

Abstract
γ-Aminobutyric acid (GABA) is the most prevalent inhibitory neurotransmitter in the mammalian brain and has been found to play an important role in the pathogenesis or the expression of many neurological diseases, including epilepsy. Although GABA can act on different receptor subtypes, the component of the GABA system that is most critical to modulation of seizure activity is the GABAA-receptor-chloride (Cl−) channel complex, which controls the movement of Cl− ions across the neuronal membrane. In the mature brain, binding of GABA to GABAA receptors evokes a hyperpolarising (anticonvulsant) response, which is mediated by influx of Cl− into the cell driven by its concentration gradient between extracellular and intracellular fluid. However, in the immature brain and under certain pathological conditions, GABA can exert a paradoxical depolarising (proconvulsant) effect as a result of an efflux of chloride from high intracellular to lower extracellular Cl− levels. Extensive preclinical and clinical evidence indicates that alterations in GABAergic inhibition caused by drugs, toxins, gene defects or other disease states (including seizures themselves) play a causative or contributing role in facilitating or maintaning seizure activity. Conversely, enhancement of GABAergic transmission through pharmacological modulation of the GABA system is a major mechanism by which different antiseizure medications exert their therapeutic effect. In this article, we review the pharmacology and function of the GABA system and its perturbation in seizure disorders, and highlight how improved understanding of this system offers opportunities to develop more efficacious and better tolerated antiseizure medications. We also review the available data for the two most recently approved antiseizure medications that act, at least in part, through GABAergic mechanisms, namely cenobamate and ganaxolone. Differences in the mode of drug discovery, pharmacological profile, pharmacokinetic properties, drug–drug interaction potential, and clinical efficacy and tolerability of these agents are discussed.
Key Points
| The inhibitory neurotransmitter γ-aminobutyric acid (GABA) has an important role in the control of neuronal excitability in the mammalian brain. Disruption of GABAergic neurotransmission exerts a causative or contributory effect in the pathogenesis or expression of many neurological diseases, including epilepsy. |
| The component of the GABA system most critical to modulation of seizure activity is the GABAA receptor, which in the mature brain, evokes a hyperpolarising Cl− current. In contrast, in the immature brain and in some neonatal pathological conditions, GABA can be depolarising (proconvulsant), owing to the presence of high intraneuronal Cl− levels (relative to the extracellular milieu) in these conditions. |
| GABA has been for many years, and continues to be, a major target for research into the design and development of newer antiseizure medications. Cenobamate and ganaxolone are the latest antiseizure medications acting on GABA that have entered the market. |
| Cenobamate acts by a dual mechanism (blockade of persistent sodium currents and GABAA receptor modulation) and has shown robust efficacy against refractory focal seizures. It is approved in Europe and the USA for the treatment of focal seizures in adults. The European indication is limited to adjunctive use in patients whose seizures are not adequately controlled despite a history of treatment with at least two other antiseizure medications. |
| Ganaxolone, an orally active neurosteroid, is a GABAA allosteric modulator that has been assessed in different types of epilepsy, and was approved in the USA in March 2022 for the treatment of seizures associated with cyclin-dependent kinase-like 5 deficiency disorder. In May 2023, the European Medicines Agency issued a positive opinion recommending its approval for the same indication. |
Introduction
Within the mature mammalian central nervous system (CNS), the amino acids γ-aminobutyric acid (GABA) and glutamate serve as the major inhibitory and excitatory neurotransmitters, respectively [1]. An imbalance in their activity has been associated with abnormal neuronal firing and can under certain conditions contribute to the pathogenesis of several CNS disorders, including epilepsy. Drugs that enhance GABAergic inhibition or decrease glutamatergic excitation have been found to exert an antiseizure effect in animal models of seizures and epilepsy [1, 2]. Similarly to treatments that inhibit excessive excitatory neurotransmission, pharmacological interventions that potentiate GABA-mediated transmission have proved to be an effective tool by which seizure activity can be prevented or suppressed in the clinical setting.
Over time, an improved understanding of the mechanisms involved in GABAergic inhibition has provided the basis for the rational design of a number of antiseizure medications (ASMs) targeting different components of the GABA system, the most notable examples being progabide [3], vigabatrin [4] and tiagabine [5]. However, many other ASMs that are known to act at least in part by potentiating GABAergic transmission (e.g. barbiturates, valproic acid, benzodiazepines, felbamate, topiramate and stiripentol) were not developed by rational target-based strategies, but were rather discovered by chance or by phenotypic screening in animal models of seizures and epilepsy [6].
In recent years, there has been an intense discovery effort to find more efficacious and better tolerated drugs that target GABA-mediated inhibition. In this review, we focus on the important role of GABA in the modulation of neuronal excitability and seizure activity and control, and we review preclinical and clinical data on recently approved drugs that target GABA-mediated neurotransmission. Drugs in development that act via the GABA system are discussed in an accompanying article [2].
Literature Search
For Sect. 3, we conducted a literature search on PubMed for studies published in English since 1 January, 2005 until 30 June, 2023, using as search terms ‘GABA AND seizures OR epilepsy’. The authors’ files and the reference lists of retrieved relevant articles were also reviewed. For Sects. 4 and 5, we searched PubMed for articles published until 30 June, 2023, using as search terms ‘cenobamate’ and ‘ganaxolone’, without language or time limitations. We selected articles that focused on the use of these medications as treatments for seizures or epilepsy. For both cenobamate and ganaxolone, we also reviewed US and European prescribing information databases and clinical trials registered in ClinicalTrials.gov targeting ‘seizures’ or ‘epilepsy’ as indications.
The GABAergic System and Its Role in the Pathogenesis of Seizures and Epilepsy
Evidence on the Role of GABA as a Modulator of Seizure Susceptibility
As described in greater detail below, the inhibitory action of GABA on neuronal excitability in the mature brain is primarily mediated by its binding to GABAA receptors, leading to the influx of chloride ions (Cl−) and cell hyperpolarisation, though actions on other GABA receptor subtypes may also contribute [1]. Multiple lines of experimental and clinical evidence over the course of three plus decades have demonstrated that disruption of GABA-mediated inhibition contributes to seizure initiation and propagation [7]. Particularly relevant in this regard is the demonstration of a reduction in GABAergic neurotransmission in multiple animal models of acquired and genetic epilepsies. For example, loss of inhibitory interneurons results in reduced release of GABA, reduced inhibition of excitatory principal cells, and facilitation of seizures and epilepsy [8]. A number of studies have shown that glutamate levels rise during a seizure, whereas GABA levels remain relatively constant [9]. This shift in the glutamate-to-GABA ratio results in an imbalance in excitation and inhibition and can contribute to the initiation and propagation of seizure activity.
Experimental research has enhanced our understanding of the GABA system and led to the development of ASMs designed to increase GABA-mediated inhibition through a variety of mechanisms, including inhibition of GABA metabolism by vigabatrin, inhibition of synaptic GABA reuptake by tiagabine and allosteric modulation of GABAA receptors by ganaxolone [10–12]. Several other ASMs, including barbiturates, benzodiazepines, cenobamate, felbamate, stiripentol, topiramate and possibly also gabapentin, pregabalin and valproic acid, act at least in part by potentiating GABAergic transmission (Fig. 1) [8]. As discussed in the accompanying article [2], there are also many innovative and repurposed treatments in clinical development that target the GABA system. The degree to which these newer therapies will improve seizure control over existing ASMs has yet to be determined.
Fig. 1.

Schematic representation of an inhibitory synapse in the central nervous system and putative major sites of action of established and investigational antiseizure medications that target the GABAA receptor. Plus (+) or minus (−) signs denote activation/potentiation or inhibition, respectively. Among positive allosteric modulators of the GABAA receptors, benzodiazepines and α2/α3/α5-selective compounds act at binding sites that differ from the binding sites of the neurosteroid ganaxolone, or from the binding sites of other allosteric modulators (see also Fig. 2). Current efforts to develop subunit-selective allosteric modulators that preferentially activate GABAA receptors devoid of the α1 subunit are based on the hypothesis that these compounds will be less sedating and less liable to develop tolerance. For more information on investigational γ-aminobutyric acid (GABA)-targeting antiseizure medications, see the accompanying article by Perucca et al. [2]. GABA-T GABA transaminase, GAD glutamic acid decarboxylase, *evidence of activation of GAD by gabapentin, pregabalin and valproic acid is inconclusive.
Modified from White and Rho [11], with permission
Other evidence in support of the important role of GABA in controlling neuronal excitation comes from animal models wherein drugs (e.g. l-allylglycine) that block the GABA-synthesising enzyme glutamate decarboxylase or drugs that block GABAA receptors (pentylenetetrazole) or GABA-activated Cl− channels (picrotoxin) are proconvulsant [12, 13]. Finally, a number of mutations in genes encoding GABA receptor subunits have been identified and found to play a pathogenic role in various epileptic syndromes, which range from generalised epilepsies to epileptic encephalopathies [14–23].
As important as GABA is in mediating inhibition, it should also be understood that an increase in GABAergic transmission can also facilitate seizure activity depending on the brain area or circuitry affected. For example, in a series of elegant studies, Iadarola and Gale [24] identified the substantia nigra as a critical midbrain site for GABA-mediated antiseizure activity. They found that activation of GABAA receptors in the midbrain but not the forebrain and hindbrain was able to block tonic extension seizures induced by maximal electroshock and tonic-clonic seizures induced by pentylenetetrazole and bicuculline [24]. In contrast, antagonism of GABAA receptors in some brain regions actually exerted an anticonvulsant rather than a proconvulsant effect, an observation that clearly demonstrates the importance of the anatomical location of GABAA receptors within the seizure network [25]. Interestingly, drugs that act by increasing the availability of GABA in the synaptic space tend to promote seizure activity in animal models of absence and myoclonic seizures, and to aggravate these seizure types in patients with generalised epilepsies. For example, inhibition of GABA-transaminase (GABA-T) by vigabatrin has been associated with increased seizure activity in the photosensitive baboon, the lethargic mouse, the Genetic Epilepsy Prone rat and the Genetic Absence Epilepsy Rat of Strasbourg, and in patients with absence or juvenile myoclonic epilepsy [26–28], an action that is presumed to result from increased GABAergic tone in the thalamo-cortical circuit that modulates spike-wave discharges. Similarly, the aggravation of absence and myoclonic seizures seen with carbamazepine and oxcarbazepine in the Genetic Absence Epilepsy Rat of Strasbourg and in patients with idiopathic generalised epilepsies is probably owing to the ability of these drugs to enhance GABA currents in the ventral basal thalamus [29, 30]. The precipitation or aggravation of absence seizures by ASMs that enhance GABA levels, i.e. vigabatrin and tiagabine, may be also due in part to activation of presynaptically located metabotropic GABAB receptors by synaptically released GABA [31–34]. Activation of presynaptic GABAB receptors results in reduced neurotransmitter release. Depending on whether these receptors reside on glutamatergic principal cells or inhibitory neurons, their activation by the GABAB receptor agonist baclofen can result in an antiseizure or a seizure-facilitating action, respectively. The effect of baclofen on seizure susceptibility is also partly dependent on age [31–34].
GABA Synthesis, Release and Metabolism
Within the CNS, the synthesis of GABA is integrally linked to the metabolism of glutamine and glutamate [7, 9, 35]. Once released from the presynaptic terminal, GABA interacts with both synaptic and extrasynaptic receptors and is then removed from the synaptic space into surrounding GABAergic neurons by the active transporter GAT1 and repackaged into vesicles by the vesicular GABA transporter vGAT for later release.
GABA is also taken up into surrounding astrocytes by GAT1 and then metabolised to succinic semialdehyde by GABA-T [35]. In turn, succinic semialdehyde is metabolised by succinic acid semialdehyde dehydrogenase to succinic acid, which then enters the tricarboxylic acid cycle. The glutamine that is synthesised from succinic acid via the tricarboxylic acid cycle is released back to the extracellular space where it can be taken up by both glutamatergic and GABAergic neurons [9, 35]. In glutamatergic neurons, glutamine is deaminated by glutaminase to glutamate, which is then packaged into vesicles by vesicular glutamate transporters for future release. GABAergic neurons also convert glutamine back to glutamate, which is decarboxylated by glutamate decarboxylase to GABA, that is then repackaged into vesicles by vGAT for subsequent release. Glutamate released from excitatory neurons can serve as another important source of GABA [9, 35]. For example, glutamate taken up by astrocytes by excitatory aminoacid transporters on the astrocytic membrane can be converted back to glutamine by glutamine synthetase. As described above, glutamine can then be recycled by both glutamatergic and GABAergic neurons and converted to glutamate and GABA, respectively [35].
GABA Receptors
Upon release from the presynaptic terminal, GABA exerts its effects through multiple receptors, including GABAA, GABAB and GABAC receptors [36]. Of these, the GABAA receptor is an important target for several drug classes including benzodiazepines, barbiturates, neurosteroids, alcohol and general anaesthetics. GABAA receptor modulators, in particular, have played an important therapeutic role as ASMs, sedative hypnotics, anxiolytics and muscle relaxants for many years [37].
GABAA receptors are pentameric ligand-gated ion channels comprising five distinct subunits that conduct a hyperpolarising Cl− current upon activation. So far, there have been 20 different GABAA receptor subunits identified, including six α (α1 to α6), four β (β1 to β4), three γ (γ1 to γ3), one δ, three ρ (ρ1 to ρ3), and one each of ε, θ and π subunits [37–40]. Importantly, these 20 subunits can come together to form a multitude of different subunit combinations that play an important role in the localisation, trafficking, kinetics and pharmacology of the GABAA receptor.
As noted above, GABA can also act on GABAB receptors, which are G protein-coupled ion channels that activate the G protein-coupled inward rectifier potassium channel (GIRK) to induce a potassium efflux from the cell [41]. Presynaptic GABAB receptors can serve as autoreceptors and their activation can result in a reduced release of GABA or other neurotransmitters, including glutamate, norepinephrine, 5-hydroxytryptamine and dopamine [41]. It is through these actions that GABAB receptors modulate neuronal excitability.
GABAC receptors are also ligand-gated ionotropic channels that are largely found in the retina. When activated by GABA, they evoke a Cl− current [42]. In contrast to GABAA receptors, GABAC receptors, defined by the presence of a ρ subunit, are insensitive to the convulsant bicuculline [42]. Unlike GABAA and GABAB receptors, GABAC receptors are not a target for any of the established ASMs or investigational ASMs in development.
GABAA Receptor Subunit Conformation and Pharmacological Response
GABAA receptor subunit heterogeneity has an important function in determining the response to GABAA receptor modulators, an observation that led to an intense effort to develop subunit-selective ligands with potentially improved efficacy and tolerability [40, 43]. Each functional GABAA receptor usually includes two α, two β, and one γ or δ subunit. GABAA receptors can be further categorised as γ-subunit-containing synaptic receptors or δ-subunit-containing extrasynaptic receptors [37]. When activated by GABA, synaptic GABAA receptors largely modulate rapid phasic inhibition. In contrast, extrasynaptic GABAA receptors largely contribute to tonic inhibition (Fig. 2) [44].
Fig. 2.
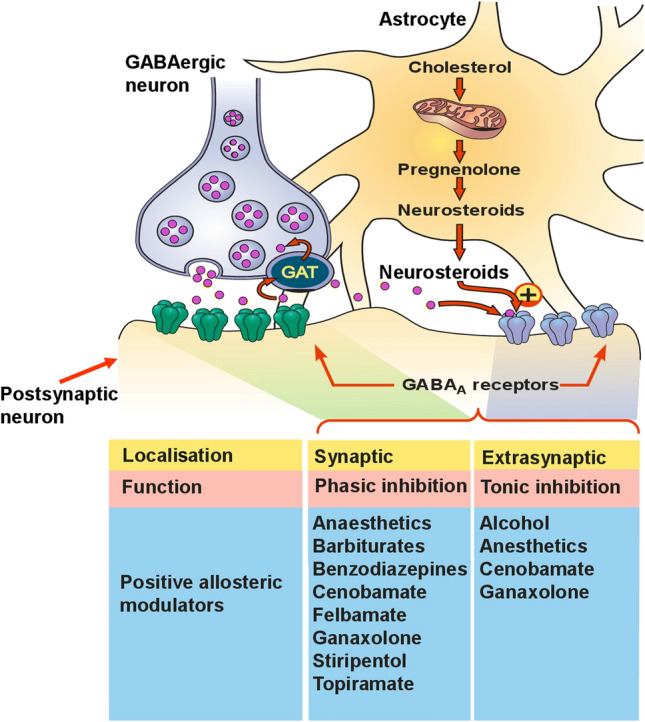
Localisation and function of GABAA receptors on postsynaptic neurons, including actions of established antiseizure medications. Although the figure also shows the synthetic pathway of endogenous neurosteroids within astrocytes, some neurosteroids are also synthesised and released by neuronal cells. There are two forms of γ-aminobutyric acid (GABA)-mediated inhibition via GABAA receptors: phasic and tonic. Phasic inhibition is triggered by synaptically released GABA acting on receptors clustered at the postsynaptic sites. These receptors contain γ2 subunits (in association with α1 to α6 subunits) and are positively modulated by benzodiazepines, barbiturates and a number of other antiseizure medications. γ2 Subunits are critical for benzodiazepine sensitivity. Tonic inhibition is mediated by “ambient” levels of extracellular GABA that have escaped presynaptic reuptake via the GABA transporter (GAT-1, or GAT in the figure) and acts primarily on receptors located extrasynaptically. These tonic inhibitory receptors contain δ subunits and are insensitive to benzodiazepines but are sensitive to modulation by cenobamate and by endogenously synthesised neurosteroids or the exogenous neurosteroid ganaxolone. Extrasynaptic receptors are also sensitive to general anaesthetics and alcohol.
