

Summary
Predicted loss of function (pLoF) variants are often highly deleterious and play an important role in disease biology, but many pLoF variants may not result in loss of function (LoF). Here we present a framework that advances interpretation of pLoF variants in research and clinical settings by considering three categories of LoF evasion: (1) predicted rescue by secondary sequence properties, (2) uncertain biological relevance, and (3) potential technical artifacts. We also provide recommendations on adjustments to ACMG/AMP guidelines’ PVS1 criterion. Applying this framework to all high-confidence pLoF variants in 22 genes associated with autosomal-recessive disease from the Genome Aggregation Database (gnomAD v.2.1.1) revealed predicted LoF evasion or potential artifacts in 27.3% (304/1,113) of variants. The major reasons were location in the last exon, in a homopolymer repeat, in a low proportion expressed across transcripts (pext) scored region, or the presence of cryptic in-frame splice rescues. Variants predicted to evade LoF or to be potential artifacts were enriched for ClinVar benign variants. PVS1 was downgraded in 99.4% (162/163) of pLoF variants predicted as likely not LoF/not LoF, with 17.2% (28/163) downgraded as a result of our framework, adding to previous guidelines. Variant pathogenicity was affected (mostly from likely pathogenic to VUS) in 20 (71.4%) of these 28 variants. This framework guides assessment of pLoF variants beyond standard annotation pipelines and substantially reduces false positive rates, which is key to ensure accurate LoF variant prediction in both a research and clinical setting.
Keywords: predicted loss-of-function variants, pLoF, PVS1, gnomAD, variant classification, population data, variant interpretation
Graphical abstract
Our study demonstrates that 27.3% of heterozygous predicted loss-of-function (pLoF) gnomAD variants, in genes associated with autosomal-recessive disease, might not undergo nonsense-mediated decay due to suspected evasion. We provide guidance on how to advance pLoF interpretation in both research and clinical settings.
Introduction
Loss-of-function (LoF) variants can have important implications in human disease biology by either partial or complete loss of gene expression depending on the zygosity of the variant.1,2 Loss of protein abundance is known to be caused by nonsense, frameshift, essential splice site, initiation codon variants, and structural variants spanning one or several exons, such as deletions and tandem partial gene duplications. Missense variants can also result in loss of protein function, though it is not straightforward to computationally predict the impact of a missense variant on protein abundance. To date, nonsense, frameshift, and essential splice site variants are the three types of single-nucleotide variants (SNVs) annotated as pLoF (predicted LoF) by standard annotation pipelines and what we define as pLoF throughout this work.3,4
Databases of human population genetic variation, such as the Genome Aggregation Database (gnomAD), enable us to refine our ability to interpret population genome-sequencing data and assess variant pathogenicity.5 pLoF variants identified through large-scale sequencing efforts like gnomAD require careful evaluation to predict their true effect.6,7 Previous studies have indicated that late truncating variants and variants that disrupt splicing at in-frame exons do not result in nonsense-mediated decay (NMD), but instead produce truncated protein products or in-frame deletions.8,9 Furthermore, all sequencing data are at risk for inclusion of sequencing artifacts, defined as variation introduced by a non-biological process such as read mis-mapping and base mis-calling.10
Current ACMG/AMP guidelines for sequence variant interpretation enable assessment of variants using criteria such as computational and predictive evidence, functional evidence, segregation evidence, de novo evidence, population evidence, and allelic data.11 These guidelines are used worldwide to classify variants as pathogenic, likely pathogenic, uncertain, likely benign, or benign. PVS1, the ACMG/AMP evidence code for LoF variation in a gene where LoF is a known mechanism of disease, is the strongest weighted evidence in the ACMG/AMP curation process, which can result in pLoF variants being classified as pathogenic with only minimal additional evidence required. As such, it is clear that pLoF variants require a more in-depth assessment to accurately predict their effect. Therefore, the ClinGen Sequence Variant Interpretation (SVI) working group developed LoF interpretation guidelines, which outline how and when to count evidence for pLoF variants and apply strength modifications to the PVS1 code.4 These additional specifications go into detail regarding how to accurately identify falsely annotated pLoF variants that are not subject to NMD, such as pLoF variants in the most 3′ exon of a gene that may produce a functional, albeit truncated, protein and therefore should not be given the full strength of PVS1.
Interpretation of pLoF variants is also important, and substantially more challenging, in the context of large population cohorts, where such variants have been consistently shown to be highly enriched for a wide variety of sequencing rescues, artifacts, and annotation errors. The driver of this enrichment is Bayesian: there is a high prior probability that a pLoF variant observed in a gene associated with disease in a rare disease patient is real, but this probability is much lower for a similar variant observed in an individual ascertained at random from the population.2,12 As a result, studies applying careful curation to population cohorts have found consistently high rates of sequencing and classification errors.5,13,14,15 High error rates complicate studies leveraging pLoF variants seen in large cohorts to explore human gene function, an approach that has proven extremely valuable for the identification and validation of potential therapeutic targets.16 While automated approaches to pLoF variant filtering remove a fraction of errors,5,17 multiple studies have demonstrated the value of deep manual curation of pLoF variants to identify evasion modes and sequencing artifacts missed by these automated tools.14,15,18
Here we present an advanced LoF curation framework for interpreting pLoF variants (nonsense, frameshift, and essential splice site) that expands on current guidelines. This framework highlights when a pLoF variant may not be subject to NMD or when it is a potential technical artifact, the latter being especially useful when assessing data from population databases like gnomAD, where sequencing validity cannot be verified with orthogonal methods. The framework considers three main categories: (1) predicted rescue by secondary sequence properties, (2) uncertain biological relevance, and (3) potential technical artifacts. We present the result from manually curating all high-quality pLoF (heterozygous) variants in 22 genes associated with autosomal-recessive (AR) disease in 141,456 individuals from gnomAD v.2.1.1 using this framework. Additionally, we provide guidance on how to utilize this framework for applying PVS1 criteria and interpreting pathogenicity, in line with, and further building on, the ACMG/AMP and ClinGen SVI recommendations for PVS1 use.4,11
Material and methods
The framework was developed by defining established mechanisms of pLoF evasion and identifying potential technical artifacts as previously reported in the literature.4,2,5,6,7,12,13 A set of 22 genes associated with AR disease with 1,113 pLoF variants was selected for manual curation. The genes were selected based on unrelated collaborations with advocacy groups to define disease prevalence. The analysis included variants passing gnomAD quality control filters, excluding low-confidence genotypes (depth < 10, genotype quality < 20, allele balance < 20% for non-reference heterozygous variants) and excluding outliers of the random forest model that considers allele-specific annotations.5 Any variant annotated as pLoF by the Variant Effect Predictor (VEP; stop-gained/nonsense, essential splice acceptor/donor [±1–2], or frameshift variants) in either exomes or genomes for any protein-coding transcript in gnomAD v.2.1.1 (VEP version 85 using GENCODE v.19 on GRCh37) was included. Variants annotated as low-confidence by the Loss-of-Function Transcript Effect Estimator5 (LOFTEE; removing variants less likely to result in LoF) were excluded. Manual curation was then independently performed by two biocurators in a custom curation interface (https://github.com/macarthur-lab/variant-curation-portal), and any discrepancies were resolved by group discussion. Resources used for curation of pLoF variants included gnomAD variant and gene pages,12 UCSC genome browser,19 and SpliceAI for essential splice site variant interpretation.20 For transcript-level flags, variants were evaluated using “Basic Gene Annotation Set from GENCODE version 19” in the UCSC genome browser. A subset of pLoF variants curated as likely not LoF/not LoF were additionally assessed for effects on PVS1 using ACMG/AMP and ClinGen SVI guidelines.4,11 The correlation between variants that are predicted to evade LoF and benign variants in ClinVar was determined using variants that had at least one submission to ClinVar (479/1,113, https://ftp.ncbi.nlm.nih.gov/pub/clinvar/, January 5, 2023).
