

Abstract
Microglia are macrophages of the brain parenchyma that exist in multiple transcriptional states and reside within a wide range of neuronal environments1–4. However, it is poorly understood how and where these states are generated. Here, using the mouse somatosensory cortex, we demonstrate that microglia density and molecular state acquisition are determined by the local composition of pyramidal neuron classes. Using single-cell and spatial transcriptomic profiling, we unveil the molecular signatures and spatial distributions of diverse microglia populations and show that certain states are enriched in specific cortical layers while others are broadly distributed throughout the cortex. Notably, conversion of deep-layer pyramidal neurons to an alternate class identity reconfigures the distribution of local, layer-enriched homeostatic microglia to match the new neuronal niche. Leveraging the transcriptional diversity of pyramidal neurons in the neocortex, we construct a ligand-receptor atlas describing interactions between individual pyramidal neuron subtypes and microglia states, revealing rules of neuron-microglia communication. Our findings uncover a fundamental role for neuronal diversity in instructing the acquisition of microglia states, as a potential mechanism for fine-tuning neuroimmune interactions within the cortical local circuitry.
Microglia (Mg), the tissue-resident macrophages of the brain, play critical roles in neuronal circuit assembly and tissue homeostasis5,6. Mg densities, morphologies, and transcriptomes vary both spatially and temporally across the brain, suggesting that external cues may drive context-dependent Mg states1–4. However, the mechanisms by which microenvironments regulate Mg states are largely unknown. Here, we show that in the neocortex, both Mg state identity and the laminar distribution of these states are determined by the specific subtypes of projection neurons (PNs) present in their local niche. Furthermore, we uncover potential mechanisms of communication defined by both neuronal class identity and layer-restricted Mg state.
Neuronal identity controls Mg density
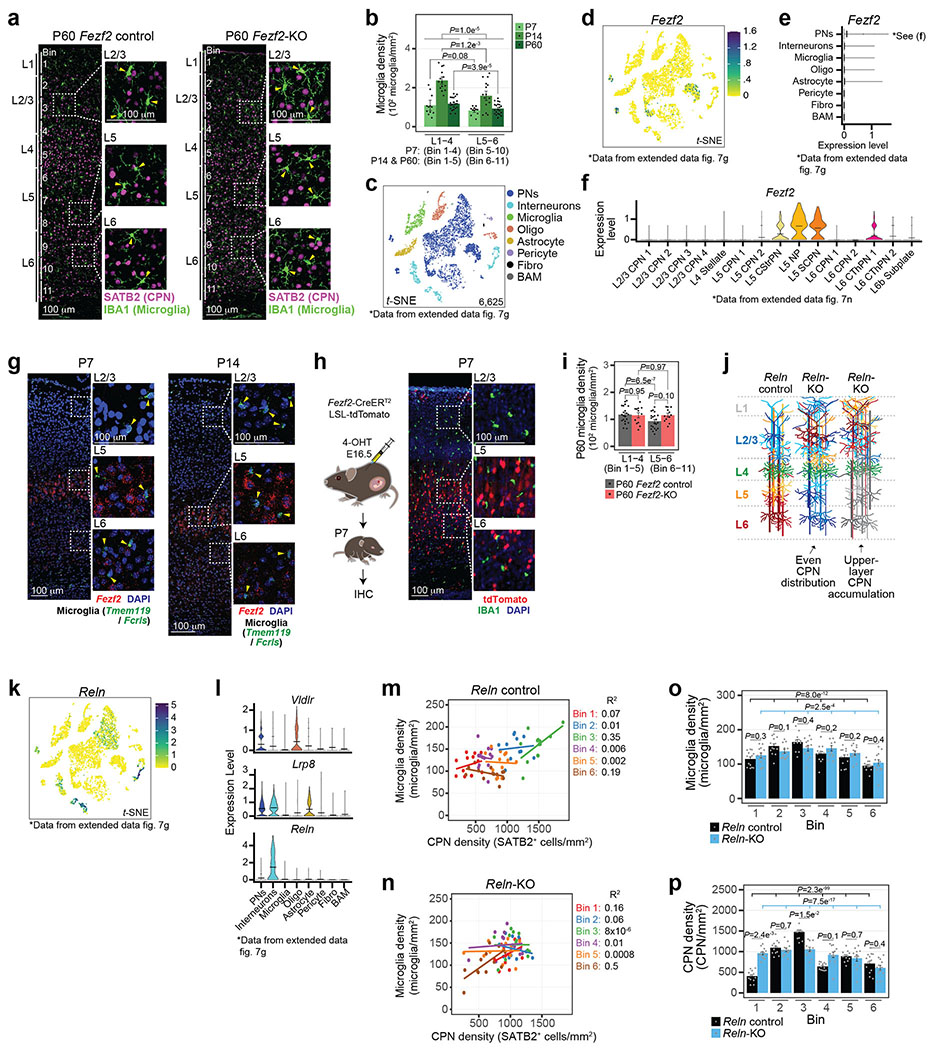
To determine whether Mg are influenced by neuronal environments, we quantified the distribution of Iba1+ Mg in primary somatosensory (S1) cortex from early postnatal development to adulthood via immunohistochemistry, using Satb2+ callosal projection neurons (CPNs) to define the upper vs. lower cortical layers (see Methods). At postnatal day 7 (P7), Mg were evenly distributed throughout all layers (Fig. 1a–d). However, by P14 and extending into adulthood (P60), Mg density was higher in layer (L) 1-4 compared to L5-6 (Fig. 1e–g, Extended Data Fig. 1a–b).
Figure 1: PN subtypes locally control Mg density in the cortex.
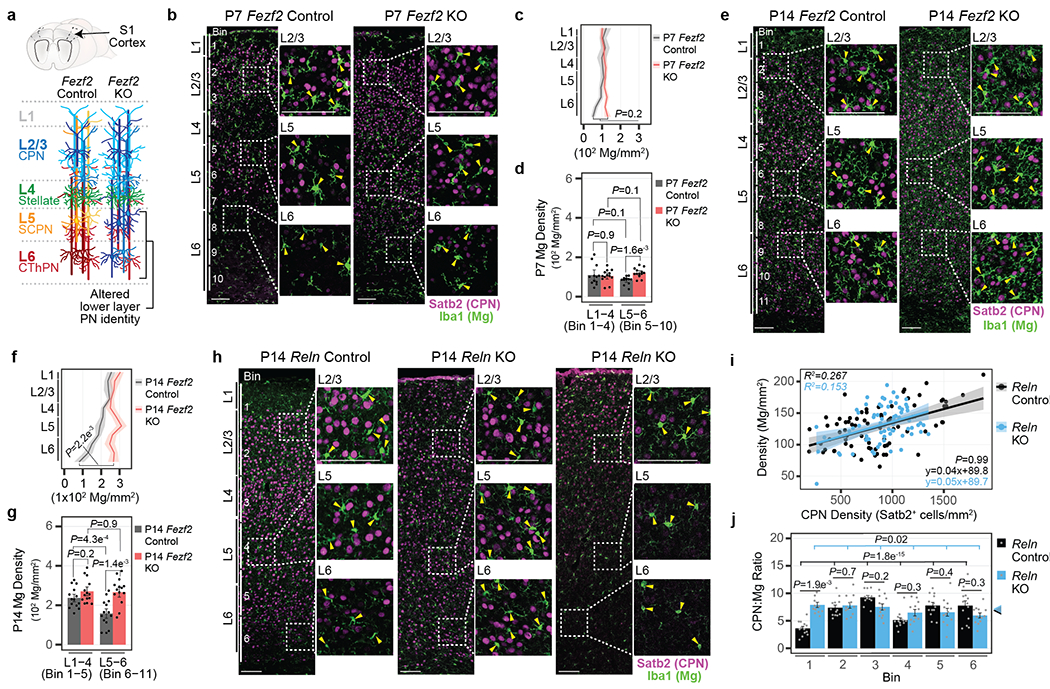
a, Schematic of Fezf2 Control and KO S1 cortex. PN classes are color-coded.
b, Representative micrograph of P7 Fezf2 Control and KO S1 cortex immunolabelled for Iba1 (green) and Satb2 (magenta). Enlarged images are boxed. Bin divisions are indicated. Arrowhead = Mg cell body. Scale = 100 μm.
c-d, Mg density across cortical layers (c) or grouped by bins corresponding to L1-4 and L5-6 (d) from P7 Fezf2 Control and KO cortices. Data are presented as mean values +/− SEM. n = 3 mice/genotype, 4 images/mouse, Statistics: linear mixed models.
e, Representative micrograph of P14 Fezf2 Control and KO S1 cortex immunolabelled for Iba1 (green) and Satb2 (magenta). Enlarged images are boxed. Bin divisions are indicated. Arrowhead = Mg cell body. Scale = 100 μm.
f-g, Mg density across cortical layers (f) or grouped by bins (g) corresponding to L1-4 and L5-6 from P14 Fezf2 Control and KO cortices. Data are presented as mean values +/− SEM. n = 4 mice/genotype, 4 images/mouse, Statistics: linear mixed models.
h, Representative micrograph of P14 Reln Control and KO S1 cortex immunolabelled for Iba1 (green) and Satb2 (magenta). Enlarged images are boxed. Bin divisions are indicated. Arrowhead = Mg cell body. Scale = 100 μm.
i, Mg density by CPN density in Reln Control and KO cortices. Dots = bin data points. Data are presented as a linear regression +/− SEM. n = 3 mice/genotype, 4 images/mouse. Statistics: Linear regression.
j, CPN:Mg ratio by bin for Reln Control and KO cortices. Colored arrowheads indicate the mean S1 cortex CPN:Mg ratio by genotype. Data are presented as mean values +/− SEM. Dots = bin ratio data points. n = 4 images/mouse, 3 mice/genotype. Statistics: linear mixed models.
These findings suggested that spatially restricted PN niches may determine Mg abundance. Indeed, the axons of L5 PN can attract Mg within the subcortical white matter tracts7. We therefore examined the cortex of Fezf2 knockout (KO) mice, in which L5-6 PN class identities are abnormally specified, while L2-4 PNs are unaffected, without a change in the total number of PNs in each layer8,9 (Fig. 1a). Fezf2 is not expressed by Mg (Extended Data Fig. 1c–h). Thus, this model permits the investigation of whether a local change in PN class identity alters the distribution of Mg.
In the P7 cortex, Fezf2 KO and Control mice displayed comparable Mg densities across all layers, with a slight increase in L6 of Fezf2 KO relative to the controls (Fig. 1b–d). However, at P14 and through P60, the density of Mg in L5-6 of the Fezf2 KO cortex was significantly higher than in controls (Fig. 1e–g, Extended Data Fig. 1i), such that Mg density in the KO L5-6 was indistinguishable from that in L1-4. This alteration was specific to L5-6, as the Mg density in L1-4, where PN class identity is not affected by the Fezf2 mutation, was similar between genotypes.
To confirm that Mg density is dependent upon the local composition of PNs, we examined the cortex of Reln KO mice, in which the radial order of neuron classes is nearly inverted (Extended Data Fig. 1j), without a change in class identity10,11. Reln and its receptors are expressed at very low levels in Mg and are unlikely to intrinsically affect Mg development (Extended Data Fig. 1k–l). In Reln Controls, Satb2+ CPNs densely populated L2-4, with a sparse distribution over L5-6 (Fig. 1h left). In Reln KOs, we found two major patterns of Satb2+ CPN distribution: 1) even distribution throughout the layers (Fig. 1h center), or 2) accumulation in L1-4, with few populating the lower layers (Fig. 1h right). We quantified Mg and CPN densities in horizontal bins of Control and KO S1 cortex (see Methods). In both Reln Control and KO mice, we found a positive correlation between CPN and Mg densities (Fig. 1i, Extended Data Fig. 1m–n). The Reln KO did not affect the overall CPN:Mg ratio, with the per bin ratio remaining similar to Control (Fig. 1j), even as CPN density fluctuated by genotype and bin (Extended Data Fig. 1o–p). This indicates that as the number of CPN in a given layer changes, so does the number of Mg, such that a consistent ratio is maintained.
Collectively, the data indicate that in two mutant models displaying altered laminar distribution of PN classes, Mg density is correlated with the PN class composition of each layer.
Mg state correlates with layer location
To determine whether Mg of different cortical layers were transcriptionally distinct, we employed the Cx3cr1GFP transgenic mouse line, which labels macrophages, including Mg12. We purified GFP+ cells from dissected L1-4, L5, and L6 of S1 cortices (Fig. 2a–b) of juvenile (P14) and adult (P60) mice by homogenization and fluorescence activated cell sorting (FACS)3 for single-cell RNA sequencing (scRNA-seq; Extended Data Fig. 2a). After removal of border associated macrophages (BAMs)13, ex vivo activated cells14, and low-quality cells (see Methods and Supplementary Information), the datasets comprised 23,156 high-quality Mg (Extended Data Fig. 2b–n).
Figure 2: Mg display cortical laminar heterogeneity.
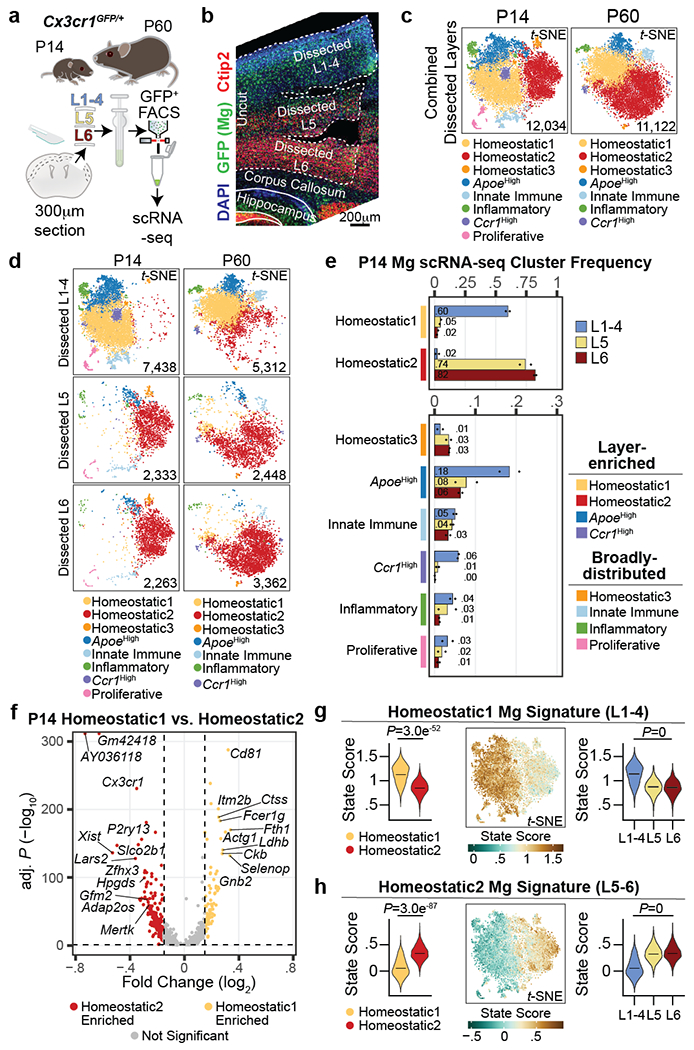
a, Schematic of S1 cortex layer dissection, GFP+ selection by FACS, and scRNA-seq from P14 and P60 Cx3cr1GFP mice. n = 2 biological replicates (pooled 1 male and 1 female) per age.
b, Representative image of a dissected Cx3cr1GFP S1 cortex, immunostained for GFP (green), Ctip2 (CFuPNs, red) and DAPI (blue). Scale = 200 μm.
c-d, t-SNE plots of combined dissected layers (c) or split by layer of origin (d) for P14 and P60 scRNA-seq datasets. Plots are color-coded by state cluster. Analyzed cell number is indicated.
e, P14 Mg cluster proportions by dissected layer (color coded). n = 2 independent biological replicates. Data are presented as normalized mean values (displayed).
f, Volcano plot of P14 Homeostatic1 versus Homeostatic2 DE genes. Genes are color-coded by enrichment (log2FC > 0.15 and −log10 BH adj. P < 0.05). Dotted lines represent enrichment cut-offs. Top signature genes are annotated. Statistics: generalized linear model with BH correction (MAST).
g-h, Mg state signature scores for Homeostatic1 (g) and Homeostatic2 (h) from P14 scRNA-seq data: (Left) Violin plot of state score split by Homeostatic1 and Homeostatic2 cluster. Data are presented as mean values with probability density. (Middle) Expression feature plot of state scores in t-SNE space. Legend: score value. (Right) Violin plot of state scores of homeostatic Mg split by dissected layer. Data are presented as mean values with probability density. Statistics: FDR adjusted p-value from linear model (Propeller).
Unsupervised clustering revealed eight and seven distinct cell clusters in the P14 and P60 datasets, respectively (Fig. 2c). All clusters expressed high levels of canonical Mg markers, confirming their Mg identity (Extended Data Fig. 2o–p). In line with previous work3,15, both homeostatic and non-homeostatic Mg clusters were present at both ages. We identified four clusters of non-homeostatic Mg, which we termed “ApoeHigh”, “Ccr1High”, “Innate Immune”, and “Inflammatory” (Fig. 2c, Extended Data Fig. 2q–v and Supplementary Information), which, combined, accounted for 27.2% and 15% of P14 and P60 Mg, respectively. We verified the presence of these four non-homeostatic states at both P14 and P60 using RNA in situ hybridization on S1 cortex (Extended Data Fig. 3).
Homeostatic Mg constituted the largest cell clusters at both ages (“Homeostatic1” and “Homeostatic2”; Fig. 2c). These clusters demonstrated high expression of canonical Mg genes, but low expression of genes associated with the non-homeostatic states (Extended Data Fig. 2o–u). A third, much smaller homeostatic cluster (“Homeostatic3”) was also present at both timepoints (Fig. 2c). Lastly, the P14 timepoint contained a small cluster (~3%) with a proliferative signature that was not observed at P60 (Extended Data Fig. 2w), consistent with published studies16.
We then investigated whether the distribution of these Mg states varied across cortical layers. Separating Mg by the dissected layer of origin (L1-4, L5, or L6) revealed a dramatic layer segregation of specific clusters (Fig. 2d). At P14 and P60, Homeostatic1 was more abundant in L1-4, whereas Homeostatic2 was more abundant in L5 and L6 (Fig. 2e, Extended Data Fig. 4a–b, see Methods), although both clusters had minor representation across all layers. The ApoeHigh cluster was enriched in L1-4 at both ages, whereas Ccr1High cluster was enriched in L1-4 only at P14; the Innate Immune, Inflammatory, and Proliferative clusters demonstrated no significant laminar bias. Together, the data indicate that homeostatic Mg are transcriptionally distinct based on their laminar position within the cortex and reveal the presence of both layer-enriched states (Homeostatic1, ApoeHigh, Ccr1High, and Homeostatic2) and broadly-distributed states (most non-homeostatic Mg; Fig. 2e).