Modified from White and Rho [11], with permission
Benzodiazepines and barbiturates are both positive allosteric modulators of the GABAA receptor. GABAA receptors with a high sensitivity to benzodiazepines typically comprise two α (α1 to 3, or α5), two β (β1 to β3) and one γ2 subunit [45]. GABAA receptors containing α4 and α6 subunits and/or lacking γ2 subunits are benzodiazepine insensitive. The classical benzodiazepine binding site resides at the α and γ interface and many of the side effects associated with the classical benzodiazepines can be explained at least in part by their nonselective binding to GABAA receptors containing α1, α2, α3, α5 and γ2 subunits. Of the various α subunits, the α1 subunits are widely distributed in the brain and modulation of α1 subunit-containing GABAA receptors by benzodiazepines is largely associated with sedative effects [37, 43, 46–48]. The α2 and α3 subunits are not considered to be involved to a major extent in sedative effects. Modulation of GABAA receptors containing α2 and α3 subunits plays an important role in the antiseizure, anxiolytic and analgesic effects of benzodiazepines [43, 49], even though actions at the α1 subunit also seem to contribute to antiseizure effects in some experimental models [50]. GABAA receptors containing α5 subunits are thought to contribute to anxiolytic effects [37, 51]. The nonbenzodiazepine Z-drugs that are used as sleep aids have high selectivity for α1-containing GABAA receptors, which as stated above are widely distributed throughout the brain [48].
Thus, the specific α subunit within a given benzodiazepine-sensitive GABAA receptor determines whether allosteric modulation results predominantly in antiseizure, sedative or anxiolytic activity. Although these findings are largely based on behavioural studies in animal models and their applicability to the clinical setting requires further study [37], GABAA receptor subunit heterogeneity has provided the medicinal chemist with an opportunity to design and develop novel ASMs with unique subunit selectivity and potentially improved efficacy and tolerability. Several of the new GABA-targeting ASMs in development and discussed in the accompanying article [2] were designed with this concept in mind.
The usefulness of benzodiazepines during long-term treatment is limited in part by the development of tolerance, which can occur as a result of decreased surface availability of GABAA receptors following prolonged drug exposure [40, 52]. Factors involved in the decreased availability and sensitivity of GABAA receptors to benzodiazepines include increased lysosomal degradation of the γ2 subunit, phosphorylation of the receptors by specific protein kinase C isoforms and activation of calcineurin-dependent dephosphorylation by diazepam leading to GABAA receptor endocytosis [40]. In addition, it has been suggested that the α1 subunit modulates tolerance to the benzodiazepines, even though the precise mechanism by which α1 subunit expression contributes to benzodiazepine-mediated tolerance has yet to be defined [40, 43]. What is clear is that drugs devoid of activity at the α1 subunit are less likely to develop tolerance [43]. Overall, these findings have stimulated efforts to develop subunit-selective allosteric modulators that lack activity at GABAA receptors containing α1 subunits, in the anticipation that these modulators would be less sedating and less likely to develop tolerance [43].
Neurosteroids differ from benzodiazepines in a couple of important ways [40]. First, they bind within the transmembrane domain between the α and β subunits, whereas the benzodiazepine binding site resides at the α and γ interface. Second, they display preferential affinity for extrasynaptic δ-containing GABAA receptors. That said, neurosteroids can also bind and modulate isoforms of synaptic GABAA receptors containing benzodiazepine-insensitive α4 and α6 subunits, and those that lack the γ2 subunit required for benzodiazepine binding. Therefore, neurosteroids might be anticipated to enhance GABAergic neurotransmission in networks wherein benzodiazepines are inactive [53].
In addition to subunit composition, location and expression patterns of GABAA receptors can influence the GABAA receptor response to modulation by GABA, drugs and disease processes [38]. One example of subunit heterogeneity within the same brain structure is provided by the hippocampus. The primary subunit conformation of phasic synaptic GABAA receptors in the dentate gyrus is α2βγ2, whereas the subunit conformation of slow tonic extrasynaptic GABAA receptors in the dentate gyrus are more likely to be α4βγ, which would be expected to be benzodiazepine insensitive [45]. Interestingly, in contrast to the dentate gyrus, extrasynaptic GABAA receptors in the hippocampal CA1 region are primarly α5βγ2 [54]. As such, a benzodiazepine with a higher affinity for α5-containing GABAA receptors is more likely to increase tonic inhibition over phasic inhibition in the CA1 region, whereas a benzodiazepine with a preference for an α2 subunit is more likely to increase phasic inhibition in the dentate gyrus.
When it comes to the kinetics of GABA-evoked Cl− channels, benzodiazepines, barbiturates and neurosteroids all display markedly different effects. In particular, benzodiazepines increase the frequency of channel opening [55], whereas phenobarbital and other barbiturates increase the duration of the channel opening [56–58]. The neurosteroids are interesting because they increase the frequency of single channel openings much like the benzodiazepines, but they also increase the duration of the channel opening much like the barbiturates [59, 60].
The Effect of Development and Disease States on GABAA Receptor Responses
In the mature brain, the binding of GABA to GABAA receptors evokes a hyperpolarising influx of Cl− driven by its concentration gradient from high extracellular levels to low intracellular Cl− levels. In contrast to the mature brain, activation of GABAA receptors in the immature brain is largely depolarising and potentially proconvulsant as a result of an outward flux of chloride from high intracellular to lower extracellular Cl− levels [61]. The direction of GABAA-mediated neurotransmission (hyperpolarising or depolarising) depends on the intracellular Cl− level, which in turn is maintained by the activity of two cotransporters. The sodium (Na+)-potassium (K+)-Cl−-cotransporter isoform 1 (NKCC1) transports Cl− into the cell, whereas the K+-Cl−-cotransporter isoform 2 (KCC2) extrudes Cl− out of the cell. The precise time during brain maturation at which GABA switches from being depolarising to hyperpolarising is not known, but it has been suggested to occur before or soon after birth [62, 63].
In both rodents and humans, the shift from a depolarising to a hyperpolarising current results from a developmentally dependent regulation of the intracellular Cl− level in postsynaptic neurons, controlled by the relative expression of NKCC1, which is high in the neonatal and early postnatal days but decreases rapidly with development. In contrast, the expression of the Cl− extruding cotransporter KCC2 is low in early postnatal days but increases rapidly within the first few days after birth [62]. Dzhala et al. [62] reported that in the human cortex, NKCC1 expression was significantly higher at 31–41 postconceptional weeks (peaking at 35 postconceptional weeks) than it was 1 year later (92 postconceptional weeks). NKCC1 expression decreased rapidly to adult levels during the first year of life. In contrast, the expression of KCC2 was substantially lower than that of the adult during the fetal and neonatal period (20–41 postconceptional weeks) and was only a fraction (2–25%) of adult levels at the time of peak NKCC1 expression. KCC2 levels rose quickly over the first year of life to adult levels. These findings suggest that at the time of birth, the GABAA receptor of the human neonate is potentially in an immature state and therefore rather than being hyperpolarising, GABA could be depolarising. Of note, there can be differences in the degree of brain maturation across brain regions and networks [64]. There is also evidence that neonatal hypoxia-ischemia, as well as a number of other pathological conditions associated with susceptibility to seizures and epilepsy, can increase NKCC1 and decrease KCC2 expression, thereby shifting Cl− homeosthasis towards the more immature state and leading to a higher probability of GABA being depolarising, at least in some brain areas [65–67]. Overall, these findings could explain why phenobarbital, the standard of care for neonatal seizures, appears to be less effective in the early neonatal period, particularly in the presence of hypoxia-asphyxia [68].
In both rodents and humans, prolonged seizure activity associated with status epilepticus has been found to result in a rapid reduction in benzodiazepine sensitivity [69, 70]. This is likely to be related to the internalisation and transient inactivation of synaptic GABAA receptors that mediate the effects of benzodiazepines and barbiturates, while the extrasynaptic δ subunit-containing GABAA receptor that mediates tonic GABA-mediated inhibition remains unaffected by this process [71]. Consistent with the latter finding, there is suggestive evidence that the efficacy of neurosteroids that bind to both synaptic and extrasynaptic GABAA receptors, unlike that of the benzodiazepines, which bind to synaptic GABAA receptors, is not reduced by status epilepticus [53]. This is most likely because extrasynaptic, unlike synaptic GABAA receptors, do not internalise [53].
The GABAergic System as a Target for ASMs
Given the critical role of GABA in modulating neuronal excitability, the GABAergic system has been, and continues to be, an important target for efforts to identify new therapies for epilepsy, convulsive status epilepticus and other CNS disorders, including anxiety, sleep and depression. As discussed above, there are three main mechanisms by which drugs can increase GABA-mediated inhibition. These include: (1) positive allosteric modulation of GABAA receptors, an action shared by benzodiazepines, barbiturates, neurosteroids and several other ASMs [2]; (2) inhibition of GABA metabolism in neuronal and glial cells, e.g. by inhibition of GABA-T, as in the case of vigabatrin [4]; and (3) inhibition of high-affinity synaptic re-uptake by the GABA tranporter GAT1, as in the case of tiagabine [5]. While these drugs provide adequate seizure control in some patients, their utility in the treatment of seizures has been limited by a number of factors, including the development of tolerance and dependence (benzodiazepines) [72] and dose-limiting adverse effects including somnolence with most of these agents [73], visual field impairment in the case of vigabatrin [74] and precipitation of nonconvulsive status epilepticus with tiagabine [75]. Hopefully, some of the GABA-targeting drugs currently in development will overcome at least some of these limitations [2].
In addition to GABA, there are a number of endogenous neurosteroids in the brain, including allopregnanolone, allotetrahydro-deoxycorticosterone and androstanediol, that have been shown to interact with synaptic and extrasynaptic GABAA receptors. These positive allosteric modulators of GABAA receptors have been shown to possess antiseizure, anxiolytic, antidepressant and neuroprotectant activity, and drug discovery efforts in recent years have identified a number of candidate neurosteroids that have demonstrated potential utility for the treatment of epilepsy and postpartum depression [76]. One of these candidates, ganaxolone, has recently been approved for the treatment of seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder, and is discussed below in greater detail.
Recently Approved ASMs Acting Via the GABAergic System
Cenobamate
Cenobamate is a carbamate derivative that was approved in 2019 by the US Food and Drug Administration for the treatment of focal seizures in adults [77] and in 2021 by the European Medicines Agency for the adjunctive treatment of focal seizures (with or without progression to bilateral tonic-clonic seizures) in adults with epilepsy not adequately controlled despite a history of treatment with at least two other ASMs [78]. Early data on the pharmacological and clinical profile of cenobamate have been reviewed in previous issues of this journal [79, 80].
Pharmacology
Cenobamate displays broad activity in a variety of models of seizures and epilepsy, including the maximal electroshock seizure test in mice and rats, seizures induced by subcutaneous pentylenetetrazole and picrotoxin in mice, focal seizures in the hippocampal kindled rat, and the 6-Hz (22-mA, 32-mA and 44-mA) mouse model of drug-resistant seizures [81]. In many of these models, cenobamate did not cause overt behavioural toxicity, with a protective index (median toxic dose/median effective dose) that compared favourably with that reported for other ASMs [81]. The antiseizure effects of cenobamate are presumed to result from a dual action. In particular, cenobamate has been shown to enhance GABA-induced phasic and tonic chloride currents through positive allosteric modulation of synaptic and extrasynaptic GABAA receptors, regardless of the α subunit composition evaluated (e.g. α1β2γ2 or α2-6β3γ2). This effect of cenobamate was not blocked by flumazenil, suggesting that it involves an interaction with a modulatory site independent of the benzodiazepine binding site [82, 83]. In addition to its ability to enhance GABA-mediated phasic and tonic inhibition, cenobamate has also been shown to block voltage-gated sodium channels through preferential inhibition of the persistent sodium current [81, 84]. Cenobamate interactions with both GABAA receptors and voltage-gated sodium channels differ from those reported for other ASMs, which might explain its broad-spectrum activity in animal models of focal and generalised seizures as well as its robust efficacy in the clinical trials conducted to date [81, 85]. Whether other properties also contribute to the antiseizure effects of cenobamate remains to be established. Of note, cenobamate was not designed using a target-based strategy to interact with the GABAergic system or with sodium channels, but its discovery was made possible by a screening approach using well-established animal models of seizures and epilepsy. Like most of the ASMs identified through a similar screening approach, the presumed mechanisms of action of cenobamate were not identified until later in its development [6].
Biopharmaceutics and Clinical Pharmacokinetics
Based on its water solubility (1.7 g/L) and lipophilicity, as indicated by a partition coefficient between octanol and water (logP) of 1.6 [86, 87], cenobamate can be classified as Biopharmaceutics Classification System Class 1, namely as a highly water-soluble and highly permeable compound [88], as well as Biopharmaceutics Drug Disposition Classification System Class 1, namely, as a highly water-soluble compoound that is eliminated by metabolism in humans [87, 89].
After oral administration of single (5–750 mg) or multiple doses (50–600 mg once daily for 14–35 days, including titration) of cenobamate in healthy subjects, maximum plasma drug concentrations (Cmax) were found to occur generally at 0.8–4.0 h after dosing [90, 91]. The area under the plasma drug concentration–time curve (AUC) of cenobamate increased more than dose proportionally after single doses, but was linearly related to dose during multiple once-daily dosing from 50 to 500 mg/day. Consistently with the classification of cenobamate as a Biopharmaceutics Classification System/Biopharmaceutics Drug Disposition Classification System Class 1 drug, gastrointestinal absorption of cenobamate is not affected by food intake or by efflux or uptake transporters in the gut [77, 92]. In a study that investigated urinary recovery after oral intake of a radiolabelled dose, ≥ 88% of the administered radioactivity was recovered in urine, mostly as metabolites, which indicates virtually complete gastrointestinal absorption [92]. Unchanged cenobamate, however, was the main entity found in the systemic circulation, representing > 98% of the total radioactivity in plasma.
The apparent volume of distribution of cenobamate has been reported to be approximately 40–50 L, and its oral clearance to be in the range of 0.4–1.4 L/h after single doses (5–750 mg) and between 0.4 and 0.5 L/h after multiple dosing (50–500 mg once daily) for 14–17 days [90]. The fraction bound to plasma proteins of cenobamate is about 60% [77, 78, 90]. Within the 100–400 mg/day dose range, the mean terminal half-life of cenobamate is approximately 50–60 h, implying that fluctuations in plasma drug concentrations during a 24-h dosing interval are quite modest [90]. Consistently with the long half-life, steady-state plasma concentrations are attained after 11–13 days of constant daily dosing [90].
Only 6% of an oral dose of cenobamate is excreted unchanged in urine. Cenobamate is metabolised extensively, primarily by glucuronidation via UGT2B7 and to a lesser extent UGT2B4, and by oxidation via cytochrome P450 (CYP) 2E1, CYP2A6, and CYP2B6 and to a lesser extent CYP2C19 and CYP3A4/5 [77, 92].
Data on the pharmacokinetic profile of cenobamate in patients receiving concurrent ASMs are limited. According to prescribing information, the pharmacokinetics of cenobamate in patients with epilepsy is similar when the drug is used as monotherapy or as adjunctive therapy, except that plasma cenobamate concentrations are decreased by an average 27–28% when it is co-administered with phenytoin [77].
Cenobamate plasma exposure (AUC) after a single dose is increased by 40–50% in patients with mild-to-moderate renal impairment (creatinine clearance 30 to < 90 mL/min) compared with healthy controls, but no differences in plama exposure compared with controls was found in patients with creatinine clearance < 30 mL/min [77]. In a similar study, cenobamate AUC was found to be 2.1-fold and 2.3-fold higher in patients with mild (Child-Pugh score 5–6) and moderate (Child-Pugh score 7–9) hepatic impairment compared with matched healthy controls [77]. No data are currently available on cenobamate pharmacokinetics in patients with severe hepatic impairment, but a pharmacokinetic and safety study in this population has been planned (ClinicalTrials.gov Identifier: NCT04791553).
