

Abstract
Objective
To describe demographic, clinical, and radiographic features of tumefactive demyelination (TD) and identify factors associated with severe attacks and poor outcomes.
Methods
Retrospective review of TD cases seen at Mayo Clinic, 1990–2021.
Results
Of 257 patients with TD, 183/257 (71%) fulfilled the 2017 multiple sclerosis (MS) McDonald criteria at the last follow‐up, 12/257 (5%) had myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD), 0 had aquaporin‐4‐IgG seropositive neuromyelitis optic spectrum disorders (AQP4+ NMOSD), and 62/257 (24%) were cryptogenic. Onset before age 18 was present in 18/257 (7%). Female to male ratio was 1.3:1. Cerebrospinal fluid oligoclonal (CSF) bands were present in 95/153 (62%). TD was the first demyelinating attack in 176/257 (69%). At presentation, 59/126 (47%) fulfilled Barkhof criteria for dissemination in space, 59/100 (59%) had apparent diffusion coefficient (ADC) restriction, and 57/126 (45%) had mass effect. Despite aggressive clinical presentation at onset, 181/257 (70%) of patients remained fully ambulatory (Expanded Disability Status Scale [EDSS] ≤4) after a 3.0‐year median follow‐up duration. Severe initial attack‐related disability (EDSS ≥4) was more common in patients with motor symptoms (81/143 vs. 35/106, p < 0.0001), encephalopathy (20/143 vs. 2/106, p < 0.0001) and ADC restriction on initial MRI (42/63 vs. 15/33, p = 0.04). Poor long‐term outcome (EDSS ≥4) was more common in patients with older onset age (41.9 ± 15 vs. 36.8 ± 15.6, p = 0.02) and motor symptoms at onset (49/76 vs. 66/171, p < 0.0001).
Interpretation
Most TD patients should be considered part of the MS spectrum after excluding MOGAD and NMOSD. Motor symptoms and older age at presentation portend a poor outcome.
Introduction
The term “tumefactive demyelination” (TD) was first coined in 1979 by van der Velden et al., when they encountered a patient with a TD lesion (TDL) on unenhanced head computerized tomography (CT) in which biopsy was consistent with multiple sclerosis (MS). 1 Since then, the nomenclature of TD has been varied in the literature and included terms such as pseudotumoral demyelinating lesions, tumefactive or tumorlike MS (TMS), and tumor‐like demyelinating lesions. 2 The adjusted incidence of TD is 0.56 per 100,000, and it occurs in 1.9% of the MS population. 3 Although TD lesions (TDL) are most commonly associated with MS, they can be seen in other inflammatory conditions such as myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD), aquaporin‐4‐IgG seropositive neuromyelitis optic spectrum disorders (AQP4+ NMOSD), and acute demyelinating encephalomyelitis (ADEM). 4 , 5 , 6 In addition, there is clinical evidence that Balo concentric sclerosis (BCS) and Marburg disease commonly occur as tumefactive variants of MS. 7 , 8 These terms have been often used interchangeably, highlighting the diagnostic challenge, elusive pathophysiology, and overlapping features of these entities. The detection of TDL frequently leads to a diagnostic dilemma, particularly in patients without previously known underlying demyelinating disease. 9 Our group has previously shown that histopathology was initially misinterpreted as a non‐demyelinating etiology in 31% of biopsy‐proven MS. 10 Consequently, misdiagnosis can lead to morbidity from brain biopsy and other procedures, delaying appropriate treatment and potentially worsening the clinical outcome. This study aims to describe the demographic, clinical, and radiological features of tumefactive demyelination and identify prognostic features to aid in the acute treatment of these conditions.
Methods
Study design and case ascertainment
The study conforms with the WMA Declaration of Helsinki. Patients or their authorized representatives provided informed written consent for using their medical information. We identified patients by searching medical records from January 1990 to December 2021 for all potentially relevant diagnostic codes using the Mayo Clinic Advanced Cohort Explorer (ACE) Tool. ACE is a clinical data extraction tool that queries multiple sources of patient information such as patient demographics, diagnosis, hospital notes, laboratory reports, flowsheets, radiology reports, and clinical notes. Study subjects were identified by using diagnostic codes related to multiple sclerosis, demyelination, transverse myelitis and optic neuritis, and searching within the free text of clinical notes and radiology reports for terms related to tumefactive multiple sclerosis, Balo concentric sclerosis, Marburg disease and Schilder disease. Over 700 patients were reviewed and patients with the final diagnosis of vasculitis, abscess or different type of CNS malignancies were excluded. Patients were included if they had a clinical history comprising a demyelinating event along with a cerebral magnetic resonance imaging (MRI) showing one or more large demyelinating plaques (minimum transverse diameter of ≥10 mm).
Baseline demographic and clinical data
For each enrolled patient, the following baseline data were collected: (i) sex; (ii) age at onset of index attack (attack leading to the first tumefactive lesion); (iii) comorbidities; (iv) the clinical presentation at onset according to patient clinical records, by verifying involvement of motor, sensitive, cerebellar, brainstem, visual, sphincter and functional cognitive systems; (v) acute treatments (i.e., high‐dose steroids, plasma exchange [PLEX], intravenous immunoglobulin[IVIGs]); (vi) Expanded Disability Status Scale (EDSS) at the index attack 11 ; and (vii) diagnosis at discharge based on the most recent criteria. 12 , 13 , 14 , 15 Patients who presented with TDL and did not meet the criteria for MS, MOGAD, or AQP4 positive NMOSD were classified as TD. TD includes clinically isolated syndrome (CIS), radiologically isolated syndrome (RIS), and ADEM. Pediatric onset tumefactive disease (POTD) was defined as the first tumefactive attack before 18 years of age.
Baseline MRI data
Neuroimaging features were reviewed regarding (i) lesion location; (ii) lesion size; (iii) lesion number on fluid‐attenuated inversion recovery (FLAIR) images; (iv) number of gadolinium (Gd)‐enhancing lesions; (v) mass effect; (vi) contrast enhancement pattern; (vii) apparent diffusion coefficient (ADC) pattern; (viii) T2‐hypointense rim; (ix) T1‐ hypointensity; and (x) fulfillment of Barkhof criteria. 16
Baseline CSF and biomarkers data
Data related to cerebrospinal fluid (CSF) analysis (biochemical analysis, cell count, isoelectrofocusing for immunoglobulin G oligoclonal bands [OCBs] detection) were collected. In a subset of cases, serum was tested for MOG antibodies by live cell‐based assay. 13 Serum in these cases was also tested for aquaporin‐4 (AQP‐4) antibody by live cell‐based assay, inactivated cell‐based assay, ELISA, or tissue immunofluorescence as previously. 17
Follow‐up characteristics
For each patient, we collected the length of follow‐up and characteristics including (i) The last available EDSS score. (ii) Disease‐modifying therapies (DMTs) that were started, continued, or modified over the follow‐up period. DMTs were categorized into: (a) mild/moderate efficacy DMTs, including interferon‐β (all formulations), and glatiramer acetate (all formulations), dimethyl fumarate, teriflunomide; or (ii) high‐efficacy DMTs, including natalizumab, fingolimod, alemtuzumab and CD20 monoclonal antibody treatments (Ocrelizumab, Rituximab).
