

Abstract
Recent work detailed the unique characteristics of fragmentation spectra derived from peptides from single human cells. This valuable report utilized an ultrahigh-field Orbitrap and directly compared the spectra obtained from high-concentration bulk cell HeLa lysates to those obtained from nanogram dilutions of the same and from nanowell-processed single HeLa cells. The analysis demonstrated marked differences between the fragmentation spectra generated at high and single-cell loads, most strikingly, the loss of high-mass y-series fragment ions. As significant differences exist in the physics of Orbitrap and time-of-flight mass analyzers, a comparison appeared warranted. A similar analysis was performed using isolated single pancreatic cancer cells compared to pools consisting of 100 cells. While a reanalysis of the prior Orbitrap data supports the author’s original findings, the same trends are not observed in time-of-flight mass spectra of peptides from single human cells. The results are particularly striking when directly comparing the matched intensity fragment values between bulk and single-cell data generated on the same mass analyzers. Instrument acquisition files, processed data, and spectrum libraries are publicly available on MASSIVE via accession MSV000090635.
Keywords: single-cell proteomics, TIMSTOF single cell, Orbitrap single cell
Introduction
Single-cell proteomics is an emerging field of study with promise in revealing absolute granularity in phenotypic response.1−4 The field is being driven from multiple fronts including the innovative analysis of historically utilized reagents and improvements in sample handling,3,5 LCMS hardware,6,7 and informatics.8,9 Excitement in this field has led to development in every front with technology that is rapidly trickling down to benefit the fields of proteomics and mass spectrometry as a whole.10,11
A recent analysis by Boekweg et al. made the surprising observation that fragmentation spectra generated on an ultra-highfield D20 Orbitrap system had unique characteristics compared to spectra obtained at higher relative peptide loads.12 These characteristics included an underrepresentation of y-series fragment ions that are not yet explained. Following an engaging conversation on #MassSpecTwitter, I sought to determine if these observations extended to fragmentation data generated using time-of-flight hardware. Files from studies in works at our lab were examined to find data that most closely matched the results in Boekweg et al. and employed TOF analyzers. Isolated single human pancreatic cancer cells treated with the KRASG12D inhibitor MRTX113313 were analyzed in August of 2022 using a data-dependent approach and 30 min of acquisition time on a TIMSTOF SCP prototype system (complete results to be published elsewhere). The numbers of MS2 spectra, peptides, and proteins were most similar to the numbers obtained from the 180 min Orbitrap single-cell files, and the same number of files was chosen and used for this brief comparison (Table 1).
Table 1. Summary of the Files Used in This Analysis.
| abbreviated file ID | description | no. of MS2 | no. of PSM | no. of peptides | no. of proteins |
|---|---|---|---|---|---|
| MRTX...A1 | 100 PANC02.03 cells, no reduction or alkylation, 30 min | 66 581 | 31 870 | 23 092 | 3474 |
| MRTX...A3–A5 | three files, each with one PANC02.03 cell as above, 30 min | 104 974 | 12 829 | 4820 | 994 |
| HeLa_200 ng_FAIMS | 200 ng of standard digest of HeLa on FAIMS 480, CV −50 and −70 | 59 184 | 20 774 | 18 933 | 3428 |
| PayneD19SC1-SC3 | three NanoPots single cells from Boekweg et al., 180 min | 68 082 | 15 800 | 6752 | 1336 |
Methods
Reanalysis of Ultrahigh-Field D20 Orbitrap Data
The original files described in Boekweg et al. were obtained from MASSIVE (MSV000087524). In this study, these authors utilized bulk HeLa data likewise deposited in MASSIVE (MSV000087689) that were deposited solely in mzML format. As this format is not compatible with the MS-Ana14 pipeline used in this work, I generated comparable data using a similarly equipped FAIMS Pro Exploris 480 system to analyze 200 ng of the same cancer cell line digest standard (Pierce 88328). Peptides were separated using an EasyNLC 1200 system using a 25 cm × 75 μm EasySpray C-18 column with a 2.0 μm particle size using a flow rate of 300 nL/min. A gradient beginning with 97% buffer A (0.1% formic acid in LCMS-grade water) and 3% B (0.1% formic acid in 80% acetonitrile) contained three separation gradient stages: first ramped to 19% B followed by a ramp to 29% B and finally to 41% B before a final step to 95% B for the remainder of the gradient. For the 60 and 120 min experiments, these ramp end times were 26, 40, 50, and 53 and 73, 101, 121, and 124 min, respectively. Two FAIMS compensation voltages were used, −50 and −70 in each experiment with MS1 scans acquired at 60 000 resolution from 350 to 1200 m/z. Fragmentation spectra were acquired on ions isolated with a 1.6 Da window using the “auto” ion accumulation time and normalized collision energy of 28 eV at 15 000 resolution.