Drug Interactions
Information on the influence of other ASMs on the pharmacokinetics of cenobamate is limited. Co-administration of phenytoin has been reported to decrease plasma cenobamate concentrations by 27–28% [77].
Extensive evidence, however, indicates that cenobamate can affect the pharmacokinetics of several concomitantly administered medications by acting as an inhibitor or an inducer of a number of CYP enzymes [80, 93]. Specifically, cenobamate has been found to inhibit in vitro CYP2B6, CYP2C19 and CYP3A (but not CYP1A2, CYP2C8, CYP2C9 or CYP2D6) and to induce CYP2B6, CYP2C8 and CYP3A4 (but not not CYP1A2, CYP2C9 or CYP2C19) [75]. In a study in healthy subjects using different CYP probe substrates, a cenobamate dose of 200 mg/day administered for 25 days after completion of titration was associated with clear evidence of inhibition of CYP2C19, and induction of CYP3A and CYP2B6 (Table 1) [94].
Table 1.
Effect of cenobamate on the pharmacokinetics of concomitantly administered medications
| Affected drug | Interaction caused by cenobamate | Probable mechanism | Comment | References |
|---|---|---|---|---|
| Antiseizure medications | ||||
| Carbamazepine | 23% decrease in carbamazepine AUC | Induction of CYP3A | Effect observed following exposure to multiple dosing with cenobamate (200 mg/day) | Prescribing information [77]; Vernillet et al. [99] |
| Clobazam | Prominent increase in plasma N-desmethylclobazam concentration | Inhibition of CYP2C19 | In a retrospective study of 5 patients, the increase in plasma levels of N-desmethylclobazam, the active metabolite of clobazam, ranged from 145 to 1852% and was associated with fatigue, which improved after reducing the clobazam dose [95]. Another study reported similar findings and provided suggestive evidence of cenobamate and clobazam may also exhibit synergic therapeutic efficacy [96] | Elakkari et al. [95]; Osborn and Abou-Khalil [96] |
| Lamotrigine | 21–51% decrease in plasma lamotrigine concentrations | Induction of lamotrigine glucuronidation | Estimates based on population pharmacokinetics analysis at a cenobamate dose of 100–400 mg/day | Prescribing information [77]; Vernillet et al. [99] |
| Phenobarbital | 37% increase in phenobarbital AUC | Inhibition of CYP2C19 (?) | Effect observed following exposure to multiple dosing with cenobamate (200 mg/day) | Prescribing information [77]; Vernillet et al. [99] |
| Phenytoin | 84% increase in phenytoin AUC | Inhibition of CYP2C19 (?) | Effect observed following exposure to multiple dosing with cenobamate (200 mg/day) | Prescribing information [77]; Vernillet et al. [99] |
| Other drugs | ||||
| Bupropion | 39% decrease in bupropion AUC | Induction of CYP2B6 | Bupropion (150 mg) was given orally as a probe substrate to test the effect of cenobamate (200 mg/day) on CYP2B6 activity. The decrease in plasma concentrations of bupropion was associated with increased plasma concentrations of the active metabolite hydroxybupropion | Prescribing information [77]; Greene et al. [94] |
| Midazolam | 72% decrease in midazolam AUC | Induction of CYP3A | Midazolam (2 mg) was given orally as a probe substrate to test the effect of cenobamate (200 mg/day) on CYP3A activity. At a dose of 100 mg/day, the mean decrease in midazolam AUC was 27% | Prescribing information [77]; Greene et al. [94] |
| Omeprazole | 107% increase in omeprazole AUC | Inhibition of CYP2C19 | Omeprazole (20 mg) was given orally as a probe substrate to test the effect of cenobamate (200 mg/day) on CYP3A activity | Prescribing information [77]; Greene et al. [94] |
AUC area under the plasma drug concentration–time curve, CYP cytochrome P450
Interactions with concomitantly administered ASMs require special consideration (Table 1). By inhibiting CYP2C19, cenobamate increases the plasma concentration of phenytoin, phenobarbital and N-desmethylclobazam, the active metabolite of clobazam [77, 95, 96]. These interactions are clinically relevant and often require a reduction of the dose of the affected ASMs [95–98]. Preliminary evidence suggests that the interaction between cenobamate and clobazam might also have a pharmacodynamic component, leading to improved seizure control when cenobamate is combined with low-dose clobazam [96]. By inducing CYP3A4 and, probably, enzymes involved in glucuronide conjugation, cenobamate can cause a modest-to-moderate reduction in the plasma concentration of carbamazepine and lamotrigine, whereas the plasma concentrations of valproic acid, levetiracetam or lacosamide are generally unaffected [77, 99].
In addition to interactions involving pharmacokinetic changes, a pharmacodynamic interaction has been reported between cenobamate and sodium channel-blocking ASMs. Patients receiving sodium channel blockers such as lacosamide, carbamazepine, oxcarbazepine, lamotrigine and phenytoin have been reported to exhibit a reduced therapeutic response to cenobamate, owing to the early appearance of dose-limiting adverse effects, especially dizziness [98, 100]. Accordingly, as discussed in a recent expert opinion paper, a reduction in the dose of sodium channel blockers may be required to improve tolerability and fully exploit the therapeutic potential of cenobamate [98]. Although the clobazam dose also often needs to be reduced after adding cenobamate to compensate for the pharmacokinetic interaction leading to increased N-desmethylclobazam levels [95, 96], a recent report has suggested that cenobamate and low-dose clobazam may also exhibit a pharmacodynamic interaction, resulting in therapeutic synergism [98].
Interactions with other medications that are substrates of CYP enzymes affected by cenobamate also need to be considered. Although no formal interaction study with steroid contraceptives has been reported, induction of steroid metabolism by cenobamate can be expected. Accordingly, prescribing information recommends that women taking oral contraceptives use additional or alternative non-hormonal birth control while taking cenobamate [77].
Clinical Efficacy
The efficacy of cenobamate in the treatment of focal seizures has been established in two randomised, double-blind, parallel-group, placebo-controlled, adjunctive-therapy trials. In the first of these trials, 222 patients with uncontrolled focal seizures aged 18–61 years were allocated to treatment with cenobamate (200 mg once daily, n = 113) or placebo (n = 109) added-on to their pre-existing therapy with one to three ASMs [101]. The double-blind period lasted 12 weeks and comprised a 6-week titration phase and a 6-week maintenance phase. The median percent reduction in seizure frequency from baseline (primary endpoint) was significantly greater for the cenobamate group compared with the placebo group (55.6% vs 21.5%; p < 0.0001). The proportion of patients attaining a ≥ 50% reduction in seizure frequency from baseline (responder rate) was 50.4% for the cenobamate group compared with 22.2% for the placebo group (p < 0.0001).
The efficacy of cenobamate was subsequently confirmed in a pivotal trial conducted in a total of 437 adults with uncontrolled focal seizures despite treatment with one to three ASMs [102]. After a prospective 8-week baseline, patients were assigned to receive add-on treatment with cenobamate 100 mg/day (n = 108), 200 mg/day (n = 110), 400 mg/day (n = 111) or placebo (n = 108) according to a dosing schedule that comprised a 6-week titration phase and a 12-week maintenance phase. For both co-primary efficacy endpoints (median percent reduction in seizure frequency from baseline over the entire treatment period and responder rate over the maintenance dose period), there was a clear dose–response relationship with a statistically significant superiority of cenobamate over placebo at each of the doses tested (Fig. 3). The proportion of patients free from seizures during the maintenance phase (calculated as a percentage of those with any maintenance phase seizure data) was 1% for the placebo group and 4% (not significantly different from placebo), 11% (p < 0.01) and 21% (p < 0.001) for the cenobamate 100-mg/day, 200-mg/day and 400-mg/day dose groups. Seizure freedom rates were also assessed by counting those patients who were free from seizures during the maintenance phase and completed the study (i.e. by excluding participants who discontinued the study treatment prematurely) as the numerator and the number of treated patients who had any post-baseline seizure data as the denominator. By using this more conservative and more clinically meaningful assessment, seizure fredom rates were 1% for the placebo group compared with 3%, 9% and 14% for the cenobamate 100-mg/day, 200-mg/day and 400 mg/day dose groups. For the 200-mg/day and 400-mg/day doses, these seizure freedom rates are considerably higher than those reported in similarly designed trials of other second-generation ASMs [103]. All seizure types assessed, namely focal aware motor seizures, focal impaired awareness seizures and focal to bilateral tonic-clonic seizures are significantly reduced by cenobamate [101, 102]. Cenobamate was found to be efficacious regardless of baseline seizure frequency, disease duration and the number of concomitant ASMs [104, 105], as well as the number of previously failed treatments [106]. Favourable responses have also been reported in patients whose epilepsy surgery was not successful [106, 107].
Fig. 3.

Efficacy outcomes for the two co-primary efficacy endpoints of the pivotal, placebo-controlled, adjunctive-therapy trial of cenobamate in adults with uncontrolled focal seizures. The median percent change in seizure frequency from the baseline endpoint was calculated in the modified intent-to-treat population (randomised patients who took at least one dose of the study medication and had any post-baseline seizure data), which included 106 patients for the placebo group and 108, 109 and 111 patients for the cenobamate 100-mg, 200-mg, and 400-mg dose groups, respectively. The responder rate endpoint (proportion of patients with at least a 50% reduction in seizure frequency from baseline) was calculated in the modified intent-to-treat maintenance phase population (randomised patients who took at least one dose of study medication in the maintenance phase and had any maintenance phase seizure data), which included 102 patients for the placebo group and 102, 98 and 95 patients for the cenobamate 100-mg, 200-mg, and 400-mg dose groups, respectively. Redrawn based on data from Krauss et al. [102]
In open-label studies, the reduction in seizure frequency attained with cenobamate was generally found to be sustained during a long-term follow-up [108–110]. Improvement in seizure frequency has also been reported to be associated with an improved quality of life [111]. In a recent open-label follow-study of 354 patients who participated in the pivotal double-blind trial, 86 individuals (24.3%) achieved a ≥ 90% seizure reduction and 36 (10%) were seizure free during > 36 to 48 months [112]. A follow-up study of 1844 patients enrolled in cenobamate trials found that retention on treatment, a combined measure of efficacy and tolerability, was 71% after 2 years, which compares favourably with retention rates reported for other ASMs [113]. A recent analysis of data from the cenobamate clinical development programme found that the all-cause mortality rate among 2132 cenobamate-treated adults (4.0 per 1000 person-years) did not differ significantly from that expected in the general population [114]. The unaltered mortality rate was considered to be probably owing to improved seizure control and, consequently, a reduced risk of sudden unexpected death in epilepsy. The analysis, however, did not include a concurrently assessed control group, and the findings require confirmation by larger studies using an improved design. Given that cenobamate possesses at least two known molecular activities, namely, inhibition of voltage-gated sodium (both phasic and persistent current) and activation of GABAA receptors (both synaptic and extrasynaptic), it is not clear which of these is primarily responsible for the antiseizure efficacy seen in pivotal trials, or whether the combination of effects on both excitatory and inhibitory mechanisms is crucial for clinical efficacy [81].
Adverse Effects
The most commonly reported treatment-emergent adverse events (TEAEs) reported in cenobamate placebo-controlled trials include somnolence, dizziness, fatigue and coordination disorders, all of which are dose related [101, 102] (Table 2). Gastrointestinal adverse events also occurred with a higher frequency among cenobamate-treated patients than among those receiving placebo. The rates of discontinuation because of adverse events were 11%, 9% and 21% for patients randomised to cenobamate 100 mg/day, 200 mg/day and 400 mg/day, respectively, compared with 4% for those randomised to placebo [77]. The most common events leading to discontinuation were ataxia, dizziness, somnolence, diplopia, nystagmus and vertigo. Adverse events reported in open-label long-term trials were consistent with those recorded in double-blind trials [109, 110, 113]. Central nervous system-related adverse events, particularly those appearing during titration or early during maintenance, can often be managed by reducing the dose of concomitant ASMs [113, 115]. In a small adjunctive-therapy study in patients with drug-refractory epilepsy, cenobamate maintenance doses between 50 and 250 mg/day were not found to be associated with impaired cognitive performance [116], but these findings require confirmation in controlled studies and assessment of higher doses.
Table 2.
Frequency of the most commonly reported adverse reactions associated with cenobamate based on a pooled analysis of tolerability data from two randomised, double-blind, placebo-controlled trials [101, 102] as reported in the prescribing information [77]. The Table includes reactions reported for any cenobamate arm with a frequency ≥5% over placebo
| Adverse reaction | Placebo (n = 216) (%) | Cenobamate 100 mg/day (n = 108) (%) | Cenobamate 200 mg/day (n = 223) (%) | Cenobamate 400 mg/day (n = 111) (%) |
|---|---|---|---|---|
| Somnolence | 11 | 19 | 22 | 37 |
| Dizziness | 15 | 18 | 22 | 33 |
| Fatigue | 7 | 12 | 14 | 24 |
| Diplopia | 2 | 6 | 7 | 15 |
| Balance disorder | 1 | 3 | 5 | 9 |
| Nausea | 3 | 6 | 6 | 9 |
| Constipation | 0 | 2 | 4 | 8 |
| Gait disturbance | 1 | 1 | 3 | 8 |
| Nystagmus | 0 | 3 | 7 | 6 |
| Dysarthria | 0 | 2 | 1 | 7 |
| Vertigo | 1 | 1 | 1 | 6 |
| Vomiting | 0 | 2 | 4 | 5 |
| Diarrhoea | 0 | 1 | 3 | 5 |
Three confirmed cases of drug rash with eosinophilia and systemic symptoms, including one fatality, were reported during the early phases of cenobamate development, following exposure to titration rate faster than currently recommended [97]. To mitigate the risk of serious idiosyncratic reactions, initiation of treatment with a very low dose (12.5 mg/day) followed by a slow dose escalation is currently recommended. To facilitate titration, cenobamate tablets are available in six different dose strengths, namely 12.5, 25, 50, 100, 150 and 200 mg [77, 78]. No cases of drug rash with eosinophilia and systemic symptoms were observed when the recommended titration schedule was applied in a prospective adjunctive-therapy open-label trial in a total of 1399 adults with focal seizures [97].
Mode of Use, Current Therapeutic Role and Future Perspectives
Cenobamate is currently approved only for use in adults with focal seizures [77, 78]. Treatment should be started at a dose of 12.5 mg/day and increased every other week over 12 weeks to 25 mg/day (weeks 3 and 4), 50 mg/day (weeks 5 and 6), 100 mg/day (weeks 7 and 8) and 150 mg/day (weeks 9 and 10) up to the recommended initial target dose of 200 mg/day (weeks 11 and 12, and onwards). The maximum recommended daily dose is 400 mg/day [77, 78]. Cenobamate is administered once daily.
If cenobamate is used in patients with renal impairment, caution should be exercised and dose adjustment should be taken into consideration [77, 78]. According to European prescribing information, the maximum recommended dose in patients with mild, moderate or severe renal impairment is 300 mg/day [78]. Use of cenobamate in patients with end-stage renal disease or patients undergoing dialysis is not recommended [77, 78]. Caution is recommended in patients with mild-to-moderate hepatic impairment, in whom the cenobamate dose should not exceed 200 mg/day, and a further dose reduction should be considered [77, 78]. Use of cenobamate in patients with severe hepatic impairment is currently not recommended. Finally, special caution is recommended in patients aged older than 65 years because of a greater frequency in these patients of renal, hepatic and cardiac dysfunction, as well as potentially interacting comedications [77, 78].
At present, cenobamate is generally used as adjunctive therapy for patients with focal epilepsy whose seizures have not been controlled by adequate doses of other ASMs. European prescribing information limits the utilisation of cenobamate to individuals who have not been adequately controlled despite a history of treatment with at least two ASMs [78]. A number of expert panel-based publications provide suggestions on the most appropriate candidates for cenobamate therapy, and its optimal use in specific populations [98, 117, 118]. It has been emphasised that an adjustment in the dose of concomitant ASMs may be needed to minimise potential adverse effects resulting from pharmacokinetic and pharmacodynamic drug interactions [98].
In general, cenobamate may not be the most appropriate choice for individuals who have a history of drug rash with eosinophilia and systemic symptoms or severe hypersensitivity reactions to other drugs [117]. Cenobamate shortens the QT interval, and should not be used in patients with familial short QT syndrome because of the risk of precipitating life-threatening arrhythmias [77, 119]. Caution is also required when using cenobamate adjunctively to other drugs that shorten the QT interval, such as digitalis, primidone and rufinamide [119].
