

Abstract
Objective
Leukodystrophies are a diverse group of rare inherited disorders that affect the white matter of the central nervous system with a wide phenotypic spectrum. We aimed to characterize the clinical and genetic features of leukodystrophies in a central‐southern Chinese cohort.
Methods
A cohort of 16 Chinese probands with leukodystrophy was recruited and performed genetic analysis by targeted panels or whole‐exome sequencing. Further functional analysis of identified mutations in the colony stimulating factor 1 receptor (CSF1R) gene was explored.
Results
A total of eight pathogenic variants (3 novel, 5 documented) were identified in genes including AARS2, ABCD1, CSF1R, and GALC. Common symptoms of leukodystrophy such as cognitive decline, behavioral symptoms, bradykinesia, and spasticity were observed in mutation carriers as well as other rare features (e.g., seizure, dysarthric, and vision impairment). Overexpressing CSF1R mutants p.M875I and p.F971Sfs*7 in vitro showed pronounced cleavage CSF1R and suppressed protein expression, respectively, and reduced transcripts of both mutants were observed. CSF1 treatment revealed deficient and suppressed CSF1R phospho‐activation with the mutants. In contrast to the plasma membrane and endoplasmic reticulum (ER) localized wild‐type CSF1R, M875I mutant showed much less membrane association and greater detainment in the ER, whereas F971Sfs*7 mutation led to aberrant non‐ER localization. Both mutations caused suppressed cell viability, which was partially resulted from deficient/suppressed CSF1R‐ERK signaling.
Interpretation
In summary, our findings expand the mutation spectrum of these genes in leukodystrophies. Supported by in vitro validation of the pathogenicity of heterozygous CSF1R mutations, our data also provide insights into the pathogenic mechanisms of CSF1R‐related leukodystrophy.
Introduction
Leukodystrophies are a diverse group of rare inherited disorders that affect the white matter of the central nervous system (CNS) with a wide phenotypic spectrum. It usually affects infants and young children, but individuals with this disease can also develop symptoms until adulthood. 1 Clinical manifestations of adult‐onset leukodystrophy consist of neuropsychiatric and cognitive changes, spasticity, ataxia, seizures, as well as visual and hearing problems. 2 Magnetic resonance imaging (MRI) often shows bilateral white matter changes in patients with leukodystrophy, although the imaging pattern may vary greatly depending on the severity and time course of the disease. 3 The diagnosis of leukodystrophy remains challenging because of the diversity of genetic etiologies and high heterogeneity in its clinical and radiological phenotypes.
Leukodystrophies can be inherited in an autosomal dominant, autosomal recessive, and X‐linked, or they may also occur spontaneously with de novo mutations. With the application of next‐generation sequencing (NGS), a growing number of studies have identified specific genes in association with leukodystrophy and unraveled the complex genetic architecture of this disease. 4 More than 60 genes linked to leukodystrophy or genetic leukodystrophy have been discovered to date, 5 , 6 but still nearly half of the patients with leukodystrophy remain undiagnosed. 7 Despite the broad clinical spectrum of leukodystrophy with multiple genes involved, the specific phenotype of these condition is strictly determined by the genetic changes and altered cell function. Many studies have explored the possible genotype–phenotype correlation in some of the specific types of leukodystrophy, such as metachromatic leukodystrophy, 8 vanishing white matter disease, 9 and CSF1R‐related leukodystrophy, 10 but unfortunately, no clear pattern of genotype–phenotype relationship was well‐established in leukodystrophies. Analysis of genotype–phenotype correlation in leukodystrophy may help in the early identification of additional modifier genes and in the development of disease‐modifying treatment strategies for leukodystrophy.
There are still very few reports on genetic analysis of leukodystrophy patients in central‐southern China. In this study, we applied whole‐exome sequencing and targeted panels in a cohort of patients with leukodystrophy from central‐southern China and analyzed the clinical features and genetic characteristics. In support, in vitro functional analysis demonstrated that CSF1R variations lead to aberrant protein expression, processing, localization, diminished/suppressed CSF1R‐ERK signaling, and ultimately inhibited cell viability.
Materials and Methods
Study population
This cohort consisted of 16 patients clinically diagnosed with leukodystrophy or genetic leukoencephalopathy, 8 males and 8 females, aged from 19 to 70 years. All patients were of central‐southern Han Chinese origin and were recruited consecutively between September 2017 and March 2022 from the Neurology Department of Wuhan Union Hospital. Written informed consent was obtained from all participants. This study was approved by the ethics committee of Wuhan Union Hospital. All affected patients were evaluated by at least two neurologists and diagnosed according to the criteria recommended by the Global Leukodystrophy Initiative (GLIA) Consortium. 11 Brain magnetic resonance imaging (MRI) was performed to identify white matter lesions and atrophy. The plasma very long chain fatty acids (VLCFAs) analysis, was also tested. Electroencephalography (EEG), visual evoked potential (VEP), and cerebrospinal fluid (CSF) were performed as well.
Genetic analysis
Genomic DNA was extracted from peripheral blood samples by reference to a standard DNA extraction method. Comparative analysis of whole‐exome sequencing and targeted panels comprising 93 genes currently associated with genetic leukodystrophies and other related disorders were performed. Sequencing was performed using Illumina Hiseq2000/2500 system as described previously. 12 Sanger sequencing was used to validate the variants after variants calling and filtering. Co‐segregation analysis was further performed to assess the variants validated by Sanger sequencing.
Construction CSF1R‐expressing vector and mutagenesis
Human CSF1R (NM_005211) cDNA clone was purchased from Youbio biotechnology (Changsha, China). The fragment of CSF1R coding sequence was amplified and was subcloned into pcDNA3.1‐FLAG vector using EcoR1 and Xho1 restriction sites and seamless cloning kit (D7010S, Beyotime biotechnology). PCR amplification was used to induce site‐directed mutagenesis (M875I) and frameshift mutagenesis (F971Sfs*7) of CSF1R‐. Insertion was then subcloned into pcDNA3.1‐FLAG vector using EcoR1 and Xho1 restriction sites and seamless cloning kit. All clones were validated by Sanger sequencing.
Cell culture and transfection
HeLa and HEK293T cells were maintained at 37°C (5% CO2) in Dulbecco's modified eagle medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (10,000 U/mL). Cells were plated onto plates/dished/wellplates for 18–24 h before transfection at 70%–80% confluence or onto coverslips as required. Plasmids and control vector (pcDNA3.1‐FLAG) were transfected using polyethylenimine (PEI, Polyscience). The transfection mixture was incubated at room temperature for 20 min before being added into each well. Cells were harvested at 24 h post‐transfection. For CSF1 treatment, the culture medium was replaced with serum‐free medium 12 h after transfection, and 12 h later, human CSF1 (MCE, HY‐P7050A) was applied at 100 ng/ml final concentration for indicated period before harvest.