Cell Isolation
A pancreatic adenocarcinoma epithelial cell line originally isolated from a female patient at the Johns Hopkins Hospital15,16 was obtained directly from a central repository that ensures cell line identity and absence of mycoplasma contamination (ATCC CRL-2553). Cells were cultured in RPMI-40 supplemented with 10% fetal bovine serum (ATCC 30-2001 and 30-2020) according to vendor protocols. After four passages, cells were removed from the plate using 3 mL of 0.25% trypsin EDTA solution (Gibco 2520014) to first wash away trypsin inhibitor followed by an incubation of approximately 5 min in the same at 37 °C to remove cells from the plate surface. Effective trypsinization was confirmed by microscopy. The trypsin was inactivated by adding 5 mL of 0.1% BSA solution (Thermo) and soybean trypsin inhibitor (Roche 10109886001). Cells were centrifuged at 300 × g for 3 min, and the remaining solution was poured off. Cells were resuspended in 0.1% BSA solution in calcium- and magnesium-free PBS by gentle tapping, and the solution was passaged by 1 mL pipet through a screen to separate clumps of cells (Falcon 5 mL FlowTube 352235). Cells were transported across the street to the Johns Hopkins University School of Public Health Cell Sorting and Sequencing Core on wet ice. Cells were stained with a propidium iodide cell viability marker and sorted directly onto microwell plates containing 2 μL of LCMS-grade acetonitrile. Cells were isolated using a DakoCytoMation MoFlo 3 analog laser cell sorter. Manual calibration of the cell sorter and cell deposition rate was performed by Dr. Hao Zhang of the sorting center. For bulk cell analysis, 100 cells were distributed into the first well in each row (A1, B1, etc.). The second column in each row was used as a method blank control with sorting buffer but no cell added. Finally, one single cell was placed in every remaining well. The deposition accuracy of cells was calculated following each sort using metrics for laser scatter previously validated in single-cell genomics studies16,17 and was estimated at greater than 95% accuracy over the approximately 8000 single cells isolated during this sort. Sorted plates were sealed immediately and placed on dry ice prior to −80 °C storage. To lyse cells and drive off remaining acetonitrile, cells were removed from the −80 °C freezer and placed directly on a 95 °C hot plate for 5 min. Cells were digested with 2 μL of 100 mM TEAB supplemented with 0.01% n-dodecyl-β-d-maltoside and containing approximately 2 ng of LCMS trypsin (Promega V5280) for 2 h at 45 °C. Following digestion, the plates were centrifuged to catch condensation and were dried by SpeedVac. Peptides were resuspended in 4 μL of 0.1% formic acid with repeated vortexing and centrifugation of the plates prior to loading on the EasyNLC1200.
nanoLC-TIMSTOF Analysis
Ninety-six well plates containing 100 cell bulk cell lysates and the lysates of single PANC 02.03 cells were directly loaded into an EasyNLC 1200 system, and peptides were separated using a method of 30 min total length using an IonOpticks Aurora 25 cm × 75 μm C-18 with 1.5 μm particle size. The 350 nL/min gradient began at 95% buffer A (0.1% formic acid in LCMS-grade water) and 5% B (0.1% formic acid in 80% acetonitrile) contained two separation gradient stages, first ramped to 25% B at 20 min followed by a ramp to 35% B at 25 min followed by a 2.5 min ramp to 95% B at 500 nL/min for the remainder of the gradient. The column temperature was maintained at 55 °C. The acquisition was performed using a TIMSTOF SCP system using the only default method included in the Compass 2022 software for the instrument. Briefly, a scan range of 0.6–1.6 1/k0 and 100–1700 m/z were employed with 10 ramps of 166 ms, resulting in an approximate 1.8 s cycle time. Ions were isolated for fragmentation with an intensity threshold of 500 counts and a target intensity of 20 000 counts. Ions that met the custom hand-drawn heat map polygon used on these instruments to help ensure low reproducibility between instruments and operators were minimum requirements for parent ion selection. In addition, an intensity threshold of 500 counts and target intensity of 20 000 counts were used for estimates for intrascan averaging parameters. Parent ion isolation was at 2.0 Th for ions up to 700 m/z and 3.0 Th for ions above 800 m/z, and the isolation windows between those points are anyone’s guess.
