. However, lower loadings (10 mol%) of iron gave the desired product in still catalytic, albeit lower yield (28%, entry 3, Figure 2). To test the role of nitrate anion in catalysis, a variety of non-nitrate-containing iron salts were tested and found to possess stoichiometric (or less) reactivity (entry 4–7, Figure 2). Having ruled out alternative iron salts, we next tested whether sodium azide could serve as a suitable nitrogen source in our catalytic reaction finding that product 1 was produced in lower, albeit still catalytic, quantities (44%, entry 8, Figure 2), potentially due to the reduced solubility of sodium azide. In an attempt to increase the conversion of the former conditions with acetonitrile/water co-solvent system improving the solubility of NaN3 in this reaction resulted in poor yield potentially due to the excess water inhibiting the reaction (12%, entry 9, Figure 2). Intriguingly, reducing the loading of TMSN3 to a single equivalent drastically hampered the reaction and resulted in a significantly lower yield (24%, entry 10, Figure 2), potentially due to RLT occurring from a polyazide species as implicated in olefin diazidation.27,36–37 We also observed a strong countercation effect of the base included in this reaction (entries 11–13, Figure 2), with Cs2CO3 unable to promote the reaction (entries 13, Figure 2) and both K2CO3 and Li2CO3 able to form azidated product 1, albeit with lower yields (entries 11–12, Figure 2) compared to Na2CO3. The absence of base somewhat reduces the yield of this reaction, suggesting that the formation of carboxylate may be relevant for the reaction (entry 14, Figure 2). Further screening on concentration showed the reaction to be slightly sensitive to dilution (entry 15, Figure 2). Control reactions revealed both iron and light irradiation are required, with full recovery of starting material in the absence of either, providing evidence for the photoreactivity of iron (entries 16–18, Figure 2).
Figure 2. Development of Iron-Photocatalyzed Decarboxylative Azidation.
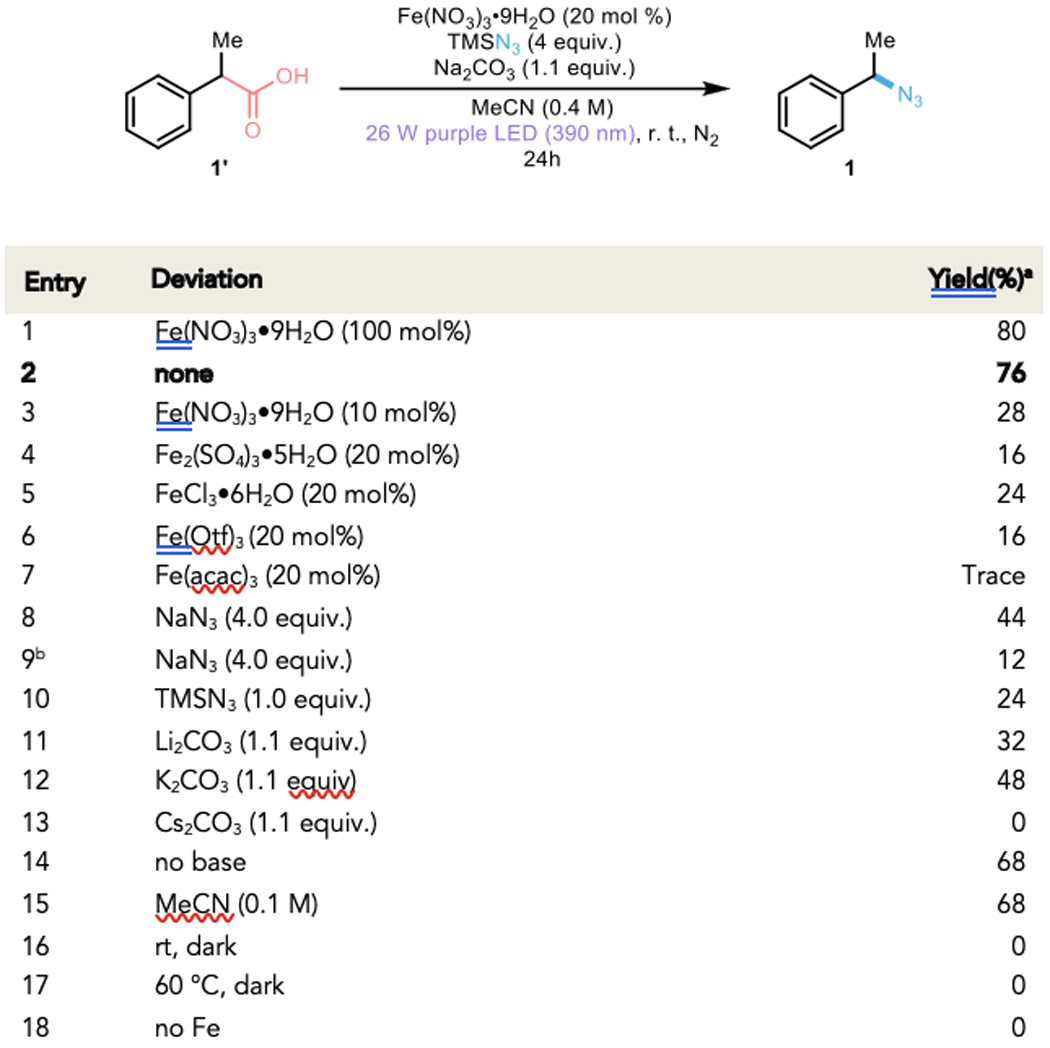
a Reactions performed on 0.1 mmol scale with CH2Br2 added as an internal standard (NMR yield). b MeCN/H2O=4/1 co-solvent system was used.
Substrate scope exploration
With the optimized conditions in hand, we next sought to assess the scope of this novel decarboxylative azidation method (Figure 3). First, arylacetic acids with different steric hindrance level of substituents from mono-alkylated (1’, 4’−5’) acids to highly hindered tertiary acids (3’, 6’) all performed well, affording desired products in yields from 64–77%. These results suggest that the azido delivery via Fe-N3 species is relatively insensitive to steric effects. Diphenylacetic acid (8’) undergoes smooth azidation with 30 mol% of iron salt under light irradiation to give 53% yield of azido product. To our delight, this protocol offers a direct way to transform a broad range of primary arylacetic acids where phenyl substituents bear functional groups such as electron-donating -OH (9’), -OMe (10’), -NH2 (11’), and -NHBz (12’) or bulky mesityl (7’) or electron-withdrawing difluoro (13’), dichloro (14’), bromo (15’), trifluoromethyl (16’), and methylsulfonyl (17’) into corresponding primary azides in moderate to good yields. Intriguingly, unprotected phenol and to a lesser extent aniline carboxylic acids leading products 9 and 11 are compatible with these reaction