Statistical analysis
Continuous variables were reported as median, range, and interquartile range (IQR), whereas categorical variables were reported as numbers and percentages. The Kruskal‐Wallis and Mann–Whitney tests were performed to test the difference between median values and the chi‐square and Fisher exact tests were used to test the difference in the distribution of categoric variables. Chi‐square and Kruskal‐Wallis were performed to test for an association between baseline characteristics and attack severity and risk of reaching an EDSS score ≥4 at the last follow‐up. For all tests, significance was set at p < 0.05. The most recent EDSS was used to compare the Olmsted County MS cohort population with our cohort.
Results
Demographic and Clinical Characteristics
We included 257 patients with TDL (Table 1). The female: male ratio was 1.3:1. A tumefactive lesion was the first clinical attack in 176/257 (69%). Some data from 75 of these patients were included in a prior publication from our group. 10 The most common clinical presentation at onset was motor dysfunction (119/257, 46%), followed by sensory dysfunction (95/257, 37%), cerebellar dysfunction (43/257, 17%), and visual field loss (23/257, 14%) (Fig. 1). Prior to the index attack, prodromal symptoms, such as malaise and headache, were present in 66/257 (26%) patients. In contrast to typical MS attacks, the duration of the clinical attack relating to the tumefactive lesion was prolonged (>12 weeks) in over a third of patients (37%). One in five patients was suspected to have a malignancy at their initial presentation. Brain biopsy was performed in 77/257 (30%), which was consistent with TD in all cases.
Table 1.
Overall characteristics of patients with tumefactive demyelination.
| Characteristic | All TD (n = 257) | TMS (n = 183) | TD (n = 62) | MOGAD (n = 12) |
|---|---|---|---|---|
| Female | 147 (57%) | 101 (55%) | 39 (63%) | 7 (58%) |
| White race | 205 (89%) | 146 (90%) | 52 (91%) | 7 (64%) |
| Mean age at index attack ± SD | 38.5 ± 15.39 | 37.3 ± 13.98 | 45.5 ± 15.74 | 20.2 ± 15.55 |
| Prodromal symptoms | ||||
| None | 191 (74%) | 140 (76%) | 49 (79.0%) | 2 (17%) |
| Fever | 20 (8%) | 9 (5%) | 5 (8%) | 6 (50.0%) |
| Malaise | 13 (5%) | 5 (3%) | 3 (5%) | 5 (42%) |
| Headache | 47 (18%) | 33 (18.0%) | 8 (13%) | 6 (50.0%) |
| Recent vaccination | 3 (1%) | 1 (0.5%) | 1 (2%) | 1 (8%) |
| Pregnancy | 2 (1%) | 2 (1%) | 0 (0.0%) | 0 (0.0%) |
| Index attack as a first demyelinating attack | 176 (69%) | 109 (60%) | 57 (93%) | 10 (83%) |
| Initial diagnosis | ||||
| Inflammatory demyelination | 182 (70%) | 136 (74%) | 37 (60%) | 9 (75%) |
| Malignancy | 54 (21%) | 35 (19%) | 18 (29%) | 1 (8%) |
| Vasculitis | 8 (3%) | 7 (4%) | 1 (1%) | 0 (0%) |
| Infection | 13 (5%) | 5 (3%) | 6 (9.5%) | 2 (16%) |
| History of pre‐existing autoimmune disorder | 20 (8%) | 16 (9.5%) | 2 (3%) | 2 (18%) |
| Duration of index attack | ||||
| <2 weeks | 24 (13%) | 18 (13%) | 4 (9%) | 2 (29%) |
| 2–6 weeks | 54 (29%) | 43 (31%) | 8 (18%) | 3 (43%) |
| 6–12 weeks | 40 (21%) | 31 (23%) | 9 (20%) | 0 (0.0%) |
| >12 weeks | 69 (37%) | 44 (32%) | 23 (52%) | 2 (29%) |
MOGAD, myelin oligodendrocytes glycoprotein antibody‐associated disease; N, number; SD, standard deviation; TD, tumefactive demyelination; TMS, tumefactive multiple sclerosis.
Figure 1.
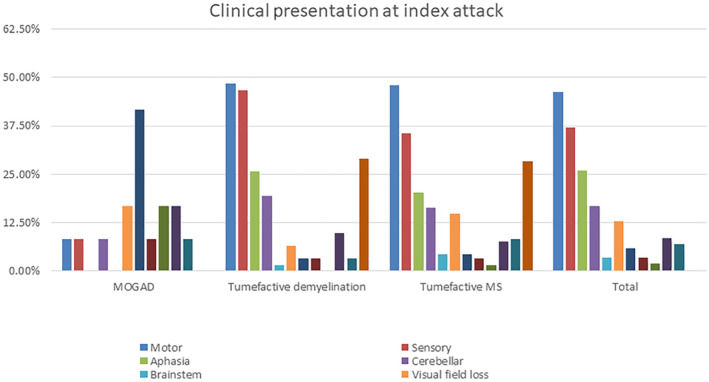
Frequency of clinical presentation at the index attack. In the TD and TMS groups, motor and sensory symptoms were the most common presentation followed by aphasia and cerebellar symptoms. In the MOGAD group, optic neuritis and visual field loss were the most common presentation followed by myelitis and encephalopathy.
Treatment and outcome are summarized in Table 2. Patients were followed for a median of 3 years (range: 0–28.9). At the last follow‐up, 183/257 (71%) fulfilled the McDonald criteria for MS, 12/257 (5%) were diagnosed as MOGAD, and 62/257 (24%) did not meet the criteria for MS, MOGAD, or NMOSD. The median (range) EDSS score at index attack was 4 (0–9.5). At the last follow‐up, the median (range) EDSS score was 2.5 (0–10) and 26/257 (10%) had died. The course of the disease was relapsing remitting (RR) in 169/257 (66%), secondary progressive (SP) 19/257 (7%), primary progressive (PP) in 2/257 (0.8%), and monophasic in 62/257 (24%). In patients for whom sufficient data were available, 46/124 (37%) had recurrent tumefactive disease. Out of 19 patients with a SP course, 11/19 (58%) evolved into the progressive phase in less than 5 years and 8/19 (42%) in more than 5 years of follow‐up. One of the PP cases was a 34‐year‐old female who presented with a seizure and a tumefactive lesion in the temporal lobe, with subsequent progressive clinical course. Another PP case was a 60‐year‐old male who presented with right progressive hemiparesis and tumefactive lesion in the left parietal lobe with enhancement on brain MRI. A brain biopsy was performed, and pathology was interpreted as demyelination. Follow‐up MRI 2 months later showed an increase in the mass size and surgical resection was performed, with pathology again being consistent with demyelination.