Comparison to Curated NIST Spectral Libraries
All Bruker .d files were converted to MGF using ProteoWizard MSConvert 3.0 using the default conversion for PASEF MGF. These steps include the merging of MS/MS spectra that possess parent ions within 0.1 1/k0, 0.05 Da and elute within a 5 s window mass of the parent ion. The TIMSTOF SCP MGF files and Thermo native. RAW files were directly imported into Proteome Discoverer 3.0 (Thermo Fisher) using the same search parameters and the MS-Ana search engine. The TIMSTOF MGF files were identified as FTMS spectra at 30 000 resolution to enable the full visualization of masses to three decimal points to enable uniform parameters for manual analysis.
The curated experimental spectral library used for all analyses was obtained from NIST (NIST_Human_Orbitrap_HCD, curated 9–23–2016) and contains approximately 1 × 106 spectra.18 Accessions were obtained from a UniProt SwissProt human FASTA library parsed in April of 2020, and FDR was estimated by allowing MS-Ana to create a randomly shuffled spectral library for each entry. The maximum number of spectra MS-Ana searched in RAM simultaneously was set to 50 000, and a 25 ppm MS1 tolerance and 0.03 Da MS2 search tolerance were used for all files for continuity. The observed coverage of 100 single PANC 03.04 cells analyzed using a 30 min experiment more closely matched the number of peptides and proteins identified in the FAIMS Orbitrap 200 ng file that was acquired using a 60 min gradient, and these files were used for all bulk comparison analyses. Likewise, the total number of proteins and peptides identified in each analysis for the single PANC03.04 cells in wells A3, A4, and A5 using the 30 min gradient were similar to the total number of peptides identified in the Boekweg et al. study, and these 3 files were used for these analyses. MS-Ana generates multiple metrics to compare experimental and library fragmentation spectra. For this analysis, the Y$, which represents the ratio of library spectrum intensity with y-ion annotation, and MatchIntensityQ, which is the ratio of matched intensity in the query spectrum appeared to provide the most insight. These values were exported from the PSMs of each output report and analyzed in GraphPad Prism 9.3.1. Frequency histograms were generated from the data from each respective mass analyzer using default parameters.
Direct Comparisons between Bulk and Single-Cell Spectra on Each Instrument
To directly assess the level of spectral distortion observed within a single hardware type, the bulk cell data from the Orbitrap and TIMSTOF instrument were converted to instrument-specific spectral libraries. The .pdresult files generated in the previous analysis were loaded into Skyline 22.2 through the “Import DDA peptide search” wizard workflow.19 All default settings were used including a 0.95 probability cutoff and referencing the original instrument files. The respective .blib files were converted to the. MSP format required for MSAna through EncyclopeDIA20 1.2.31 by first converting each .blib file to “Library” and then converting the resulting .dlib file to .MSP. The resulting .MSP was loaded into Proteome Discoverer 3.0, and spectral library mass cutoffs were set at 25 ppm MS1 and 0.03 Da MS2 tolerance for both library files. Decoy spectral libraries were constructed in MSAna using the “Random Sequence” decoy model. In this model, the terminal amino acids are maintained and the internal amino acid sequences are randomly shuffled.18,21 The single-cell data from each respective instrument was then searched against the bulk library data identically as described for the curated NIST library. Histograms demonstrating the PSM MatchIntensityQ values were generated in Proteome Discoverer and exported for direct comparative visualization in GraphPad Prism 9.3.1. Mirror plot spectra for publication purposes were generated by exporting peptide spectral match data and importing into the Universal Spectrum Annotator.22 The output .SVG files for the TOF and Orbitrap mirror plots were combined in Adobe Illustrator 2022.