conditions and not with the LMCT/RPC protocol, demonstrating the complementarity of this approach.3 Aniline product 11, sulfonamide product 12, and dihydrobenzofuran product 18 are formed in reduced yield compared to other arylacetic acids. We hypothesize that the more strongly coordinating moieties in these species impede the coordination of azide and carboxylic acid to generate our key LMCT and RLT complexes, preventing efficient reaction. Next, we explored the ability of our protocol to directly azidate more challenging aliphatic carboxylic acids (19’−22’), a substrate class that was incompatible with the copper(II)-mediated LMCT/RPC protocol.3 First, the transformation of lauric acid (19’) into desired product was observed, though in low yield. Importantly, the remaining mass balance for this reaction is unreacted starting material, suggesting the low yield is due to reduced reactivity and not intrinsic incompatibility. Two secondary carboxylic acids (20’, 21’) were also tolerated, providing corresponding products in moderate yield, again with the mass balance being unreacted starting material. Adamantane carboxylic acid (22’) was also found to be compatible with our protocol; however, its limited solubility in acetonitrile may have significantly hamstrung its yield. To improve the yields in aliphatic carboxylic acids, we next sought to screen a series of nitrate sources (see supporting information for more details). To our delight, we found unactivated compounds 19-22 were able to be synthesized in higher yields with magnesium nitrate as an additive; however, the primary lauric acid (19’) still exhibited low overall reactivity. Encouraged by the performance of our photocatalytic decarboxylative azidation protocol, we next endeavored to explore a different array of commercially-available active pharmaceutical ingredients (APIs). First, we derivatized several APIs including Ibuprofen (24’), Fluribiprofen (25’), Loxoprofen (26’), Naproxen (27’), and Zaltoprofen (29’) into their corresponding azidated products in moderate to good yields. Importantly, this strategy is able to be utilized for the late-stage derivatization of the aliphatic carboxylic acid Gemfibrozil (28’), to give compound (28) in moderate yield, again demonstrating the ability of our photocatalytic approach to selectively form C–N bonds over elimination to form alkenes. Interestingly, our updated protocol for challenging aliphatic carboxylic acids worked well to enable compound (28) to be afforded in 71% yield. Additionally, the heterocyclic substrate N-Boc-protected proline proceeded smoothly to the α-azido product (23’) in 57% yield. Finally, Indomethacin (30’), a derivative of indole-3-acetic acid (IAA, 3-IAA), the most common naturally occurring plant hormone of the auxin class, was also tolerated under less base, delivering 3-azidomethylated indole compound (30) in 33% yield. This compound can be readily derivatized into a novel triazole product (36) via Huisgen cycloaddition and other functional groups as well (see Supporting Information (SI) for more details). The simplicity of this method combined with the low cost, earth-abundance, and low toxicity of iron and the use of nitrate as a simple, included external oxidant, makes this method a mild, economical and straightforward approach to transform both activated and unactivated carboxylic acids into azide compounds on both feedstock and complex molecules.
