

Abstract
We report a system and assay for performing fully-automated measurement of 6 proteins simultaneously with single molecule sensitivity. The system combines handling of samples, reagents, and consumables, with a module for imaging single molecule arrays (Simoa) to enable immunoassays that have high sensitivity (~fg/mL), are multiplexed, and are fully-automated. A 6-plex cytokine Simoa assay for IL-6, TNF-α, GM-CSF, IL-10, IL-1β, and IL-1α was developed on the system. The assays had limits of detection in the range 0.01–0.03 pg/mL, and the average imprecision (CV) of the Simoa signal was 4.2%. This assay was used to measure the concentrations of these cytokines in the plasma of patients with Crohn‘s Disease (CD), before and after treatment with anti-TNF-α antibody drugs, and in the serum of Type 1 diabetics. Concentrations of TNF-α and IL-6 in the CD samples determined using the fully-automated, multiplex Simoa assay had good correlation with the manual, single-plex assays previously reported. Drug treatment caused reductions in the mean concentration of all 6 cytokines in the plasma of CD patients. The concentrations of 4 cytokines were significantly higher in diabetics compared to healthy controls. The system could enable the widespread, multiplexed measurement of protein biomarkers with low abundance.
Keywords: Automation, Simoa, digital ELISA, multiplexed immunoassays
Graphical abstract
Introduction
The sensitive and multiplex detection of protein biomarkers has emerged as a key component of clinical measurements in biomedicine, especially in the pharmacodynamic and pharmacokinetic characterizations of candidate drug therapies.1 Immunoassays with high analytical sensitivity offer the potential to provide quantitative information on the clinical state of all diseased and healthy subjects, and to directly quantify the biochemical impact of candidate therapies on the protein target. Multiplex measurements provide the ability to understand biochemically complex physiological states, and responses to those states after administration of a drug, using a single measurement. In order for sensitive and multiplex assays to have a direct impact on drug development and clinical diagnostics, however, they must be implemented on instrumentation that is reliable, reproducible, scalable, and cost effective. Here we report an assay performed on a fully-automated immunoanalyzer for simultaneously measuring six cytokines in blood with femtomolar sensitivity.
Platforms that run ligand binding assays—especially immunoassays—represent essential technologies in the drug development process. A consortium of technologists and drug development scientists from the American Association of Pharmaceutical Scientists (AAPS) recently defined the “ideal” ligand binding assay platform.2 They highlighted 9 attributes and quantitative performance targets that contribute to the idealized platform: sensitivity; dynamic range; precision; ruggedness; total assay time; multiplex; throughput; multi-modality; and life cycle support. The attributes and performance targets identified by these authors provide an excellent guideline for the characteristics needed for automated immunoassay platforms. Assay sensitivity, multiplexing and automation are three key capabilities of immunoassay systems that address many aspects of this idealized list. In recent years, several immunoassay technologies have emerged that greatly enhance analytical sensitivity over conventional immunoassays,3,4,5,6 including single molecule arrays (Simoa) developed in our group.3 The ability to measure proteins at much lower concentrations than previously has led to new clinical and scientific insights in several fields, including inflammatory disease, neurology, cardiovascular disease, cancer, and infectious diseases.7,8 Platforms that allow multiplexing of protein quantification have also emerged, most notably electrochemiluminescence detection on planar spotted arrays9 and fluorescence detection of suspended, multiplexed bead populations.10 These technologies have been successful in allowing multiplex protein measurements in drug development processes, although these technologies have sensitivities comparable to conventional immunoassays. Automated immunoanalyzers have been available for more than 2 decades and have enabled high throughput assays with short time to first results, with excellent precision and ruggedness in laboratories across the world. These automated systems, however, have not provided high sensitivity or, in general, multiplexing capabilities. A platform that offers high sensitivity, multiplex immunoassays with full automation has not been reported.
