

SUMMARY
Loss of antimicrobial proteins such as REG3 family members compromises the integrity of the intestinal barrier. Here, we demonstrate that overproduction of REG3 proteins can also be detrimental by reducing a protective species in the microbiota. Patients with inflammatory bowel disease (IBD) experiencing flares displayed heightened levels of secreted REG3 proteins that mediated depletion of Enterococcus faecium (Efm) from the gut microbiota. Efm inoculation of mice ameliorated intestinal inflammation through activation of the innate immune receptor NOD2, which was associated with the bacterial DL-endopeptidase SagA that generates NOD2-stimulating muropeptides. NOD2 activation in myeloid cells induced IL-1β secretion to increase the proportion of IL-22-producing CD4+ T helper cells and innate lymphoid cells that promote tissue repair. Finally, Efm was unable to protect mice carrying a NOD2 gene variant commonly found in IBD patients. Our findings demonstrate that inflammation self-perpetuates by causing aberrant antimicrobial activity that disrupts symbiotic relationships with gut microbes.
Keywords: Enterococci, antimicrobial proteins, REG3, NOD2, inflammatory bowel disease
eTOC Blurb
Jang et al. demonstrate that antimicrobial REG3 proteins overproduced during inflammatory bowel disease perpetuate inflammation by depleting Enterococcus faecium from the microbiota. DL-endopeptidase SagA secreted by E. faecium activates myeloid NOD2 signaling to produce IL-1β, which induces the protective cytokine IL-22 from lymphoid cells.
Graphical Abstract:
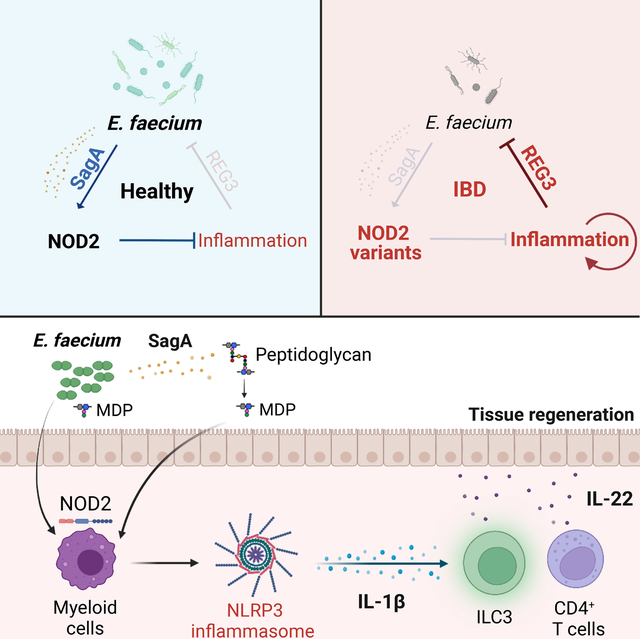
INTRODUCTION
Secretion of antimicrobial proteins (AMPs) at barrier surfaces represents a ubiquitous and evolutionarily ancient defense strategy. AMPs such as defensins and C-type lectins are abundant in the mammalian gastrointestinal tract where they limit the overgrowth of microbes, typically by disrupting membrane integrity of their targets1–3. Although certain AMPs recognize structures of invasive bacteria such as flagellin4, many display broad-spectrum activity, reflecting the diversity of pathogenic and non-pathogenic microbes in the gut. Regenerating family member 3 (REG3) C-type lectins REG3A and REG3G bind to the peptidoglycan cell wall of Gram-positive (+) bacteria in the gut5,6. Reg3γ in mice mediates spatial segregation between the host and the microbiota7–9. REG3A (known as HIP/PAP), the main paralog in human, may have a similar role as murine Reg3γ because its transgenic expression in mice alters microbiota composition10. Additionally, REG3 proteins regulate intestinal crypt regeneration and scavenge reactive oxygen species10,11
Dysregulated AMP production is associated with human inflammatory bowel disease (IBD)12–17, a chronic immune-mediated disorder of the gut involving complex interactions between genetic and environmental factors. Although immunomodulatory medications can offer substantial relief to patients, maintaining disease remission remains a major challenge. Consistent with the link between AMPs and intestinal inflammation, an altered microbiota composition is a common feature of IBD18–20. Also, population genetic studies have implicated genes associated with host-microbe interactions21,22. Variants of the intracellular microbial sensor nucleotide-binding oligomerization domain-containing protein 2 (NOD2) are among the strongest genetic risk factors for IBD23,24. NOD2 mediates production of AMPs and cytokines in the presence of peptidoglycan byproducts23. Nod2-deficient mice are susceptible to enteric bacterial infections and display an altered microbiota composition depending on the animal facility25–34, and IBD patients harboring NOD2 variants display impaired production of AMPs35–37. However, NOD2 variants are found in only a subset of IBD patients, indicating that additional mechanisms may be involved. Also, IBD is associated with increased REG3 expression38,39, suggesting a different role for these AMPs in disease pathogenesis.
Enterococcus species, Gram+ bacteria in the gut microbiota that activate and are controlled by NOD240–42, are highly sensitive to killing by REG3A and REG3G6,9. The relationship between enterococci and IBD is debatable. Enterococcus species are opportunistic pathogens in hospital settings43 and induce intestinal inflammation in Il10-deficient mice44,45. Yet, enterococci are consumed as probiotics for treatment of diarrheal diseases46,47. Recent studies identified a DL-endopeptidase, SagA, secreted by certain enterococci including Enterococcus faecium (Efm)48–50. Processing of peptidoglycan by SagA generates muropeptides that activate NOD2 to enhance colonization resistance towards enteric pathogens and antitumor immunity41,50,51. Interventions that promote antitumor immunity exacerbate intestinal inflammation52. Therefore, it is unclear whether NOD2 activation by enterococci would worsen or improve IBD.
