

Abstract
Objective
Hereditary spastic paraplegias (HSPs) are a group of inherited neurodegenerative disorders characterized by slowly progressive lower limb spasticity and weakness. HSP type 54 (SPG54) is autosomal recessively inherited and caused by mutations in the DDHD2 gene. This study investigated the clinical characteristics and molecular features of DDHD2 mutations in a cohort of Taiwanese patients with HSP.
Methods
Mutational analysis of DDHD2 was performed for 242 unrelated Taiwanese patients with HSP. The clinical, neuroimaging, and genetic features of the patients with biallelic DDHD2 mutations were characterized. A cell‐based study was performed to assess the effects of the DDHD2 mutations on protein expression.
Results
SPG54 was diagnosed in three patients. Among them, two patients carried compound heterozygous DDHD2 mutations, p.[R112Q];[Y606*] and p.[R112Q];[p.D660H], and the other one was homozygous for the DDHD2 p.R112Q mutation. DDHD2 p.Y606* is a novel mutation, whereas DDHD2 p.D660H and p.R112Q have been reported in the literature. All three patients manifested adult onset complex HSP with additional cerebellar ataxia, polyneuropathy, or cognitive impairment. Brain proton magnetic resonance spectroscopy revealed an abnormal lipid peak in thalamus of all three patients. In vitro studies demonstrated that all the three DDHD2 mutations were associated with a considerably lower DDHD2 protein level.
Interpretation
SPG54 was detected in approximately 1.2% (3 of 242) of the Taiwanese HSP cohort. This study expands the known mutational spectrum of DDHD2, provides molecular evidence of the pathogenicity of the DDHD2 mutations, and underlines the importance of considering SPG54 as a potential diagnosis of adult‐onset HSP.
Introduction
Hereditary spastic paraplegias (HSPs) are a group of inherited neurodegenerative disorders mainly presenting with progressive lower extremity spasticity and weakness. 1 , 2 Clinically, HSPs are classified into pure or complex forms. Pure HSPs are characterized by progressive lower limb spastic weakness, hypertonic urinary bladder, and mild diminution of vibrational sensation in the lower limbs, whereas complex HSPs have additional neurological or non‐neurological manifestations, such as ataxia, seizure, cognitive impairment, and peripheral neuropathy. 3 , 4 Currently, more than 89 genes or genetic loci have been implicated in HSPs and the inheritance pattern of HSP subtypes can be autosomal dominant (AD), autosomal recessive (AR), or X‐linked inherited. 3 , 4 , 5 , 6
Spastic paraplegia type 54 (SPG54; MIM# 615033) is a rare AR complex HSP caused by biallelic mutations in the DDHD2 gene. Thirty‐four variants of DDHD2 mutations have been reported in SPG54 patients. 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 Most of the variants result in loss‐of‐function of the protein product (pLoF variants), including nonsense or frameshift truncating mutations, and a minor group of variants are missense mutations, including p.Y56C, p.W103R, p.R112Q, p.T186M, p.G197R, p.V220F, pR242C, pR242H, p.P269L, p.P339S, p.Q487H, p.F502S, pG510E, p.D660H, p.C683S, and p.Y699C. 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 Among these mutations, only p.R287* and p.D660H have been identified in more than two families. 8 , 15 , 16 Most SPG54 patients have early‐onset disease, with symptoms presenting in the first decade of life. 10 , 17 , 18 Adult‐onset SPG54 has only been reported in four patients from three families; three of these patients presented with spastic‐ataxia syndrome. 7 , 10 The common neuroimaging features of SPG54 include a thin corpus callosum (TCC), white matter hyperintensity (WMH) in magnetic resonance imaging (MRI), and lipid accumulation in the thalami, and basal ganglia detected by proton magnetic resonance spectroscopy (1H‐MRS). 10 , 15 , 17 , 19
The protein encoded by DDHD2 is DDHD‐domain‐containing 2 protein (DDHD2), which belongs to the intracellular phospholipase A1 (PLA1) family and hydrolyzes fatty acid substrates from the sn‐1 position of phospholipids. 5 , 20 DDHD2 also exhibits triglyceride hydrolase activities, which modulate triglyceride metabolism and lipid accumulation in the brain. 21 , 22 , 23 DDHD2 is ubiquitously located and expressed in subcellular membrane compartments in the perinuclear area or proximal to the cis‐Golgi apparatus, indicating potential regulation of membrane trafficking. 22 , 24 Although molecular mechanisms of SPG54 remained unclear, previous functional studies demonstrated markedly decreased PLA1 activities in cells expressing DDHD2 W103R, V220F, and D660H mutant proteins, 7 suggesting that DDHD2 mutations may cause SPG54 through a loss‐of‐function effect.
In this study, we investigated the mutational spectrum and clinical phenotypes of SPG54 in a cohort of 242 unrelated Taiwanese patients with HSP. The neuropsychological, electrophysiological, and neuroimaging features of the patients carrying biallelic DDHD2 mutations were investigated. We further assessed the effects of the identified DDHD2 mutations on protein expression with in vitro study.