Because of the need for slow titration, cenobamate is not an appropriate choice when a rapid onset of action is required [117]. The need for slow titration, however, is not a significant problem in patients with long-standing drug-resistant epilepsy, and cenobamate may be a preferred choice in these individuals because the probability of achieving seizure freedom appears to be higher with cenobamate than with other available ASMs [93, 117, 118, 120]. Of note, an improvement in seizure control during cenobamate treatment can be expected before completion of the titration phase because a significant reduction in seizure frequency can occur at doses as low as 50 mg/day [118, 121]. In a recent study from the UK, cenobamate was reported to have superior cost effectiveness to other recently introduced ASMs based on indirect estimates of comparative efficacy [122]. Yet, its utilisation to date appears to have been surprisingly limited, as indicated by the fact that 2 years after its introduction in the US market less than 5% of adults with drug-resistant focal epilepsy were found to be receiving the drug [123]. Lack of awareness about the therapeutic potential of cenobamate and restrictions to access created by healthcare payors, hospitals and regulatory agencies were considered to have contribute to its low utilisation.
Despite promising preliminary results from small studies in patients aged 10–18 years [124, 125], there is at present insufficient information to make recommendations for use in paediatric populations. A dose-escalation study to evaluate the pharmacokinetics and safety profile of cenobamate in children aged 2–18 years with focal seizures is ongoing (NCT04903314), together with a paediatric safety and efficacy open-label study (NCT05067634). Based on results from animal studies suggesting broad-spectrum antiseizure activity [80], there is also considerable interest in assessing potential effectiveness against seizure types other than focal. A randomised, double-blind, placebo-controlled trial to assess the efficacy of cenobamate in the treatment of generalised tonic-clonic seizures in patients aged 12 years and above with idiopathic generalised epilepsy is in progress (NCT03678753). Participants in the latter studies are offered the opportunity to continue treatment in an open-label extension trial (NCT03961568). There have also been preliminary case reports suggestive of potential usefulness in the management of adults with Dravet syndrome [126, 127], Lennox–Gastaut syndrome [127, 128], other forms of combined and generalised epilepsy [127], and generalised tonic-clonic seizures associated with idiopathic generalised epilepsy [127]. Cenobamate has been reportedly effective in controlling super-refractory status epilepticus in two patients [129]. Although a slow titration was used, the dose was increased at a faster rate than recommended, and related safety concerns should be carefully considered before using cenobamate in this situation.
Ganaxolone
Over the years, extensive evidence has accrued on the role of endogenous neurosteroids such as allopregnanolone in modulating neurotransmission, and the possibility of using these compounds or various synthetic derivatives for the treatment of various CNS disorders [43, 130]. Although several properties can contribute to the actions of neurosteroids [43, 131, 132], positive allosteric modulation of synaptic and extrasynaptic GABAA receptors appears to be the major mechanism underlying their antiseizure effects [43, 133–135]. Allopregnanolone itself has anticonvulsant and possibly antiepileptogenic activity in animal models of seizures and epilepsy [53, 136–138], but its use in the long-term oral treatment of epilepsy has been prevented by its low oral bioavailability, owing to its high presystemic metabolic clearance. An intravenous formulation of allopregnanolone (brexanolone, SAGE-547) underwent clinical studies as a potential treatment for super-refractory status epilepticus [138–140] but this indication was not pursued after the compound failed to meet the primary efficacy endpoint in a phase III trial [141]. Brexanolone was eventually developed and marketed as a treatment for postpartum depression [142]. Different approaches to circumvent the oral bioavailability limitations of allopregnanolone include the use of prodrugs, such as LIT-300, or synthetic derivatives with improved pharmacokinetic properties, such as ganaxolone, zuranolone (SAGE-217) and ETX-155 [43, 140]. Among these compounds, ganaxolone has been the most extensively characterised preclinically and clinically in the treatment of seizure disorders.
Ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one) is a methyl analogue of allopregnanolone. The addition of a methyl moiety at position 3β of the steroid skeleton transforms the endogenous compound allopregnanolone into the exogenous compound ganaxolone, a switch that has pharmacokinetic and biopharmaceutic implications, including the feasibility of oral administration. Ganaxolone is currently approved in the USA for the treatment of seizures associated with CDKL5 deficiency disorder in patients 2 years of age and older [143].
Pharmacology
Ganaxolone and allopregnanolone belong to the group of neurosteroids that lack hormonal activity and act as positive allosteric modulators at GABAA receptors [6, 138]. While benzodiazepines bind only to synaptic GABAA receptors, ganaxolone acts at both synaptic and extrasynaptic sites [138, 144]. Synaptic GABAA receptors are internalised during prolonged seizures, becoming functionally inactive and resistant to benzodiazepines as seizure duration increases. Ganaxolone-responsive extrasynaptic GABAA receptors, containing α and δ subunits, do not internalise, and do not become functionally inactive during prolonged seizure activity, which makes ganaxolone an attractive candidate for the treatment of status epilepticus [145]. Ganaxolone is not known to possess any other molecular activities and its antiseizure properties are thought to reside primarily in its ability to potentiate the actions of GABA at synaptic and extrasynaptic GABAA receptors.
Early studies on the pharmacological profile of ganaxolone demonstrated its effectiveness against pentylenetetrazole-induced clonic convulsions in mice and rats with median effective dose (ED50) values of 4.3 and 7.8 mg/kg intraperitoneally (i.p.), respectively, and against seizures induced by bicuculline (ED50 = 4.6 mg/kg, i.p.), tert-butylbicyclophosphorothionate (ED50 = 11.7 mg/kg, i.p.) and aminophylline (ED50 = 11.5 mg/kg, i.p.) in mice [146]. In the maximal electroshock model in mice, ganaxolone ED50 (29.7 mg/kg, i.p.) was similar to its median neurotoxic dose (TD50) in the rotarod test (33.4 mg/kg, i.p.) [146].
Further experiments have confirmed the protective activity of ganaxolone in a wide range of models of seizures and epilepsy [147], including the amygdala/hippocampal kindling [148, 149], the pentylenetetrazole kindling [150] and cocaine kindling [151] models in mice, the corneal kindling model in rats [146], cocaine-induced convulsions in mice [152] flurothyl-induced seizures in rats [153], N-methyl-D-aspartate-induced seizures in a rat model of cryptogenic, infantile, epileptic infantile spasms syndrome [154], seizures induced by ethanol withdrawal in mice [155] and seizures induced by 6-Hz stimulation in mice [156]. Ganaxolone has also been found to be effective in counteracting the reduction in the seizure threshold induced by neurosteroid withdrawal in a rat model of catamenial epilepsy [157], and in protecting against seizures induced by perinatal asphyxia in neonatal lambs [158]. In some models, ganaxolone interacts synergistically with the GABAergic agents tiagabine and midazolam and with cannabidiol in protecting against seizures [149, 159, 160]. However, exacerbation of absence-like seizures by ganaxolone has been reported in the low-dose pentylenetetrazole and the gamma-hydroxybutyric acid models of absence seizures in rats [161]. In another animal model of absence seizures, the WAG/Rij rat, ganaxolone caused seizure aggravation or seizure protection depending on the dose and the site of the brain into which it was injected [162].
The potential value of ganaxolone in the treatment of status epilepticus has been evaluated in several animal models. Specifically, ganaxolone has been found to be active against status epilepticus induced by tetramethylenedisulfotetramine in mice [163], diazepam-resistant lithium-pilocarpine-induced status epilepticus in rats [164], pilocarpine-induced status epilepticus in mice [145], and status epilepticus induced by the organophosphate compounds soman and diisopropylfluorophosphate in rats [165]. Interestingly, the potency of ganaxolone in the pilocarpine-induced status epilepticus model, the kindling model and the 6-Hz model in mice was greater in female animals than in male animals [145]. This finding was ascribed to a greater abundance in female animals of extrasynaptic GABAA receptors containing the δ subunit (δGABA receptors), consistent with the observation that no sex differences in seizure protection were recorded in animals lacking extrasynaptic δ-containing GABAA receptors.
There is suggestive evidence for ganaxolone having anti-apoptotic and neuroprotective activity [158, 166, 167] and antiepileptogenic effects [150] in some models. Ganaxolone has also been reported to protect against audiogenic seizures and improve behavioural deficits in a mouse model of Angelman syndrome [168] and to ameliorate neurobehavioural outcomes in male guinea pigs born preterm [169].
Biopharmaceutics and Clinical Pharmacokinetics
Because ganaxolone has very low estimated water solubility of 0.71 mg/L [170] and a high lipophilicity (logP = 4.0) [87], it classifies by both Biopharmaceutics Classification System and Biopharmaceutics Drug Disposition Classification System criteria as a Class 2 drug, namely as a poorly water-soluble, highly-permeable drug eliminated by metabolism in humans [10, 87–89].
In single-dose pharmacokinetic studies in healthy adults, a linear and approximately dose-proportional increase in AUC and Cmax values of ganaxolone have been reported over the 50–600 mg dose range [171]. At doses between 900 and 1500 mg, AUC also increased linearly with dose, whereas the increase in Cmax was less than dose proportional. In these studies, Cmax was generally reached within 1–3 h after dosing. Thereafter, plasma ganaxolone concentrations declined biexponentially, with an early-phase half-life of about 7–11 h and a late-phase terminal half-life of 35–40 h [172, 173]. Pharmacokinetic parameters during multiple dosing using a variety of dosing paradigms were consistent with those reported after intake of single doses [171, 172].
In general, the oral bioavailability of Biopharmaceutics Drug Disposition Classification System Class 2 drugs increases after co-administration with a high-fat meal, owing to inhibition of efflux transporters in the intestine and facilitated solubilisation of the drug in the intestinal lumen [89]. In the early studies discussed above, ganaxolone was formulated as a β-cyclodextrin complex and exhibited a very prominent food effect, with increases in AUC of five-fold to 15-fold when the product was given with food compared with the fasting state [172]. Since then, the manufacturer has developed a variety of formulations, including the currently marketed suspension form and a capsule form, which do not contain β-cyclodextrin [138, 172]. These formulations show improved bioavailability when taken in the fasting state, and a reduced food effect when taken with a meal [172]. When the currently marketed oral suspension was co-administered with a high-fat meal, Cmax and AUC values increased by three-fold and two-fold, respectively, compared with administration under fasted conditions [143]. These findings indicate that the oral bioavailability of ganaxolone is incomplete, at least when the drug is taken in the fasting state. The absolute oral bioavailability of ganaxolone has not been established because no pharmacokinetic studies following intravenous and oral doses appear to have been performed.
In the efficacy studies that led to regulatory approval, ganaxolone was taken with food to maximise its oral bioavailability. Accordingly, as per prescribing information, ganaxolone should be taken with food because its efficacy ‘when administered in the fasting state is unknown’ [143]. Steady-state plasma ganaxolone concentrations were reached within 48–72 hours when dosed with food [138].
The fraction of ganaxolone bound to plasma protein is approximately 99%. Ganaxolone undergoes a highly complex metabolism [174]. No less than 59 different primary and secondary metabolites have been identified. The inducible enzyme CYP3A4/5 appears to be the main enzyme responsible for ganaxolone metabolism [172]. Other enzymes involved in ganaxolone metabolism include CYP2B6, CYP2C19 and CYP2D6 [143] as well as phase II enzymes [174]. Following a single oral dose of 300 mg of [14C]-ganaxolone to healthy male subjects, 55% and 18% of the total radioactivity was recovered in faeces and in urine, respectively, almost exclusively in the form of metabolites [143, 174, 175].
Details of the pharmacokinetics of ganaxolone in special populations have not been published. According to prescribing information, after accounting for body weight, pharmacokinetic exposures among patients included in the safety and efficacy trial of ganaxolone in CDKL5 deficiency disorder ‘were comparable across the age groups 2 to less than 6 years of age (n = 45), 6 to less than 12 years of age (n = 28), and 12 to less than 18 years of age (n = 16)’ [143].
Drug Interactions
In in vitro studies, ganaxolone was not found to be an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4/5 or an inducer of CYP1A2, CYP2B6 or CYP3A4/5 at clinically relevant concentrations [143]. In line with these findings, co-administration of ganaxolone at steady state (400 mg twice daily) with midazolam, a sensitive CYP3A4 substrate, in healthy subjects did not result in clinically relevant changes in plasma midazolam exposure [138, 143]. Ganaxolone is not a substrate of breast cancer resistance protein, P-glycoprotein, organic cation transporter 1, organic cation transporter 2, organic anion transporter polypeptide B1, or organic anion transporter polypeptide B3 and does not inhibit these transporters nor the multidrug and toxic compound extrusion 1 transporter, multidrug and toxic compound extrusion 2K transporter or the bile salt export pump at clinically relevant concentrations [143].
In healthy subjects, co-administration of ganaxolone with rifampicin (a strong inducer of CYP2C19 and CYP3A4, and a moderate inducer of CYP2B6) decreased ganaxolone Cmax and AUC by 57% and 68%, respectively, an interaction that is likely to be clinically significant. Co-administration of ganaxolone with the strong CYP3A4 inhibitor itraconazole in healthy subjects increased ganaxolone AUC by 17% but did not change its Cmax. Changes in ganaxolone exposure when co-administered with strong, moderate or weak CYP3A4 inhibitors are not expected to be clinically significant [143]. Clinical studies assessing the interaction between ganaxolone and other ASMs have not been reported to date, although serum drug measurements in a controlled trial conducted in patients with focal seizures found that plasma ganaxolone concentrations were approximately 40% lower in patients taking enzyme-inducing ASMs compared with patients not taking enzyme inducers [176].
Clinical Efficacy
CDKL5 Deficiency Disorder
CDKL5 deficiency disorder is a severe developmental and epileptic encephalopathy caused by pathogenic variants of the CDKL5 gene located on the X-chromosome [177]. It is an ultra-rare condition, with an estimated incidence of 2.36 per 100,000 livebirths [178]. Clinical manifestations generally appear in the first 3 months of life, may affect the function of a wide a variety of organs and systems, and typically include prominent hypotonia, severe intellectual disability, cortical visual impairment, and clusters of infantile spasms and tonic seizures, though other seizure types may also occur [177–179]. Seizures are typically drug resistant.
Following encouraging findings from an open-label exploratory trial of ganoxolone in a small group of children with CDKL5 deficiency disorder [180], a randomised, double-blind, placebo-controlled, adjunctive-therapy trial was successfully completed [181]. The MARIGOLD trial enrolled 101 patients with a pathogenic or probably pathogenic CDKL5 variant and uncontrolled seizures despite treatment with up to four concomitant ASMs. The mean age of participants was 6.8 years (range 2–19 years) and approximately 78% were female [143]. After a 6-week prospective baseline, patients were randomised 1:1 to receive ganaxolone (up to a maximum dose 63 mg/kg/day for patients weighing ≤ 28 kg, or 1800 mg/day for patients weighing > 28 kg) or placebo for a 6-week titration period, which was followed by a 13-week maintenance phase. Study medications were administered as a suspension, dosed three times daily with food. The median percentage change from baseline in the 28-day frequency of major motor seizures (bilateral tonic, generalised tonic-clonic, bilateral clonic, atonic, or focal to bilateral tonic-clonic seizures) over the 17-week treatment period (primary endpoint) was − 30.7% for the ganaxolone group compared with − 6.9% for the placebo group (p = 0.004) (Fig. 4). The responder rate for major motor seizure frequency was 24% in the ganaxolone group versus 10% in the placebo group (p = 0.064). Preliminary data from an open-label extension study suggest that seizure reduction during ganaxolone treatment is maintained over a longer term follow-up [182].
Fig. 4.

Efficacy outcomes of the pivotal, placebo-controlled, adjunctive-therapy trial of ganaxolone in patients with cyclin-dependent kinase-like 5 deficiency disorder. The median percent change in major motor seizure frequency from baseline (primary efficacy endpoint) was calculated in a modified intent-to-treat population (randomised patients who took at least one dose of the study medication and had evaluable baseline seizure data), which included 51 patients for the placebo group and 49 patients for the ganaxolone group. The same population was included in the calculation of the responder rate (proportion of patients with at least a 50% reduction in seizure frequency from baseline), which was was a key secondary efficacy endpoint. Drawn based on data from Knight et al. [181]
Drug-Resistant Focal Epilepsy
In an early double-blind, placebo-controlled, proof-of-concept study that used a presurgical design, ganaxolone (1500 mg/day on day 1 followed by 1875 mg/day on days 2–8) failed to reach the primary efficacy endpoint, even though other endpoints provided signals suggestive of an antiseizure effect [183]. A subsequent phase II, double-blind, adjunctive-therapy trial randomised 147 adults with uncontrolled focal seizures to receive either ganaxolone (n = 98) or placebo (n = 49) over a 10-week period, which included a 2-week titration phase (up 1500 mg/day) and an 8-week maintenance phase [176]. The mean percent change in seizure frequency from baseline was − 17.6% in the ganaxolone group, compared with 2.0% in the placebo group (p = 0.014). The responder rate (24% ganaxolone, 15% placebo) did not differ significantly between the two groups (p = 0.19). Although the effect size in this study was modest, the results were considered to support continued development of ganaxolone for this indication. In an open-label extension of the same trial, 24% of patients were reported to have experienced a 50% reduction in seizure frequency compared with baseline after treatment for a mean of 274 days and doses that ranged between 900 and 1500 mg/day [138].