Table 2.
Clinical and CSF findings of patients with tumefactive demyelination.
| Characteristic | All TD (n = 257) | TMS (n = 183) | TD (n = 62) | MOGAD (n = 12) |
|---|---|---|---|---|
| Median (range) duration of follow‐up, years | 3.0 (0–28.9) | 3.4 (0–28.9) | 2.2 (0.1–28.7) | 2.8 (0.1–10.1) |
| Median EDSS at index attack (range) | 4 (0–9.5) | 4 (0–9.5) | 4 (0–9) | 4.5 (0–9) |
| Median (range) EDSS at the last follow‐up | 2.5 (0–10) | 2.5 (0–10) | 3 (0–10) | 1 (0–10) |
| CSF findings (n = 153) | ||||
| Elevated protein, No (%) | 71 (46%) | 50 (48%) | 15 (37%) | 6 (60%) |
| Elevated cell count, No (%) | 52 (34%) | 36 (35%) | 9 (22%) | 7 (78%) |
| Positive oligoclonal band, No (%) | 95 (62%) | 76 (62%) | 18 (42%) | 1 (9%) |
| Presence of MOG‐IgG, No (%) | 14 (5%) | 1 (0.5%) | 1 (1%) | 12 (100%) |
| Index attack treatment | ||||
| No treatment | 41 (16%) | 31 (17%) | 9 (14%) | 1 (8%) |
| Corticosteroid | 210 (82%) | 149 (81%) | 51 (82%) | 10 (83%) |
| PLEX | 55 (21%) | 39 (21%) | 14 (22%) | 2 (17%) |
| Cyclophosphamide | 14 (5%) | 11 (6%) | 3 (5%) | 0 (0.0%) |
| Mitoxantrone | 6 (2%) | 4 (2%) | 1 (1%) | 1 (8%) |
| IVIG | 11 (4%) | 5 (3%) | 3 (5%) | 3 (25%) |
| Disease‐modifying therapy | ||||
| Mild/moderate efficacy DMTs | 113 (44.0%) | 107 (58%) | 5 (8%) | 1 (8%) |
| High‐efficacy DMTs | 56 (22%) | 45 (24%) | 9 (14%) | 2 (17%) |
Mild/moderate efficacy DMTs: interferon‐β (all formulations), and glatiramer acetate (all formulations), dimethyl fumarate, teriflunomide. High‐efficacy DMTs: natalizumab, fingolimod, alemtuzumab and CD20 monoclonal antibody treatments (Ocrelizumab, Rituximab).
DMT, disease‐modifying therapy; MOGAD, myelin oligodendrocytes glycoprotein antibody‐associated disease; N, number; PLEX, plasma exchange; SD, standard deviation; TD, tumefactive demyelination; TMS, tumefactive multiple sclerosis.
CSF findings
CSF analysis was performed on 153/257 (59%) patients. CSF Protein was >45 mg/dl in 71/153 (46%), nucleated cell count was >5 cells per mm3 in 52/153 (34%). Two or more CSF unique oligoclonal bands (OCBs) were demonstrated in 95/153 (62%) patients. Serum AQP4‐IgG was tested in 106/153 (69%) patients with no positive result and MOG‐IgG in 63/153 (41%) patients with 14/63 (22%) positive results (Table 2).
Pediatric onset tumefactive demyelination (POTD)
Table 3 summarizes the clinical and imaging features of POTD patients. And 18/257 (7%) patients presented with TDL before the age of 18 years. The mean age at onset was 9.3 ± 5.7 years. Prodromal symptoms were present on 11/18 (61%). The tumefactive lesion was the first demyelinating event in 16/18 (89%). In 9/18 (50%) the onset was polysymptomatic. At the index attack, the most common presentation was motor symptoms 7/18 (39%), followed by sensory dysfunction 5/18 (28%), encephalopathy 5/18 (28%), and cerebellar dysfunction 4/18 (22%). The median duration of follow‐up was 3.4 years (range: 0.1–28.7). The median (range) EDSS at the index attack was 4 (0–9) and improved to 0 (0–10) at the last follow‐up. And 10/18 (56%) were diagnosed as MS, 6/18 (33%) had MOGAD and 2/18 (11%) had TD. CSF analysis showed elevated protein in 5/15 (33%), elevated cell count in 7/15 (47%), and positive OCBs in 3/15 (20%). The duration of the index attack and the time to the first sign of improvement were less than 2 weeks on 7/14 (50%) and 10/14 (71%), respectively. And 14/18 (78%) were initially commenced on high‐dose corticosteroid. 6/18 (33%) were treated with second‐line treatments such as PLEX, IVIG, and mitoxantrone.
Table 3.
Demographic, clinical and neuroimaging features of patients with pediatric‐onset tumefactive demyelination (n = 18).