Molecular signature of layer-enriched Mg
Next, we sought to establish the transcriptional signatures of these cortical Mg states. We calculated differentially-expressed (DE) genes for each Mg state cluster by age (see Methods; Extended Data Fig. 4c, Supplementary Tables 2–3). Given the strong layer enrichment of homeostatic Mg, we focused on Homeostatic1 and Homeostatic2. At each timepoint (P14 and P60), more than 450 genes were differentially expressed (Bonferroni adj. P < 0.05, log2FC > 0.15) between these populations, which were associated with distinct gene ontology terms (Fig. 2f, Extended Data Fig. 4d–h, Supplementary Table 4). Importantly, all DE genes showed some level of expression in both homeostatic states, indicating that these clusters represent states distinguished by variation in expression across large gene sets rather than the binary expression of “marker” genes. Canonical Mg genes showed no or only slight differential expression between Homeostatic1 and Homeostatic2, indicating that core Mg identity does not vary (Extended Data Fig. 4i). We found only a few genes that were differentially expressed by sex (Extended Data Fig. 4j, see Methods)17, indicating that sex-specific differences in Mg did not influence these results.
Finally, we defined the transcriptional fingerprint of layer-enriched homeostatic Mg. For this, we contrasted Homeostatic1 and Homeostatic2 using a more stringent cut-off (log2FC > 0.25, Bonferroni adj. P < 0.05). This resulted in 10 signature genes defining Homeostatic1 and 32 signature genes defining Homeostatic2 in the P14 dataset; similar signature genes were obtained at P60 (Supplementary Table 5). To assess the specificity of these fingerprints across Mg populations, we used the P14 signature genes to calculate upper- and lower-layer niche state “scores” for each cell (see Methods). These state signatures divided homeostatic Mg by cluster and layer (Fig. 2g–h, Extended Data Fig. 4k–l); as expected, the non-homeostatic clusters showed no clear bias toward either of these states (Extended Data Fig. 4m). As a complementary approach, we employed PLIER18, which identified similar gene expression modules (Supplementary Table 6, Extended Data Fig. 4n–p).
Mg states parallel local neuron type
We next sought to define the spatial distribution of these Mg states in relation to PN subtypes, using multiplex error-robust fluorescent in situ hybridization (MERFISH19) on P14 S1 cortex. Our probe library (Supplementary Table 7) targeted 75 cell type- and Mg state-enriched genes (Extended Data Fig. 5a–b), taken from published work20 and our profiling data (see Extended Data Fig. 7 for PNs, Fig. 2 for Mg) These 75 transcripts were imaged by MERFISH in 20,253 cells from three biological replicates of P14 Cx3cr1GFP brains (Extended Data Fig. 5c–h and Supplementary Information).
Unsupervised clustering produced six clusters, which included all major cell types of the parenchyma (Fig. 3a–b left, Extended Data Fig. 6a–c). Each cell type demonstrated variation in its spatial distribution (Fig. 3c), most notably PNs, which as expected, were mostly absent from L1, and Mg, which were more abundant in L2/3 than in L6, consistent with the scRNA-seq data (Fig. 1g). We sub-clustered the PNs to characterize their diversity and define the cortical layers. Unsupervised clustering of all Slc17a7+/Neurod2+ cells (Fig. 3a center) identified 8 classes of PNs annotated by their gene expression and laminar location (Fig. 3b center, Extended Data Fig. 6d–f; see Methods and Supplementary Information)21. PN subtypes demonstrated spatial sequestration corresponding to their expected laminar location (Fig. 3d, Extended Data Fig. 6g).
Figure 3: Cortical Mg display spatial heterogeneity.
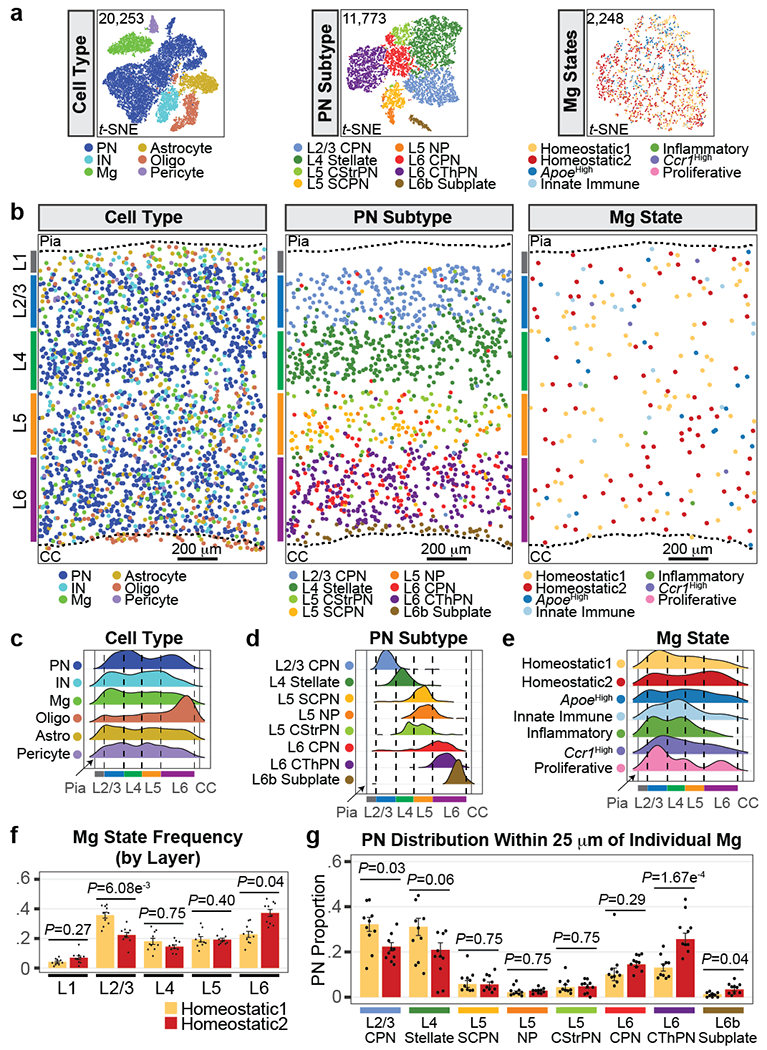
a, t-SNE plots of MERFISH data split by cell type (left), PN subtype (middle), and Mg state (right). Clusters are color-coded. Analyzed cell number is indicated. Mg states did not result in separate clusters likely because the MERFISH library only interrogates 32 genes, compared to the thousands of genes detected by scRNA-seq.
b, Representative spatial MERFISH map of S1 cortex split by cell type (left), PN subtype (middle), and Mg state (right). Cells labels are color-coded as indicated. Cortical layers (colored bars), pia, and corpus callosum (CC) are annotated. Scale = 200 μm.
c-e, Ridge plots of cell type (c), PN subtype (d), and Mg state (e) distributions along the dorsal-ventral axis of S1 cortex captured by MERFISH. Distributions are normalized as distance from the pia (left black solid line) to the CC (right black solid line). Cell types and states are color-coded. Cortical layer boundaries are indicated with dashed lines. n = 10 ROIs from 3 mice.
f, Bar graph of homeostatic Mg state frequency by cortical layer. Mg states are color-coded. Data are normalized by the total Mg state count and presented as mean values +/− SEM. n = 10 ROIs from 3 mice. Statistics: linear models.
g, PN subtype composition in the neighborhood of individual Mg (within 25 μm radius of the center of the cell body), by state (color-coded as in f). Mg for each state are pooled by sample (dots). Data are presented as mean values +/− SEM. n = 10 ROIs from 3 mice. Statistics: FDR-adj. Propeller.
We then characterized the heterogeneity and spatial localization of Mg states. Tmem119+/Fcrls+ Mg were annotated using the state-enriched signature genes (Extended Data Fig. 6h–k; see Methods). Homeostatic1 Mg were enriched in the upper layers, whereas Homeostatic2 Mg were more abundant in the deep layers (Fig. 3a–b right, 3e). Additionally, Ccr1High Mg were found at a higher abundance in L1-4. Comparing the layer boundaries defined via the PN subtypes confirmed that Homeostatic1 was enriched in L2/3 while Homeostatic2 was enriched in L6 (Fig. 3f), consistent with the scRNA-seq data (Fig. 2e).
With PN and Mg identities established, we leveraged the spatial resolution of the MERFISH dataset to identify the PN neighbors for each homeostatic Mg. Individual Mg occupy non-overlapping territories, with their processes spanning a maximum radius of 25 μm22. We therefore calculated the composition of PN subtypes located within the territory of each Mg (see Methods). Homeostatic1 Mg were statistically more likely to be found in proximity to L2/3 CPN neighbors compared to Homeostatic2 Mg. In contrast, Homeostatic2 Mg were significantly more likely to be found in proximity to L6 Corticothalamic PNs (CThPN) or L6b Subplate PNs (Fig. 3g). Taken together, these MERFISH data confirm that the spatial distribution of Mg states is correlated with the subclass identity of neighboring PNs.
Mg state location is altered in Fezf2 KO
We predicted that a change in PN identity within a specific cortical layer would alter the transcriptional signature of layer-enriched homeostatic Mg. To test this, we employed Fezf2 KO mice. We first FACS-purified nuclei from unfractionated P14 S1 cortices of Fezf2 Control and KO mice to extract the transcriptional identities of PNs by single nucleus RNA-seq (snRNA-seq) from 13,048 total nuclei (Extended Data Fig. 7a–k). Sub-clustering Slc17a7+/Neurod2+ PNs identified 19 clusters (Extended Data Fig. 7l). The Control dataset included all expected cortical PN subtypes21,23 (Extended Data Fig. 7m–n). Fezf2 KOs showed similar numbers of L2-4 PNs compared to Control (Extended Data Fig. 7o), but all L5-6 Corticofugal (CFuPN) subtype clusters were absent. Instead, the Fezf2 KO cortex had increased numbers of L5-6 CPNs, as well as four KO-specific clusters (termed “Mismatch” 1-4; Extended Data Fig. 7l–o). To gain insight into the identity of the Fezf2 KO Mismatch PNs, we used Azimuth24, a reference-based mapping pipeline. In the Fezf2 KO L2-4 PN and L5-6 CPN clusters, more than 90% of cells strongly matched the respective populations from the Fezf2 Control (Extended Data Fig. 7p–q). Mismatch 1 and 2 had a transcriptional signature resembling deep-layer CPN, while Mismatch 4 resembled CThPN, similar to our prior findings8. Mismatch 3 showed an intermediate identity between L5-6 CPN and CThPN (Extended Data Fig. 7o–q). In sum, the deep layer PNs of the Fezf2 KO are disproportionately composed of deep-layer CPN and CPN-like cells, at the expense of bona fide CFuPN populations, whereas the L2-4 PNs of the Fezf2 KO are produced normally.
We then investigated Mg state heterogeneity in response to altered PN niches using scRNA-seq of Mg isolated from layer-dissected cortices of Fezf2 Control and KO mice crossed to the Tcerg1l-CreERT2/LSL-tdTomato reporter line25, which marks L5b PNs following administration of tamoxifen (Fig. 4a–c, Extended Data Fig. 8a–b). After removing low quality cells, BAMs13, and ex vivo-activated Mg14, the dataset comprised 16,108 cells, which expressed markers of Mg identity (Extended Data Fig. 8c–m and Supplementary Information).
Figure 4: Conversion of PN class identity alters the laminar proportion of homeostatic Mg.
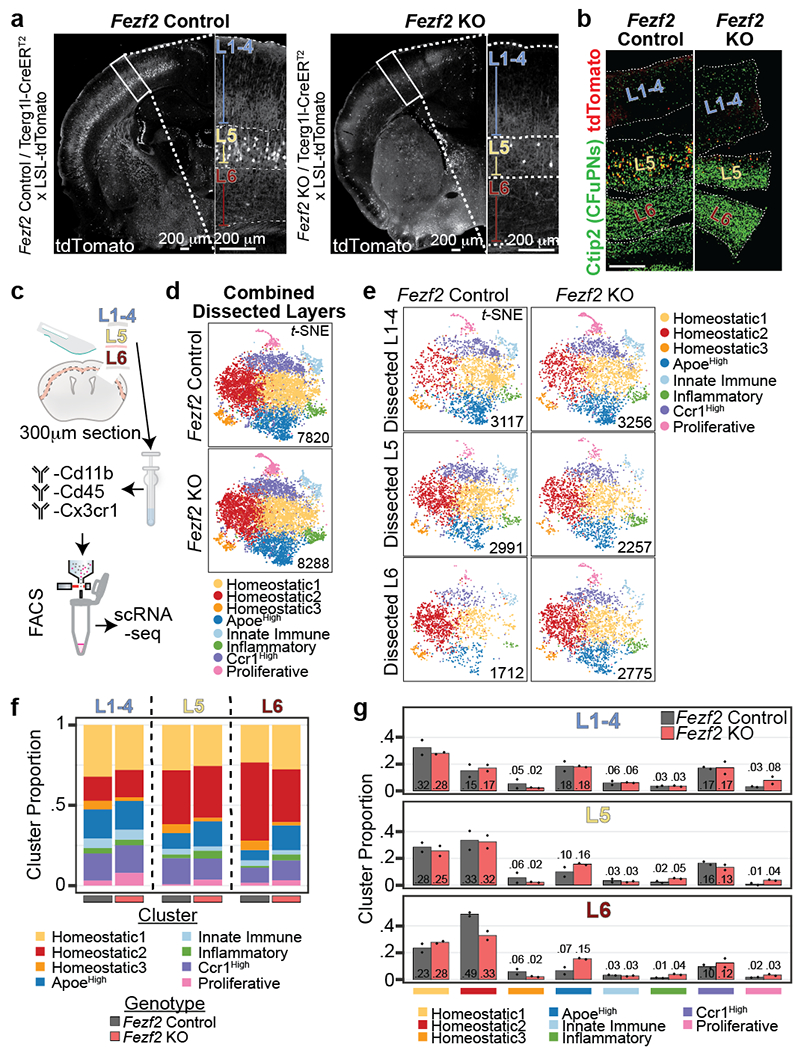
a, Representative micrographs of tdTomato signal in Fezf2 Control and KO P14 cortical hemisphere (left and right, respectively). Enlarged S1 cortex is boxed at right with layers indicated. L5b of Fezf2 KO contains few remaining tdTomato+ cells. Scale = 200 μm.
b, Representative micrograph of immunohistochemistry for tdTomato (red) and Ctip2 (green) in Fezf2 Control (left) and KO (right) P14 dissected sections. Scale = 200 μm.
c, Schematic of experimental design for layer-specific scRNA-seq from P14 Fezf2 Control and KO S1 cortex. n = 2 biological replicates (pooled 1 male and 1 female).
d, t-SNE of Fezf2 Control (top) and KO (bottom) Mg scRNA-seq clusters color-coded by state. The number of analyzed Mg by genotype is indicated.
e, t-SNE of Fezf2 Control (left) and KO (right) Mg scRNA-seq color-coded clusters split by dissected layer. The number of analyzed Mg from each layer by genotype is indicated.
f-g, Stacked bar plot (f) and bar plot (g) of Mg state cluster proportions split by dissected layer and genotype from Fezf2 Control and KO mice. Data are normalized to the total cell count in each dissected layer by genotype. For (g) Data are presented as normalized mean values (displayed). n = 2 biological replicates (pooled 1 male and 1 female each) per genotype.
Unsupervised processing of Mg from both genotypes together yielded eight clusters (Fig. 4d), corresponding to the same clusters identified in the Cx3cr1GFP dataset (Fig. 2c), including the five non-homeostatic clusters (ApoeHigh, Innate Immune, Inflammatory, Ccr1High, and Proliferative Mg), and the three clusters of Homeostatic Mg (Extended Data Fig. 9a–e, Supplementary Table 8). Azimuth24 confirmed that the Mg clusters identified in this dataset were similar to the clusters identified in the Cx3cr1GFP dataset (Extended Data Fig. 9f–g). We analyzed the data by dissected layer of origin and genotype. The Fezf2 Control clusters were enriched among the dissected layers similar to the Cx3cr1GFP dataset (Fig. 4e–g). There was no variance in cluster proportion between dissected L1-4 and L5 of Fezf2 Control and KO. However, dissected L6 demonstrated a decrease in the proportion of Homeostatic2 in the Fezf2 KO mice, while ApoeHigh was increased in L6, approaching levels similar to those observed in L1-4. This shows that Mg states normally enriched in the deep layers (Homeostatic 2) have reduced layer bias in the Fezf2 KO cortex, where deep-layer neuron identity is altered. The data confirm that layer enrichment of homeostatic Mg states is dependent upon the local layer composition of PNs.
Subtype-specific Mg-PN interactome
Given our finding that PNs control the transcriptional signature of nearby homeostatic Mg, we aimed to uncover their molecular mediators. We reasoned that layer-enriched Mg and PN subtypes would signal through cognate cell-surface or secreted ligands and receptors (L-R). We employed the molecular database CellPhoneDB26 to identify L-R pairs in the Fezf2 KO and Control sc/snRNA-seq datasets (see Methods). We tested each PN subtype against the two layer-enriched homeostatic Mg states (Homeostatic1 and 2; Extended Data Fig. 10a; BH adj. P < 0.05). The Control dataset returned 90 L-R pairs with significant expression (Fig. 5a–e, Extended Data Fig. 10b–c), including signaling molecules with well-established roles in neuron-Mg signaling, such as Il34-Csf1r27 (Extended Data Fig. 10c–f). However, most of the predicted L-R pairs have no identified roles in Mg. Receptor expression (Fig. 5a–e, bolded) was found on both Mg (69/90 pairs) and PNs (39/90 pairs), suggesting bidirectional signaling between PNs and Mg.
Figure 5: L-R interactome of PN-Mg communication in the neocortex.