We have previously reported our approach to high sensitivity immunoassays using Simoa, also known as digital ELISA.3,8,11,12 In this approach, single protein molecules are captured on antibody-coated, paramagnetic beads, the captured proteins are labeled with an enzyme label, and single beads are isolated and sealed in arrays of femtoliter wells in the presence of enzyme substrate. The sealing step confines the fluorescent product of the enzyme-substrate reaction to ~40 fL volume, and within 30 s the fluorescence generated by a single enzyme can be detected on an uncooled CCD camera using a white light excitation source.3 The fraction of beads associated with at least one enzyme (fon), and the intensity from each well (Ibead) is then determined by image analysis, and the average number of enzymes per bead (AEB) is calculated using either fon and the Poisson distribution equation (at low concentrations) or using the average Ibead (at high concentrations).11 The ability to count single immunocomplexes reduced the concentrations of labeling reagents used, lowered the backgrounds of the assays, and resulted in an average of a 1000-fold improvement in sensitivity over analog immunoassays.3 Subsequent to the initial demonstration of Simoa, we developed a low cost array consumable (Simoa disc) for loading and sealing the beads in arrays of 216,000 wells,12 and an optical system was developed to image these arrays at multiple wavelengths.13 Based on the consumable and optical imager, we developed a method to multiplex the digital ELISA approach, and demonstrated a manual 4-plex cytokine assay.14 Here, we demonstrate the integration of these capabilities into a fully automated system, and describe how we used this system to develop and test an assay for quantifying 6 cytokines simultaneously with single molecule resolution. The multiplex detection of cytokines is particularly important in understanding the underlying molecular basis of chronic inflammatory diseases, and for quantifying the impact of candidate drugs on the physiological concentrations of these molecules. We have illustrated the use of the fully automated 6-plex by analyzing blood samples of patients with Crohn‘s disease (CD) and diabetes.
Materials and Methods
Materials
2.7-μm diameter carboxyl-functionalized paramagnetic beads were purchased from Agilent Technologies. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) was purchased from Thermo Scientific. Phosphate buffered saline (PBS) was purchased from Amresco. Alexa Fluor 488 hydrazide and Resorufin-β-D-galactopyranoside (RGP) were purchased from Life Technologies. Cy5 Mono Hydrazide was purchased from GE Healthcare. Hilyte Fluor 750 Hydrazide was purchased from Anaspec. Antibodies and proteins standards were all purchased from commercial vendors. All other chemicals were purchased from Sigma-Aldrich. Detection antibodies were biotinylated using standard methods described previously.11 Streptavidin-β-galactosidase (SβG) was conjugated in house using methods described previously.11 Simoa discs comprised of 24 arrays of 216,000 40-fL-sized microwells molded into cyclic olefin copolymer (COC) and bonded to a microfluidic manifold were obtained from Sony DADC.12 Fluorocarbon oil (Krytox®) was obtained from DuPont.
Preparation of populations of protein-specific multiplex encoded beads
Six distinct dye-encoded bead populations were prepared by reacting different levels of a single hydrazide dye with carboxyl-functionalized paramagnetic beads, as described previously:14 1 level of Alexafluor 488 (L1), 3 levels of Cy5 (L1, 2, 3), and 2 levels of HyLite Fluor 750 (L1 and 2). Antibodies to IL-6, TNF-α, GM-CSF, IL-10, IL-1β, and IL-1α were conjugated to Alexafluor 488 L1, Cy5 L1, Cy5 L2, Cy5 L3, HyLite Fluor 750 L1, and HyLite Fluor 750 L2, respectively, as described previously.14 The 6 antibody-coated encoded bead populations were then mixed in equal volumes in a 15 mL opaque bottle to give 1,000,000 beads/plex/mL or, 6,000,000 total beads/mL, and stored at 2–8 °C. Just prior to use, the beads were vortexed for 30 s to ensure proper bead resuspension.
Simoa Measurements
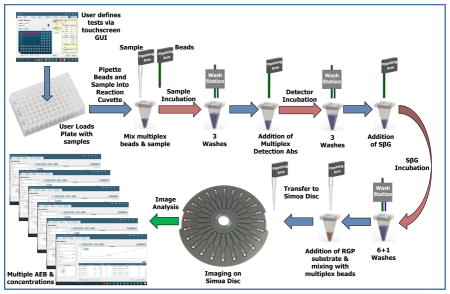
All Simoa measurements were performed on a Simoa HD-1 Analyzer (Quanterix Corporation). The Simoa measurements occurred in three keys steps on the instrument (Figure 1): liquid handling for the formation of immunocomplexes on multiplexed beads; imaging of the beads sealed in the Simoa disc; and, analysis of the images to yield AEB and concentrations for each protein. Each of these steps are described in detail below.
Figure 1.

Schematic illustrating the work flow of “sample-in to result-out” on the automated, multiplexSimoa system.