Immunoprecipitation and western blotting
Transfected and treated cells were immediately washed with cold PBS and lysed in lysis buffer (Beyotime Biotechnology, P0013). Anti‐FLAG antibody (Beyotime biotechnology, AF0036, 1:80) and protein A/G magnetic beads (MCE) were incubated with the lysate at 4°C overnight. Immunoprecipitates were washed and boiled in 2× Laemmli sample buffer (Bio‐Rad) followed by heat denaturation and western blotting. For routine western blotting, cells were lyzed in 2× Laemmli sample buffer followed by sonication and heat denatured at 95°C for 5 min before loading onto SDS‐PAGE gels. Transferred nitrocellulose membranes were blocked and then reacted with primary antibodies overnight at 4°C under gentle rocking. Primary antibodies are as follows: anti‐pan p‐Tyrosine (clone PY20) antibody (Millipore, 05‐947, 1:2000 dilution), anti‐FLAG antibody (Beyotime biotechnology, AF519‐1, 1:2000 dilution), anti‐Actin antibody (Proteinab Biotech, Cat# 10003‐M01, 1:3000 dilution), anti‐p‐ERK1/2 antibody (Cell signaling Technology, #4370, 1:2000 dilution), and anti‐ERK1/2 antibody (Cell signaling Technology, #4696, 1:2000 dilution). The corresponding horseradish peroxidase (HRP)‐conjugated anti‐rabbit secondary antibodies (Beyotime biotechnology, 1:1000 dilution) was used, followed by probing with enhanced chemiluminescence (ECL) western blotting substrate (Beyotime biotechnology P0018AM) and visualized using Tanon scanner system (Tanon5200).
Immunofluorescence staining
Transfected cells grown on coverslips were washed with PBS and fixed in 4% PFA, followed by permeabilization with 0.1% Triton X‐100 in PBS. The cells were then blocked for 1 h in 5% goat serum, followed by incubation overnight with primary antibodies at 4°C. Anti‐FLAG (Beyotime biotechnology, AF519, 1:200 dilution) and anti‐Calnexin antibody (Abcam, ab22595, 1:500 dilution) were used. Secondary antibodies were Dylight (488, 594)‐labeled goat anti‐mouse and goat anti‐rabbit antibodies (Abbkine Scientific Co., 1:500 dilution). Actin‐tracker Red‐Rhodamine (Beyotime Biotechnology, C2207S, 1:200 dilution) was incubated for 45 min in dark after the secondary antibody to visualize actin. DAPI was used to visualize the nuclei. Images were captured at 40× objective with inverted fluorescence microscope (ICX41, Sunny optical technology).
Cell Counting Kit‐8 (CCK‐8) assay
HeLa cell suspension was inoculated at 15,000 cells per well in a 96‐well plate for 24 h culture prior to transfection. Prepared expression plasmids were transfected using PEI reagent. For each well, 10 μl transfection mixture including 200 ng of total DNA and 0.5 μg PEI was prepared. After 24 h, 10 μL of CCK‐8 solution (Biosharp Life Science, BS350A) was added to each well of the plate for 1 h‐incubation before absorbance was measured at 450 nm.
Quantitative real‐time PCR
Transfected HeLa cells were collected in RNA isolator (Vazyme Biotech, R401) for total RNA extraction, followed by reverse‐transcription using HiScript II first strand cDNA synthesis kit (Vazyme Biotech, R22‐01‐AB). cDNA was used for qRT‐PCR using SYBR Green Master mix (Vazyme Biotech, Q311‐02‐AA) in QUANTAGENE q225 real‐time PCR machine (KUBO biotechnology). The FLAG‐encoding sequence were included in the primers: CSF1R forward, 5′‐GCAGCAGCAGTGAGCTGG‐3′, CSF1R reverse, 5′‐ CGTCATGGTCTTTGTAGTCG‐3′; GAPDH forward, 5′‐ TGCACCACCAACTGCTTAGC‐3′, GAPDH reverse, 5′‐ GGCATGGACTGTGGTCATGAG‐3′. The ΔΔCt method has been used for fold change analysis using GAPDH as the internal control.
Statistical analysis
Statistical analysis was performed using the Prism 9 software (GraphPad). The biochemical analysis was performed using a minimum of three biological replicates per condition, and randomization of experimental groups was not required. Quantification of immunoblots was conducted with ImageJ. Densities of bands were normalized to individual control before being normalized to the average of the wild‐type group. Two groups were compared using Student's t‐test. Three or more groups were assessed with one‐way ANOVA followed by a post hoc test for Sidak's multiple comparisons. All comparisons were performed whenever possible with all significant changes being indicated on the graphs. Values are presented as the mean ± standard error of mean (SEM).
Results
Genetic findings and pathogenicity classification of variants
Overall, about 97.41% of the target bases were covered with at least 20× per individual, and the mean depth of coverage for all target regions was 184.28. Only those variants that may affect protein function with extremely low allele frequency (minor allele frequency below 0.1%) reported by dbSNP, the Genome Aggregation Database (GnomAD), NHLBI Exome Sequencing Project (ESP6500), and Exome Aggregation Consortium (ExAC) database, were retained after filtering and assumed to be likely pathogenic. Finally, we identified three novel potentially pathogenic variants (AARS2: c.2146‐2A>G, ABCD1: p.S149Hfs*44, GALC: p.G256A) and five known pathogenic variants (AARS2: p.M151T, ABCD1: p.R554H, CSF1R: p.F971Sfs*7, CSF1R: p.M875I) in six unrelated families with leukodystrophy (Fig. 1). All of the three novel variants were confirmed by Sanger sequencing and were predicted as harmful effects by computational tools (Table 1).
Figure 1.