Figure 3. Scope of photocatalytic decarboxylative azidation of carboxylic acidsa.

aStandard conditions: Substrate (0.1 mmol), Fe(NO3)3·9H2O (0.02 mmol), TMSN3 (0.4 mmol), Na2CO3 (1.1 mmol), MeCN (0.25 mL), 390 nm LED light, rt, 24 h. bCH2Br2 was added as an internal standard (NMR yield).
cFe(NO3)3·9H2O (0.03 mmol) was used.
dNa2CO3 (0.04 mmol) was used.
eReaction performed on 0.3 mmol scale with (2 X 52W) purple LEDs (390 nm).
fReaction was performed under the following conditions: Substrate (0.1 mmol), Fe(NO3)3·9H2O (0.03 mmol), TMSN3 (0.4 mmol), Na2CO3 (0.02 mmol), MeCN/DCM=1/1 (0.25 mL), Mg(NO3)2·6H2O (0.1 mmol), 390 nm LED light, rt, 24 h. NMR yield is in the parentheses (CH2Br2 as an internal standard).
gsame conditions as f but MeCN (0.25 mL) was used. NMR yield is in the parentheses (CH2Br2 as an internal standard).
hsame conditions as f but Fe(NO3)3·9H2O (0.02 mmol) was used. NMR yield is in the parentheses (CH2Br2 as an internal standard).
iReaction performed on 500 mg scale and yield is in the parentheses.
j Same conditions as f but Fe(NO3)3·9H2O (0.02 mmol) and MeCN (0.25 mL) were used. NMR yield is in the parentheses (CH2Br2 as an internal standard).
Mechanistic Studies and Possible Mechanism
To acquire preliminary mechanistic insights into the photocatalytic decarboxylative azidation, a series of mechanistic experiments were performed (Figure 4). First, the sodium salt of substrate 1’ was subjected to the standard reaction conditions (Figure 4, Panel A1). With no acidic proton, the desired azidated product was accessed in decent yield, ruling out a HAT- decarboxylation process initiated by free azido radical.38 Next, addition of 1.0 equivalent of radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) in the system completely suppressed the reaction and TEMPO adduct 34 was afforded in 31%, strongly indicating the intermediacy of a carbon-centered radical in our reaction (Figure 4, Panel A2). With the scavenger result in hand, we next sought to provide more experimental support for nitrate counteranion as an internal oxidant in this reaction. Instead of using Fe(NO3)3·9H2O, FeCl3·6H2O was employed due to its stoichiometric reactivity in our initial screen, as well as to exclude internal oxidative counteranion (Figure 4, Panel A3-i). Intriguingly, by treating this reaction with 1.0 equivalent of NaNO3 as external oxidant to mimic the standard conditions, the yield of desired decarboxylative azide product increased from 24% to 60%, largely restoring the activity of the iron. Next, to investigate the photochemical oxidation ability of nitrate anion at ambient temperature, FeCl2 was utilized as the iron source in our conditions (Figure 4, Panel A3-ii). The yield of desired decarboxylative azide product was provided in 58% yield under these species, suggesting that active ferric species needed for dearboxylation might be generated first via an internal redox process of ferrous species with nitrate anion under the reaction conditions. From these results and previous studies where the redox ability of nitrate and nitrite were shown, we postulate the nitrate counteranion in Fe(NO3)3·9H2O also serves as the terminal oxidant in this catalytic decarboxylative azidation. In order to explore the possibility of a radical-polar crossover (RPC) mechanism in analogy to the work of Yoon, we designed substrate (31’) with an adjacent tert-butyl group and substrate (32’), a primary carboxylic acid with an adjacent cumyl group, which might be able to undergo ‘1,2-methyl shift’ upon the generation of adjacent carbon cation.3 In both cases, we might expect to see some rearranged azide if a discrete carbocation was formed prior to azide capture. However, unrearranged azide 31 and 32 were obtained in 79% yield and 9%, respectively, without the observation of the carbocation rearrangement product, suggesting that RPC is less likely. This result is in line with the azide being delivered via radical ligand transfer (RLT) from an iron-azide species (Figure 4, Panel A4-i,-ii). This result is consistent with the formation of terminal azidation product 19 with no rearrangement (Figure 3). We next sought to explore the rate of eventual radical ligand-transfer (RLT) process using cyclopropyl-containing radical clock substrates (33’). Subjecting this substrate to the reaction conditions resulted in generation of ring-opened product 33 in 20% yield. As only rearrangement product and no terminal azidated product were found, we postulate that iron-catalyzed, azido-ligand-transfer takes place after migrational ring-opening, meaning that the RLT rate must be slower than the rate constant of 1.8 × 1011 s−1 (approx. for the ring-opening of (2-phenylcyclopropyl)carbinyl radical) in our system (Figure 4, Panel A5).39 The UV−vis absorption spectra (Figure 4, Panel B) show that iron nitrate, substrate, or TMSN3 separately have optical absorption in the UV-B range. Interestingly, addition of substrate or TMSN3 to the Fe(III)-containing MeCN solution results in substantial changes to the absorbance spectrum (a mild absorbance 340 – 350 nm appears in each case, a mild absorbance 420–430 nm appears in the former case, and a broad, weak absorbance 460–480 nm appears in the latter case), indicating the formation of iron(III) carboxylates40–42 and iron(III) azides.43–46 A mixture of iron nitrate, substrate, and TMSN3 exhibits a similar, but shifting band near 350 nm compared to the spectra of iron(III) carboxylates and iron(III) azides. This new feature suggests that an azido-iron carboxylate species might be forming in our reaction. Importantly, the absorbance between 340 and 380 nm slightly overlaps with the emission of the purple LED lamps used in our process, consistent with excitation of the Iron(III) carboxylates under our reaction conditions. After these substrate and UV-vis studies, we next performed variable time normalization analysis (VTNA), a form of visual kinetic analysis, to gain more insights into the kinetic character of this reaction (see supporting information for more details).47–48 The reaction was found to exhibit positive, approximately first-order dependence on iron, first-order dependence on the aliphatic carboxylic acid, zeroth-order dependence on nitrate oxidant, and zeroth-order dependence on azide source under our reaction conditions. Additionally, we observed a slower reaction profile compared to the standard reaction with weaker intensity of light (25%W, 50% intensity corresponding to ~50% reactivity, see supporting information for more details), suggesting that the iron-catalyzed decarboxylation might be a light-dependence process. Together, these results are consistent with the first LMCT decarboxylation being the rate-determining step (RDS) while RLT should be readily effected once the alkyl radical is generated.
Figure 4. Mechanistic studies on the photocatalytic decarboxylative azidation.

a NMR yield with CH2Br2 added as internal standard. b yield without NaNO3 as additive (Figure 2, entry 1). cReaction performed on a 0.26 mmol scale.