Methods
Study subjects
A continuous series of 242 unrelated patients with a clinical diagnosis of HSP were recruited at the Department of Neurology, Taipei Veterans General Hospital from January 1998 to January 2022. The patients had either (1) pure spastic paraplegia, (2) spastic quadriparesis with earlier and more severe involvement of lower limbs or (3) spastic paraplegia as an early and prominent sign of a neurodegenerative disease involving multiple parts of the nervous system. Other disease etiologies were excluded through brain and spine imaging studies and basic biochemical tests. 25 , 26 , 27 Among the 242 patients, 133 were men and 109 were women, and 95 patients (39%) had a family history of HSP phenotype, including 72 AD families, 19 AR families, and 4 families with X‐linked inheritance. The other 147 (61%) were apparently sporadic cases. The average age of disease onset was 29.2 ± 17.7 years (range: 0–69 years). All the participants were Taiwanese of Han Chinese ethnicity. Peripheral blood samples were collected from participants after their written informed consent was obtained. The protocols of this study were approved by the Institutional Review Board of Taipei Veterans General Hospital.
Mutational analysis
Mutational analysis of DDHD2 was performed with a targeted resequencing panel covering 76 confirmed HSP disease genes as well 57 genes associated with diseases manifesting HSP‐like phenotype on an Illumina HiSeq2500 platform (Table S1). Sequenced read alignment and variant calling were performed with the reference Human Genome version 38 (hg38/GRCh38). The DDHD2 pathogenic variants were verified with Sanger sequencing and named according to the reference DDHD2 sequence (NM_015214.3). The pathogenicity of the novel DDHD2 mutation was assessed according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG‐AMP) guidelines. 28
Clinical, neuropsychological, and electrophysiological evaluations and imaging studies
The probands and their families received complete neurological examinations. Disease severity was evaluated with Spastic Paraplegia Rating Scale (SPRS) 27 and SPATAX‐EUROSPA disability score. 29 SPRS is a validated HSP‐specific severity scale with 13 items and is used to measure functional impairment of walking ability, muscle strength, spasticity, pain, and urinary function. Each item was scored from 0 (full function) to 4 (most severe impairment). The SPATAX‐EUROSPA disability score grades functional impairment as 0 (no functional handicap), 1 (showing signs at examination), 2 (able to run, walking unlimited), 3 (unable to run, limited walking without aid), 4 (walking with one stick), 5 (walking with two sticks), 6 (requiring wheelchair), and 7 (confined to bed). The Scale for Assessment and Rating of Ataxia (SARA) were also used to quantify disease severity in patients with cerebellar manifestations. 30 Mini‐Mental State Examination (MMSE) was performed to assess cognitive function. 31 Nerve conduction studies (NCSs) were conducted by standard techniques utilizing a Medelec MS25 electromyograph (Mistro, Surrey, UK) with surface electrode stimulations and recordings. 32 Brain and spine MRI were performed with 3‐Tesla MR system (Signa EXCITE, GE Medical Systems, Milwaukee, WI, USA). T1‐weighted images (T1WI), T2‐weighted images (T2WI) and 1H‐MRS were reviewed.
Expression plasmids, cell culture, and transfection
A wild‐type (WT) human DDHD2 expression plasmid with FLAG‐tagged in the C‐terminal region (backbone: pcDNA3.1+/c‐(K)‐DYK; clone ID: OHu23306) was purchased from GenScript (Piscataway, NJ, USA). The mutations, including c.335G>A (p.R112Q), c.1818C>A (p.Y606*), and c.1978G>C (p.D660H), were incorporated into the WT construct, respectively, by PCR‐based site‐directed mutagenesis method using Pfu Turbo DNA polymerase (Agilent, Santa Clara, CA, USA). Given that the p.Y606* mutation may lead to premature truncation of the protein product, the C‐terminal FLAG‐tag (DYKDDDDK) of all DDHD2 constructs (WT and mutants) was moved to the N‐terminal using Q5 Site‐Directed Mutagenesis Kit according to the manufacturer's protocol (New England Biolabs, Ipswich, MA, USA).
Human embryonic kidney 293T (HEK293T) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) and supplemented with 10% fetal bovine serum at 37°C under 5% CO2. Transient transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Reverse‐transcription polymerase chain reaction (RT‐PCR)
The total RNA of the transfected cells was harvested 24 h after transfection using Qiagen RNeasy mini kit (Qiagen, Valencia, CA, USA). The cDNA synthesis was conducted using SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The DDHD2 mRNA expression was normalized against GAPDH mRNA expression. Data analysis and plots were generated using Prism (GraphPad Software, San Diego, CA, USA).
Analysis of DDHD2 protein levels and cycloheximide (CHX)‐chase assays
Twenty‐four hours post‐transfection, cells were harvested and analyzed with western blotting. The steady‐state DDHD2 protein levels were analyzed using anti‐FLAG antibody (#8146, Cell Signaling Technology, Danvers, MA, USA). β‐Actin was used as a loading control and detected with anti‐β‐actin antibody (#8226; Abcam, Cambridge, UK). To determine the stability of the WT and mutant DDHD2 proteins, CHX‐chase assays were conducted. At 24 h post‐transfection, the transfected cells were treated with 100 mg/μl cycloheximide (Sigma‐Aldrich, St. Louis, MO, USA) for 0–8 h and harvested at the indicated time points. Cell lysates were prepared and then subjected to western blotting with the anti‐FLAG antibody. Actin was used as loading control. The ratios of DDHD2 to actin were calculated densitometrically. Data analysis and plots were generated using Prism (GraphPad Software, San Diego, CA, USA).