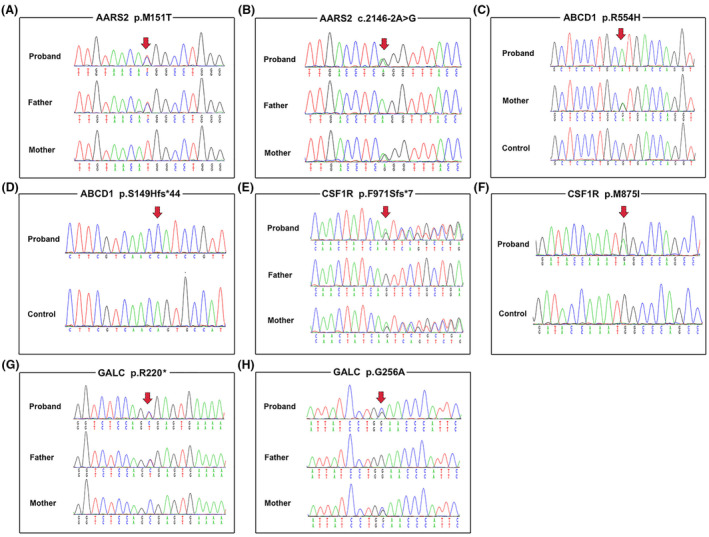
Segregation of identified mutations in the AARS2, ABCD1, CSF1R, and GALC genes. (A and B) Sequence chromatogram of the c.452T>C (p.M151T) and c.2146‐2A>G mutations in the AARS2 gene in family 1, respectively. (C) Sequence chromatogram of the c.1661G>A (p.R554H) missense mutation in the ABCD1 gene in family 3. (D) Sequence chromatogram of the c.445_449del (p.S149Hfs*44) frameshift mutation in the ABCD1 gene in family 2. (E) Sequence chromatogram of the c.2909_2910insATCA (p.F971Sfs*7) frameshift mutation in the CSF1R gene in family 4. (F) Sequence chromatogram of the c.2625G>A (p.M875I) missense mutation in the CSF1R gene in family 5. (G and H) Sequence chromatogram of the c.658C>T (p.R220*) and c.767G>C (p.G256A) mutations in the GALC gene in family 6, respectively.
Table 1.
Summary of the mutations identified in this study.
| Mutations | Bioinformatics prediction | Population frequency | ACMG | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Refseq NM | Nucleotide | Amino acid | Polyphen2 | SIFT | ESP6500 | GnomAD | ExAC | ||
| AARS2 | NM_020745.4 | c.452T>C | p.M151T | Probably damaging | Deleterious | 0 | 0 | 0 | Likely pathogenic | [13] |
| AARS2 | NM_020745.4 | c.2146‐2A>G | NA | NA | NA | 0 | 0.000014 | 0 | Pathogenic | This study |
| ABCD1 | NM_000033.4 | c.1661G>A | p.R554H | Deleterious | Deleterious | 0 | 0 | 0 | Pathogenic | [14] |
| ABCD1 | NM_000033.4 | c.445_449del | p.S149Hfs*44 | NA | NA | 0 | 0 | 0 | VUS | This study |
| CSF1R | NM_005211.3 |
c.2909_291 0insATCA |
p.F971Sfs*7 | NA | NA | 0 | 0.000040 | 0.000068 | VUS | [15] |
| CSF1R | NM_005211.3 | c.2625G>A | p.M875I | Probably damaging | Deleterious | 0 | 0 | 0 | Likely pathogenic | [16] |
| GALC | NM_000153.4 | c.658C>T | p.R220* | NA | NA | 0 | 0 | 0.000008 | Pathogenic | [17] |
| GALC | NM_000153.4 | c.767G>C | p.G256A | Possibly damaging | Tolerated | 0 | 0 | 0 | Likely pathogenic | This study |
ACMG, American College of Medical Genetics; ESP6500, Exome Sequencing Project v.6500; ExAC, Exome Aggregation Consortium; GnomAD, Genome Aggregation Database; NA, not applicable; SIFT, Sorting Intolerant From Tolerant; VUS, variants of uncertain significance.
Clinical and imaging features of the patients with potentially pathogenic variants
In the present study, we identified 8 potentially pathogenic mutations in 6 (37.5%) of 16 patients, including a patient with AARS2‐related leukoencephalopathy (proband 1), two patients (probands 2 and 3) with Adrenoleukodystrophy (ALD), two patients with CSF1R‐related leukoencephalopathy (probands 4 and 5), and a patient with Krabbe disease (proband 6). Among those patients with potentially pathogenic variants, the mean age at onset was 31.83 ± 13.92 years, and 33.3% (2/6) of patients were females. The clinical characteristics of the six probands are summarized in Table 2 and described in detail below.
Table 2.
Clinical characteristics and genetic analysis of six probands carrying mutations.
| Proband no | Sex | Age at onset (years) | Age at present (years) | Predominant symptoms | MRI features | Gene | Variants |
|---|---|---|---|---|---|---|---|
| 1 | M | 28 | 33 | Cognitive impairment; changes in personality and behavior; unsteady gait; bladder and bowel dysfunction | Cerebellum vermis and callosal atrophy, WM abnormality in the periventricular and CC, restricted diffusion on DWI in the CC | AARS2 |
c.452T>C (p.M151T) c.2146‐2A>G |
| 2 | M | 46 | 47 | Rapid cognitive decline; behavioral symptoms; dysarthric; severe apraxia; rigidity; seizures; bladder and bowel dysfunction |
WM abnormality involving the periventricular WM, CC, internal and external capsules |
ABCD1 | c.445_449del (p.S149Hfs*44) |
| 3 | M | 47 | 53 | Progressive bilateral lower extremity weakness; spasticity; bladder and bowel dysfunction | WM abnormalities in bilateral cerebellar hemispheres | ABCD1 | c.1661G>A (p.R554H) |
| 4 | M | 37 | 49 | Seizures; vision impairment |
MRI at age 37 years old: multiple confluent T2/FLAIR hyperintense in the subcortical, juxtacortical and periventricular MRI at age 48 years old: WM abnormality in the periventricular and subcortical, bilateral optic nerve changes, CC atrophy |
CSF1R | c.2909_2910insATCA (p.F971Sfs*7) |
| 5 | F | 27 | 29 | Personality and behavioral changes; cognitive impairments; slurred speech; bradykinesia; rigidity; urinary incontinence | Bilaterally periventricular WM hyperintensity, ventricular enlargement and CC atrophy | CSF1R | c.2625G>A (p.M875I) |
| 6 | F | 6 | 19 | Unsteady gait; spasticity; urinary incontinence; constipation | Symmetric T2 hyperintensity of the corticospinal tracts and the periventricular WM | GALC |
c.658C>T (p.R220*) c.767G>C (p.G256A) |
AAO, age at onset; CC, corpus callosum; F, female; M, male; NA, not applicable; WM, white matter.