i) Automated capture of multiple proteins on subpopulations of paramagnetic beads and formation of enzyme-labeled immunocomplexes
The instrument was first loaded with the necessary reagents, consumables, and samples needed to perform the assays. The reagents were: a bottle containing a mixture of 6 protein-specific, encoded beads; a bottle containing a mixture of 6 protein-specific, biotinylated detection antibodies; a bottle of enzyme conjugate (SβG); and a bottle of enzyme substrate (RGP). The consumables were: stacks of Simoa discs (up to 2 × 16 discs/stack); disposable tips (up to 6 × 96 tips/box) for pipetting the sample into the reaction cuvette, and beads onto the Simoa disc; and stacks of reaction cuvettes (up to 10 × 50 cuvettes per stack) for performing sample mixing, washing, and incubation of beads. Samples (150 μL) were loaded onto the instrument in a 96-well microtiter plate. The following bead manipulation steps and incubations were then performed on the instrument. First, 100 μL of multiplex bead mixture (100,000 beads per plex = 600,000 beads total) was pipetted by a fixed tip pipettor from the bead bottle into a reaction cuvette. The beads were pelleted by a magnet in the washer module and the supernatant liquid was aspirated. A sample or calibrator (100 μL) was pipetted into the cuvette using a disposable tip pipettor, and the beads were dispersed in the sample or calibrator by a shaker. The mixture of sample and beads was incubated in an incubator module for 35 min at 25 °C. The beads were then magnetically separated and washed three times in 5× PBS and 0.1% Tween 20 in the washer module. 100 μL of a solution containing mixtures of biotinylated detection antibodies (anti-IL-6 at 0.125 μg/mL; anti-TNF-α at 0.4 μg/mL; anti-GM-CSF at 0.1 ug/mL; anti-IL-10 at 0.1 ug/mL; anti-IL-1β at 0.1 ug/mL; anti-IL-1α at 0.3 ug/mL) was added by the fixed tip pipettor to the reaction cuvette, shaken to disperse the beads, and incubated for 5 min at 25 °C. The beads were then magnetically separated and washed three times in 5× PBS and 0.1% Tween 20 in the washer module. 100 μL of a solution containing 50 pM of SβG was added by the fixed tip pipettor to the reaction cuvette, shaken to disperse the beads, and incubated for 5 min at 25 °C. The beads were then magnetically separated and washed six times in 5× PBS and 0.1% Tween 20, and washed once in PBS in the washer module. 25 μL of 100 μM RGP in PBS was added by a disposable tip pipettor to the reaction cuvette, the beads were mixed, and 15 μL of this bead-substrate mixture was transferred into an inlet port of an array on a Simoa disc for analysis.
ii) Automated imaging of multiplex beads in arrays of femtoliter wells
A Simoa disc composed of 24, 3 mm × 4 mm arrays of ~216,000 femtoliter wells and individually addressable microfluidic manifolds was loaded by a pneumatic arm onto a platen above the optical imaging system.13 The Simoa disc was used for the load, seal, and imaging of the encoded beads in the presence of RGP in the microwells as described previously.12,14 As described above, 15 μL of the solution containing the mixture of bead subpopulations and RGP was pipetted into the inlet port of an array the disc. Vacuum pressure was then applied to the outlet port, and drew the bead solution over the arrays of femtoliter wells. The beads were allowed to settle via gravity into the wells of the array for 90 seconds. After the beads had settled, 40 μL of fluorocarbon oil was dispensed into the inlet port, and vacuum was simultaneously applied to the outlet port to pull the oil over the array. The oil pushed the aqueous solution and the beads that were not in the wells off of the array surface, and formed a liquid-tight seal over the wells containing beads and enzyme substrate. Once the wells were sealed, the disc was rotated to be aligned with the optics that image the array. The fluorescence optical system has been described before13 and was composed of: a white light illumination source; a custom, 12-element, wide field of view (3 × 4 mm object) microscope lens system; a CCD camera (Allied Vision, Prosilica GT3300 8 Mp). The imaging process took 45 s in total for each array, and was composed of the following sequential steps. Initially, the array was indexed to be aligned with the CCD chip and the array was clamped against a reference plane to which the rest of the optical system was aligned. Next, the imaging system automatically focused to the array by taking successive images at different focus positions using “dark field” images of the array using the 622 nm/615 nm excitation/emission filters (exposure time = 0.3 ms), and setting focus to the highest contrast image. After focusing, a seven step image acquisition process occurred. First, a “dark field” image of the array was acquired by using the 622 nm/615 nm excitation/emission filters (exposure time = 0.3 ms). Second, an image at 574 nm/615 nm excitation/emission (exposure time = 3 s) was acquired; this image is the t = 0 image (F1) of the single molecule resorufin signal. Third, an image at excitation/emission of 740 nm/800 nm (exposure time = 3s) was acquired to identify beads labelled with the HyLite Fluor 750 dye. Fourth, an image at excitation/emission of 680 nm/720 nm (exposure time = 3 s) was acquired; this image was not used in this work. Fifth, an image at excitation/emission of 622 nm/667 nm (exposure time = 3 s) was acquired to identify beads labelled with the cy5 dye. Sixth, an image at excitation/emission of 574 nm/615 nm excitation/emission (exposure time = 3 s) was acquired 30 s after the image F1; this image is the t = 30 s image (F2) of the single molecule resorufin signal. Seventh, an image at excitation/emission of 490 nm/530 nm (exposure time = 2 s) was acquired to identify beads labelled with the Alexa Fluor 488 dye. Images were saved as a single IPL file.