Results and Discussion
Due to the intrinsic challenges in converting Bruker TIMSTOF files to mzML I was unable to directly utilize the Jupyter notebooks developed by Boekweg et al. MS-Ana was used for this analysis as it generates similar metrics for each peptide spectral match. Most notably, the MatchIntensityQ value provides a total matched intensity ratio between experimental and library spectra, which serves as an inverse approximation of the fragment ion signal loss when compared to the library spectra. Using this metric, a marked shift between the bulk cell lysate sample and the single HeLa cell lysates can be observed in this reanalysis (Figure 1A). An unpaired t test found a significant difference between these two sets of values (p < 0.001). However, when comparing the same value between the 100 cell PANC 02.03 cell lysates and those of single cells, no comparable shift in distribution is observed, (p > 0.05, Figure 1B).
Figure 1.
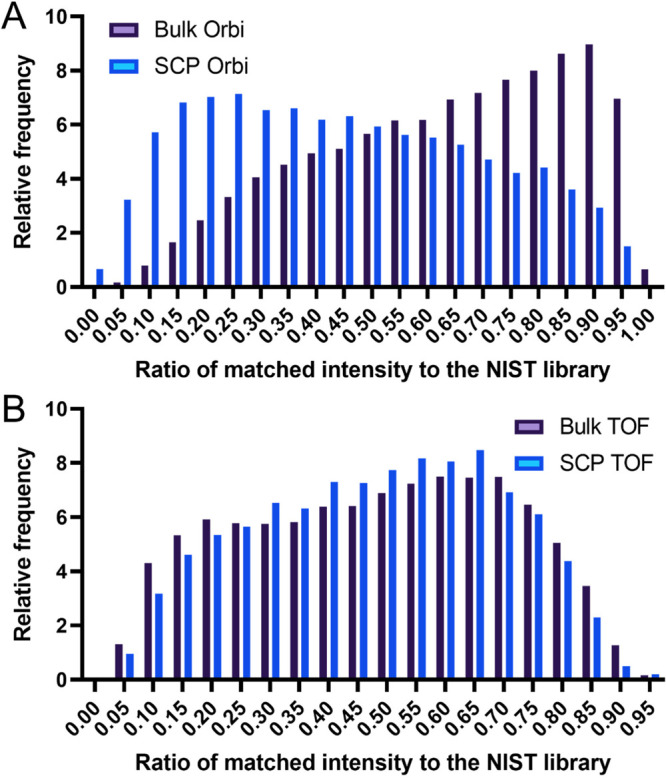
Histogram of the matched intensity distribution of each analysis to the curated fragmentation spectra in the NIST library. (A) Bulk Orbitrap fragmentation data compared to single-cell fragmentation data. (B) Same for time-of-flight mass spectra.
Due to suggestions made during peer review, I performed a second analysis to expand on this observation. The bulk cell data from the Orbitrap and TIMSTOF instruments were used to construct instrument-specific spectral libraries for each platform. The same analysis with MSAna was performed to directly compare single-cell spectra to these new instrument-specific bulk cell libraries. Figure 2 is a representative histogram overlaying the results from the two instruments. As shown, Orbitrap single-cell spectra exhibit a marked shift toward a lower number of matched fragment ions versus bulk data, while TIMSTOF SCP spectra appear to possess a more similar level of match to bulk TIMSTOF spectra overall.
Figure 2.
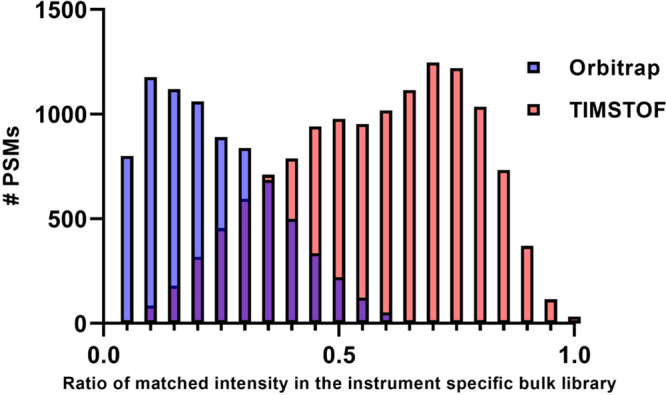
Histogram demonstrating the ratio of matched fragment intensity values when comparing single-cell fragmentation spectra to bulk data from each instrument. In this metric, 1.0 would represent spectra with a 100% fragment intensity match between bulk and single-cell spectra.