Presentations of patient with AARS2‐related leukodystrophy
The proband 1 in family 2, carrying 2 heterozygous mutations (c.2146‐2A>G and p.M151T) in AARS2, was a 33‐year‐old male patient and presented with cognitive decline accompanied by changes in personality and behavior since age 28. As the disease progressed, he became taciturn with uncontrollable laughing, and soon needed assistance getting dressed and completing toileting. His physical examination revealed altered mental status with a Glasgow Coma Scale score of 13, along with unsteady gait and positional horizontal nystagmus. Brain MRI demonstrated multiple hyperintensity signals in the periventricular white matter and body of the corpus callosum (Fig. 2A–C). Interestingly, hyperintensity lesions with restricted diffusion in the corpus callosum were also observed on diffusion‐weighted imaging (DWI). Computed tomography (CT) scan revealed no calcification in the brain. Genetic analysis showed that the proband carried compound heterozygous mutations in the AARS2 gene (c.2146‐2A>G and p.M151T), and his unaffected parents were all heterozygous.
Figure 2.
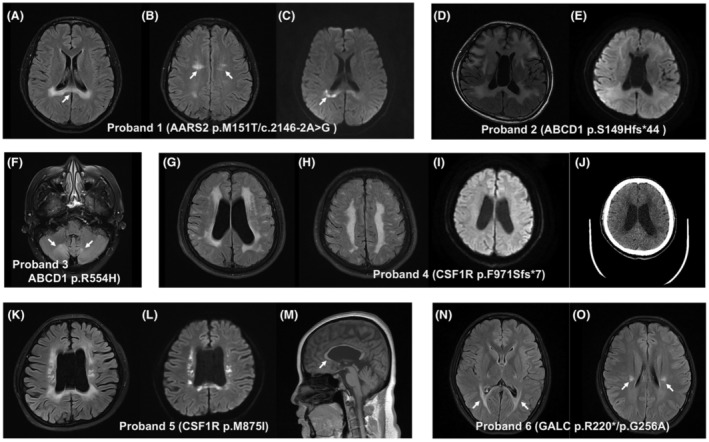
Brain magnetic resonance imaging (MRI) and computerized tomography (CT) findings in patients with genetic leukodystrophies. Hyperintensities in the body of the corpus callosum (A, arrow) and periventricular white matter (B, arrows) on T2 FLAIR images, and restricted diffusion on diffusion‐weighted MR images (C, arrow), were observed in the patient (proband 1) with AARS2 mutations. (D) Axial T2 FLAIR images in the patient (proband 2) with ABCD1 p.S149Hfs*44 mutation show hyperintense signal changes involving not only the periventricular white matter and corpus callosum, but also internal and external capsules. (E) There was no restricted diffusion detected on diffusion‐weighted MR images of the same patient. (F) Axial T2 FLAIR MR images showing nonspecific hyperintensities of the white matter in bilateral cerebellar hemispheres (arrows) in the patient (proband 3) with ABCD1 p.R554H mutation. (G and H) Axial T2 FLAIR MR images in the patient (proband 4) with CSF1R p.F971Sfs*7 mutation show hyperintensities in the periventricular and deep subcortical white matter. There was no restricted diffusion observed on diffusion‐weighted MR images of the same patient (I). (J) Axial noncontrast brain CT of the same patient demonstrated no calcification. Ventricular enlargement and corpus callosal atrophy (M, arrow), with hyperintensities in periventricular white matter on T2 FLAIR images (K) and restricted diffusion on diffusion‐weighted MR images (L), are observed in the patient (proband 5) with CSF1R p.M875I mutation. (N and O) Axial T2 FLAIR MR images in the patient (proband 6) with GALC mutations show involvement of the corticospinal tracts and periventricular white matter (arrows).
Presentations of ALD patients with ABCD1 mutations
Proband 2 was a 47‐year‐old male, carrying the p.S149Hfs*44 variant in ABCD1, who presented with a 15‐month history of cognitive impairment and behavioral symptoms. He was struggling to follow a conversation and became apathetic, irritable, and emotional lability. As the disease progressed, he had speech dysfunction including slurred speech and dysarthria, together with bladder and bowel dysfunction. During hospitalization, by the age of 47, the patient developed generalized tonic–clonic seizures. His seizures were well controlled by sodium valproate monotherapy. The Mini‐Mental State Examination (MMSE) assessment of the patient yields a low score of 14, indicating severe cognitive impairment. On examination, he exhibited severe limb apraxia, rigidity, spotty skin pigmentation, along with episodic memory deficits and executive dysfunction. The brain MRI revealed hyperintensity signals in the periventricular white matter and corpus callosum, as well as the internal and external capsules (Fig. 2D). The plasma ACTH concentrations were normal, but serum VLCFA levels were elevated. On follow‐up, the patient continued to experience severe psycho‐behavioral symptoms and chronic urinary incontinence. Genetic screen showed that the proband had a novel heterozygous missense mutation (c.445_449del, p.S149Hfs*44) in the ABCD1 gene. His mother was considered to be asymptomatic (without any symptoms of the disease) and died in an accident when she was 68 years old.
Proband 3 was a 53‐year‐old male, carrying a heterozygous missense mutation (c.1661G>A, p.R554H) in ABCD1, who experienced bilateral lower extremity weakness at age 47. As the condition progressed, he exhibited severe lower limbs spasticity and gait abnormality. Neurological examination revealed hyperactivity of the upper and lower extremity deep tendon reflexes, together with bilateral positive Babinski sign. Based on the results of both clinical and neurological examination, the patient was initially diagnosed with hereditary spastic paraplegia (HSP). Over the past 2 years, the patient's limbs weakness symptoms continued to progress slowly, and he was struggling with bladder and bowel dysfunction. Brain MRI showed white matter abnormalities predominantly in bilateral cerebellar hemispheres (Fig. 2F). The plasma of ACTH concentration was in the normal range. The electromyography (EMG) recording showed no abnormalities. However, the serum VLCFA levels were not available in the proband. The diagnosis of HSP was no longer inappropriate at this time, and therefore, adult‐onset leukodystrophy was suspected. Genetic testing revealed a known pathogenic mutation, p.R554H, in the ABCD1 gene.
