

Summary
Plant breeding is constrained by trade‐offs among different agronomic traits by the pleiotropic nature of many genes. Genes that contribute to two or more favourable traits with no penalty on yield are rarely reported, especially in wheat. Here, we describe the editing of a wheat auxin response factor TaARF12 by using CRISPR/Cas9 that rendered shorter plant height with larger spikes. Changes in plant architecture enhanced grain number per spike up to 14.7% with significantly higher thousand‐grain weight and up to 11.1% of yield increase under field trials. Weighted Gene Co‐Expression Network Analysis (WGCNA) of spatial–temporal transcriptome profiles revealed two hub genes: RhtL1, a DELLA domain‐free Rht‐1 paralog, which was up‐regulated in peduncle, and TaNGR5, an organ size regulator that was up‐regulated in rachis, in taarf12 plants. The up‐regulation of RhtL1 in peduncle suggested the repression of GA signalling, whereas up‐regulation of TaNGR5 in spike may promote GA response, a working model supported by differential expression patterns of GA biogenesis genes in the two tissues. Thus, TaARF12 complemented plant height reduction with larger spikes that gave higher grain yield. Manipulation of TaARF12 may represent a new strategy in trait pyramiding for yield improvement in wheat.
Keywords: CRISPR/Cas9, TaARF12, grain yield, plant height, spike length, green revolution
Introduction
Wheat (Triticum aestivum L.) is one of the most widely grown crops and a staple food for 35% of the world's population (FAO, http://www.fao.org/faostat/). Plant architecture is the main factor that affects cultivation adaptability, harvesting efficiency and final grain yield (Guo et al., 2020). Among these, plant height and spike morphology are two of the main determinants for plant architecture. The “Green Revolution” (GR) of the 1960s, for example, boosted rice and wheat yield by adopting semi‐dwarf varieties that were resistant to yield‐reducing ‘lodging’ (flattening of plants by wind and rain; Pingali, 2012). In rice, the GR gene sd1 reduced plant height by affecting gibberellin (GA) biogenesis (Sasaki et al., 2002), whereas the wheat GR genes Rht‐B1b (Reduced height 1, Rht1) and Rht‐D1b (Rht2) are insensitive to GA due to mutations in the DELLA domain responsible for GA signalling (Peng et al., 1999; Van De Velde et al., 2021). Genes to improve wheat spike development for grain yield increase have also been identified, such as the major domestication gene Q (Liu et al., 2018; Zhang et al., 2020), TEOSINTE BRANCHED1 (TB1) (Dixon et al., 2018), and CONSTANS‐like B5 (TaCOL‐B5) (Zhang et al., 2022).
Plant height is especially affected by plant hormones like GA (Eshed and Lippman, 2019), auxin (Guo et al., 2020), and strigolactone (Chesterfield et al., 2020). Among them, GA is the most important hormone for plant height regulation. In addition to the rice sd1 and the wheat Rht1 and Rht2 that are involved in GA signalling, GA biogenesis and metabolizing genes, such as the barley gibberellin 3‐oxidase 1 (Cheng et al., 2023), the wheat Rht12 (Sun et al., 2019), Rht18 (Ford et al., 2018) and Rht24b (Tian et al., 2022), have also been identified to participate in plant height development. Auxin signalling genes, such as auxin response factors (ARFs), played important roles in this process too. In rice, for example, overexpression of OsARF19 reduced plant height with smaller grain size (Zhang et al., 2015), whereas the mutation of OsARF11 produced plants with shorter plant height and fewer grains per panicle (Sims et al., 2021). OsARF12 could reduce plant height with aborted apical spikelets when mutated (Zhao et al., 2022). Unfortunately, the reduced plant height is often accompanied by shorter inflorescence or other negative trade‐offs on grain yield. The GR genes, for instance, also confer such negative effects: the rice sd1 reduced plant height but with smaller panicles (Su et al., 2021), and the wheat Rht1 and Rht2 mutants reduced plant height but with significantly reduced stem diameter, spike length and thousand‐grain weight (Figure S1), and eventually reduced grain yield per plant (Van De Velde et al., 2017). Thus, genes that reduce plant height but increase spike size are scarce.
Here, we report the genome editing of the wheat TaARF12 gene that produced shorter plant height, thicker stem and larger spikes. The change in plant morphology increased grain number per spike, grain size and final grain yield in the field. We further identified hub genes by Weighted Gene Co‐Expression Network Analysis (WGCNA) of spatial–temporal transcriptome profiles that revealed differential GA signalling responses in peduncle and rachis, possibly via hub genes like RhtL1, a DELLA domain‐free Rht‐1 paralog, and TaNGR5, an organ size regulator respectively. Our work proposed a model that takes advantage of two contrasting functions of one pleiotropic gene that counterbalances conventional negative trade‐offs in wheat breeding, providing a new strategy to develop high‐yield varieties for future food security.
Results
Editing of the wheat TaARF12 gene by using CRISPR/Cas9
The wheat auxin response factor (ARF) gene TaARF12 was identified by genome‐wide association studies (GWAS) to be responsible for plant architecture (Li et al., 2022). To study the functions of the gene, we designed two guide RNAs (gRNAs) to simultaneously target the coding regions of the three homoeologs (TaARF12‐A/TraesCS2A03G1263300, TaARF12‐B/TraesCS2B03G1444700 and TaARF12‐D/TraesCS2D03G1217700) by using CRISPR/Cas9 (Figure S2). Among a total of 12 transformed plants (named #1 to #12), 11 (91.7%) showed the integration of the CRISPR/Cas9 vector into the genome by detecting the Bar gene. To characterize the mutation types, we designed subgenome‐specific primers to sequence the target regions of TaARF12 in A, B and D subgenomes (Table S16). We tracked these transgenic plants from T0 to T4 generations and confirmed that 11 lines were edited as shown in Table S1. Three homozygous transgenic lines were used for subsequent analyses: #5 with all three loss‐of‐function homoeologs, #11 with two loss‐of‐function homoeologs (B and D) and #9 with only the A homoeolog mutated (Figure S2).
TaARF12 mutations reduced plant height but increased spike length
Morphological observation of TaARF12 transgenic plants showed that the three lines #5, #11 and #9 reduced plant height by 14.9%, 14.3% and 6.3% respectively (Figure 1a,c) that were suitable for agronomic application. Further observation found that every stem internode became shorter in mutant plants when compared with those of the wild type (Figure S3). However, stem diameters of the transgenic plants were increased (Figure 1b,d). We then used peduncle, the longest stem internode, for stem analyses. Longitudinal section showed that taarf12 peduncle cells were significantly shorter, up to 11.2% (P value < 0.001) (Figure 1k–m), while the number of cells decreased up to 15.6% (P < 0.001, Figure 1n). Transversely, there were more cell layers in mutant lines relative to the wild type, rendering much thicker peduncles in transgenic plants (Figure 1o,p; Figure S4). On the other hand, spikes of the transgenic plants were significantly larger, with the lengths of spikes for lines #5, #11 and #9 increasing by 15.1%, 12.3% and 4.9% respectively (Figure 1e,g). For spikes, we studied the morphology of rachis, the axis where spikelets attached. We found that rachis internodes of the longer rachis were longer with increased internode numbers (Figure 1f,h–j; Figure S5). Dissection of the rachis showed significant cell number increase longitudinally from stage W5.5. By stage W9.0, cell length was increased by 8.5% (P value < 0.001), while the cell number was increased by 10.9% (P value < 0.01; Figure 1q–t; Figure S6). Cross‐sectional observation showed that the thicker rachis was composed of more and wider cells (Figure 1u,v). The larger spikes indeed conferred higher biomasses for rachis and spikelet (Figure S7). Thus, taarf12 mutations had caused repression of cell division and cell elongation in peduncle elongation but promoted two‐dimensional cell division and cell expansion in rachis development, leading to shorter plant stature but larger spikes.