A phase III, placebo-controlled, double-blind, adjunctive-therapy trial has been completed, but results have been published only in abstract form [184]. In this study, adults with drug-resistant focal epilepsy were randomised to receive ganaxolone (1800 mg/day, n = 179) or placebo (n = 180). The double-blind phase consisted of a 2-week titration period and a 12-week maintenance period. The median percent reduction in seizure frequency from baseline (primary endpoint) was 21.3% for the ganaxolone group compared with 10.2% for the placebo group, a non-statistically significant difference (p = 0.15). Responder rates also did not differ significantly between groups (ganaxolone 28.7% vs placebo 22.7%, p = 0.21).
Protocadherin 19 (PCDH19)-Clustering Epilepsy
PCDH19-clustering epilepsy is an X-linked developmental and epileptic encephalopathy affecting predominantly female individuals, with the onset of symptoms generally in the first 3 years of life [177]. The syndrome is characterised by the occurrence of focal seizures (often associated with typical fearful screaming) and, in some cases, also tonic-clonic seizures and atypical absences. Seizures often occur in clusters, which may be precipitated by fever. Associated manifestations may include intellectual disability and behavioural and psychiatric disorders [177].
Based on evidence that impaired steroidogenesis may play a pathogenic role in PCDH19-clustering epilepsy [185], ganaxolone has been tested in a double-blind, randomised, adjunctive-therapy trial in 21 girls with this rare condition [186, 187]. The trial consisted of a 12-week prospective baseline followed by a 17-week double-blind phase, that included a 4-week titration phase and a 13-week maintenance phase. Ganaxolone was titrated up to a dose of 63 mg/kg/day (maximum 1800 mg/day), given in three divided administrations with food. The median percent reduction on seizure frequency from baseline was 61.5% in the ganaxolone group (n = 10) and 24.0% in the placebo group (n = 11), a difference that failed to reach statistical significance probably owing to the wide variation in intervals between seizure clusters [186, 187]. Results did not change after a re-assessment of seizure outcomes according to pre-treatment blood levels of allopregnanolone sulphate, which was included as a potential responsiveness biomarker based on results from an earlier exploratory study [147, 173].
Drug-Resistant Status Epilepticus
Because of the efficacy of ganaxolone in animal models of drug-resistant status epilepticus [145, 163–165] and retained effectiveness when tolerance develops to benzodiazepines [188], there has been considerable interest in assessing its potential clinical effectiveness in the treatment of refractory status epilepticus. As ganaxolone has low water solubility, an ad hoc intravenous formulation had to be developed utilising as an excipient Captisol®, a sulfobutylether-β-cyclodextrin [175, 189]. Preliminary studies with this formulation in patients with refractory convulsive and nonconvulsive status epilepticus yielded promising results [189, 190]. Of note, in a phase II dose-finding study in 17 patients aged 12 years and older, no patient required escalation to intravenous anaesthetic treatment within 24 hours of ganaxolone initiation, which was the study’s primary endpoint [190]. The median time to seizure cessation was 5 minutes after ganaxolone initiation. A randomised, double-blind, placebo-controlled trial (NCT04391569) in patients aged 12 years and older with refractory status epilepticus is currently ongoing.
Other Seizure Disorders
Because of evidence relating GABAergic dysfunction to seizure activity in epilepsy associated with tuberous sclerosis complex [191], a formal phase II, exploratory, adjunctive-therapy open-label study of ganaxolone (up to 63 mg/kg/day, with a maximum of 1800 mg/day) has been conducted in 23 patients with tuberous sclerosis complex aged 2–32 years. The median percent reduction in seizure frequency during the 12 weeks of ganaxolone treatment compared with baseline was 16.6% [192]. A phase III, double-blind, randomised, placebo-controlled, adjunctive-therapy trial (NCT05323734) to assess the efficacy of ganaxolone in children and adults with tuberous sclerosis complex-related epilepsy is ongoing, with the opportunity for participants to be enrolled in an open-label extension study (NCT05604170).
Open-label exploratory studies in paediatric patients with focal and generalised epilepsies [172, 193, 194], including patients with epileptic spasms [172, 195], reported an improvement in seizure frequency with ganaxolone, but these findings are difficult to interpret because of the lack of a control group. A randomised, double-blind, placebo-controlled, crossover, adjunctive-therapy trial in 56 patients aged 4–24 months with drug-resistant infantile epileptic spasms syndrome has been completed. The primary outcome was the epileptic spasm frequency assessed by a 24-hour video electroencephalogram at day 9. The results have not been published in detail, but ganaxolone treatment reportedly did not result in a significant reduction in epileptic spasm frequency in this study [147].
Studies in Non-Epilepsy Indications
In small, randomised, double-controlled trials, ganaxolone could not be differentiated from placebo in the treatment of anxiety, hyperactivity and attention deficits in children and adolescents with Fragile X syndrome [196], and in the treatment of symptoms of post-traumatic stress-disorder in veteran or civilian adult outpatients [197]. In a recent open-label pilot study in ten postmenopausal women with persistent depression despite adequate antidepressant therapy, treatment with ganaxolone (up to 450 mg twice daily if tolerated) was associated with an improvement in depression scores [198], but the uncontrolled design complicates the interpretation of these results. Ganaxolone on a twice-daily dosing regimen was not well tolerated in this study, with sleepiness, fatigue and dizziness being commonly reported.
Adverse Effects
In the MARIGOLD trial in CDKL5 deficiency disorder, ganaxolone was generally well tolerated, the most common TEAEs being somnolence, pyrexia and upper-respiratory tract infection (Table 3). Treatment-emergent adverse events leading to a dose reduction or temporary discontinuation of the study medication were reported in 22% of patients in the ganaxolone group and in 16% of patients in the placebo group [181]. Overall, 48 of 50 patients randomised to ganaxolone (96%) and 47 of 51 randomised to placebo (92%) completed the double-blind phase. In the large phase III trial in adults with drug-resistant focal seizures, the most common adverse events reported in the ganaxolone group (n = 179) and placebo group (n = 180) were somnolence (23.5% vs 4.5%), dizziness (19.6% vs 4.5%) and fatigue (11.7% vs 6.8%) [184].
Table 3.
Frequency of most commonly reported adverse reactions associated with ganaxolone in the randomised, double-blind, placebo-controlled trial in patients with cyclin-dependent kinase-like 5 deficiency disorder [181] as reported in the US prescribing information [143]. The Table includes reactions reported for the ganaxolone arm with a frequency ≥ 4% over placebo
| Adverse reaction | Placebo (n = 51) (%) | Ganaxolone (n = 50) (%) |
|---|---|---|
| Somnolence | 20 | 38 |
| Pyrexia | 8 | 18 |
| Upper respiratory tract infection | 6 | 10 |
| Seasonal allergy | 0 | 6 |
| Salivary hypersecretion | 2 | 6 |
| Bronchitis | 0 | 4 |
The safety profile of ganaxolone was further evaluated in a pooled analysis of data from placebo-controlled trials in epilepsy and other neuropsychiatric disorders, including 1101 participants allocated to ganaxolone and 743 allocated to placebo [199]. The most common TEAEs reported with a higher frequency in ganaxolone-treated subjects were somnolence (ganaxolone 22.4%, placebo 8.1%), dizziness (ganaxolone 12.6%, placebo 3.9%) and fatigue (ganaxolone 9.3%, placebo 4.8%). Central nervous system-related TEAEs appeared to be dose related. No significant changes were recorded in body weight and there were no clinically significant trends for electrocardiogram parameters or vital signs. The frequency of serious TEAS was 2.8% in the ganaxolone-treated group and 3.8% in the placebo-treated group.
Mode of Use, Current Therapeutic Role and Future Perspectives
Ganaxolone oral suspension (50 mg/mL) is currently approved in the USA for the treatment of seizures associated with CDKL5 deficiency disorder in patients 2 years of age and older. The recommended titration schedule and maintenance dosage are based on body weight, and the maximum total daily dose after titration is 63 mg/kg/day for patients weighing ≤ 28 kg, and 1800 mg/day for those weighing > 28 kg [143]. Ganaxolone should be taken three times daily with food. The same product should become commercially available in Europe in the near future because on 25 May, 2023, the European Medicines Agency’s Committee for Medicinal Products for Human Use adopted a positive opinion recommending its approval for the treatment of seizures associated with CDKL5 deficiency disorder in patients 2–17 years of age, with the opportunity to continue treatment in patients 18 years of age and older [200].
The treatment of CDKL5 deficiency disorder is challenging because seizures are usually resistant to ASMs. Management usually involves polypharmacy with a variety of ASMs, and any improvement in seizure control after treatment changes is often short-lived [178, 201]. Ganaxolone is the first medication approved specifically for this condition, and it represents a welcome addition to the existing armamentarium. Further data from postmarketing experience are required to establish its place in the treatment algorithm. At least in some settings, utilisation of this newer medication is likely to be limited by cost considerations. According to US prescribing information, concomitant use of enzyme-inducing ASMs (e.g. carbamazepine, phenytoin, phenobarbital and primidone) should be preferably avoided in patients taking ganaxolone because the reduction in plasma ganaxolone concentrations caused by these agents may decrease its therapeutic efficacy. If enzyme-inducing ASMs are combined, the ganaxolone dose may need to be increased, without however exceeding the maximum recommended dose [143]. Because ganaxolone undergoes hepatic metabolism, its dose may need to be reduced in patients with hepatic impairment [143].
Ganaxolone is currently undergoing further investigations for potential additional indications. Randomised controlled trials are scheduled or ongoing in patients with CDLK5 deficiency disorder aged 6 months to less than 2 years (NCT05249556), in patients with seizures associated with TSC-related epilepsy (NCT05323734) and in patients with refractory status epilepticus (NCT04391569).
Conclusions
The latest additions to the list of ASMs that act on the GABA system illustrate different approaches to drug discovery in epilepsy. Cenobamate originated from the structural modification of compounds known to possess antiseizure activity, and was selected ultimately for clinical development based on its pharmacological profile in rodent models of seizures and epilepsy. Its ability to enhance GABA-mediated inhibition was discovered at a later time, when development was already advanced [6]. Ganaxolone, in contrast, was selected for development as a potential treatment for epilepsy with the precise objective of targeting the GABA system through positive allosteric modulation of GABAA receptors, consistent with its known properties as an orally active neurosteroid [156].
Cenobamate and ganaxolone show important differences in their mechanisms of action. Cenobamate exerts it antiseizure activity by at least two distinct primary mechanisms, namely the blockade of voltage-gated sodium channels and the potentiation of GABAergic responses at benzodiazepine-insensitive synaptic and extrasynaptic GABAA receptor sites. The antiseizure effects of ganaxolone, however, are primarily ascribed to allosteric modulation of synaptic and extrasynaptic GABAA receptors. These differential pharmacological profiles translate into differences in antiseizure activity in the clinical setting. Although comparisons of results across trials should be interpreted cautiously, the efficacy of cenobamate against focal seizures [101, 102] appears to be far more robust than that of ganaxolone [176, 184]. Accordingly, only cenobamate is currently approved for the treatment of focal seizures, and current regulatory approval for ganaxolone is limited to the treatment for CCDKL5 deficiency disorder, an orphan indication. Both drugs are currently under investigation for additional epilepsy-related indications, and their ultimate role in the management of seizure disorders will be defined after completion of further studies and the accumulation of broader postmarketing experience.
After decades of preclinical and clinical studies, extensive evidence has emerged that drugs acting on the GABA system and used in the treatment of epilepsy are not a homogeneous therapeutic class. In fact, there are profound differences among many of these agents in their activity against different seizure types, and tolerability profiles [6]. These differences can be explained not only by interactions with other targets and pharmacokinetic factors, but also by their specific modalities of interaction with different components of the GABAergic system. An improved understanding of these interactions provides opportunities for the design of novel therapeutic agents with potentially improved efficacy and adverse effect profiles. As discussed in an accompanying article [2], the pipeline of investigational treatments for seizures and epilepsy targeting GABAergic neurotransmission is vast and diverse, and includes highly innovative approaches that could lead to important therapeutic breakthroughs.
Acknowledgements
We thank Ms. Natalia Erenburg for her kind assistance with the editing of the Figures.
Declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The preparation of this article was not supported by any funding source.
Conflicts of interest/competing interests
Emilio Perucca received speaker fees or fees from consulting or participation in advisory boards/data safety monitoring boards from Angelini, Arvelle, Biopas, Eisai, GW Pharma, Janssen, PMI Life Sciences, Sanofi group of companies, Shackelford Pharma, SKL Life Science, Sun Pharma, Takeda, UCB Pharma, Xenon Pharma and Zogenix, and royalties from Wiley, Elsevier and Wolters Kluwers, all outside the submitted work. Meir Bialer received speaker’s or consultancy fees from Boehringer Ingelheim, Clexio Therapeutics, Guidepoint, Pharma2B, Shackelford Pharma and US WorldMeds. H. Steve White has received grant funding from UCB Pharma and Eisai Pharmaceuticals, Neurelis, Inc. and consultant fees from GW Pharmaceuticals, Neurelis, Inc., Takeda Pharmaceuticals, SK Life Sciences and Jazz Pharmaceuticals and speaker honoraria from SK Pharmaceuticals, Takeda Pharmaceuticals and UCB Pharma.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ contributions
This article is an invited review. All authors contributed to the literature search. EP produced the initial draft of the Summary, Key Points, Introduction, Sect. 4.1 and Conclusions. HSW produced the initiat draft of Sect. 3. MB produced the initial draft of Sect. 4.2. All authors participated equally in the finalisation of the manuscript and they all met ICMJE criteria for authorship. All authors have read and approved the final submitted manuscript, and agree to be accountable for the work.