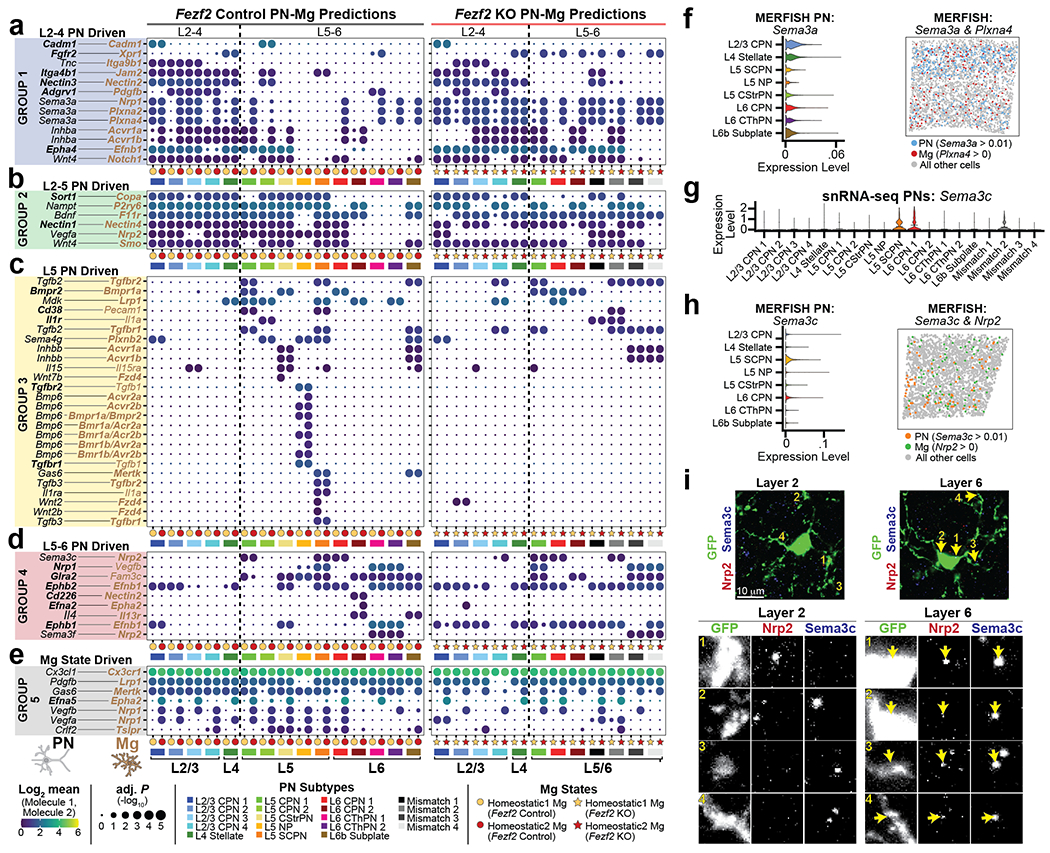
a-e, Dot plot of Ligand-Receptor (L-R) expression between PN subtypes and homeostatic Mg states in Fezf2 Control (left) and KO (right) cortices. PN data are from Extended Data Fig. 7. Mg data are from Fig. 4. L-R pairs are grouped by PN class interaction (a-e, colored boxes). Receptor genes are bolded. Legend: PN subtype = colored boxes, homeostatic Mg = colored circles (Fezf2 Control) and colored stars (Fezf2 KO). Circle size = −log10 BH adj. P values. Dot color = log2 mean expression level of the interacting L-R pairs. Dotted vertical line divides upper- and lower layers of neocortex. Statistics: permutation-based BH adj. P.
f, (Left) Violin plot of Sema3a expression in PN subtypes. Data are presented as mean values with probability density. (Right) Thresholded spatial expression feature plot of PN Sema3a (blue) and Mg Plxna4 (red) from a representative MERFISH ROI. Gene expression thresholding is indicated. PNs and Mg not meeting threshold and all other cells are greyed out. Data originate from Fig. 3.
g, Violin plot of Sema3c expression in PN subtypes (color-coded). Data originate from Extended Data Fig. 7l. Data are presented as mean values with probability density.
h, (Left) Violin plot of Sema3c expression in PN subtypes. (Right) Spatial expression feature plot of thresholded PN Sema3c (orange) and thresholded Mg Nrp2 (green) from a representative MERFISH ROI. Gene expression thresholding is indicated. PNs and Mg not meeting threshold, and all other cell types, are greyed out. Data originate from Fig. 3.
i, Top, representative micrographs of P14 Cx3cr1GFP L2 and L6 immunostained for Sema3c (blue), Nrp2 (red) and GFP (green). Bottom, enlarged numbered inserts split by channel, shown in grayscale. Arrows indicate co-localized Nrp2 and Sema3c puncta. Images are max-projection Z-stacks of 3 consecutive optical sections. Scale bar: 10 μm.
We stratified the Fezf2 Control L-R interaction results into two categories: 1) those showing signaling between Mg and all PN subtypes (Extended Data Fig. 10c) and 2) those with specificity for particular PN or Mg categories (Fig. 5a–e). We focused on the latter, as they are most likely to represent novel subtype-restricted PN-Mg signaling. L-R pairs that showed specificity among PN subtypes were subdivided into four groups.
Group 1 was driven predominantly by L2-4 CPNs (Fig. 5a, Extended Data Fig. 10g–i). L-R pairs in this group were predicted for CPNs and Mg of both superficial and deep layers, indicating that the identity of the PN type, rather than the strict laminar location, may drive the interaction. To confirm the interactome predictions, we included selected L-R pairs in our MERFISH probe library. We confirmed that Plxna4-expressing Mg accumulate near Sema3a-expressing L2/3 CPNs (Fig. 5f). In the Fezf2 KO, these CPN-driven interactions were similar to Controls (Fig. 5a). However, the KO Mismatch clusters were also predicted to use these upper-layer L-R pairs, consistent with the callosal nature of the deep-layer mis-specified PNs.
Group 2 included six L-R pairs that were specific to L2 through L5 PNs and Mg (Fig. 5b). These interactions were entirely excluded from L6 CThPNs. The Fezf2 KO data indicated significant interactions of these L-R pairs between nearly all Mismatch PN types and Mg.
The remaining two groups were driven by deep-layer PN subtypes: Group 3 by L5 PN subtypes (Fig. 5c) and Group 4 by L5-6 CFuPNs (Fig. 5d). For example, L5 Subcerebral PNs (SCPN) and L6 CPNs were predicted to interact via Sema3c with Nrp2-expressing Mg. Our snRNA-seq and MERFISH data confirmed that among PNs, only L5 SCPN and L6 CPNs express Sema3c, and that Mg express Nrp2 (Fig. 5g–h). Immunodetection of Sema3c and Nrp2 in P14 cortices showed co-localization around L6 Mg, but not L2 Mg (Fig. 5i), demonstrating the specific distribution of these signaling molecules at the protein level. In the Fezf2 KO, most Group 3 interactions were not significant (Fig. 5c).
In addition, we identified a set of L-R pairs influenced by Mg state (Group 5). These were primarily predicted to involve Homeostatic2 over Homeostatic1 Mg (Fig. 5e, Extended Data Fig. 10j–l). These pairs generally showed expression of the receptor on Mg, suggesting differential capacity of layer-enriched homeostatic Mg to receive different PN signals. Several of these L-R pairs did not reach significance in the Fezf2 KO data, such as Crlf2-Tslpr, suggesting that expression of the Mg-tuned L-R pairs is altered when the local composition of PNs is modified.
Collectively, these differential L-R pairs suggest multiple modes of PN-Mg signaling in the cortex: 1) generic signals between all PNs and Mg, 2) signals driven by specific PN classes, and 3) differential receptivity of layer-enriched homeostatic Mg. The pattern of these signals are predictably modified when PN subtypes are mis-specified, pointing to unique neuroimmune interactions modulated by PN identity. Most PN-Mg groupings showed multiple overlapping combinations of L-R pairs, suggesting that acquisition of laminar-enriched Mg states requires coordinated sets of PN-derived stimuli rather than a single signal. This data identifies candidate signals from PNs that may be responsible for “tuning” local Mg to layer-enriched states, and serves as a resource for future functional testing of PN-Mg communication.
Discussion
Emerging evidence shows that Mg directly regulate the activity of neurons through feedback mechanisms28–30, while neuronal features, such as electrophysiological activity, influence Mg biology31. These complex processes point to the need for precisely-regulated interactions between Mg and their neuronal partners. We find that cortical Mg states can be broadly divided into PN subtype-responsive Mg, and Mg which are not sensitive to the local PN niche. The presence of PN-responsive Mg is interesting, given that PN diversity influences the distribution and development of other cell types, including inhibitory interneurons32–34 and oligodendrocytes35. These findings point at finely controlled interactions between multiple cell types of the local cortical circuit, orchestrated at least in part by the specific classes of neurons within the laminar niche. The presence of type-specific neuron-Mg interactions is supported by recent work which identified a GABA-receptive Mg population that selectively prunes GABAergic rather than glutamatergic synapses36. Interestingly, subsets of autism risk genes are enriched in PNs and Mg of the upper layers of the human cortex37, suggesting a role for niche-specific interactions in disease, and motivating further investigation into the bidirectional communication between PN subtypes and Mg states in health and disease.
METHODS
Data and Code Availability
Raw FASTQ files and counts matrices from scRNA-seq data of Mg from dissected P14 and P60 Cx3cr1GFP layers, Mg from dissected Fezf2 Control and KO/Tcerg1l-CreERT2/LSL-tdTomato layers, and Fezf2 Control and KO S1 nuclei are available at GEO accession number GSE158096. Raw MERFISH images are available at gs://pyramidal_neurons_microglia. Processed MERFISH counts matrix is available at GEO accession number GSE193760. Tool needed to open raw images can be found at https://github.com/ZhuangLab/storm-analysis/tree/master/imagej_plugins. All source code can be found at https://github.com/kimkh415/MicrogliaLayers. All data are available upon reasonable request from Lead Contact.
Experimental Models
Mouse Lines
Fezf2 KO –
Fezf2 Control (WT and heterozygotes) and KO mice were generated by Hirata et. al.38 and maintained on a C57Bl/6 background.
Reln KO –
The Reln KO mouse line was procured from Jackson Laboratories (stock #000235) and was maintained on a C57Bl/6N background from Charles River (strain code 027). Reln WT were used as Controls.
Cx3cr1GFP –
This transgenic knock-in reporter mouse, first published by Jung et al. 200012, was obtained from Jackson Laboratories (stock #005582) and maintained as homozygous breeders. All experimental Cx3cr1GFP mice were heterozygous for the reporter and generated by crossing Cx3cr1GFP homozygous males with C57Bl/6N Control females from Charles River (strain code 027).
Fezf2 Control and KO/Tcerg1l-CreERT2/LSL-tdTomato –
The tamoxifen-responsive cre-recombinase knock-in (Tcerg1l-CreERT2) was produced by the lab of Josh Huang at Cold Spring Harbor and maintained on a mixed C57Bl/6 background25. The transgenic line was bred to homozygosity carrying the ROSA26loxSTOPlox-tdTomato reporter allele obtained from Jackson Laboratories (stock # 007914). The Fezf2 null allele was introduced into the line by mating Fezf2 heterozygous males with double-homozygous Tcerg1l-CreERT2/tdTomato females. The resulting Fezf2 heterozygous pups were then bred until both Tcerg1l-CreERT2 and ROSA26loxSTOPlox-tdTomato alleles returned to homozygosity. Experimental mice were injected on P2 with 50 mg/kg 4-hydroxytamoxifen (Sigma, H6278) to induce Cre-mediated recombination. Fezf2 WT and heterozygous littermates were used as Controls.
Mouse husbandry
All animal procedures were approved by the Harvard University IACUC and conducted in compliance with institutional and federal guidelines. Mice were housed in individually ventilated cages using a 12-hour light/dark cycle and had access to water and food ad libitum. Male and female mice were used for each experiment. Equal ratios of male and female mice were used in single-cell and single-nucleus RNA-seq experiments, with the exception of one replicate of the Fezf2 Control and KO layer dissected Mg experiment, which used only females due to animal availability.
Method Details
Immunohistochemistry
Brains for immunohistochemistry were prepared by transcardial perfusion of ice-cold PBS, then ice-cold 4% paraformaldehyde (Electron Microscopy Sciences, 15710) diluted in PBS. Brains were stored overnight in 4% paraformaldehyde at 4°C, followed by sequential washes with PBS, then incubated in 30% sucrose in PBS at 4°C for cryoprotection. Prepared brains were frozen in a solution of 2-parts 30% sucrose, 1-part OCT (Tissue-Tek, 4583) in Peel-A-Way embedding molds (VWR, 15160-215) and stored at −80°C. Brains were cryosectioned at 20 μm on a Leica cryostat, then the sections were stored at −20°C in 1:1 PBS/glycerol until use. Matched sections of primary somatosensory cortex (S1 cortex) were used for immunohistochemistry. Briefly, sections were washed twice with PBST (1x PBS with 0.2% Triton X-100), then incubated in blocking buffer consisting of PBST with 8% (v:v) normal goat serum (Invitrogen, 16210-072) or normal donkey serum (Sigma, D9663). Primary antibody solutions were prepared by diluting the antibodies in blocking buffer and incubating the tissues overnight at 4°C. Tissues were washed in PBST, then incubated with Alexa Fluor-conjugated secondary antibodies diluted in blocking buffer for 2 hours at room temperature. Finally, sections were washed in PBST, mounted with DAPI-Fluoromount-G (Southern Biotech 0100-20) onto glass slides, then coverslipped (VWR, 48393-106). Primary antibodies used targeted: Satb2 (Abcam, ab51502), Iba1 (Wako, 019-19741), Ctip2 (Abcam, ab18465), GFP (Millipore, AB16901), RFP (Rockland Antibodies, 600-401-379S), Sema3c (Sigma, SAB1304782), and Nrp2 (R&D Systems, AF2215). Secondary antibodies used were: Goat anti-Mouse Alexa 488 (Thermo Fisher, A-11029), Goat anti-Rabbit Alexa 546 (Thermo Fisher, A-11035), Goat anti-Rat (Thermo Fisher, A-21247), Donkey anti-Rat Alexa 647 (Sigma, ab150155), Donkey anti-Chicken Alexa 488 (Jackson ImmunoResearch, 703-545-155), Donkey anti-Rabbit Alexa 546 (Thermo Fisher, A-10040), and Donkey anti-Goat Alexa 647 (Thermo Fisher, A21447).
RNA Fluorescent in situ Hybridization
FISH experiments were performed using the RNAscope Multiplex Fluorescent V2 Assay kit (ACDBio, 323100). RNAscope Catalog Target Probes used: Abcg1 (ACDBio, 422221), Apoe (ACDBio, 313271-C3), Ccr1 (ACDBio, 402721), Fcrls (ACDBio, 441231-C2), Fezf2 (ACDBio, 313301-C2), Ifit3 (ACDBio, 508251), and Tmem119 (ACDBio, 472901-C2). PFA-fixed tissues were cryosectioned at 16 μm and heated at 70°C prior to use to adhere tissues to glass slides. Sections were hydrated with PBS for 5 minutes, dried and re-hydrated 5 minutes to remove residual OCT. Target retrieval solution (ACDBio 322000) was heated to a rolling boil and incubated on the slides for 5 minutes followed by two quick rinses of water and dehydration with 100% ethanol. Protease III (<P14 tissues) or Protease IV (P60 tissues) reagent (ACDBio, 322340) was applied to the slides for 15-60 minutes, depending on age of the tissue, at 40°C. Slides were washed 2x in water, followed by pooled probe hybridization and signal amplification according to manufacturer’s protocols.
Confocal Imaging
Confocal imaging of immunohistochemically-labelled and RNAscope in situ hybridization-stained tissue sections was performed using a Zeiss 700 inverted confocal microscope. Tile scan images spanning the width of the S1 cortex, from pia to corpus callosum, were captured using a 20x air objective and stitched using Zeiss Zen Software (version 2.6). Z-stack images were acquired using the 63x oil-immersion objective with a 0.5 μm Z-step. All confocal imaging was captured using Zen Black software (Zeiss, v2.3).
Widefield Imaging
Widefield fluorescence imaging of immunohistochemically-labelled tissue sections was performed using a Zeiss Axio Imager 2 upright microscope. Tile scan images spanning the width of the S1 cortex, from pia to corpus callosum, were captured using a 10x air objective and stitched using Zeiss Zen Blue Software.
Image Analysis
Quantification of Mg Density by Cortical Layer
Mg density measurements of Fezf2 Control and KO tissues were performed on confocal single-optical plane tile scan images spanning the entire thickness of S1 primary cortex. Tissues were section-matched to ensure consistency and reduce the effects of arealization. Immunohistochemistry was performed to identify Mg (Iba1+) and CPNs (Satb2+). Images were thresholded such that only the Iba1+ Mg cell bodies were masked, using FIJI (version 2.0.0-rc-69/1.52p). The density of masked Mg cell bodies was quantified in ~100 μm-thick non-overlapping bins beginning at the pia and proceeding ventrally until the corpus callosum. Mg/mm2 quantifications were averaged per bin, for at least 3 images per mouse-genotype-timepoint combination. The individual performing the counts was blinded to the identity of the image.
Quantification of CPN:Mg Ratio
Mg and Satb2+ CPN density and ratio quantifications for Reln Control and KO tissues were performed on confocal single-optical plane tile scan images spanning the entire thickness of the S1 primary cortex. Tissues were immunostained to identify Mg (Iba1+) and PN layers (Satb2+). Images were binned into 6 equal, non-overlapping bins, beginning at the pia and proceeding ventrally until the corpus callosum. Iba1+ Mg and Satb2+ CPN cell bodies were counted independently using the FIJI/ImageJ multipoint tool and converted to a density measurement by normalizing to the bin size. CPN:Mg ratios were averaged per bin in >4 images per mouse per genotype.
Sema3c-Nrp2 Colocalization on Microglia
P14 Cx3cr1GFP cortices were immunostained with antibodies against Sema3c, Nrp2, and GFP and imaged on the Zeiss 700 inverted confocal microscope with the 63x oil-immersion objective. Individual Mg were imaged at random from cortical L2 and L6 as Z-stacks (0.5 μm Z-steps) spanning the thickness of the soma, while capturing major and minor branch processes. Images were processed in FIJI/ImageJ and Photoshop to remove background signal (applied equally to all images). Three consecutive Z-slices were merged to create Z-projections suitable for visualizing colocalization of Nrp2 and Sema3c on GFP+ Mg.
S1 Cortex Layer Dissection
Cx3cr1GFP (P14 and P60) and Fezf2 Control and KO/Tcerg1l-CreERT2/tdTomato mice were used for S1 cortex layer dissections. P14 mice were anesthetized with isofluorane, followed by quick decapitation and brain resection into ice-cold Hibernate-A (Brain Bits, HALF). Tissue and cells were kept on ice and/or in ice-cold media for the entirety of the protocol to minimize aberrant Mg activation3,14. Brains were coronally sectioned at 300 μm in Hibernate-A on a Leica vibratome, and sections were selected from anterior-posterior regions between the points where the corpus callosum crosses the midline and when the hippocampus dips below the dentate gyrus. S1 cortical slices were dissected with a scalpel in Hibernate-A using one of the following two methods:
For Cx3cr1GFP tissues, sections were dissected under a compound light microscope. S1 cortex was selected for the region containing barrel cortex. L1-4 was demarcated by the base of the barrels in L4 up through and including the pia. L5 was determined as the layer containing large cell bodies beginning at the base of the barrels in L4, while L6 was everything from the base of L5 until the start of the corpus callosum.
For Fezf2 Control and KO/Tcerg1l-CreERT2/tdTomato tissues, sections were dissected under a fluorescence dissection microscope. Tcerg1l-CreERT2 reliably labels L5 cortical-spinal motor neurons of S1 plus a few subtypes of interneurons. S1 cortex was selected by the region containing barrel cortex and where the thickness of the tdTomato positive cells in L5 thins (roughly 750-1000 μm from the midline). L1-4 was demarcated as the region of S1 from the pia to the beginning of the tdTomato-labelled cells. L5 was determined by the band of tdTomato positive cells, while L6 began at the base of tdTomato cells until the corpus callosum. See Fig. 4b for representative dissections.