Figure 1.
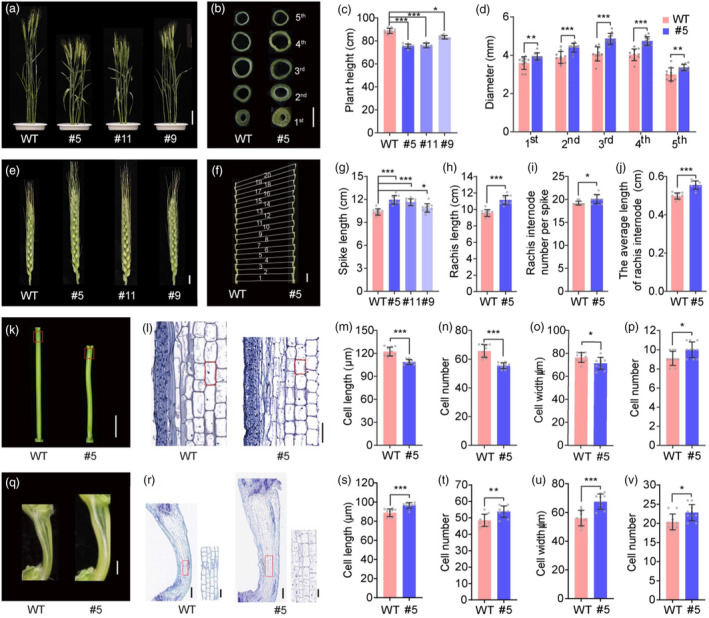
Genome editing of TaARF12 leads to reduced plant height and increased spike length. (a) Plant height. Scale bar, 10 cm. (b) Transverse section stems morphology of each internode. Scale bar, 5 mm. (c, d) Statistical analyses of plant height (c) and internode diameter (d). (e) Spike length. Scale bar, 1 cm. (f) Rachis and rachis internode numbers. Scale bar, 1 cm. Statistical analyses of spike length (g), rachis length (h), rachis number per spike (i) and the average length of rachis internode (j). (k) Peduncles at Waddington developmental stage 9.0 (W9.0). Scale bar, 1 cm. (l) Longitudinal sections of W9.0‐peduncles derived from regions indicated by red rectangles in (k). Scale bar, 100 μm. (m–p) Quantification of longitudinal cell length (m) and cell number (n), and horizontal cell width (o) and cell number (p) of the peduncles. (q) Rachis internodes at W9.0. Scale bar, 1 mm. (r) Longitudinal sections of W9.0 rachis internodes. A cell profile was shown on the right that corresponds to the red rectangle on the left. Scale bar, 500 μm for the left and 100 μm for the right of each panel. (s–v) Quantification of longitudinal cell length (s) and cell number (t), and horizontal cell width (u) and cell number (v) of the rachis internodes were shown. Data were mean ± SD; P values were determined by two‐tailed Student's t‐test. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
We then tested whether such improvement in wheat plant architecture may enhance grain yield by planting taarf12 lines in multiple plots for field testing. As shown in Figure 2, the grain number per spike was increased by 14.7% (P value < 0.001) with longer grain length and significantly higher thousand‐grain weight (up to 3.9%, P value < 0.05) (Figure 2a–l). Field tests also showed higher harvest index, and up to 9.7%–11.1% (P value < 0.05) increase in total grain yield relative to the Rht‐B1b‐carrying control, with no significant effect on tiller number and flowering time, despite increased tiller angle and enlarged flag leaf (Figure 2m,n; Figures S8 and S9). Thus, the contrasting morphological improvements in plant height and spike length were beneficial for yield potential.
Figure 2.
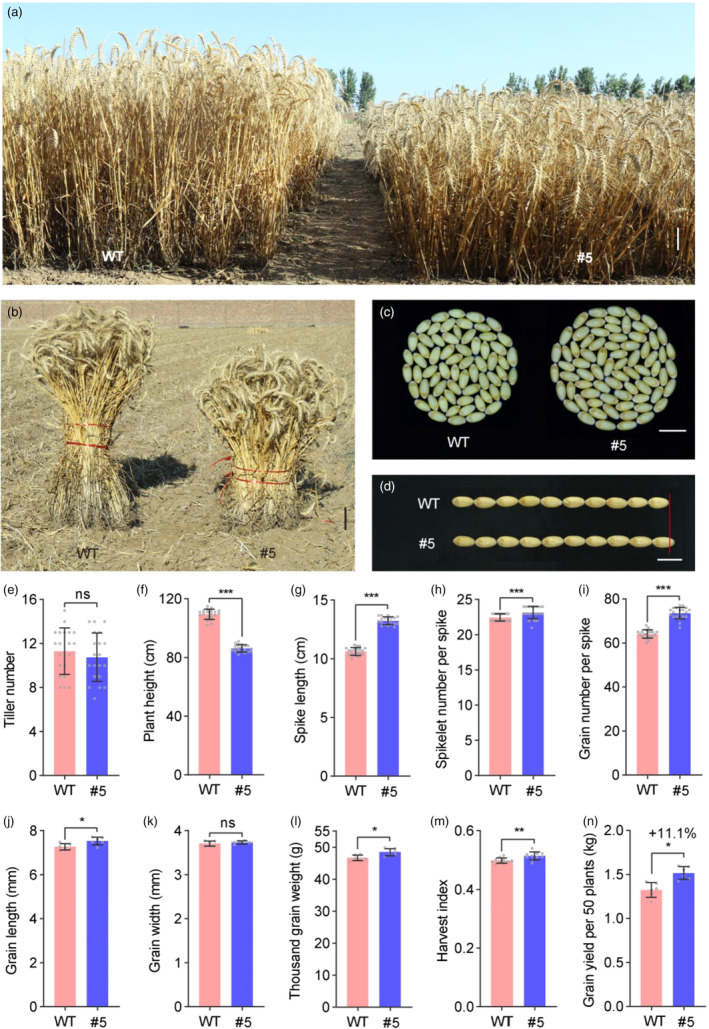
Genome editing of TaARF12 enhances wheat grain yield. (a) Wheat plants in the field. Scale bar, 10 cm. (b) The harvest of 50 wheat plants. Scale bar, 10 cm. (c) Grains harvested from main tillers of one plant. Scale bar, 1 cm. (d) Grain length comparison. Scale bar, 1 cm. (e–n) Statistics of tiller number (e), plant height (f), spike length (g), spikelet number per spike (h), grain number per spike (i), grain length (j), grain width (k), thousand‐grain weight (l), harvest index (m) and grain yield per 50 plants (n) of the wild‐type (WT) Fielder and transgenic line #5. Data were mean ± SD; P values were determined by two‐tailed Student's t‐test. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
Differential expression patterns of TaARF12 in peduncle and rachis
To investigate possible mechanisms of TaARF12 for peduncle and rachis (RA) development, we studied the expression patterns of the gene in the two tissues. We found that TaARF12 was preferentially expressed in young stem internode (IN) and spike (SP; Figure 3a). Its expression was higher at early stages of organ development than at later stages (Figure 3b,c). In peduncle, its expression at lower part (designated as IB for the bottom of internode), where cell division persisted during stem development, was much higher than that in the upper part (designated as internode upper part, or IU) where cell elongation occurred (Figure 3b). In situ hybridization assay showed that TaARF12 was mainly expressed in epidermis cells, sclerenchyma cells and vascular bundles in the peduncle (Figure 3d–g). In spikes, in addition to lodicule, glume, lemma and palea, hybridization signals were particularly found at the base of rachilla that connected spikelets to the rachis (Figure 3h,i). Together, TaARF12 exhibited spatial, temporal and tissue specificity in gene expression patterns that may suggest different regulatory modes in peduncle and rachis.
Figure 3.