References
- 1.Akyuz E, Polat AK, Eroglu E, Kullu I, Angelopoulou E, Paudel YN. Revisiting the role of neurotransmitters in epilepsy: an updated review. Life Sci. 2021;265:118826. doi: 10.1016/j.lfs.2020.118826. [DOI] [PubMed] [Google Scholar]
- 2.Perucca E, White HS, Bialer M. New GABA-targeting therapies for the treatment of seizures and epilepsy. II. Treatments in clinical development. CNS Drugs. 10.1007/s40263-023-01025-4. [DOI] [PMC free article] [PubMed]
- 3.Bergmann KJ. Progabide: a new GABA-mimetic agent in clinical use. Clin Neuropharmacol. 1985;8:13–26. doi: 10.1097/00002826-198503000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Grant SM, Heel RC. Vigabatrin: a r review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in epilepsy and disorders of motor control. Drugs. 1991;41:889–926. doi: 10.2165/00003495-199141060-00007. [DOI] [PubMed] [Google Scholar]
- 5.Schachter SC. A review of the antiepileptic drug tiagabine. Clin Neuropharmacol. 1999;22:312–317. [PubMed] [Google Scholar]
- 6.Löscher W. Single-target versus multi-target drugs versus combinations of drugs with multiple targets: preclinical and clinical evidence for the treatment or prevention of epilepsy. Front Pharmacol. 2021;12:730257. doi: 10.3389/fphar.2021.730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treiman DM. GABAergic mechanisms in epilepsy. Epilepsia. 2001;42(Suppl. 3):8–12. doi: 10.1046/j.1528-1157.2001.042suppl.3008.x. [DOI] [PubMed] [Google Scholar]
- 8.Liu YQ, Yu F, Liu WH, He XH, Peng BW. Dysfunction of hippocampal interneurons in epilepsy. Neurosci Bull. 2014;30:985–998. doi: 10.1007/s12264-014-1478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarlo GL, Holton KF. Brain concentrations of glutamate and GABA in human epilepsy: a review. Seizure. 2021;91:213–227. doi: 10.1016/j.seizure.2021.06.028. [DOI] [PubMed] [Google Scholar]
- 10.Nohria V, Giller E. Ganaxolone. Neurotherapeutics. 2007;4:102–105. doi: 10.1016/j.nurt.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White HS, Rho JM, editors. Mechanisms of action of antiepileptic drugs. New York: Professional Communications, Inc.; 2010. [Google Scholar]
- 12.Squires RF, Saederup E, Crawley JN, Skolnick P, Paul SM. Convulsant potencies of tetrazoles are highly correlated with actions on GABA/benzodiazepine/picrotoxin receptor complexes in brain. Life Sci. 1984;35:1439–1444. doi: 10.1016/0024-3205(84)90159-0. [DOI] [PubMed] [Google Scholar]
- 13.Newland CF, Cull-Candy SG. On the mechanism of action of picrotoxin on GABA receptor channels in dissociated sympathetic neurones of the rat. J Physiol. 1992;447:191–213. doi: 10.1113/jphysiol.1992.sp018998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Absalom NL, Liao VWY, Johannesen KMH, Gardella E, Jacobs J, Lesca G, et al. Gain-of-function and loss-of-function GABRB3 variants lead to distinct clinical phenotypes in patients with developmental and epileptic encephalopathies. Nat Commun. 2022;13:1822. doi: 10.1038/s41467-022-29280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chuang SH, Reddy DS. Genetic and molecular regulation of extrasynaptic GABA-A receptors in the brain: therapeutic insights for epilepsy. J Pharmacol Exp Ther. 2018;364:180–197. doi: 10.1124/jpet.117.244673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Y, Wei ZH, Liu C, Li GY, Qiao XZ, Gan YJ, et al. Genetic variations in GABA metabolism and epilepsy. Seizure. 2022;101:22–29. doi: 10.1016/j.seizure.2022.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Cossette P, Lachance-Touchette P, Rouleau GA. Mutated GABAA receptor subunits in idiopathic generalized epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s basic mechanisms of the epilepsies. 4th ed. Bethesda (MD): National Center for Biotechnology Information (US); 2012: p. 827–46. [PubMed]
- 18.Hernandez CC, Tian X, Hu N, Shen W, Catron MA, Yang Y, et al. Dravet syndrome-associated mutations in GABRA1, GABRB2 and GABRG2 define the genetic landscape of defects of GABAA receptors. Brain Commun. 2021;3:fcab033. doi: 10.1093/braincomms/fcab033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang J-Q. Defects at the crossroads of GABAergic signaling in generalized genetic epilepsies. Epilepsy Res. 2017;137:9–18. doi: 10.1016/j.eplepsyres.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macdonald RL, Kang JQ, Gallagher MJ. GABAA receptor subunit mutations and genetic epilepsies. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies. 4th ed. Bethesda (MD): National Center for Biotechnology Information (US); 2012: p. 857–68. [PubMed]
- 21.Maillard PY, Baer S, Schaefer É, Desnous B, Villeneuve N, Lépine A, et al. Molecular and clinical descriptions of patients with GABAA receptor gene variants (GABRA1, GABRB2, GABRB3, GABRG2): a cohort study, review of literature, and genotype-phenotype correlation. Epilepsia. 2022;63:2519–2533. doi: 10.1111/epi.17336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riaz M, Abbasi MH, Sheikh N, Saleem T, Virk AO. GABRA1 and GABRA6 gene mutations in idiopathic generalized epilepsy patients. Seizure. 2021;93:88–94. doi: 10.1016/j.seizure.2021.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Symonds JD, Zuberi SM, Johnson MR. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr Opin Neurol. 2017;30:193–199. doi: 10.1097/WCO.0000000000000433. [DOI] [PubMed] [Google Scholar]
- 24.Iadarola MJ, Gale K. Substantia nigra: site of anticonvulsant activity mediated by gamma-aminobutyric acid. Science. 1982;218:1237–1240. doi: 10.1126/science.7146907. [DOI] [PubMed] [Google Scholar]
- 25.Gale K. Subcortical structures and pathways involved in convulsive seizure generation. J Clin Neurophysiol. 1992;9:264–277. doi: 10.1097/00004691-199204010-00007. [DOI] [PubMed] [Google Scholar]
- 26.Snodgrass SR. GABA and epilepsy: their complex relationship and the evolution of our understanding. J Child Neurol. 1992;7:77–86. doi: 10.1177/088307389200700114. [DOI] [PubMed] [Google Scholar]
- 27.Hosford DA, Wang Y. Utility of the lethargic (lh/lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures. Epilepsia. 1997;38:408–414. doi: 10.1111/j.1528-1157.1997.tb01729.x. [DOI] [PubMed] [Google Scholar]
- 28.Berkovic SF. Aggravation of generalized epilepsies. Epilepsia. 1998;39(Suppl. 3):S11–S14. doi: 10.1111/j.1528-1157.1998.tb05115.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu L, Zheng T, Morris MJ, Wallengren C, Clarke AL, Reid CA, et al. The mechanism of carbamazepine aggravation of absence seizures. J Pharmacol Exp Ther. 2006;319:790–798. doi: 10.1124/jpet.106.104968. [DOI] [PubMed] [Google Scholar]
- 30.Zheng T, Clarke AL, Morris MJ, Reid CA, Petrou S, O'Brien TJ. Oxcarbazepine, not its active metabolite, potentiates GABAA activation and aggravates absence seizures. Epilepsia. 2009;50:83–87. doi: 10.1111/j.1528-1167.2008.01759.x. [DOI] [PubMed] [Google Scholar]
- 31.Hosford DA, Clark S, Cao Z, Wilson WA, Jr, Lin FH, Morrisett RA, et al. The role of GABAB receptor activation in absence seizures of lethargic (lh/lh) mice. Science. 1992;257:398–401. doi: 10.1126/science.1321503. [DOI] [PubMed] [Google Scholar]
- 32.Motalli R, Louvel J, Tancredi V, Kurcewicz I, Wan-Chow-Wah D, Pumain R, et al. GABA(B) receptor activation promotes seizure activity in the juvenile rat hippocampus. J Neurophysiol. 1999;82:638–647. doi: 10.1152/jn.1999.82.2.638. [DOI] [PubMed] [Google Scholar]
- 33.Mares P, Tabashidze N. Contradictory effects of GABA-B receptor agonists on cortical epileptic afterdischarges in immature rats. Brain Res Bull. 2008;75:173–178. doi: 10.1016/j.brainresbull.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Avoli M, Lévesque M. GABAB Receptors: are they missing in action in focal epilepsy research? Curr Neuropharmacol. 2022;20:1704–1716. doi: 10.2174/1570159X19666210823102332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98:641–653. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- 36.Chebib M, Johnston GA. The 'ABC' of GABA receptors: a brief review. Clin Exp Pharmacol Physiol. 1999;26:937–940. doi: 10.1046/j.1440-1681.1999.03151.x. [DOI] [PubMed] [Google Scholar]
- 37.Sieghart W, Savić MM. International Union of Basic and Clinical Pharmacology. CVI: GABAA receptor subtype- and function-selective ligands: key issues in translation to humans. Pharmacol Rev. 2018;70:836–78. [DOI] [PubMed]
- 38.Olsen RW, Tobin AJ. Molecular biology of GABAA receptors. FASEB J. 1990;4:1469–1480. doi: 10.1096/fasebj.4.5.2155149. [DOI] [PubMed] [Google Scholar]
- 39.Sigel E, Steinmann ME. Structure, function, and modulation of GABA(A) receptors. J Biol Chem. 2012;287:40224–40231. doi: 10.1074/jbc.R112.386664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castellano D, Shepard RD, Lu W. Looking for novelty in an "old" receptor: recent advances toward our understanding of GABAARs and their implications in receptor pharmacology. Front Neurosci. 2021;14:616298. doi: 10.3389/fnins.2020.616298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bowery NG, Bettler B, Froestl W, Gallagher JP, Marshall F, Raiteri M, et al. International Union of Pharmacology. XXXIII. Mammalian gamma-aminobutyric acid(B) receptors: structure and function. Pharmacol Rev. 2002;54:247–64. [DOI] [PubMed]
- 42.Enz R, Cutting GR. GABAC receptor rho subunits are heterogeneously expressed in the human CNS and form homo- and heterooligomers with distinct physical properties. Eur J Neurosci. 1999;11:41–50. doi: 10.1046/j.1460-9568.1999.00423.x. [DOI] [PubMed] [Google Scholar]
- 43.Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, et al. GABAkines: advances in the discovery, development, and commercialization of positive allosteric modulators of positive allosteric modulators of GABAA. Pharmacol Ther. 2022;234:10830. doi: 10.1016/j.pharmthera.2021.108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004;27:569–575. doi: 10.1016/j.tins.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 45.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- 46.Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy JM, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 47.McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 48.Nutt DJ, Stahl SM. Searching for perfect sleep: the continuing evolution of GABAA receptor modulators as hypnotics. J Psychopharmacol. 2010;24:1601–1612. doi: 10.1177/0269881109106927. [DOI] [PubMed] [Google Scholar]
- 49.Rivas FM, Stables JP, Murphree L, Edwankar RV, Edwankar CR, Huang S, et al. Antiseizure activity of novel gamma-aminobutyric acid (A) receptor subtype-selective benzodiazepine analogues in mice and rat models. J Med Chem. 2009;52:1795–1798. doi: 10.1021/jm801652d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Möhler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- 51.Botta P, Demmou L, Kasugai Y, Markovic M, Xu C, Fadok JP, et al. Regulating anxiety with extrasynaptic inhibition. Nat Neurosci. 2015;18:1493–1500. doi: 10.1038/nn.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gallager DW, Lakoski JM, Gonsalves SF, Rauch SL. Chronic benzodiazepine treatment decreases postsynaptic GABA sensitivity. Nature. 1984;308:74–77. doi: 10.1038/308074a0. [DOI] [PubMed] [Google Scholar]
- 53.Rogawski MA, Loya CM, Reddy K, Zolkowska D, Lossin C. Neuroactive steroids for the treatment of status epilepticus. Epilepsia. 2013;54(Suppl. 6):93–98. doi: 10.1111/epi.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nusser Z, Sieghart W, Benke D, Fritschy JM, Somogyi P. Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells. Proc Natl Acad Sci USA. 1996;93:11939–11944. doi: 10.1073/pnas.93.21.11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi DW, Farb DH, Fischbach GD. Chlordiazepoxide selectively augments GABA action in spinal cord cell cultures. Nature. 1977;269:342–344. doi: 10.1038/269342a0. [DOI] [PubMed] [Google Scholar]
- 56.MacDonald RL, Rogers CJ, Twyman RE. Barbiturate regulation of kinetic properties of the GABAA receptor channel of mouse spinal neurones in culture. J Physiol. 1989;417:483–500. doi: 10.1113/jphysiol.1989.sp017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacDonald RL, Twyman RE. Kinetic properties and regulation of GABAA receptor channels. Ion Channels. 1992;3:315–343. doi: 10.1007/978-1-4615-3328-3_10. [DOI] [PubMed] [Google Scholar]
- 58.Rogers CJ, Twyman RE, Macdonald RL. Benzodiazepine and beta-carboline regulation of single GABAA receptor channels of mouse spinal neurones in culture. J Physiol. 1994;475:69–82. doi: 10.1113/jphysiol.1994.sp020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Twyman RE, Macdonald RL. Neurosteroid regulation of GABAA receptor single-channel kinetic properties of mouse spinal cord neurons in culture. J Physiol. 1992;456:215–245. doi: 10.1113/jphysiol.1992.sp019334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belelli D, Hogenkamp D, Gee KW, Lambert JJ. Realising the therapeutic potential of neuroactive steroid modulators of the GABAA receptor. Neurobiol Stress. 2019;12:100207. doi: 10.1016/j.ynstr.2019.100207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, et al. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4:490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- 62.Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 63.Vanhatalo S, Palva JM, Andersson S, Rivera C, Voipio J, Kaila K. Slow endogenous activity transients and developmental expression of K+-Cl- cotransporter 2 in the immature human cortex. Eur J Neurosci. 2005;22:2799–2804. doi: 10.1111/j.1460-9568.2005.04459.x. [DOI] [PubMed] [Google Scholar]
- 64.Kirmse K, Zhang C. Principles of GABAergic signaling in developing cortical network dynamics. Cell Rep. 2022;38:110568. doi: 10.1016/j.celrep.2022.110568. [DOI] [PubMed] [Google Scholar]
- 65.Briggs SW, Galanopoulou AS. Altered GABA signaling in early life epilepsies. Neural Plast. 2011;2011:527605. doi: 10.1155/2011/527605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang S, Kadam S. Pre-clinical models of acquired neonatal seizures: differential effects of injury on function of chloride cotransporters. Austin J Cerebrovasc Dis Stroke. 2014;1:1026. [PMC free article] [PubMed] [Google Scholar]
- 67.Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014;55:806–818. doi: 10.1111/epi.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Donovan MD, Griffin BT, Kharoshankaya L, Cryan JF, Boylan GB. Pharmacotherapy for neonatal seizures: current knowledge and future perspectives. Drugs. 2016;76:647–661. doi: 10.1007/s40265-016-0554-7. [DOI] [PubMed] [Google Scholar]
- 69.Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wasterlain C. Fifty years of research on status epilepticus: seizures use hippocampal memory circuits to generate status epilepticus and disrupt brain development. Epilepsy Behav. 2023;141:109142. doi: 10.1016/j.yebeh.2023.109142. [DOI] [PubMed] [Google Scholar]
- 72.Engin E. GABAA receptor subtypes and benzodiazepine use, misuse, and abuse. Front Psychiatry. 2023;13:1060949. doi: 10.3389/fpsyt.2022.1060949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perucca P, Gilliam FG. Adverse effects of antiepileptic drugs. Lancet Neurol. 2012;11:792–802. doi: 10.1016/S1474-4422(12)70153-9. [DOI] [PubMed] [Google Scholar]
- 74.Heim MK, Gidal BE. Vigabatrin-associated retinal damage: potential biochemical mechanisms. Acta Neurol Scand. 2012;126:219–228. doi: 10.1111/j.1600-0404.2012.01684.x. [DOI] [PubMed] [Google Scholar]
- 75.Kellinghaus C, Dziewas R, Lüdemann P. Tiagabine-related non-convulsive status epilepticus in partial epilepsy: three case reports and a review of the literature. Seizure. 2002;11:243–249. doi: 10.1053/seiz.2001.0594. [DOI] [PubMed] [Google Scholar]
- 76.Reddy DS. Neurosteroid replacement therapy for catamenial epilepsy, postpartum depression and neuroendocrine disorders in women. J Neuroendocrinol. 2022;34:e13028. doi: 10.1111/jne.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xcopri (cenobamate tablets). Prescribing information (revised June 2022). https://www.xcopri.com/pdf_file/xcopri_cenobamate_prescribing_information_medication_guide_combined.pdf. Accessed 30 Jun 2023.
- 78.Ontozry (cenobamate tablets). Summary of product characteristics. European Medicine Agency Cenobamate authorization. https://www.ema.europa.eu/en/documents/product-information/ontozry-epar-product-information_en.pdf. Accessed 30 Jun 2023.
- 79.Lattanzi S, Trinka E, Zaccara G, Striano P, Del Giovane C, Silvestrini M, et al. Adjunctive cenobamate for focal-onset seizures in adults: a systematic review and meta-analysis. CNS Drugs. 2020;34:1105–1120. doi: 10.1007/s40263-020-00759-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roberti R, De Caro C, Iannone LF, Zaccara G, Lattanzi S, Russo E. Pharmacology of cenobamate: mechanism of action, pharmacokinetics, drug-drug interactions and tolerability. CNS Drugs. 2021;35:609–618. doi: 10.1007/s40263-021-00819-8. [DOI] [PubMed] [Google Scholar]
- 81.Guignet M, Campbell A, White HS. Cenobamate (XCOPRI): can preclinical and clinical evidence provide insight into its mechanism of action? Epilepsia. 2020;61:2329–2339. doi: 10.1111/epi.16718. [DOI] [PubMed] [Google Scholar]
- 82.Sharma R, Nakamura M, Neupane C, Jeon BH, Shin H, Melnick SM, et al. Positive allosteric modulation of GABAA receptors by a novel antiepileptic drug cenobamate. Eur J Pharmacol. 2020;879:173117. doi: 10.1016/j.ejphar.2020.173117. [DOI] [PubMed] [Google Scholar]
- 83.Song WS, Cho YS, Oh SP, Yoon SH, Kim YS, Kim MH. Cognitive and behavioral effects of the anti-epileptic drug cenobamate (YKP3089) and underlying synaptic and cellular mechanisms. Neuropharmacology. 2022;221:109292. doi: 10.1016/j.neuropharm.2022.109292. [DOI] [PubMed] [Google Scholar]
- 84.Nakamura M, Cho JH, Shin H, Jang IS. Effects of cenobamate (YKP3089), a newly developed anti-epileptic drug, on voltage-gated sodium channels in rat hippocampal CA3 neurons. Eur J Pharmacol. 2019;855:175–182. doi: 10.1016/j.ejphar.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 85.Löscher W, Sills GJ, White HS. The ups and downs of alkyl-carbamates in epilepsy therapy: how does cenobamate differ? Epilepsia. 2021;62:596–614. doi: 10.1111/epi.16832. [DOI] [PubMed] [Google Scholar]
- 86.Pubchem. Cenobamate. https://pubchem.ncbi.nlm.nih.gov/#query=Cenobamate. Accessed 21 Mar 2023.
