

KRAS-mutant pancreatic cancer models, including KRAS inhibitor–resistant models, show exquisite sensitivity to combined pan-ERBB and KRAS targeting, which provides the rationale for testing this drug combination in clinical trials.
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a lethal cancer with a low survival rate. Recently, new drugs that target KRASG12D, a common mutation in PDAC, have been developed. We studied one of these compounds, MRTX1133, and found it was specific and effective at low nanomolar concentrations in patient-derived organoid models and cell lines harboring KRASG12D mutations. Treatment with MRTX1133 upregulated the expression and phosphorylation of EGFR and HER2, indicating that inhibition of ERBB signaling may potentiate MRTX1133 antitumor activity. Indeed, the irreversible pan-ERBB inhibitor, afatinib, potently synergized with MRTX1133 in vitro, and cancer cells with acquired resistance to MRTX1133 in vitro remained sensitive to this combination therapy. Finally, the combination of MRTX1133 and afatinib led to tumor regression and longer survival in orthotopic PDAC mouse models. These results suggest that dual inhibition of ERBB and KRAS signaling may be synergistic and circumvent the rapid development of acquired resistance in patients with KRAS mutant pancreatic cancer.
Significance:
KRAS-mutant pancreatic cancer models, including KRAS inhibitor–resistant models, show exquisite sensitivity to combined pan-ERBB and KRAS targeting, which provides the rationale for testing this drug combination in clinical trials.
Graphical Abstract
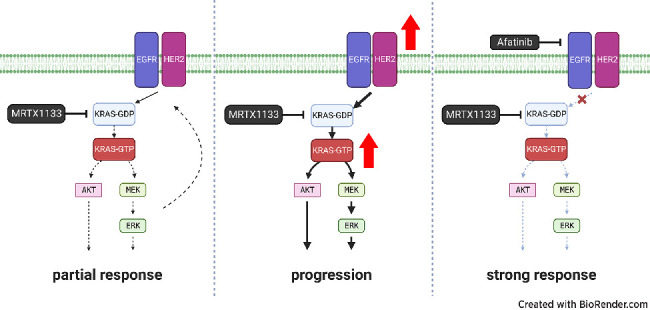
Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for more than 90% of all pancreatic malignancies, and PDAC patients have a 5-year survival rate of 11% (1). More than 90% of PDACs harbor oncogenic KRAS mutations. Approaches to drug KRAS were unsuccessful until the recent development of KRASG12C inhibitors such as sotorasib (AMG510) and adagrasib (MRTX849), which are now both FDA approved (2, 3). However, only 2% of patients with PDAC harbor the KRASG12C mutation, limiting the potential applicability in this setting. Instead, KRASG12D (∼40%) and KRASG12V (∼32%) mutations are most common in PDAC (4). Recently, researchers have taken advantage of activating conformational changes present in KRAS mutants to identify compounds that bind noncovalently to GDP-bound KRASG12D with high affinity (5). One such agent, MRTX1133 (Mirati Therapeutics), has shown preclinical efficacy in KRASG12D mutated colon, lung, and pancreas cancer (5, 6). Most recently, Kemp and colleagues thoroughly tested MRTX1133 in mouse models of PDAC and demonstrated tumor regression after monotherapy, at least in part due to immunomodulation (7). However, these studies did not evaluate how acquired resistance impacts the efficacy of MRTX1133.
In clinical testing of KRAS-targeted therapy, researchers found that KRASG12C inhibition in non–small cell lung cancer (NSCLC) and colorectal cancer resulted in relatively short durations of response due to the rapid development of genetic and nongenetic mechanisms of resistance (8). Interestingly, Amodio and colleagues demonstrated that EGFR blockade can reverse resistance to KRASG12C inhibition in colorectal cancer models (9). This underlies the importance of understanding the kinetics and feedback signaling mechanisms of upstream receptor tyrosine kinases (RTK), KRAS, and their effector pathways when testing novel KRAS inhibitors. Indeed, the combination of EGFR-targeted antibody cetuximab potentiates the activity of MRTX1133 in xenograft models of PDAC (6). Yet, it remains unclear if this strategy will combat the development of MRTX1133 resistance in PDAC. Furthermore, in a previous study, we found that inhibition of KRAS downstream signaling using MEK1/2 and AKT inhibitors in PDAC patient-derived organoids (PDO) led to the hyperactivation of EGFR, HER2, and HER3 (10). Importantly, targeting EGFR was not effective in organoids and did not lead to synergy with KRAS effector blockade due to continued signaling via HER2 and HER3. However, irreversible pan-ERBB inhibition was synergistic with MEK1/2 inhibition both in PDOs as well as in xenograft models of PDAC, leading to tumor regressions in vivo. We hypothesized that inhibition of KRASG12D may lead to a similar feedback activation of ERBB signaling and therefore dual ERBB/KRAS blockade may synergize in PDAC and lead to durable response in vivo. The optimization of treatment strategies combining novel KRAS inhibitors and targeted therapeutics that intercept acquired resistance could advance PDAC treatment and promote response in patients.
Materials and Methods
Cell culture
Human PDAC lines were obtained from ATCC and maintained in Roswell Park Memorial Institute (RPMI) 1640 (Sigma-Aldrich, R8758) with 10% fetal bovine serum (FBS) and penicillin/streptomycin at 37°C with 5% CO2. Murine KPC cell lines were received from the Tuveson Laboratory and maintained in RPMI1640 with 10% FBS and penicillin/streptomycin at 37°C with 5% CO2. All cells and organoids were routinely tested for Mycoplasma using PCR (Universal Mycoplasma Detection Kit, ATCC, 30–1012K). MRTX1133-resistant models were developed by continuously treating cells in 10-cm dishes with sub-IC50 doses until the cells could tolerate 100-fold higher doses than the IC50. Resistant lines were maintained at their highest tolerated dose until the end of the study.
Organoid culture
Patient-derived and mouse organoids were received from the Tuveson Laboratory and maintained in a complete human/murine medium at 37°C with 5% CO2. The protocol to propagate and sustain organoid cultures was described previously (11, 12).