We found IBD patients displayed overproduction of REG3 proteins that deplete enterococci from the gut microbiota. Efm or a Lactococcus strain engineered to express sagA protected mice from intestinal injury through NOD2 activity in myeloid cells, which mediated an increase in lymphoid cells producing the regenerative cytokine IL-22. Efm colonization was associated with heightened IL-22 levels in human stool, and this beneficial effect of Efm colonization was abrogated in mice harboring the R702W variant of NOD2 associated with IBD. Our findings uncover a mechanism of perpetuating intestinal inflammation initiated by a harmful feedback loop involving excess AMP production and depletion of protective enterococci, which renders the host functionally NOD2-deficient.
RESULTS
REG3 proteins overproduced by IBD patients inhibit enterococci
Although REG3A mRNA is increased in inflamed intestinal tissues from IBD patients38,39, it is unclear whether gene expression is associated with REG3 protein secretion and activity. We collected stool from 56 IBD patients experiencing disease flares (Table S1). An equal number of non-IBD (NIBD) patients experiencing gastrointestinal symptoms, such as diarrhea, were used as a control cohort (Table S2). Stool extracts were used to detect the two human REG3 family members, REG3A and REG3G (Figure 1A), and two additional paralogs: REG1A, a pancreatic growth factor that should not be co-regulated with REG3 proteins53, and REG4, a less-characterized potential AMP54,55. As expected, REG1A was equally present in NIBD and IBD (Figures 1B and 1C). In contrast, REG3A was detected in a higher proportion of IBD patients than NIBD patients, and REG3G and REG4 were exclusively present in IBD samples (Figures 1B and 1C). An ELISA confirmed higher REG3A concentration in stool extracts from IBD than those of NIBD (Figure 1D). Thus, the presence of REG3 proteins is more prevalent and enriched in IBD patients.
Figure 1. Increased REG3A and REG3G in IBD patient stool inhibit Enterococcus.
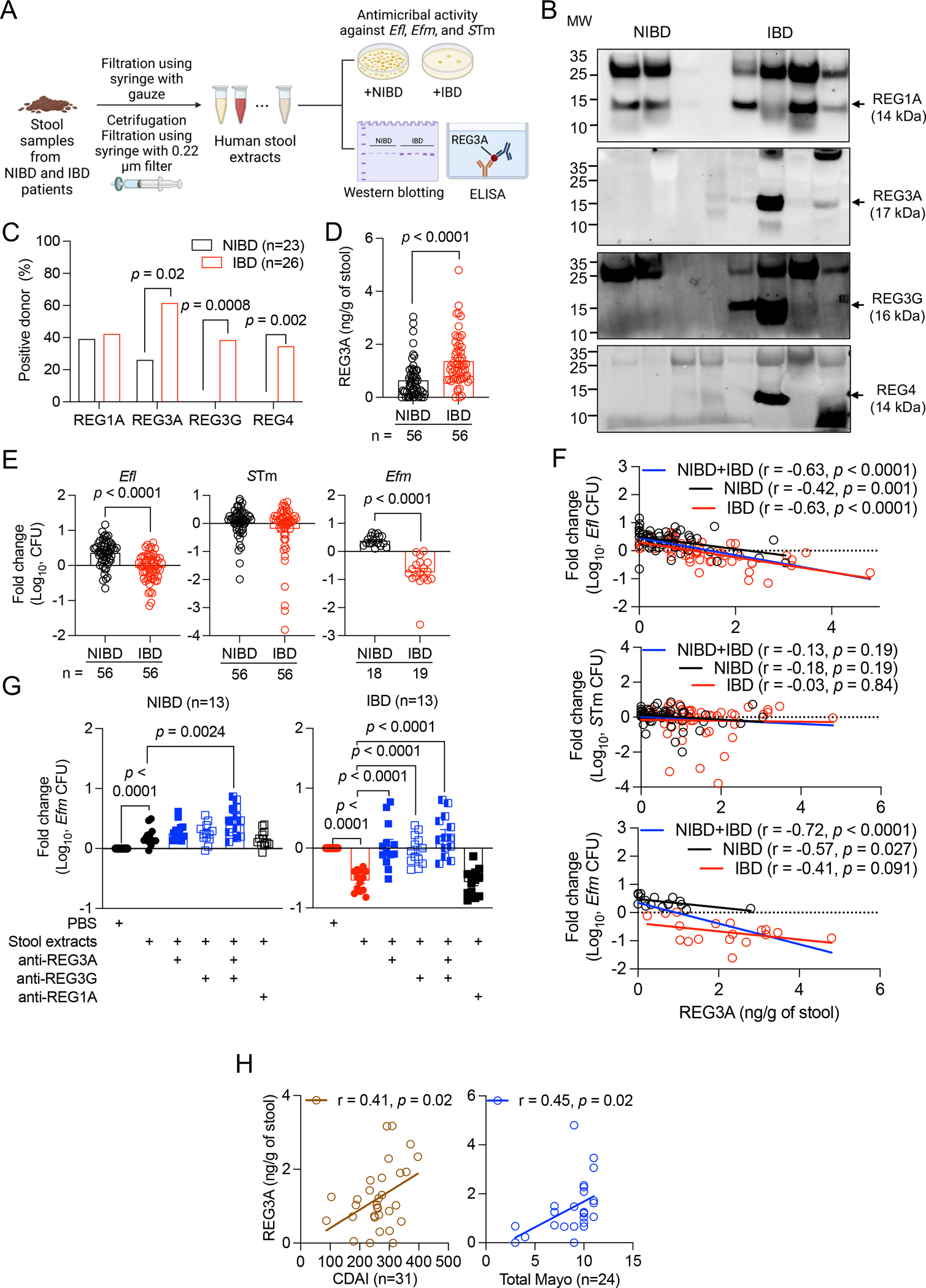
A) Schematic of stool extract preparation and analysis. B) Western blots of REG1A, REG3A, REG3G, and REG4 in stool extracts from representative 3 non-IBD (NIBD) and 5 IBD patients. C) Proportion of specimens from NIBD and IBD patients in which REG1A, REG3A, REG3G, or REG4 were detectable by western blot. D) Quantification of REG3A in NIBD and IBD stool extracts by ELISA. E) Fold changes in colony forming unit (CFU) of Enterococcus faecalis (Efl) and Salmonella Typhimurium (STm) cultured in NIBD and IBD stool extracts. Results obtained with Efl were confirmed with E. faecium (Efm). F) Correlation between REG3A concentration and CFU fold changes of Efl (upper), STm (middle), and Efm (lower) cultured in NIBD and IBD stool extract. G) Fold change in Efm CFU cultured in PBS or NIBD (left) and IBD (right) stool extracts in the presence of indicated antibodies. H) Correlation between REG3A concentration and Crohn’s disease activity index (CDAI) for CD patients or total Mayo score for ulcerative colitis (UC) patients. Data points in D-H represent individual patients. Bars represent mean ± SEM and at least three independent experiments were performed. r, Pearson correlation coefficient. Indicated p values by Fisher’s exact test in C, unpaired t test, two-tailed in D, E, and G, and simple linear regression analysis in F and H. See also Figure S1 and Tables S1–S3.