Each variant was assessed for evidence suggesting LoF evasion and potential technical artifacts by the rules that define the final verdict of LoF, likely LoF, uncertain LoF, likely not LoF, or not LoF (Table 1). The thresholds can be modified (conservative or lenient) depending on the overall aim of a curation project (Table 2). A conservative cut-off generates fewer false positive LoF/likely LoF but more false negatives (variants called as not LoF while in fact they cause true LoF), while lenient rules are more inclusive and will discard fewer variants as likely not LoF/not LoF but instead result in more false positive pLoF variants. Flags were applied conservatively to identify any variant potentially not causing LoF given that gnomAD, like any population database of genome and exome research data, is likely to be enriched for pLoF variants that do not result in LoF, especially in genes associated with disease.
Table 1.
Rules that define LoF verdicts
| LoF | Likely LoF | Uncertain | Likely not LoF | Not LoF |
|---|---|---|---|---|
| Predicted rescue by secondary sequence properties | ||||
| Splice rescue that introduces stop codon | translation re-initiation removing >25% coding sequence |
weak translation reinitiationa | splice rescue with weak predictionsa | splice rescue with strong predictionsa |
| variant falls within overhang exon | MNV resulting in a missense/synonymous variant | |||
| Intron retention | in-frame and out-of-frame rescue events | strong translation re-initiationa | frame-restoring indel | |
| in-frame exon skipping (according to splicing prediction) | ||||
| Uncertain biological relevance | ||||
| Minority of transcripts with pext at maximum for genea | minority of transcripts with pext close to maximum for genea | minority of transcripts with pext < 50% of the maximum of the gene (pext > 20% of maximum)a |
weak exon conservation with pext < 50% of the maximum of the gene (pext > 20% of maximum)a | Pext < 20% of maximum for the genea |
| Weak exon conservation with pext at maximum for genea | weak exon conservation with pext close to maximum for genea | nonsense variant in overprinted transcript | splice variant not supported by pexta | |
| variant terminates within the last exon or last 50 bp of the penultimate exona | ||||
| Potential technical artifacts | ||||
| No read data for splice and nonsense variants | genotyping errors above thresholda | no read data for frameshift variants | genotyping errors below thresholda | |
| GC-rich region | ||||
| Strand bias | low complexity sequence | homopolymera | ||
| minor mapping errorsa | complex mapping errorsa | |||
When multiple rules apply to a pLoF variant, the most impactful consequence is assigned. LoF, loss of function; MNV, multi-nucleotide variant; pext, proportion expressed across transcripts; bp, basepairs.
Specific thresholds and additional recommendations for a subset of these rules are found in Table 2.
Table 2.
LoF curation rules details
| Rule | Conservative rules | Lenient rules | Other factors to consider |
|---|---|---|---|
| Predicted rescue by secondary sequence properties | |||
| Essential splice rescuea,b | SpliceAI predicts rescue at above threshold score of 0.2. strong rescue determined by rescue event prediction at score within 0.2 of donor/acceptor loss | distance from canonical splice site | |
| out-of-frame cryptic splice sites | |||
| strength of splicing predictors | |||
| type of splicing predictor used | |||
| inclusion of termination codon | |||
| differences for donor vs. acceptor splice sites | |||
| Translation reinitiationa | downstream methionine within first exon removes >25% of coding sequence and is fairly well conserved | downstream methionine within first exon removes >10% of coding sequence and is fairly well conserved | alternative methionine start sites beyond first exon, specifically for genes with small first exons |
| functional domain at 5′ end of gene | |||
| other pathogenic variants in same region | |||
| In-frame exon | SpliceAI predicts in-frame exon skipping that removes <25% coding sequence | SpliceAI predicts in-frame exon skipping that removes <10% coding sequence | functional domain contained within exon |
| Uncertain biological relevance | |||
| Low pexta | pext ≤ 20% max for gene | pext ≤ 10% max for gene | tissue-specific pext |
| multiple biologically relevant transcripts | |||
| low pext for whole gene | |||
| long genes (3′ bias) | |||
| Minority of transcripts/weak exon conservation/low pexta | pext < 50% max for gene | pext < 30% max for gene | same as above |
| Last exona | termination event removes <25% coding sequence | termination event removes <10% coding sequence | other pathogenic variants in 3′ end of gene |
| functional domain at 3′ end of gene | |||
| Potential technical artifacts | |||
| Genotyping errors (DP, AB, GQ)a | read depth < 15 | read depth < 10 | combinations of multiple technical errors |
| allele balance < 35% | allele balance < 25% | ||
| genotype quality < 30 | genotype quality < 20 | ||
| Mapping errorsa | UCSC repeat masker tracks > 5 | UCSC repeat masker tracks > 3 | |
| Homopolymera | number of nucleotide repeats in reference ≥ 5 | number of nucleotide repeats in reference ≥ 7 | indels vs. SNVs at homopolymer repeats |
| known pathogenic variants occurring at this position | |||
Each rule includes a list of other factors to consider when defining a framework for LoF curation. SNVs, single-nucleotide variants; pext, proportion expressed across transcripts; bp, base pairs.
LoF verdicts that are defined by these thresholds can be found in Table 1.
Essential splice rescue flag relies on the prediction of in silico tools, thus the recommended threshold for that tool applies. For SpliceAI that is 0.2, with no lenient threshold.