Based on these preliminary mechanistic experiments and prior literature studies,32–34, 37we propose a possible mechanism for the iron-mediated photochemical nucleophilic decarboxylative azidation (Figure 5). Initial deprotonation of substrate I allows the coordination of the carboxylate intermediate to dissolved iron salt, generating an iron carboxylate species II,14,15 which can be homolyzed via photoinduced LMCT process to afford carboxylic radical III (Step A) and release reduced iron (II) species. Based on the preliminary kinetic data, we propose that this homolysis is the rate limiting step in our standard conditions. The generated intermediate III can then undergo facile decarboxylation (Step B) to form carbon-centered radical intermediate IV, which is sequestered by reactive iron-azide species via a Radical-Ligand-Transfer (RLT) process to deliver azidated product V (Step C) and another equivalent of reduced iron (II) species. Finally, both released iron (II) species from the steps above could be oxidized to iron (III) species by nitrogen oxide species derived from the nitrate counterion (Step D).
Figure 5. Proposed Mechanism.

Conclusions
In summary, we have developed the first photocatalytic, nucleophilic decarboxylative azidation of aliphatic carboxylic acids using a ligand-to-metal-charge-transfer (LMCT) manifold. Importantly, avoidance of an explicit radical-polar crossover (RPC) step allows for non-benzylic unactivated carboxylic acids, especially secondary and tertiary carboxylic acids, to be engaged in decarboxylative azide formation, providing a new protocol employing nucleophilic azide as a N-source for decarboxylative C-N bond formation in LMCT methods. This method takes advantage of the ability of iron to serve as both a LMCT and radical ligand transfer (RLT) species and represents an intriguing example of the nitrate anion as a cheap and selective terminal oxidant. Together, this method allows for C–N bonds to be formed selectively under mild photocatalytic conditions and provides useful lessons for design of future catalytic decarboxylative functionalization reactions using LMCT.
EXPERIMENTAL PROCEDURES
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Julian G. West (jgwest@rice.edu).
Materials availability
All other data supporting the findings of this study are available within the article and the supplemental information or from the lead contact upon reasonable request.
Data and code availability
There is no dataset or code associated with this publication. Full experimental procedures and experimental data are provided in the supplemental information.
Supplementary Material
Hightlights.
The first nucleophilic decarboxylative azidation via earth abundant iron catalysis
Direct decarboxylation of unactivated alkyl carboxylic acids to obtain desired azides
Reaction performed with without additional external oxidant
Nitrate counteranion functions as a cheap and selective terminal oxidant
ACKNOWLEDGMENTS
We acknowledge financial support from CPRIT (RR190025), NIH (R35GM142738), and the Welch Foundation (C-2085). J.G.W. is a CPRIT Scholar in Cancer Research. Dr. Yohannes H. Rezenom (TAMU/LBMS), Dr. Ian M Riddington (UT Austin Mass Spectrometry Facility) and Dr. Christopher L.
Pennington (Rice University Mass Spectrometry Facility) are acknowledged for assistance with mass spectrometry analysis.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.checat.2023.100603.
DECLARATION OF INTERESTS