We examined the antimicrobial activity of NIBD and IBD stool extracts against Enterococcus faecalis (Efl) and Salmonella Typhimurium (STm) as Gram+ and − enteric bacteria, respectively. Bacteria displayed modest growth in NIBD stool extracts compared with PBS control, indicating the presence of nutrients in stool (Figure 1E). Efl colony forming units (CFUs) after culturing in IBD stool extracts was lower compared with NIBD samples, whereas STm numbers were similar in both groups (Figure 1E). We confirmed this antimicrobial activity of IBD stool extracts with another Enterococcus species, Efm (Figure 1E). REG3A concentration negatively correlated with Efl and Efm CFUs following treatment with NIBD and IBD stool extracts, but not with STm CFUs (Figure 1F). Additionally, antimicrobial activity against Efm in IBD stool extracts was inhibited by antibodies against REG3A or REG3G, individually or together, but not REG1A control antibodies (Figure 1G). Increased numbers of Efm were recovered in NIBD stool extracts compared with PBS control, and adding anti-REG3A and REG3G antibodies together enhanced growth, suggesting low levels of antimicrobial activity in these samples (Figure 1G). Thus, Efm growth impairment in human stool extracts is attributed to REG3A and REG3G.
IBD stool extracts from both males and females displayed higher amounts of REG3A than NIBD samples (Figure S1A). REG3A concentrations were similar for patients with Crohn’s disease (CD) and ulcerative colitis (UC), the two major types of IBD (Figure S1B). REG3A concentration did not differ among age groups in NIBD patients, however, it displayed a modest inverse relationship with age among IBD patients (Figure S1C). We further observed that IBD patients younger than age 60 displayed increased REG3A concentration compared with matched NIBD controls, and this difference was lost in the older age group (Figure S1D). Disease severity displayed a positive correlation with REG3A concentrations (Figure 1H). Thus, REG3A protein overproduction is common to CD and UC flares as described previously for RNA39.
Enterococcus and Efm are lost from the gut microbiota in IBD patients
The above results raise the possibility that Enterococcus is depleted in the gut microbiota of IBD patients. 16S rRNA sequencing indicated that the microbiota composition of NIBD and IBD were similar according to alpha and beta diversities (Figures 2A and 2B), confirming that our NIBD cohort is an appropriate control for IBD patients. However, >80% of the sequencing reads aligned to Enterococcus for one of the NIBD samples. Thus, we excluded this patient from subsequent analyses. Consistent with our in vitro findings (Figure 1), the relative abundance of Enterococcus in IBD patients was lower than that in NIBD patients (Figure 2C).
Figure 2. Enterococcus and Efm are depleted in gut microbiota from IBD patients.
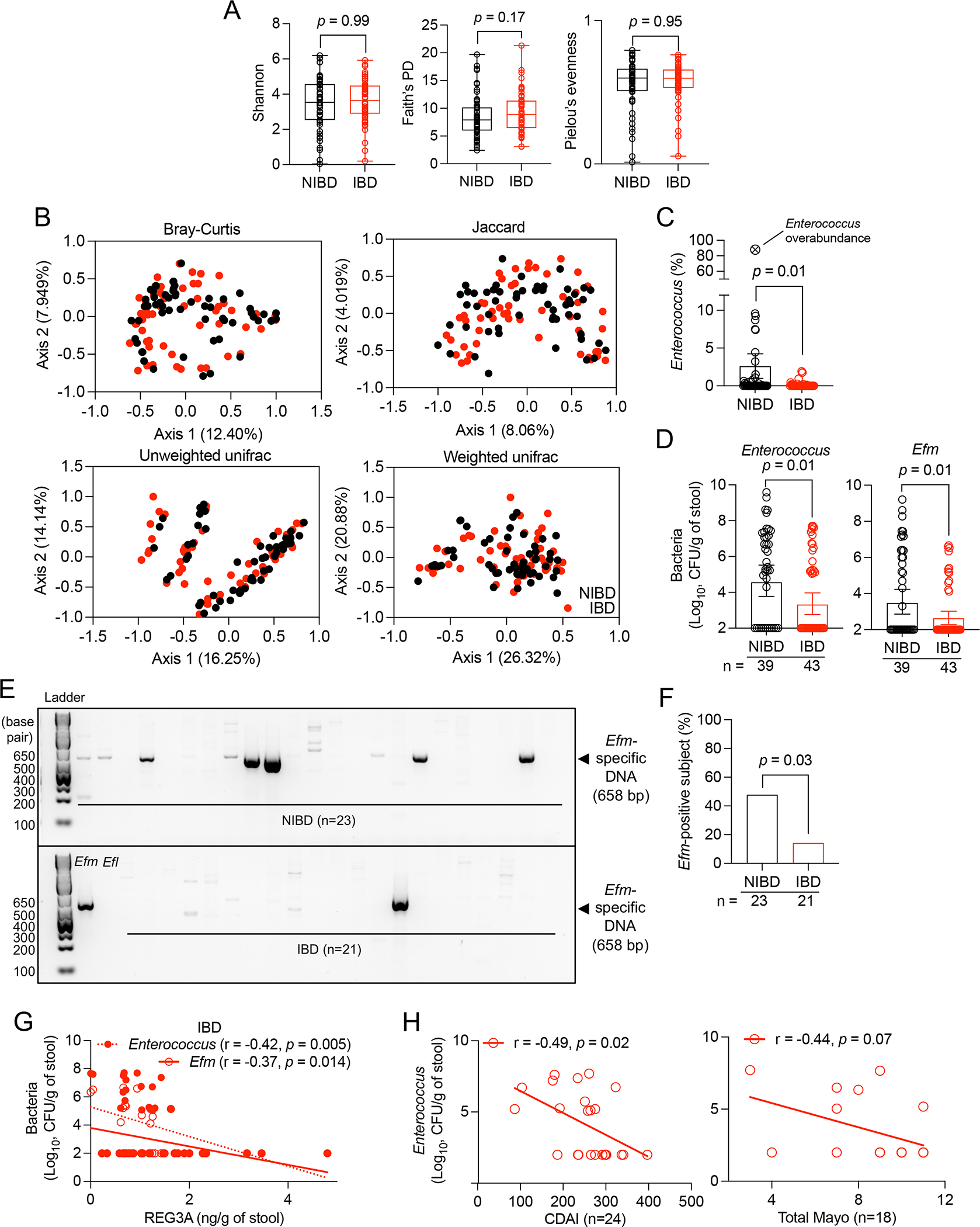
A and B) 16S rRNA sequencing of stool from NIBD and IBD patients from Figure 1. Alpha diversity calculated by Shannon, Faith’s phylogenetic diversity (PD), and Pielou’s evenness indices (A). Principle coordinate analyses of beta diversity determined by Bray-Curtis, Jaccard, and Unweighted and Weighted unifrac methods (B). C) Proportion of sequencing reads representing Enterococcus in NIBD and IBD patient stool. One NIBD sample contained >80% Enterococcus, which is shown as a reference on the graph but excluded from statistical analysis and downstream assays. D) Total Enterococcus and Efm CFUs in NIBD and IBD specimens. E) Gel image of Efm detected by PCR in stool DNA. Genomic DNA isolated from Efm and Efl in the first two lanes of the bottom panel serves as positive and negative controls, respectively. F) Proportion of NIBD and IBD patients in which Efm was detectable by PCR. G) Correlation between REG3A concentration and total Enterococcus and Efm CFUs in IBD stool. H) Correlation between Enterococcus CFUs and CDAI and total Mayo score. Data points in A-D, G, and H represent individual patients. Bars represent mean ± SEM from at least three independent experiments. r, Pearson correlation coefficient. Indicated p values by Kruskal-Wallis test in A, unpaired t test, two-tailed in C and D, Fisher’s exact test in F, and simple linear regression analysis in G and H. See also Figure S1 and Tables S1–S3.