Animals
All animal experiments were conducted under the procedures approved by the Institutional Animal Care and Use Committee at the University of California San Diego (protocol number: S09158). Mice were maintained at a maximum of 5 mice per cage with 12-hour light/dark cycles. C57BL/6J mice were purchased from The Jackson Laboratory while NOD SCID gamma (NSG) mice were purchased from Animal Care Program (ACP) at UCSD. Briefly, 5,000 KPC FC1242 or 100,000 SUIT2 cells in 20 μL Matrigel [Corning Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix, Corning, 354230] were orthotopically injected into the tail of pancreas of mice anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine. Mice were monitored daily for five days after surgery for abnormal behavior. Tumor sizes were measured twice weekly using ultrasonography (Convex L20 HD3, Clarius). When the largest tumor volume reached 200 mm3, mice were treated with 5 mg/kg afatinib (Afatinib group), 6 mg/kg MRTX1133 (low dose group), 12 mg/kg MRTX1133 (high dose group), or 5 mg/kg afatinib and 12 mg/kg MRTX1133 (Combo group) intraperitoneally. Mice were sacrificed with isoflurane at day 10 (for endpoint studies) or when the largest tumor reached 1.5 cm in diameter or when mice exhibited abnormal behavior (survival study).
Therapeutic experiments with cells and organoids
All the drugs used in this study including MRTX1133 (Chemgood, C-1420) and afatinib (Selleckchem, S1011) were dissolved in dimethyl sulfoxide (DMSO) at 10 mmol/L stock concentration. The maximum concentration of DMSO per well was 1%. PDAC organoids and cell lines were plated in 384-well plates at the density of 1,000 cells per well in 20 or 30 μL media, respectively. Drugs were dispensed in each well using the D300e Digital Dispenser (Tecan). Seventy-two hours after treatment, cell viability was measured using a luminescent ATP-based assay (CellTiter-Glo, Promega) and Synergy H1 plate reader. The Combenefit software was used to determine synergy and calculate the Bliss, Loewe, and highest single agent (HSA) synergy scores (13). The list of drugs used in this study is in Supplementary Table S1. Bliss independence models the effects of individual drugs in a combination as independent competing events (14). Loewe models expected effects as if a drug was combined with itself (15). The HSA model expects the combination effect to equal the highest individual drug effect (16). As a result, the Bliss and Loewe models are more stringent than the HSA model in identifying synergy.
Histology and immunohistochemistry
Tissues were fixed in 10% neutral-buffered formalin, embedded in paraffin, prepared as 5-μm sections, and stained with hematoxylin and eosin according to standard procedures. For immunohistochemistry (IHC), heat-induced antigen retrieval was performed in citric acid buffer (pH 6.8) in a pressure cooker for 20 minutes. Endogenous peroxidases were quenched with 3% H2O2 in tris-buffered saline (TBS) for 20 minutes at room temperature. Tissue sections were blocked with 200 μL of 10% goat and 2.5 horse sera. Afterward, sections were incubated with the primary anti-Ki67 antibody (ab15580) overnight at 4°C. Sections were washed with TBS with 1% Tween 20 (TBST) three times for 5 minutes each time before incubating with Goat anti-Rabbit IgG ImmPRESS secondary antibody for 30 minutes at room temperature. Sections were washed again with TBST three times for 5 minutes each before developing the signals with ImmPACT DAB HRP Substrate. Slides were counterstained, dehydrated, and coverslipped following standard protocol.
Quantification of IHC staining
Histologic slides were scanned and processed in QuPath ver 0.3.2. Scanned slides were opened as brightfield (H-DAB) in QuPath, and the intensity of the DAB stain and the hematoxylin counterstain were adjusted using Estimate Stain Vectors Function. Cells were annotated based on their morphologies and locations to allow QuPath to classify each cell type automatically. Three healthy tumor regions were randomly selected per slide and positive cells were detected using the Cell Detection function. The number of positive cells from three different mice was averaged to get the average number of positive cells per group.
Western blotting
Western blotting was performed as described previously with minor modifications. Briefly, 2% SDS lysis buffer with complete Protease (Roche, 11697498001) and PhosSTOP phosphatase (Roche, 4906845001) inhibitors were added directly to the cell pellet after washing thoroughly with Dulbecco PBS. Cell lysates were sonicated using a sonicator (QSonica, Q125) for three seconds. Protein concentrations were measured with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225) before adding NuPAGE LDS Sample Buffer (4×; Thermo Fisher Scientific, NP0008) and denaturing at 98°C for 10 minutes. For human PDO and 2D cell lines, and murine 2D cell lines, three microgram proteins were separated by SDS-PAGE and electrotransferred onto 0.2 μm PVDF membranes (Bio-Rad Laboratories, Inc., 1704156) using the Trans-Blot Turbo Transfer System (Bio-Rad Laboratories, Inc.). Membranes were blocked with 5% Skim milk in Tris-buffered saline with 5% TBST or 5% bovine serum albumin (Sigma-Aldrich, 126593) in TBST for 1 hour at room temperature and incubated with the primary antibody in the blocking solution overnight at 4°C. The membranes were washed with TBST three times for 5 minutes each before incubating with the corresponding secondary anti-mouse (Thermo Fisher Scientific, 31430) or anti-rabbit IgG antibody (Thermo Fisher Scientific, 31460) in blocking solution. Signals were developed with Immobilon Western Chemiluminescent HRP substrate (MilliporeSigma, WBKLS0500) and visualized in ChemiDoc Imaging System (Bio-Rad Laboratories, Inc). For murine organoids, 30 to 50 μg proteins were separated by SDS-PAGE and electrotransferred onto 0.45 μm PVDF membranes (Thermo Fisher Scientific, 88518) using the Mini Trans-Blot Cell system (Bio-Rad Laboratories, Inc.). Membranes were blocked with 5% BSA (Thermo Fisher Scientific, 1605100) in TBST for 1 hour at room temperature and incubated with primary antibody in blocking solution overnight at 4°C. The membranes were washed with TBST three times for 5 minutes each before incubating with the corresponding secondary anti-rabbit IgG antibody (Cell Signaling Technology, 7074) in blocking solution. Signals were developed with Amersham ECL Western Blotting System (VWR, RPN2209), were exposed to autoradiography film, and visualized using a film processor. Captured data were processed and analyzed using ImageJ and Image Lab software (17). The list of antibodies used for Western blotting can be found in Supplementary Table S2A.