Single-cell Suspension Preparation and FACS
Live Mg suspensions from dissected tissues were prepared for fluorescence activated cell sorting (FACS) and maintained under ice-cold conditions. Briefly, layer dissected tissues were transferred to a 5 mL microcentrifuge tube with Hibernate-A and pelleted at 500 g for 5 min, and then the supernatant was decanted. The pellet was resuspended in 1 mL dissociation buffer (20 mL PBS (Gibco, 15630-080) + 20 μL RNasin (Promega, 40 units/μl, N2515) + 400 μL DNAse (Worthington, 12,500 units/mL, LS002006)) and transferred to a 2 mL Dounce homogenizer (Sigma D8938-1SET). The tissue was homogenized with 15 plunges of pestle “A”. Homogenate was passed through a 70 μm cell strainer (Miltenyi, 130-110-916), then pelleted at 500 g for 5 min. The pellet was resuspended in 775 μL PBS, then the debris was removed with Debris Removal Solution (Miltenyi, 130-109-398) as per the manufacturer’s protocol with small volume adaptations. The cell suspension was centrifuged at 3000 g for 7 min (slow ramp-down), followed by removal of the upper 2 phases (liquid and debris). The lower cell phase was washed with 2 mL PBS, centrifuged at 500 g for 5 min, then resuspended in 500 μL FACS buffer (PBS with 0.04% BSA (NEB, B9000S)). Experiments using the Fezf2 Control and KO/Tcerg1l-CreERT2/tdTomato lines required antibody labelling to fluorescently label Mg. A combination of three primary antibodies, anti-Cd11b-PE (Biolegend, 101208), anti-Cd45-APC/Cy7 (Biolegend, 103116), and anti-Cx3cr1-APC (Biolegend 149008) were diluted at 1:200 concentration in 500 μL FACS buffer and incubated with the cells at 4°C for 15 minutes. The labelled cells were washed with 5 mL PBS, pelleted at 500 g for 5 min, resuspended in 500 μL of FACS buffer, and finally passed through a 40 μm filter (Corning, 352235). Hoechst (Thermo Fisher, H3570) and propidium iodide (Thermo Fisher, P3566) at a volume of 1 μL each were added to the final cell suspension to identify live and dead cells, respectively.
Mg (GFP+ from the Cx3cr1GFP mouse line, or Cx3cr1+/Cd45low/Cd11b+ cells from the Fezf2 Control and KO/Tcerg1l-CreERT2/tdTomato mouse line) were sorted on the Beckman Coulter MoFlo Astrios EQ Cell Sorter pre-chilled to 4°C, using Summit Software (version 6.3.1). Cell suspensions were sorted using a 100 μm nozzle and collected into wells of a 96-well plate pre-filled with 10 μL of FACS buffer.
For experiments involving Cx3cr1GFP Mg, S1 cortical dissections from 1 male and 1 female mouse were pooled, homogenized, sorted, and processed for scRNA-seq. Two biological replicates for each age (P14 and P60) were obtained. For experiments with Fezf2 Control and KO/Tcerg1l-CreERT2/tdTomato Mg, S1 cortical dissections from 2 female (replicate 1) or 1 male and 1 female (replicate 2) mice were pooled, homogenized, sorted, and processed for scRNA-seq. Two biological replicates for each genotype were obtained.
Single-nucleus Suspension Preparation and FACS
Whole S1 cortices from Fezf2 Control and KO mice (1 male and 1 female for each genotype) were flash-frozen in liquid nitrogen and stored at −80°C. Frozen tissue was homogenized (Sigma D8938-1SET) in 2 mL EZ Lysis Buffer (Sigma, NUC101) with 25 strokes of pestle A, followed by 20 strokes of pestle B under ice-cold conditions. The homogenate was transferred to a 5 mL conical tube, adjusted to a volume of 4 mL with EZ Lysis Buffer, then pelleted at 500 g for 5 min. The pellet was resuspended in 4 mL EZ Lysis Buffer and incubated on ice for 3 minutes, followed by 500 g centrifugation for 5 min. The pellet was resuspended in 500 μL of Nuclei Solubilization Buffer (10 mL PBS (Gibco, 15630-080) + 50 μL of 20 mg/mL BSA (NEB, B9000S) + 10 μL RNasin (Promega, N26515)) spiked with 1 μL of Hoechst and passed through a 40 μm filter (Corning 352235). Nuclei were sorted on a Beckman Coulter MoFlo Astrios EQ Cell Sorter, pre-chilled to 4°C, using a 100 μm nozzle, and collected into wells of a 96-well plate pre-filled with 10 μL of Nuclei Solubilization Buffer.
Single-cell RNA Sequencing (scRNA-seq) and Single-nucleus RNA Sequencing (snRNA-seq)
scRNA-seq libraries were prepared with the Chromium Single Cell 3’ Kit v3 (10x Genomics, 1000075) for all samples except the final replicate of Fezf2 Control and KO/Tcerg1l-CreERT2/tdTomato, which was prepared with Chromium Single Cell 3’ Kit v3.1 (10x Genomics, PN-1000121), due to version phase-out. Sorted live, dissected Mg (470-15,000 cells/sample) suspended in FACS buffer or sorted nuclei (15,000 nuclei/sample) suspended in Nuclei Solubilization Buffer were loaded onto a v3 Chromium Single Cell 3’ B Chip (10x Genomics, 1000073) or v3.1 Chromium Single Cell 3’ G Chip (10x Genomics, PN-1000127) and processed through the Chromium controller. scRNA-seq libraries from different dissected samples were pooled based on molar concentrations and expected captured cell counts, then sequenced on a NextSeq 500 instrument (Illumina) with 26 bases for read 1, 57 bases for read 2, and 8 bases for the i7 index. The mean reads per cell in each sample was >30,000 with a minimum sequencing saturation of 66% (in most cases >80%). snRNA-seq libraries were sequenced on a NovaSeq (Illumina) instrument with 28 bases for read 1, 91 bases for read 2, and 8 bases for the i7 index. The mean reads per nucleus was >56,000 with a minimum sequencing saturation of >48%.
scRNA-seq Data Analysis Pipeline
Single-cell RNA-seq binary cell call (BCL) files for experiments from P14 Cx3cr1GFP, P60 Cx3cr1GFP, P14 Fezf2 Control and KO mice were converted to FASTQs using the mkfastq function from Cell Ranger39 software version 3.0.1 with the default parameters. FASTQs were aligned to mm10 (Ensembl 93) reference transcriptome, and gene expression matrices were obtained using the count function of the Cell Ranger with the default parameters. R version 3.6.0 and Seurat40 package version 3.2.0 were used to perform all downstream analyses except Azimuth (see below).
For initial quality filtering, genes expressed in more than three cells were kept, and cells expressing more than 500 genes were kept. Cells with greater than 10% mitochondrial gene expression (percent.mt) were removed. The SCTransform41 function from Seurat (sctransform package version 0.2.1) with vars.to.regress parameter (to account for batch effects) set to percent.mt and sample (two biological replicates for each dissected cortical layer) was used to normalize, find variable genes, and scale the dataset. The identified list of variable genes was used to perform the principal component analysis (RunPCA function with the default parameters). Using the top 30 principal components and Louvain clustering algorithm, cell clusters were identified (FindNeighbors function with dims=1:30 and FindClusters function with resolution=0.5 unless otherwise indicated). For visualization, the top 30 PC dimensions were further reduced to two t-distributed stochastic neighbor embedding (t-SNE) dimensions (RunTSNE function with dims=1:30). The procedure, from SCTransform to dimensionality reduction for visualization, was repeated each time unwanted cell populations were removed, allowing more-accurate capture of the variation among the remaining cells, until we were left with only the cells of interest. Visualization of scRNA-seq- and snRNA-seq data was performed with ggplot2 (version 3.2.1, https://ggplot2.tidyverse.org).
P14 Cx3cr1GFP Mg scRNA-seq Dataset Clean-up
Initial cell clustering identified 13 different clusters. Cells in clusters expressing BAM and Oligo-specific genes (for example F13a1 and Lyve1; 297 cells, and Mog and Mbp; 65 cells, respectively), low-quality signals (low UMI/cell, low genes/cell and high percent.mt; 735 cells), and mixed signatures of different cell types (60 cells) were also removed. After processing the remaining 12,034 cells, 10 different cell clusters were identified. These clusters were assigned Mg subtype labels based on the cluster-specific marker genes. G2m-Phase and S-Phase proliferative clusters were combined as one cluster, termed “Proliferative” as determined by Nestorowa et al42.
P60 Cx3cr1GFPMg scRNA-seq Dataset Clean-up
Initial cell clustering identified 13 different clusters. Clusters expressing BAM- and astrocyte-specific genes (for example F13a1 and Lyve1; 205 cells, and Aldh1l1 and Gfap; 235 cells, respectively) and low-quality signals (low UMI/cell, low gene/cell, and high percent.mt; 1,817 cells) were also removed. After processing the remaining 11,332 cells, 9 different cell clusters were identified. These clusters were then assigned Mg subtype labels based on the cluster-specific marker genes. Two clusters were labeled as “Homeostatic2”, because they showed very similar gene expression profiles, having only 6 DE genes between them—three of which were the sex-linked genes Xist, Eif2s3y, and Ddx3y, the other three being Sgk1, Gpx1, and Hspa5.
P14 Fezf2 Control Mg scRNA-seq Dataset Clean-up
For the Fezf2 Control and KO dataset, we took additional measures to account for batch effect. PCA identified that major drivers separating biological replicates were ribosomal and mitochondrial genes. Accordingly, we removed mitochondrial (mt-^) and ribosomal (rpl^ and rps^) genes from the list of variable genes identified from SCTransform and proceeded with the downstream analysis. Initial cell clustering identified 15 distinct clusters. Cells in clusters expressing neuron-specific genes (for example Neurod2 and Slc17a7; 308 cells), BAM-specific genes (for example F13a1 and Lyve1; 61 cells), ex vivo activation signatures14 (for example Junb and Dusp1; 1256 cells), apoptosis signatures (for example Cdkn1a and Bax; 191 cells), and low-quality signals (low UMI/cell, low genes/cell and high percent.mt (> 10%); 1666 cells) were also removed. One biological replicate displayed high fractions of cells (19% from L1-4, 38% of L5 and 52% from L6) with significant enrichment for ex vivo activation signatures and clustered separately, likely due to a FACS issue. However, the remaining cells from that replicate clustered well with the other replicates and showed expected gene expression for Mg subtypes; thus, we kept those cells rather than removing the entire replicate. After processing the remaining 16,108 cells, 9 different cell clusters were identified. These clusters were assigned Mg subtype labels based on the cluster specific marker genes. G2m-Phase and S-Phase proliferative clusters were combined as one cluster termed “Proliferative” as determined by Nestorowa et al42.
snRNA-seq Data Analysis Pipeline
The snRNA-seq dataset (Fezf2 Control and KO) was processed similarly to the scRNA-seq dataset, using the identical software, packages, and parameters (unless otherwise mentioned). snRNA-seq FASTQs were aligned to the mm10 (Ensembl 93) pre-mRNA reference. The pre-mRNA reference was built using the guidelines provided by the Cell Ranger. snRNA-seq data were normalized using SCTransform; however, the samples were not regressed, in order to maintain the variation between the genotypes. Initial cell clustering resulted in 23 different cell clusters. One cluster (914 cells) that had very low UMI/cell and genes/cell and expressed mixed signatures of different cell types was removed (see circled cluster in Extended Data Fig. 7e).
When identifying marker genes for each cluster, percent.mt was omitted from the latent variable parameter of FindAllMarkers function. After processing the remaining 13,048 cells, 23 different cell clusters were identified, and were classified into 8 major cell types: pyramidal neurons (PN), interneurons (IN), Mg, BAM, oligodendrocyte-lineage cells (Oligo), astrocyte (Astro), fibroblast, and pericyte, based on expression of known signature genes20. PNs were subsetted from this object and processed further to identify subtypes. Processing of the PN subset (with increased clustering resolution to 2) resulted in 26 different cell clusters. These clusters were assigned a subtype assignment, based on the known PN subtype signatures8,21. Clusters that shared near-identical expression profiles were grouped, leaving 15 clusters of PN subtypes, plus an additional 4 clusters specific to the Fezf2 KO genotype, termed Mismatch 1-4.
Differential Expression Analyses
Differential expression analyses for scRNA-seq and snRNA-seq data clusters were performed using the FindAllMarkers function with the following parameters: test.use=‘MAST’43, only.pos=TRUE and latent.vars=c(‘percent.mt’, ‘nCount_RNA’, ‘Replicate’). Differentially expressed (DE) genes having an absolute value average log2 fold change > 0.15 and an FDR-adjusted P < 0.05 (accounting for multiple testing) were flagged as DE. Signature gene lists (for Homeostatic1 and Homeostatic2 Mg) are filtered lists of DE genes with an average log2 fold change cutoff set to 0.25 and an FDR adjusted P cutoff at 0.05. To account for sample-to-sample variability, DEG between Homeostatic1 and Homeostatic2 were additionally computed using Nebula44 (R package version 1.1.7) taking sample into account. Genes that appeared in both lists (MAST and Nebula) between Homeostatic1 and Homeostatic2 comparisons were considered DE.
Differences in the gene expression profile between male and female Mg were identified by first classifying all Mg from P14 and P60 Cx3cr1GFP scRNA-seq datasets into the two groups: Any Mg with >0 Xist count were considered to be female, and the rest classified as male; male cells also expressed Ddx3y. The accuracy of these assignments may have been limited by the ability of scRNA-seq to capture the gene expression of these key marker genes. DE genes between the two groups were found using the same method and parameters as described above, with FindAllMarkers and test.use=‘MAST’43.
Gene Ontology (GO) Analysis
GO analyses were performed on P14 Cx3cr1GFP Mg scRNA-seq data clusters using clusterProfiler45 (v3.14.3). A list of DE genes (log2 fold change > 0.15 and adjusted P < 0.05) from target clusters were calculated using MAST (see scRNA-seq analysis pipeline above) and used as the “gene =” input for enrichGO within clusterProfiler. A background list of all genes expressed by the P14 Cx3cr1GFP Mg scRNA-seq dataset was used for the argument “universe =”. Specific code arguments used were: keyType = “SYMBOL”, OrgDb = the genome-wide annotation for Mouse (https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html), ont = “BP”, pAdjustMethod = “BH”, qvalueCutoff = 0.05. The top 10 significant GO terms (Benjamini-Hochberg adjusted) were plotted using the dotplot function.
State Score Calculation
State scores from scRNA-seq data were computed using the AddModuleScore function in Seurat with the ctrl parameter set to 50. Scores were calculated for Mg identity14, BAM identity13, ex vivo Activation14, Cell cycle42, Homeostatic1 signature, and Homeostatic2 signature. The complete list of genes used to compute state scores can be found in Supplementary Table 1 (Mg and BAM identity, ex vivo Activation), Table 5 (Homeostatic1 and Homeostatic2), and Table 6 (PLIER LVs, see below). Cell cycle scoring was computed following the Seurat vignette (https://satijalab.org/seurat/archive/v3.1/cell_cycle_vignette.html) demonstrated by Nestorowa et al42.
Multiplex Error-Robust Fluorescent in-situ Hybridization (MERFISH)
Design of MERFISH Encoding Probes
In order to map major cell types, PN subtypes and Mg states (clusters identified in P14 Cxc3r1GFP scRNA-seq data) of the S1 cortex, a MERFISH panel of 75 target genes was designed. We included an additional 29 target genes chosen for their specificity in the L-R interactome of Fig. 5. The MERFISH genes selected and their target cell type or Mg state can be found in Supplementary Table 7. The 104-plex target gene probe library was constructed and purchased in coordination with Vizgen (VZG123).
MERFISH Staining and Imaging
P14 Cx3cr1GFP mice were euthanized with isofluorane. Brains were transferred to a sterile 5 mL conical tube (VWR, 10002-731), flash frozen in liquid nitrogen and stored at −80°C until further processing. Brains were cryosectioned at 10 μm, trimmed to expose only S1 cortex, and mounted onto 40 mm coverslips (Vizgen VZG123) coated with Polyethyleneimine (Sigma, P3143) and yellow green fluorescent microspheres (Polysciences, 17149-10). Thin sections were 4% PFA-fixed onto the coverslips for 15 minutes at RT, washed 3x in PBS for 5 min each and dehydrated in 70% EtOH. Tissue-mounted coverslips were processed according to Vizgen’s protocol and imaged on a MERSCOPE alpha scope (Vizgen), imaging seven Z-planes per region of interest (ROI).
Cell Segmentation and Generation of Count Tables
Encoded MERFISH images of RNA spots were de-multiplexed using MERlin (https://github.com/emanuega/MERlin), using the codebook provided by Vizgen. This pipeline outputs a table with the coordinates and identity of all detected transcripts. S1 cortex regions were selected using DAPI and PN subtype localization as guides. Out of 12 imaged ROIs, two were excluded from the analyses because they did not include S1 cortex. Decoded data were used to generate binary mosaics of the tissue in which transcripts represented a single pixel.
Mg were segmented using methods similar to those described by Favuzzi et al.36. Briefly, binary images of Tmem119/Fcrls signals were used to identify Mg. A dilation disk 9 pixels in diameter surrounding Tmem119/Fcrls signals was used to set Mg segments. Segmented elements < 500 pixels were removed to eliminate isolated transcripts. Each segmentation was dilated again using an 8-pixel diameter disk, followed by a removal of objects < 1000 pixels to eliminate regions with a low density of Tmem119/Fcrls. To maximize the capture of RNA transcript species of Mg, a final dilation using an 8-pixel diameter was created to set the final Mg segmentation boundary. To segment Mg along all of the Z-planes of the tissue, we set a threshold such that Tmem119+/Fcrls+ must be > 6 transcripts in each Z layer.
Images of binary DAPI-identified nuclei were used to segment all other cell types. To avoid double-counting Mg, we masked Mg segmentations onto the DAPI images and removed those DAPI nuclei from down-stream analyses. We segmented cells by serially running morphological dilations on the nuclei masks to retrieve typical-sized nuclei while discouraging segmented cell clumping (multiplets). Briefly, a dilation disk diameter of 3 pixels set the structuring element around each DAPI-identified nucleus. Objects which were considered to be either too big (> 1500 pixels2; for example: clumped DAPI-identified nuclei) or too small (< 600 pixels2; for example: fluorophore aggregates) were removed. The resulting nuclei were morphologically dilated again with a 6-pixel diameter disk, followed by an exclusion of objects with an area > 3000 pixel2 to remove merged cells. This conservative approach restricted the capture of RNA transcripts to single cells while excluding the RNA signal from the cytoplasm of a neighboring cell. The pipeline was performed on all Z-slices to segment cells in three-dimensional space.
Counts tables were produced by summing the number of each transcript inside the area of segmented cells (Mg and DAPI-identified nuclei). Mg- and DAPI-segmentation counts were combined by concatenating their respective counts tables.
Cell Clustering, Cell Scoring and DE analysis of MERFISH Data
The MERFISH count matrices were processed replicating the procedure described in Moffitt et. al19. All cell type-specific or Mg state-enriched target genes (not including the L-R target genes) were used as variable genes. Gene counts for each cell were normalized by the total count/cell, without performing log transformation or introducing a scaling factor. Z-scores were calculated on the normalized counts for each gene across all cells. Data dimensionality was reduced with PCA, followed by identification of cell clusters, and visualized by t-SNE dimensions similar to the approach described for scRNA-seq data analysis. The number of PC dimensions used in downstream analyses was determined using JackStraw significance test (JackStraw function of Seurat with prop.freq parameter set to 0.5) and standard deviations.