- 87.Odi R, Bibi D, Wagr T, Bialer M. A Perspective into the physicochemical and biopharmaceutic properties of marketed antiepileptic drugs: from phenobarbital to cenobamate and beyond. Epilepsia. 2020;61:1543–1552. doi: 10.1111/epi.16597. [DOI] [PubMed] [Google Scholar]
- 88.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 89.Wu C-Y, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 90.Vernillet L, Greene SA, Kamin M. Pharmacokinetics of cenobamate: results from single and multiple oral ascending-dose studies in healthy subjects. Clin Pharmacol Drug Dev. 2020;9:428–443. doi: 10.1002/cpdd.769. [DOI] [PubMed] [Google Scholar]
- 91.Yang E, Sunwoo J, Huh KY, Kim YK, Lee S, Jang IJ, et al. Pharmacokinetics and safety of cenobamate, a novel antiseizure medication, in healthy Japanese, and an ethnic comparison with healthy non-Japanese. Clin Transl Sci. 2022;15:490–500. doi: 10.1111/cts.13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vernillet L, Greene SA, Kim HW, Melnick SM, Glenn K. Mass balance, metabolism, and excretion of cenobamate, a new antiepileptic drug, after a single oral administration in healthy male subjects. Eur J Drug Metab Pharmacokinet. 2020;45:513–522. doi: 10.1007/s13318-020-00615-7. [DOI] [PubMed] [Google Scholar]
- 93.Barbieri MA, Perucca E, Spina E, Rota P, Franco V. Cenobamate: a review of its pharmacological properties, clinical efficacy and tolerability profile in the treatment of epilepsy. CNS Neurol Disord Drug Targ. 2023;22:394–403. doi: 10.2174/1871527321666220113110044. [DOI] [PubMed] [Google Scholar]
- 94.Greene SA, Kwak C, Kamin M, Vernillet L, Glenn KJ, Gabriel L, et al. Effect of cenobamate on the single-dose pharmacokinetics of multiple cytochrome P450 probes using a cocktail approach in healthy subjects. Clin Transl Sci. 2022;15:899–911. doi: 10.1111/cts.13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Elakkary S, Hagemann A, Klimpel D, Bien CG, Brandt C. A retrospective non-interventional study evaluating the pharmacokinetic interactions between cenobamate and clobazam. Epilepsia. 2023 doi: 10.1111/epi.17515. [DOI] [PubMed] [Google Scholar]
- 96.Osborn M, Abou-Khalil B. The cenobamate-clobazam interaction: evidence of synergy in addition to pharmacokinetic interaction. Epilepsy Behav. 2023;142:109156. doi: 10.1016/j.yebeh.2023.109156. [DOI] [PubMed] [Google Scholar]
- 97.Sperling MR, Klein P, Aboumatar S, Gelfand M, Halford JJ, Krauss GL, et al. Cenobamate (YKP3089) as adjunctive treatment for uncontrolled focal seizures in a large, phase 3, multicenter, open-label safety study. Epilepsia. 2020;61:1099–1108. doi: 10.1111/epi.16525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith MC, Klein P, Krauss GL, Rashid S, Seiden LG, Stern JM, Rosenfeld WE. Dose adjustment of concomitant antiseizure medications during cenobamate treatment: expert opinion consensus recommendations. Neurol Ther. 2022;11:1705–1720. doi: 10.1007/s40120-022-00400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vernillet L, Kamin M. Drug–drug interactions between cenobamate and other antiepileptic drugs: results from phase I studies with carbamazepine, phenobarbital, phenytoin, and divalproex sodium [poster]. Presented at American Society for Clinical Pharmacology & Therapeutics Annual Meeting, March 21–24, 2018, Orlando (FL).
- 100.Brandt C, Sánchez-Álvarez JC, Steinhoff BJ, Milanov I, Serratosa JM. Efficacy and safety of adjunctive cenobamate: Post-hoc analysis of study C017 in patients grouped by mechanism of action of concomitant antiseizure medications. Seizure. 2022;96:86–93. doi: 10.1016/j.seizure.2022.02.003. [DOI] [PubMed] [Google Scholar]
- 101.Chung SS, French JA, Kowalski J, et al. Randomized phase 2 study of adjunctive cenobamate in patients with uncontrolled focal seizures. Neurology. 2020;94:e2311–e2322. doi: 10.1212/WNL.0000000000009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krauss GL, Klein P, Brandt C, Lee SK, Milanov I, Milovanovic M, et al. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. 2020;19:38–48. [correction in Lancet Neurol. 2020;19:e3]. [DOI] [PubMed]
- 103.Gazzola DM, Balcer LJ, French JA. Seizure-free outcome in randomized add-on trials of the new antiepileptic drugs. Epilepsia. 2007;48:1303–1307. doi: 10.1111/j.1528-1167.2007.01136.x. [DOI] [PubMed] [Google Scholar]
- 104.Rosenfeld WE, Nisman A, Ferrari L. Efficacy of adjunctive cenobamate based on number of concomitant antiseizure medications, seizure frequency, and epilepsy duration at baseline: a post-hoc analysis of a randomized clinical study. Epilepsy Res. 2021;172:106592. doi: 10.1016/j.eplepsyres.2021.106592. [DOI] [PubMed] [Google Scholar]
- 105.Aboumatar S, Biton V, Wechsler R, Ferrari L, Rosenfeld WE. Post hoc analysis of a phase 3 study for treatment of uncontrolled focal seizures: adjunctive cenobamate dose and seizure reduction by baseline seizure frequency. Epilepsy Res. 2022;186:107014. doi: 10.1016/j.eplepsyres.2022.107014. [DOI] [PubMed] [Google Scholar]
- 106.Peña-Ceballos J, Moloney PB, Munteanu T, Doyle M, Colleran N, Liggan B, et al. Adjunctive cenobamate in highly active and ultra-refractory focal epilepsy: a 'real-world' retrospective study. Epilepsia. 2023;64:1225–1235. doi: 10.1111/epi.17549. [DOI] [PubMed] [Google Scholar]
- 107.Abou-Khalil B, Aboumatar S, Klein P, Krauss GL, Sperling MR, Rosenfeld WE. Efficacy of cenobamate for uncontrolled focal seizures in patients with previous epilepsy-related surgery: post hoc analysis of a phase 3, multicenter, open-label study. Epilepsy Res. 2022;184:106952. doi: 10.1016/j.eplepsyres.2022.106952. [DOI] [PubMed] [Google Scholar]
- 108.Sperling MR, Abou-Khalil B, Aboumatar S, Bhatia P, Biton V, Klein P, et al. Efficacy of cenobamate for uncontrolled focal seizures: post hoc analysis of a phase 3, multicenter, open-label study. Epilepsia. 2021;62:3005–3015. doi: 10.1111/epi.17091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rosenfeld WE, Ferrari L, Kamin M. Efficacy of cenobamate by focal seizure subtypes: post-hoc analysis of a phase 3, multicenter, open-label study. Epilepsy Res. 2022;183:106940. doi: 10.1016/j.eplepsyres.2022.106940. [DOI] [PubMed] [Google Scholar]
- 110.Villanueva V, Santos-Carrasco D, Cabezudo-García P, Gómez-Ibáñez A, Garcés M, Serrano-Castro P, et al. Real-world safety and effectiveness of cenobamate in patients with focal onset seizures: outcomes from an Expanded Access Program. Epilepsia Open. 2023 May 7. 10.1002/epi4.12757. [Online ahead of print]. [DOI] [PMC free article] [PubMed]
- 111.Elizebath R, Zhang E, Coe P, Gutierrez EG, Yang J, Krauss GL. Cenobamate treatment of focal-onset seizures: quality of life and outcome during up to eight years of treatment. Epilepsy Behav. 2021;116:107796. doi: 10.1016/j.yebeh.2021.107796. [DOI] [PubMed] [Google Scholar]
- 112.Klein P, Aboumatar S, Brandt C, Dong F, Krauss GL, Mizne S, et al. Long-term efficacy and safety from an open-label extension of adjunctive cenobamate in patients with uncontrolled focal seizures. Neurology. 2022;99:e989–e998. doi: 10.1212/WNL.0000000000200792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sander JW, Rosenfeld WE, Halford JJ, Steinhoff BJ, Biton V, Toledo M. Long-term individual retention with cenobamate in adults with focal seizures: pooled data from the clinical development program. Epilepsia. 2022;63:139–149. doi: 10.1111/epi.17134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rosenfeld WE, Ferrari L, Kerr WT, Sperling MR. Sudden unexpected death in epilepsy during cenobamate clinical development. Epilepsia. 2023 May 23. 10.1111/epi.17662. [Online ahead of print]. [DOI] [PubMed]
- 115.Rosenfeld WE, Abou-Khalil B, Aboumatar S, Bhatia P, Biton V, Krauss GL, et al. Post hoc analysis of a phase 3, multicenter, open-label study of cenobamate for treatment of uncontrolled focal seizures: effects of dose adjustments of concomitant antiseizure medications. Epilepsia. 2021;62:3016–3028. doi: 10.1111/epi.17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schuetz E, Wagner K, Metternich B, Papadopoulou G, Kravalis K, Heers M, et al. Effects of cenobamate on cognitive performance of epilepsy patients. Seizure. 2022;102:129–133. doi: 10.1016/j.seizure.2022.10.004. [DOI] [PubMed] [Google Scholar]
- 117.Steinhoff BJ, Rosenfeld WE, Serratosa JM, Brandt C, Klein P, Toledo M, et al. Practical guidance for the management of adults receiving adjunctive cenobamate for the treatment of focal epilepsy-expert opinion. Epilepsy Behav. 2021;123:108270. doi: 10.1016/j.yebeh.2021.108270. [DOI] [PubMed] [Google Scholar]
- 118.Villani F, Cianci V, Di Bonaventura C, Di Gennaro G, Galimberti CA, Guerrini R, et al. Use of cenobamate for the treatment of focal epilepsy: an Italian expert opinion paper. Expert Rev Neurother. 2022;22:935–940. doi: 10.1080/14737175.2023.2171291. [DOI] [PubMed] [Google Scholar]
- 119.Darpo B, Sager PT, Xue H, Kamin M. A Phase 1 clinical study evaluating the effects of cenobamate on the QT interval. Clin Pharmacol Drug Dev. 2022;11:523–534. doi: 10.1002/cpdd.1077. [DOI] [PubMed] [Google Scholar]
- 120.Lattanzi S, Trinka E, Zaccara G, Striano P, Russo E, Del Giovane C, et al. Third-generation antiseizure medications for adjunctive treatment of focal-onset seizures in adults: a systematic review and network meta-analysis. Drugs. 2022;82:199–218. doi: 10.1007/s40265-021-01661-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Steinhoff BJ, Ben-Menachem E, Brandt C, García Morales I, Rosenfeld WE, Santamarina E, et al. Onset of efficacy and adverse events during cenobamate titration period. Acta Neurol Scand. 2022;146:265–275. doi: 10.1111/ane.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Laskier V, Agyei-Kyeremateng KK, Eddy AE, Patel D, Mulheron S, James S, et al. Cost-effectiveness of cenobamate for focal seizures in people with drug-resistant epilepsy. Epilepsia. 2023;64:843–856. doi: 10.1111/epi.17506. [DOI] [PubMed] [Google Scholar]
- 123.Klein P, Krauss GL, Steinhoff BJ, Devinsky O, Sperling MR. Failure to use new breakthrough treatments for epilepsy. Epilepsia. 2023;64:1458–1465. doi: 10.1111/epi.17564. [DOI] [PubMed] [Google Scholar]
- 124.Makridis KL, Bast T, Prager C, Kovacevic-Preradovic T, Bittigau P, Mayer T, et al. Real-world experience treating pediatric epilepsy patients with cenobamate. Front Neurol. 2022;13:950171. doi: 10.3389/fneur.2022.950171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Varughese RT, Shah YD, Karkare S, Kothare SV. Adjunctive use of cenobamate for pediatric refractory focal-onset epilepsy: a single-center retrospective study. Epilepsy Behav. 2022;130:108679. doi: 10.1016/j.yebeh.2022.108679. [DOI] [PubMed] [Google Scholar]
- 126.Makridis KL, Friedo AL, Kellinghaus C, Losch FP, Schmitz B, Boßelmann C, et al. Successful treatment of adult Dravet syndrome patients with cenobamate. Epilepsia. 2022;63:e164–e171. doi: 10.1111/epi.17427. [DOI] [PubMed] [Google Scholar]
- 127.Agashe S, Worrell G, Britton J, Noe K, Ritaccio A, Wirrell EC, et al. Cenobamate in generalized epilepsy and combined generalized and focal epilepsy. Neurol Clin Pract. 2023;13:e200133. doi: 10.1212/CPJ.0000000000200133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Falcicchio G, Lattanzi S, Negri F, de Tommaso M, La Neve A, Specchio N. Treatment with cenobamate in adult patients with Lennox-Gastaut syndrome: a case series. J Clin Med. 2022;12:129. doi: 10.3390/jcm12010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Carlson JM, Molyneaux BJ, Lee JW. Safe use of cenobamate in super refractory status epilepticus: a case series. Neurohospitalist. 2023;13:169–172. doi: 10.1177/19418744221147083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Morrow AL. Recent developments in the significance and therapeutic relevance of neuroactive steroids: introduction to the special issue. Pharmacol Ther. 2007;116:1–6. doi: 10.1016/j.pharmthera.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wendler A, Wehling M. Many or too many progesterone membrane receptors? Clinical implications. Trends Endocrinol Metab. 2022;33:850–868. doi: 10.1016/j.tem.2022.10.001. [DOI] [PubMed] [Google Scholar]
- 132.Tang W, Beckley JT, Zhang J, Song R, Xu Y, Kim S, et al. Novel neuroactive steroids as positive allosteric modulators of NMDA receptors: mechanism, site of action, and rescue pharmacology on GRIN variants associated with neurological conditions. Cell Mol Life Sci. 2023;80:42. doi: 10.1007/s00018-022-04667-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABA(A) receptors. Pharmacol Ther. 2007;116:20–34. doi: 10.1016/j.pharmthera.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 134.Seljeset S, Laverty D, Smart TG. Inhibitory neurosteroids and the GABAA receptor. Adv Pharmacol. 2015;72:165–187. doi: 10.1016/bs.apha.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 135.Lévesque M, Biagini G, Avoli M. Neurosteroids and focal epileptic disorders. Int J Mol Sci. 2020;21:9391. doi: 10.3390/ijms21249391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Biagini G, Panuccio G, Avoli M. Neurosteroids and epilepsy. Curr Opin Neurol. 2010;23:170–176. doi: 10.1097/WCO.0b013e32833735cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Biagini G, Rustichelli C, Curia G, Vinet J, Lucchi C, Pugnaghi M, et al. Neurosteroids and epileptogenesis. J Neuroendocrinol. 2013;25:980–990. doi: 10.1111/jne.12063. [DOI] [PubMed] [Google Scholar]
- 138.Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS, et al. Progress report on new antiepileptic drugs: a summary of the Twelfth Eilat Conference (EILAT XII) Epilepsy Res. 2015;111:85–141. doi: 10.1016/j.eplepsyres.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 139.Rosenthal ES, Claassen J, Wainwright MS, Husain AM, Vaitkevicius H, Raines S, et al. Brexanolone as adjunctive therapy in super-refractory status epilepticus. Ann Neurol. 2017;82:342–352. doi: 10.1002/ana.25008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Witkin JM, Lippa A, Smith JL, Jin X, Ping X, Biggerstaff A, et al. The imidazodiazepine, KRM-II-81: an example of a newly emerging generation of GABAkines for neurological nd psychiatric disorders. Pharmacol Biochem Behav. 2022;213:173321. doi: 10.1016/j.pbb.2021.173321. [DOI] [PubMed] [Google Scholar]
- 141.BusinessWire. Sage Therapeutics reports top-line results from phase 3 STATUS trial of brexanolone in super-refractory status epilepticus, September 12, 2017. https://www.businesswire.com/news/home/20170912005509/en/. Accessed 5 Mar 2023.
- 142.Kaufman Y, Carlini SV, Deligiannidis KM. Advances in pharmacotherapy for postpartum depression: a structured review of standard-of-care antidepressants and novel neuroactive steroid antidepressants. Ther Adv Psychopharmacol. 2022;12:20451253211065859. doi: 10.1177/20451253211065859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ztalmy (ganaxolone), oral suspension. Prescribing information (revised March 2022). https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215904s000lbl.pdf. Accessed 30 Jun 2023.