Characteristics of patients with CSF1R‐related leukodystrophy
Proband 4 was a 49‐year‐old male, carrying the p.F971Sfs*7 mutation in CSF1R, who had a nearly 12‐year history of recurrent and stereotyped generalized motor seizures. He was prescribed the antiepileptic drug carbamazepine and eventually stopped taking it after 1 year without a seizure, but he developed seizures again around 4 years after symptom onset. The MRI imaging scans at the first onset of seizure showed multiple confluent T2/FLAIR hyperintense lesions involving the subcortical, juxtacortical, and periventricular white matter, although the lesions remained grossly unchanged in the patient's subsequent annual MRI screening. Based on the results of both clinical and neuroimaging findings, the initial diagnosis of multiple sclerosis (MS) was suspected at that time. As the condition progressed, he experienced reduced vision in both eyes (0.15 in the right and 0.3 in the left) at 48 years old. However, his visual field, color perception, and ocular fundus examination were normal at presentation. Because of his worsening vision, he was referred to our hospital. At admission, the proband had severe visual impairment. His cognitive abilities including memory and other cognitive domains were relatively intact. There were no other positive findings in the neurological examination. The brain MRI revealed multiple small patches of hyperintensity on T2‐weighted sequences, running parallel to the walls of the lateral ventricle, predominantly in periventricular and deep subcortical white matter (Fig. 2G,H). Brain computed tomography (CT) did not show any evidence of calcification (Fig. 2J). The laboratory tests, VEP, EEG, and CSF analysis were all normal. The patient received methylprednisolone treatment (intravenous injection of 1000 mg/day for 5 days) followed by oral prednisolone and showed an improvement in visual acuity. After 3 months, the patient's visual acuity continued to deteriorate and recovered again after a period of high‐dose methylprednisolone treatment. Whole‐exome sequencing revealed that the proband carried a known heterozygous missense mutation (c.2909_2910insATCA, p.F971Sfs*7) in the CSF1R gene. Genetic testing of the patient's parents showed that his mother was an asymptomatic carrier of the same CSF1R mutation. Moreover, we obtained the brain MRI scan of the patient's mother at age 78, without any imaging features of leukoencephalopathy.
The proband in family 5, carrying the c.2625G>A (p.M875I) mutation in CSF1R, was a 29‐year‐old female patient and referred to us for the evaluation of psychomotor symptoms and cognitive impairments. Two years before referral, she first noticed significant personality and behavioral changes, characterized by apathy, taciturnity, social disinhibition, and depression. Six months later, she developed slurred speech and speaking problems. The cognitive decline was exacerbated as the condition progressed. She developed bradykinesia, rigidity, and urinary incontinence, and required a wheelchair for mobility. She scored 12/30 on the MMSE. No relevant family history was found in the family, and both her parents were healthy. Neurological examination showed slurred speech, generalized bradykinesia, rigidity, as well as severe impairment in episodic memory and executive functions. Brain MRI showed corpus callosal atrophy and ventricular enlargement, together with hyperintensity of bilaterally periventricular white matter on T2/FLAIR images with restricted diffusion (Fig. 2K–M).
Characteristics of Krabbe patient with GALC mutations
Proband 6 was a 19‐year‐old female, carrying 2 heterozygous missense mutations (p.R220* and p.G256A) in GALC, who had noticed an unsteady gait at age 6. As the disease progressed, she began to experience severe spasticity in the lower limbs, together with delayed walking and frequent falls. At the age of 10, she had urinary incontinence and constipation. She now needs support for walking. Neurological examination revealed hyperactive deep tendon reflexes in both upper and lower extremities and bilateral positive Babinski sign. The plasma VLCFAs analysis showed that the activity of galactosylceramidase was decreased. Brain MRI showed bilateral symmetrical T2 hyperintensity of the corticospinal tracts and the periventricular white matter without any significant diffusion restriction (Fig. 2N,O). Genetic testing of the patient's parents revealed that his parents were heterozygous carriers of the p.R220* and p.G256A mutations in the GALC gene, respectively. Her elder brother was found to be an unaffected heterozygous carrier of the GALC p.R220* mutation.
Functional characterization of CSF1R mutants in vitro
We first constructed the CSF1R mutants and overexpressed these transgenes in HeLa cells. A 175 kDa full‐length protein and a weaker cleaved band were observed in wild‐type (WT) CSF1R transfected cell lysate, consistent with the size validated by many antibody manufacturers. M875I mutation caused pronounced cleavage of the full‐length protein, while the F971Sfs*7 showed much less expression of the full‐length protein with visible cleavage similar to the WT (Fig. 3A). Quantification suggested significant reduction of full‐length CSF1R in both mutants and surprisingly increased cleavage only for M875I mutation (Fig. 3B). Transcriptional analysis confirmed reduced CSF1R‐FLAG transcripts in both mutants transfected cells (Fig. 3C).
Figure 3.
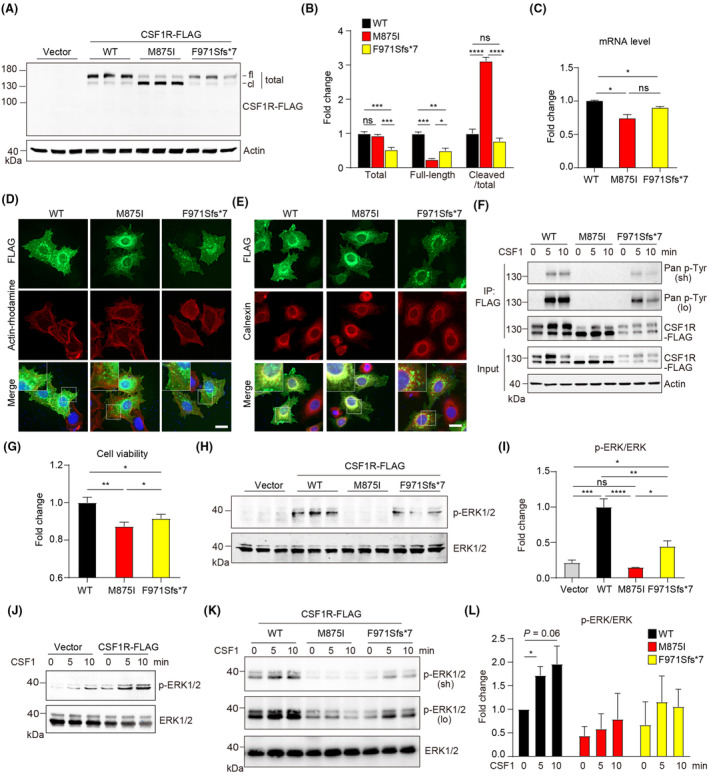
Characterization of CSF1R variants in vitro. (A) Immunoblotting of CSF1R‐FLAG in transfected HeLa cells as indicated. fl, Full‐length form; cl, cleaved form. (B) Quantification of A. n = 3 per group. (C) Quantitative analysis of mRNA expression after indicated transfection. n = 4 per group. WT, wild‐type. (D) Immunofluorescence staining of CSF1R‐FLAG overexpressing cells and counterstaining with actin‐tracker Rhodamine. DAPI labels the nuclei. Augmented area in the dashed boxes was shown. Scale bar, 20 μm. (E) Immunofluorescence staining of CSF1R‐FLAG and endoplasmic reticulum marker Calnexin in transfected HeLa cells. DAPI labels the nuclei. Augmented area in the dashed boxes was shown. Scale bar, 20 μm. (F) Immunoblotting detection of the level of tyrosine phosphorylation of CSF1R in transfected HEK293T cells after CSF1 treatment as indicated. IP, immunoprecipitation; sh, short exposure; lo, long exposure. (G) CCK8 assay suggests suppressed cell viability induced by the mutants. n = 8 per group. (H) Immunoblotting of pERK1/2 and total ERK1/2 in transfected HeLa cells. (I) Quantification of H. n = 3 per group. ns, not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (J) ERK1/2 activation after CSF1 treatment in CSF1R‐transfected HEK293T cells. (K) Diverse degree of ERK1/2 activation after acute CSF1 treatment in HEK293T cells transfected with indicated mutants. lo, long exposure; sh, short exposure. (L) Quantification of K. Fold changes were normalized to untreated CSF1R (WT)‐transfected group. n = 3 per group. *p < 0.05.