This section is required for all research articles, resources, reviews, and perspectives. Please use it to disclose any competing interests in accordance with Cell Press’s Declaration of Interests policy. If there are no interests to declare, please write, “The authors declare no competing interests.” The text in this section should match the text provided in the Declaration of Interests form.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Vitaku E, Smith DT, and Njardarson JT (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 2.Brown D. and Boström J. (2016) Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 59, 4443–4458. 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- 3.Li QY, Gockel SN, Lutovsky GA, DeGlopper KS, Baldwin NJ, Bundesmann MW, Tucker JW, Bagley SW, and Yoon TP (2022). Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of Cu(ii) carboxylates. Nature Chemistry 14, 94–99. 10.1038/s41557-02100834-8. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juliá F. Ligand-to-Metal Charge Transfer (LMCT) Photochemistry at 3d-Metal Complexes: An Emerging Tool for Sustainable Organic Synthesis. ChemCatChem n/a, e202200916. 10.1002/cctc.202200916. [DOI]
- 5.Hossain A, Bhattacharyya A, and Reiser O. (2019) Copper’s rapid ascent in visible-light photoredox catalysis. Science 364, eaav9713. 10.1126/science.aav9713. [DOI] [PubMed] [Google Scholar]
- 6.Abderrazak Y, Bhattacharyya A, and Reiser O. (2021) Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis. Angew. Chem. Int. Ed 60, 21100–21115. 10.1002/anie.202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu P, López-Rojas P, and Ritter T. (2021) Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination. J. Am. Chem. Soc 143, 5349–5354. 10.1021/jacs.1c02490 [DOI] [PubMed] [Google Scholar]
- 8.Su W, Xu P, and Ritter T. (2021) Decarboxylative Hydroxylation of Benzoic Acids. Angew. Chem. Int. Ed 60, 24012–24017. 10.1002/anie.202108971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen TQ, Pedersen PS, Dow NW, Fayad R, Hauke CE, Rosko MC, Danilov EO, Blakemore DC, Dechert-Schmitt A-M, Knauber T, et al. (2022). A Unified Approach to Decarboxylative Halogenation of (Hetero)aryl Carboxylic Acids. J. Am. Chem. Soc 144, 8296–8305. 10.1021/jacs.2c02392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S, Singh J, and Sharma A. (2021). Visible Light Assisted Radical-Polar/Polar-Radical Crossover Reactions in Organic Synthesis. Adv. Synth. Catal 363, 3146–3169. 10.1002/adsc.202100205. [DOI] [Google Scholar]
- 11.Bacha JD, and Kochi JK (1968). Alkenes from acids by oxidative decarboxylation. Tetrahedron 24, 2215–2226. 10.1016/00404020(68)88124-4. [DOI] [Google Scholar]
- 12.Bian K-J, Nemoto D, Kao S-C, He Y, Li Y, Wang X-S, and West JG (2022). Modular Difunctionalization of Unactivated Alkenes through Bio-Inspired Radical Ligand Transfer Catalysis. Journal of the American Chemical Society 144, 1181011821. 10.1021/jacs.2c04188. [DOI] [PubMed] [Google Scholar]
- 13.Akira S, and Tetsuo Y. (1986). VISIBLE LIGHT- AND GAMMA RAY-INDUCED ALKYLATION IN PYRIDINE RING. EFFECTIVE ALKYLATION WITH VISIBLE LIGHT IN THE PRESENCE OF IRON(III) SULFATE. Chemistry Letters 15, 409–412. 10.1246/cl.1986.409. [DOI] [Google Scholar]
- 14.Feng G, Wang X, and Jin J. (2019). Decarboxylative C–C and C–N Bond Formation by Ligand-Accelerated Iron Photocatalysis. European Journal of Organic Chemistry 2019, 6728–6732. 10.1002/ejoc.201901381. [DOI] [Google Scholar]
- 15.Li Z, Wang X, Xia S, and Jin J. (2019). Ligand-Accelerated Iron Photocatalysis Enabling Decarboxylative Alkylation of Heteroarenes. Organic Letters 21, 4259–4265. 10.1021/acs.orglett.9b01439. [DOI] [PubMed] [Google Scholar]
- 16.Xia S, Hu K, Lei C, and Jin J. (2020). Intramolecular Aromatic C–H Acyloxylation Enabled by Iron Photocatalysis. Organic Letters 22, 1385–1389. 10.1021/acs.orglett.0c00002. [DOI] [PubMed] [Google Scholar]
- 17.Kang YC, Treacy SM, and Rovis T. (2021). Iron-Catalyzed Photoinduced LMCT: A 1° C–H Abstraction Enables Skeletal Rearrangements and C(sp3)–H Alkylation. ACS Catal. 11, 7442–7449. 10.1021/acscatal.1c02285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang YC, Treacy SM, and Rovis T. (2021). Iron-Catalyzed C(sp3)–H Alkylation through Ligand-to-Metal Charge Transfer. Synlett 32, 1767–1771. 10.1055/s-0040-1720388. [DOI] [Google Scholar]