Results
Evidence suggesting LoF evasion determines final verdict
Each pLoF variant is assessed for evidence of LoF evasion and assigned flags according to rules (Table 1) subdivided into three categories: (1) predicted rescue by secondary sequence properties, (2) uncertain biological relevance, and (3) potential technical artifacts (Figure 1). The combination of flags is used to determine pLoF verdict: (1) LoF, (2) likely LoF, (3) uncertain LoF, (4) likely not LoF, or (5) not LoF. The visualization of read data to assess variant quality and potential rescues (such as frame-restoring indels) is essential to this protocol. Thus, frameshift variants without read data were classified as “uncertain,” unless additional evidence of the variant suggested LoF evasion or predicted the variant as a potential technical artifact. Alternatively, lack of nearby rescues could be confirmed using vcf files if access to visualization tools is not possible. However, when a nearby secondary variant is present, a vcf is insufficient to predict phasing.
Figure 1.

Schematic showing the main categories of evidence in pLoF interpretation
(A) LoF evasion as a result of a predicted rescue by secondary sequence properties. A termination event near a source of rescue allows for translation of the sequence into protein and escapes NMD.
(B) A termination event within an exon of uncertain biological relevance is predicted to evade loss of protein abundance. Uncertain biological relevance can be identified here by a combination of the location of the termination event within a minority of transcripts, weakly conserved exon relative to surrounding region, and low mean pext score, suggesting that the affected exon is in fact of low biological importance.
(C) Potential technical artifacts where analytical confirmation is needed to confirm the variant.
Predicted rescue by secondary sequence properties
Rescue flags are assigned to pLoF variants that are predicted to be rescued by a secondary sequence property such as an in-phase multi-nucleotide variant (MNV), frame-restoring indel, essential splice site rescue, in-frame exon skipping, translation reinitiation, and overhanging exon.21,22,23,24,25,26,27,28 Since standard variant annotation pipelines do not assess the variant in the context of the surrounding sequence, these variants will be annotated as pLoF despite nearby rescues.
MNVs refer to multiple SNVs found within the same codon and haplotype that have arisen either as a single mutational event or as multiple coincidental mutations. MNVs often have a different effect on the protein sequence in aggregate than the same variants considered individually,22,29,30 but existing variant annotation pipelines consider all SNVs as independent events, resulting in errors in impact prediction of MNVs (Figure S1). Frame-restoring indels can also be rescued by the presence of another variant on the same haplotype; for example one variant annotated as causing a frameshift can be rescued by one or several nearby indels, with the aggregate impact being an in-frame indel (potentially with some intervening sequence resulting in multiple missense substitutions) rather than a frameshift (Figure S2).
Essential splice site variants cause LoF by disrupting splicing, typically resulting in usage of a cryptic splice site and/or exon skipping leading to introduction of an early termination codon.31,32 Using established splice predictors like SpliceAI can help predict the effect of variation at an essential site, i.e., using an up- or downstream cryptic splice site that is either in-frame or out-of-frame, exon skipping, intron retention, or a combination of these events. Out-of-frame cryptic splice sites will in most cases result in introduction of an early termination codon and NMD, while in-frame cryptic sites result in an in-frame indel without a loss of protein abundance. Of note, for in-frame cryptic splice sites resulting in partial intron retention, the intronic sequence needs to be assessed for inclusion of termination codons that could result in early truncation and NMD. Essential splice site variants at the border of an in-frame exon can also result in an in-frame deletion of that exon rather than introducing an early termination codon and NMD, which can also be predicted by SpliceAI.
Other types of transcript rescue include translation reinitiation and overhang exons. The translation reinitiation flag is assigned to variants that have a nearby in-frame methionine downstream of the termination event that may re-initiate translation. The overhang exon flag is assigned to variants that fall in an exon extension (Figure S3). Overhang exons are often weakly conserved, have a lower pext score, and fall in a minority of coding transcripts. Variants that fall within overhang exons are considered rescued by splicing out the overhanging sequence through essential splice sites of other transcripts and thus are predicted to evade LoF.
Uncertain biological relevance
Variants of uncertain biological relevance are expected to result in NMD within at least one transcript, but their effect on the overall protein abundance is not predicted to have a biological impact. Specific flags include minority of transcripts, weak exon conservation, low pext, and overprinting. We also include variants in the last exon where NMD is not expected but the functional impact of the pLoF variant is unclear. These flags highlight the requirement for in-depth interpretation of the variant, transcript, and conservation.33,34,35,36
Pext, proportion expressed across transcripts, scores are available on the gnomAD browser gene page and inform the relative per-base expression across transcripts in GTEx tissues.17 Variants that fall in low pext regions, defined as an exon with mean pext value < 20% of the maximum pext across the gene, are often in biologically dispensable alternative transcripts (Figure S4). It is noteworthy that most splice variants fall outside the coding region, and in those cases the pext score will always be 0. However, the biological relevance of the splice site can be interpreted by looking at the score of the adjacent exon; if the adjacent exon has a low pext score it is likely biologically dispensable. We also used the absence of a drop in the pext score within an exon at an annotated splice site as evidence that it is likely a splice site (and transcript) of low biologic relevance (Figure S5).
Variants that are annotated as pLoF in a minority of coding transcripts across the gene need to be assessed for whether or not they fall in the most biologically relevant transcript (e.g., MANE Select and MANE Plus Clinical are recommended for GRCh38). In genes with multiple transcripts, transcript expression in a specific tissue or the mean expression across tissues might be a useful indicator of biological relevance. In this protocol, we flagged variants occurring in <50% of coding transcripts. Likewise, a pLoF variant located in a weakly conserved exon may indicate that loss of that exon does not impact gene function. Some variants are located in exons with slightly lower pext scores (∼50% of gene maximum), a minority of transcripts, and/or weakly conserved exons, implying that the exon itself is less relevant to gene function. The combination of these flags (minority of transcript, weak exon conservation, and low pext) can be used to assess the biological relevance of the transcripts in which the variant is annotated as pLoF. Of note, lack of a pext score should not be used as evidence for or against LoF evasion, as this is likely due to the variant being located in a transcript that was not included when pext scores were generated. It is also important to consider that GTEx gene-expression data used to generate pext is derived from adult postmortem tissues, which may not accurately represent gene expression during early development37 and not all disease-relevant tissues are available (e.g., inner ear).
The overprinting flag is applicable for a variant in a transcript with an unconserved alternate open reading frame (ORF; Figure S6). Overprinting has been described as a means for de novo gene birth and is widely reported in viral DNA, and more recently in plants and animals.38,39,40 If a variant is annotated as pLoF only in the “novel” overprinted transcript and has a different annotation (missense/synonymous) in the ancestral frame, it is not considered to cause LoF in the primary gene annotated at the locus.