It is worth noting, however, that the bulk cell lysate data generated on the D20 Orbitrap has a considerably higher fragment intensity match distribution than any of the TOF data generated when compared to the curated NIST library. A number of factors likely contribute to this, including the fact that the NIST library was primarily acquired using Orbitrap systems18,24 as well as the required fragment isolation windows on each instrument. Due to the physical constraints of transfer time and prepulse storage effects, low-mass fragment ions are lost in TIMSTOF data unless a second optimized step is performed.
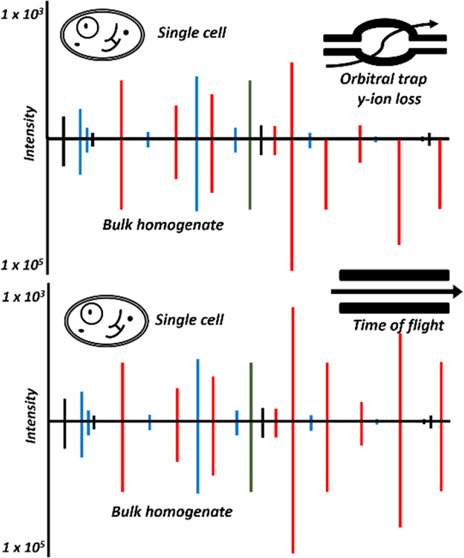
As such, the loss of single and diamino acid fragment ions or low-mass diagnostic ions is currently unavoidable without doubling the instrument cycle time with TIMS stepping, as previously described.2 In order to provide an illustration of these results in a single peptide, a custom filter was used to identify MS2 spectra in Orbitrap SCP data with no measurable coisolation interference, zero missed cleavages, yet low MatchIntensityQ values. Once peptides were identified I searched the TIMSTOF SCP data for the same PSMs and filtered for sorted spectra by their highest relative intensity signal match (Figure 3). As shown, this peptide appears to have the underrepresentation of high-mass y-series fragment ions as previously described in Orbitrap data (Figure 3A). This effect appears less pronounced in the TOF spectrum despite a similar relative sequence intensity, but the loss of low-mass fragment ions can be clearly observed (Figure 3B).
Figure 3.

Fragmentation spectra from single cells observed for the peptide SVPTSTVFYPSDGVATEK from transketolase. (A) Orbitrap SCP data (top) compared to the library reference. (B) TIMSTOF SCP data (top) compared to the same reference.
Conclusions
Single-cell proteomics is a rapidly maturing field that is beginning to step out from beneath the shadow of LCMS proteomics. The analysis of single cells has both unique opportunities and challenges that have no real parallel in traditional proteomics. As such, we may need to question all previous conclusions drawn in the past for LCMS-based shotgun proteomics. While this study is in no way conclusive or comprehensive, it does suggest some obvious strengths and weaknesses in the two dominant hardware platforms used for the application today.
Acknowledgments
This study was the result of a weekend conversation by #MassSpecTwitter, and this specific analysis resulted from a comment by Sebastian Paez (@jspaezp1) of the Purdue University Department of Medicinal Chemistry and Molecular Pharmacology. I must also thank Sebastian Dorl, Karl Mechtler, and Viktoria Dorfer for assistance with MSAna and access to unpublished results. Funding for this study was provided by the NIH through R01AG064908 (B.C.O.) and R01GM103853 (B.C.O.).
Data Availability Statement
All new data and Proteome Discoverer 3.0 output files have been deposited and made publicly available at MASSIVE23 as accession (MSV000090635, doi:10.25345/C5ZK55R59).
The author declares no competing financial interest.