Enterococci are resistant to lysis techniques56–59. High-throughput sequencing techniques yield inaccurate measurements of taxa resistant to lysis60, potentially explaining why prior studies did not observe reduced Enterococcus in IBD patients61–63. Plating stool on selective agar confirmed our 16S sequencing result and showed that the burden and detection rate of total Enterococcus and Efm in IBD specimens were lower than in NIBD (Figures 2D and S1E). Efm-specific DNA was also detected in stool from NIBD patients more frequently than IBD patients (Figures 2E and 2F). Efm accounted for 15% of the total enterococci in IBD compared with 36% in NIBD, indicating that Efm was particularly vulnerable to depletion in IBD patients. (Figure S1F). Total Enterococcus and Efm burden in IBD stool displayed a negative correlation with REG3A concentration (Figure 2G) and with disease severity (Figure 2H).
To increase taxonomical resolution, we sequenced the genomes of 79 and 122 Enterococcus isolates collected from 8 NIBD and 11 IBD patients (Table S3). Efm represents a higher proportion of the total Enterococcus isolates from NIBD stool than IBD stool (Figure S1G). Phylogenetic tree analysis revealed a comparable distribution of Enterococcus isolates among NIBD and IBD patients (Figure S1H). Isolates within a species were resolved by grouping into clusters of species variants having less than 100 single nucleotide polymorphisms (SNPs) different from each other (Figure S1I). NIBD patients displayed a higher number of Enterococcus variants than IBD patients (Figures S1J and S1K). Half of NIBD patients possessed more than 3 different Enterococcus variants whereas less than 2 variants were detected in most IBD patients (Figure S1K). These results are consistent with a loss of diversity of enterococci due to a bottleneck imposed by the increased REG3 proteins in IBD patients.
Enterococci protect against intestinal injury in mice through NOD2
We next examined the relationship between Enterococcus and intestinal inflammation. While optimizing conditions for administering dextran sulfate sodium (DSS) in drinking water, a model of intestinal chemical injury, we observed extreme differences in susceptibility between wild-type (WT) C57BL/6J (B6) mice bred in two rooms within the same vivarium. Mice bred in room 6 (Rm 6) receiving 5% DSS displayed higher lethality, weight loss, and disease score compared with mice raised in room 13 (Rm 13) (Figures 3A–3C). Lipocalin-2 (LCN2), a marker of inflammation64, was more abundant in stool from DSS-treated Rm 6 mice compared with Rm 13 mice (Figure 3D). Shortening of the colon, another marker of intestinal inflammation, was more pronounced in Rm 6 than Rm 13 mice on day 9 (Figure 3E). Susceptibility to DSS diverges due to differences in microbiota composition that arise from separate parental lineages65,66. Because we were investigating enterococci, we examined their presence in the microbiota and observed a 2-log higher burden of endogenous enterococci in Rm 13 mice than Rm 6 mice (Figure 3F). Similar to the human samples, Enterococcus levels decreased following DSS-induced intestinal inflammation in mice (Figure 3G). In all subsequent experiments, we used mice from Rm 6.
Figure 3. Enterococcus protects against intestinal injury through NOD2.