Variants that terminate in the last exon of a gene or the last 50–55 bp of the penultimate coding exon typically result in truncated protein products due to NMD escape.41,42 Therefore, pLoF variants assigned with the last exon flag are not predicted to result in lost protein abundance but rather the presence of a truncated protein, which may or may not have a deleterious effect on protein function and needs further assessment. LOFTEE (v.1.0.3) does not flag by NMD location but instead by GERP score of the affected region of the protein, so a number of pLoF variants predicted to escape NMD are not annotated as such by LOFTEE.5 Furthermore, not all genes are subject to NMD, and those that are not subject to NMD need to be interpreted differently. This is unknown for most genes and in the absence of other knowledge, our default assumption is to expect NMD.
Potential technical artifacts
Technical flags are assigned to pLoF variants that are likely artifacts from sequencing data rather than true variants and include genotyping, mapping, and homopolymer flags. Technical flags are used for variants in regions where the confidence of finding a real LoF variant is decreased by the region quality and where there is a higher rate of false positive variant calls in exome and genome capture.43,44,45,46,47,48
Genotyping flags are assigned to variants with a skewed allele balance, low read depth, or low genotype quality; or that fall in low complexity or GC-rich regions; or demonstrate strand bias (variants called predominantly on the forward or reverse strand; Figure S7). Mapping flags are assigned to variants that fall in a region of the genome where there are known mapping difficulties due to repetitive genome-wide sequences, and variation in these regions might be a result of mis-mapped reads (Figure S8). UCSC genome browser’s repeats tracks can be used to identify regions that are likely to be mis-mapped. Homopolymers, a sequence of consecutive identical bases, are enriched for false positive indels due to polymerase slippage during PCR amplification. Slippage results in inaccurate reports of repeat length, which are then incorrectly annotated as frameshift variants.49,50,51,52,53
Curation framework predicts that 27% of pLoF variants do not result in LoF
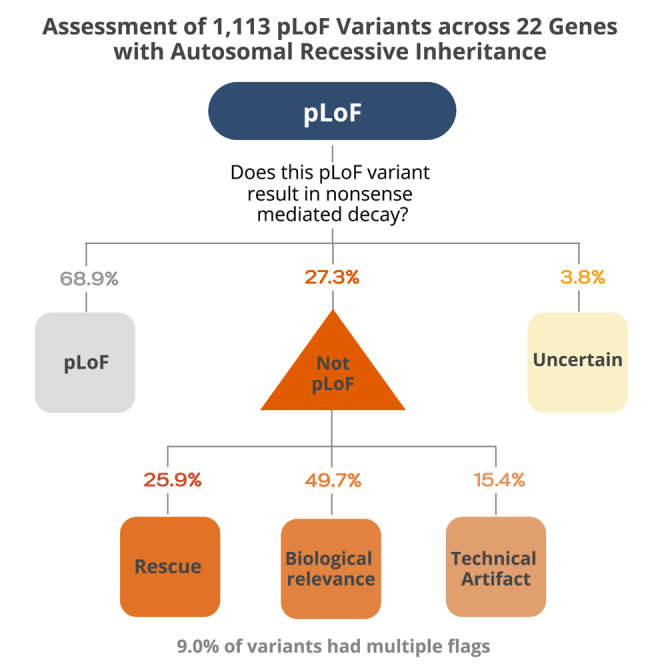
Curation was performed on 1,113 pLoF high-confidence by LOFTEE variants in 22 genes associated with AR disease. LOFTEE pre-filtered 143 variants as low confidence mostly due to their location in the end-truncating region of the gene (Figure S9).5 Of the 1,113 LOFTEE high-confidence pLoF variants, 304 (27.3%) were interpreted as likely not LoF/not LoF, 42 (3.8%) were interpreted as uncertain LoF, and 767 variants (68.9%) remained LoF/likely LoF after curation (Figure 2A; Table S1). The frequency of pLoF evasion and potential technical artifacts in AR genes was significantly lower than the 66.5% evasion rate observed for heterozygous pLoF variants in genes associated with dominant disease (n = 403),17 where LoF variants are expected to be absent or depleted from gnomAD (p = 5.44 × 10−43; Figure S10).
Figure 2.

Evaluation of 1,113 heterozygous high-confidence predicted loss-of-function (pLoF) variants in 22 genes associated with autosomal recessively inherited disease in gnomAD predict 27.3% do not result in true LoF
(A and B) Distribution of LoF verdicts in whole set (A) and per gene (B).
(C) Reasons for variants predicted as likely not LoF/not LoF (n = 304 variants). Combination of reasons refers to variants with more than one reason for likely not LoF/not LoF verdict.
(D) The number of pLoF variants assigned with each flag within each classification category, colored by categories: potential technical artifacts (blue), uncertain biological relevance (purple), and rescue by secondary sequence properties (red). Tx, transcript; multi, multiple.
A variable proportion of LoF evasion and potential technical artifacts was observed between genes, explained by gene-specific properties (Figures 2B and S9). HADH displayed the highest degree of LoF evasion and potential technical artifacts, 72.7% (24/33), mainly due to several regions with low pext scores, while GBE1 displayed the lowest degree, 5.0% (2/40) of LoF evasion and potential technical artifacts. Across the 1,113 variants, the most common variant class was frameshift, with 470 variants (42.2%), followed by 360 (32.3%) stop-gained/nonsense variants, 283 (25.4%) essential splice variants, 153 donors (13.8%), and 130 acceptor variants (11.7%) (Figures S11A–S11C). The proportion of variants that were predicted to not result in LoF depended on variant class (Figure S11D). Stop-gained/nonsense variants were most likely to be predicted as true LoF compared to other variant types (p = 4.01 × 10−6; post hoc 2 × 2 chi-squared test; Bonferroni significance threshold p < 0.0125 for 4 post hoc tests) with only 19.1% predicted as likely not LoF/not LoF, whereas 40.0% of essential splice acceptor variants were predicted as likely not LoF/not LoF (p = 2.81 × 10−4, post hoc 2 × 2 chi-squared test). Frameshift variants also had a slightly elevated proportion of predicted likely not LoF/not LoF (30.9%, p = 0.0102, post hoc 2 × 2 chi-squared test). Essential splice donor variants did not significantly deviate from the mean across other variant types (24.8%, p = 0.463, post hoc 2 × 2 chi-squared test).