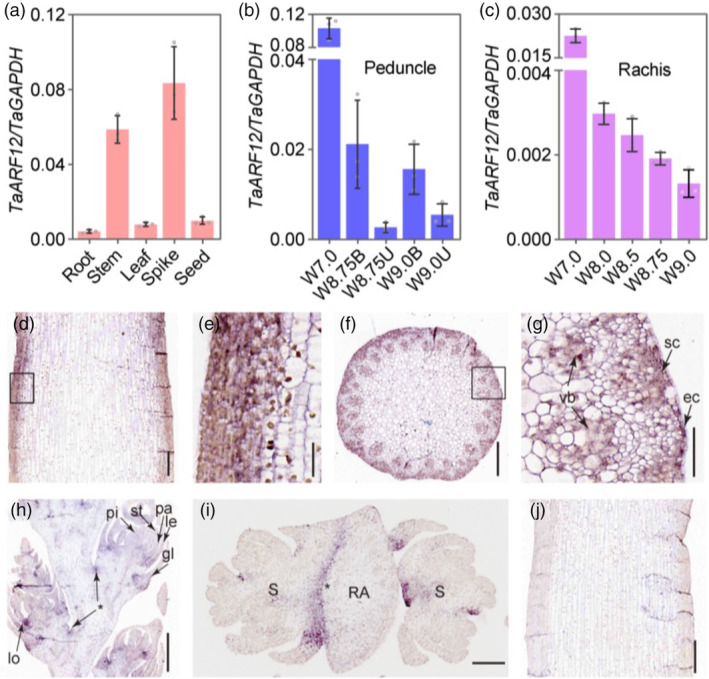
TaARF12 expression patterns. (a–c) Tissue specificity (a), in developing peduncle (b) and in developing rachis (c) by qRT–PCR. (d–i) In situ hybridization signals in longitudinal section of a peduncle (d) with (e) as the enlarged region corresponding to the rectangle in (d), in a cross‐section of a peduncle (f) with (g) corresponding to an enlarged picture of the rectangle region in (f), in a longitudinal section of a developing spike with floral organs indicated (h) and cross‐section of rachis showing the signal (asterisk) at the joint between rachis and rachilla (i). (j) The negative control by a sense probe. ec, epidermal cell; gl, glume; le, lemma; lo, lodicule; pa, palea; pi, pistil; sc, sclerenchyma cell; st, stamen; vb, vascular bundle; RA, rachis; S, spikelet. Scale bars, 50 μm in (g), 100 μm in (e), 200 μm in (d, f, i, j) and 500 μm in (h).
Contrasting transcriptome profiles in peduncle and rachis in taarf12 plants
To further dissect the regulatory mechanisms of TaARF12 in reducing plant height but making larger spikes when mutated, we performed a time‐series RNA‐seq analysis of peduncle and rachis at key developmental stages of W5.5, 7.0, 7.5, 8.0, 8.5, 8.75 and 9.0 (Patil et al., 2019), from both the wild‐type and taarf12 plants (Figure 4a,b; Figure S7a and Table S2). Principal component analysis (PCA) showed that transcriptome profiles in peduncle were clearly distinguished from those of rachises, despite the physical continuity of peduncle and rachis (Figure 4c). Consistently, expression profiles at earlier developmental stages (W7.0‐W8.75 for peduncle and pre‐W8.75 for rachis) were differentiable from those at later stages (W9.0 for internode upper part, or IU, and W8.75 for rachis, or RA). Moreover, differential responses to taarf12 mutation were observed in peduncle and rachis as well as in IU and IB once they reached stage W8.75 (Figure 4c). The relationship among gene expression profiles in different organs, developmental stages, and genotypes was supported by the pairwise Pearson correlation coefficients (Figure 4d).
Figure 4.
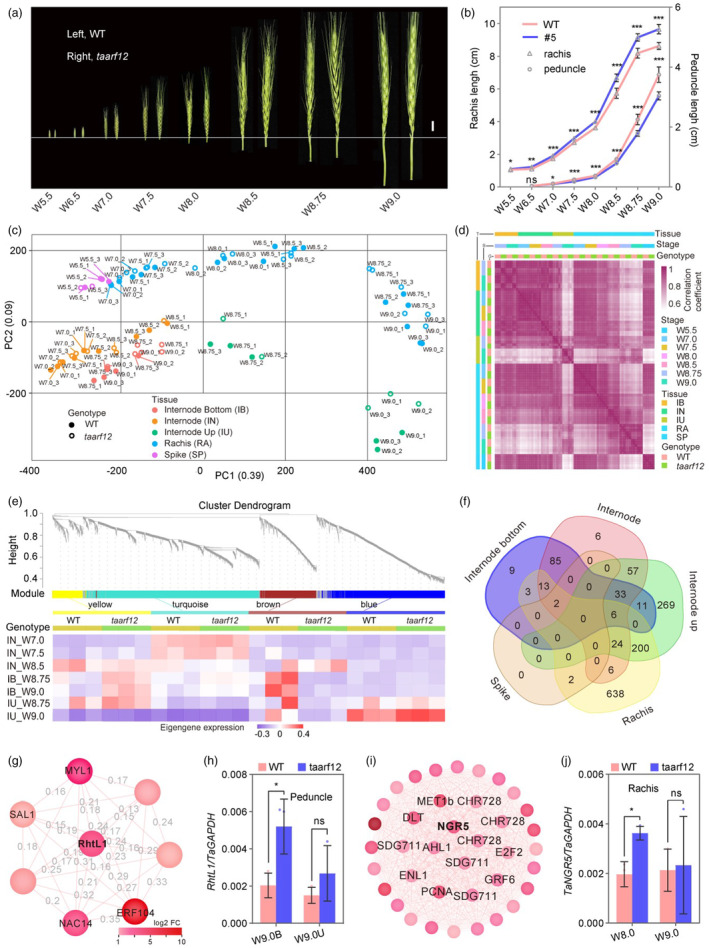
Contrasting transcriptome profiles in peduncle and rachis. (a) Spikes with peduncles of WT and taarf12 plants at various developmental stages were indicated. Scale bar, 1 cm. (b) Comparative statistics of peduncle and rachis lengths. (c) Principal component analysis (PCA) of spatial–temporal transcriptome profiles of different tissues at various developmental stages in WT and taarf12 plants. (d) Heatmap of pairwise Pearson correlation coefficients of all transcriptome profiles. (e, top) Gene co‐expression modules (CMs) generated in WGCNA among internodes. (e, bottom) Differential eigengene expression patterns of CMs between the WT and taarf12 peduncles across developmental stages W7.0‐W9.0. Colour squares below each genotype were from three biological replicates. (f) Venn diagram showing numbers of hub genes (signed KME > 0.9) shared among young stem internode (IN), the bottom of internode, the upper of internode (IU), spike and rachis (RA). (g) The expression weight network for eight genes in the brown module from the peduncle. (g) Relative expression levels of RhtL1 during peduncle development. (i) The expression weight network of 38 genes with known functions from the turquoise module of the rachis. (j) Relative expression levels of TaNGR5 during rachis development. Data were mean ± SD; P values were determined by two‐tailed Student's t‐test. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
WGCNA revealed RhtL1 and TaNGR5 as two hub genes in peduncle and rachis respectively
We then performed Weighted Gene Co‐Expression Network Analysis (WGCNA) across experiments and samples. A total of 53 976 non‐redundant differentially expressed genes (DEGs) were identified from 52 270 tissue‐specific DEGs (IN, IB, IU, RA), 48 008 development‐associated DEGs and 17 231 genotype‐associated DEGs (Figure S10a, Tables S3 and S4). To make the analysis more effective, we focused on 6687 DEGs that were annotated as transcription factors, hormone‐ and photosynthesis‐related genes in MapMan Bins (Figure S10b and Table S5). WGCNA revealed four peduncle modules with 343–1880 genes and six spike/rachis modules with 162–2095 genes (Figure 4e; Figures S11, S12 and Tables S6, S7). Eigengene expression patterns were different for these modules that showed distinctive gene ontology (GO) enrichment terms (Figure 4e; Figures S11, S12 and Tables S8, S9). For example, in the brown module, eigengene expression level was up‐regulated at W8.75 and W9.0 in the taarf12‐IB that was most significantly enriched for regulation of jasmonic acid (JA)‐mediated signalling pathway (Figure 4e; Table S8). In contrast, the eigengene expression level in the yellow module was down‐regulated at W8.75 and W9.0 in taarf12‐IB, which was significantly enriched with genes for positive regulation of development. WGCNA, thus, demonstrated that taarf12 mutation caused gene expression changes in core regulatory networks in both tissue‐specific and developmental stage‐specific manners.