iii) Automated Analysis of Simoa Images to determine AEB and concentrations
A custom image analysis software program integrated into the software used to control the Simoa HD-1 Analyzer was used to determine from the captured images the enzyme activity associated with each bead within each subpopulation. The image analysis method has been described previously,14 and is summarized briefly here. A masking method was applied to the dark field image to define the locations and boundaries of the wells. The resulting well mask was applied to each fluorescence image to determine the presence of specific beads and enzymes within the wells.14 Wells that had been classified as containing a single bead from a particular bead subpopulation were then classified as: a) associated with enzyme activity (“on” or active), if the fluorescence from resorufin within that well increased beyond a known threshold (40 fluorescence counts), or; b) not associated with enzyme activity (“off” or inactive), if the fluorescence from resorufin within that well did not increase beyond the threshold.12 The increase in fluorescence was determined for each “on” bead (Ibead).11 For each bead subpopulation, the fraction of “on” beads (fon) was determined. In the digital range (fon < 0.7), fon was converted to average number of enzymes per bead (AEB) using the Poisson distribution equation.11 In the analog range (fon > 0.7), AEB was determined from the average Ibead of all the beads in an array (Ībead).11 From the resulting AEB values of calibrators of known concentration, the concentrations of samples of unknown concentration were determined from interpolation using fitting software built into the system software.
Clinical Samples
Plasma samples from 32 patients—16 case subjects exhibiting inflammatory bowel disease and 16 age- and gender-matched control subjects—were obtained from a clinical study performed at the Mayo Clinic (Rochester, MN) under IRB as described previously.7 All case subjects had clinically active Crohn‘s Disease (CD) and started a course of anti-TNF-α therapy (infliximab, adalimumab, or certolizumab pegol). Two plasma collections were planned for each case subject; the first sample collection occurred prior to initiation of anti-TNF-α therapy (n = 16), and the second occurred 12 weeks post initial infusion therapy (n = 8). Plasma samples were processed and frozen at −70 °C. Full details of the clinical study are provided elsewhere.7 Serum samples from diabetic patients and healthy individuals collected using the same method were obtained from Bioreclamation (Westbury, NY). Prior to testing, all clinical samples were pre-treated by passing them over a Protein G column. This process removes from the sample interfering antibodies, e.g., those produced by the immune response to the anti-TNF-α therapy, and also any possible interference by the antibody drug itself as described previously.7
Results and Discussion
Process for fully-automated, multiplex single molecule immunoassays
The measurements of single3 and multiple14 proteins using digital ELISA have been described previously. The process that we developed for fully automating multiplex digital ELISA is shown in Figure 1.
The full details of the process are described in the Methods and Materials section. In brief, consumables, reagents, and samples (in a 96-well microtiter plate) were first loaded onto the system. Onboard software was used to define the assays to be performed by the system (incubation times, assay steps, etc.), to ensure that sufficient resources were available for the assays, and to control the execution of the automated assays. The instrument automated the processing and analysis of the samples to produce the Simoa measurements for 6 different proteins simultaneously for each sample. Beads were incubated sequentially with sample, a mixture of detection antibodies, and enzyme conjugate, with washes in between each incubation. After the formation of immunocomplexes on the beads, the beads were resuspended in enzyme substrate, and transferred to the Simoa disc to be loaded, sealed, and imaged in a microwell array. The images at the characteristic wavelengths of enzyme product and bead dyes were then analyzed to determine the average number of enzymes per bead (AEB) for each of the different 6 encoded bead types.14 The samples were processed from the microtiter plate in serial manner with a fixed cadence, i.e., a new sample was picked up by the disposable tip and mixed with beads in a reaction cuvette every 45 s. After the time to first result for the first sample (83 min), a new sample result was produced, therefore, every 45 s. A detailed description of the system architecture is provided elsewhere.15
Assay Performance
Figure 2 shows representative curves for 6 human cytokines spiked at known concentrations into bovine serum in 3 runs over 3 days in the fully-automated 6-plex immunoassay. Tables S1–S3 in the Supplemental Material show the corresponding AEB values and coefficients of variation (CVs) for the three calibration runs. In these tests, recombinant forms of all 6 human proteins were spiked at the same concentration. Based on extrapolating concentrations at signals that are 3 sd above the assay backgrounds, the average limits of detection (LODs) in these calibration tests were 10 fg/mL for IL-6, 24 fg/mL for TNF-α, 16 fg/mL for GM-CSF, 26 fg/mL for IL-10, 13 fg/mL for IL-1β, and 26 fg/mL for IL-1α. The LODs for each run and average are summarized in Table S4. The sensitivities of the IL-6, TNF-α, IL-1β, and IL-1α are comparable (10–30 fg/mL) to those previously reported for 4-plex Simoa assays (based on a semi-automated assay using a Tecan liquid handler and imaging Simoa discs on the same imager submodule in the automated instrument)14 and for assays run in single-plex (using a Tecan robot and microwell arrays etched into glass fiber bundles),7 using the same antibody pairs.