Variants curated as likely not LoF/not LoF had a median of two flags per variant (Figure S12), though for the vast majority (91.1%, 277/304 variants) a single flag was sufficient to label them as likely not LoF/not LoF: last exon (32.6%), homopolymer (13.8%), low pext (13.2%), splice rescue (11.8%), in-frame exon (7.9%), MNV or frame-restoring indels (4.6%), overprinting (3.9%), other transcript rescues (e.g., translation reinitiation, overhang exon) (1.6%), mapping errors (1.3%), and multiple technical artifacts (0.3%) (Figure 2D). Variants interpreted as likely not LoF/not LoF due to their location, either in the last exon or at the border of an in-frame exon, were only considered likely not LoF/not LoF if they removed less than 25% of the coding sequence. Of 99 variants interpreted as likely not LoF/not LoF due to location in the last exon, 75.7% (75/99) terminated in the last 10% of the coding sequence and the remaining 24.2% (24/99) of variants terminated in the last 10%–25% of the coding sequence. A similar pattern was observed for splice variants at the border of in-frame exons; 75.0% (18/24) resulted in a deletion of less than 10% of the protein coding sequence, and 25.0% (6/24) of variants resulted in a deletion spanning 10%–25% of the protein-coding sequence. Although the vast majority of variants had a single primary explanation for evasion (91.1%), they often had additional less impactful flags (Figure S13). Only 8.9% (27/304) of variants were assigned multiple flags that are sufficient for a prediction of likely not LoF/not LoF, with the most prevalent combination being other transcript rescues (overhang exon) and low pext.
An analysis on the effect of all splice variants (n = 283) using SpliceAI revealed that 25.8% (73/283) of splice variants are predicted to lead to a potential LoF rescue by an in-frame cryptic splice event, in-frame exon skipping, or location at a non-essential exon (Figure S14). Of note, 2.1% (6/283) were predicted to splice an in-frame intronic sequence that included a termination codon. These were expected to result in LoF, highlighting the need to consider several rescue mechanisms in parallel when assessing a final verdict. Other categories of splicing effects that retained LoF/likely LoF interpretations were out-of-frame rescues, out-of-frame exon skipping, multiple out-of-frame events, and presumed intron retention.
Variants given a verdict of LoF/likely LoF either had no flags (white, Figure 2C) or had potential technical artifacts and/or uncertain biological relevance flags (light blue and purple, Figure 2C) not considered strong enough evidence for the variant to be predicted as likely not LoF/not LoF (Table 1). The 42 variants (3.8%) that were given a verdict of uncertain LoF were mainly uncertain because of unavailable read data for visualization of potential frame-restoring indels in the surrounding region for variants in gnomAD. Of the 42 variants, 5 (11.9%) had no flags assigned to them but were marked uncertain LoF mostly due to unclear splicing mechanisms.
ACMG/AMP-guided pLoF interpretation
The LoF curation protocol presented above predicts a variant’s likelihood to result in LoF but does not assess the variant’s pathogenicity. A pLoF variant curated as likely not LoF/not LoF may still be pathogenic via other mechanisms besides complete loss of gene expression, such as an in-frame deletion of a functional domain resulting in a catalytically inactive protein. Here we build upon the previous ClinGen SVI guidelines by Abou Tayoun et al.4 and provide a framework for further adjusting PVS1 for pLoF variants with a verdict of uncertain LoF, likely not LoF, or not LoF (Figure 3).
Figure 3.

Framework for adjusting ACMG/AMP PVS1 criteria for variants curated as uncertain LoF, likely not LoF, or not LoF
For analytically confirmed uncertain LoF, likely not LoF, or not LoF variants, PVS1 should be modified accordingly; no changes to PVS1 (gray), downgrade PVS1 by one level (light pink, PVS1_strong max), downgrade PVS1 by two levels (purple, PVS1_moderate max), or not to use PVS1 at any level (dark pink). Downgrading is done by worst consequence and not in an additive manner.
The assessment of variants as technical artifacts is important for the accurate return of individual patients results, as well as the review of evidence from population databases such as gnomAD to ensure that variant occurrence and population allele frequencies are accurately represented. All variants assigned technical flags are by definition located in a region with quality concerns, and therefore allele frequencies in these regions in gnomAD may be higher than expected and thus, need to be interpreted with care.12 Variants with quality concerns should be confirmed by an orthogonal method before assessing pathogenicity, as only a real variant will confer a disease risk. However, the analytic validity of a variant is a separate step from pathogenicity classification and therefore technical artifact consideration is not used to modify PVS1 strength in the context of a variant classification framework.
Figure 3 highlights modifications to the application of PVS1 following use of this framework for further pLoF interpretation. If a variant has been assigned several flags that suggest downgrading PVS1, the flag resulting in the most substantial downgrade should be applied to the curation (and not the sum of different consequences). For example, if a curation has resulted in both a splice rescue flag (downgrade PVS1) and a low pext flag (do not use PVS1), then PVS1 should not be applied, instead of downgrading.
Evaluation of the 479 variants (of the 1,113 assessed) that had ClinVar entries demonstrated that the 125/479 pLoF variants that were predicted as likely not LoF/not LoF were more likely to be classified as B/LB (16/125, 12.8%) in ClinVar compared to the 346/479 pLoF variants predicted as LoF/likely LoF (2/346, 0.6%) (p < 0.0001, Fisher’s exact test). We further assessed the 89 variants predicted as likely not LoF/not LoF that had been submitted as pathogenic/likely pathogenic in ClinVar (Figure S15; Table S2). The majority, 63 of 89 variants, were pathogenic/likely pathogenic in ClinVar by multiple submitters (2-star submission and above), and 45 of 63 variants had multiple reported cases and functional studies. The 2-star and above submissions were mostly likely not LoF/not LoF due to last exon flag and homopolymer flag (Figure S15). Of note, the accuracy of the ClinVar pathogenicity classifications was not formally evaluated.
To investigate the concordance between variants predicted to evade LoF and the effect on PVS1, we assessed a subset of 200 out of 304 pLoF variants curated as likely not LoF/not LoF (Figure 3; Table S3). Variants flagged as potential artifacts cannot be assessed using PVS1 unless analytically confirmed, so 37 variants with a technical artifact flag resulting in likely not LoF/not LoF verdict were excluded, leaving 163 variants. Including the updates to PVS1 presented in this manuscript and the ACMG/AMP guidelines for interpretation of loss-of-function variants, the PVS1 criteria was downgraded by at least 1 level, to PVS1_strong or lower, for 162 of 163 variants (99.4%). Of the 162 downgraded variants, PVS1 was affected due to updated guidelines of this framework in 17.2% (28 variants), with an effect on the final ACMG/AMP classification for 20 of those 28 variants (19 variants downgraded from likely pathogenic to VUS and one variant from pathogenic to likely pathogenic).