Results
Identification of the DDHD2 mutations
Three patients in the HSP cohort carried biallelic DDHD2 mutations, including two patients with compound heterozygous mutations, p.[R112Q];[Y606*] (c.[335G>A];[1818C>A]) and p.[R112Q];[D660H] (c.[335G>A];[1978G>C]), and one with homozygous p.R112Q (c.335G>A) mutation (Fig. 1A–C). Among these mutations, DDHD2 p.Y606* is novel, whereas p.R112Q and p.D660H have been reported to be associated with SPG54 in previous studies. 9 The p.Y606* mutation is a nonsense mutation that may disrupt the DDHD domain and lead to DDHD2 protein truncation (Fig. 1D). This mutation was absent in the Genome Aggregation Database (gnomAD v2.1.1; http://gnomad.broadinstitute.org/), as well as the 1517 ethnically matched control genomes from the Taiwan Biobank database (http://www.twbiobank.org.tw/). According to the ACMG‐AMP guidelines, 28 the DDHD2 p.Y606* mutation was predicted to be a null variant (PVS1 criterion); absent in population databases (PM2), capable of disrupting the DDHD domain (PM1); capable of shortening the length of DDHD2 protein (PM4); detected in trans with a known pathogenic mutation, p.R112Q (PM3), and identified in a patient with HSP (PP4). Thus, DDHD2 p.Y606* is classified as a pathogenic variant.
Figure 1.

Genetic analysis of the patients with spastic paraplegia type 54 (SPG54). (A–C) Pedigrees of the three SPG54 families and the Sanger sequencing traces of DDHD2 mutations including DDHD2 c.[335G>A];[1978G>C] (p.[R112Q];[D660H]) and DDHD2 c.[335G>A];[1818C>A] (p.[R112Q];[Y606*]) in compound heterozygous form and DDHD2 c.335G>A (p.R112Q) in homozygous form in families A, B, and C, respectively. The probands are indicated by arrows. The “M” represents a mutant DDHD2 allele, and the “W” refer to a wild‐type allele. The squares and circles denote for males and females, separately. The filled and open symbols represent affected and unaffected members, respectively. Dotted symbols indicate obligate carriers. The altered amino acid residues are labeled in red. (D) Scheme of the DDHD2 protein and its domains (WWE, lipase, sterile‐alpha‐motif (SAM), and DDHD), labeling with SPG54‐related mutations identified in the literatures (marked in black) and the present study (the novel mutation marked in red, and the recurred mutations marked in green).
Clinical information and other evaluations
The clinical information of patients with SPG54 was summarized in Table 1. The proband of family A (A‐II‐2) carrying the DDHD2 p.[R112Q];[D660H] mutation was a 61‐year‐old woman born to non‐consanguineous parents with no family history of neurological diseases. Her motor developmental milestones in childhood were normal, but she had insidious motor function regression and unsteady gait since age 35, which indicated pyramidal and cerebellar manifestations. The patient was able to stand and walk independently until age 55. Neurological examination at age 61 revealed lower limb spasticity, mild weakness of hip flexors, and increased deep tendon reflexes (DTRs) of biceps and knee but decreased DTRs of ankle. She also had bilateral extensor plantar responses, and prominent limb, and truncal ataxia. Impaired sensation of all modalities over distal limbs were also noted, indicating a length‐dependent peripheral neuropathy. Her SPRS score was 12 (Table S2) and her SARA score was 11. The patient could walk without aid for a short distance (disability score: 3; Video S1). Her MMSE score at age 61 was 19 out of 30, which indicated cognitive impairment. The NCS showed axonal sensorimotor polyneuropathy (Table S3). Brain and spine MRIs were normal. However, her brain 1H‐MRS revealed an abnormal lipid peak in bilateral thalami (Fig. 2A). The patient's sister and daughter were heterozygous to DDHD2 p.R112Q mutation and presented normal findings in neurological examinations at age 59 and 38, respectively.
Table 1.
Clinical characteristics of Taiwanese SPG54 patients.
| Patients | A‐II‐2 | B‐II‐3 | C‐II‐6 |
|---|---|---|---|
| Genetic variants | c.[335G>A];[1978G>C] | c.[335G>A];[1818C>A] | c.[335G>A];[c.335G>A] |
| Protein | p.[R112Q];[D660H] | p.[R112Q];[p.Y606*] | p.[R112Q];[p.R112Q] |
| Gender | Female | Female | Female |
| Age at onset (years) | 35 | 45 | 50 |
| Disease duration (years) | 26 | 23 | 12 |
| SPRS score (decline rate) | 12 (0.46/year) | 32 (1.39/year) | 22 (1.83/year) |
| SARS score (decline rate) | 11 (0.42/year) | 12 (0.52/year) | 9 (0.75/year) |
| Disability score (0–7) a | 3 | 5 | 5 |
| Spasticity (Knee extension) | + | + | + |
| Muscle strength (Hip flexors) b | 4 | 4 | 4 |
| DTR c | |||
| Biceps | +++ | +++ | ++ |
| Knee | +++ | +++ | +++ |
| Ankle | + | ++ | + |
| Babinski sign | + | + | + |
| Sensory deficits | |||
| Surface | + | − | + |
| Position/vibration | + | + | + |
| Urinary symptoms | − | + | − |
| Cerebellum ataxia | + | − | − |
| Cognitive impairment | + | + | − |
| Peripheral neuropathy | + | − | + |
−, absence; +, presence; DTR, deep tendon reflex; SARA, Scale for Assessment and Rating of Ataxia; SPRS, Spastic Paraplegia Rating Scale.