Figure 2.

Plots of average number of enzymes per bead (AEB) against concentration (0–1 pg/mL) of 6 cytokines spiked into bovine serum from the first run of three measured using the 6-plex assay performed on the automated Simoa system. The inset plots show the curves across the entire range (0–30 pg/mL). The solid lines are fits to the data using a 4 parameter logistic equation; all fits had R2 values > 0.99. The 10 pg/mL data point for GM-CSF was omitted from that fit to improve the fit quality of the low concentration data. As for all plots, error bars are shown based on one standard deviation of triplicate measurements. If the error is smaller than the symbol plotted, error bars are not shown.
The average imprecision (CV) in AEB of the calibrator samples (excluding the blank or zero concentration calibrator that has significant Poisson noise based on the low number of active beads) across the three days was 4.2% (n = 108), with a range from 0.4–11.8%. This imprecision compares favorably with that reported for the semi-automated assay based on Tecan robot and microwell arrays etched into glass fiber bundles that had an average CV of 7.1%.11 Assay imprecision is likely dominated by stacking of imprecision in liquid handling steps, and the improvement shown over the manual assay is likely due to the greater precision of the integrated system. The inter-run imprecision between the three runs is shown in Table S5. The average inter-run imprecision in AEB of the calibrator samples (excluding the blank) was 8.3%, and ranged from 1% to 15%. While these calibration runs were not designed statistically to determine the limit of quantitation (LOQ) of the assays—defined as the concentration at which the imprecision of the concentration determined is ≤20%—we were able to provide a bounded estimate of LOQ. Figure S1 shows plots of the imprecision and inaccuracy of the concentration determined of calibrators read back off the calibration curve. For all calibrators including the lowest at 0.1 pg/mL, the imprecision of the concentration determined was <20% (Figure S1A) showing that the LOQ in all cases was <0.1 pg/mL. These values are consistent with more formal LOQ determinations for single-plex assays.15 The error in the read back was <10% in all cases (Figure S1B) further indicating the quantitative quality of the measurements.
We have shown previously by spiking in individual proteins in a 4-plex at high concentrations (100 pg/mL) that specificities of the remaining 3-plexes were maintained over ~1000-fold in concentration.14 At higher concentrations, false positive signals started to appear on beads that were specific to proteins that were not present in the samples because of antibody cross-reactivity.14 We performed similar experiments to verify the specificity of the fully-automated 6-plex Simoa assay (Figure S2). As for the 4-plex, small increases in background are observed in some specific beads once the concentrations of “off target” proteins reaches 100 pg/mL, indicating good specificity and low cross-reactivity over the dynamic range of concentrations.
Clinical sample results
To demonstrate the potential utility of this system for clinical diagnostics and drug screening, we evaluated the fully automated 6-plex Simoa assay by testing two sets of clinical samples: the plasma of patients with CD and the serum of diabetics (and healthy controls).
Patients with Crohn’s disease
We previously performed a prospective study measuring the concentrations of TNF-α and IL-6 in the plasma of patients with CD using manual, single-plex Simoa assays.7 The study showed that Simoa provided an approximately 1000-fold improvement in the sensitivity of standard immunoassays for these two analytes (LOD ~ 0.01 pg/mL). This greater sensitivity allowed both proteins to be measured in all patients before and after treatment, and in all healthy controls with good precision (physiological concentrations ~1–10 pg/mL). Here, we retested the same samples using the 6-plex Simoa assay on the fully-automated instrument to evaluate the potential for providing information on more cytokines simultaneously in high throughput. In this study, the concentrations of the 6 cytokines were determined on the instrument for 16 patients with CD and 16 age-matched healthy subjects. Concentrations were also determined for the 8 CD patients that returned for follow up after treatment with anti-TNF-α drugs. All plasma samples were first passed over a Protein G column to remove any interfering antibodies as described previously,7 diluted 4-fold, and then tested in triplicate using Simoa.
Figure 3 shows scatter plots of the concentrations of the 6 cytokines determined for CD patients before treatment, and healthy controls. The concentrations of the 6 cytokines for all samples are shown in Table S6, and the mean concentrations of the 6 cytokines in the plasma of CD patients and controls are summarized in Table S7. Of the 6 cytokines, only the mean concentration of IL-6 in CD patients was statistically different than healthy controls (P = 0.022). These data are consistent with the previously reported single-plex assessment of these samples, where IL-6 showed a significant difference in concentrations, but TNF-α did not.7
Figure 3.