Active GTPase pulldown
Active GTPase pulldown was done according to the manufacturer's instructions (Active Ras Detection Kit, Cell Signaling Technology). Briefly, cells in an 80% confluent 10-cm culture dish were trypsinized, washed three times with ice-cold PBS, and then lysed with 500 μL of lysis/binding/wash buffer (25 mmol/L Tris-HCl, pH 7.2, 150 mmol/L NaCl, 5 mmol/L MgCl2, 5% glycerol, 1% NP40) supplemented with Complete Protease (Roche, 11697498001) and PhosSTOP phosphatase inhibitors (Roche, 4906845001). Protein samples were measured with the Pierce BCA Protein Assay Kit and adjusted to 1 mg/mL with the lysis/binding/wash buffer. Protein samples were subsequently added to glutathione agarose beads in the spin columns provided in the kit for 1 hour at 4°C under constant rocking. The beads were washed three times with the lysis/binding/wash buffer and eluted with 50 μL of 1× SDS-PAGE sample buffer. Fifteen microliters of eluted protein was used for each replicate of a Western blot analysis.
RNA sequencing
Parental SUIT2 cells and MRTXR SUIT2 cells were cultured in 10-cm dishes in duplicate. Cells were left untreated or treated with 60 nmol/L MRTX1133 for 24 hours. Total RNA was extracted Nucleospin RNA Isolation Kit (Macherey-Nagel GmbH & Co.) following the manufacturer's instructions. RNA samples were submitted to the University of California San Diego Institute of Genomic Medicine Core for further analysis. Quality testing was carried out by measuring RNA integrity and OD readings (260/280 and 260/230). All samples had RIN scores of 9.4 or higher, and OD readings were within the 1.8 to 2.2 range. Differential expression analyses were analyzed by gene set enrichment analysis (GSEA).
Reverse-phase protein array
Reverse-phase protein array (RPPA) was performed as described previously (18). Briefly, 2 × 106 SUIT2 cells were seeded in 10 cm dishes and treated with the DMSO control, 120 nmol/L afatinib, 60 nmol/L MRTX1133, or a combination of 120 nmol/L afatinib and 60 nmol/L MRTX1133. After 24 hours, cells were dissociated by scraping with a cell lifter. Cell pellets were flash frozen and were analyzed and quantified by the Department of Bioinformatics and Computational Biology at MD Anderson Cancer Center (Houston, TX).
RNA sequencing and RPPA analysis
The raw counts were analyzed using the limma-voom pipelines (19–21) in the R environment (4.2.1) using standard default parameters. For the RPPA data, the normalized log scale abundance data was analyzed using simple linear model in R, and significance levels were assessed using appropriate contrasts. GSEA was done using the R package fgsea and the MSigDB v 7.5 from the R package msigdbr (22).
Linear models for microarray (Limma, using R-limma package) were used to compare groups regarding log2 expression values. The Benjamini–Hochberg procedure was applied to control the false discovery rate (FDR). A gene was considered significantly changed if FDR <0.05. If the log2-fold change in expression for a test compound versus control was greater than 0, it was said to be upregulated; otherwise, it was downregulated. In addition, log2-fold changes between two groups of interest for all the genes included in the analysis set were extracted for GSEA (R-fgsea package). Enrichment in Hallmark or Reactome pathways was examined. For each gene set, the number of expressed genes in the analysis data set was calculated along with normalized enrichment scores (NES), the P value and q value for testing enrichment significance, and the number of core enrichment genes. The list of antibodies used for RPPA analysis can be found in Supplementary Table S2B.
Whole exome sequencing
DNA was extracted from cells using Quick-DNA Miniprep Plus Kit (Zymo Research, D4068) following the manufacturer's instructions. Library preparation, sequencing reactions, and bioinformatic analysis were conducted at Azenta US. Genomic DNA samples were quantified using Qubit 2.0 Fluorometer (Thermo Fisher Scientific). Enrichment probes were designed against the region of interest and synthesized through Twist Biosciences – Twist Human Comprehensive Panel. Library preparation was performed according to the manufacturer's guidelines. The sequencing libraries were multiplexed and clustered onto multiple lanes of a flow cell and loaded onto the Illumina HiSeq instrument according to the manufacturer's instructions. The samples were sequenced using a 2× 150 bp Paired End configuration. Image analysis and base calling were conducted by the HiSeq Control Software. Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and de-multiplexed using Illumina bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification. Sequencing adapters and low-quality bases in raw reads were trimmed using Trimmomatic 0.39. Cleaned reads were then aligned to the GRCh38 reference genome using Sentieon 202112.01. Alignments were then sorted, and PCR/Optical duplicates were marked. BAM files were generated because of this step. Somatic SNVs and small INDELs were called by using Sentieon 202112 (TNSeq algorithm). The VCF files generated by the pipeline were then normalized (left alignment of INDELs and splitting multiallelic sites into multiple sites) using bcftools 1.13. Overlapped transcripts were identified for each variant, and the effects of the variants on the transcripts were predicted by Ensembl Variant Effect Predictor (VEP) v104.
Statistical analysis
Determination of IC50 and statistical analysis was performed using GraphPad Prism version 9 (GraphPad) and R 4.2.1. All results were expressed as mean ± SD unless otherwise stated. The Student t test was used to compare statistical differences between groups, while Tukey test was used to analyze differences between multiple groups. Kaplan–Meier curves and the log-rank test were used to analyze differences in survival time between groups. Linear models for microarray (Limma, using R-limma package) were used to compare groups regarding log2 expression values. The Benjamini–Hochberg procedure was applied to control the FDR. A gene was considered significantly changed if FDR < 0.05. If the log2-fold change in expression for a test compound versus control was greater than 0, it was said to be upregulated; otherwise, it was downregulated. In addition, log2-fold changes between two groups of interest for all the genes included in the analysis set were extracted and preranked for GSEA (R-fgsea package). Enrichments in Hallmark or Reactome pathways were examined. For each gene set, the number of expressed genes in the analysis data set was calculated along with normalized enrichment scores (NES), the P value and q value for testing enrichment significance, and the number of core enrichment genes.
Data availability
The data, RNA sequencing (RNA-seq) analyses and whole exome sequencing, generated in this study are publicly available in GEO (GSE234449) and SRA (PRJNA978606) databases. All other raw data are available upon request from the corresponding authors.