| Characteristic | All TD (n = 18) | TMS (n = 10) | TD (n = 2) | MOGAD (n = 6) |
|---|---|---|---|---|
| Female | 9 (50%) | 6 (60%) | 0 (0.0%) | 3 (50%) |
| White race | 11 (69%) | 5 (56%) | 2 (100%) | 4 (80%) |
| Mean age at index attack ± SD | 9.3 ± 5.6 | 9.6 ± 6.4 | 14.4 ± 4.8 | 7.2 ± 3.7 |
| Prodromal symptoms | 11 (61%) | 5 (56%) | 2 (100%) | 4 (80%) |
| Median (range) of follow‐up, years | 3.4 (0.1–28.7) | 3.4 (0.4–12.8) | 14.6 (0.5–28.7) | 3.6 (0.1–10.1) |
| Median EDSS at index attack ± SD | 4 (0–9) | 4 (2.5–6) | 5 (4–6.5) | 4.5 (0–9) |
| Median EDSS at the last follow‐up | 0 (0–10) | 1 (0–7) | 2.5 (0–5) | 0 (0–10) |
| Index attack as a first demyelinating attack Yes (%) | 16 (89%) | 8 (80%) | 2 (100%) | 6 (100%) |
| CSF findings | ||||
| Elevated protein | 5 (33%) | 2 (22%) | 1 (100%) | 2 (40%) |
| Elevated cell count | 7 (47%) | 3 (33%) | 0 (0.0%) | 4 (80%) |
| Positive oligoclonal band | 3 (20%) | 2 (22%) | 1 (100%) | 0 (0.0%) |
| Presence of MOG‐IgG | 4 (33%) | 1 (14%) | 0 (0.0%) | 3 (75%) |
| Focus of index lesion | ||||
| Frontal | 1 (8%) | 0 (0.0%) | 0 (0.0%) | 1 (20%) |
| Parietal | 3 (25%) | 2 (33%) | 1 (100%) | 0 (0.0%) |
| Occipital | 1 (8%) | 1 (17%) | 0 (0.0%) | 0 (0.0%) |
| Cerebellum | 4 (33%) | 2 (33%) | 0 (0.0%) | 2 (40%) |
| Brainstem | 2 (17%) | |||
| Barkhof criteria fulfilled | 7 (64%) | 3 (60%) | 1 (100%) | 3 (60%) |
| Presence of ADC restriction | 3 (30%) | 3 (60%) | 0 (0%) | 0 (0%) |
ADC, apparent diffusion coefficient; EDSS, Expanded Disability Status Scale; MOGAD, myelin oligodendrocytes glycoprotein antibody‐associated disease; N, number; TD, tumefactive demyelination; TMS, tumefactive multiple sclerosis.
Neuroimaging
Good‐quality initial MRI imaging was available in 126 patients. The remainder were identified by radiology report and/or pathology report, when the initial MRI was of insufficient quality or not available. All subjects had at least one MRI to review. A solitary TDL was present in 56/126 (44%), BCS in 21/126 (17%), and multiple tumefactive lesions in 49/126 (39%) (Table 4, Fig. 2). Lesions were mainly located in the frontal lobe 54/126 (43%), followed by parietal 39/126 (31%), occipital 15/126 (12%), temporal lobes 12/126 (9%), cerebellum 9/126 (7%), and brainstem 7/126 (5%). At the initial presentation, 59/126 (47%) patients met the Barkhof criteria for dissemination in space. “Butterfly lesions,” bihemispheric lesions crossing the corpus callosum, were present in 9/126 (7%) (Fig. 2a‐c). A hypointense T2W rim was observed in 30/126 (24%) patients. T1 hypointensity in the acute lesion was seen in 106/126 (84%) patients. ADC imaging was available in 100 patients. ADC restriction was seen in 59/100 (59%). Post‐gadolinium enhancement was present in 106/126 (84%) TDL, with central enhancement in 30/126 (24%), incomplete open‐ring enhancement in 26/126 (21%), heterogeneous pattern in 47/126 (37%), and concentric ring enhancement in 8/126 (6%).
Table 4.
Radiological characteristics of initial MRI imaging in patients with tumefactive demyelination.
| Characteristics | All TD | TMS | TD | MOGAD | p value 1 |
|---|---|---|---|---|---|
| Solitary lesion | 56 (44%) | 33 (42%) | 20 (56%) | 3 (27%) | 0.06 |
| Balo lesion | 21 (17%) | 17 (21%) | 4 (11%) | 0 (0%) | |
| Multiple tumefactive lesion | 49 (39%) | 29 (37%) | 12 (33%) | 8 (73%) | |
| Butterfly lesion | 9 (7%) | 3 (4%) | 4 (11%) | 2 (20%) | 0.1 |
| Mean index lesion size ± SD (mm) | 20.3 ± 7.0 | 20.0 ± 6.8 | 21.4 ± 8 | 19.2 ± 5.7 | 0.7 |
| Focus of index lesion | |||||
| Frontal | 54 (42%) | 37 (47%) | 16 (43%) | 1 (9%) | 0.06 |
| Parietal | 39 (31%) | 27 (34%) | 11 (30%) | 1 (9%) | 0.2 |
| Temporal | 12 (9%) | 5 (6%) | 4 (11%) | 3 (27%) | 0.08 |
| Occipital | 15 (12%) | 8 (10%) | 6 (16%) | 1 (9%) | 0.6 |
| Cerebellum | 9 (7%) | 6 (8%) | 1 (3%) | 2 (18%) | 0.2 |
| Brainstem | 7 (5%) | 4 (5%) | 1 (3%) | 2 (18%) | 0.1 |
| Mass effect | 0.5 | ||||
| None | 68 (54%) | 44 (56%) | 16 (43%) | 8 (80%) | |
| Mild | 43 (34%) | 25 (32%) | 16 (43%) | 2 (20%) | |
| Moderate | 13 (10%) | 8 (10%) | 5 (13%) | 0 (0%) | |
| Severe | 1 (1%) | 1 (1%) | 0 (0%) | 0 (0%) | |
| Number of enhancing lesions ± SD | 2.9 ± 3.8 | 3.3 ± 4.1 | 2.4 ± 3.2 | 1.7 ± 2.5 | 0.13 |
| Number of FLAIR lesion ± SD | 4.7 ± 3.3 | 5.1 ± 3.6 | 3.4 ± 2.5 | 5.6 ± 2.34 | 0.01 |
| Fulfillment of Barkhof criteria | 59 (48%) | 41 (53%) | 13 (35%) | 5 (50%) | 0.2 |
| T2 hypointense rim | 30 (24%) | 8 (22%) | 20 (26%) | 2 (20%) | 0.87 |
| T1 hypointensity | 106 (88%) | 68 (89%) | 31 (89%) | 7 (70%) | 0.2 |
| ADC restriction | 59 (59%) | 38 (59%) | 19 (68%) | 2 (25%) | 0.09 |
ADC, apparent diffusion coefficient; FLAIR, Fluid‐attenuated inversion recovery; MOGAD, myelin oligodendrocytes glycoprotein antibody‐associated disease; N, number; TD, tumefactive demyelination; TMS, tumefactive multiple sclerosis.
Chi‐Square p‐value and Kruskal‐Wallis between TMS, TD and MOGAD groups.
Figure 2.