Scatter plots of the concentrations determined using Simoa of 6 cytokines in the plasma of CD patients and matched healthy controls. The thick and thin solid lines are mean concentrations and standard error means (SEM) for that population. The dotted lines represent the average LOD of each cytokine in the 6-plex Simoa assay.
Figure S3 in the Supplemental Material shows bar charts of the concentrations the 6 cytokines in the plasma of CD patients before and after treatment with anti-TNF-α drugs; concentration values are provided in Table S6. Table S8 shows the percentage change in concentration of each cytokine upon administration of an anti-TNF-α antibody drug for each of the 8 patients, along with the mean % change and the mean concentrations before and after treatment. Three of the cytokines (IL-6, TNF-α and GM-CSF) showed a consistent reduction in plasma concentration across most patients after the drug treatment, with mean reductions of 52%, 34%, and 25%, respectively. The mean changes in concentrations of IL-6 and TNF-α using the automated, multiplexed Simoa assay were consistent with the changes determined previously using the manual, single-plex assay,7 as shown in Table S8. We note that the mean concentrations of all 6 cytokines were lower after treatment than before treatment, despite the greater patient-to-patient changes observed for IL-10, IL-1β, and IL-1α (Table S8). This interesting observation is explained by the fact that the greater the concentration of the cytokine before treatment, the greater the reduction in its concentration after treatment (Figure S4). We believe that this observation has not been reported previously (because of lack of sufficiently sensitive cytokine assays), and may provide an indication of which patients will respond most to anti-cytokine antibody therapies, at least at the biochemical level. We did not observe this correlation in the clinical symptoms,7 although the cohort was small.
Figure 4 shows plots comparing the concentrations determined for IL-6 and TNF-α in the same plasma samples from CD patients using the multiplexed Simoa assay and the manual, single-plex Simoa assay.7 For IL-6, there was an excellent correlation between the concentrations determined using the two methods (r2 = 0.94 and slope of linear regression = 0.97). The correlation for TNF-α (r2 = 0.57 and slope of linear regression = 0.55) was not as good as IL-6. One possible explanation for the differences in concentrations of TNF-α determined using these two measurements is a difference in sample preparation. In this multiplex Simoa study, we pre-treated all samples using a protein G column before testing to remove interfering antibodies, whereas in the previous single-plex Simoa study, we only performed the sample treatment on patient samples who had been on one of the anti-TNF-α drugs (infliximab) that was shown to cause interference in the TNF-α Simoa assay. As the measurement of TNF-α may be impacted by the exact composition of antibodies in the sample, the protein G treatment may have caused differences in concentrations determined in the two methods. We also note that the concentrations of TNF-α measured are over a narrow range (2–8 pg/mL) that might impact the power of the linear regression. Overall, these data indicate that the concentrations determined by the automated system in multiplex Simoa are comparable to those of earlier, manual incarnations of the technology.
Figure 4.

Plots showing correlation between concentration of IL-6 and TNF-α in the plasma of CD patients determined using the fully-automated, 6-plex Simoa assay (Table S6) and the manual, single-plex Simoa assay previously reported.7
As described above, the imprecision of the Simoa signal (AEB) using the multiplex Simoa assay was good (mean = 4.2%, excluding blank calibrators). When determining the imprecision in the concentrations determined in samples (i.e., concentration CVs), the imprecision is determined by the signal precision, response of the assay (slope), and the concentration of the samples themselves. The mean concentration imprecision CVs (Table S6) for all of the samples tested here were 5.4% (range 0.3–32%), 7.2% (0.5–32%), 13% (0.2–61%), 8.1% (0.4–40%), 12% (0.2–42%), and 20% (0.7–53%) for IL-6, TNF-α, GM-CSF, IL-10, IL-1β, and IL-1α, respectively. While this imprecision is acceptable for quantifying concentrations (CV ≤ 20%), the imprecision of the GM-CSF, IL-1α, and IL-1β were higher because the average concentrations were lower than the other three cytokines with concentration CVs < 10% (Table S7). As the concentrations of analyte approaches the limit of detection, imprecision increases.
Patients with Diabetes
Figure 5 shows scatter plots comparing the concentrations of the 6 cytokines in the serum of patients with Type 1 diabetes and healthy controls, determined using multiplexed Simoa. The mean concentrations are summarized in Table S9, along with P values from an unpaired t-test. TNF-α, GM-CSF, IL-10, and IL-1β all had statistically higher concentrations in the serum of diabetics compared to the serum of healthy controls. The treatment of Type 1 diabetes—an autoimmune disease of the pancreas—with immununotherapies is under active investigation,16,17 and the ability to measure cytokine response to these developmental therapies in diabetics could provide valuable information on their efficacy.