Since hub genes are highly connected with other genes and may play central roles in gene networks, we then studied their functions in view of their tissue specificity. A total of 232, 162, 600, 4 and 894 hub genes (kME > 0.90) were identified in IN, IB, IU, SP, and RA respectively (Figure 4f). Among them, 200 were shared between IU and RA, while 638, 269 and 9 were specific to RA, IU and IB respectively (Figure 4f; Tables S10–S13). These tissue‐specific hub genes displayed distinctive functions as shown by GO enrichment analysis, especially the nine IB‐specific hub genes that were enriched for negative regulation of the gibberellic acid‐mediated signalling pathway (Table S14). Further scrutiny of these IB‐specific hub genes identified TraesCS3D03G0554000, an ortholog of the rice SLR1‐like gene (SLRL1) that encoded a DELLA protein lacking the DELLA domain (Itoh et al., 2005), and hence was named RhtL1 (Figure 4g; Figure S13a,c and Table S15). RhtL1 was specifically up‐regulated in IB, but not in IU and RA in the taarf12 mutant, suggesting that probable GA signalling reduction in IB that may be responsible for plant height reduction (Figure 4h; Figure S14a–c). Meanwhile, we found among 200 RA‐IU‐shared hub genes TraesCS1D03G0590300, the ortholog of the rice NGR5 (NITROGEN‐MEDIATED TILLER GROWTH RESPONSE 5) gene that was up‐regulated in RA at W8.0 stage that may contribute to rapid elongation in rachis (Figure 4i,j; Figures S13b,d, S14d–f). The rice NGR5 encodes an APETALA2‐domain transcription factor and is a target of gibberellin receptor GIBBERELLIN INSENSITIVE DWARF1 (GID1)‐promoted proteasomal destruction and competes with the DELLA protein (Wu et al., 2020). Specific up‐regulation of TaNGR5 in taarf12 rachis may promote GA signalling that contributed to larger spikes. Thus, it appeared that genome editing of TaARF12 may have caused differential regulation of GA signalling in peduncle and rachis.
RhtL1 and TaNGR5 for differential GA signalling in peduncle and rachis
To further confirm the roles of RhtL1 and TaNGR5 in GA signalling, we firstly studied the microsynteny of the two genes in the wheat and rice genomes. Indeed, the two genes were all located in collinear regions in the two genomes, with eight and nine genes out of 11 being correspondent homologs respectively (Figure S13a,b). Phylogenetic analysis supported the orthologous relationship of the two genes with their rice counterparts (Figure S13c,d and Table S15), consistent with protein sequence similarity comparison where RhtL1 showed more similarity to SLRL1 than to Rht‐1 and SLR1 (Figure 5a; Figure S15a). These data supported the idea of the existence of the two types of DELLA‐like genes in the common ancestor of wheat and rice.
Figure 5.
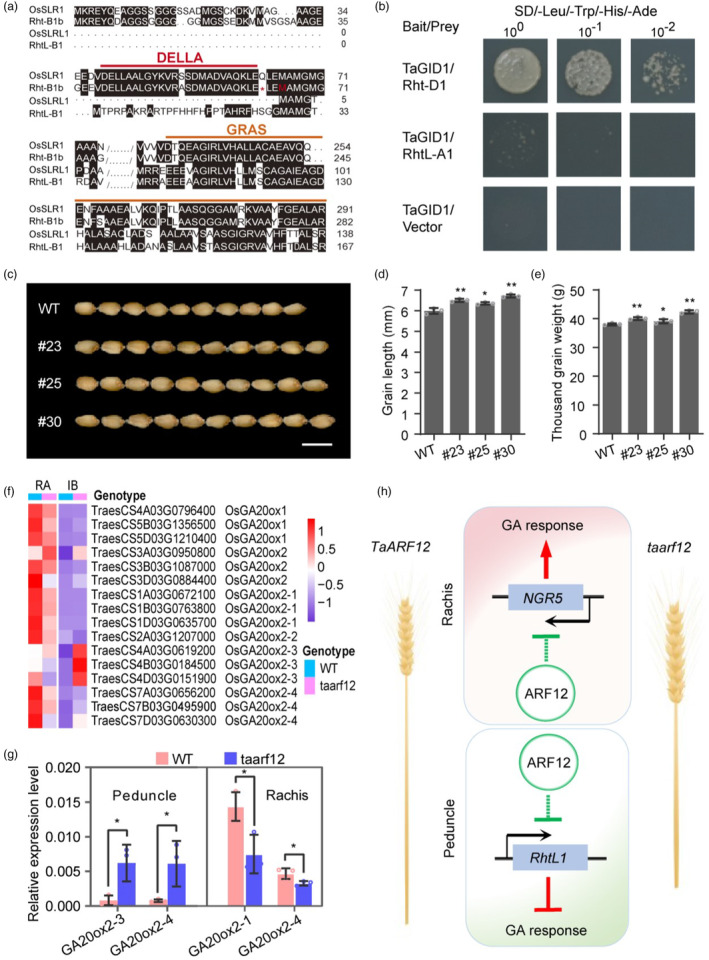
A working model for the functions of TaARF12 on wheat plant architecture. (a) Sequence alignment of Rht‐1, RhtL1, SLR1 and SLRL1. (b) TaGID1 only interacts with Rht‐1 in yeast cells, not RhtL1. (c–e) Over‐expression of TaNGR5 increased grain length (d) and thousand‐grain weight (e). Scale bar, 1 cm. (f) Heatmap showing spatial–temporal expression patterns of DEGs in the GA pathway. (g) GA biosynthesis genes expression patterns in the peduncle and rachis. (h) A working model for TaARF12. Lines with arrows at the end indicate positive regulation and lines ended with a perpendicular line represent negative regulation. Data were mean ± SD; P values were determined by two‐tailed Student's t‐test. *P < 0.05; **P < 0.01; ***P < 0.001.
Based on the roles of their orthologs in rice, we deduce that in peduncle, up‐regulation of RhtL1 by taarf12 may provide cumulative repression on GA signalling that further reduced the peduncle length of the transgenic receptor line cv. Fielder which carries the Rht‐B1b gene. Moreover, unlike Rht‐1, RhtL1 was unable to interact with GID1 as shown by yeast two‐hybrid assay in wheat (Figure 5b), demonstrating conserved functions of the gene in rice and wheat. These observations were further supported by the fact that two GA20 oxidase 2 homologs TraesCS4A03G0619200 (TaGA20ox2‐3) and TraesCS7A03G0656200 (TaGA20ox2‐4) for GA biosynthesis were specifically up‐regulated starting W8.75 in the peduncle of taarf12 plants, especially in IB (Figure 5f,g; Figure S16), indicative of GA signalling repression. The lack of a DELLA domain voided the degradation of RhtL1 by the proteasomal pathway but provided an alternative route to modulate GA signalling by transcription regulation.