References
- Slavov N. Unpicking the Proteome in Single Cells. Science 2020, 367, 512. 10.1126/science.aaz6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsburn B. C.; Yuan Y.; Bumpus N. N. Insights into Protein Post-Translational Modification Landscapes of Individual Human Cells by Trapped Ion Mobility Time-of-Flight Mass Spectrometry. Nat. Commun. 2022, 13 (1), 7246. 10.1038/s41467-022-34919-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R.; Zhu Y.; Liang Y.; Cong Y.; Piehowski P.; Dou M.; Zhao R.; Qian W.-J.; Burnum-Johnson K.; Ansong C. Single Cell Proteome Mapping of Tissue Heterogeneity Using Microfluidic Nanodroplet Sample Processing and Ultrasensitive LC-MS. J. Biomol. Technol. 2019, 30, S61. [Google Scholar]
- Schoof E. M.; Furtwangler B.; Uresin N.; Rapin N.; Savickas S.; Gentil C.; Lechman E.; Keller U. a. d.; Dick J. E.; Porse B. T. Quantitative Single-Cell Proteomics as a Tool to Characterize Cellular Hierarchies. Nat. Commun. 2021, 12 (1), 3341. 10.1038/s41467-021-23667-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlmayr D.; Ctortecka C.; Seth A.; Mendjan S.; Tourniaire G.; Mechtler K.. An Automated Workflow for Label-Free and Multiplexed Single Cell Proteomics Sample Preparation at Unprecedented Sensitivity. bioRxiv 2021, 10.1101/2021.04.14.439828. [DOI] [Google Scholar]
- Mund A.; Coscia F.; Hollandi R.; Kovács F.; Kriston A.; Brunner A.-D.; Bzorek M.; Naimy S.; Rahbek Gjerdrum L. M.; Dyring-Andersen B.; Bulkescher J.; Lukas C.; Gnann C.; Lundberg E.; Horvath P.; Mann M.. AI-Driven Deep Visual Proteomics Defines Cell Identity and Heterogeneity. bioRxiv 2021, 10.1101/2021.01.25.427969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner A.-D.; Thielert M.; Vasilopoulou C. G.; Ammar C.; Coscia F.; Mund A.; Hoerning O. B.; Bache N.; Apalategui A.; Lubeck M.; Richter S.; Fischer D. S.; Raether O.; Park M. A.; Meier F.; Theis F. J.; Mann M.. Ultra-High Sensitivity Mass Spectrometry Quantifies Single-Cell Proteome Changes upon Perturbation. bioRxiv 2021, 10.1101/2020.12.22.423933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderaa C.; Gatto L.. Replication of Single-Cell Proteomics Data Reveals Important Computational Challenges. bioRxiv 2021, 10.1101/2021.04.12.439408. [DOI] [PubMed] [Google Scholar]
- Kalxdorf M.; Müller T.; Stegle O.; Krijgsveld J. IceR Improves Proteome Coverage and Data Completeness in Global and Single-Cell Proteomics. Nat. Commun. 2021, 12 (1), 4787. 10.1038/s41467-021-25077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks J.; Leduc A.; Wallmann G.; Huffman R. G.; Willetts M.; Khan S.; Specht H.; Ralser M.; Demichev V.; Slavov N.. Increasing the Throughput of Sensitive Proteomics by PlexDIA. Nat. Biotechnol. 2022, 10.1038/s41587-022-01389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassem S.; van der Pan K.; de Jager A. L.; Naber B. A. E.; de Laat I. F.; Louis A.; van Dongen J. J. M.; Teodosio C.; Díez P. Proteomics for Low Cell Numbers: How to Optimize the Sample Preparation Workflow for Mass Spectrometry Analysis. J. Proteome Res. 2021, 20 (9), 4217–4230. 10.1021/acs.jproteome.1c00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekweg H.; Van Der Watt D.; Truong T.; Johnston S. M.; Guise A. J.; Plowey E. D.; Kelly R. T.; Payne S. H. Features of Peptide Fragmentation Spectra in Single-Cell Proteomics. J. Proteome Res. 2022, 21 (1), 182–188. 10.1021/acs.jproteome.1c00670. [DOI] [PubMed] [Google Scholar]