- 19.Meunier B. (1992). Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chemical Reviews 92, 1411–1456. 10.1021/cr00014a008. [DOI] [Google Scholar]
- 20.Groves JT (2014). Using push to get pull. Nature Chemistry 6, 8991. 10.1038/nchem.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang X, and Groves JT (2017). Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C–H activation. J. Biol. Inorg. Chem 22, 185–207. 10.1007/s00775-0161414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Que L. (2007). The road to nonheme oxoferryls and beyond. Acc. Chem. Res 40, 493–500. 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 23.Ge L, Chiou M-F, Li Y, and Bao H. (2020). Radical azidation as a means of constructing C(sp3)-N3 bonds. Green Synth. Catal 1, 86–120. 10.1016/j.gresc.2020.07.001. [DOI] [Google Scholar]
- 24.Huang X, Bergsten TM, and Groves JT (2015). Manganese-catalyzed late-stage aliphatic C-H azidation. J. Am. Chem. Soc 137, 5300–5303. 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]
- 25.Fu N, Sauer GS, Saha A, Loo A, and Lin S. (2017). Metal-catalyzed electrochemical diazidation of alkenes. Science 357, 575–579. 10.1126/science.aan6206. [DOI] [PubMed] [Google Scholar]
- 26.Rui J, Zhao Q, Huls AJ, Soler J, Paris JC, Chen Z, Reshetnikov V, Yang Y, Guo Y, Garcia-Borràs M, et al. (2022). Directed evolution of nonheme iron enzymes to access abiological radical-relay C(sp3)−H azidation. Science 376, 869–874. 10.1126/science.abj2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan Y-A, Lu D-F, Chen Y-R, and Xu H. (2016). Iron-Catalyzed Direct Diazidation for a Broad Range of Olefins. Angew. Chem. Int. Ed Engl 55, 534–538. 10.1002/anie.201507550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nyfeler E, and Renaud P. (2008). Decarboxylative Radical Azidation Using MPDOC and MMDOC Esters. Organic Letters 10, 985–988. 10.1021/ol702832x. [DOI] [PubMed] [Google Scholar]
- 29.Liu C, Wang X, Li Z, Cui L, and Li C. (2015). Silver-Catalyzed Decarboxylative Radical Azidation of Aliphatic Carboxylic Acids in Aqueous Solution. Journal of the American Chemical Society 137, 9820–9823. 10.1021/jacs.5b06821. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Y, Li X, Wang X, Huang X, Shen T, Zhang Y, Sun X, Zou M, Song S, and Jiao N. (2015). Silver-Catalyzed Decarboxylative Azidation of Aliphatic Carboxylic Acids. Organic Letters 17, 4702–4705. 10.1021/acs.orglett.5b02155. [DOI] [PubMed] [Google Scholar]
- 31.Marcote DC, Street-Jeakings R, Dauncey E, Douglas JJ, Ruffoni A, and Leonori D. (2019). Photoinduced decarboxylative azidation of cyclic amino acids. Organic & Biomolecular Chemistry 17, 1839–1842. 10.1039/C8OB02702A. [DOI] [PubMed] [Google Scholar]
- 32.Wang K, Li Y, Li X, Li D, and Bao H. (2021). Iron-Catalyzed Asymmetric Decarboxylative Azidation. Organic Letters 23, 8847–8851. 10.1021/acs.orglett.1c03355. [DOI] [PubMed] [Google Scholar]
- 33.Stowers KJ, Kubota A, and Sanford MS (2012). Nitrate as a redox co-catalyst for the aerobic Pd-catalyzed oxidation of unactivated sp3-C–H bonds. Chemical Science 3, 3192–3195. 10.1039/C2SC20800H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenzel MN, Owens PK, Bray JTW, Lynam JM, Aguiar PM, Reed C, Lee JD, Hamilton JF, Whitwood AC, and Fairlamb IJS (2017). Redox Couple Involving NOx in Aerobic Pd-Catalyzed Oxidation of sp3-C–H Bonds: Direct Evidence for Pd– NO3–/NO2– Interactions Involved in Oxidation and Reductive Elimination. Journal of the American Chemical Society 139, 1177–1190. 10.1021/jacs.6b10853. [DOI] [PubMed] [Google Scholar]