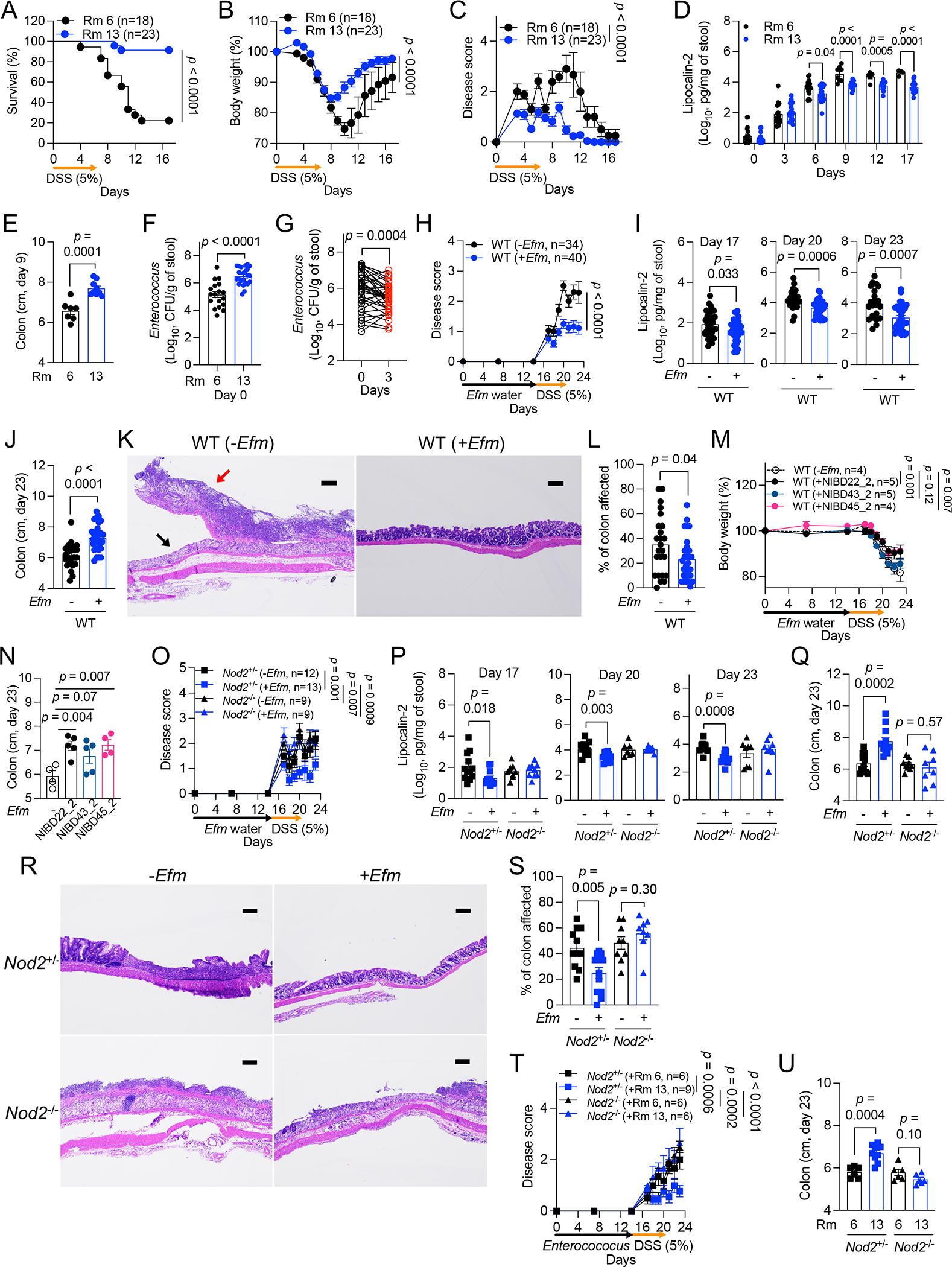
A-G) Dextran sulfate sodium (DSS)-induced intestinal injury of wild-type (WT) B6 mice bred in room 6 (Rm 6) or 13 (Rm 13). Mice were examined for survival (A), changes in body weight (B) disease score (C), fecal lipocalin-2 (LCN2) (D), colon length on day 9 (E), endogenous Enterococcus burden on day 0 (F), and changes in Enterococcus burden on day 3 (G). H-L) DSS treatment of WT mice from Rm 6 following administration of Efm or control. Mice were examined for changes in disease score (H), fecal LCN2 (I), and colon length on day 23 (J). Representative images of hematoxylin and eosin (H&E)-stained sections of the colon (K) and quantification of the proportion of colon affected (L). Black arrow indicates destroyed colonic epithelium replaced with a diffuse ulcer and pus; red arrow indicates acute inflammation with primary neutrophils, lymphocytes, plasma cells, and dead cell debris. Breakdown of histological measurements by individual mice is provided in Table S4 for this and other figures. M and N) Body weight change (M) and colon length on day 23 (N) after DSS treatment of WT mice from Rm 6 following administration of Efm isolates from NIBD patients. O-S) DSS treatment of Nod2+/− and Nod2−/− mice from Rm 6 following administration of Efm or control. Mice were examined for changes in disease score (O), fecal LCN2 at indicated time points (P), and colon length on day 23 (Q). Representative images of H&E-stained sections of the colon (R) and quantification of the proportion of colon affected (S). T and U) Disease score (T) and colon length on day 23 (U) after DSS treatment of Nod2+/− and Nod2−/− mice following administration of Enterococcus isolated from Rm 6 or Rm 13. Bars, 200 μm. Data points in D-G, I, J, L, N, P, Q, S, and U represent individual mice. Data points in B, C, H, M, O, and T represent mean ± SEM. Bars represent mean ± SEM from at least two independent experiments. Indicated p values by log-rank Mantel-Cox test in A, two-way ANOVA test in B, C, H, M, O, and T, unpaired t test, two-tailed in D-F, I, J, L, N, P, Q, S, and U, and paired t test, two-tailed in G. See also Figures S2 and S3 and Table S4.