Discussion
There are several mechanisms by which pLoF variants can escape LoF, which is why careful assessment beyond standard annotation pipelines are key to reduce false positive rates and ensure accurate prediction in both research and clinical settings. We present a new framework that refines the interpretation of pLoF variants’ predicted impact and introduces a structured methodology to predict variants as LoF, likely LoF, uncertain LoF, likely not LoF, or not LoF. Further, we expand on how this evidence ties into the assessment of pathogenicity by current ACMG/AMP guidelines, and specifically how PVS1 should be modified, in line with and further building upon standards provided by the ClinGen SVI working group.4
The LoF curation protocol introduces three different categories of evidence that should be assessed: rescue by secondary sequence properties, uncertain biological relevance, and potential technical artifacts. Of 1,113 high-confidence pLoF variants in 22 genes associated with AR disease investigated here, 304 variants (27.3%) were predicted as likely not LoF/not LoF. The main reasons were truncation in the 3′ end of the gene, location in a homopolymer region, location in a low-pext region, and essential splice variants with in-frame cryptic rescues, highlighting the importance of detailed assessment to accurately interpret a pLoF variant. Only 47/304 variants (15.5%) were predicted as likely not LoF/not LoF because of potential technical artifacts, highlighting that this is a minor category and the majority (84.5%) are predicted likely not LoF/not LoF due to rescue by secondary sequence properties or uncertain biological relevance. A similar rate of LoF evasion and potential technical artifacts is observed for homozygous pLoF variants in gnomAD, in line with the expected enrichment of rescue mechanisms and artifacts seen for pLoF variants in general.5,54
In addition to the expected difference in evasion between gene sets, there were differences in frequency of evasion between LoF variant classes. Nonsense variants were more likely to be predicted as true LoF with a lower evasion rate of 19%, while 40% of essential splice acceptor variants were predicted likely not LoF/not LoF. The elevated evasion rate for essential splice acceptor variants was mainly driven by location in the last exon and cryptic splice site rescue, with the cryptic splice acceptor variant rescues suggestively being due to less consensus site conservation for splice acceptor than donor sites.32,55 This can guide how we think of splice acceptor variants as escape seems more common, whereas nonsense variants to a large extent are predicted as true LoF (Figure S11).
The LoF curation protocol does not determine variant pathogenicity, but rather predicts the likelihood of a pLoF variant evading LoF or acting as a technical artifact. Additional information regarding variant classification is required to determine a pLoF variant’s pathogenicity, including if there is a non-NMD or other less damaging predicted effect of a pLoF variant, as well as segregation data, case-level evidence, functional evidence, de novo evidence, and population evidence. Variants predicted as likely not LoF/not LoF were enriched for variants classified as benign/likely benign in ClinVar, highlighting that our framework can identify variants that potentially evade LoF and do not cause disease. The two variants predicted as likely LoF by our framework but classified as benign/likely benign in ClinVar (indicating potential missed evasion of LoF) are annotated as pLoF in one non-MANE Select IDUA transcript (Ensembl: ENST00000247933.4) (three transcripts reported in Gencode v.19), with a mean pext score of 0.4 (max for the gene is 0.6–0.7), which suggests some biological relevance and the variants were therefore not excluded as likely not LoF/not LoF across all tissues. One can speculate that the affected transcript is non-essential for the enzymatic function of IDUA that is disrupted in the IDUA-associated metabolic condition mucopolysaccharidosis (MIM: 607014).
Importantly, variants predicted as likely not LoF/not LoF by our framework may still be pathogenic via mechanisms other than loss of protein abundance. In some genes, truncating variants within the last exon may alter the function of the gene rather than result in LoF. For example, truncating variants in the last exon of CCND2 can remove a ubiquitination site and prevent normal cyclin degradation.56 In particular, the 89 variants predicted as likely not LoF/not LoF but classified as pathogenic/likely pathogenic in ClinVar were assessed for alternative pathogenicity evidence. For +2-star ClinVar submissions with multiple reported cases and functional studies (63/89), the majority were defined as likely not LoF/not LoF due to the location of the variant in the last exon (which can be pathogenic via impact on the protein function) or due to location in an homopolymer (which would be reclassified as LoF if the variant was confirmed by an orthogonal method). Those variants with 0–1 stars in ClinVar had a larger number of reasons for LoF evasion, highlighting the utility of this framework. Of note, 3/89 previously classified as pathogenic in ClinVar changed classification during the period of revising this paper, demonstrating that ClinVar variants, especially those classified by a single submitter, warrant further scrutiny to confirm their pathogenicity. We established an effect on PVS1 in 99.4% (162 of 163) of assessed variants predicted as likely not LoF/not LoF using the existing PVS1 guidelines4,11 in combination with our framework. Importantly, PVS1 was affected in 17.2% (28 of 163) of variants as a result of the updated guidelines provided in this report, mostly due to essential splice site variants predicted to be rescued by in-frame cryptic splice events, MNVs, or frame-restoring indels. This result highlights the importance of considering the new properties presented here when assessing pLoF variants and their pathogenicity. Further, for 71.4% (20/28) variants, the effect on PVS1 resulted in a downgraded ACMG/AMP variant classification, mostly from likely pathogenic to uncertain significance, highlighting the clinical impact of this framework. Overlooking these mechanisms of escape confers a risk of overestimating the pathogenicity of pLoF variants. As a future direction, we plan to incorporate lessons learned through this advanced framework of pLoF interpretation into LOFTEE to improve automated LoF prediction, though we anticipate that manual evaluation will continue to serve a critical role.
One important consideration during LoF curation is that the general rate of evasion and potential technical artifacts (27.3% in this variant set) will vary depending on how the variants were ascertained (from affected individuals or from population data), as well as methods used for identifying the variants (large-scale sequencing or standardized clinical sequencing including orthogonal confirmation). Since LoF variants as a group are under negative selection and the proportion of artifacts from genotyping will be constant across the genome and variant classes, pLoF variants in population data will be more enriched for artifacts, especially in genes associated with disease that are constrained for LoF.13,54 In cohorts enriched with individuals affected by severe disease, the contrary is true, with an expected enrichment for pLoF variants that are true positives and also pathogenic. Therefore, it is expected that the evasion rate and number of technical artifacts are much lower in a cohort of affected individuals. Thus, the source of the variant data should impact the conservative or lenient threshold set for the different flags presented in this protocol, and we recommend a conservative approach for any curation of population data.
This protocol aims to include any of the community-established and -accepted mechanisms of LoF evasion and predictions of technical artifacts to improve pLoF predictions. However, additional mechanisms resulting in LoF evasion have been suggested and are likely to be established in the future. For example, it has been suggested that pLoF variants in an exon longer than 400 bp or a variant located in the first 150 coding bp will escape nonsense-mediated decay.7 Clonal hematopoiesis should be considered when assessing pLoF variants in genes associated with this phenomenon.57,58 Genes susceptible to clonal hematopoiesis due to proliferative advantage from haploinsufficiency (monoallelic LoF) is an aspect not within the scope of this protocol (beyond hard filtering variants with an allele balance of less than 20%).