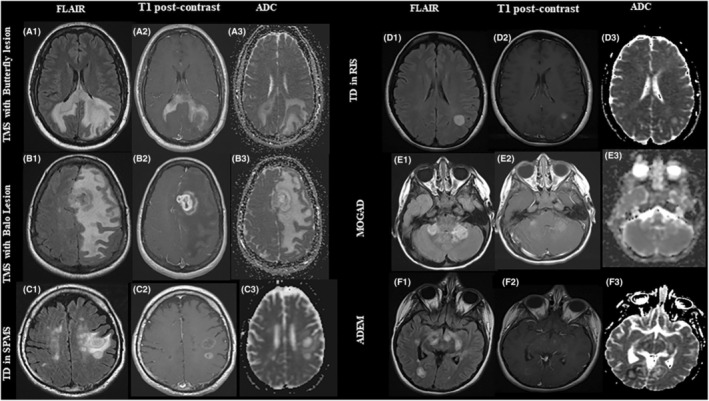
Examples of tumefactive brain lesions. A 35‐year‐old male presented with seizure and visual field loss (A1–A3). Brain MRI axial FLAIR demonstrates a large heterogeneous left hemisphere lesion with extensive edema and prominent involvement of the corpus callosum (A1) with heterogenous contrast enhancement on axial‐ T1 post‐contrast MRI (A2). Apparent diffusion coefficient (ADC) indicated mild diffusion restriction, predominantly along the margin of the T2 hyperintensity in the frontal and parietal lobes (A3). (B) A 38‐year‐old female presented with aphasia (B1–B3). MRI brain axial FLAIR sequence indicates concentric rings with severe edema (B1) and irregular rim enhancement in the left frontal lobe on axial‐T1‐post contrast (B2). No associated restriction on ADC sequence (B3). (C) A 61‐ year‐old male with SPMS presented with cognitive decline developed multiple tumefactive lesions on brain MRI axial FLAIR images in frontal lobe and temporal lobe (C1) with complete and incomplete ring enhancement on axial T1‐post contrast images (C2) and peripheral ADC restriction (C3). (D) A 40‐year‐old female presented with veered feeling in her arm and accidentally found to have a tumefactive lesion in the frontal lobe on brain MRI axial FLAIR images (D1) with minimal enhancement on axial T1‐post contrast image (D2) and no associated restricted diffusion (D3). (E) A 22‐year‐old male developed weakness in his legs following mononucleosis. Patchy area of increased signal on brain MRI axial and FLAIR images in both cerebral hemispheres (E1) with minimal heterogenous enhancement (E2) and no associated restricted diffusion (E3). (F) A 13‐ year‐old female presented with difficulty walking and multiple tumefactive lesion. Brain MRI FLAIR demonstrates a large tumefactive T2‐hyperintense lesion in the temporal and occipital lobes (F1) with an incomplete ring enhancement on axial T1‐post contrast image (F2) and a peripheral restricted diffusion (F3).
Acute phase treatments
Treatments included corticosteroids (210/257, 82%) plasma exchange (PLEX) (55/257, 21%) and IVIG (11/257, 4%). Patients who did not respond to corticosteroids or PLEX were treated with cyclophosphamide 14/257 (5%) or mitoxantrone 6/257 (2%), as described in our previous report (Table 2). 18
Disease‐modifying therapies
Disease‐modifying therapies (DMTs) were initiated in 124/257 (48%) after index attack. Mild to moderate efficacy DMTs were prescribed in 113/257 (44%), while 56/257 (22%) were treated with high efficacy therapies. 11/257 (4%) were on treatment with DMTs at the time of the index attack.
Characteristics associated with severe attacks and long‐term outcome
Severe deficit at the initial attack (EDSS ≥4) was more common in patients with motor symptoms at onset (81/143 [57%] vs. 35/106 [33%], p < 0.0001), encephalopathy (20/143 [14%] vs. 2/106 [2%], p < 0.0001), multiple TD lesions compared with solitary or Balo lesions (35/75 [47%] vs. 12/46 [26%], p = 0.04), and ADC restriction on the initial brain MRI (42/63 vs. 15/33, p = 0.04).
Poor long‐term outcome (EDSS ≥4 at last follow‐up) was more common in patients with an older age of onset (41.9 ± 15 vs. 36.8 ± 15.6, p = 0.02), and in patients with motor symptoms at onset (49/76 vs. 66/171, p < 0.0001). Treatment with high‐efficacy DMTs was associated with better motor outcomes (EDSS <4) (24/76 [32%] vs. 30/171 [17%], p = 0.01). No patient with tumefactive MOGAD had an EDSS ≥4 at last follow up. Other characteristics evaluated were not significantly associated with either severe attacks or worse long‐term outcomes (Table 5 ).
Table 5.
Features associated with EDSS score.
| Characteristics | EDSS ≥4 at index attack (n = 143) | EDSS <4 at the index attack (n = 106) | p‐value | EDSS ≥4 at the last follow‐up (n = 76) | EDSS <4 at the follow‐up (n = 171) | p‐value |
|---|---|---|---|---|---|---|
| Gender | ||||||
| Female | 82 (57%) | 61 (57%) | 0.9 | 46 (60%) | 75 (44%) | 0.5 |
| Male | 61 (43%) | 45 (42%) | 30 (39%) | 96 (56%) | ||
| Mean age at the onset ± SD | 38.4 ± 16.6 | 38.7 ± 14.3 | 0.7 | 41.9 ± 15 | 36.8 ± 15.6 | 0.02 |
| Index attack as a first demyelinating attack | 93 (65%) | 78 (75%) | 0.09 | 54 (71%) | 117 (70%) | 0.8 |
| First neurological symptoms at index attack | ||||||
| Motor | 81 (57%) | 35 (33%) | <0.0001 | 49 (64%) | 66 (38%) | <0.0001 |
| Sensory | 49 (34%) | 42 (40%) | 0.4 | 26 (34%) | 38 (22%) | 0.51 |
| Aphasia | 32 (22%) | 18 (17%) | 0.3 | 15 (20%) | 27 (16%) | 0.66 |
| Cerebellar | 26 (18%) | 16 (15%) | 0.5 | 12 (16%) | 4 (2%) | 1.00 |
| Brainstem | 3 (2%) | 6 (6%) | 0.1 | 4 (5%) | 20 (12%) | 0.23 |
| Visual field loss | 18 (13%) | 15 (14%) | 0.7 | 13 (17%) | 11 (6%) | 0.24 |
| Optic neuritis | 9 (6%) | 5 (5%) | 0.6 | 3 (4%) | 4 (2%) | 0.43 |
| Bladder/bowel/sexual | 7 (5%) | 1 (1%) | 0.08 | 4 (5%) | 4 (2%) | 0.23 |
| Myelitis | 3 (2%) | 2 (2%) | 0.90 | 1 (1%) | 4 (2%) | 0.6 |
| Encephalopathy | 20 (14%) | 2 (2%) | <0.0001 | 8 (10%) | 14 (8%) | 0.55 |
| Seizure | 6 (4%) | 10 (9%) | 0.09 | 6 (8%) | 11 (6%) | 0.67 |
| Positive oligoclonal band | 51 (54%) | 41 (53%) | 0.82 | 31 (61%) | 59 (50%) | 0.21 |
| Presence of MOG‐IgG | 7 (21 T) | 3 (11%) | 0.26 | 0 (0.0%) | 12 (26%) | 0.04 |