To determine whether Enterococcus colonization protects against intestinal injury, we inoculated mice by administering Efm in the drinking water67 and then switched to 5% DSS. Body weight and fecal LCN2 were unchanged in Efm-colonized mice without DSS treatment (Figures S2A and S2B). In mice receiving DSS, both total Enterococcus and Efm levels decreased during the treatment and partially recovered after cessation (Figures S2C and S2D). Although it is unclear whether REG3 proteins in mice and humans are equivalent in their function68,69, they both impact Enterococcus colonization. The expression of Reg3α, Reg3β, Reg3γ, Reg3δ, and Reg4 reached the peak on day 20 and then gradually decreased, and their expression was inversely associated with both total Enterococcus and Efm burden (Figures S2E–S2G).
Efm-colonized WT B6 mice treated with DSS displayed less body weight reduction, disease score, and fecal LCN2 compared with DSS-treated mice that did not receive Efm (Figures 3H, 3I, S2H, and S2I). On day 23, Efm-colonized mice displayed less colon shortening and histopathologic changes (Figures 3J–3L; see Table S4). Inoculating mice with Efm after DSS treatment also mitigated these signs of disease (Figures S3A–S3G; see Table S4). To link the Efm-mediated protection in mice with our findings in human specimens, we inoculated mice with 3 Efm isolates from NIBD patients (Table S3) and confirmed colonization (Figure S2J). Mice colonized with any of the 3 isolates displayed less body weight reduction and colon shortening than those that were untreated (Figures 3M, 3N, and S2K). Therefore, Efm confers protection against DSS-induced inflammation.
Given that Efm protects against enteric pathogens in a NOD2-dependent manner41, Efm should lose its beneficial properties in Nod2-deficient mice during intestinal injury. The colonization patterns of Efm in Nod2+/− and Nod2−/− mice were consistent with those in WT mice (Figure S2L). In contrast to Nod2+/− mice in which we reproduced the protective effect of the bacterium, Efm administration to Nod2−/− mice did not ameliorate signs of disease (Figures 3O–3S, S2M, and S2N; see Table S4). Lastly, we inoculated Nod2+/− and Nod2−/− mice with Enterococcus strains collected from Rm 6 or Rm 13 mice and confirmed they achieved a similar degree of colonization (Figure S2O). Rm 13 Enterococcus, but not Rm 6 Enterococcus, protected Nod2+/− mice from weight reduction, disease score, and colon shortening following DSS treatment, whereas Nod2−/− mice remained susceptible with either source of Enterococcus (Figures 3T, 3U and S2P). Thus, certain Enterococcus species including Efm confer protection against intestinal injury in a Nod2-dependent manner.
SagA mediates NOD2-dependent protection against intestinal injury
SagA secreted by Efm generates NOD2-stimulating muropeptides41,50. Because SagA is essential for growth of Efm70, we tested the role of SagA by orally inoculating mice with Lactococcus lactis (Lls) expressing wild-type SagA (Lls-sagAWT) or catalytically inactive (C443A) SagA (Lls-sagAC443A) (Figure S4A). A lower concentration of DSS (3%) was used because mice were pre-treated with antibiotics (abx) to facilitate Lls colonization, and abx increases susceptibility to DSS71,72. Inoculation with PBS and the parental Lls strain were used as controls for effects of abx treatment and Lls colonization, respectively. Lls strains displayed a similar degree of colonization (Figures S4B and S4D). Mice given Lls-sagAWT displayed enhanced survival compared with all the other conditions, and other disease parameters were generally improved (Figures 4A–4E and S4C). Lls-sagAWT ameliorated disease in Nod2+/− mice but not Nod2−/− mice (Figures 4F–4J and S4E). If protection is mediated through peptidoglycan processing by secreted SagA, bacterial colonization would potentially be dispensable. To test this, we administered filtered culture supernatants from Lls strains to mice without abx treatment and then switched to 5% DSS (Figure S4F). In contrast to supernatant from the parental Lls and Lls-sagAC443A cultures, or sterile broth, supernatant from Lls-sagAWT was sufficient for reducing mortality, weight loss, disease score, fecal LCN2, and colon shortening (Figures 4K–4M, S4G, and S4H). These protective effects of Lls-sagAWT culture supernatant were NOD2-dependent (Figures 4N–4P, S4I, and S4J). These findings indicate that activating NOD2 downstream of SagA protects against intestinal injury.
Figure 4. SagA mediates NOD2-dependent protection against intestinal injury.