Results
MRTX1133 selectively inhibits KRASG12D in mutant human and mouse PDAC models
To determine the potency and specificity of MRTX1133, a small-molecule inhibitor of KRASG12D, we performed an in vitro dose-response assay with several three-dimensional (3D) patient-derived organoids (PDO) harboring the KRASWT, KRASG12V, and KRASG12D mutations. We have previously shown that PDOs are predictive models of patient tumor drug response (11). We found that MRTX1133 specifically inhibited the growth of KRASG12D PDOs with IC50s less than 20 nmol/L. Meanwhile, PDOs carrying KRASWT or KRASG12V alleles were not affected by MRTX1133 at nanomolar concentrations (Fig. 1A). Reduced levels of pMEK1/2, pERK1/2, and pAKT were observed in KRASG12D, but not KRASWT, PDOs within 3 hours of 60 nmol/L MRTX1133 treatment (Fig. 1B). After an initial reduction at 3 and 24 hours, pAKT levels started to recover at 72 hours. Interestingly, pEGFR and pHER2 increased during treatment in KRASG12D PDOs (Fig. 1B and C). To determine the cross-model consistency of these findings, we next examined MRTX1133 dose response using two-dimensional (2D) KRASWT and KRASG12D human PDAC cell lines. KRASG12D mutant cell lines were responsive to treatment, while KRASWT BXPC3 cells were not, similar to published findings (Fig. 1D; ref. 6). After treatment with 60 nmol/L MRTX1133, pMEK1/2 was dramatically decreased, while pERK1/2 was partially reduced in the SUIT2 KRASG12D cell line (Fig. 1E). As observed in the organoid models, pAKT levels were initially decreased and then recovered at later time points in SUIT2 cells but were unchanged in BXPC3. The levels of pEGFR and pHER2 decreased by 24 hours and recovered at 72 hours (Fig. 1E and F). Phosphorylated levels of EGFR and HER2 remained unaffected by MRTX1133 in BXPC3 cells. To delineate the cross-species applicability of these results, we next tested the effectiveness of MRTX1133 in three mouse KRASLSL-G12D/+; P53LSL-R172H/+; PDX-Cre (KPC) organoid lines (12). The mouse organoids had a partial response to sub-micromolar MRTX1133 concentrations (Fig. 1G). Treatment with 60 nmol/L MRTX1133 led to a decrease in pERK, pMEK1/2, and pAKT in mT9 organoids but did not significantly affect signaling cascades in mT3 organoids (Fig. 1H). In line with these findings, mT9 organoids were more sensitive to MRX1133. This highlights the model-to-model heterogeneity among genetically engineered mouse PDAC cells. Levels of pEGFR and pHER2 remained unaffected during treatment (Fig. 1I). We also tested MRTX1133 efficacy in three mouse KRASLSL-G12D/+; P53LSL-R172H/+; PDX-Cre (KPC) cell lines (23). Notably, these mouse monolayer PDAC cell lines displayed heterogenous responses to MRTX1133 (Fig. 1J). In these mouse 2D cells, MRTX1133 treatment decreased pERK1/2 and pAKT acutely at 3 hours, but phosphorylation levels recovered by 72 hours. (Fig. 1K). As seen in human models, pEGFR and pHER2 increased during treatment, especially in FC1245 cells (Fig. 1L). We find that response to MRTX1133 is heterogenous in human and mouse models. Overall, our experiments in mouse organoids and monolayer cells revealed they are generally somewhat less sensitive to MRTX1133 in vitro than human models. Taken together, these results demonstrate that MRTX1133 specifically and effectively targets KRASG12D mutant PDAC cells in vitro. Our results demonstrate that MRTX1133 decreases signaling downstream of mutant KRAS in a variety of models. Further, inhibition of mutant KRAS leads to increased priming of ERBB receptors, potentially leading to adaptive resistance similar to what has been observed with MEK inhibitors (24).
Figure 1.

MRTX1133 specifically targets oncogenic KRASG12D human and mouse PDAC models and leads to the upregulation of ERBB activation. A, Dose–response analysis of PDOs harboring the KRASWT, KRASG12V, or KRASG12D alleles 5 days after treatment with MRTX1133. B, Immunoblot analyses of the upstream and downstream targets of KRAS signaling in hT102 (KRASWT) and hM1F (KRASG12D) at 0, 3, 24, and 72 hours using 60 nmol/L MRTX1133. C, Quantification of B. D, Dose–response analysis of PDAC cell lines harboring the KRASWT or KRASG12D alleles 3 days after treatment with MRTX1133. E, Immunoblot analyses of the upstream and downstream targets of KRAS in BXPC3 (KRASWT) and SUIT2 (KRASG12D) at 0, 3, 24, and 72 hours using 60 nmol/L MRTX1133 for BXPC3 and SUIT2. F, Quantification of E. G, Dose–response analysis of mouse KRASG12D KPC organoids 3 days after treatment with MRTX1133. H, Immunoblot analyses of the upstream and downstream targets of KRAS in two different KPC mouse organoids (mT3 and mT9) at 0, 3, 24, and 72 hours using 60 nmol/L MRTX1133. I, Quantification of H. J, Dose–response analysis of mouse KRASG12D KPC cell lines 3 days after treatment with MRTX1133. K, Immunoblot analyses of the upstream and downstream targets of KRAS in two different KPC lines at 0, 3, 24, and 72, hours using 60 nmol/L MRTX1133. L, Quantification of K. Data, mean values ± SD. Immunoblots are representative of at least three independent experiments.
MRTX1133 induces expression of ERBB receptors
We observed that downstream KRASG12D signaling is reduced upon treatment, while activation of upstream receptor tyrosine kinases, EGFR and HER2, is increased. To further examine the effects of MRTX1133 on pathway activation in MRTX1133-treated human PDAC cells, we performed RPPA and transcriptomic analysis. We found that after 24 hours, total HER2 and HER3 protein levels were higher, while total EGFR was lower in MRTX1133-treated SUIT2 cells (Supplementary Fig. S1A and Supplementary Table S3). Furthermore, EGFR, HER2, and HER3 transcripts were significantly increased in MRTX1133-treated SUIT2 cells (LogFC = 0.24, 1.07, 1.55; Padj = 1.51E−03, 2.55E−10, 2.76E−12, respectively; Supplementary Table S3). Using RPPA, we also found that p-S6 was increased in the treated SUIT2 cells after 24 hours, suggesting that the PI3K/AKT/mTOR pathway is active after MRTX1133 treatment, as was recently shown in PDAC (7). Interestingly, both YAP protein and pYAP were increased after treatment, which recapitulated a known resistance mechanism to targeting oncogenic KRAS signaling (25). GSEA revealed that MRTX1133 decreased pathways promoting cell proliferation while epithelial-to-mesenchymal transition (EMT) and immune pathways were concomitantly increased (Supplementary Fig. S1B and S1C). Reassuringly, a significant positive correlation was found between transcript and protein levels comparing the RPPA and RNA-seq datasets (Supplementary Fig. S1D). Taken together, these analyses suggest that MRTX1133 promotes the expression of ERBB receptors. Hyperactivation of these RTKs could provide a putative acute resistance mechanism to MRTX1133 akin to what has been described in colorectal cancer (9).