The disability stage 3 means “unable to run, limited walking without aid” and stage 5 means “walking with two sticks.”
Medical Research Council (MRC) scale for muscle strength: 0–5.
MRC scale for tendon reflex: − to ++++.
Figure 2.
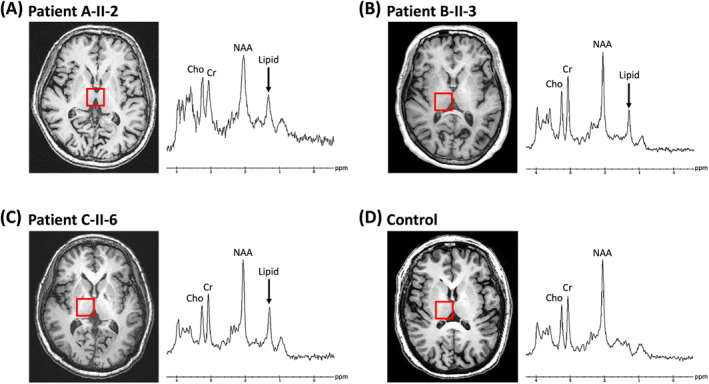
Proton magnetic resonance spectroscopy obtained from the patient A‐II‐2 (A), patient B‐II‐3 (B), patient C‐II‐6 (C) and healthy control (D) at a magnetic field of 3‐Tesla (echo time 35 ms). The arrows indicate the pathologic lipid peak at 1.3 ppm detected at bilateral thalami. Cho, choline; Cr, creatine; NAA, N‐acetylaspartate.
The proband of family B (B‐II‐3), who carried DDHD2 p.[R112Q];[Y606*] mutation, was a 68‐year‐old woman born to non‐consanguineous parents without family history of neurological diseases. Her early motor steps were normal. Initial symptoms included lower extremity stiffness and frequent falls beginning at age 45. Her motor function deteriorated gradually, and from age 60 onward, she only could walk fewer than 10 steps at a time. She also experienced urinary urgency and occasional incontinence. A neurological examination at age 68 revealed lower limb spasticity, mild weakness of hip flexors and knee flexors, general hyperreflexia and bilateral extensor plantar responses. Impaired vibration sensation of the distal lower limbs was also present. Her SPRS score was 32 (Table S2). The patient required two assistive canes to walk (disability score: 5; Video S1). Her MMSE score was 23 out of 30, indicating cognitive impairment. Her NCS (Table S3) and conventional brain and spine MRIs were normal. However, her brain 1H‐MRS demonstrated an abnormal lipid peak in bilateral thalami (Fig. 2B). Her two daughters, who carried heterozygous DDHD2 p.R112Q mutation, both presented healthy with normal findings in neurological examinations at age 46 and 43.
The proband of family C (C‐II‐6) was a 62‐year‐old woman carrying homozygous DDHD2 p.R112Q mutation. She was from a non‐consanguineous family with normal early motor development and began to have gait disturbance at age 50. Then, her motor ability worsened continuously and she could walk only with bilateral assistance at age 59. A neurological examination at age 62 revealed lower limb spasticity, mild weakness of hip flexors, increased DTRs of knee but decreased in ankle jerks, and bilateral extensor plantar responses. Impaired sensation of all modalities over the distal lower limbs indicated a length‐dependent peripheral neuropathy. Her SPRS score was 22 (Table S2). The patient required two assistive canes for walking (disability score: 5; Video S1). The NCS showed axonal sensorimotor polyneuropathy (Table S3). Brain and spine MRIs were normal. The cerebral 1H‐MRS also revealed an abnormal lipid peak in bilateral thalami (Fig. 2C).
DDHD2 mutations decreased steady‐state protein levels and protein stability
To investigate the effects of the three DDHD2 mutations, we examined the mRNA and protein expression using HEK293T cells transfected with FLAG‐tagged WT or mutated forms of DDHD2 constructs. An RT‐PCR analysis showed that DDHD2 p.R112Q, p.Y606*, and p.D660H mutations did not change the mRNA expression of DDHD2 (Fig. 3A). However, a western blot analysis showed a dramatic decrease in protein levels of the R112Q and D660H mutants at 24 h post‐transfection (Fig. 3B and C). As expected, the p.Y606* mutant construct produced truncated protein products, which level was only minimal at steady‐state compared to that of DDHD2‐WT (Fig. 3B and C). Because the reduced levels of mutant DDHD2 protein were independent from mRNA levels, we further tested whether the stabilities of mutant proteins changed. The cycloheximide chase assay showed that p.R112Q and p.D660H mutations had significantly accelerated the degradation of DDHD2 proteins (Fig. 3D and E). Similar results were found in C‐terminal FLAG‐tagged DDHD2 proteins (Figure S1). The p.Y606* was not assessed because the mutant protein level was too low.
Figure 3.