- 144.Nik AM, Pressly B, Singh V, Antrobus S, Hulsizer S, Rogawski MA, et al. Rapid throughput analysis of GABAA receptor subtype modulators and blockers using DiSBAC1(3) membrane potential red dye. Mol Pharmacol. 2017;92:88–99. doi: 10.1124/mol.117.108563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Reddy DS, Carver CM, Clossen B, Wu X. Extrasynaptic γ-aminobutyric acid type A receptor-mediated sex differences in the antiseizure activity of neurosteroids in status epilepticus and complex partial seizures. Epilepsia. 2019;60:730–743. doi: 10.1111/epi.14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carter RB, Wood PL, Wieland S, Hawkinson JE, Belelli D, Lambert JJ, et al. Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3α-hydroxy-3β-methyl-5α-pregnan-20-one), a selective, high-affinity, steroid modulator of the γ-aminobutyric acid A receptor. J Pharmacol ExpTher. 1997;280:1284–1295. [PubMed] [Google Scholar]
- 147.Lattanzi S, Riva A, Striano P. Ganaxolone treatment for epilepsy patients: from pharmacology to place in therapy. Expert Rev Neurother. 2021;21:1317–1332. doi: 10.1080/14737175.2021.1904895. [DOI] [PubMed] [Google Scholar]
- 148.Reddy DS, Rogawski MA. Ganaxolone suppression of behavioral and electrographic seizures in the mouse amygdala kindling model. Epilepsy Res. 2010;89:254–260. doi: 10.1016/j.eplepsyres.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chuang SH, Reddy DS. Isobolographic analysis of antiseizure activity of the GABA type A receptor-modulating synthetic neurosteroids brexanolone and ganaxolone with tiagabine and midazolam. J Pharmacol Exp Ther. 2020;372:285–298. doi: 10.1124/jpet.119.261735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gasior M, Ungard JT, Beekman M, Carter RB, Witkin JM. Acute and chronic effects of the synthetic neuroactive steroid, ganaxolone, against the convulsive and lethal effects of pentylenetetrazol in seizure-kindled mice: comparison with diazepam and valproate. Neuropharmacology. 2000;39:1184–1196. doi: 10.1016/S0028-3908(99)00190-2. [DOI] [PubMed] [Google Scholar]
- 151.Kaminski RM, Gasior M, Carter RB, Witkin JM. Protective efficacy of neuroactive steroids against cocaine kindled-seizures in mice. Eur J Pharmacol. 2003;474:217–22. [DOI] [PubMed]
- 152.Gasior M, Carter RB, Goldberg SR, Witkin JM. Anticonvulsant and behavioral effects of neuroactive steroids alone and in conjunction with diazepam. J Pharmacol Exp Ther. 1997;282:543–553. [PubMed] [Google Scholar]
- 153.Liptáková S, Velísek L, Velísková J, Moshé SL. Effect of ganaxolone on flurothyl seizures in developing rats. Epilepsia. 2000;41:788–793. doi: 10.1111/j.1528-1157.2000.tb00244.x. [DOI] [PubMed] [Google Scholar]
- 154.Yum MS, Lee M, Ko TS, Velíšek L. A potential effect of ganaxolone in an animal model of infantile spasms. Epilepsy Res. 2014;108:1492–1500. doi: 10.1016/j.eplepsyres.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 155.Nipper MA, Jensen JP, Helms ML, Ford MM, Crabbe JC, Rossi DJ, et al. Genotype differences in sensitivity to the anticonvulsant effect of the synthetic neurosteroid ganaxolone during chronic ethanol withdrawal. Neuroscience. 2019;397:127–137. doi: 10.1016/j.neuroscience.2018.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kaminski RM, Livingood MR, Rogawski MA. Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice. Epilepsia. 2004;45:864–867. doi: 10.1111/j.0013-9580.2004.04504.x. [DOI] [PubMed] [Google Scholar]
- 157.Reddy DS, Rogawski MA. Enhanced anticonvulsant activity of ganaxolone after neurosteroid withdrawal in a rat model of catamenial epilepsy. J Pharmacol Exp Ther. 2000;294:909–915. [PubMed] [Google Scholar]
- 158.Miller SL, Bennet L, Sutherland AE, Pham Y, McDonald C, Castillo-Melendez M, et al. Ganaxolone versus phenobarbital for neonatal seizure management. Ann Neurol. 2022;92:1066–1079. doi: 10.1002/ana.26493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Golub V, Ramakrishnan S, Reddy DS. Isobolographic analysis of adjunct antiseizure activity of the FDA-approved cannabidiol with neurosteroids and benzodiazepines in adult refractory focal onset epilepsy. Exp Neurol. 2023;360:114294. doi: 10.1016/j.expneurol.2022.114294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Reddy DS, Mbilinyi RH, Ramakrishnan S. Efficacy of the FDA-approved cannabidiol on the development and persistence of temporal lobe epilepsy and complex focal onset seizures. Exp Neurol. 2023;359:114240. doi: 10.1016/j.expneurol.2022.114240. [DOI] [PubMed] [Google Scholar]
- 161.Snead OC., 3rd Ganaxolone, a selective, high-affinity steroid modulator of the gamma-aminobutyric acid-A receptor, exacerbates seizures in animal models of absence. Ann Neurol. 1998;44:688–691. doi: 10.1002/ana.410440417. [DOI] [PubMed] [Google Scholar]
- 162.Citraro R, Russo E, Di Paola ED, Ibbadu GF, Gratteri S, Marra R, et al. Effects of some neurosteroids injected into some brain areas of WAG/Rij rats, an animal model of generalized absence epilepsy. Neuropharmacology. 2006;50:1059–1071. doi: 10.1016/j.neuropharm.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 163.Zolkowska D, Wu CY, Rogawski MA. Intramuscular allopregnanolone and ganaxolone in a mouse model of treatment-resistant status epilepticus. Epilepsia. 2018;59(Suppl. 2):220–227. doi: 10.1111/epi.13999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Saporito MS, Gruner JA, DiCamillo A, et al. Intravenously administered ganaxolone blocks diazepam-resistant lithium-pilocarpine induced status epilepticus in rats: comparison with allopregnanolone. J Pharmacol Exp Ther. 2019;368:326–337. doi: 10.1124/jpet.118.252155. [DOI] [PubMed] [Google Scholar]
- 165.Barker BS, Spampanato J, McCarren HS, Smolik M, Jackson CE, Hornung EN, et al. Screening for efficacious anticonvulsants and neuroprotectants in delayed treatment models of organophosphate-induced status epilepticus. Neuroscience. 2020;425:280–300. doi: 10.1016/j.neuroscience.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thomas P, Pang Y. Anti-apoptotic actions of allopregnanolone and ganaxolone mediated through membrane progesterone receptors (PAQRs) in neuronal cells. Front Endocrinol (Lausanne). 2020;11:417. doi: 10.3389/fendo.2020.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Crombie GK, Palliser HK, Shaw JC, Hodgson DM, Walker DW, Hirst JJ. Evaluating changes in GABAergic and glutamatergic pathways in early life following prenatal stress and postnatal neurosteroid supplementation. Psychoneuroendocrinology. 2022;139:105705. doi: 10.1016/j.psyneuen.2022.105705. [DOI] [PubMed] [Google Scholar]
- 168.Ciarlone SL, Wang X, Rogawski MA, Weeber EJ. Effects of the synthetic neurosteroid ganaxolone on seizure activity and behavioral deficits in an Angelman syndrome mouse model. Neuropharmacology. 2017;116:142–150. doi: 10.1016/j.neuropharm.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 169.Shaw JC, Berry MJ, Dyson RM, Crombie GK, Hirst JJ, Palliser HK. Reduced neurosteroid exposure following preterm birth and its' contribution to neurological impairment: a novel avenue for preventative therapies. Front Physiol. 2019;10:599. doi: 10.3389/fphys.2019.00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.DrugBank. Ganaxolone. https://go.drugbank.com/drugs/DB05087. Accessed 8 Apr 2023.
- 171.Monaghan EP, Navalta LA, Shum L, Ashbrook DW, Lee DA. Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity. Epilepsia. 1997;38:1026–1031. doi: 10.1111/j.1528-1157.1997.tb01486.x. [DOI] [PubMed] [Google Scholar]
- 172.Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS, et al. Progress report on new antiepileptic drugs: a summary of the Ninth Eilat Conference (EILAT IX) Epilepsy Res. 2009;83:1–43. doi: 10.1016/j.eplepsyres.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 173.Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Perucca P, et al. Progress report on new antiepileptic drugs: a summary of the Fifteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XV). II. Drugs in more advanced clinical development. Epilepsia. 2020;61:2365–85. [DOI] [PubMed]
- 174.Fitch WL, Smith S, Saporito M, Busse G, Zhang M, Ren J, et al. Complex metabolism of the novel neurosteroid, ganaxolone, in humans: a unique challenge for metabolites in safety testing assessment. Drug Metab Dispos. 2023;51:753–763. doi: 10.1124/dmd.122.001218. [DOI] [PubMed] [Google Scholar]
- 175.Lamb YN. Ganaxolone: first approval. Drugs. 2022;82:933–940. doi: 10.1007/s40265-022-01724-0. [DOI] [PubMed] [Google Scholar]
- 176.Sperling MR, Klein P, Tsai J. Randomized, double-blind, placebo-controlled phase 2 study of ganaxolone as add-on therapy in adults with uncontrolled partial-onset seizures. Epilepsia. 2017;58:558–564. doi: 10.1111/epi.13705. [DOI] [PubMed] [Google Scholar]
- 177.Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63:1349–1397. doi: 10.1111/epi.17239. [DOI] [PubMed] [Google Scholar]
- 178.Leonard H, Downs J, Benke TA, Swanson L, Olson H, Demarest S. CDKL5 deficiency disorder: clinical features, diagnosis, and management. Lancet Neurol. 2022;21:563–576. doi: 10.1016/S1474-4422(22)00035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Amin S, Monaghan M, Aledo-Serrano A, Bahi-Buisson N, Chin RF, Clarke AJ, et al. International consensus recommendations for the assessment and management of individuals with CDKL5 deficiency disorder. Front Neurol. 2022;13:874695. doi: 10.3389/fneur.2022.874695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Specchio N, Chez M, Tarquinio D. Ganaxolone in children with CDKL5 gene-related epileptic encephalopathy: preliminary analysis from an open-label trial. Epilepsia. 2021;58:s163–s164. [Google Scholar]
- 181.Knight EMP, Amin S, Bahi-Buisson N, Benke TA, Cross JH, Demarest ST, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2022;21:417–427. doi: 10.1016/S1474-4422(22)00077-1. [DOI] [PubMed] [Google Scholar]
- 182.Specchio N, Amin S, Aimetti A, Hulihan J. Extended duration safety and efficacy of ganaxolone for the treatment of CDKL5 deficiency disorder: preliminary open-label extension analysis (Marigold Study). Poster presented at the Annual Meeting of the American Epilepsy Society (virtual meeting), December 4–8, 2020. https://marinuspharma.com/wp-content/uploads/2020/12/5_AES2020_Marigold-Prelim-OpenLabel-Analysis.pdf. Accessed 4 Mar 2023.
- 183.Laxer K, Blum D, Abou-Khalil BW, Morrell MJ, Lee DA, Data JL, et al. Assessment of ganaxolone's anticonvulsant activity using a randomized, double-blind, presurgical trial design. Ganaxolone Presurgical Study Group. Epilepsia. 2000;41:1187–1194. doi: 10.1111/j.1528-1157.2000.tb00324.x. [DOI] [PubMed] [Google Scholar]
- 184.Lappalainen J, Tsai J, Amerine W, Patroneva A. A multicenter, double-blind, randomized, placebo-controlled phase 3 trial to determine the efficacy and safety of ganaxolone as adjunctive therapy for adults with drug-resistant focal-onset seizures [P5237]. Neurology. 2017;88:P5.237.
- 185.Trivisano M, Lucchi C, Rustichelli C, Terracciano A, Cusmai R, Ubertini GM, et al. Reduced steroidogenesis in patients with PCDH19-female limited epilepsy. Epilepsia. 2017;58:e91–e95. doi: 10.1111/epi.13772. [DOI] [PubMed] [Google Scholar]
- 186.Sullivan J, Gunning B, Zafar M, Guerrini R, Zolnowska M, Gecz J, et al. Phase 2, placebo-controlled clinical study of oral ganaxolone in PCDH19-clustering epilepsy. Poster presented at the Annual Meeting of the American Epilepsy Society, Chicago, IL, December 3–7, 2021. https://marinuspharma.com/wp-content/uploads/2021/12/83008-GNX-in-PCDH19-AES-Poster_2021-11-05_FINAL.pdf/. Accessed 3 Mar 2023.
- 187.Sullivan J, Gunning B, Zafar M, Guerrini R, Gecz J, Kolc KL, et al. Phase 2, placebo-controlled clinical study of oral ganaxolone in PCDH19-clustering epilepsy. Epilepsy Res. 2023;191:107112. doi: 10.1016/j.eplepsyres.2023.107112. [DOI] [PubMed] [Google Scholar]
- 188.Reddy DS, Rogawski MA. Chronic treatment with the neuroactive steroid ganaxolone in the rat induces anticonvulsant tolerance to diazepam but not to itself. J Pharmacol Exp Ther. 2000;295:1241–1248. [PubMed] [Google Scholar]
- 189.Singh RK, Singh R, Stewart A, Van Poppel K, Klinger S, Hulihan J, et al. Intravenous ganaxolone in pediatric super-refractory status epilepticus: a single hospital experience. Epilepsy Behav. 2022;20:100567. doi: 10.1016/j.ebr.2022.100567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vaitkevicius H, Ramsay E, Swisher CB, Husain AM, Aimetti A, Gasior M. Phase 2 open-label, dose-fnding study of intravenous ganaxolone for the treatment of refractory status epilepticus. Epilepsia. 2021;62(Suppl. 3):348. doi: 10.1111/epi.17343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Aronica E, Specchio N, Luinenburg MJ, Curatolo P. Epileptogenesis in tuberous sclerosis complex-related developmental and epileptic encephalopathy. Brain. 2023;146:2694–2710. doi: 10.1093/brain/awad048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Perucca P, et al. Progress report on new antiepileptic drugs: a summary of the Sixteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XVI): II. Drugs in more advanced clinical development. Epilepsia. 2022b;63:2883–910. [DOI] [PubMed]
- 193.Pieribone VA, Tsai J, Soufflet C, Rey E, Shaw K, Giller E, Dulac O. Clinical evaluation of ganaxolone in pediatric and adolescent patients with refractory epilepsy. Epilepsia. 2007;48:1870–1874. doi: 10.1111/j.1528-1167.2007.01182.x. [DOI] [PubMed] [Google Scholar]
- 194.Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Tomson T, et al. Progress report on new antiepileptic drugs: a summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). II. Drugs in more advanced clinical development. Epilepsia. 2018;59:1842–66. [DOI] [PubMed]
- 195.Kerrigan JF, Shields WD, Nelson TY, Bluestone DL, Dodson WE, Bourgeois BF, et al. Ganaxolone for treating intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res. 2000;42:133–139. doi: 10.1016/S0920-1211(00)00170-4. [DOI] [PubMed] [Google Scholar]
- 196.Ligsay A, Van Dijck A, Nguyen DV, Lozano R, Chen Y, Bickel ES, et al. A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J Neurodev Disord. 2017;9:26. doi: 10.1186/s11689-017-9207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rasmusson AM, Marx CE, Jain S, Farfel GM, Tsai J, Sun X, et al. A randomized controlled trial of ganaxolone in posttraumatic stress disorder. Psychopharmacology. 2017;234:2245–2257. doi: 10.1007/s00213-017-4649-y. [DOI] [PubMed] [Google Scholar]
- 198.Dichtel LE, Nyer M, Dording C, Fisher LB, Cusin C, Shapero BG, et al. Effects of open-label, adjunctive ganaxolone on persistent depression despite adequate antidepressant treatment in postmenopausal women: a pilot study. J Clin Psychiatry. 2020;81:19m12887. [DOI] [PMC free article] [PubMed]
- 199.Gasior M, Hulihan J, Miller I. Aggregated safety and tolerability experience from the ganaxolone development program (P1-8.002) [abstract]. Neurology. 2022;98:18 (supplement).
- 200.European Medicines Agency. Ztalmy. Ganaxolone. https://www.ema.europa.eu/en/medicines/human/summaries-opinion/ztalmy. Accessed 2 Jul 2023.
- 201.Vossler DG. Ganaxolone: a new treatment for CDKL5 deficiency disorder. Epilepsy Curr. 2022;22:348–350. doi: 10.1177/15357597221125238. [DOI] [PMC free article] [PubMed] [Google Scholar]