Irreversible pan-ERBB inhibitors synergize with MRTX1133 in vitro
To improve the efficacy of MRTX1133 and overcome the rapid development of resistance to the drug, we screened clinically approved and preclinical small molecules that inhibit upstream and downstream KRAS signaling. In multiple PDO models, we found striking synergy between the approved irreversible pan-ERBB inhibitor afatinib and MRTX1133 across multiple drug concentrations (Fig. 2A; Supplementary Fig. S2). The reversible EGFR and HER2 inhibitor lapatinib and EGFR inhibitor erlotinib also synergized with MRTX1133, but in a less consistent manner and over more restricted dose ranges. MEK1/2 and ERK1/2 inhibitors, trametinib and ulixertinib, exhibited largely additive effects with little to no synergy observed when combined with MRTX1133. Similar results were obtained using human KRASG12D SUIT2 and ASPC1 cells but not KRASWT BXPC3 cells (Fig. 2B and C; Supplementary Figs. S3–S5). Similarly, we found remarkable synergy between MRTX1133 and afatinib in mouse KPC organoid and cell lines, indicating that this combination is effective in both human and mouse PDAC cell culture models (Supplementary Fig. S6). Additional pan-ERBB irreversible inhibitors, neratinib and canertinib, were potently synergistic with MRTX1133 (Supplementary Figs. S4–S6). Notably, we found intermediate synergy combining the anti-EGFR clinical antibody cetuximab with MRTX1133 in SUIT2 cells (Supplementary Fig. S5). These results recapitulate recently published preclinical results from Hallin and colleagues (6). We also found that MRTX1133 synergizes well with afatinib in a KRASG12D, but not KRAS wild-type, colon cancer cell line (Supplementary Fig. S7). We did not observe synergy when combining commonly used chemotherapeutics gemcitabine or 5-FU with MRTX1133 (Supplementary Figs. S3–S5). Biochemical dissection of signaling cascades revealed that the combination of MRTX1133 and afatinib dramatically reduced pEGFR and pHER2 as well as pERK, in most PDO, human 2D and mouse 2D and 3D models (Fig. 2D and I; Supplementary Fig. S8A–S8C). These results indicate that the KRASG12D inhibitor synergizes with pan-ERBB inhibitors in vitro and successfully attenuates signaling through these pathways.
Figure 2.

Irreversible Pan-ERBB inhibitors work synergistically with MRTX1133 in vitro. A, Evaluation of the synergistic effect of MRTX1133 with afatinib (panERBB inhibitor), lapatinib (EGFR/HER2 inhibitor), erlotinib (EGFR inhibitor), trametinib (MEK1/2 inhibitor), and ulixertinib (ERK1/2 inhibitor) using the BLISS synergy model in hF44 PDO, B and C, SUIT2 (B), and ASPC1 (C) show the potent and consistent synergy of MRTX1133 with afatinib. D–F, Western blot analyses of the upstream and downstream pathway targets of KRAS show a synergistic downregulation of the expression of EGFR and HER2 in hM1F (D), SUIT2 (E), and ASPC1 (F). G–I, Quantification of D, E, and F, respectively. Data, mean values ± SD. Immunoblots are representative of at least three independent experiments.
Irreversible pan-ERBB inhibition can overcome MRTX1133 resistance in vitro
Acquired resistance to KRASG12C inhibitors has been well documented (8, 9, 26). Therefore, we sought to determine if KRASG12D and pan-ERBB dual inhibition remains synergistic in MRTX1133-resistant cells. We developed a human SUIT2 cell line with profound resistance to MRTX1133 after repeated exposure to increasing concentrations of the inhibitor. MRTX1133 dose-response assays confirmed these cells (now termed MRTXR for MRTX1133-resistant) had an increased MRTX1133 IC50 compared with parental cells SUIT2 cells (Fig. 3A). Interestingly, MRTXR SUIT2 was slightly more sensitive to afatinib (Fig. 3B). We then tested the combination of MRTX1133 and afatinib and observed that synergy was preserved, albeit reduced, in the resistant cells (Fig. 3C). Cetuximab displayed intermediate synergy in the MRTXR-SUIT2 cells, suggesting that pan-ERBB irreversible inhibition is necessary to preserve synergy with KRASG12D inhibition in the resistant setting (Supplementary Fig. S9A and S9B). MRTXR SUIT2 cells treated with 60 nmol/L MRTX1133 had decreased pMEK1/2 and pAKT levels at 3 hours, but phosphorylation returned to basal levels within 24 hours (Fig. 3D). Notably, pHER2 dramatically increased 72 hours posttreatment. The combination of afatinib and MRTX1133 for 24 hours dramatically reduced pEGFR, pHER2, pMEK1/2, and pAKT, indicating that this treatment strategy remained effective at shutting down protumorigenic signaling in the resistant cells (Fig. 3E). We noted, however, that pERK1/2 was not reduced in these conditions, and signaling was not completely inhibited using these drug concentrations at 24 hours.
Figure 3.

MRTX1133 and afatinib combination remains effective in cells that have acquired MRTX1133 resistance. A and B, Cell viability of parental and MRTXR SUIT2 lines after treatment with MRTX1133 (A) or afatinib (B). C, Evaluation of the synergistic effect of MRTX1133 with afatinib using the BLISS and HSA synergy models in MRTXR SUIT2. D and E, Immunoblot analysis for the upstream and downstream signaling of KRAS after single-agent (D) or combination therapy with MRTX1133 and afatinib (E). F, qRT-PCR analysis KRAS expression in parental and MRTXR SUIT2 cells. G, Immunoblot analysis for RASG12D in parental or MRTXR SUIT2. H, Immunoblot analysis of RASG12D and pan-RAS in parental SUIT2, untreated MRTXR SUIT2, and MRTX1133-treated MRTXR SUIT2 after RAS-GTP pulldown. I, Quantification of H. Values are quantifications from two different Western blots normalized to 0.6% GAPDH input. Data, mean values ± SD. Immunoblots are representative of at least three independent experiments.