In vitro functional study of the identified DDHD2 variants in HEK293T cells. (A) Representative reverse‐transcription polymerase chain reaction analysis of DDHD2 mRNA levels in HEK293T cells transfected with different DDHD2 constructs. GAPDH mRNA was used as a loading control. Densitometric quantification is shown below. The error bars indicate standard error of the mean (SEM) from three independent experiments. (B) Representative western blot analysis for steady‐state expression of DDHD2 proteins in HEK293T cells transfected with different DDHD2 constructs. The band of the truncated DDHD2‐Y606* protein was labeled by a red arrow. β‐actin was used as a loading control. (C) Relative DDHD2 mutant protein expression at steady‐state was normalized to β‐actin and calculated in reference to DDHD2‐WT/β‐actin. Data were obtained from four independent experiments. Statistical analysis was performed by one‐way ANOVA followed by post hoc analysis (n = 4). (D) HEK293 cells were transfected with either DDHD2‐WT, R112Q, or D660H constructs for 24 h and subsequently subjected to cycloheximide (CHX)‐chase assays. Data were obtained from three independent experiments. Representative western blot analysis is shown. (E) The quantified results of the CHX‐chase assays. Two‐way ANOVA and post hoc Tukey tests were performed at every time point (n = 3). All data are presented as mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 for DDHD2 mutants versus Wild‐type groups.
Discussion
We screened 242 unrelated Taiwanese patients with HSP and identified three SPG54 patients carrying biallelic DDHD2 mutations. Among them, two patients carried compound heterozygous DDHD2 mutations, p.[R112Q];[D660H] and p.[R112Q];[Y606*], and one patient was homozygous for the DDHD2 p.R112Q mutation. Among these mutations, DDHD2 Y606* was novel. There are several interesting findings. First, DDHD2 mutations accounted for 1.2% (3 out of 242) of all HSP patients and 2% (3 out of 147) of patients with AR or sporadic HSP in the Taiwanese cohort. Second, SPG54 in our study presented as adult‐onset complex HSP with additional manifestations including ataxia, neuropathy, or cognitive impairment. Third, 1H‐MRS showing abnormal lipid peak in thalamus was a common and distinctive neuroradiological characteristic of SPG54. Finally, in vitro studies showed that three DDHD2 mutations (p.R112Q, p.Y606*, and p.D660H) resulted in severely decreased steady‐state protein levels, which were independent of the mRNA levels but may be associated with accelerated degradation of the mutant proteins.
SPG54 is an uncommon HSP subtype. An Italian study investigated 239 unrelated patients with HSP and found that SPG54 accounted for 1.7% of overall cases and 2.4% of the patients with AR or apparently sporadic HSP. 33 In a large cohort of French people with HSP, 7 out of 1550 index patients carried biallelic DDHD2 mutations, which represented 0.45% (7 out of 1550) of all patients with HSP and 3.8% (7 out of 186) of patients with AR‐HSP. 12 In a Chinese cohort with 99 HSP patients and the Japan Spastic Paraplegia Research Consortium (JASPAC) cohort with 383 HSP patients, both studies reported that fewer than 1% of their respective patients carried DDHD2 mutations. 9 , 34 However, in pediatric‐onset HSP cohorts, SPG54 was responsible for approximately 2% of HSP cases, which indicated that SPG54 more frequently caused HSP symptoms in early childhood. 13 , 14 , 35 Concordant to the results observed in other ethnic groups, SPG54 accounted for only 1.2% (3 out of 242) of all patients with HSP and 2% (3 out of 147) of patients with AR or apparently sporadic HSP in our Taiwanese cohort. These findings suggest that SPG54 is a rare HSP subtype and should be considered only in patients with AR or apparently sporadic HSP.
SPG54 typically presents with pediatric‐onset HSP. Previous reports have indicated that over 90% of SPG54 patients had congenital‐ or infantile‐onset delay in motor and cognitive milestones, followed by lower limb predominant spastic weakness. Additional features included cerebellar ataxia, abnormal ocular movements, strabismus, optic nerve hypoplasia, color vision deficiency, and short stature. 10 , 15 , 17 , 18 , 36 Adult‐onset SPG54 is relatively rare in the literatures. Only four adult‐onset cases have been reported. Three of the patients presented with ataxic‐spastic syndrome since 40s and none of the patients had cognitive impairment. 7 , 10 Surprisingly, we found three adult‐onset SPG54 patients and they all demonstrated complex HSP phenotypes with variable extra characteristics. All three patients manifested lower limb spasticity beginning at middle age. Patient A‐II‐2 had prominent cerebellar ataxia, axonal sensorimotor polyneuropathy and cognitive impairment. Patients B‐II‐3 and C‐II‐6 mainly demonstrated severe spasticity accompanied with cognitive impairment or polyneuropathy. These findings suggest that both infantile‐onset and adult‐onset SPG54 may present with complex HSP. There was no clear association between the onset age and the core features of neurological defects.