| Focus of index lesion | ||||||
| Frontal | 35 (46%) | 15 (32%) | 0.12 | 18 (46%) | 33 (40%) | 0.5 |
| Parietal | 26 (34%) | 13 (28%) | 0.45 | 12 (31%) | 25 (30%) | 0.9 |
| Temporal | 8 (10%) | 4 (8%) | 0.71 | 4 (10%) | 8 (10%) | 0.9 |
| Occipital | 6 (8%) | 9 (19%) | 0.06 | 3 (8%) | 12 (14%) | 0.3 |
| Cerebellum | 4 (5%) | 5 (11%) | 0.26 | 4 (10%) | 5 (6%) | 0.4 |
| Brainstem | 6 (8%) | 1 (2%) | 0.18 | 4 (10%) | 3 (4%) | 0.1 |
| Type of lesion | ||||||
| Solitary lesion | 27 (36%) | 27 (57%) | 0.4 | 19 (50%) | 36 (43%) | 0.2 |
| Balo lesion | 13 (17%) | 8 (17%) | 3 (8%) | 17 (20%) | ||
| Multiple tumefactive lesion | 35 (47%) | 12 (26%) | 16 (42%) | 30 (36%) | ||
| Presence of ADC restriction | 42/63 (67%) | 15/33 (45%) | 0.04 | 20/30 (67%) | 36/66 (54%) | 0.3 |
| Barkhof criteria fulfilled | 39/75 (52%) | 18/45 (40%) | 0.2 | 22/39 (56%) | 34/81 (42%) | 0.15 |
| Index attack treatment | ||||||
| No treatment | 17 (12%) | 23 (22%) | 0.03 | 5 (7%) | 33 (19%) | 0.01 |
| Steroid | 123 (86%) | 81 (76%) | 0.06 | 66 (87%) | 137 (80%) | 0.2 |
| PLEX | 42 (33%) | 8 (7%) | <0.001 | 31 (41%) | 22 (13%) | <0.001 |
| IVIG | 11 (7.7%) | 0 (0%) | <0.001 | 8 (10%) | 3 (2%) | <0.001 |
| Disease‐modifying treatment (DMT) | ||||||
| Mild‐to‐moderate | 67 (47%) | 41 (39%) | 0.1 | 36 (47%) | 73 (43%) | 0.5 |
| High | 34 (24%) | 19 (18%) | 0.2 | 24 (32%) | 30 (17%) | 0.01 |
ADC, apparent diffusion coefficient; DMT, disease‐modifying treatment; EDSS, Expanded Disability Status Scale; MOGAD, myelin oligodendrocytes glycoprotein antibody‐associated disease; N, number; SD, standard deviation; TD, tumefactive demyelination; TMS, tumefactive multiple sclerosis.
Comparison with a population‐based MS cohort
Data from the clinic‐based cohort were compared with a previously published population‐based cohort of patients from Olmsted County, MN, USA. 19 , 20 The population‐based cohort included 297 patients with MS and an average follow‐up of 19 years. EDSS scores were compared between groups and binned according to follow‐up duration of 5 years or less and more than 5 years (Fig. 3). In patients with 5 or fewer years follow up, there were no significant differences between EDSS in the Olmsted County cohort (median 2.8) versus TMS (median 2.8, p = 0.9), TD (median 3, p = 0.2), and MOGAD (median 2.5, p = 0.6). In cases with more than 5 years of follow‐up, there were no significant differences between the Olmsted County cohort (median 3) versus TMS (median 2.5, p = 0.08) and TD (median 2, p = 0.07). The median (range) EDSS was significantly lower in MOGAD group compared to the Olmsted County cohort (0 versus 3, p = 0.008). In addition, when TMS and TD were combined into one group, the median EDSS was lower in TMS/TD group when compared to the Olmsted County MS cohort (2.5 vs. 3, p = 0.02).
Figure 3.
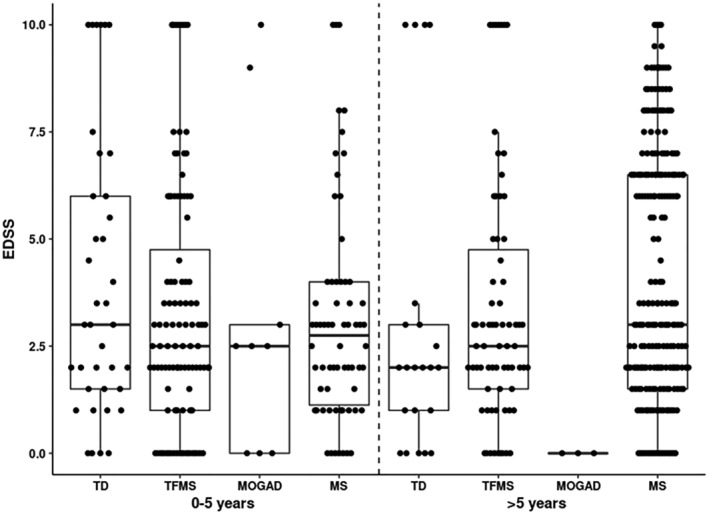
Follow‐up EDSS stratified by disease duration. In patients with 5 or fewer years of follow up, there were no significant differences in the median EDSS between the population‐based MS cohort (MS) (median 2.8) and the cohorts of Tumefactive MS (TMS) (median 2.8, p = 0.9), Tumefactive demyelination (TD) (median 3, p = 0.2), or myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD) (median 2.5, p = 0.7). In patients with more than five years of follow‐up, there were no significant differences between the population‐based MS cohort (median EDSS 3) and TMS (median 2.5, p = 0.08) or TD (median 2, p = 0.07). When the TMS and TD groups were combined, the median EDSS was lower in the TMS/TD combined group, when compared with the population‐based MS cohort (2.5 vs. 3, p = 0.02). EDSS was significantly lower in MOGAD group compared to population‐based cohort (median 0 versus 3; p = 0.008).
Discussion
Tumefactive demyelination represents an aggressive form of central nervous system demyelination and most commonly occurs due to multiple sclerosis. This case series describes the acute and long‐term prognostic features associated with worse motor outcomes. Despite an aggressive clinical presentation at the index attack, our findings suggest a relatively milder long‐term clinical motor outcome and progression conversion in TD compared with a population based MS cohort. 19 A monophasic course was observed in a quarter of patients, and the majority developed RRMS. Over two thirds of patients remained fully ambulatory (EDSS ≤4) after a mean follow‐up of 6 years. These favorable clinical outcomes are consistent with previous reports. 3 , 10 , 21 , 22 , 23 , 24
Motor symptoms at index attack were associated with a worse severity of index attack and worse long‐term outcomes. These findings may be partly explained by the fact that the outcome assessed by EDSS is heavily biased toward ambulation, particularly at higher EDSS. We have previously demonstrated the efficacy of cyclophosphamide for the treatment of tumefactive multiple sclerosis. 18 However, appropriate selection of patients for cyclophosphamide use is challenging. The presence of features associated with the poor long‐term outcome should prompt discussion of cyclophosphamide use or an alternative induction therapy if the patient does not have a sufficient clinical response to corticosteroids or plasma exchange.