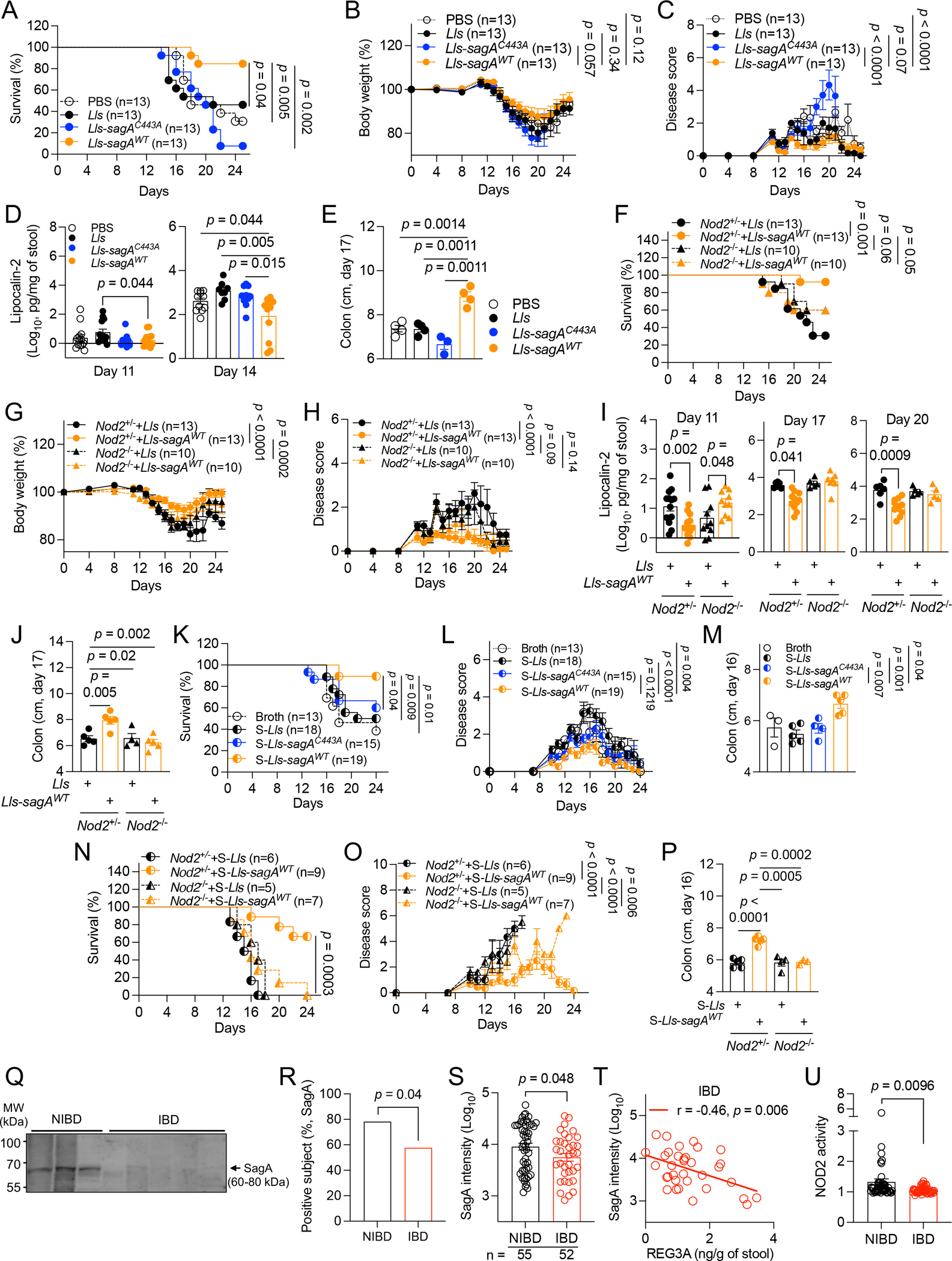
A-E) Antibiotics (abx)-treated WT mice orally inoculated with PBS, Lactococcus lactis (Lls), Lls-sagAWT, or Lls-sagAC443A and given DSS were examined for survival (A), changes in body weight (B), disease score (C), fecal LCN2 (D), and colon length on day 17 (E). F-J) Abx-treated Nod2+/− and Nod2−/− mice inoculated with Lls or Lls-sagAWT and given DSS were examined for survival (F), changes in body weight (G) and disease score (H), fecal LCN2 (I), and colon length on day 17 (J). K-M) DSS-treated WT mice receiving broth or culture supernatants of Lls (S-Lls), Lls-sagAWT (S-Lls-sagAWT), or Lls-sagAC443A (S-Lls-sagAC443A) in drinking water were examined for survival (K), changes in disease score (L), and colon length on day 16 (M). N-P) DSS-treated Nod2+/− and Nod2−/− mice receiving S-Lls or S-Lls-sagAWT were examined for survival (N), changes in disease score (O), and colon length on day 16 (P). Q) Western blots of SagA in stool extracts from representative 3 NIBD and 5 IBD patients. R and S) Proportion of NIBD and IBD patients in which SagA was detectable by western blot (R) and band intensity (S). T) Correlation between SagA and REG3A. U) NOD2 activity detected by HEK-Blue NOD2 reporter cells incubated with NIBD and IBD stool extracts. Values represent fold change over background control cells. Data points in D, E, I, J, M, and P represent individual mice. Data points in S-U represent individual patients. Data points in B, C, G, H, L, and O represent mean ± SEM. Bars represent mean ± SEM from at least two independent experiments. r, Pearson correlation coefficient. Indicated p values by log-rank Mantel-Cox test in A, F, K, and N, two-way ANOVA test in B, C, G, H, L, and O, unpaired t test, two-tailed in D, E, I, J, M, P, S, and U, Fisher’s exact test in R, and simple linear regression analysis in T. See also Figure S4 and Tables S1 and S2.
The decreased Efm burden in IBD patients suggests that SagA-mediated NOD2 activation would be reduced in these individuals. Indeed, SagA was detected in NIBD specimens at both a higher frequency and levels compared with IBD (Figures 4Q–4S). Also, SagA levels negatively correlated with REG3A concentration in IBD patients (Figure 4T). We found higher NOD2 activity using reporter cells stimulated with NIBD stool extracts than IBD samples (Figure 4U). Therefore, the decrease in Efm observed in IBD is associated with reduced availability of NOD2 ligands.
NOD2 in myeloid cells is indispensable for Efm-mediated protection
NOD2 has been extensively investigated in myeloid and intestinal epithelial cells73–75. Therefore, we developed Nod2fl/fl;LysM-Cre and Villin-Cre mice that are Nod2-deficient in myeloid and intestinal epithelial cells, respectively, and confirmed the presence of Efm in stool following inoculation (Figures 5A and 5G). Unlike Nod2fl/fl controls in which signs of disease were ameliorated, Efm colonization of Nod2fl/fl;LysM-Cre+ mice did not improve disease parameters (Figures 5B–5F). In contrast, Efm reduced mortality, body weight reduction, disease score, fecal LCN2, and colon length in Nod2fl/fl;Villin-Cre+ mice to a similar extent as Nod2fl/fl controls (Figures 5H–5L). Thus, NOD2 is required in myeloid cells and dispensable in the intestinal epithelium for Efm-mediated protection.
Figure 5. NOD2 in myeloid cells is required for Efm-mediated protection.