Amplification of mutant KRAS is a reported mechanism of resistance to KRAS G12C inhibition. Accordingly, we assessed the gene expression levels of KRAS in the MRTXR cells. Using qPCR, we found an increase in KRAS transcripts (P = 0.064; Fig. 3F). Using a RASG12D-specific antibody, we found a dramatic accumulation of mutant protein by immunoblot analysis (Fig. 3G). We next sought to determine the levels of GTP-bound KRASG12D mutant protein. Using a pulldown assay, we found that MRTXR SUIT2 cells had increased active KRASG12D-GTP at a steady state compared to parental cells (Fig. 3H and I). Together, these data reveal a substantial increase in KRAS activity in the MRTX1133-resistant setting. MRTX1133 (1 μmol/L) treatment was sufficient to effectively inhibit GTP-binding of mutant KRAS in the resistant cells. This suggests that resistance to MRTX1133 in SUIT2 is at least partially driven by an increase of KRASG12D mutant protein. To determine if MRTXR SUIT2 cells acquired new KRAS mutations to circumvent MRTX1133 treatment, we performed whole exome sequencing. Comparing parental and MRTXR cells, we detected KRASG12C mutation in the KRASWT allele of the resistant cell with a mutant allele frequency of 4% (Supplementary Fig. S9C). These preliminary results indicate that a new population of SUIT2 cells may be selected by the continuous MRTX1133 treatment.
Using a similar strategy, we developed a mouse MRTX1133-resistant cell model, MRTXR FC1242 (Supplementary Fig. S10A). This resistant murine model did not display increased sensitivity to afatinib, and the combination of afatinib and MRTX1133 was no longer synergistic (Supplementary Fig. S10B and S10C). Indeed, when treated with MRTX1133, MRTXR FC1242 cells did not upregulate pEGFR/pHER2, and therefore, the combination did not perform better than the single agents (Supplementary Fig. S10D and S10E). This result suggests that resistance can develop independent of ERBB signaling upregulation.
To further understand what pathways and signaling changes drive resistance in the human and mouse MRTXR cell models, we performed transcriptome and RPPA analysis. GSEA of resistant cells compared to parental revealed that EMT (in human and mouse cells) and immune pathways (interferon alpha and gamma – in human models) were upregulated among others (Supplementary Fig. S11A and S11B). These pathways have been previously associated with increased drug resistance and cancer stem cell phenotypes (27, 28). Conversely, MYC-driven gene transcription and oxidative phosphorylation programs were downregulated in resistant cells. The RPPA GSEA supported these results as they positively correlated with the transcriptional analysis, thereby increasing our confidence in these findings (Supplementary Fig. S11C).
Irreversible pan-ERBB inhibitor, afatinib, potentiates MRTX1133 response in vivo
To establish if the in vitro synergy observed using MRTX1133 and afatinib translates to improved in vivo efficacy, we performed endpoint and survival studies. We orthotopically injected SUIT2 cells into immunocompromised mice and followed tumor growth longitudinally using ultrasound imaging. Once the largest tumor volume reached 200 mm3, we treated the mice with vehicle, MRTX1133, afatinib, or both for 10 days. At the endpoint, mice treated with vehicle or afatinib had similar tumor growth, indicating that the low dose of afatinib used in this preclinical experiment did not provide meaningful tumor control as a monotherapy (Fig. 4A–C). Both MRTX1133 monotherapy and combination controlled tumor growth effectively in this model. We next performed the experiment with MRTXR SUIT2 orthotopic tumors and found that only the combination of both afatinib and MRTX1133 led to a statistically significant decrease in tumor burden compared with vehicle-treated mice (Fig. 4D–F). These data indicate that combined KRAS G12D and ERBB inhibition remains effective in these human orthotopic xenografts following the acquisition of MRTX1133 resistance.
Figure 4.

Irreversible Pan-ERBB inhibitor, afatinib, potentiates MRTX1133 in vivo. A, Posttreatment tumor volumes normalized to pretreatment volumes of NSG mice orthotopically injected with SUIT2 and treated for 10 days with vehicle, MRTX1133, afatinib, or combo. B, Tumor weights of mice in A. C, Tumors of mice in A imaged at necropsy. D, Posttreatment tumor volumes normalized to pretreatment volumes of NSG mice orthotopically injected with MRTXR SUIT2 and treated for 10 days with vehicle, MRTX1133, afatinib, or combo. E, Tumor weights of mice in D. F, Tumors of mice in D imaged at necropsy. G, Posttreatment tumor volumes normalized to pretreatment volumes of C57BL/6J mice orthotopically injected with parental KPC 1242 cells and treated for 10 days with vehicle, MRTX1133, afatinib, or combo. H, Tumor weights of mice in G. I, Tumors of mice in G imaged at necropsy. J, Posttreatment tumor volumes normalized to pretreatment volumes of C57BL/6J mice orthotopically injected with MRTXR KPC 1242 cells and treated for 10 days with vehicle, MRTX1133, afatinib, or combo. K, Tumor weights of mice in J. L, Tumors of mice in J imaged at necropsy. M, Survival study of C57BL/6J mice orthotopically injected with parental KPC 1242 cells and treated with vehicle, MRTX1133, afatinib, or combo. †, censored data; *, P < 0.05; **, P < 0.01; ***, P < 0.001, log-rank test. Data, mean values ± SD.
In parallel, we orthotopically injected FC1242 parental and MRTXR KPC cells into the tail of the pancreas of immunocompetent syngeneic mice. Mice were enrolled in study when the largest tumor volume reached 200 mm3 and mice were treated for 10 days. In the parental FC1242 tumors, afatinib alone did not provide meaningful tumor control as a monotherapy (Fig. 4G–I). In this model, we tested two dosing of MRTX1133. As expected, we observed better growth control with a higher dose of MRTX1133. In contrast to the monotherapy groups, three of four animals had significant growth inhibition in the combination cohort, with tumor weights that were significantly decreased compared with vehicle-treated mice. This suggests that the combination of afatinib with MRTX1133 is effective in vivo. In tumors generated from MRTXR FC1242 KPC cells, we found a significant tumor burden decrease with afatinib alone, suggesting that acquisition of resistance to MRTX1133 potentiates response to ErbB inhibition despite lack of overt biochemical activation of this pathway in our in vitro assays. A decrease in tumor weight was also observed with MRTX1133 monotherapy (Fig. 4J–L). However, we found no additional benefit to combination therapy as compared to MRTX1133 monotherapy in this model of resistance in this short-term intervention study, exactly mirroring the in vitro results we observed. Next, we performed immunohistochemical analyses of the FC1242 parental and MRTXR tumors at endpoint for a cell division marker (Ki67), and a cell death marker (cleaved caspase-3; CC3). These studies did not reveal any significant differences in proliferation or cell death among the groups in either model (Supplementary Fig. S12A–S12D). Interestingly, the MRTXR-derived tumors displayed more well-defined glandular structures and increased stroma as compared with their parental KPC cell–derived counterparts (Supplementary Fig. S12E).