Figure 5.

Scatter plots of the concentrations determined using Simoa of 6 cytokines in the serum of patients with Type 1 diabetes and healthy controls. The thick and thin solid lines are mean concentrations and SEM for that population. The dotted lines represent the average LOD of each cytokine in the 6-plex Simoa assay.
In summary, the data presented in this work indicate that fully-automated, multiplexed Simoa assays can enable the accurate and precise measurement of cytokine concentrations in clinical samples. As described previously,7 current multiplexed immunoassays are not sufficiently sensitive to provide precise information on the change in concentration of cytokines upon drug administration, or simply do not detect them in blood at all.7 The capability described in this manuscript would, therefore, enable researchers to quantify the effect that candidate anti-cytokine drugs have on the systemic concentration of drug targets and related proteins. To provide an objective viewpoint of the capabilities of the system presented here, in Table 1 we compare the “ideal” ligand binding system—referenced above and described by Spriggs et al.2—with the automated Simoa system. While some of the attributes were not available in the Simoa system (e.g., 384-well plate format) and some were not assessed in this study (i.e., varied laboratory conditions), the automated Simoa system compares favorably to most of the requirements of the idealized system. We note that these authors did not consider specificity, accuracy, or linearity in their criteria, attributes that are important for immunoassay systems. In this manuscript, we have addressed the specificity of the multiplex Simoa assay; in other publications, we address linearity and accuracy.15 We conclude from this comparison that the technology should be of value in helping the scientists that are endeavoring to develop effective and safe protein-based drugs.
Table 1.
Comparison of “Ideal” ligand binding assay system proposed by Spriggs et al.,2 and the automated Simoa instrument presented here.
| Platform Attribute | Measure of Success (Sprigg et al. AAPS J., 2012)2 | Automated Simoa performance |
|---|---|---|
| Sensitivity | Capable of quantifying low analyte concentrations of drug and biomarkers in a variety of matrices | 1000-fold more sensitive than standard immunoassaysa,3,8 fg/mL sensitivitya |
| Dynamic Range | Greater than 3 log range | >4 log rangea,11 |
| Precision | Less than 2% variability for the instrument signal of internal standard | 4.2% variability in AEBa |
| Ruggedness | Consistent performance under varied laboratory conditions | Full automation has the potential to minimize lab-to-lab variabilityc |
| Tolerance to biological interferences | Standard immunoassay approaches to reduce interferences8 | |
| Total Assay time | Results in 1 h or less | Time to first result = 83 mina (45 min also demonstrated)b |
| Multiplexing | Should have capabilities | 6-plex immunoassay demonstrated;a 10-plex decoding demonstratedb |
| Minimal cross-talk due to detection mechanisms | Optical cross-talk <0.1%14 | |
| Flexibility/throughput | Automation compatible | Fully automateda |
| Capable of assay miniaturization (e.g., 384+ well plates or other substrate) | 96-well plates and primary tubes as sample inputa | |
| Utilization of various solid supports | Standard immobilization chemistries compatible with beadsa | |
| Ability to run in low and high throughput environments | Yesa | |
| Possible to use throughout the life cycle of drug development | Yes. Available for researchers, contract-research organizations, and under development for regulatory approvala | |
| Multi-modality | Measure wide variety of therapeutics including proteins, peptides, antibodies, etc. | Proteins,a antibodies,b DNA18 demonstrated |
| Life Cycle Support | Multiple sources for reagent availability | Yes. Many commercial sources of antibody pairs.8 |
| Ability to label reagents in-house | Yes. Open platform via “homebrew” kits.a |
This work.
Unpublished work.
Not tested in this work.
Supplementary Material
Highlights.