In conclusion, we present a framework that aids in the interpretation of pLoF variants by considering mechanisms of LoF evasion and indications of potential technical artifacts, alongside updated guidelines for applying PVS1 for classifying pLoF variant pathogenicity. The results presented here highlight how inadequate pLoF variant assessment stands a risk of overinterpreting the effect and pathogenicity of pLoF variants and that this framework can substantially reduce the false positive rate of pLoF in both research and clinic settings.
Acknowledgments
We thank the individuals and researchers whose data are in gnomAD for their contributions to research. We thank the ClinGen Sequence Variant Interpretation Working Group for useful discussion of PVS1 modifications and Raymond Walters for valuable advice on statistical analysis. This work was supported in part by NIH grants NHGRI U24HG011450, NHGRI U24HG006834, NHGRI U01HG011755, NIGMS R01GM104371, and NHLBI R01HL143295, and by research funding from the Chan Zuckerberg Initiative to H.L.R. and from Genzyme Corporation to D.G.M. S.G. was supported by The Knut and Alice Wallenberg Foundation scholarship program for postdoctoral studies at the Broad Institute. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH, CZI, or Genzyme.
Declaration of interests
E.E. is a current employee of Clalit Research Institute and a graduate student at the Tel Aviv University Faculty of Medicine; her partner is an employee of GSK. K.J.K. is a paid advisor of Tome Biosciences and Vor Biosciences and is a member of the scientific advisory board of Nurture Genomics. S.M.H. is an employee of Ambry Genetics. D.G.M. is a paid advisor to GlaxoSmithKline (GSK), Insitro, Variant Bio, and Overtone Therapeutics and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Google, Merck, Microsoft, Pfizer, and Sanofi-Genzyme. H.L.R. has received support from Illumina and Microsoft to support rare disease gene discovery and diagnosis. A.O’D.L. is a paid advisor of Tome Biosciences and member of the scientific advisory board for Congenica and the Simons Foundation SPARK for Autism study.
Published: August 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.08.005.
Supplemental information
Data and code availability
The curation results generated during this study are available as a supplemental table, for download at https://gnomad.broadinstitute.org/downloads, or can be viewed at the respective gene page at https://gnomad.broadinstitute.org.
References
- 1.Alkuraya F.S. Human knockout research: new horizons and opportunities. Trends Genet. 2015;31:108–115. doi: 10.1016/j.tig.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 2.MacArthur D.G., Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum. Mol. Genet. 2010;19:R125–R130. doi: 10.1093/hmg/ddq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mort M., Ivanov D., Cooper D.N., Chuzhanova N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 4.Abou Tayoun A.N., Pesaran T., DiStefano M.T., Oza A., Rehm H.L., Biesecker L.G., Harrison S.M., ClinGen Sequence Variant Interpretation Working Group ClinGen SVI Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018;39:1517–1524. doi: 10.1002/humu.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dyle M.C., Kolakada D., Cortazar M.A., Jagannathan S. How to get away with nonsense: Mechanisms and consequences of escape from nonsense-mediated RNA decay. Wiley Interdiscip. Rev. RNA. 2020;11 doi: 10.1002/wrna.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindeboom R.G.H., Vermeulen M., Lehner B., Supek F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019;51:1645–1651. doi: 10.1038/s41588-019-0517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy E., Maquat L.E. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 9.Rivas M.A., Pirinen M., Conrad D.F., Lek M., Tsang E.K., Karczewski K.J., Maller J.B., Kukurba K.R., DeLuca D.S., Fromer M., et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348:666–669. doi: 10.1126/science.1261877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metzker M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 11.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gudmundsson S., Singer-Berk M., Watts N.A., Phu W., Goodrich J.K., Solomonson M., Genome Aggregation Database Consortium. Rehm H.L., MacArthur D.G., O’Donnell-Luria A. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2021;43:1012–1030. doi: 10.1002/humu.24309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narasimhan V.M., Hunt K.A., Mason D., Baker C.L., Karczewski K.J., Barnes M.R., Barnett A.H., Bates C., Bellary S., Bockett N.A., et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352:474–477. doi: 10.1126/science.aac8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saleheen D., Natarajan P., Armean I.M., Zhao W., Rasheed A., Khetarpal S.A., Won H.-H., Karczewski K.J., O’Donnell-Luria A.H., Samocha K.E., et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–239. doi: 10.1038/nature22034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minikel E.V., Karczewski K.J., Martin H.C., Cummings B.B., Whiffin N., Rhodes D., Alföldi J., Trembath R.C., van Heel D.A., Daly M.J., et al. Evaluating drug targets through human loss-of-function genetic variation. Nature. 2020;581:459–464. doi: 10.1038/s41586-020-2267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cummings B.B., Karczewski K.J., Kosmicki J.A., Seaby E.G., Watts N.A., Singer-Berk M., Mudge J.M., Karjalainen J., Satterstrom F.K., O’Donnell-Luria A.H., et al. Transcript expression-aware annotation improves rare variant interpretation. Nature. 2020;581:452–458. doi: 10.1038/s41586-020-2329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whiffin N., Armean I.M., Kleinman A., Marshall J.L., Minikel E.V., Goodrich J.K., Quaife N.M., Cole J.B., Wang Q., Karczewski K.J., et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020;26:869–877. doi: 10.1038/s41591-020-0893-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B., et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Hu J., Ng P.C. Predicting the effects of frameshifting indels. Genome Biol. 2012;13:R9. doi: 10.1186/gb-2012-13-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q., Pierce-Hoffman E., Cummings B.B., Alföldi J., Francioli L.C., Gauthier L.D., Hill A.J., O’Donnell-Luria A.H., et al. Genome Aggregation Database Production Team. Genome Aggregation Database Consortium Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nat. Commun. 2020;11:2539. doi: 10.1038/s41467-019-12438-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei L., Liu L.T., Conroy J.R., Hu Q., Conroy J.M., Morrison C.D., Johnson C.S., Wang J., Liu S. MAC: identifying and correcting annotation for multi-nucleotide variations. BMC Genom. 2015;16:569. doi: 10.1186/s12864-015-1779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenfeld J.A., Malhotra A.K., Lencz T. Novel multi-nucleotide polymorphisms in the human genome characterized by whole genome and exome sequencing. Nucleic Acids Res. 2010;38:6102–6111. doi: 10.1093/nar/gkq408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson K.K., Green M.R. Mechanism for cryptic splice site activation during pre-mRNA splicing. Proc. Natl. Acad. Sci. USA. 1990;87:6253–6257. doi: 10.1073/pnas.87.16.6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green M.R. Pre-mRNA splicing. Annu. Rev. Genet. 1986;20:671–708. doi: 10.1146/annurev.ge.20.120186.003323. [DOI] [PubMed] [Google Scholar]