- 35.Neidig ML, Carpenter SH, Curran DJ, DeMuth JC, Fleischauer VE, Iannuzzi TE, Neate PGN, Sears JD, and Wolford NJ (2019). Development and Evolution of Mechanistic Understanding in Iron-Catalyzed Cross-Coupling. Accounts of Chemical Research 52, 140–150. 10.1021/acs.accounts.8b00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lv D, Sun Q, Zhou H, Ge L, Qu Y, Li T, Ma X, Li Y, and Bao H. (2021). Iron-Catalyzed Radical Asymmetric Aminoazidation and Diazidation of Styrenes. Angew. Chem. Int. Ed 60, 12455–12460. 10.1002/anie.202017175. [DOI] [PubMed] [Google Scholar]
- 37.Zhu C-L, Wang C, Qin Q-X, Yruegas S, Martin CD, and Xu H. (2018). Iron(II)-Catalyzed Azidotrifluoromethylation of Olefins and N-Heterocycles for Expedient Vicinal Trifluoromethyl Amine Synthesis. ACS Catalysis 8, 5032–5037. 10.1021/acscatal.8b01253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Hu X, Morales-Rivera CA, Li G-X, Huang X, He G, Liu P, and Chen G. (2018). Epimerization of Tertiary Carbon Centers via Reversible Radical Cleavage of Unactivated C(sp3)–H Bonds. Journal of the American Chemical Society 140, 9678–9684. 10.1021/jacs.8b05753. [DOI] [PubMed] [Google Scholar]
- 39.Valentine AM; LeTadiac-Badiatti M-H; Toy PH; Newcomb M; Lippard SJ (1999) Oxidation of Ultrafast Radical Clock Substrate Probes by the Soluble Methane Monooxygenase from Methylococcus capsulatus (Bath). J. Biol. Chem 274, 10771–10776. 10.1074/jbc.274.16.10771 [DOI] [PubMed] [Google Scholar]
- 40.Dekkiche BA; Seraghni N; Debbache N; Ghoul I; Sehili T. (2018) Effect of Natural and Artificial Light on Fe(III) Organic Complexes Photolysis: Case of Fe (III)-Malonate and Fe(III)-Malate. Int. J. Chem. React. Eng, 17. 10.1515/ijcre-2018-0106 [DOI] [Google Scholar]
- 41.Wang L; Zhang C; Mestankova H; Wu F; Deng N; Pan G; Bolte M; Mailhot G. (2009). Photoinduced degradation of 2,4-dichlorophenol in water: influence of various Fe(III) carboxylates. Photochem. Photobiol. Sci, 8, 1059–1065. 10.1039/b902607j. [DOI] [PubMed] [Google Scholar]
- 42.Ohyoshi E. (1984). Spectrophotometric determination of formation constants of 1:1 complexes of lanthanides with 4-(2-pyridylazo)resorcinol (par). Talanta, 31(12), 1129–1132. 10.1016/00399140(84)80264-7. [DOI] [PubMed] [Google Scholar]
- 43.Sabenya G; Lázaro L; Gamba I; Martin-Diaconescu V; Andris E; Weyhermüller T; Neese F; Roithova J; Bill E; Lloret-Fillol J; Costas M. (2017). Generation, Spectroscopic, and Chemical Characterization of an Octahedral Iron(V)-Nitrido Species with a Neutral Ligand Platform. J. Am. Chem. Soc, 139(27), 9168–9177. 10.1021/jacs.7b00429 [DOI] [PubMed] [Google Scholar]
- 44.Day CS; Fawcett A; Chatterjee R; Hartwig JF (2021). Mechanistic Investigation of the Iron-Catalyzed Azidation of Alkyl C(sp3)-H Bonds with Zhdankin’s λ3-Azidoiodane. J. Am. Chem. Soc 143(39), 16184–16196. 10.1021/jacs.1c07330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer K; Bill E; Mienert B; Weyhermüller T; Wieghardt K. (1999) Photolysis of cis- and trans-[FeIII(cyclam)(N3)2]+ Complexes: Spectroscopic Characterization of a Nitridoiron(V) Species. J. Am. Chem. Soc, 121(20), 4859–4876. 10.1021/ja983454t [DOI] [Google Scholar]
- 46.Grapperhaus CA; Mienert B; Bill E; Weyhermüller T; Wieghardt K. (2000). Mononuclear (nitrido)iron(V) and (oxo)iron(IV) complexes via photolysis of [(cyclam-acetato) FeIII(N3)]+ and ozonolysis of [(cyclam-acetato) FeIII(O3SCF3)]+ in water/acetone mixtures. Inorg. Chem 39(23), 5306–5317. 10.1021/ic0005238 [DOI] [PubMed] [Google Scholar]
- 47.Blackmond DG (2005). Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angewandte Chemie International Edition 44, 4302–4320. 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- 48.Nielsen CDT, and Burés J. (2019). Visual kinetic analysis. Chemical Science 10, 348–353. 10.1039/C8SC04698K. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There is no dataset or code associated with this publication. Full experimental procedures and experimental data are provided in the supplemental information.