When finding cell neighbors, k.param parameter of the FindNeighbors function was set to 10. Cluster visualization was performed similar to scRNA-seq by reducing the top 30 PC dimensions to two t-distributed stochastic neighbor embedding (t-SNE) dimensions (RunTSNE function with dims=1:30). Cell clustering of all segmented cells (Mg and DAPI-identified cells) resulted in 11 different clusters. We removed one cluster containing 584 cells which demonstrated close to 0 UMI/cell and 0 genes/cell. We also removed one additional cluster with 1,699 cells, to which we were unable to accurately assign a cell type, given the limited selection of genes in the MERFISH probe library. Major cell types (Mg, PN, IN, Oligo, Astro, and Pericytes) were assigned to the remaining 9 clusters (20,253 cells), based upon available signature genes included in the MERFISH codebook and in accordance with known cell type markers. PNs (Slc17a7+ and Neurod2+) were sub-clustered (as described for all segmented cells), yielding 10 different cell clusters that were assigned PN subtype labels according to the expression of specific marker genes included in the MERFISH codebook and in agreement with the snRNA-seq data from Extended Data Fig. 7.
To identify the Mg states, we subset Tmem119+/Fcrls+ cells and used a Z-scoring approach to score each Mg based on state-enriched gene expressions as identified in our P14 Cx3cr1GFP scRNA-seq data (Fig. 2). For each gene count, we subtracted the average count of that gene and divided by the variance of these counts. This results in a score that has an average of zero and a variance of one for each gene; negative scores reflect a cell that demonstrates lower expression of the gene, whereas a positive score reflects an increased expression of the gene. Each cell was scored for the 7 target Mg states by computing the average Z-score for all the genes from each state module present in the MERFISH codebook. We applied minimal thresholds on “key genes” for non-homeostatic state signatures. “Key genes” are genes from the P14 Cx3cr1GFP scRNA-seq dataset that are significantly and always up-regulated by cells in the considered state. The thresholds applied for calling Mg states were as follows: 1) for ApoeHigh Mg, a cell must have a Z-score > 1 for Apoe, 2) for Ccr1High, a cell must have a Z-score above 0.5 for Ccr1, 3) for Proliferative Mg, a cell must have a Z-score > 0 for all three proliferative genes Mcm4, Ube2c and Mki67, 4) for Innate Immune Mg, a cell must have a Z-score > −0.2 for both Oasl2 and Rtp4, and finally, 5) for Inflammatory Mg, a cell must have a Z-score > 0.1 for Cd63. All remaining Mg were considered homeostatic and were called Homeostatic1 or Homeostatic2, depending on the state for which they scored highest.
Note: two MERFISH target genes produced no counts, namely Epo and Lyz2. These two genes were predicted to fail by Vizgen due to transcript length requirements. All other MERFISH target genes produced signal.
Cell Type Distribution Maps and Analyses from MERFISH Data
Cell type and Mg state distributions across the cortical layers were computed from MERFISH data. ROIs were manually rotated to have the pial surface (determined by DAPI) facing right. The horizontal distance (in pixels) between every segmented cell and the pial surface was computed. Cell distances were normalized per ROI (0-1), with the cell displaying the furthest distance from the pia set to 1. Cell type and Mg state distributions were plotted using the ggplot2 geom_density_ridges function with a scale = 1.5 and a rel_min_height = 0.01.
The boundaries between the cortical layers were calculated by the distribution of PN subtypes in the MERFISH dataset. The normalized PN distribution maps (on a scale of 0-1), were manually thresholded to segment the different cortical layers following well-established localizations of PN classes, as follows:
L1 –region (normalized distance of 0-0.09) between pial surface and the beginning of L2/3 CPNs; contains very few PNs,
L2/3 – region (normalized distance of 0.09-0.28) containing the vast majority of upper-layer CPNs, excluding L4 stellate neurons,
L4 – region (normalized distance of 0.28-0.45) containing the vast majority of L4 stellate neurons, with a limited abundance of surrounding L2/3 CPNs (dorsal) and L5 SCPN (ventral) subtypes,
L5 – region (normalized distance of 0.45-0.63) between L4 Stellate neurons (dorsal) and L6 CThPNs (ventral), including the majority of multiple subtypes, including SCPN, NP, and CStrPN.
L6 – region (normalized distance of 0.63-1.0) housing the vast majority of L6 CThPNs, starting at the base of L5 until the end, including L6b Subplate PNs.
The bar plot in Extended Data Fig. 6g reflects the accuracy of the layer cutoffs as a function of the frequency of each PN subtype. The layer cutoffs, determined by the PN subtypes, were used to calculate the Mg state frequencies by layer (Fig. 3f) and to depict the layer boundaries in the RidgePlots of Fig. 3c–e.
Microglia Neighbor Counts
The proportion of PN subtypes residing within the territories of individual Mg were quantified from MERFISH data. Mg are positioned in non-overlapping domains (a.k.a. territories) and display a max cell (soma plus processes) radius of roughly 25 μm22. The number of PNs, by subtype, were calculated residing within the 25 μm radius circle emanating from the center point of every microglia in our MERFISH dataset. Only PNs whose center point were localized within the ~1963 μm2 (area = πr2) territory circle of each homeostatic Mg state were counted (see Fig. 3g). “Neighborhoods” were computed by averaging the proportion of PN subtypes within Homeostatic1 and Homeostatic2 Mg territories by ROI, then across the whole dataset.
Pathway-Level Information ExtractoR (PLIER)
We applied PLIER18 version 0.99.0 to unbiasedly capture the Homeostatic1 and Homeostatic2 signatures from the P14 Cx3cr1GFP Mg scRNA-seq dataset using the cluster-enriched DE genes. PLIER reduces the dimension of the input gene expression data into a low dimensional space of latent variables (LVs). While doing so, it finds a decomposition that best aligns the gene loadings of LVs to the input prior knowledge (in our case the signature gene lists). Among the output LVs, only LVs with high confidence (area under the curve (AUC) > 0.5 and FDR adj. P < 0.05) were annotated for Homeostatic1 or Homeostatic2. Annotated LVs were considered as gene modules by taking the top 50 genes based on the gene loadings. The list of genes in the LVs can be found in Supplementary Table 6. The LV gene modules were used as input to score all Mg from the P14 Cx3cr1GFP Mg scRNA-seq dataset as described under “state score calculation”.
Azimuth Cell Type Predictions
Azimuth24 (included in Seurat version 4.0.0; R version 4.0.3) was run on Fezf2 Control and KO scRNA-seq data to map Mg subtypes to the P14 Cx3cr1GFP scRNA-seq reference dataset. Additionally, Azimuth was run on Fezf2 KO snRNA-seq data to map PN subtypes to the Fezf2 Control snRNA-seq data. The reference datasets (prepared as described in the scRNA-seq and snRNA-seq analysis), were additionally analyzed via supervised principal component analysis (sPCA) to better capture the structure defined in the nearest neighbor graph and down-sampled to have equal number of cells for each Mg and PN subtype identified. Specific code arguments used are as follows: FindTransferAnchors function was used to identify “anchors” between the reference and the query datasets with normalization.method = ‘SCT’ and reference.reduction = ‘sPCA’, with all other arguments set to default; TransferData function was used to transfer the reference cell types to the query cells, with all arguments set to default. Accuracy of the predictions was assessed by visualizing the prediction scores (ranging from 0-1, 0 being the least confident and 1 being the most confident) and correspondence to the query dataset manual annotations (for Mg predictions see Fig. 2c, for PN predictions see Extended Data Fig. 7l).
CellPhoneDB
Differential ligand-receptor (L-R) expression and interaction predictions were performed between PN subtypes and homeostatic Mg. We filtered scRNA-seq and snRNA-seq datasets to systematically compare the Fezf2 Control and KO brains by selecting clusters of interest as follows: “Homeostatic1” and “Homeostatic2” Mg from Fezf2 Control scRNA-seq dataset, “Homeostatic1” and “Homeostatic2” Mg from Fezf2 KO scRNA-seq dataset, and all PN subtypes from Fezf2 Control and KO snRNA-seq datasets. We applied CellphoneDB26 version 2.0 to obtain the interactomes of the two conditions (Fezf2 Control and Fezf2 KO) which were independently assembled by comparing Mg from 1) Fezf2 Control Mg with Fezf2 Control PNs together and 2) Fezf2 KO Mg with Fezf2 KO PNs together. CellphoneDB was executed by invoking the statistical_analysis method of CellphoneDB with the following parameters: --subsampling --subsampling-log=true –threads=8. Since CellphoneDB generates raw P-values as output, the Benjamini and Hochberg method was applied to correct for multiple testing. Output interactions were filtered using an adjusted P threshold of 0.05 and a log2 mean cutoff of 0. Predicted interactions are inferred based on the mean expression of the cognate ligands and receptors from RNA-seq data of each PN sub-type and Mg state. This approach captures high-confidence interaction predictions; however, weakly expressed transcripts or drop-out expression values may result in missed predictions. Because CellphoneDB is built upon human references, we converted mouse gene symbols to human Ensembl IDs using useMart function from biomaRt package version 2.40.5. Human gene names were then returned to mouse gene names for figure building.
Statistics and Reproducibility
Significance of differences in Mg density between the Fezf2 Control and KO brains (Fig. 1c, d, f, g and Extended Data Fig. 1i) was computed using linear mixed-effect model (LMM; lmer function from lme4 R package version 1.1-27). LMM was fit on the data in question, with mouse sample as a random effect, and the genotype as a predictor. P values were obtained using the lmerTest R package version 3.1-3. Significance for the difference within a genotype between the two layers L1-4 and L5-6 was computed similarly using the layer as a predictor.
Significance of the Mg density differences between the upper and lower layers of the Fezf2 Control brain (Extended Data Fig. 1b) was computed using LMM. LMMs were fit on the density data using biological replicate as a random effect, age as a fixed effect and with or without the layers as another fixed effect. The fitted models were compared using the ANOVA function to evaluate the use of an additional predictor. Significance of Mg density difference across ages in the same data by layer was computed using a similar approach, with biological replicate as a random effect and with or without age as a fixed effect. Significance between the two layers was also computed for each individual time point. For this purpose, we used lmerTest as described above, fitting LMM on the data with the image (L1-4 and L5-6 are segmented from each image) as a random effect and layer as a fixed effect. Biological replicate was omitted here to address the model fit being singular.
For the Reelin Control and KO datasets, we computed the significance of the differences in Mg density, CPN density and Mg:CPN ratio (Fig. 1j and Extended Data Fig. 1o–p) between the genotypes within each bin. This was computed using the lmerTest method described above with biological replicate as a random effect and genotype as a fixed effect. Note for Mg density data: to avoid model fit being singular, we fit a liner model (LM) instead, treating biological replicate as a fixed effect. On the same dataset, the significance of the differences across bins within each genotype was also computed. For this purpose, we fitted LMM on the data with image and biological replicate as random effects and with or without bin as a fixed effect. We then used ANOVA to compare the models. Note for CPN density: LM was used instead to avoid model fit being singular. For LM, both image and biological replicate were treated as fixed effects.
To calculate significance of state score differences in scRNA-seq data (for example Fig. 2g), we used LMM. LMMs were fit on each state score data using biological replicate as a random effect, and with or without the Mg subtype as a fixed effect. The fitted models were then compared using the ANOVA function. P-values were adjusted across different state scores using the BH method.
Statistics for compositional data (i.e., proportions of cells in clusters) were computed using the Propeller function from R package speckle version 0.0.2 (https://github.com/Oshlack/speckle). Unlike other classical statistical tests for compositional data (e.g., Chi-squared or Fisher exact test), the Propeller function takes into consideration the variability of the cell type proportion in different biological replicates, making it suitable for use in single-cell experiments. The input Seurat objects was prepared, having cluster identity in the active.ident slot, and “group” and “sample” columns in the metadata. The “group” was set to the dissected layer of origination (L1-4, L5 and L6) in the cases of P14 and P60 Cx3cr1GFP scRNA-seq datasets and Fezf2 Control and KO scRNA-seq dataset (Fig. 2e, Extended Data Fig. 4a), Mg state (Homeostatic1 and Homeostatic2) in the case of Mg neighborhood PN composition (Fig. 3g), and the genotype in the case of P14 Fezf2 Control and KO snRNA-seq dataset (Fig. 4g and Extended Data Fig. 7k, 7o). The “sample” contained biological replicates for each group. With the prepared Seurat object as input, the Propeller function was run with the default parameters (arcsine square root transformation by the default). P-values are adjusted for multiple testing with a BH-correction. Note: in keeping with the publisher’s standard, Propeller statistics for compositional data were not calculated for data in Fig. 2e, 4g, and Extended Data Fig. 4a, 7k, 7o, as these experiments had an n<3.
One Fezf2 KO scRNA-seq sample (sample #3) was excluded from the dataset. The data from this sample did not match that for the other two Fezf2 KO samples. Re-genotyping of the pooled mice for Sample #3 revealed it was a mixture of one Fezf2 KO and one Fezf2 Heterozygote. The sample could therefore not serve as a Fezf2 KO replicate. No other animals were excluded from the analyses. Two MERFISH ROI regions were excluded as described above. No statistical methods were used to predetermine sample sizes.
All micrographs depicted in the figures are representative and were reproducible in all experiments with the following sample sizes: Figures: 1b (n = 3 mice/genotype, 4 images/mouse); 1e (n = 4 mice/genotype, 4 images/mouse); 1h (n = 3 mice/genotype, 4 images/mouse); 2b (n = 3 mice/age, 2 images/mouse); 4a–b (n = 3 mice/age, 2 images/mouse); 5i (3 mice, 3 images/mouse); and Extended Data Figures: 1a (n = 4 mice/genotype, 6 images/mouse); 1g–h (n = 2 mice/age, 2 images/mouse); 3b (n = 3 mice/age, 4 images/mouse); 3d (n = 3 mice/age, 4 images/mouse); 3f (n = 3 mice/age, 4 images/mouse); 3g (n = 3 mice/age, 4 images/mouse); 5d (n = 3 mice, 10 ROI).
Extended Data
Extended Data Figure 1: Control experiments associated with Fezf2 and Reln KO and control cortices.
Related to Fig. 1
a, Representative micrograph of P60 Fezf2 Control and KO S1 cortex immunolabelled for Iba1 (green) and Satb2 (magenta). Enlarged representative layer images are boxed. Bin divisions for cell counts are indicated. Arrowhead = Mg cell body. Scale = 100 μm.
b, Mg density grouped by bins corresponding to L1-4 and L5-6 for P7, P14 and P60 Control S1 cortices. Data are presented as mean values +/− SEM. n = 3 mice, 4 images/mouse (P7), 4 mice, 4 images/mouse (P14), and 4 mice, 6 images/mouse (P60). Statistics: Linear mixed models.
c, t-SNE of snRNA-seq from P14 S1 cortex color-coded by cell type. PN = Pyramidal Neuron, IN = Inhibitory Neuron, Oligo = Oligodendrocyte-lineage cell, Fibro = Fibroblast, BAM = Border Associated Macrophage. Data is reproduced from Extended Data Fig. 7g (Fezf2 Control only).
d-e, Expression feature plot (d) and violin plot (e) for Fezf2 transcript levels from snRNA-seq data. Fezf2 is excluded from Mg. Cell cluster labels are as in (c). Legend = expression value. Data is from Extended Data Fig. 7g.
f, Violin plot of the expression level of Fezf2 among PN subtypes. Clusters are color-coded. Lines = mean. Data are presented as mean values with probability density. Data is from Extended Data Fig. 7n.
g, Representative micrograph of RNA fluorescence in situ hybridization images from P7 (left) and P14 (right) S1 cortices marking Fezf2 (red), Fcrls/Tmem119 (Mg, green), and DAPI. Enlarged inserts show that Fezf2 is excluded from all Mg. Arrowheads = Mg. Scale = 100 μm.
h, (Left) Schematic of Fezf2-CreERT2/LSL-tdTomato mice injected with 4-Hydroxytamoxifen at embryonic day 16.5 to mark and lineage-trace Fezf2-expressing cells. (Right) Representative widefield tile scan image of a P7 coronal brain section immunolabelled for Mg (Iba1, green), endogenous tdTomato (red) and DAPI. Enlarged inserts show that tdTomato signal is excluded from all Mg. Scale = 100 μm.
i, Mg density grouped by bins corresponding to L1-4 and L5-6 of P60 Fezf2 Control and KO cortices. Data are presented as mean values +/− SEM. n = 4 mice, 6 images/mouse (P60). Statistics: linear mixed models.
j, Cartoon schematic of S1 cortices of Reln Control and KO mice. Observed phenotypic variations among Reln KO cortices are schematized, with PN subytpes color-coded.
k-l, Expression feature plot for Reln (k) and violin plots (l) for Reln and the genes that encode the cell surface receptors of Reln, Lrp8 and Vldlr. Reln, Lrp8 and Vldlr expression is excluded from nearly all Mg. Cell cluster labels are as in (c). Legend = expression value. Data are presented as mean values with probability density (l only). Data is from Extended Data Fig. 7g.
m-n, Scatter plot of Mg density (y-axis) by CPN density (x-axis) per bin from Reln Control (m) and KO (n) S1 cortices. Dots = bin data points. Data are presented as a linear regression color-coded by bin. R2 is listed for each bin, n = 4 images/mouse, 3 = mice/genotype.
o-p, Mg density (o) and CPN density (p) by cortical bin for Reln Control and KO S1 cortices. Data are presented as mean values +/− SEM. n = 4 images/mouse, 3 mice/genotype. Statistics: linear models.
Extended Data Figure 2: Quality control and analysis of scRNA-seq of Mg from Cx3cr1GFP layer dissected cortices.