ADC restriction on brain MRI was associated with increased severity of the index attack. The etiology of the restricted diffusion in these MS lesions is unknown, and multiple hypotheses have been raised. First, the myelin fragments and reduced fiber tract organization may restrict water diffusion 25 ; Second, based on immunopathology studies of acute MS lesions, restricted diffusion might reflect the activated microglia and oligodendrocyte apoptosis before infiltration of blood‐borne inflammatory cells. 26 , 27 Third, physical clustering of microglia and release of excitotoxins lead to oligodendrocyte injury with associated cytotoxic edema. 28 Finally, proinflammatory cascades in acute MS inflammation can result in cell death through mitochondrial dysfunction and decreased energy production. Since cell death is an irreversible process and ADC restriction at the index attack was not associated with the poor outcome at the last follow‐up; therefore, the first and the third hypothesis might explain our observation in TD population.
Our data support prior reports from our center that showed a slight female preponderance. 3 , 10 , 29 , 30 The average age at TD onset in the present cohort was 38, similar to previous reports and consistent with typical MS. 3 , 10 , 29 , 31 Older age at TD onset was associated with a worse prognosis, similar to that seen in typical MS. 32 , 33 In our cohort, 69% presented with their first demyelinating attack highlighting the diagnostic challenge at presentation. In addition to brain MRI, spinal fluid analysis, spinal cord imaging, serum MOG and AQP4 antibody testing, and if practical, OCT can be performed to aid in clinical diagnosis. A trial of plasma exchange with a short‐interval MRI can also be considered, as non‐inflammatory etiologies, such as glial tumor or lymphoma, are not expected to respond to plasma exchange. Ring‐enhancing lesions on brain MRI with peripheral diffusion restriction and rapid ADC pattern changes are more common in TD than in tumors or abscesses. 25 Brain FDG PET has also been proposed to discriminate malignant lesions from inflammatory brain lesions. 34 In 61%, TD onset was characterized by the involvement of more than one functional system, with motor and sensory predominance. Also, despite the typical symptoms and signs of MS in the present series, atypical presentations, including seizure, abulia, and aphasia, were observed. In contrast, typical MS more frequently presents with the involvement of a single functional system. This supports previous reports showing the clinical presentation of TD is often polysymptomatic given the size, location, and potential mass effect of the lesion. 3 , 10 , 22 , 29 , 35
Our results suggest most patients with TDL should be considered a part of the MS spectrum with other inflammatory CNS demyelinating conditions, such as MOGAD and NMOSD, representing a smaller subset. Despite the early aggressive course in our cohort, its long‐term course was comparable to typical MS, and more than 70% of patients remain fully ambulatory at 6 years. Motor and encephalopathy symptoms at the index attack, ADC restriction, and brainstem lesions were associated with severe index attack. Younger age at onset, non‐motor clinical presentation, and MOG‐IgG presence are favorable prognostic factors, consistent with prior studies. 36 , 37
The main limitation of this study is its retrospective nature. It is possible that some cases of MOGAD were misclassified as MS, due to the lack of availability of MOG‐IgG testing. All cases were reviewed, and diagnosis was confirmed based on the most recent diagnostic criteria for each of multiple sclerosis, NMOSD and MOGAD. NMO‐IgG and MOG‐IgG were tested retrospectively in all patients for whom had samples available. MOGAD was the most likely to be misidentified due to the recency of the availability of the clinical antibody testing and the published criteria, however the MRI features of tumefactive MOGAD appear quite distinct from tumefactive MS. Although EDSS‐assessed disability assessed seems promising, the long‐term cognitive outcome is uncertain. The EDSS focuses on ambulation and may not measure MS‐related disabilities in the upper limb or cognitive functions (e.g., executive function). Additionally, changes on the EDSS are nonlinear and may not necessarily relate to actual differences in patient‐reported outcomes. Future prospective studies are warranted to highlight the best treatment approach with long‐term cognitive and disability assessments.
Author Contributions
M. Fereidhan Esfahani conducted research and wrote the first draft. Messrs. Decker and Weigand analyzed the data. Drs. Lopez Chiriboga, Flanagan, Tillema, and Lucchinetti identified study participants. Drs. Eckel Passow and Tobin developed the study concept and supervised the project. All authors contributed to critical revision of the final draft.
Conflict of Interest
None.
Acknowledgements
Mahboubeh Fereidan‐Esfahani was funded by a fellowship from the Mayo Clinic Center for MS and Autoimmune Neurology. W. Oliver Tobin has received research funding from Mallinckrodt Inc., the Mayo Clinic Center for MS and Autoimmune Neurology and the National institutes of Health. This study was supported by grant funding from the National Institutes of Health 1R01NS121928. The funders played no role in the preparation of this manuscript.
Funding Statement
This work was funded by National Institute of Neurological Disorders and Stroke grant 1R01NS121928.
Data Availability Statement
De‐identified patient data will be shared upon request to the corresponding author.