Among our three patients with SPG54 and the aforementioned four adult‐onset SPG54 cases, 7 , 10 more than half of the adult‐onset HSP cases (4 out of 7) exhibited additional symptoms such as cerebellar ataxia and polyneuropathy. These findings suggest that adult‐onset SPG54 usually manifests as ataxia‐spasticity spectrum disease (ASSD), which is a large group of inherited neurodegenerative diseases characterized by spasticity and ataxia. 37 The differential diagnoses of ASSD include autosomal recessive ataxia of Charlevoix‐Saguenay, late‐onset Friedreich ataxia, adult‐onset Alexander disease, spinocerebellar ataxia types 3, 7, 17, 23, 28, 40, and 42, Spastic ataxia types 1–9, and some AR‐HSP (SPG5, SPG7, SPG11, SPG15, SPG20, and SPG21). 38 Our finding indicates that SPG54 is also a member of ASSD and should be considered in patients with overlapping spasticity and ataxia of unknown cause, especially in those with adulthood onset disease, AR or apparently sporadic inheritance, and superimposed polyneuropathy.
MRI and 1H‐MRS are useful diagnostic tools for SPG54. In MRI studies of SPG54, TCC and WMH both were detected in over 70% of SPG54 patients. 10 , 17 However, TCC and WMH are not features specific to SPG54, and are also commonly found in patients with other AR‐HSP including SPG7, SPG11, SPG15, SPG18, SPG21, SPG35, SPG46, SPG47, SPG49, SPG50, and SPG63. 17 , 19 , 39 An abnormal lipid peak of 1.3 ppm identified in basal ganglia and thalami with 1H‐MRS was regarded as an important imaging marker for SPG54. 15 , 40 The mechanism underlying abnormal lipid peak may be related to the function of DDHD2 protein, which is both a phospholipase and a triglyceride lipase in the central nerve system. A recent study showed that depletion of Ddhd2 in mice resulted in massive accumulation of triglyceride and lipid droplets in the brain and spinal cord. 21 It was still unclear whether other HSP subtypes (including SPG5, SPG28, SPG31, SPG38, SPG39, SPG46, and SPG56) with disease genes related to lipid metabolism could also exhibit abnormal lipid peaks in 1H‐MRS. 5 , 23 In our study, the brain and spine MRIs of three adult‐onset SPG54 patients were grossly normal, despite the extended duration of the disease (26, 23, and 12 years). However, the 1H‐MRS of all our three SPG54 patients demonstrated an abnormal lipid peak in the bilateral thalami (Fig. 2). This suggested that 1H‐MRS is more sensitive than conventional MRI in detecting neuroradiological abnormalities in SPG54.
We provided in vitro evidence supporting the pathogenicity of DDHD2 p.R112Q, p.Y606*, and p.D660H mutations. SPG54 is an AR disorder more frequently caused by nonsense or frameshift truncating mutations. Markedly decreased PLA1 activities of DDHD2 mutant proteins have been demonstrated in functional studies of many pathogenic DDHD2 missense variants, including p.W103R, p.V220F, and p.D660H. 7 These studies have indicated that SPG54 is caused by the loss‐of‐function effect of DDHD2 mutations. Through cell transfection studies, we demonstrated that the p.R112Q, p.Y606*, and p.D660H mutations led to vastly decreased DDHD2 protein levels (Fig. 3B and C). Moreover, we showed that DDHD2‐R112Q and DDHD2‐D660H mutant proteins had faster protein degradation than DDHD2‐WT (Fig. 3D and E). These findings suggest that these DDHD2 mutations may lead to SPG54 with a loss‐of‐function effect through compromising protein stability and greatly decreasing DDHD2 protein expression levels.
In conclusion, SPG54 is a differential diagnosis of AR‐HSP or sporadic HSP in Taiwan, and DDHD2 mutations account for 2% of AR or sporadic HSP cases. DDHD2 Y606* is a novel pathogenic mutation of SPG54. Our study broadens the spectrum of DDHD2 mutations, provides molecular evidence supporting the pathogenicity of the DDHD2 mutations, and highlights the potential for SPG54 as an diagnosis of adult‐onset HSP.
Author Contributions
Ying‐Tsen Chou, Shao‐Lun Hsu, Yi‐Chu Liao, Yi‐Chung Lee contributed to conception and design of the study. Ying‐Tsen Chou, Shao‐Lun Hsu, Yu‐Shuen Tsai, Yi‐Jiun Lu, Kai‐Wei Yu, Hsiu‐Mei Wu contributed to acquisition and analysis of data. Ying‐Tsen Chou, Shao‐Lun Hsu, Yi‐Chu Liao, Yi‐Chung Lee contributed to drafting of the manuscript.
Conflict of Interest
The authors declared no conflict of interests.
Supporting information
Table S1. The gene list of the targeted resequencing panel for HSP.
Table S2. The score of Spastic Paraplegia Rating Scale in Taiwanese SPG54 patients.
Table S3. Nerve conduction studies of the Taiwanese patients with spastic paraplegia type 54.
Figure S1. Cycloheximide‐chase assay of the C‐terminal FLAG‐tagged DDHD2 variants in HEK293T cells. (A) HEK293 cells were transfected with vectors carrying either C‐terminal FLAG‐tagged DDHD2‐WT, R112Q, or D660H for 24 h and subsequently subjected to cycloheximide (CHX)‐chase assays for indicative times. Representative western blot analysis is shown. β‐Actin was used as a loading control. (B) The quantified results of the CHX‐chase assays from three independent experiments. One‐way ANOVA and post hoc Dunnett's tests were performed at every time point. All data are presented as mean ± SEM. *P < 0.05 and **P < 0.01 for DDHD2 mutants versus wild‐type groups.