Next, we assessed the subcellular localization of the mutants by counterstaining the actin cytoskeleton and the plasma membrane. FLAG‐tag labeling showed broad distribution of CSF1R‐WT at the plasma membrane and the cytoplasm, with the majority of signal at the perinuclear region; CSF1R‐M875I showed less cell membrane association whereas CSF1R‐F971Sfs*7 indicated attenuated signal of expression (Fig. 3D). Furthermore, counterstaining of CSF1R and endoplasmic reticulum (ER) specific marker protein Calnexin indicated a highly overlapped pattern of CSF1R‐M875I and Calnexin, while non‐ER localization of CSF1R were also observed for CSF1R‐F971Sfs*7, suggesting that both mutants spatially act differently from the WT (Fig. 3E).
Functional activities and consequences of the variations were then tested. After acute stimulation with human macrophage colony stimulating factor (CSF1), CSF1R‐WT showed drastic tyrosine phosphorylation of CSF1R at 5 min and 10 min, indicative of robust and consistent CSF1R activation. M875I mutant showed completely abolished activation while F971Sfs*7 mutant was activated 5 min after treatment then activity started to decay 10 min after treatment (Fig. 3F). Cell viability in both mutant groups indicated by CCK‐8 assay was significantly reduced, and F971Sfs*7 mutant showed slightly higher cell activity than the M875I group (Fig. 3G). We then wondered if CSF1R‐activated MAPK/ERK signaling pathway is involved in altered cell activity. CSF1R‐WT transfection induced drastic ERK activation compared with the vector, which was completely abolished by M875I mutation, and F971Sfs*7 mutation showed 53% reduction of the activation compared to the WT (Fig. 3H,I). Furthermore, CSF1 treatment induced a basal level of ERK activation in the vector group and enhanced ERK activation above the baseline in CSF1R‐expressing cells, and further activation was induced by CSF1 stimulation (Fig. 3J). In line with the phospho‐activation of CSF1R, downstream ERK activation showed similar trends after CSF1 stimulation, that is, CSF1 induced the most robust ERK activation in the WT group, no activation in the M875I group, and intermediate level of activation in the F971Sfs*7 group, respectively (Fig. 3K,L). In summary, in vitro validation suggested the two mutations induced dramatic depletion of full‐length CSF1R expression, partially due to compromised transcription, in addition to the enormous protein cleavage found with M875I mutant. Dysfunction of CSF1R/ERK activation by both mutations may lead to depressed cell viability, with the detainment of CSF1R‐M875I in the ER likely resulting in a more severe deficiency in ERK activation.
Discussion
Using next‐generation sequencing, we identified three novel pathogenic variants and five previously identified pathogenic variants among the patients with genetic leukodystrophy in central‐southern China. The identification of three novel variants within AARS2, ABCD1, and GALC genes expands the mutation spectrum of these genes in genetic leukodystrophy.
AARS2 and CSF1R are the two genes responsible for genetic leukodystrophy with similar clinical and radiological phenotypes. 18 Both CSF1R‐ and AARS2‐related leukoencephalopathy patients show a combination of frontal‐predominant cognitive impairment and motor dysfunction, accompanied by the mostly confluent and asymmetric white matter involvement predominantly in the frontoparietal, periventricular, as well as the corpus callosum, and often with restricted diffusion on DWI. 2 The calcifications observed in bilateral frontal periventricular white matter are often used to distinguish CSF1R‐related leukodystrophy from AARS2‐related leukodystrophy. 19 In the present study, we found two unrelated CSF1R‐related leukodystrophy cases and one case associated with AARS2 mutations. The patient with AARS2 mutations showed typical clinical and imaging features and prominent cognitive decline and behavior problems consistent with cases previously reported. 20 The two patients with CSF1R‐related leukodystrophy shared similar image features but presented with a large variation in clinical phenotypes, indicating the remarkable clinical heterogeneity of the disease. The most frequent initial manifestation of adult‐onset leukodystrophy, including cognitive deterioration, gait problems, and neuropsychiatric symptoms, 21 were not observed in the patient (proband 4) with mutation CSF1R‐F971Sfs*7 who presented a prominent manifestation of seizures and vision impairment. Decreased visual acuity and optic nerve atrophy are rare in CSF1R‐related leukodystrophy. Shu et al. reported a case of a 30‐year‐old female patient who experienced decreased visual acuity and optic nerve impairment. 22 Abnormal optic nerve myelin metabolism and axonal degeneration of retinal ganglion cells may be the primary ocular pathology process, independent of typical white matter pathologies in CSF1R‐related leukoencephalopathy. 22 Moreover, the p.F971Sfs*7 mutation has previously been reported in a 43‐year‐old male patient who experienced rapidly progressive memory deficit and personality change, 23 while a slower disease progression (more than 10 years) was significantly observed in our patient with the same mutation.