On the other hand, NGR5 overexpression in wheat resulted in significant increase in grain length, grain width and thousand‐grain weight (Figure 5c–e; Figure S17), similar to its roles in rice (Hirano et al., 2017; Qiao et al., 2017). The interaction of NGR5 with GID1 was confirmed in wheat (Figure S15b). Moreover, genes for chromatin modification complex were found in the same co‐expression module as NGR5 which is known in rice to be integral for NGR5 functions (Figures S18 and S19). In contrast to that in the peduncle, GA20 oxidase 2 homologs TraesCS1A03G0672100 (TaGA20ox2‐1) and TraesCS7A03G0656200 (TaGA20ox2‐4) were down‐regulated in rachis (Figure 5f,g; Figure S16), suggesting a promotion of GA signalling. These data proposed a working model for the functions of TaARF12 in wheat plant height development and yield increase. As shown in Figure 5h, we hypothesized that TaARF12 mutation exerted its function by de‐repressing the transcription of RhtL1 and NGR5 in stem and spike, respectively, that caused differential responses of GA signalling and resulting in reduced plant height and enlarged spikes that may contribute to the significant yield increase in wheat.
Discussion
The “Green Revolution” greatly alleviated the problem of food shortage at the time. The reduced plant height enhances plants against lodging and allows dense plant growth and hence higher grain yield (Pingali, 2012). Despite this, the genes reduced stem diameter, spike length and thousand‐grain weight, leaving space for yield improvement by further modifying plant architecture (Van De Velde et al., 2017). Such effects can be achieved by multiple biological functions of the same gene, so‐called gene pleiotropy, which in more cases than not produces one advantage trait at the expense of the other. Therefore, efforts to seek better semidwarf genes have never stopped (Pearce, 2021; Tang et al., 2021; Tian et al., 2022). We showed here a new semidwarf gene TaARF12 that may compensate for the yield potential left since the Green Revolution. TaARF12 mutation can further reduce plant height in green revolution varieties in an agriculturally applicable manner that may re‐enforce lodging resistance. More excitingly, the degree of plant height reduction appeared to be programmable because the effect was correlated with the copy number of genes edited. Despite the wider tiller angle and the larger flag leaf that may negatively impact plant growing density, the harvest index was still increased, which was confirmed by the final grain gain in field trials. Thus, TaARF12 represented a new source of semidwarf genes that confer further yield potential with minimum negative effects.
The GR genes Rht1 and Rht2 encode the DELLA‐type proteins involved in GA signalling throughout wheat plants, making both stem and spike shorter and smaller (Peng et al., 1999; Van De Velde et al., 2017). In contrast, TaARF12 appeared to work oppositely in peduncle and rachis. Such genetic effects may represent a new gene regulatory strategy for further yield improvement in wheat. In this work, a comprehensive WGCNA of transcriptome profiles from multiple tissues and organs at various developmental stages from the wild‐type and taarf12 mutant plants was performed. We identified contrasting transcriptome regulation modes in peduncle and rachis. Two distinctive hub genes, RhtL1 and TaNGR5, were identified through WGCNA. Genome co‐linearity, phylogenetic analysis and protein–protein interaction evidence supported largely conserved functions of these two genes in rice and wheat.
In rice, the DELLA‐free SLRL1 acted as an offshoot of the canonical GA signalling pathway and lost its interaction with the GA receptor GID1, allowing GA signalling regulation by transcription regulation (Itoh et al., 2005). In wheat taarf12 plants, up‐regulation of RhtL1 potentially repressed GA signalling which was highly possible to be responsible for plant height reduction. The second hub gene TaNGR5 was confirmed by overexpression to increase organ size that may contribute to larger spikes and grains in taarf12. Moreover, multi‐omics database search showed that TaNGR5 was indeed a predicted downstream gene of TaARF12 (Chen et al., 2023).
Despite significant progress in understanding the molecular mechanisms for crop yield, most of them studied hormone‐related genes (Eshed and Lippman, 2019). Our work showed that the pleiotropic effects of the auxin response factor TaARF12 were dependent on differential regulation of GA signalling and its elegant feedback mechanism. Defects in GA signalling usually resulted in elevated GA synthesis due to increased expression of GA biosynthetic genes, or vice versa (Schwechheimer, 2008). Indeed, our results indicated that RhtL1 acts as a repressor of GA signalling, while TaNGR5 acts as a promoter of GA signalling. Therefore, the repressed GA signalling in peduncle and the promoted GA signalling in rachis were hypothesized to be underlining the concerted regulation of two important agronomic traits to increase grain yield. Genome editing of TaARF12 may produce wheat varieties with semidwarf statures and enlarged spikes and hence higher yield and represents valuable resources for crop improvement. New strategies such as cis‐element editing provide novel opportunities to effectively pyramid multiple traits using single or multiple genes without much adverse penalty (Song et al., 2022). The continuous effort to seek more optimal plant architecture with maximum yield potential should, in its own way, contribute to the world food security.
Materials and methods
Plant materials and field growth conditions
Wheat plants were grown in experimental stations in Beijing, China (N 40.23°, E 116.56°) from February to June during 2020–2021 and Hebei, China (N 37.84°, E 114.83°) from October to June during 2021–2022 for phenotyping and grain yield assessment. For experiments using spikes and internodes, plants were grown in a greenhouse under the conditions of 16‐h day, 22 °C and 8‐h night, 18 °C.
Constructs and wheat transformation
The CRISPR/Cas9 construct was developed as reported previously (Xing et al., 2014). Two gRNA target sites were selected in conserved exon regions of the TaARF12‐A, ‐B and ‐D. OsU3ter‐TaU3p sequence was amplified from the pCBC‐MT1T2 vector using specific primers and guide RNA sequences and cloned into the pBUE411 vector via digestion with BsaI and ligation with T4 DNA ligase. The full‐length open reading frame (ORF) of TaNGR5 was amplified and used to construct the pUbi::TaNGR5 overexpression transgene, as described elsewhere (Kong et al., 2022). The above constructs were used to transform wheat cv. Fielder by the Agrobacterium‐mediated transformation method (Wang et al., 2017). Primers are listed in Table S16.
In situ hybridization
Wheat spikes and peduncles of stage W7.5 were collected in 3.7% formol‐acetic‐alcohol (FAA). The samples were infiltrated for 0.5–1 h by applying vacuum and fixed with fresh FAA at 4 °C overnight, then, dehydrated, infiltrated, embedded in Paraplast Plus and sectioned at 8 μm using a Leica RM2235 microtome (Leica, Wetzlar, Germany). Digoxigenin‐labelled RNA antisense and sense probes of TaARF12 were synthesized in vivo under T7 RNA polymerase with a DIG RNA labelling kit (Roche, Basel, Switzerland). RNA in situ hybridization was performed as described previously (Liu et al., 2014). Images were obtained using a ZEISS AXIO IMAGER Z2. Primers for probe synthesis are listed in Table S16.
Histological analysis
Peduncles, spikes and rachises of the wild‐type Fielder and taarf12 plants stored in paraffin were analysed as described previously (Liu et al., 2014). The prepared tissue sections were stained with 0.05% (w/v) toluidine blue for 5 s and images were collected with a light microscope (AXIO IMAGER Z2; ZEISS, Oberkochen, Germany). Cell size and cell number were calculated using the Image J software.
Yeast two‐hybrid assay
The yeast two‐hybrid vectors pGBKT7 and pGADT7 were constructed according to the MatchMaker GAL4 Two‐Hybrid System 3 (Clontech, CA, USA) manufacturer's manual. The ORF of TaGID1 and TaNGR5 were cloned into the pGADT7 vector to obtain the pGADT7‐TaGID1 and pGADT7‐TaNGR5. The full‐length ORF of TaNGR5, RhtL1 and Rht1 was cloned into the pGBKT7 vector to obtain the pGBKT7‐TaNGR5, pGBKT7‐RhtL1 and pGBKT7‐Rht1. PCR was performed using Phanta Super‐Fidelity DNA Polymerase (Vazyme Biotech, Nanjing, China). Primers are listed in Table S16.