References
- 1. van der Velden M, Bots GT, Endtz LJ. Cranial CT in multiple sclerosis showing a mass effect. Surg Neurol. 1979;12:307‐310. [PubMed] [Google Scholar]
- 2. Algahtani H, Shirah B, Alassiri A. Tumefactive demyelinating lesions: a comprehensive review. Mult Scler Relat Disord. 2017;14:72‐79. [DOI] [PubMed] [Google Scholar]
- 3. Fereidan‐Esfahani M, Decker PA, Passow JEE, et al. Population‐based incidence and clinico‐radiological characteristics of tumefactive demyelination in Olmsted County, Minnesota, United States. Eur J Neurol. 2022;29:782‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hardy TA, Reddel SW, Barnett MH, Palace J, Lucchinetti CF, Weinshenker BG. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. 2016;15:967‐981. [DOI] [PubMed] [Google Scholar]
- 5. Sánchez P, Chan F, Hardy TA. Tumefactive demyelination: updated perspectives on diagnosis and management. Expert Rev Neurother. 2021;21:1005‐1017. [DOI] [PubMed] [Google Scholar]
- 6. Villarreal JV, Abraham MJ, Acevedo JAG, et al. Tumefactive multiple sclerosis (TMS): a case series of this challenging variant of MS. Mult Scler Relat Disord. 2021;48:102699. [DOI] [PubMed] [Google Scholar]
- 7. Balo J. Encephalitis periaxialis concentrica. Arch Neuro Psych. 1928;19:242‐264. [Google Scholar]
- 8. Johnson MD, Lavin P, Whetsell WO Jr. Fulminant monophasic multiple sclerosis, Marburg's type. J Neurol Neurosurg Psychiatry. 1990;53:918‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vakrakou AG, Tzanetakos D, Evangelopoulos ME, et al. Clinico‐radiologic features and therapeutic strategies in tumefactive demyelination: a retrospective analysis of 50 consecutive cases. Ther Adv Neurol Disord. 2021;14:17562864211006503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lucchinetti CF, Gavrilova RH, Metz I, et al. Clinical and radiographic spectrum of pathologically confirmed tumefactive multiple sclerosis. Brain. 2008;131:1759‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33:1444‐1452. [DOI] [PubMed] [Google Scholar]
- 12. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162‐173. [DOI] [PubMed] [Google Scholar]
- 13. Sechi E, Buciuc M, Pittock SJ, et al. Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing. JAMA Neurol. 2021;78:741‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jarius S, Paul F, Aktas O, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. 2018;15:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sastre‐Garriga J, Tintoré M, Rovira A, et al. Specificity of Barkhof criteria in predicting conversion to multiple sclerosis when applied to clinically isolated brainstem syndromes. Arch Neurol. 2004;61:222‐224. [DOI] [PubMed] [Google Scholar]
- 17. Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin‐4‐IgG assays. Neurology. 2012;78:665‐671. discussion 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fereidan‐Esfahani M, Tobin WO. Cyclophosphamide in treatment of tumefactive multiple sclerosis. Mult Scler Relat Disord. 2021;47:102627. [DOI] [PubMed] [Google Scholar]
- 19. Mayr WT, Pittock SJ, McClelland RL, et al. Incidence and prevalence of multiple sclerosis in Olmsted County, Minnesota, 1985–2000. Neurology. 2003;61:1373‐1377. [DOI] [PubMed] [Google Scholar]
- 20. Tobin WO, Kalinowska‐Lyszczarz A, Weigand SD, et al. Clinical correlation of multiple sclerosis immunopathologic subtypes. Neurology. 2021;97:e1906‐e1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kalinowska‐Lyszczarz A, Tillema JM, Tobin WO, et al. Long‐term clinical, MRI, and cognitive follow‐up in a large cohort of pathologically confirmed, predominantly tumefactive multiple sclerosis. Mult Scler. 2022;28:441‐452. [DOI] [PubMed] [Google Scholar]
- 22. Sánchez P, Meca‐Lallana V, Barbosa A, Manzanares R, Palmí I, Vivancos J. Tumefactive demyelinating lesions of 15 patients: Clinico‐radiological features, management and review of the literature. J Neurol Sci. 2017;381:32‐38. [DOI] [PubMed] [Google Scholar]
- 23. Kaeser MA, Scali F, Lanzisera FP, Bub GA, Kettner NW. Tumefactive multiple sclerosis: an uncommon diagnostic challenge. J Chiropr Med. 2011;10:29‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brod SA, Lindsey JW, Nelson F. Tumefactive demyelination: clinical outcomes, lesion evolution and treatments. Mult Scler J Exp Transl Clin. 2019;5:2055217319855755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abou Zeid N, Pirko I, Erickson B, et al. Diffusion‐weighted imaging characteristics of biopsy‐proven demyelinating brain lesions. Neurology. 2012;78:1655‐1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lassmann H. Hypoxia‐like tissue injury as a component of multiple sclerosis lesions. J Neurol Sci. 2003;206:187‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karonen JO, Partanen PL, Vanninen RL, Vainio PA, Aronen HJ. Evolution of MR contrast enhancement patterns during the first week after acute ischemic stroke. AJNR Am J Neuroradiol. 2001;22:103‐111. [PMC free article] [PubMed] [Google Scholar]
- 28. Su KG, Banker G, Bourdette D, Forte M. Axonal degeneration in multiple sclerosis: the mitochondrial hypothesis. Curr Neurol Neurosci Rep. 2009;9:411‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Altintas A, Petek B, Isik N, et al. Clinical and radiological characteristics of tumefactive demyelinating lesions: follow‐up study. Mult Scler. 2012;18:1448‐1453. [DOI] [PubMed] [Google Scholar]
- 30. Wallner‐Blazek M, Rovira A, Fillipp M, et al. Atypical idiopathic inflammatory demyelinating lesions: prognostic implications and relation to multiple sclerosis. J Neurol. 2013;260:2016‐2022. [DOI] [PubMed] [Google Scholar]
- 31. Pittock SJ, McClelland RL, Achenbach SJ, et al. Clinical course, pathological correlations, and outcome of biopsy proved inflammatory demyelinating disease. J Neurol Neurosurg Psychiatry. 2005;76:1693‐1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langer‐Gould A, Popat RA, Huang SM, et al. Clinical and demographic predictors of long‐term disability in patients with relapsing‐remitting multiple sclerosis: a systematic review. Arch Neurol. 2006;63:1686‐1691. [DOI] [PubMed] [Google Scholar]
- 33. Runmarker B, Andersen O. Prognostic factors in a multiple sclerosis incidence cohort with twenty‐five years of follow‐up. Brain. 1993;116(Pt 1):117‐134. [DOI] [PubMed] [Google Scholar]
- 34. Völk S, Unterrainer M, Albert NL, et al. TSPO PET with 18F‐GE‐180 to differentiate variants of multiple sclerosis: relapsing‐remitting multiple sclerosis, Tumefactive demyelination, and Baló's concentric sclerosis. Clin Nucl Med. 2020;45:e447‐e448. [DOI] [PubMed] [Google Scholar]
- 35. Di Gregorio M, Torri Clerici VLA, Fenu G, et al. Defining the course of tumefactive multiple sclerosis: a large retrospective multicentre study. Eur J Neurol. 2021;28:1299‐1307. [DOI] [PubMed] [Google Scholar]
- 36. Deschamps R, Pique J, Ayrignac X, et al. The long‐term outcome of MOGAD: an observational national cohort study of 61 patients. Eur J Neurol. 2021;28:1659‐1664. [DOI] [PubMed] [Google Scholar]
- 37. Lopez‐Chiriboga AS, Sechi E, Buciuc M, et al. Long‐term outcomes in patients with myelin oligodendrocyte glycoprotein immunoglobulin G‐associated disorder. JAMA Neurol. 2020;77:1575‐1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
De‐identified patient data will be shared upon request to the corresponding author.