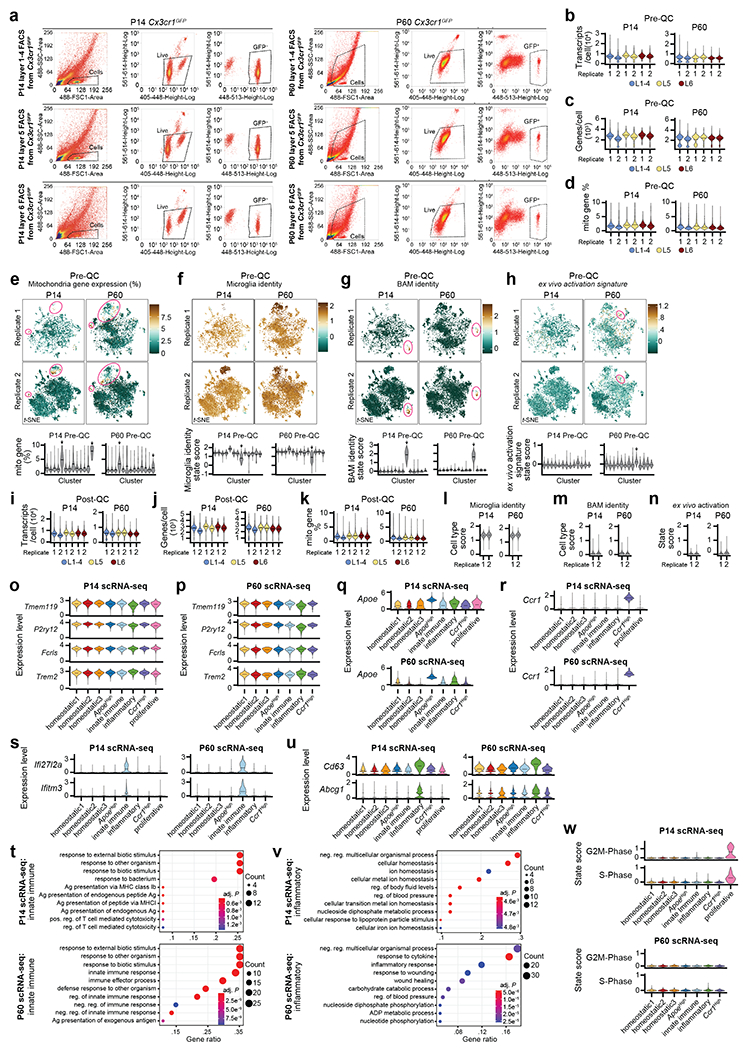
Related to Fig. 2
a, FACS histogram plots of GFP+ cell sorting from P14 (left) and P60 (right) Cx3cr1GFP mice. S1 cortices were dissected and sorted separately for L1-4 (top), L5 (middle) and L6 (bottom). FACS gating for whole cells (left), live cells (Hoechst+, middle), and GFP+ cells (right) are plotted as dashed boxes. Only live GFP+ cells were used for scRNA-seq experiments.
b-d, Violin plots of initial quality control (Pre-QC) metrics of the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Transcripts/cell (b), genes/cell (c), and percent of mitochondrial gene expression/cell (d) are plotted. Data are split by replicate and color-coded by dissected layer. Data are presented as mean values with probability density.
e, (Top) Expression feature plots for percent mitochondrial gene expression for the Pre-QC P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Feature plots are split by replicate for each age. Low-quality cells (mitochondrial gene > 10%) removed are circled. Legend = percentage. (Bottom) Violin plot of Pre-QC percent mitochondrial gene expression by initial clustering for the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Low-quality clusters (mitochondrial gene > 10%) removed are denoted with arrows. Data are presented as mean values with probability density (bottom only).
f-g, (Top) Expression feature plots for Mg (f) and BAM (g) cell type identity for the Pre-QC P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Feature plots are split by replicate for each genotype. BAMs removed are circled. (Bottom) Violin plot of Pre-QC Mg (f) and BAM (g) cell type identity by initial clustering for the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. BAM clusters removed are denoted with arrows. Data are presented as mean values with probability density (bottom only). Cell type scores (legend) are defined as a function of gene expression for core Mg and BAM genes identified in Marsh et al14 and Jordao et al13 (Supplementary Table 1).
h, (Top) Expression feature plots for ex vivo activation signature for the Pre-QC P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Feature plots are split by replicate for each genotype. ex vivo-activated cells that were removed are circled. (Bottom) Violin plot of Pre-QC ex vivo activation signature by initial clustering for the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. ex vivo-activated clusters that were removed are denoted with arrows. Cell scores (legend) are defined as a function of gene expression for core ex vivo activation genes14 (Supplementary Table 1).
i-k, Violin plots of final quality control (Post-QC) metrics of the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets following the removal of BAMs, low-quality cells and ex vivo-activated cells. Transcripts/cell (i), genes/cell (j), and percent of mitochondrial gene expression/cell (k) are plotted. Data are split by replicate and color-coded by layer sample. Data are presented as mean values with probability density.
l-m, Violin plots of the core Mg (l) and BAM (m) cell type scores split by replicate and age for the Post-QC P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Cell type scores are defined as a function of gene expression for core Mg14 and BAM13 genes (Supplementary Table 1). The final data strongly identify Mg. Data are presented as mean values with probability density.
n, Violin plots of the ex vivo activation state score of all Post-QC cells from the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Data are displayed as a state score, defined as a function of the core genes that are up-regulated in Mg through aberrant ex vivo activation14 (Supplementary Table 1). Data are split by replicate. Data are presented as mean values with probability density.
o-p, Violin plots for canonical Mg genes plotted by color-coded cluster from the P14 (o) and P60 (p) Cx3cr1GFP scRNA-seq experiments. Data are presented as mean values with probability density.
q-r, Violin plots for Apoe (q), and Ccr1 (r) expression in P14 (top) and P60 (bottom) Mg Cx3cr1GFP scRNA-seq clusters. Data are presented as mean values with probability density.
s, Violin plots depicting expression of the interferon-induced genes Ifi27l2a (top) and Ifitm3 (bottom) in P14 (left) and P60 (right) Mg Cx3cr1GFP scRNA-seq clusters. Data are presented as mean values with probability density.
t, Dot plot of the top 10 significant gene ontology terms enriched from the DE genes in the Innate Immune cluster compared to all other scRNA-seq clusters at P14 (top) and P60 (bottom). Legend: color = adj. P, dot size = number of genes. Statistics: hypergeometric test with BH procedure.
u, Violin plots depicting expression of the inflammation-induced genes Cd63 (top) and Abcg1 (bottom) in P14 (left) and P60 (right) Mg Cx3cr1GFP scRNA-seq clusters. Data are presented as mean values with probability density.
v, Dot plot of the top 10 significant gene ontology terms enriched from the DE genes in the Inflammatory cluster compared to all other scRNA-seq clusters at P14 (top) and P60 (bottom). Legend: color = adj. P, dot size = number of genes. Statistics: hypergeometric test with BH procedure.
w, Violin plots for G2M (top) and S-phase (bottom) cell cycle phase scores42 for P14 (top) and P60 (bottom) Mg Cx3cr1GFP scRNA-seq clusters. Data are presented as mean values with probability density.
Extended Data Figure 3: Identification of cortical non-homeostatic Mg in situ.
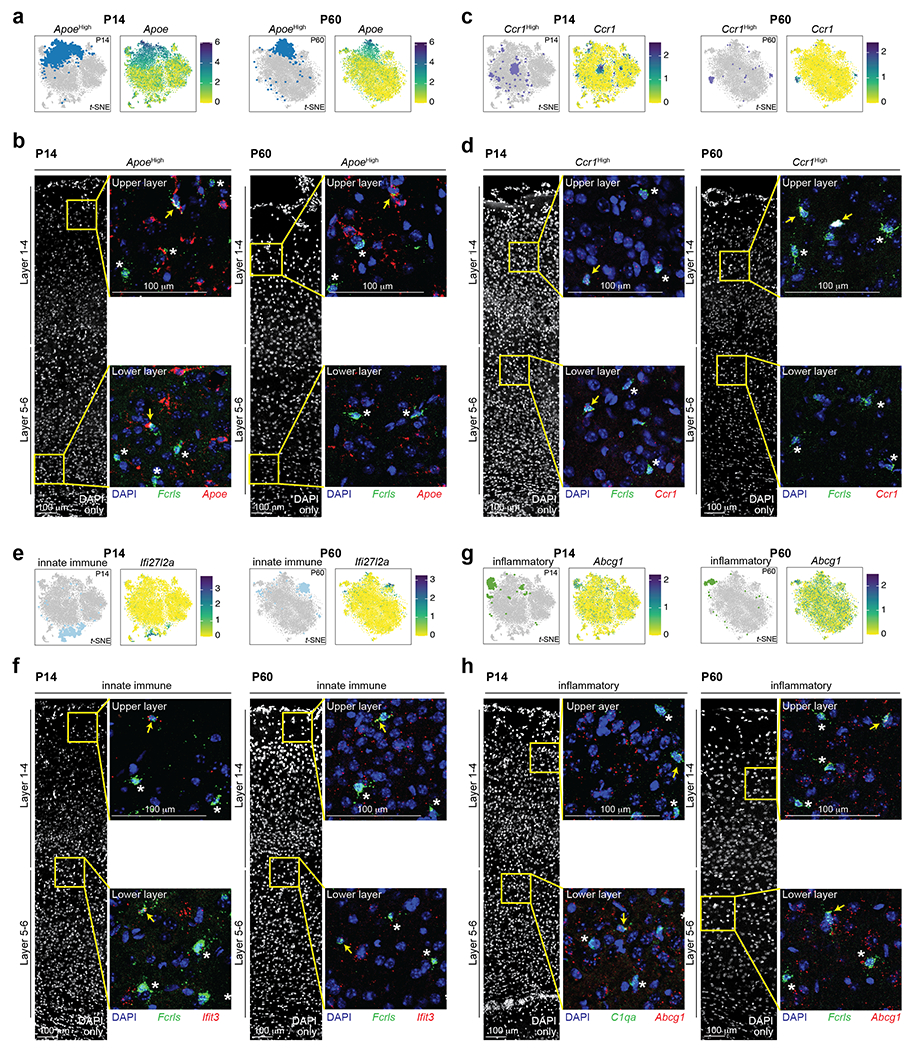
Related to Fig. 2
a, t-SNE plot highlighting the ApoeHigh cluster (left) and feature plot of Apoe expression (right) in the P14 and P60 Cx3cr1GFP scRNA-seq datasets. Legend = expression value.
b, Representative tile scan confocal RNA fluorescence in situ hybridization images from P14 (left) and P60 (right) S1 cortices marking Apoe (red), Fcrls (Mg, green) and DAPI. Enlarged inserts (yellow boxes) show Apoe-positive (arrow) and Apoe-negative (*) Mg at upper- and lower-layers of the cortex. Scale = 100 μm.
c, t-SNE plot highlighting the Ccr1High cluster (left) and feature plot of Ccr1 expression (right) in the P14 and P60 Cx3cr1GFP scRNA-seq Mg datasets. Legend = expression value.
d, Representative tile scan confocal RNA fluorescence in situ hybridization images from P14 (left) and P60 (right) S1 cortices marking Ccr1 (red), Fcrls (Mg, green) and DAPI. Enlarged inserts (yellow boxes) show Ccr1-positive (arrow) and Ccr1-negative (*) Mg at upper- and lower-layers of the cortex. Scale = 100 μm.
e, t-SNE plot highlighting the Innate Immune cluster (left) and feature plot of Ifi27l2a expression (right) in the P14 and P60 Cx3cr1GFP scRNA-seq Mg datasets. Legend = expression value.
f, Representative tile scan confocal RNA fluorescence in situ hybridization images from P14 (left) and P60 (right) S1 cortices marking Innate Immune Mg with Ifit3 (red), Fcrls (Mg, green) and DAPI. Enlarged inserts (yellow boxes) show Ifit3-positive (arrow) and Ifit3-negative (*) Mg at upper- and lower-layers of the cortex. Scale = 100 μm.
g, t-SNE plot highlighting the Inflammatory cluster (left) and feature plot of Abcg1 expression (right) in the P14 and P60 Cx3cr1GFP scRNA-seq Mg datasets. Legend = expression value.
h, Representative tile scan confocal RNA fluorescence in situ hybridization images from P14 (left) and P60 (right) S1 cortices marking Inflammatory Mg with Abcg1 (red), Fcrls or C1qa (Mg, green) and DAPI. Enlarged inserts (yellow boxes) show Abcg1-positive (arrow) and Abcg1-negative (*) Mg at upper- and lower-layers of the cortex. Scale = 100 μm.
Extended Data Figure 4: Characterization of the transcriptional signature of Mg states.
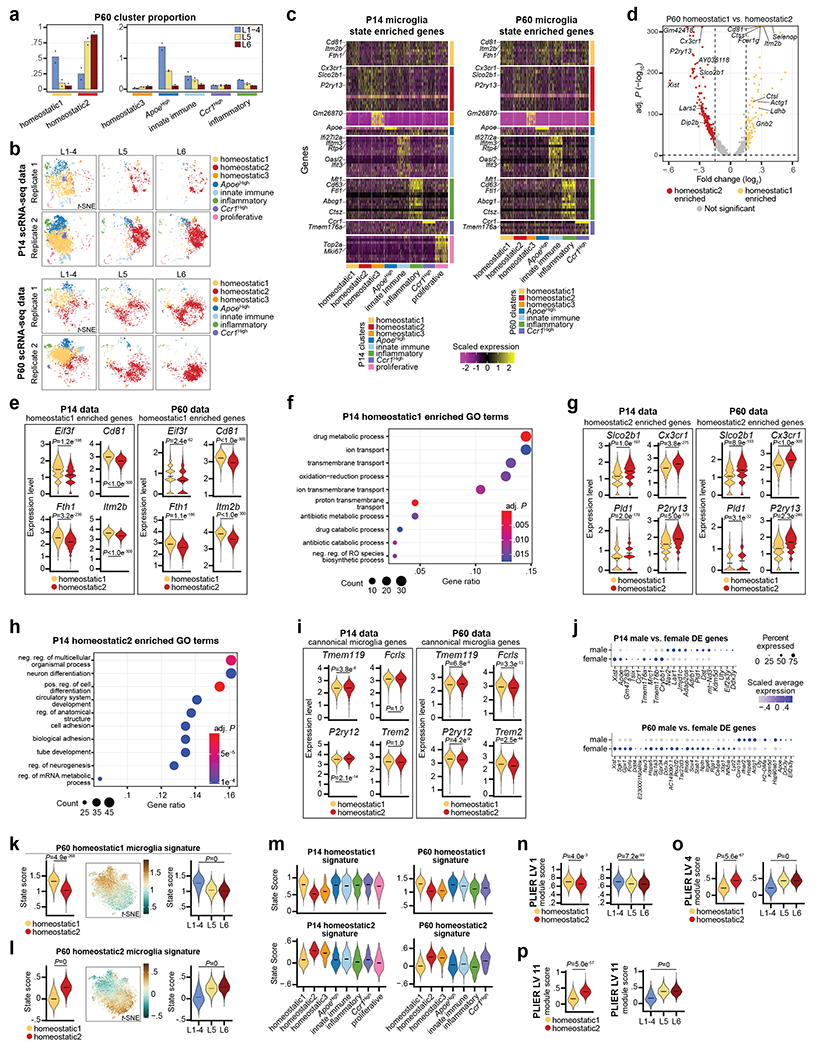
Related to Fig. 2
a, Bar plot of P60 Cx3cr1GFP scRNA-seq Mg cluster proportions normalized by dissected layer. Color coding indicates the layer from which the Mg were isolated. Data are presented as normalized mean values. n = 2 independent biological replicates.
b, t-SNE of P14 (top) and P60 (bottom) Cx3cr1GFP scRNA-seq color-coded Mg state clusters split by dissected layer of origin and replicate.
c, Heatmap of the top DE genes by Mg cluster in the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Each row is a gene. Color-coded clusters are grouped. Legend = scaled expression value.
d, Volcano plot of the DE genes between Homeostatic1 versus Homeostatic2 clusters in the P60 Cx3cr1GFP scRNA-seq dataset. Genes are color-coded by enrichment (log2 Fold Change > 0.15 and −log10 BH adj. P < 0.05). Homeostatic1-enriched (gold), Homeostatic2-enriched (red), not significant (grey). Several homeostatic state signature genes for each state are annotated. Vertical and horizontal dotted lines represent enrichment cut-offs. Statistics: generalized linear model with BH correction (MAST).
e, Violin plots of Homeostatic1-enriched genes expressed in Homeostatic1 and Homeostatic2 clusters from P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Data are presented as mean values with probability density.; Statistics: generalized linear model with BH correction (MAST).
f, Dot plot of the top 10 significant gene ontology terms enriched from the DE genes of the Homeostatic1 cluster compared to all other clusters in the P14 Cx3cr1GFP scRNA-seq dataset. Legend: color = BH adj. P, dot size = number of genes. Statistics: hypergeometric test with BH procedure.
g, Violin plots of Homeostatic2-enriched Mg genes expressed in Homeostatic1 and Homeostatic2 clusters from P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq dataset. Data are presented as mean values with probability density. Statistics: generalized linear model with BH correction (MAST).
h, Dot plot of the top 10 significant gene ontology terms enriched from the DE genes of Homeostatic2 cluster compared to all other clusters in the P14 Cx3cr1GFP scRNA-seq dataset. Legend: color = BH adj. P, dot size = number of genes. Statistics: hypergeometric test with BH procedure.
i, Violin plots of canonical Mg genes expressed in Homeostatic1 and Homeostatic2 clusters from the P14 (left) and P60 (right) Cx3cr1GFP scRNA-seq datasets. Data are presented as mean values with probability density. Statistics: generalized linear model with BH correction (MAST).
j, Dot plot of the DE genes between male and female Mg from the P14 (top) and P60 (bottom) Cx3cr1GFP scRNA-seq datasets. The data represent analysis from the combined dissected layers. Legend: color = scaled average expression, dot size = percent of the cluster cells expressing the gene. Most sex-specific DE genes do not match the list of DE gene between Homeostatic state clusters. Statistics: generalized linear model with BH correction (MAST).
k-l, Homeostatic1 (k) and Homeostatic2 (l) Mg signature state scoring of P60 Cx3cr1GFP scRNA-seq data: (Left) Violin plot of state scores for Mg in clusters Homeostatic1 and Homeostatic2. (Middle) Expression feature plot of state scores in t-SNE space. Legend: score value. (Right) Violin plot of state scores of homeostatic Mg split by dissected layer. Violin plot data are presented as mean values with probability density. Statistics: linear mixed models. Genes pertaining to the Homeostatic1 signature are listed in Supplemental Table 5.
m, Violin plots of the Homeostatic1 (top row) and Homeostatic2 (bottom row) state scores split by all clusters from P14 (left) and P60 (bottom) Cx3cr1GFP scRNA-seq datasets. Data are presented as mean values with probability density. State scores are defined as a function of gene expression pertaining to the genes enriched in the Homeostatic signatures listed in Supplementary Table 5.
n-p, Violin plots of gene module scores for PLIER latent variable (LV) 1 (n), LV 4 (o), and LV 11 (p) applied to Homeostatic1 and 2 Mg from the P14 scRNA-seq data. The data are split by state (left) and by dissected layer (right). Data are presented as mean values with probability density. Statistics: linear mixed models. Genes pertaining to the LV modules are listed in Supplementary Table 6.
Extended Data Figure 5: Quality control metrics for the MERFISH datasets.