We present a fully automated instrument for multiplex, single molecule immunoassays
A multiplex assay for IL-6, TNF-α, GM-CSF, IL-10, IL-1β, and IL-1α was developed
Sensitivities were in the range 10–30 fg/mL
Inflammatory markers were measured in the blood of Crohn‘s patients and diabetics
Acknowledgments
This project was supported by Award Number R43CA133987 from the National Cancer Institute. We thank our colleagues at Quanterix and Stratec Biomedical for their creativity and endeavor during the collaboration to develop the Simoa HD-1 Analyzer. We thank Dr. David H. Wilson for his help in estimating LOQ. We acknowledge Dr. David W. Hanlon (Quanterix Corporation), Dr. William A. Faubion, and Ms. Mary A. Till (Mayo Clinic) for their contributions to the original Crohn‘s Disease clinical study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yu X, Schneiderhan-Marra N, Joos TO. Protein microarrays for personalized medicine. Clin Chem. 2010;56:376–87. doi: 10.1373/clinchem.2009.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spriggs FP, Don Zhong Z, Safavi A, et al. Ligand binding assays in the 21st Century laboratory: platforms. AAPS J. 2012;14:113–118. doi: 10.1208/s12248-012-9321-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rissin DM, Kan CW, Campbell TG, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;28:595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Todd J, Freese B, Lu A, et al. Ultrasensitive flow-based immunoassays using single-molecule counting, Clin. Chem. 2007;53:1990–1995. doi: 10.1373/clinchem.2007.091181. [DOI] [PubMed] [Google Scholar]
- 5.Kelley SO, Mirkin CA, Walt DR, et al. Advancing the speed, sensitivity and accuracy of biomolecular detection using multi-length-scale engineering. Nat Nanotechnol. 2014;9:969–80. doi: 10.1038/nnano.2014.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barletta JM, Edelman DC, Constantine NT, et al. Lowering the detection limits of HIV-1 viral load using real-time immuno-PCR for HIV-1 p24 antigen. Am J Clin Pathol. 2004;122:20–7. doi: 10.1309/529T-2WDN-EB6X-8VUN. [DOI] [PubMed] [Google Scholar]
- 7.Song L, Hanlon DW, Chang L, et al. Single molecule measurements of tumor necrosis factor α and interleukin-6 in the plasma of patients with Crohn’s disease. J Immunol Methods. 2011;372:177–86. doi: 10.1016/j.jim.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 8.Rissin DM, Wilson DH, Duffy DC. Measurement of Single Protein Molecules Using Digital ELISA. In: Wild D, editor. The Immunoassay Handbook: Theory and applications of ligand binding, ELISA and related techniques. 4. Elsevier; Oxford, UK: 2013. [Google Scholar]
- 9.Chowdhury F, Williams A, Johnson P. Validation and comparison of two multiplex technologies, Luminex and Mesoscale Discovery for human cytokine profiling. J Immunol Methods. 2009;340:55–64. doi: 10.1016/j.jim.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Hsu HY, Joos TO, Koga H. Multiplex microsphere-based flow cytometric platforms for protein analysis and their application in clinical proteomics - from assays to results. Electrophoresis. 2009;30:4008–19. doi: 10.1002/elps.200900211. [DOI] [PubMed] [Google Scholar]
- 11.Rissin DM, Fournier DR, Piech T, et al. Simultaneous detection of single molecules and singulated ensembles of molecules enables immunoassays with broad dynamic range. Anal Chem. 2011;83:2279–2285. doi: 10.1021/ac103161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kan CW, Rivnak AJ, Campbell TG, et al. Isolation and detection of single molecules on paramagnetic beads using sequential fluid flows in microfabricated polymer array assemblies. Lab Chip. 2012;12:977–95. doi: 10.1039/c2lc20744c. [DOI] [PubMed] [Google Scholar]
- 13.McGuigan W, Fournier DR, Watson GW, et al. The optics inside an automated single molecule array analyzer. In: Vo-Dinh T, Mahadevan-Jansen A, Grundfest WS, editors. Proc SPIE 8935, Advanced biomedical and clinical diagnostic systems XII. 2014. p. 89350X. [Google Scholar]
- 14.Rissin DM, Kan CW, Song L, et al. Multiplexed single molecule immunoassays. Lab Chip. 2013;13:2902–2911. doi: 10.1039/c3lc50416f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson DH, Rissin DM, Kan CW, et al. The Simoa HD-1 Analyzer: a novel fully automated digital immunoassay analyzer with single-molecule sensitivity and multiplexing. 2015 doi: 10.1177/2211068215589580. submitted. [DOI] [PubMed] [Google Scholar]
- 16.Kim JH, Jin SM, Kim HS, et al. Immunotherapeutic treatment of autoimmune diabetes. Crit Rev Immunol. 2013;33:245–81. doi: 10.1615/critrevimmunol.2013006913. [DOI] [PubMed] [Google Scholar]
- 17.Baumann B, Salem HH, Boehm BO. Anti-inflammatory therapy in type 1 diabetes. Curr Diab, Rep. 2012;12:499–509. doi: 10.1007/s11892-012-0299-y. [DOI] [PubMed] [Google Scholar]
- 18.Song L, Shan D, Zhao M, et al. Direct Detection of Bacterial Genomic DNA at Sub-Femtomolar Concentrations Using Single Molecule Arrays. Analytical Chemistry. 2013;85:1932–1939. doi: 10.1021/ac303426b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.