The disease penetrance is age‐dependent in CSF1R mutation carriers, reaching 10% by age 27 years, 50% by age 43 years, and nearly 95% by age 60 years. 5 Of note, apparent incomplete penetrance was observed in our study. Both the p.F971Sfs*7 and p.M875I CSF1R mutation carriers were severely affected since the age of 37 years and 27 years respectively, whereas their mothers with the same mutation remain asymptomatic. Interestingly, the p.M875I CSF1R mutation has previously been reported in a family in which the proband's asymptomatic mother was a carrier and had a history of rheumatoid arthritis with long‐term prednisone treatment. 16 The authors also hypothesized that the clinical features of CSF1R‐related leukoencephalopathy might be minimized by chronic immunosuppression in the patient's mother, which resulted in significant correction of altered microglial phenotype and physiologic balance between CSF‐1R and CSF‐2R signaling. 16 Our findings indicate that variants of specific genes confer a predisposition to develop HDLS, with additional genetic and/or environmental factors that might interact and determine clinical manifestations of the disease. In this study, we first confirmed the pathogenicity of the two CSF1R mutations (p.F971Sfs*7 and p.M875I), which had never been reported as pathogenic with functionally testified.
To date, more than 100 different CSF1R mutations have been identified, most of which occur in the tyrosine kinase domain and affect kinase activity. 15 , 24 , 25 , 26 CSF1R is primarily expressed in the microglia in the CNS where it mediates cell proliferation, differentiation, and survival. Upon ligand engagement, CSF1R induces dimerization and autophosphorylation of the tyrosine residues that serve as docking sites for multiple downstream kinase signaling pathways. 27 , 28 Recently, a dominant‐negative mechanism of kinase‐deficient CSF1R has been postulated in CSF1R‐related leukoencephalopathy. 29 , 30 Additionally, microglial CSF1R haploinsufficiency has been reported to provoke a significant reduction in microglia density and widespread depletion of microglia in both humans and zebrafish. 31 Together, these data suggest that CSF1R mutations result in partial loss of function in CSF1R signaling, probably via both mechanisms of dominant‐negative effect and haploinsufficiency. In our study, the M875I mutant caused pronounced protein cleavage and F971Sfs*7 mutation showed only 50% of protein expression, suggesting that these mutations may cause leukodystrophy due to haploinsufficiency or loss of function. The M875I mutation located in the cytoplasmic region may alter the dimensional conformation, exposing the processing site for proteases. Pronounced cleavage of M875I mutant may lead to aberrant ER detainment and deficient transportation to the plasma membrane. Dysfunction of CSF1R/ERK activation by both of the mutations, demonstrated at both baseline condition and CSF1‐stimulated condition, may directly lead to depressed cell viability, with the detainment of CSF1R‐M875I in the ER likely resulting a more severe deficiency in ERK activation. In summary, both mutants cause depressed cell activity due to deficiencies in gene transcription, protein processing, translocation, and signaling transduction.
Krabbe disease (also called globoid cell leukodystrophy, GLD) is a rare autosomal recessive disorder caused by a deficiency of the lysosomal enzyme galactocerebrosidase. Typical clinical features of late‐onset Krabbe disease include progressive spastic paraparesis, cognitive decline, ataxia, peripheral neuropathy, and visual loss. 2 In this study, we identified the two variants (p.R220* and p.G256A) within GALC in the proband of family 6. The proband presented with juvenile‐onset spastic paraplegia phenotypes, which was consistent with the clinical presentations of previously reported patients with juvenile‐onset Krabbe disease. To date, more than 140 different mutations in the GALC gene have been reported worldwide. 32 There is no clear correlation between genotype and phenotype in patients with Krabbe disease. Interestingly, the common GALC 30‐kb deletion of European origin (c.1161 + 6532_polyA+9kbdel) has been reported to cause the infantile Krabbe disease if present in the homozygous or compound heterozygous state along with another mutation. 33 While the mutation p.Gly286Asp in exon 8 was found in association with a disease course with a late onset if occurring as a homozygous or compound heterozygous together with another mutation. In this case, the juvenile‐onset phenotype was probably due to the contribution of novel missense mutation p.G256A, in trans to the nonsense p.R220* mutation (expected to introduce a premature termination codon).
Adult‐onset adrenoleukodystrophy (ALD) is the most common adult‐onset leukodystrophy that affects the adrenal gland and the white matter of the nervous system. In the adult‐onset form, symptoms typically start at 30 to 40 years of age and include progressive stiffness or rigidity, weakness or paralysis of the lower limbs, ataxia, seizures, urinary incontinence, speech disturbances, and peripheral neuropathy. In our cohort, the p.S149Hfs*44 mutation in ABCD1 caused typical clinical and neuroimaging features of adult‐onset ALD, while the patient with p.R554H mutation presented a predominant spastic paraplegia phenotype. Our results reinforce the notion that ALD accounts for a significant proportion of spastic paraplegia entities and should be considered in patients with proposed HSP.
In summary, our findings expand the mutation spectrum of these genes in genetic leukodystrophy and provide a better understanding of the phenotypic and genotypic profiles of this disease. Our data also demonstrate that the use of NGS has great promise as a tool for discovering unknown or rare genetic mutations associated with leukoencephalopathy. In addition, we have performed in vitro functional assays to confirm the pathogenicity of the two CSF1R mutations (p.F971Sfs*7 and p.M875I). Despite the limitations, our findings provide insights into pathogenetic mechanisms of CSF1R‐related leukodystrophy.
Author Contributions
LZJ conceived the ideas. LZJ, WY, and LCZ designed the study, analyzed and interpreted data, and edited the manuscript. LYJ, XJM, and XY performed the experiments, collected and analyzed data, performed statistical analysis, and wrote the original draft manuscript. All the authors have read and approved the final version for publication. This work was supported by grants from the National Natural Science Foundation of China to Zhijun Liu (82101504), and grants from the National Natural Science Foundation of China (32070961) to Chuanzhou Li.
Conflict of Interest
None of the authors has any conflict of interest to disclose.
Consent for Publication
All authors have read and approved the content and agreed to submit for consideration for publication in the journal.
Acknowledgements
The authors thank the participants for their help and support.
Funding Statement
This work was funded by National Natural Science Foundation of China grants 32070961 and 82101504.
Contributor Information
Chuanzhou Li, Email: chuanzhouli@hust.edu.cn.
Yan Wu, Email: wuyan_120@163.com.
Zhijun Liu, Email: liuzhijunhubei@163.com.