Finally, we asked if the tumor control we observed after 10 days of treatment in the parental FC1242 model would translate to a survival advantage for combination-treated mice. In fact, the mice treated with combination of MRTX1133 and afatinib did survive significantly longer, while high-dose MRTX1133 alone provided a lesser survival benefit (Fig. 4M). These results highlight the promising potential of dual KRAS and ERBB inhibition to treat PDAC and intercept the acquisition of resistance.
Discussion
KRASG12D-mutant PDAC cells are exquisitely vulnerable to MRTX1133 and afatinib combination
In this study, we demonstrate that the KRASG12D inhibitor MRTX1133 is specific and effective in vitro in patient-derived organoid models, as well as human and mouse PDAC cell lines. Our results demonstrate that low nanomolar MRTX1133 can effectively abolish KRASG12D signaling through the MAPK cascade while slightly downregulating signaling through PI3K/AKT, which parallels recently published data (7). We observed that human and mouse PDAC cell models display a heterogenous response to MRTX1133, with mouse models generally displaying lower sensitivity to the inhibitor both in dose-response and immunoblot assays. Furthermore, we found that MRTX1133 treatment promotes increased expression and phosphorylation of multiple ERBB receptors, which is reminiscent of feedback signaling observed using MEK inhibitors (29). Therefore, we hypothesized that combining MRTX1133 with an irreversible pan-ERBB inhibitor could enhance anticancer activity. We observed dramatic synergy when combining approved small-molecule inhibitors afatinib, neratinib, and canertinib with MRTX1133, suggesting this combination approach could be beneficial to future PDAC patients. Importantly the dual inhibition of ERBB and KRASG12D signaling was highly effective at diminishing both MAPK and PI3K/AKT signaling. We validated this combination approach in vivo using aggressive human and mouse transplant models of PDAC. MRTX1133 monotherapy resulted in some inhibition of tumor growth, consistent with prior reports (6, 7), while the combination of afatinib and MRTX1133 resulted in a greater degree of growth inhibition and increased the survival of treated animals.
Pan-ERBB inhibition is an optimal and synergistic pairing with MRTX1133
We note that pan-ERBB inhibition is essential for maximizing synergy with MRTX1133 in our PDAC models. MRTX1133 treatment caused increased expression of several ERBB genes. Further, our results indicate that activating phosphorylation of EGFR, as well as HER2, increases during KRASG12D blockade with some heterogeneity observed across a variety of PDAC models. Accordingly, the greatest synergy was observed with irreversible pan-ERBB inhibitors as opposed to EGFR or HER2 monotherapy such as cetuximab, lapatinib, or erlotinib. These results are supported by recently published data using a KRASG12C inhibitor and a clinical trial is underway to validate the approach of combining adagrasib with afatinib in lung cancer (KRYSTAL-1; ref. 3).
Combining MRTX1133 with downstream inhibitors such as MEK and ERK inhibitors did not provide meaningful synergy. These results suggest that MRTX1133 can control MAPK signaling and drugs targeting this pathway will not likely provide additional benefit. Surprisingly, in our experiments upstream targeting of KRAS signaling using a SHP2 inhibitor was not synergistic in the human cell lines we tested, and future experiments will explore additional inhibitors. Therefore, our study results suggest that potent KRAS inhibition is ideally combined with direct targeting of ERBBs.
MRTX1133 and afatinib combination remains effective in cells that have acquired MRTX1133 resistance
We have established a human model of acquired resistance to MRTX1133. While KRASG12D inhibition is not effective in the MRTXR cells, the combination of afatinib and MRTX1133 remains synergistic and effectively downregulates KRAS signaling cascades. The combination is the most effective at controlling tumor growth in an in vivo model of resistance. In a separate experiment using a mouse model of MRTX1133-resistance, pan-ERBB inhibition was not efficacious as the resistance mechanism(s) did not appear to leverage increased ERBB signaling. This prompts the hypothesis that cotargeting pan-ERBB with MRTX1133 in PDAC may provide synergistic antitumor activity in some patients and the combination could overcome the acquisition of ERBB-mediated resistance to KRAS inhibitors. Importantly, these data suggest that tumor expression of pEGFR/pHER2 could be leveraged as a predictive biomarker for dual KRAS/ERBB inhibition.
To prevent toxic adverse events, we focused our in vivo study on lower doses of MRTX1133 and afatinib and indeed did not observe adverse toxicity in animals treated with monotherapy or combination therapy. Higher doses of MRTX1133 monotherapy have now been tested in animal models (7), and future studies will test if increasing the dosing of MRTX1133 and afatinib can lead to complete responses by leveraging the synergistic combination effect.
Limitations of the study
Our combinatorial drug study is not exhaustive; therefore, we hope and suspect that additional synergistic combinations will be uncovered and characterized, especially those that are unaffected by acquired MRTX1133 resistance. In our cell model, resistance can also be driven by the acquisition of mesenchymal and cancer-stem cell transcriptional programs suggesting novel targetable vulnerabilities to be explored in the future. Reliance on ERBB-driven mechanisms of resistance to KRAS inhibition appears to be heterogeneous across the model systems tested, suggesting that future studies should investigate cohorts of human and murine models rather than singular model systems. However, our data also suggest that combined inhibition potentiates initial response and may increase the durability of response. Additional studies will be necessary to define a comprehensive list of resistance-promoting factors. These studies are necessary as they will provide actionable predictive biomarkers to personalize combinations with KRAS inhibitors in patients in upcoming clinical trials. We also observed early indications of genetic resistance mechanisms to MRTX1133, which will require further study to understand the prevalence of these resistance mechanisms and identify strategies to effectively restore treatment response. To this point, it will be interesting to study how resistance to upcoming pan-KRAS inhibitors develops.