RNA extraction and qRT–PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen, CA, USA) and reverse transcribed into cDNA using the PrimeScript RT reagent Kit (TaKaRa, Tokyo, Japan) according to the manufacturer's instructions. qRT‐PCR assays were performed using SYBR Premix EX Taq (Takara) on an ABI 7500 real‐time PCR system in a total volume of 25 μL and TaGAPDH was used as the internal control. Three biological replicates were performed for each experiment. All PCR primers used are listed in Table S16.
Statistical analyses
Statistical analysis was performed in GraphPad Prism 8.0.2. Hollow plots represent individual data points. P values were tested using two‐tailed Student's t‐tests and are given in the figure legends.
RNA sequencing and data processing
Samples used for RNA‐seq analysis were prepared from spike (W5.5), rachis (W7.0, W7.5, W8.0, W8.5, W8.75, W9.0; Waddington et al., 1983), the whole internode (peduncle; W7.0, W7.5, W8.5), the bottom part of the peduncle (IB) and the upper part of the peduncle (IU) at stages W8.75 and W9.0 in the wild‐type and taarf12 plants in the greenhouse, with two or three biological replicates. Total RNA was extracted using the TRIzol reagent (Invitrogen). mRNA sequencing libraries were generated following the Illumina manufacturer's recommendations. An average of 57.0 million 150‐bp pair‐end raw reads were generated for each sample on the Illumina HiSeq 2000 platform. Quality control was conducted using the fastp version 0.20.1 with the parameter “‐u 30 ‐q 20 ‐l 120” to generate approximately 55.8 million high‐quality pair‐end clean reads for each sample. The wheat reference genome sequence (IWGSC RefSeq v2.1) and annotations were downloaded from URGI (https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/v2.1/). Reads were aligned to the genome using HISAT2 (v 2.1.0; Kim et al., 2019), with default parameters. Reads with multiple hits and mapping quality <60 were removed. As a result, average of 85.67% of reads were aligned to the genome concordantly exactly one time (Table S2). HTSeq (v 0.13.5) was used to count the read numbers mapped to the gene models (Anders et al., 2015). For all comparisons, read counts were normalized to the aligned FPKM (fragments per kilobase of transcript per million mapped reads) to obtain the relative levels of expression. Differential expression analyses between different developmental stages or tissues were performed using the DESeq2 (v 1.36.0) R packages. We used adjusted P‐value < 0.05 and |log2FC| ≥ 1 to identify differentially expressed genes (DEGs; Appels et al., 2018).
Principal component analysis (PCA) and sample correlation analysis
All expression genes with expression values (FPKM) > 1 in at least one sample were used for PCA using the prcomp function in R, with default settings. We calculated Pearson correlation coefficients between pairwise samples with the FPKM values of all expressed genes.
Weighted gene co‐expression network analysis (WGCNA)
WGCNA was performed on DEGs encoding transcription factors, hormone and photosynthesis‐related as annotated by MapMan (v 3.5.1R2; Langfelder and Horvath, 2008; Liu et al., 2021). Gene expression dataset of spike (SP), rachis (RA), internode (IN), IB (the bottom of internode) and IU (the upper part of internode) individually using the functions in the R package WGCNA (v 1.70.3). The initial gene set was further filtered by FPKM with an average ≥1 in at least one of the three samples for the corresponding genotype and was within the top 75% of the above genes that had the highest median absolute deviation of FPKM across different development stages. The network analysis was performed using a soft threshold (power/β) that was determined to produce a scale‐free network with optimal scale‐free topology model fit and mean connectivity. The WGCNA blockwiseModules function was used to construct a signed network. Briefly, gene co‐expression relationships were calculated as bi‐weight mid‐correlation coefficients raised to the soft threshold, transforming the gene expression correlation adjacency matrix to a TOM, which was then converted to a dissimilarity matrix that was used to generate a hierarchical cluster tree. To identify the co‐expressed gene modules, we used the dynamic tree cut method with the following parameters: deepSplit level 2, detectCutHeight of 0.995, minModuleSize of 100 and tree mergeCutHeight of 0.25. The connectivity of each gene to its corresponding module was calculated using a module membership value that was defined as the bi‐weight mid‐correlation between the gene expression and the corresponding ME. Hub genes within each module were calculated using the WGCNA R package function signedKME6. The RNA‐seq analysis strategy is shown in Figure S20.
Conflict of interest
The authors declare no conflict of interest.
Authors' contributions
L.A. and M.L. designed the experiments. K.X., W.F., G.S., D.Z., F.M., C.D., C.Y. and L.R. performed the experiments; W.Z. contributed to RNA‐seq data analysis; G.X., Z.S., W.K. and Y.X. helped in generating the transgenic materials. Y.L., L.S., Z.P. and L.D. provided valuable advice about the project. L.A., K.X. W.F. and W.Z. drafted the manuscript. M.L. and F.X. revised the manuscript.
Supporting information
Figure S1 The genetic effect of the wheat Green Revolution gene Rht‐B1b that reduced plant height but produced smaller spikes.
Figure S2 CRISPR/Cas9 mediated mutagenesis of TaARF12.
Figure S3 Morphological comparison of stem internodes of the wild‐type and taarf12 transgenic lines.
Figure S4 Longitudinal dissection illustrated that taarf12 peduncles were shorter with fewer cells which may be responsible for reduced peduncle length while more cell layers presented in stem may make it stronger.
Figure S5 Comparison of the wild‐type and taarf12 plants showed that the longer spikes in the latter were caused by rachis internode elongation.
Figure S6 Longitudinal dissection of taarf12 rachis demonstrates that the enlarged rachis was caused by increased cell number and cell size relative to that of the wild type.
Figure S7 Phenotypic comparison of wild‐type and taarf12 peduncle and rachis during their development.
Figure S8 Statistics from field trials proved that the taarf12 plants boost grain yield.
Figure S9 Grain traits from field plants revealed increased grain yield in taarf12 plants.
Figure S10 Venn diagrams showing DEGs shared by different spatial, temporal and tissue comparisons.
Figure S11 Heatmap of enriched gene ontology (GO) genes in each co‐expression module of peduncle.
Figure S12 Gene co‐expression modules of spike (SP) and rachis (RA) at W5.5‐W9.0 of the wild‐type and taarf12.
Figure S13 Rice–wheat genome collinearity at RhtL1 and TaNGR5 regions and phylogenetic trees of related genes from related species.
Figure S14 Spatial–temporal expression patterns of RhtL1 and TaNGR5 in wheat.
Figure S15 Sequence similarity of wheat and rice, DELLA and DELLA‐like proteins and their protein–protein interaction patterns showing their functional conservation.
Figure S16 Heatmap showing spatial–temporal expression patterns of DEGs in the GA pathway.
Figure S17 TaNGR5 overexpression led to wider grains and higher thousand‐grain weight.
Figure S18 Heatmap showing spatial–temporal expression patterns of DEGs related to epigenetic modification.
Figure S19 Confirmation of expression patterns of epigenetic modification genes in the wild‐type and taarf12 plants.
Figure S20 The RNA‐seq analysis pipeline.
Table S1 Genome editing site information of TaARF12.
Table S2 RNA‐seq data output and the alignment rate.
Table S3 Numbers of DEGs from various comparisons.
Table S4 DEGs list from various type.
Table S5 The selected gene set with MapMan annotations for WGCNA.
Table S6 The gene list of peduncles WGCNA modules.
Table S7 The gene list of spikes/rachises WGCNA modules.
Table S8 List of enriched GO terms for internode WGCNA modules.
Table S9 List of enriched GO terms for rachis WGCNA modules.
Table S10 List of rachis‐specific DEG hub genes (638).
Table S11 List of DEG hub genes (269) specific to the upper internode.