References
- 1. Helman G, Venkateswaran S, Vanderver A. The spectrum of adult‐onset heritable white‐matter disorders. Handb Clin Neurol. 2018;148:669‐692. [DOI] [PubMed] [Google Scholar]
- 2. Lynch DS, Wade C, Paiva ARB, et al. Practical approach to the diagnosis of adult‐onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry. 2019;90(5):543‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Resende LL, de Paiva ARB, Kok F, da Costa LC, Lucato LT . Adult leukodystrophies: a step‐by‐step diagnostic approach. Radiographics. 2019. Jan‐Feb;39(1):153‐168. [DOI] [PubMed] [Google Scholar]
- 4. Vanderver A, Simons C, Helman G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol. 2016;79(6):1031‐1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lynch DS, Rodrigues Brandao de Paiva A, Zhang WJ, et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain. 2017;140(5):1204‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vanderver A, Prust M, Tonduti D, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab. 2015;114(4):494‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonkowsky JL, Keller S, AAP Section on Neurology, Council on Genetics . Leukodystrophies in children: diagnosis, care, and treatment. Pediatrics. 2021;148(3):e2021053126. [DOI] [PubMed] [Google Scholar]
- 8. Biffi A, Cesani M, Fumagalli F, et al. Metachromatic leukodystrophy—mutation analysis provides further evidence of genotype‐phenotype correlation. Clin Genet. 2008;74(4):349‐357. [DOI] [PubMed] [Google Scholar]
- 9. van der Lei HD, van Berkel CG, van Wieringen WN, et al. Genotype‐phenotype correlation in vanishing white matter disease. Neurology. 2010;75(17):1555‐1559. [DOI] [PubMed] [Google Scholar]
- 10. Guerreiro R, Kara E, Le Ber I, et al. Genetic analysis of inherited leukodystrophies: genotype‐phenotype correlations in the CSF1R gene. JAMA Neurol. 2013;70(7):875‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parikh S, Bernard G, Leventer RJ, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab. 2015;114(4):501‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu ZJ, Lin HX, Wei Q, et al. Genetic Spectrum and variability in Chinese patients with amyotrophic lateral sclerosis. Aging Dis. 2019;10(6):1199‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee JM, Yang HJ, Kwon JH, et al. Two Korean siblings with recently described ovarioleukodystrophy related to AARS2 mutations. Eur J Neurol. 2017;24(4):e21‐e22. [DOI] [PubMed] [Google Scholar]
- 14. Zhan ZX, Liao XX, Du J, et al. Exome sequencing released a case of X‐linked adrenoleukodystrophy mimicking recessive hereditary spastic paraplegia. Eur J Med Genet. 2013;56(7):375‐378. [DOI] [PubMed] [Google Scholar]
- 15. Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK. CSF1R‐related leukoencephalopathy: a major player in primary microgliopathies. Neurology. 2018;91(24):1092‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tipton PW, Stanley ER, Chitu V, Wszolek ZK. Is pre‐symptomatic immunosuppression protective in CSF1R‐related leukoencephalopathy? Mov Disord. 2021;36(4):852‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu C, Sakai N, Taniike M, Inui K, Ozono K. Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype‐phenotype correlation. J Hum Genet. 2006;51(6):548‐554. [DOI] [PubMed] [Google Scholar]
- 18. Lynch DS, Zhang WJ, Lakshmanan R, et al. Analysis of mutations in AARS2 in a series of CSF1R‐negative patients with adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol. 2016;73(12):1433‐1439. [DOI] [PubMed] [Google Scholar]
- 19. Lakshmanan R, Adams ME, Lynch DS, et al. Redefining the phenotype of ALSP and AARS2 mutation‐related leukodystrophy. Neurol Genet. 2017;3(2):e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X, Wang Q, Tang H, et al. Novel alanyl‐tRNA synthetase 2 pathogenic variants in leukodystrophies. Front Neurol. 2019;10:1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dulski J, Heckman MG, White LJ, Zur‐Wyrozumska K, Lund TC, Wszolek ZK. Hematopoietic stem cell transplantation in CSF1R‐related leukoencephalopathy: retrospective study on predictors of outcomes. Pharmaceutics. 2022;14(12):2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shu Y, Long L, Liao S, et al. Involvement of the optic nerve in mutated CSF1R‐induced hereditary diffuse leukoencephalopathy with axonal spheroids. BMC Neurol. 2016;16:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu L, Liu J, Sha L, et al. Sporadic cases with novel mutations and pedigree in hereditary leukoencephalopathy with axonal spheroids. J Alzheimers Dis. 2017;56(3):893‐898. [DOI] [PubMed] [Google Scholar]
- 24. Chitu V, Gokhan S, Stanley ER. Modeling CSF‐1 receptor deficiency diseases—how close are we? FEBS J. 2022;289(17):5049‐5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo L, Ikegawa S. From HDLS to BANDDOS: fast‐expanding phenotypic spectrum of disorders caused by mutations in CSF1R. J Hum Genet. 2021;66(12):1139‐1144. [DOI] [PubMed] [Google Scholar]
- 26. Ishiguro T, Konno T, Hara N, et al. Novel partial deletions, frameshift and missense mutations of CSF1R in patents with CSF1R‐related leukoencephalopathy. Eur J Neurol. 2023. Mar;21:1861‐1870. [DOI] [PubMed] [Google Scholar]
- 27. Chitu V, Gokhan S, Nandi S, Mehler MF, Stanley ER. Emerging roles for CSF‐1 receptor and its ligands in the nervous system. Trends Neurosci. 2016;39(6):378‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elmore MR, Najafi AR, Koike MA, et al. Colony‐stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stables J, Green EK, Sehgal A, et al. A kinase‐dead Csf1r mutation associated with adult‐onset leukoencephalopathy has a dominant inhibitory impact on CSF1R signalling. Development. 2022;149(8):dev200237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leng C, Lu L, Wang G, et al. A novel dominant‐negative mutation of the CSF1R gene causes adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia. Am J Transl Res. 2019;11(9):6093‐6101. [PMC free article] [PubMed] [Google Scholar]
- 31. Oosterhof N, Kuil LE, van der Linde HC, et al. Colony‐stimulating factor 1 receptor (CSF1R) regulates microglia density and distribution, but not microglia differentiation in vivo. Cell Rep. 2018;24(5):1203‐1217.e6. [DOI] [PubMed] [Google Scholar]
- 32. Irahara‐Miyana K, Enokizono T, Ozono K, Sakai N. Exonic deletions in GALC are frequent in Japanese globoid‐cell leukodystrophy patients. Hum Genome Var. 2018;5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szymanska K, Lugowska A, Laure‐Kamionowska M, et al. Diagnostic difficulties in Krabbe disease: a report of two cases and review of literature. Folia Neuropathol. 2012;50(4):346‐356. [DOI] [PubMed] [Google Scholar]