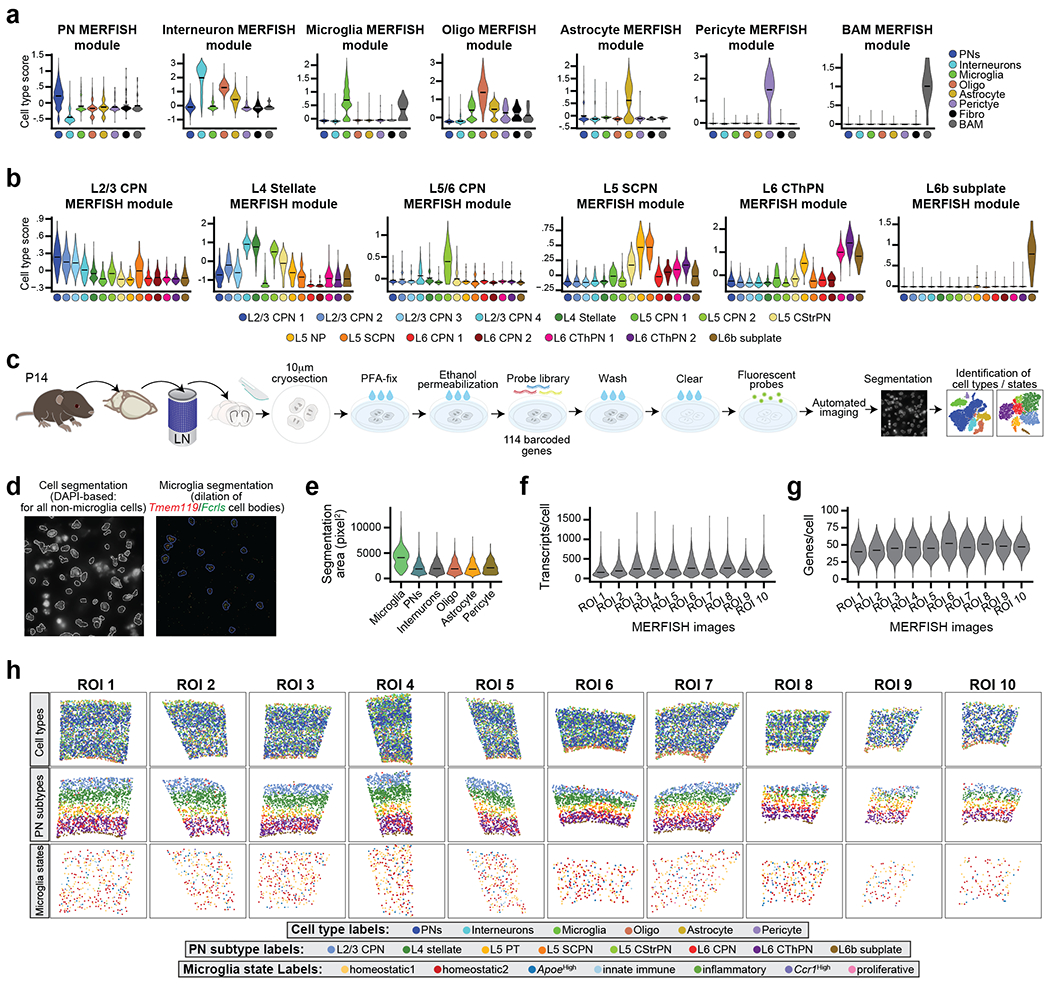
Related to Fig. 3
a-b, Violin plots of cell-type scores for MERFISH gene modules targeting cell types (a) and PN subtypes (b). The data are from P14 Fezf2 Control snRNA-seq in Extended Data Fig. 7. Data are presented as mean values with probability density. State scores are defined as a function of gene expression for the genes grouped by cell type or PN subtype listed in Supplementary Table 7.
c, Cartoon schematic of the MERFISH pipeline to map cell types and Mg states in the P14 S1 cortex.
d, Representative micrographs depicting the automated segmentation of cells for MERFISH analysis. (Left) All nuclei were automatically segmented (white outlines) based on DAPI signal. (Right) Mg were separately segmented based on Tmem119 (red) and Fcrls (green) RNA puncta with a small dilation (blue outline) in order to maximize RNA detection and counts. See Methods for full cell segmentation details.
e, Violin plots of the size of segmented cells split by the cell types identified in the MERFISH dataset. Data are presented as mean values with probability density.
f-g, Violin plots representing the number of RNA transcripts/cell (f) and genes/cell (g) for the MERFISH dataset split by region of interest (ROI) samples. Data are presented as mean values with probability density.
h, Spatial distribution MERFISH maps of cell types (top row), PN subtypes (middle row), and Mg states (bottom row) for each ROI from all imaged samples. Cropped images displayed are the analyzed regions of interest (S1 cortex) for each sample. Cell types and states are color-coded as indicated in the legend.
Extended Data Figure 6: Identification of cell type and Mg state from MERFISH data.
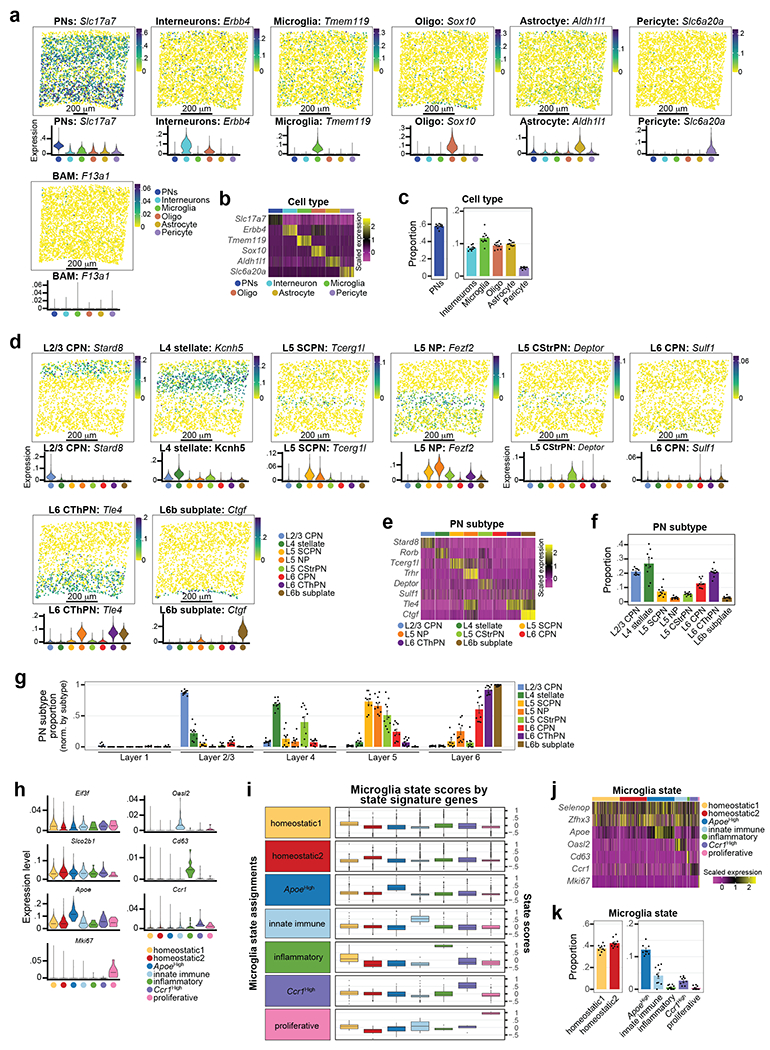
Related to Fig. 3
a, Spatial distribution feature (top row) and violin plots (bottom row) for the genes targeting cell types, via MERFISH. The spatial distribution feature plots are from a single representative image (ROI 1). Violin plots are split by cell type cluster. Spatial distribution feature plot legend = expression value. Violin plot legend = cell type.
b, Heatmap of gene expression specific to cell types as captured by MERFISH. Cell type clusters are color coded (subset of 200 cells/cluster). Legend: scaled expression value.
c, Mean cell type cluster proportion across MERFISH ROIs. Cell types are color coded. Data are presented as mean values +/− SEM. Dots = individual ROI frequencies. n = 10 ROI from 3 mice.
d, Spatial distribution feature (top row) and violin plots (bottom row) for the genes targeting PN subtypes by MERFISH. Spatial distribution feature plots shown are a single representative image (ROI 1). Violin plots are split by PN subtype cluster. Spatial distribution feature plot legend = expression value. Violin plot legend = cell type.
e, Heatmap of gene expression specific to PN subtypes as captured by MERFISH. PN subtype clusters are color coded (subset of 200 cells/cluster). Legend: scaled expression value.
f, Mean PN subtype cluster proportion across MERFISH ROIs. PN subtypes are color-coded. Data are normalized to the total PN count. Dots = individual ROI proportions. Data are presented as mean values +/− SEM. n = 10 ROI from 3 mice.
g, Mean layer proportion for each PN subtype identified by MERFISH. Data are normalized to the total count for each PN subtype. See methods for the calculation strategy for layer distinctions. PN subtypes are color-coded. Dots = individual sample proportions for each PN subtype by cortical layer. Data are presented as mean values +/− SEM. n = 10 ROI from 3 mice.
h, Violin plots for representative genes targeting different Mg states by MERFISH. Mg states are color-coded and split by state. Data are presented as mean values with probability density.
i, Box and whisker plots for MERFISH Mg state scoring. Mg state assignments are color coded and labelled at the left. See Methods for full details on MERFISH Mg state scoring. Mg state-enriched genes used for scoring are listed in Supplementary Table 7. Plots indicate median, 1st and 3rd quartiles, 1.5 inter-quartile range plus outlying points. n = all Tmem119+/Fcrls+ Mg from 10 ROI of 3 mice.
j, Heatmap of gene expression enriched in Mg states captured by MERFISH. Mg state assignments are color coded (subset of 200 cells/cluster). Legend: scaled expression value.
k, Mean Mg state assignment proportions across MERFISH ROIs. Mg state assignments are color coded. Dots = individual ROI proportions. Data are presented as mean values +/− SEM. n = 10 ROI from 3 mice.
Extended Data Figure 7: Molecular profiles of cell types and PN subtypes in Fezf2 Control and KO cortices by snRNA-seq.
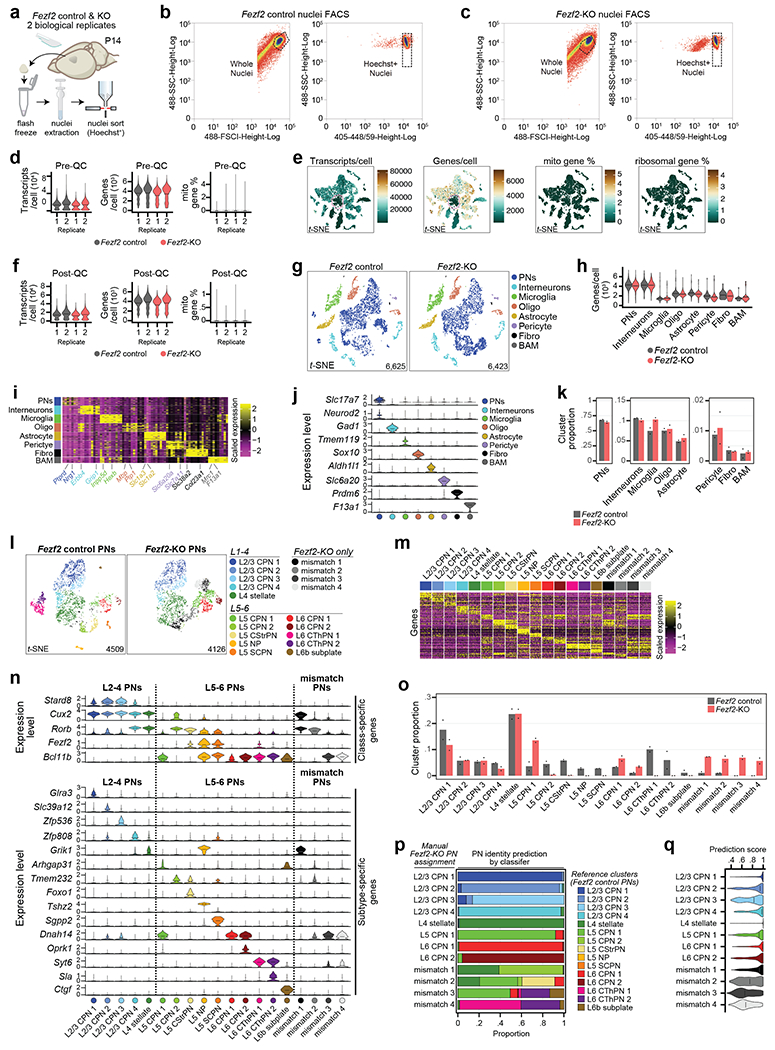
Related to Fig. 4
a, Cartoon of S1 cortex dissection, nuclei extraction, and purification by FACS from P14 Fezf2 Control and KO mice. n = 2 biological replicates for each genotype (1 male and 1 female for each genotype).
b-c, FACS scatter plots sorting nuclei from P14 Fezf2 Control (b) and KO (c) S1 cortices. FACS gating for whole nuclei (left) and Hoechst+ nuclei (right) are plotted as dashed boxes. Only Hoechst+ nuclei were used for snRNA-seq experiments.
d, Violin plots of initial quality control (Pre-QC) metrics for the Fezf2 Control and KO snRNA-seq dataset. Data are split by replicate and color-coded by genotype. Transcripts/cell (left), genes/cell (middle), and percent of mitochondrial gene expression/cell (right) are plotted. Data are presented as mean values with probability density.
e, Expression feature plots for Pre-QC metrics: transcripts/cell, genes/cell, percent mitochondrial gene expression/cell, and percent ribosomal gene expression/cell for the Pre-QC P14 Fezf2 Control and KO snRNA-seq data sets. Low-quality nuclei (circled) were those expressing very low genes/nucleus and did not return DE genes specific to known cell types. Legend: number (transcripts/nucleus and genes/nucleus), percent expression (mitochondrial and ribosomal gene %).
f, Violin plots of quality control (Post-QC) metrics of the P14 Fezf2 Control and KO snRNA-seq datasets following the removal of low-quality nuclei. Transcripts/nucleus (left), genes/nucleus (middle), and percent of mitochondrial gene expression/nucleus (right) are plotted, split by replicate and genotype. Data are presented as mean values with probability density.
g, t-SNE of Post-QC P14 Fezf2 Control (left) and KO (right) snRNA-seq datasets color-coded by cell type cluster. PN = Pyramidal Neurons, IN = Inhibitory Neuron, Oligo = Oligodendrocyte-lineage cells, Fibro = Fibroblast, BAM = Border Associated Macrophage. The number of sequenced nuclei is annotated for each genotype is noted.
h, Violin plot of the number of genes/nuclei split by cell type cluster and genotype for the P14 Fezf2 Control and KO snRNA-seq dataset. Data are presented as mean values with probability density.
i, Heatmap of gene expression for the top DE genes by cell type cluster from the P14 Fezf2 Control and KO snRNA-seq dataset. Each row is a gene and grouped by cell type cluster (color-coded). Legend = scaled expression value.
j, Violin plots depicting the expression level of marker genes for each cell type cluster. Cell type clusters are color coded. Data are presented as mean values with probability density.
k, Bar plot of mean cluster proportions by genotype for the P14 Fezf2 Control and KO snRNA-seq dataset. Data are normalized to the total cell count for each genotype. Color-coding indicates the genotype. n = 2 biological replicates for each genotype (1 male and 1 female for each genotype).
l, t-SNE of PN subtype clusters from the P14 Fezf2 Control and KO snRNA-seq dataset split by genotype. PNs were subsetted Slc17a7+/Neurod2+ nuclei from (g). PN subtypes are color-coded and labelled by their cortical layer localization. The number of sequenced nuclei for each genotype is annotated.
m, Heatmap of gene expression for the top 10 DE genes for each PN subtype cluster in the P14 Fezf2 Control and KO snRNA-seq datasets. Each row is a gene and grouped by PN subtype cluster (color-coded). Legend = scaled expression value.
n, Violin plots depicting the expression of select markers genes (split by cluster): PN class-specific (top) and PN subtype-specific (bottom). Clusters are color-coded. Data are presented as mean values with probability density. Dashed line splits upper- and lower- cortical layer PN subtypes, as well as Fezf2 KO mismatch PNs.
o, Bar plot of the mean PN subtype cluster proportion by genotype from P14 Fezf2 Control and KO snRNA-seq datasets. Data are normalized to the total PN count for each genotype. Color coding indicates the genotype. n = 2 biological replicates for each genotype (1 male and 1 female for each genotype).
p, Stacked bar plots depicting the prediction results of the Fezf2 KO PN subtype clusters using Azimuth24. PNs of the manually assigned subtype clusters (rows) from the P14 Fezf2 KO snRNA-seq data were re-classified based on the PN subtypes defined from the Control snRNA-seq data (color-coded). The re-classified data clusters are shown as proportions of the manual cluster size.
q, Violin plots of the Azimuth prediction scores for the P14 Fezf2 KO re-classified clusters. Re-classified clusters are color-coded. Data are presented as mean values with probability density.
Extended Data Figure 8: Cell sorting and quality control analysis for scRNA-seq of Mg from Fezf2 Control and KO layer dissected cortices.
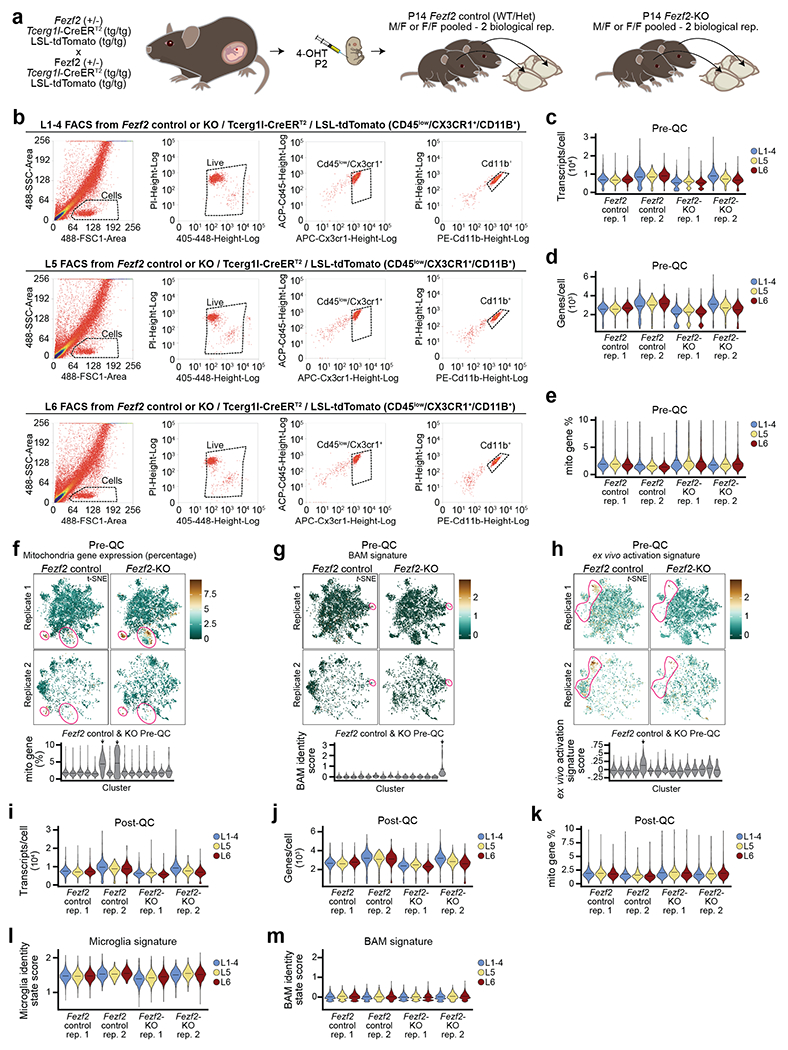
Related to Fig. 4
a, Cartoon schematic of the compound transgenic mouse line used to fluorescently label cortical L5b of Fezf2 Control and KO mice. 4-Hydroxytamoxifen (4-OHT, 50 mg/kg) was administered to pups on P2. Pups were sacrificed at P14 for dissection of the S1 cortex. n = 2 biological replicates (pooled 1 male and 1 female).
b, Representative FACS scatter plots sorting Cd45low/Cx3cr1+/Cd11b+ Mg from P14 Fezf2 Control or KO/Tcerg1l-CreERT2/LSL-tdTomato mice. The S1 cortex was dissected and sorted separately for L1-4 (top), L5 (middle) and L6 (bottom). FACS gating for whole cell (far left), live cells (Hoechst+ middle left), Cd45low/Cx3cr1+ (middle right) and Cd11b+ (far right) is plotted as dashed boxes. Only Cd45low/Cx3cr1+/Cd11b+ Mg were used for scRNA-seq experiments.
c-e, Violin plots of initial quality control (Pre-QC) metrics for the P14 Fezf2 Control (left) and KO (right) scRNA-seq datasets. Data are split by replicate and genotype, and color coded by dissected layer. Transcripts/cell (c), genes/cell (d), and percent of mitochondrial gene expression/cell (e) are plotted. Data are presented as mean values with probability density.
f, (Top) Expression feature plots for percent mitochondrial gene expression for the Pre-QC P14 Fezf2 Control and KO scRNA-seq dataset. Feature plots are split by replicate for each genotype. Low-quality cells (mitochondrial gene > 10%) that were removed are circled. Legend = percentage. (Bottom) Violin plot of Pre-QC percent mitochondrial gene expression by initial clustering for the P14 Fezf2 Control and KO scRNA-seq dataset. Low-quality clusters (mitochondrial gene > 10%) removed are denoted with arrows. Violing plot data are presented as mean values with probability density.
g-h, (Top) Expression feature plots for BAM cell type identity (g) and ex vivo activation signature (h) from the Pre-QC P14 Fezf2 Control and KO scRNA-seq dataset. Feature plots are split by replicate for each genotype. BAMs and ex vivo activated cells that were removed are circled. (Bottom) Violin plot of Pre-QC BAM (g) and ex vivo activated (h) signature scores by initial clustering for the P14 Fezf2 Control and KO scRNA-seq dataset. BAM and ex vivo activated clusters that were removed are denoted with arrows. Violin plot data are presented as mean values with probability density. Scores are calculated as a function of gene expression for core BAM13 and ex vivo activated14 genes, respectively (Supplementary Table 1).
i-k, Violin plots of quality control (Post-QC) metrics of the P14 Fezf2 Control and KO scRNA-seq dataset following the removal of BAMs, low-quality, and ex vivo activated cells. Data are split by replicate, genotype, and dissected layer. Transcripts/cell (i), genes/cell (j), and percent of mitochondrial gene expression/cell (k) are plotted. Data are presented as mean values with probability density.
l-m, Violin plots of the core Mg (l) and BAM (m) cell type scores from the post-QC Fezf2 Control and KO scRNA-seq dataset. Data are split by replicate and genotype, and color coded by dissected layer. The final data strongly identify Mg. Cell type scores are defined as a function of gene expression for core Mg14 and BAM13 genes (Supplementary Table 1). Data are presented as mean values with probability density.