Table S12 List of DEG hub genes (9) specific to the internode bottom portion.
Table S13 List of 200 DEG hub genes shared in the upper internode and rachis.
Table S14 List of enriched GO terms among hub genes.
Table S15 Gene IDs for phylogenetic trees of SLRL1 and NGR5.
Table S16 Primers used in this study.
Acknowledgements
We acknowledge the financial support from National Natural Science Foundation of China (31971930, 32172050, 31921005), National major agricultural science and technology project (NK2022060101), National Key Research and Development Program of China (2021YFF1000204), Hainan Yazhou Bay Seed Lab (B21HJ0215), CAAS Agricultural Science and Technology Innovation Program (CAAS‐ZDRW202002, CAAS‐ZDRW202201), Hebei Natural Science Foundation (C2021205013) and the Youth Innovation of Chinese Academy of Agricultural Sciences (Y2023QC38). We thank Dr. Xigang Liu of Hebei Normal University for valuable advice. We thank the bioinformatics facility at ICS‐CAAS for computing support. LM is a “Yellow River Delta Scholar” at Sino‐Agro Experimental Station for Salt Tolerant Crops, Dongying, Shandong Province, China.
Contributor Information
Xiangdong Fu, Email: xdfu@genetics.ac.cn.
Long Mao, Email: maolong@caas.cn.
Aili Li, Email: liaili@caas.cn.
Data availability
All data supporting the findings of this study are available in the article and its Supplemental figures and tables can be obtained from the corresponding authors upon reasonable request. The sequencing data have been deposited in a National Center for Biotechnology Information BioProject database under accessions PRJNA869883.
References
- Anders, S. , Pyl, P.T. and Huber, W. (2015) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appels, R. , Eversole, K. , Stein, N. , Feuillet, C. , Keller, B. , Rogers, J. , Pozniak, C.J. et al. (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 361, eaar7191. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Guo, Y. , Guan, P. , Wang, Y. , Wang, X. , Wang, Z. , Qin, Z. et al. (2023) A wheat integrative regulatory network from large‐scale complementary functional datasets enables trait‐associated gene discovery for crop improvement. Mol. Plant, 16, 393–414. [DOI] [PubMed] [Google Scholar]
- Cheng, J. , Hill, C. , Han, Y. , He, T. , Ye, X. , Shabala, S. , Guo, G. et al. (2023) New semi‐dwarfing alleles with increased coleoptile length by gene editing of gibberellin 3‐oxidase 1 using CRISPR‐Cas9 in barley (Hordeum vulgare L.). Plant Biotechnol. J. 21, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesterfield, R.J. , Vickers, C.E. and Beveridge, C.A. (2020) Translation of strigolactones from plant hormone to agriculture: achievements, future perspectives, and challenges. Trends Plant Sci. 25, 1087–1106. [DOI] [PubMed] [Google Scholar]
- Dixon, L.E. , Greenwood, J.R. , Bencivenga, S. , Zhang, P. , Cockram, J. , Mellers, G. , Ramm, K. et al. (2018) TEOSINTE BRANCHED1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum). Plant Cell, 30, 563–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed, Y. and Lippman, Z.B. (2019) Revolutions in agriculture chart a course for targeted breeding of old and new crops. Science, 366, eaax0025. [DOI] [PubMed] [Google Scholar]
- Ford, B.A. , Foo, E. , Sharwood, R. , Karafiatova, M. , Vrána, J. , MacMillan, C. , Nichols, D.S. et al. (2018) Rht18 semidwarfism in wheat is due to increased GA 2‐oxidaseA9 expression and reduced GA content. Plant Physiol. 177, 168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W. , Chen, L. , Herrera‐Estrella, L. , Cao, D. and Tran, L.‐S.P. (2020) Altering plant architecture to improve performance and resistance. Trends Plant Sci. 25, 1154–1170. [DOI] [PubMed] [Google Scholar]
- Hirano, K. , Yoshida, H. , Aya, K. , Kawamura, M. , Hayashi, M. , Hobo, T. , Sato‐Izawa, K. et al. (2017) SMALL ORGAN SIZE 1 and SMALL ORGAN SIZE 2/DWARF AND LOW‐TILLERING form a complex to integrate auxin and brassinosteroid signaling in rice. Mol. Plant, 10, 590–604. [DOI] [PubMed] [Google Scholar]
- Itoh, H. , Shimada, A. , Ueguchi‐Tanaka, M. , Kamiya, N. , Hasegawa, Y. , Ashikari, M. and Matsuoka, M. (2005) Overexpression of a GRAS protein lacking the DELLA domain confers altered gibberellin responses in rice. Plant J. 44, 669–679. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Paggi, J.M. , Park, C. , Bennett, C. and Salzberg, S.L. (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat. Biotechnol. 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, X. , Wang, F. , Geng, S. , Guan, J. , Tao, S. , Jia, M. , Sun, G. et al. (2022) The wheat AGL6‐like MADS‐box gene is a master regulator for floral organ identity and a target for spikelet meristem development manipulation. Plant Biotechnol. J. 20, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, A. , Hao, C. , Wang, Z. , Geng, S. , Jia, M. , Wang, F. , Han, X. et al. (2022) Wheat breeding history reveals synergistic selection of pleiotropic genomic sites for plant architecture and grain yield. Mol. Plant, 15, 504–519. [DOI] [PubMed] [Google Scholar]
- Liu, D. , Wang, D. , Qin, Z. , Zhang, D. , Yin, L. , Wu, L. , Colasanti, J. et al. (2014) The SEPALLATA MADS‐box protein SLMBP 21 forms protein complexes with JOINTLESS and MACROCALYX as a transcription activator for development of the tomato flower abscission zone. Plant J. 77, 284–296. [DOI] [PubMed] [Google Scholar]
- Liu, P. , Liu, J. , Dong, H. and Sun, J. (2018) Functional regulation of Q by microRNA172 and transcriptional co‐repressor TOPLESS in controlling bread wheat spikelet density. Plant Biotechnol. J. 16, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , He, G. and Deng, X. (2021) Biological pathway expression complementation contributes to biomass heterosis in Arabidopsis. Proc. Natl Acad. Sci. USA, 118, e2023278118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, V. , McDermott, H.I. , McAllister, T. , Cummins, M. , Silva, J.C. , Mollison, E. , Meikle, R. et al. (2019) APETALA2 control of barley internode elongation. Development, 146, dev170373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce, S. (2021) Towards the replacement of wheat ‘Green Revolution’ genes. J. Exp. Bot. 72, 157–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, J. , Richards, D.E. , Hartley, N.M. , Murphy, G.P. , Devos, K.M. , Flintham, J.E. , Beales, J. et al. (1999) ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature, 400, 256–261. [DOI] [PubMed] [Google Scholar]
- Pingali, P.L. (2012) Green revolution: impacts, limits, and the path ahead. Proc. Natl Acad. Sci. USA, 109, 12302–12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, S. , Sun, S. , Wang, L. , Wu, Z. , Li, C. , Li, X. , Wang, T. et al. (2017) The RLA1/SMOS1 transcription factor functions with OsBZR1 to regulate brassinosteroid signaling and rice architecture. Plant Cell, 29, 292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, A. , Ashikari, M. , Ueguchi‐Tanaka, M. , Itoh, H. , Nishimura, A. , Swapan, D. , Ishiyama, K. et al. (2002) A mutant gibberellin‐synthesis gene in rice. Nature, 416, 701–702. [DOI] [PubMed] [Google Scholar]
- Schwechheimer, C. (2008) Understanding gibberellic acid signaling—are we there yet? Curr. Opin. Plant Biol. 11, 9–15. [DOI] [PubMed] [Google Scholar]