Concluding remarks
Our study provides robust evidence supporting the combination of MRTX1133 with pan-ERBB inhibition. We used advanced human patient-derived organoid models as well as in vivo models to show that synergy between afatinib and MRTX1133 results in reduced tumor burden and prolonged survival. This study provides the rationale for testing this drug combination in clinical trials for patients with pancreatic cancer.
Supplementary Material
Table S1. List of drugs used in this study.
Table S2. List of antibodies used in this study.
Table S3. Results of RPPA and transcriptomic analyses.
Supplemental Figure and Table Legends
Figure S1 - S12
Acknowledgments
This work was made possible by a grant from The Lustgarten Foundation (A.M. Lowy and H. Tiriac) as well as generous donations from the Alexandrina M. McAfee Trust Foundation (A.M. Lowy) and the Research for a Cure of Pancreatic Cancer Fund (A.M. Lowy). V. Pantazopoulou was supported by a Postdoctoral Fellowship from the Swedish Research Council. S. Ogawa was supported by JSPS KAKENHI Grant Number JP21J01035. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). RNA sequencing was conducted at the IGM Genomics Center, University of California, San Diego, La Jolla, CA (P30CA023100). RNA sequencing analysis was conducted by the Center for Computational Biology and Bioinformatics, which is funded by the Altman Clinical and Translational Research Institute (ACTRI; UL1TR001442). The RPPA core at the MD Anderson Cancer Center is supported by NCI Grant #CA16672 and Dr. Yiling Lu's NIH R50 Grant #R50CA221675: Functional Proteomics by Reverse Phase Protein Array in Cancer.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
D.D. Engle reports personal fees from Cold Spring Harbor Laboratory outside the submitted work. A.M. Lowy reports personal fees from Kinnate, Corcept, Rafael, Bluestar Genomics, Pfizer, and Fibrogen outside the submitted work. H. Tiriac reports grants from Lustgarten Foundation during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
K.C.M. Gulay: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. X. Zhang: Data curation, software, formal analysis, visualization, writing–review and editing. V. Pantazopoulou: Formal analysis, validation, investigation, methodology, writing–review and editing. J. Patel: Investigation, writing–review and editing. E. Esparza: Investigation, writing–review and editing. D.S. Pran Babu: Investigation, writing–review and editing. S. Ogawa: Formal analysis, supervision, validation, investigation, methodology, writing–review and editing. J. Weitz: Data curation, writing–review and editing. I. Ng: Conceptualization, resources, data curation, supervision, funding acquisition, investigation, project administration, writing–review and editing. E.S. Mose: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, project administration, writing–review and editing. M. Pu: Conceptualization, resources, data curation, supervision, funding acquisition, writing–review and editing. D.D. Engle: Conceptualization, resources, data curation, supervision, funding acquisition, writing–review and editing. A.M. Lowy: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, methodology, project administration, writing–review and editing. H. Tiriac: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 2. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther 2021;6:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ou S-HI, Jänne PA, Leal TA, Rybkin II, Sabari JK, Barve MA, et al. First-in-human phase I/IB dose-finding study of adagrasib (MRTX849) in patients with advanced KRASG12C solid tumors (KRYSTAL-1). J Clin Oncol 2022;40:2530–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012;72:2457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem 2022;65:3123–33. [DOI] [PubMed] [Google Scholar]
- 6. Hallin J, Bowcut V, Calinisan A, Briere DM, Hargis L, Engstrom LD, et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat Med. 2022;28:2171–82. [DOI] [PubMed] [Google Scholar]
- 7. Kemp SB, Cheng N, Markosyan N, Sor R, Kim I-K, Hallin J, et al. Efficacy of a small molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov 2023;13:298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med 2021;384:2382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR blockade reverts resistance to KRASG12C inhibition in colorectal cancer. Cancer Discov 2020;10:1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ponz-Sarvise M, Corbo V, Tiriac H, Engle DD, Frese KK, Oni TE, et al. Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin Cancer Res 2019;25:6742–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boj SF, Hwang C-I, Baker LA, Chio IIC, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016;32:2866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bliss CI. The toxicity of poisons applied jointly1. Ann Appl Biol 1939;26:585–615. [Google Scholar]
- 15. Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 1953;3:285–90. [PubMed] [Google Scholar]
- 16. Berenbaum MC. What is synergy? Pharmacol Rev 1989;41:93–141. [PubMed] [Google Scholar]
- 17. Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang Y-J, Frazier ML, Zhang N, Liu Q, Wei C. Reverse-phase protein array analysis to identify biomarker proteins in human pancreatic cancer. Dig Dis Sci 2014;59:968–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roy I, McAllister DM, Gorse E, Dixon K, Piper CT, Zimmerman NP, et al. Pancreatic cancer cell migration and metastasis is regulated by chemokine-biased agonism and bioenergetic signaling. Cancer Res 2015;75:3529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown WS, McDonald PC, Nemirovsky O, Awrey S, Chafe SC, Schaeffer DF, et al. Overcoming adaptive resistance to KRAS and MEK inhibitors by co-targeting mTORC1/2 complexes in pancreatic cancer. Cell Rep Med 2020;1:100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158:185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021;599:679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Angelis C, Fu X, Cataldo ML, Nardone A, Pereira R, Veeraraghavan J, et al. Activation of the IFN signaling pathway is associated with resistance to CDK4/6 inhibitors and immune checkpoint activation in ER-positive breast cancer. Clin Cancer Res 2021;27:4870–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hong S-K, Lee H, Kwon O-S, Song N-Y, Lee H-J, Kang S, et al. Large-scale pharmacogenomics based drug discovery for ITGB3 dependent chemoresistance in mesenchymal lung cancer. Mol Cancer 2018;17:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger RD, et al. Allele-specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov 2018;8:648–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of drugs used in this study.
Table S2. List of antibodies used in this study.
Table S3. Results of RPPA and transcriptomic analyses.
Supplemental Figure and Table Legends
Figure S1 - S12
Data Availability Statement
The data, RNA sequencing (RNA-seq) analyses and whole exome sequencing, generated in this study are publicly available in GEO (GSE234449) and SRA (PRJNA978606) databases. All other raw data are available upon request from the corresponding authors.