- Wang X.; Allen S.; Blake J. F.; Bowcut V.; Briere D. M.; Calinisan A.; Dahlke J. R.; Fell J. B.; Fischer J. P.; Gunn R. J.; Hallin J.; Laguer J.; Lawson J. D.; Medwid J.; Newhouse B.; Nguyen P.; O’Leary J. M.; Olson P.; Pajk S.; Rahbaek L.; Rodriguez M.; Smith C. R.; Tang T. P.; Thomas N. C.; Vanderpool D.; Vigers G. P.; Christensen J. G.; Marx M. A. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65 (4), 3123–3133. 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- Stadlmann J.; Hoi D. M.; Taubenschmid J.; Mechtler K.; Penninger J. M. Analysis of PNGase F-Resistant N-Glycopeptides Using SugarQb for Proteome Discoverer 2.1 Reveals Cryptic Substrate Specificities. Proteomics 2018, 18, 1700436. 10.1002/pmic.201700436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Zhou W.; Velculescu V. E.; Kern S. E.; Hruban R. H.; Hamilton S. R.; Vogelstein B.; Kinzler K. W. Gene Expression Profiles in Normal and Cancer Cells. Science (80-.) 1997, 276 (5316), 1268–1272. 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]
- Kuboki Y.; Fischer C. G.; Beleva Guthrie V.; Huang W.; Yu J.; Chianchiano P.; Hosoda W.; Zhang H.; Zheng L.; Shao X.; Thompson E. D.; Waters K.; Poling J.; He J.; Weiss M. J.; Wolfgang C. L.; Goggins M. G.; Hruban R. H.; Roberts N. J.; Karchin R.; Wood L. D. Single-Cell Sequencing Defines Genetic Heterogeneity in Pancreatic Cancer Precursor Lesions. J. Pathol. 2019, 247 (3), 347–356. 10.1002/path.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Gemenetzis G.; Kinny-Köster B.; Habib J. R.; Groot V. P.; Teinor J.; Yin L.; Pu N.; Hasanain A.; van Oosten F.; Javed A. A.; Weiss M. J.; Burkhart R. A.; Burns W. R.; Goggins M.; He J.; Wolfgang C. L. Pancreatic Circulating Tumor Cell Detection by Targeted Single-Cell next-Generation Sequencing. Cancer Lett. 2020, 493, 245–253. 10.1016/j.canlet.2020.08.043. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Burke M.; Mirokhin Y. A.; Tchekhovskoi D. V.; Markey S. P.; Yu W.; Chaerkady R.; Hess S.; Stein S. E. Reverse and Random Decoy Methods for False Discovery Rate Estimation in High Mass Accuracy Peptide Spectral Library Searches. J. Proteome Res. 2018, 17, 846. 10.1021/acs.jproteome.7b00614. [DOI] [PubMed] [Google Scholar]
- Schilling B.; Rardin M. J.; MacLean B. X.; Zawadzka A. M.; Frewen B. E.; Cusack M. P.; Sorensen D. J.; Bereman M. S.; Jing E.; Wu C. C.; Verdin E.; Kahn C. R.; MacCoss M. J.; Gibson B. W. Platform-Independent and Label-Free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline. Mol. Cell. Proteomics 2012, 11, 202. 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle B. C.; Pino L. K.; Egertson J. D.; Ting Y. S.; Lawrence R. T.; MacLean B. X.; Villén J.; MacCoss M. J. Chromatogram Libraries Improve Peptide Detection and Quantification by Data Independent Acquisition Mass Spectrometry. Nat. Commun. 2018, 9, 5128. 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias J. E.; Gygi S. P. Target-Decoy Search Strategy for Increased Confidence in Large-Scale Protein Identifications by Mass Spectrometry. Nat. Methods 2007, 4, 207. 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Schmidt T.; Samaras P.; Dorfer V.; Panse C.; Kockmann T.; Bichmann L.; van Puyvelde B.; Perez-Riverol Y.; Deutsch E. W.; Kuster B.; Wilhelm M. Universal Spectrum Explorer: A Standalone (Web-)Application for Cross-Resource Spectrum Comparison. J. Proteome Res. 2021, 20 (6), 3388–3394. 10.1021/acs.jproteome.1c00096. [DOI] [PubMed] [Google Scholar]
- Choi M.; Carver J.; Chiva C.; Tzouros M.; Huang T.; Tsai T.-H.; Pullman B.; Bernhardt O. M.; Hüttenhain R.; Teo G. C.; Perez-Riverol Y.; Muntel J.; Müller M.; Goetze S.; Pavlou M.; Verschueren E.; Wollscheid B.; Nesvizhskii A. I.; Reiter L.; Dunkley T.; Sabidó E.; Bandeira N.; Vitek O. MassIVE.Quant: A Community Resource of Quantitative Mass Spectrometry–Based Proteomics Datasets. Nat. Methods 2020, 17 (10), 981–984. 10.1038/s41592-020-0955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam H.; Deutsch E. W.; Eddes J. S.; Eng J. K.; King N.; Stein S. E.; Aebersold R. Development and Validation of a Spectral Library Searching Method for Peptide Identification from MS/MS. Proteomics 2007, 7, 655. 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All new data and Proteome Discoverer 3.0 output files have been deposited and made publicly available at MASSIVE23 as accession (MSV000090635, doi:10.25345/C5ZK55R59).