Video S1. The videos of gait of the SPG54 patients, A‐II‐2, B‐II‐3, and C‐II‐6 at age 61, 68, and 62 years, respectively.
Acknowledgments
The authors thank the patients who participated in this study. The authors also acknowledge the Genomics Center for Clinical and Biotechnological Applications of National Core Facility for Biopharmaceuticals, Taiwan (MOST 109‐2740‐B‐010‐002) and Compass Bioinformatics, Taiwan for the sequencing and genetic analysis service. This work was supported by the grants from Ministry of Science and Technology, Taiwan, ROC (MOST 107‐2314‐B‐075‐014‐MY3), Taipei Veterans General Hospital (V112C‐001) and Brain Research Center, National Yang‐Ming University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. This work was financially supported by the “Center for Intelligent Drug Systems and Smart Bio‐devices (IDS2B)” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Funding Statement
This work was funded by Brain Research Center, National Yang‐Ming University; Genomics Center for Clinical and Biotechnological Applications of National Core Facility for Biopharmaceuticals, Taiwan grant MOST 109‐2740‐B‐010‐002; Ministry of Science and Technology, Taiwan, ROC grant MOST 107‐2314‐B‐075‐014‐MY3; Taipei Veterans General Hospital grant V112C‐001; Center for Intelligent Drug Systems and Smart Bio‐devices (IDS2B).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Fink JK. Advances in the hereditary spastic paraplegias. Exp Neurol. 2003;184(Suppl 1):S106‐S110. [DOI] [PubMed] [Google Scholar]
- 2. Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18(12):1136‐1146. [DOI] [PubMed] [Google Scholar]
- 3. Meyyazhagan A, Orlacchio A. Hereditary spastic paraplegia: an update. Int J Mol Sci. 2022;23(3):1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elsayed LEO, Eltazi IZ, Ahmed AE, Stevanin G. Insights into clinical, genetic, and pathological aspects of hereditary spastic paraplegias: a comprehensive overview. Front Mol Biosci. 2021;8:690899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panza E, Meyyazhagan A, Orlacchio A. Hereditary spastic paraplegia: genetic heterogeneity and common pathways. Exp Neurol. 2022;357:114203. [DOI] [PubMed] [Google Scholar]
- 6. Deng R, Medico‐Salsench E, Nikoncuk A, et al. AMFR dysfunction causes autosomal recessive spastic paraplegia in human that is amenable to statin treatment in a preclinical model. Acta Neuropathol. 2023;10.1007/s00401‐023‐02579‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doi H, Ushiyama M, Baba T, et al. Late‐onset spastic ataxia phenotype in a patient with a homozygous DDHD2 mutation. Sci Rep. 2014;4:7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Magariello A, Citrigno L, Zuchner S, et al. Further evidence that DDHD2 gene mutations cause autosomal recessive hereditary spastic paraplegia with thin corpus callosum. Eur J Neurol. 2014;21(3):e25‐e26. [DOI] [PubMed] [Google Scholar]
- 9. Dong EL, Wang C, Wu S, et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol Neurodegener. 2018;13(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nicita F, Stregapede F, Tessa A, et al. Defining the clinical‐genetic and neuroradiological features in SPG54: description of eight additional cases and nine novel DDHD2 variants. J Neurol. 2019;266(11):2657‐2664. [DOI] [PubMed] [Google Scholar]
- 11. Xu X, Lu F, Du S, et al. Case report: novel compound heterozygous missense mutations in the DDHD2 gene in a Chinese patient associated with spastic paraplegia type 54. Front Pediatr. 2022;10:997274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mereaux JL, Banneau G, Papin M, et al. Clinical and genetic spectra of 1550 index patients with hereditary spastic paraplegia. Brain. 2022;145(3):1029‐1037. [DOI] [PubMed] [Google Scholar]
- 13. Wang J, Fang F, Ding C, et al. Clinical and genetic spectrum of hereditary spastic paraplegia in Chinese children. Dev Med Child Neurol. 2023;65(3):416‐423. [DOI] [PubMed] [Google Scholar]
- 14. Schiavoni S, Spagnoli C, Rizzi S, et al. Paediatric‐onset hereditary spastic paraplegias: a retrospective cohort study. Dev Med Child Neurol. 2020;62(9):1068‐1074. [DOI] [PubMed] [Google Scholar]
- 15. Schuurs‐Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, et al. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet. 2012;91(6):1073‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Citterio A, Arnoldi A, Panzeri E, et al. Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J Neurol. 2014;261(2):373‐381. [DOI] [PubMed] [Google Scholar]
- 17. Erfanian Omidvar M, Torkamandi S, Rezaei S, et al. Genotype‐phenotype associations in hereditary spastic paraplegia: a systematic review and meta‐analysis on 13,570 patients. J Neurol. 2021;268(6):2065‐2082. [DOI] [PubMed] [Google Scholar]
- 18. Fereshtehnejad SM, Saleh PA, Oliveira LM, et al. Movement disorders in hereditary spastic paraplegia (HSP): a systematic review and individual participant data meta‐analysis. Neurol Sci. 2023;44(3):947‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dosi C, Pasquariello R, Ticci C, et al. Neuroimaging patterns in paediatric onset hereditary spastic paraplegias. J Neurol Sci. 2021;425:117441. [DOI] [PubMed] [Google Scholar]