- Sims, K. , Abedi‐Samakush, F. , Szulc, N. , Macias Honti, M.G. and Mattsson, J. (2021) OsARF11 promotes growth, meristem, seed, and vein formation during rice plant development. Int. J. Mol. Sci. 22, 4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X. , Meng, X. , Guo, H. , Cheng, Q. , Jing, Y. , Chen, M. , Liu, G. et al. (2022) Targeting a gene regulatory element enhances rice grain yield by decoupling panicle number and size. Nat. Biotechnol. 40, 1403–1411. [DOI] [PubMed] [Google Scholar]
- Su, S. , Hong, J. , Chen, X. , Zhang, C. , Chen, M. , Luo, Z. , Chang, S. et al. (2021) Gibberellins orchestrate panicle architecture mediated by DELLA–KNOX signalling in rice. Plant Biotechnol. J. 19, 2304–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, L. , Yang, W. , Li, Y. , Shan, Q. , Ye, X. , Wang, D. , Yu, K. et al. (2019) A wheat dominant dwarfing line with Rht12, which reduces stem cell length and affects gibberellic acid synthesis, is a 5AL terminal deletion line. Plant J. 97, 887–900. [DOI] [PubMed] [Google Scholar]
- Tang, T. , Botwright, A.T. , Spielmeyer, W. and Richards, R.A. (2021) Effect of gibberellin‐sensitive Rht18 and gibberellin‐insensitive Rht‐D1b dwarfing genes on vegetative and reproductive growth in bread wheat. J. Exp. Bot. 72, 445–458. [DOI] [PubMed] [Google Scholar]
- Tian, X. , Xia, X. , Xu, D. , Liu, Y. , Xie, L. , Hassan, M.A. , Song, J. et al. (2022) Rht24b, an ancient variation of TaGA2ox‐A9, reduces plant height without yield penalty in wheat. New Phytol. 233, 738–750. [DOI] [PubMed] [Google Scholar]
- Van De Velde, K. , Chandler, P.M. , Van Der Straeten, D. and Rohde, A. (2017) Differential coupling of gibberellin responses by Rht‐B1c suppressor alleles and Rht‐B1b in wheat highlights a unique role for the DELLA N‐terminus in dormancy. J. Exp. Bot. 68, 443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Velde, K. , Thomas, S.G. , Heyse, F. , Kaspar, R. , Van Der Straeten, D. and Rohde, A. (2021) N‐terminal truncated RHT‐1 proteins generated by translational reinitiation cause semi‐dwarfing of wheat Green Revolution alleles. Mol. Plant, 14, 679–687. [DOI] [PubMed] [Google Scholar]
- Waddington, S. , Cartwright, P. and Wall, P. (1983) A quantitative scale of spike initial and pistil development in barley and wheat. Ann. Bot. 51, 119–130. [Google Scholar]
- Wang, K. , Liu, H. , Du, L. and Ye, X. (2017) Generation of marker‐free transgenic hexaploid wheat via an Agrobacterium‐mediated co‐transformation strategy in commercial Chinese wheat varieties. Plant Biotechnol. J. 15, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, K. , Wang, S. , Song, W. , Zhang, J. , Wang, Y. , Liu, Q. , Yu, J. et al. (2020) Enhanced sustainable green revolution yield via nitrogen‐responsive chromatin modulation in rice. Science, 367, eaaz2046. [DOI] [PubMed] [Google Scholar]
- Xing, H. , Dong, L. , Wang, Z. , Zhang, H. , Han, C. , Liu, B. , Wang, X. et al. (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 14, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Wang, S. , Xu, Y. , Yu, C. , Shen, C. , Qian, Q. , Geisler, M. et al. (2015) The auxin response factor, OsARF19, controls rice leaf angles through positively regulating OsGH 3‐5 and OsBRI 1. Plant Cell Environ. 38, 638–654. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Li, A. , Song, G. , Geng, S. , Gill, B.S. , Faris, J.D. and Mao, L. (2020) Comprehensive analysis of Q gene near‐isogenic lines reveals key molecular pathways for wheat domestication and improvement. Plant J. 102, 299–310. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Jia, H. , Li, T. , Wu, J. , Nagarajan, R. , Lei, L. , Powers, C. et al. (2022) TaCol‐B5 modifies spike architecture and enhances grain yield in wheat. Science, 376, 180–183. [DOI] [PubMed] [Google Scholar]
- Zhao, Z. , Yin, X. , Li, S. , Peng, Y. , Yan, X. , Chen, C. , Hassan, B. et al. (2022) miR167d‐ARFs module regulates flower opening and stigma size in rice. Rice, 15, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The genetic effect of the wheat Green Revolution gene Rht‐B1b that reduced plant height but produced smaller spikes.
Figure S2 CRISPR/Cas9 mediated mutagenesis of TaARF12.
Figure S3 Morphological comparison of stem internodes of the wild‐type and taarf12 transgenic lines.
Figure S4 Longitudinal dissection illustrated that taarf12 peduncles were shorter with fewer cells which may be responsible for reduced peduncle length while more cell layers presented in stem may make it stronger.
Figure S5 Comparison of the wild‐type and taarf12 plants showed that the longer spikes in the latter were caused by rachis internode elongation.
Figure S6 Longitudinal dissection of taarf12 rachis demonstrates that the enlarged rachis was caused by increased cell number and cell size relative to that of the wild type.
Figure S7 Phenotypic comparison of wild‐type and taarf12 peduncle and rachis during their development.
Figure S8 Statistics from field trials proved that the taarf12 plants boost grain yield.
Figure S9 Grain traits from field plants revealed increased grain yield in taarf12 plants.
Figure S10 Venn diagrams showing DEGs shared by different spatial, temporal and tissue comparisons.
Figure S11 Heatmap of enriched gene ontology (GO) genes in each co‐expression module of peduncle.
Figure S12 Gene co‐expression modules of spike (SP) and rachis (RA) at W5.5‐W9.0 of the wild‐type and taarf12.
Figure S13 Rice–wheat genome collinearity at RhtL1 and TaNGR5 regions and phylogenetic trees of related genes from related species.
Figure S14 Spatial–temporal expression patterns of RhtL1 and TaNGR5 in wheat.
Figure S15 Sequence similarity of wheat and rice, DELLA and DELLA‐like proteins and their protein–protein interaction patterns showing their functional conservation.
Figure S16 Heatmap showing spatial–temporal expression patterns of DEGs in the GA pathway.
Figure S17 TaNGR5 overexpression led to wider grains and higher thousand‐grain weight.
Figure S18 Heatmap showing spatial–temporal expression patterns of DEGs related to epigenetic modification.
Figure S19 Confirmation of expression patterns of epigenetic modification genes in the wild‐type and taarf12 plants.
Figure S20 The RNA‐seq analysis pipeline.
Table S1 Genome editing site information of TaARF12.
Table S2 RNA‐seq data output and the alignment rate.
Table S3 Numbers of DEGs from various comparisons.
Table S4 DEGs list from various type.
Table S5 The selected gene set with MapMan annotations for WGCNA.
Table S6 The gene list of peduncles WGCNA modules.
Table S7 The gene list of spikes/rachises WGCNA modules.
Table S8 List of enriched GO terms for internode WGCNA modules.
Table S9 List of enriched GO terms for rachis WGCNA modules.
Table S10 List of rachis‐specific DEG hub genes (638).
Table S11 List of DEG hub genes (269) specific to the upper internode.
Table S12 List of DEG hub genes (9) specific to the internode bottom portion.
Table S13 List of 200 DEG hub genes shared in the upper internode and rachis.
Table S14 List of enriched GO terms among hub genes.
Table S15 Gene IDs for phylogenetic trees of SLRL1 and NGR5.
Table S16 Primers used in this study.
Data Availability Statement
All data supporting the findings of this study are available in the article and its Supplemental figures and tables can be obtained from the corresponding authors upon reasonable request. The sequencing data have been deposited in a National Center for Biotechnology Information BioProject database under accessions PRJNA869883.