- 27.Padgett R.A., Grabowski P.J., Konarska M.M., Seiler S., Sharp P.A. Splicing of messenger RNA precursors. Annu. Rev. Biochem. 1986;55:1119–1150. doi: 10.1146/annurev.bi.55.070186.005351. [DOI] [PubMed] [Google Scholar]
- 28.Biba D., Klink G., Bazykin G.A. Pairs of Mutually Compensatory Frameshifting Mutations Contribute to Protein Evolution. Mol. Biol. Evol. 2022;39 doi: 10.1093/molbev/msac031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaplanis J., Akawi N., Gallone G., McRae J.F., Prigmore E., Wright C.F., Fitzpatrick D.R., Firth H.V., Barrett J.C., Hurles M.E., Deciphering Developmental Disorders study Exome-wide assessment of the functional impact and pathogenicity of multinucleotide mutations. Genome Res. 2019;29:1047–1056. doi: 10.1101/gr.239756.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chabot B., Shkreta L. Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol. 2016;212:13–27. doi: 10.1083/jcb.201510032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anna A., Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J. Appl. Genet. 2018;59:253–268. doi: 10.1007/s13353-018-0444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schoch K., Tan Q.K.-G., Stong N., Deak K.L., McConkie-Rosell A., McDonald M.T., Undiagnosed Diseases Network. Goldstein D.B., Jiang Y.-H., Shashi V. Alternative transcripts in variant interpretation: the potential for missed diagnoses and misdiagnoses. Genet. Med. 2020;22:1269–1275. doi: 10.1038/s41436-020-0781-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiStefano M.T., Hemphill S.E., Cushman B.J., Bowser M.J., Hynes E., Grant A.R., Siegert R.K., Oza A.M., Gonzalez M.A., Amr S.S., et al. Curating Clinically Relevant Transcripts for the Interpretation of Sequence Variants. J. Mol. Diagn. 2018;20:789–801. doi: 10.1016/j.jmoldx.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lareau L.F., Green R.E., Bhatnagar R.S., Brenner S.E. The evolving roles of alternative splicing. Curr. Opin. Struct. Biol. 2004;14:273–282. doi: 10.1016/j.sbi.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Frankish A., Uszczynska B., Ritchie G.R.S., Gonzalez J.M., Pervouchine D., Petryszak R., Mudge J.M., Fonseca N., Brazma A., Guigo R., Harrow J. Comparison of GENCODE and RefSeq gene annotation and the impact of reference geneset on variant effect prediction. BMC Genom. 2015;16(Suppl 8):S2. doi: 10.1186/1471-2164-16-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neme R., Tautz D. Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genom. 2013;14:117. doi: 10.1186/1471-2164-14-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delaye L., Deluna A., Lazcano A., Becerra A. The origin of a novel gene through overprinting in Escherichia coli. BMC Evol. Biol. 2008;8:31. doi: 10.1186/1471-2148-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter C.W., Jr. Simultaneous codon usage, the origin of the proteome, and the emergence of de-novo proteins. Curr. Opin. Struct. Biol. 2021;68:142–148. doi: 10.1016/j.sbi.2021.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Chang Y.-F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 42.Lewis B.P., Green R.E., Brenner S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA. 2003;100:189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen R., Paul J.S., Albrechtsen A., Song Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xuan J., Yu Y., Qing T., Guo L., Shi L. Next-generation sequencing in the clinic: promises and challenges. Cancer Lett. 2013;340:284–295. doi: 10.1016/j.canlet.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy S., LaFramboise W.A., Nikiforov Y.E., Nikiforova M.N., Routbort M.J., Pfeifer J., Nagarajan R., Carter A.B., Pantanowitz L. Next-Generation Sequencing Informatics: Challenges and Strategies for Implementation in a Clinical Environment. Arch. Pathol. Lab Med. 2016;140:958–975. doi: 10.5858/arpa.2015-0507-RA. [DOI] [PubMed] [Google Scholar]
- 46.Treangen T.J., Salzberg S.L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alkan C., Sajjadian S., Eichler E.E. Limitations of next-generation genome sequence assembly. Nat. Methods. 2011;8:61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lunter G., Rocco A., Mimouni N., Heger A., Caldeira A., Hein J. Uncertainty in homology inferences: assessing and improving genomic sequence alignment. Genome Res. 2008;18:298–309. doi: 10.1101/gr.6725608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang Y., Turinsky A.L., Brudno M. The missing indels: an estimate of indel variation in a human genome and analysis of factors that impede detection. Nucleic Acids Res. 2015;43:7217–7228. doi: 10.1093/nar/gkv677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang H., Wu Y., Narzisi G., O’Rawe J.A., Barrón L.T.J., Rosenbaum J., Ronemus M., Iossifov I., Schatz M.C., Lyon G.J. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89. doi: 10.1186/s13073-014-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shin S., Park J. Characterization of sequence-specific errors in various next-generation sequencing systems. Mol. Biosyst. 2016;12:914–922. doi: 10.1039/c5mb00750j. [DOI] [PubMed] [Google Scholar]
- 52.Su Z., Ning B., Fang H., Hong H., Perkins R., Tong W., Shi L. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev. Mol. Diagn. 2011;11:333–343. doi: 10.1586/erm.11.3. [DOI] [PubMed] [Google Scholar]
- 53.Feng W., Zhao S., Xue D., Song F., Li Z., Chen D., He B., Hao Y., Wang Y., Liu Y. Improving alignment accuracy on homopolymer regions for semiconductor-based sequencing technologies. BMC Genom. 2016;17:521. doi: 10.1186/s12864-016-2894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gudmundsson S., Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., et al. Addendum: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2021;597:E3–E4. doi: 10.1038/s41586-021-03758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Divina P., Kvitkovicova A., Buratti E., Vorechovsky I. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur. J. Hum. Genet. 2009;17:759–765. doi: 10.1038/ejhg.2008.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mirzaa G., Parry D.A., Fry A.E., Giamanco K.A., Schwartzentruber J., Vanstone M., Logan C.V., Roberts N., Johnson C.A., Singh S., et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat. Genet. 2014;46:510–515. doi: 10.1038/ng.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carlston C.M., O’Donnell-Luria A.H., Underhill H.R., Cummings B.B., Weisburd B., Minikel E.V., Birnbaum D.P., Exome Aggregation Consortium. Tvrdik T., MacArthur D.G., Mao R. Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Hum. Mutat. 2017;38:517–523. doi: 10.1002/humu.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gudmundsson S., Carlston C.M., O’Donnell-Luria A. Interpreting variants in genes affected by clonal hematopoiesis in population data. Hum. Genet. 2023:1–5. doi: 10.1007/s00439-023-02526-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The curation results generated during this study are available as a supplemental table, for download at https://gnomad.broadinstitute.org/downloads, or can be viewed at the respective gene page at https://gnomad.broadinstitute.org.