- 20. Yaginuma S, Kawana H, Aoki J. Current knowledge on mammalian phospholipase A(1), brief history, structures, biochemical and pathophysiological roles. Molecules. 2022;27(8):2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inloes JM, Hsu KL, Dix MM, et al. The hereditary spastic paraplegia‐related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci USA. 2014;111(41):14924‐14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inloes JM, Kiosses WB, Wang H, Walther TC, Farese RV Jr, Cravatt BF. Functional contribution of the spastic paraplegia‐related triglyceride hydrolase DDHD2 to the formation and content of lipid droplets. Biochemistry. 2018;57(5):827‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tadepalle N, Rugarli EI. Lipid droplets in the pathogenesis of hereditary spastic paraplegia. Front Mol Biosci. 2021;8:673977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sato S, Inoue H, Kogure T, Tagaya M, Tani K. Golgi‐localized KIAA0725p regulates membrane trafficking from the Golgi apparatus to the plasma membrane in mammalian cells. FEBS Lett. 2010;584(21):4389‐4395. [DOI] [PubMed] [Google Scholar]
- 25. Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1(8334):1151‐1155. [DOI] [PubMed] [Google Scholar]
- 26. Fink JK, Heiman‐Patterson T, Bird T, et al. Hereditary spastic paraplegia: advances in genetic research. Hereditary Spastic Paraplegia Working Group. Neurology. 1996;46(6):1507‐1514. [DOI] [PubMed] [Google Scholar]
- 27. Schule R, Holland‐Letz T, Klimpe S, et al. The Spastic Paraplegia Rating Scale (SPRS): a reliable and valid measure of disease severity. Neurology. 2006;67(3):430‐434. [DOI] [PubMed] [Google Scholar]
- 28. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chrestian N, Dupre N, Gan‐Or Z, et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol Genet. 2017;3(1):e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmitz‐Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717‐1720. [DOI] [PubMed] [Google Scholar]
- 31. Shyu Y‐IL, Yip P‐K. Factor structure and explanatory variables of the Mini‐Mental State Examination (MMSE) for elderly persons in Taiwan. J Formos Med Assoc. 2001;100(10):676‐683. [PubMed] [Google Scholar]
- 32. Lin KP, Chan MH, Wu ZA. Nerve conduction studies in healthy Chinese: correlation with age, sex, height and skin temperature. Zhonghua Yi Xue Za Zhi (Taipei). 1993;52(5):293‐297. [PubMed] [Google Scholar]
- 33. D'Amore A, Tessa A, Casali C, et al. Next generation molecular diagnosis of hereditary spastic paraplegias: an Italian cross‐sectional study. Front Neurol. 2018;9:981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koh K, Ishiura H, Tsuji S, Takiyama Y. JASPAC: Japan spastic paraplegia research consortium. Brain Sci. 2018;8(8):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Travaglini L, Aiello C, Stregapede F, et al. The impact of next‐generation sequencing on the diagnosis of pediatric‐onset hereditary spastic paraplegias: new genotype‐phenotype correlations for rare HSP‐related genes. Neurogenetics. 2018;19(2):111‐121. [DOI] [PubMed] [Google Scholar]
- 36. Bogdanova‐Mihaylova P, Austin N, Alexander MD, et al. Spastic ataxia associated with colour vision deficiency due to DDHD2 mutations. Eur J Neurol. 2020;27(1):e9‐e10. [DOI] [PubMed] [Google Scholar]
- 37. Synofzik M, Schule R. Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord. 2017;32(3):332‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pedroso JL, Vale TC, Franca Junior MC, et al. A diagnostic approach to spastic ataxia syndromes. Cerebellum. 2022;21(6):1073‐1084. [DOI] [PubMed] [Google Scholar]
- 39. da Graca FF, de Rezende TJR, Vasconcellos LFR, Pedroso JL, Barsottini OGP, Franca MC Jr. Neuroimaging in hereditary spastic paraplegias: current use and future perspectives. Front Neurol. 2018;9:1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vavla M, Montanaro D, Pizzighello S, et al. Brain magnetic spectroscopy imaging and hereditary spastic paraplegia: a focused systematic review on current landmarks and future perspectives. Front Neurol. 2020;11:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The gene list of the targeted resequencing panel for HSP.
Table S2. The score of Spastic Paraplegia Rating Scale in Taiwanese SPG54 patients.
Table S3. Nerve conduction studies of the Taiwanese patients with spastic paraplegia type 54.
Figure S1. Cycloheximide‐chase assay of the C‐terminal FLAG‐tagged DDHD2 variants in HEK293T cells. (A) HEK293 cells were transfected with vectors carrying either C‐terminal FLAG‐tagged DDHD2‐WT, R112Q, or D660H for 24 h and subsequently subjected to cycloheximide (CHX)‐chase assays for indicative times. Representative western blot analysis is shown. β‐Actin was used as a loading control. (B) The quantified results of the CHX‐chase assays from three independent experiments. One‐way ANOVA and post hoc Dunnett's tests were performed at every time point. All data are presented as mean ± SEM. *P < 0.05 and **P < 0.01 for DDHD2 mutants versus wild‐type groups.
Video S1. The videos of gait of the SPG54 patients, A‐II‐2, B‐II‐3, and C‐II‐6 at age 61, 68, and 62 years, respectively.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.