Extended Data Figure 9: Mg transcriptional signatures in the Fezf2 KO cortex.
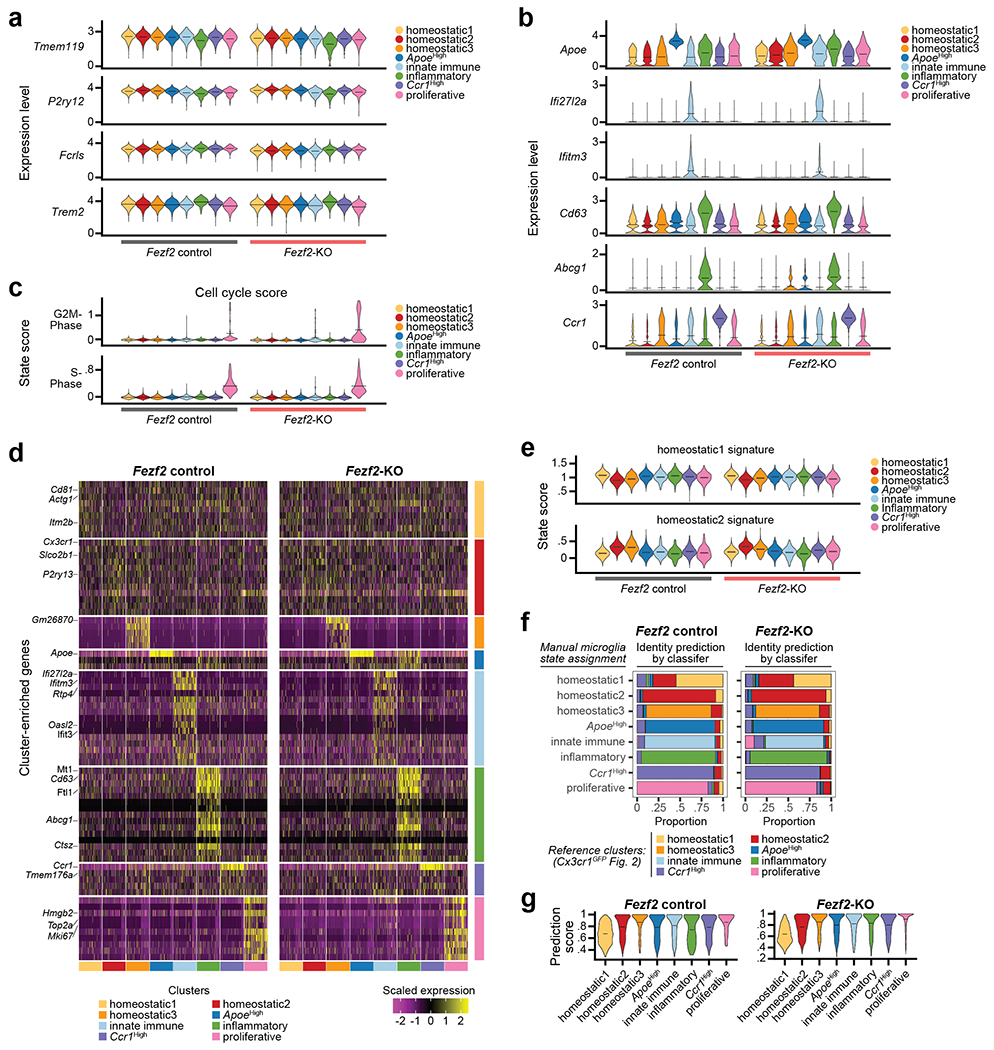
Related to Fig. 4
a-b, Violin plots for canonical Mg (a) and state-enriched (b) genes plotted by cluster and split by genotype from the P14 Fezf2 Control and KO layer dissected scRNA-seq dataset. Clusters are color coded. Data are presented as mean values with probability density.
c, Violin plots for G2M (top) and S-phase (bottom) scores for the P14 Fezf2 Control and KO scRNA-seq data split by cluster. Cells are scored by cell cycle phase42. Data are presented as mean values with probability density.
d, Heatmap of gene expression for the top DE genes for each Mg state cluster in the P14 Fezf2 Control (left) and KO (right) scRNA-seq dataset. Each column is a gene and grouped by Mg state cluster (color-coded). Legend = scaled expression value.
e, Violin plots of the Homeostatic1 (top row) and Homeostatic2 (bottom row) state scores split by all clusters from the P14 Fezf2 Control and KO scRNA-seq dataset. Clusters are color-coded. Data are presented as mean values with probability density. State scores are defined as a function of gene expression pertaining to the genes enriched in the Homeostatic signatures listed in Supplementary Table 5.
f, Stacked bar plots depicting Mg state prediction results using Azimuth24. Mg of the manually assigned clusters (rows) from the P14 Fezf2 Control (left) and KO (right) scRNA-seq dataset were re-classified based on the cell types defined in the P14 Cx3cr1GFP scRNA-seq dataset (color coded). The re-classified data are shown as proportions of the total cell count per manually assigned cluster.
g, Violin plots of the Azimuth prediction scores for the P14 Fezf2 Control (top) and KO (bottom) re-classified clusters. Re-classified clusters are color coded. Data are presented as mean values with probability density.
Extended Data Figure 10: Extended data for the ligand-receptor, PN-Mg interactome of the neocortex.
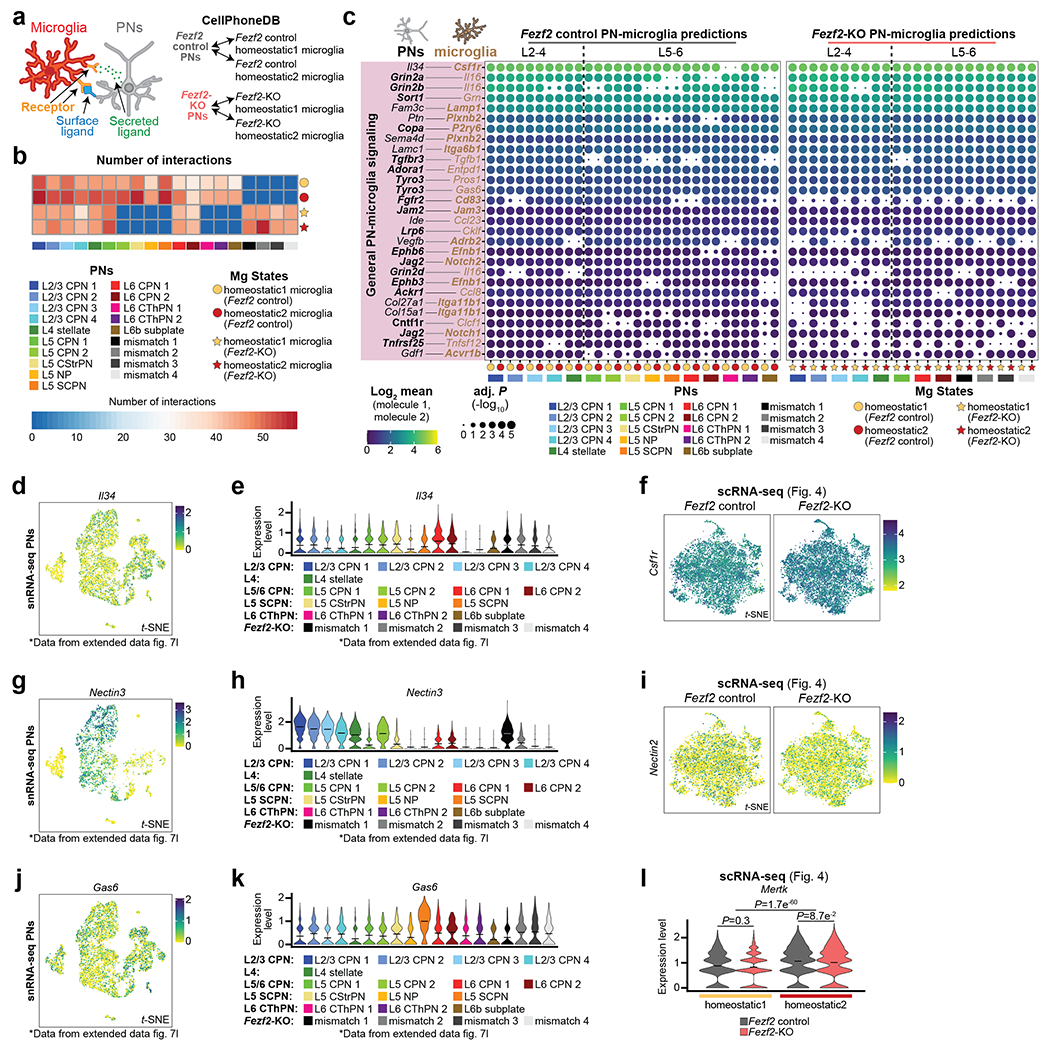
Related to Fig. 5
a, Cartoon schematic of the ligand-receptor analysis to uncover novel PN-Mg subtype interactions using CellPhoneDB26. The interactome predicts interactions between PN subtypes (snRNA-seq data, Extended Data Fig. 7) and homeostatic Mg (scRNA-seq data, Fig. 4) from P14 Fezf2 Control and KO mice.
b, Heatmap of the number of significant ligand-receptor (L-R) interaction pairs between each PN subtype (column) and Mg state (row) cluster. Legend: PN subtype (colored boxes) and homeostatic Mg state (color-coded circles for the Fezf2 Control Mg, stars for the Fezf2 KO Mg). Scale = number of interactions.
c, Dot plot for L-R expression between PN subtypes and homeostatic Mg states in Fezf2 Control (left) and KO (right) cortices. Legend: PN subtype (colored boxes) and layered homeostatic Mg (color-coded circles for the Fezf2 Control Mg, stars for the Fezf2 KO Mg) used in each pairwise comparison. −log10 adj. P values are indicated by circle size. Dot color indicates the average expression level of the interacting molecules from PN subtypes and Mg state. Receptor genes are bolded. Dotted vertical line divides upper- and lower-layer PN subtypes. Statistics: permutation-based BH adj. P.
d-e, Expression feature (d) and violin plots (e) of Il34 levels in PN subtypes from the P14 Fezf2 Control and KO snRNA-seq dataset. Data originate from Extended Data Fig. 7l. Legend = expression (d) and color-coded PN-subtypes (e). Violin plot data are presented as mean values with probability density.
f, Expression feature plot of Csf1r levels in Mg from the P14 Fezf2 Control (left) and KO (right) scRNA-seq datasets. Data originate from Fig. 4. Legend = Expression value.
g-h, Expression feature (g) and violin plots (h) of Nectin3 levels in PN subtypes from the P14 Fezf2 Control and KO snRNA-seq dataset. Data originate from Extended Data Fig. 7l. Legend = expression (g) and color-coded PN subtypes (h). Violin plot data are presented as mean values with probability density.
i, Expression feature plot of Nectin2 levels in Mg from the P14 Fezf2 Control (left) and KO (right) scRNA-seq dataset. Data originate from Fig. 4. Legend = Expression value.
j-k, Expression feature (j) and violin plots (k) of Gas6 levels in PN subtypes from the P14 Fezf2 Control and KO snRNA-seq dataset. Data originate in Extended Data Fig. 7l. Legend = expression (j) and color-coded PN-subtypes (k). Violin plot data are presented as mean values with probability density.
l, Violin plot of Mertk levels in Mg from the P14 Fezf2 Control and KO scRNA-seq dataset. Data denote the differential expression of Mertk between Homeostatic1 and Homeostatic2 and between genotypes. Data are presented as mean values with probability density. Statistics: generalized linear model with BH correction (MAST).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Arlotta and Levin laboratories for insightful discussions and editing of the manuscript. We thank Juliana Brown for the dedicated revision work on the manuscript. We thank Sean Simmons for the expert review of the bioinformatic work contained within the manuscript. This work was supported by grants from the Broad Institute of MIT and Harvard and the US National Institute of Health to P.A., the Stanley Center for Psychiatric Research to P.A. and J.Z.L, and The Klarman Cell Observatory to J.Z.L. J.A.S. was supported by a career developmental award from HHMI.
Footnotes
COMPETING INTEREST
P.A. is a SAB member in System 1 Biosciences and Foresite Labs and is a co-founder of FL60. J.A.S. is an employee of Sana Biotechnology, Inc. as of 09/2022.
MATERIALS AND CORRESPONDENCE
Extended Data and Supplementary Information are available for this paper.
DATA AVAILABILITY
The data that support the findings of this study are available at gs://pyramidal_neurons_microglia with permissions from the corresponding author (public access restriction in place due to storage cost).
REFERENCES
- 1.Bennett FC et al. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biase LMD et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 95, 341–356.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammond TR et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawson LJ, Perry VH, Dri P & Gordon S Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Colonna M & Butovsky O Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 35, 1–28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prinz M, Jung S & Priller J Microglia Biology: One Century of Evolving Concepts. Cell 179, 292–311 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Fujita Y, Nakanishi T, Ueno M, Itohara S & Yamashita T Netrin-G1 Regulates Microglial Accumulation along Axons and Supports the Survival of Layer V Neurons in the Postnatal Mouse Brain. Cell Reports 31, 107580 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Bella DJD et al. Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 595, 554–559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molyneaux BJ, Arlotta P, Hirata T, Hibi M & Macklis JD Fezl Is Required for the Birth and Specification of Corticospinal Motor Neurons. Neuron 47, 817–831 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Kwan KY, Šestan N & Anton ES Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139, 1535–1546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartfuss E et al. Reelin signaling directly affects radial glia morphology and biochemical maturation. Development 130, 4597–4609 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Jung S et al. Analysis of Fractalkine Receptor CX 3 CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol Cell Biol 20, 4106–4114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jordão MJC et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, (2019). [DOI] [PubMed] [Google Scholar]
- 14.Marsh SE et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat Neurosci 25, 306–316 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masuda T et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Askew K et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Reports 18, 391–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villa A et al. Sex-Specific Features of Microglia from Adult Mice. Cell Reports 23, 3501–3511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao W, Zaslavsky E, Hartmann BM, Sealfon SC & Chikina M Pathway-level information extractor (PLIER) for gene expression data. Nat Methods 16, 607–610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tasic B et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kongsui R, Beynon SB, Johnson SJ & Walker FR Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. J Neuroinflamm 11, 182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matho KS et al. Genetic dissection of the glutamatergic neuron system in cerebral cortex. Nature 598, 182–187 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Easley-Neal C, Foreman O, Sharma N, Zarrin AA & Weimer RM CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia Development and Maintenance in White and Gray Matter Brain Regions. Front Immunol 10, 2199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badimon A et al. Negative feedback control of neuronal activity by microglia. Nature 586, 417–423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiyoshi R et al. Microglia Enhance Synapse Activity to Promote Local Network Synchronization. Eneuro 5, ENEURO.0088-18.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cserép C et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367, 528–537 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Liu YU et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat Neurosci 22, 1771–1781 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Z et al. Instructing Perisomatic Inhibition by Direct Lineage Reprogramming of Neocortical Projection Neurons. Neuron 88, 475–483 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lodato S et al. Excitatory Projection Neuron Subtypes Control the Distribution of Local Inhibitory Interneurons in the Cerebral Cortex. Neuron 69, 763–779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wester JC et al. Neocortical Projection Neurons Instruct Inhibitory Interneuron Circuit Development in a Lineage-Dependent Manner. Neuron 102, 960–975.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomassy GS et al. Distinct Profiles of Myelin Distribution Along Single Axons of Pyramidal Neurons in the Neocortex. Science 344, 319–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Favuzzi E et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 184, 4048–4063.e32 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velmeshev D et al. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 364, 685–689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirata T et al. Zinc finger gene fez‐like functions in the formation of subplate neurons and thalamocortical axons. Dev Dynam 230, 546–556 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Zheng GXY et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nestorowa S et al. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finak G et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He L et al. NEBULA is a fast negative binomial mixed model for differential or co-expression analysis of large-scale multi-subject single-cell data. Commun Biology 4, 629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw FASTQ files and counts matrices from scRNA-seq data of Mg from dissected P14 and P60 Cx3cr1GFP layers, Mg from dissected Fezf2 Control and KO/Tcerg1l-CreERT2/LSL-tdTomato layers, and Fezf2 Control and KO S1 nuclei are available at GEO accession number GSE158096. Raw MERFISH images are available at gs://pyramidal_neurons_microglia. Processed MERFISH counts matrix is available at GEO accession number GSE193760. Tool needed to open raw images can be found at https://github.com/ZhuangLab/storm-analysis/tree/master/imagej_plugins. All source code can be found at https://github.com/kimkh415/MicrogliaLayers. All data are available upon reasonable request from Lead Contact.
The data that support the findings of this study are available at gs://pyramidal_neurons_microglia with permissions from the corresponding author (public access restriction in place due to storage cost).