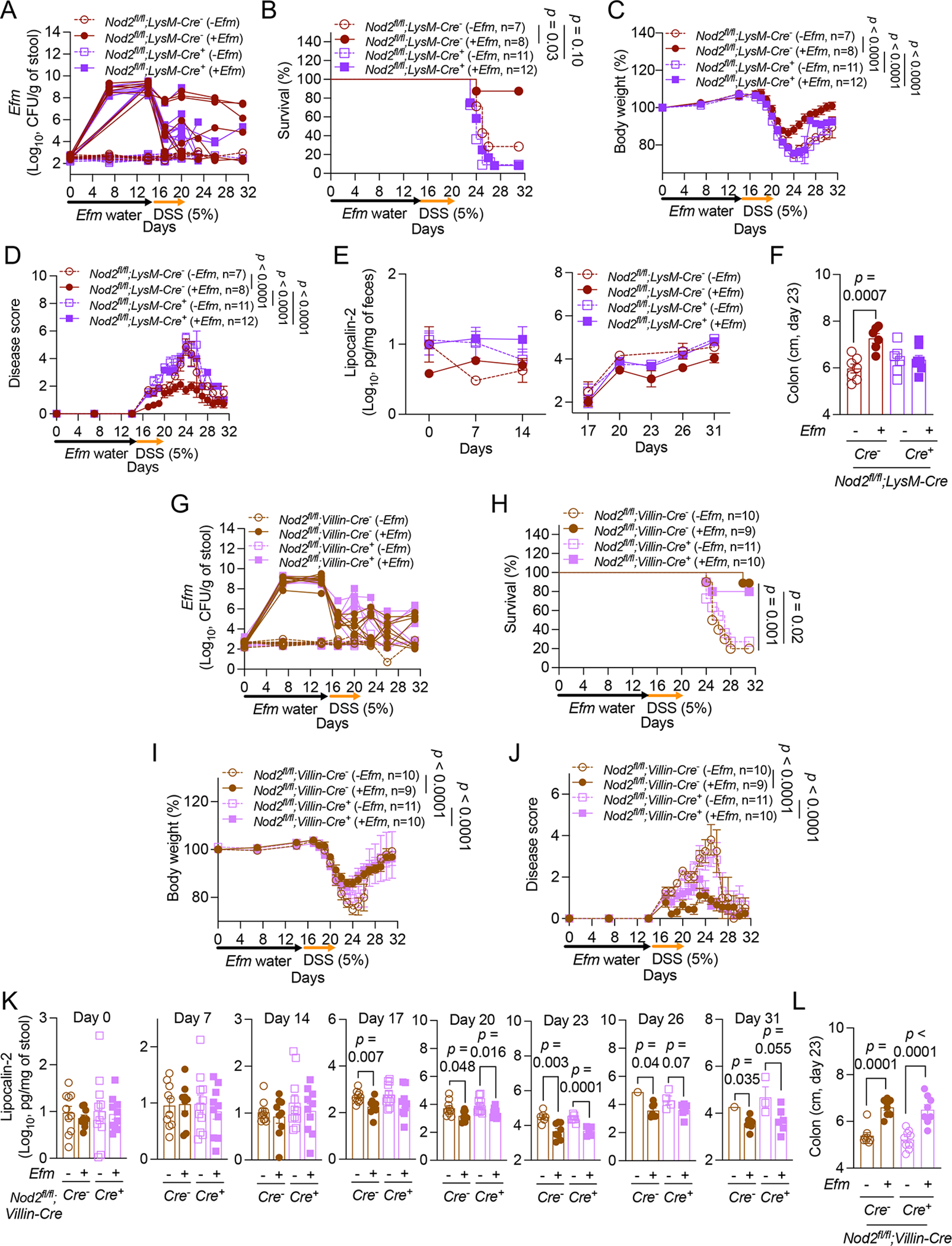
A-L) DSS-treated Nod2fl/fl;LysM-Cre− and Cre+ mice (A-F) or Nod2fl/fl;Villin-Cre− and Cre+ mice (G-L) following administration of Efm or control were examined for Efm burden in stool (A and G), survival (B and H), changes in body weight (C and I), disease score (D and J), fecal LCN2 (E and K), and colon length on day 23 (F and L). Lines in A and G and data points in F, K, and L represent Individual mice. Data points in C-E, I, and J represent mean ± SEM. Bars represent mean ± SEM from at least three independent experiments. Indicated p values by log-rank Mantel-Cox test in B and H, two-way ANOVA test in C, D, I, and J, and unpaired t test, two-tailed in F, K, and L.
Efm induces IL-22 production by lymphoid cells downstream of myeloid NOD2
NOD2 activation in myeloid cells is frequently associated with pro-inflammatory cytokine production73–75. One way to reconcile this established function with our observations would be if NOD2-activated myeloid cells act on other cell types to produce factors involved in tissue repair such as the cytokine IL-22, which is protective in DSS models76,77. We observed increased IL-22 production in gut explants harvested from WT mice on day 14 following Efm administration (prior to DSS) and day 20 (with DSS) compared with uncolonized mice (Figures 6A and 6B). IL-22 secretion was not enhanced by Efm in Nod2−/− and Nod2fl/fl;LysM-Cre+ mice on days 14 and 20 (Figures 6A and 6B). Group 3 innate lymphoid cells (ILC3s) and CD4+ T cells produce IL-22 in the gut. Efm increased the proportion and number of both IL-22+ ILCs and CD4+ T cells in WT mice, and not Nod2−/− and Nod2fl/fl;LysM-Cre+ mice (Figures 6C–6F, S5A–S5D). NOD2 in antigen-presenting cells can induce IL-10 production from regulatory T cells (Tregs)78. However, Efm did not alter the proportion of IL10+ CD4+ T cells, CD4+ T helper 1 (Tbet+) and 2 (GATA3+) cells, and Tregs (Figures S5F–S5I). Consistent with ILC3s being a source of IL-22, Efm increased the proportion and number of ILC3s and not the proportion of ILC1s and ILC2s in a NOD2-dependent manner (Figure 6G, 6H, S5E, S5J, and S5K). Therefore, Efm induces expansion of IL-22-producing CD4+ T cells and ILCs in a myeloid NOD2-dependent manner.
Figure 6. IL-1β induced by Efm protective IL-22-producing lymphoid cells.
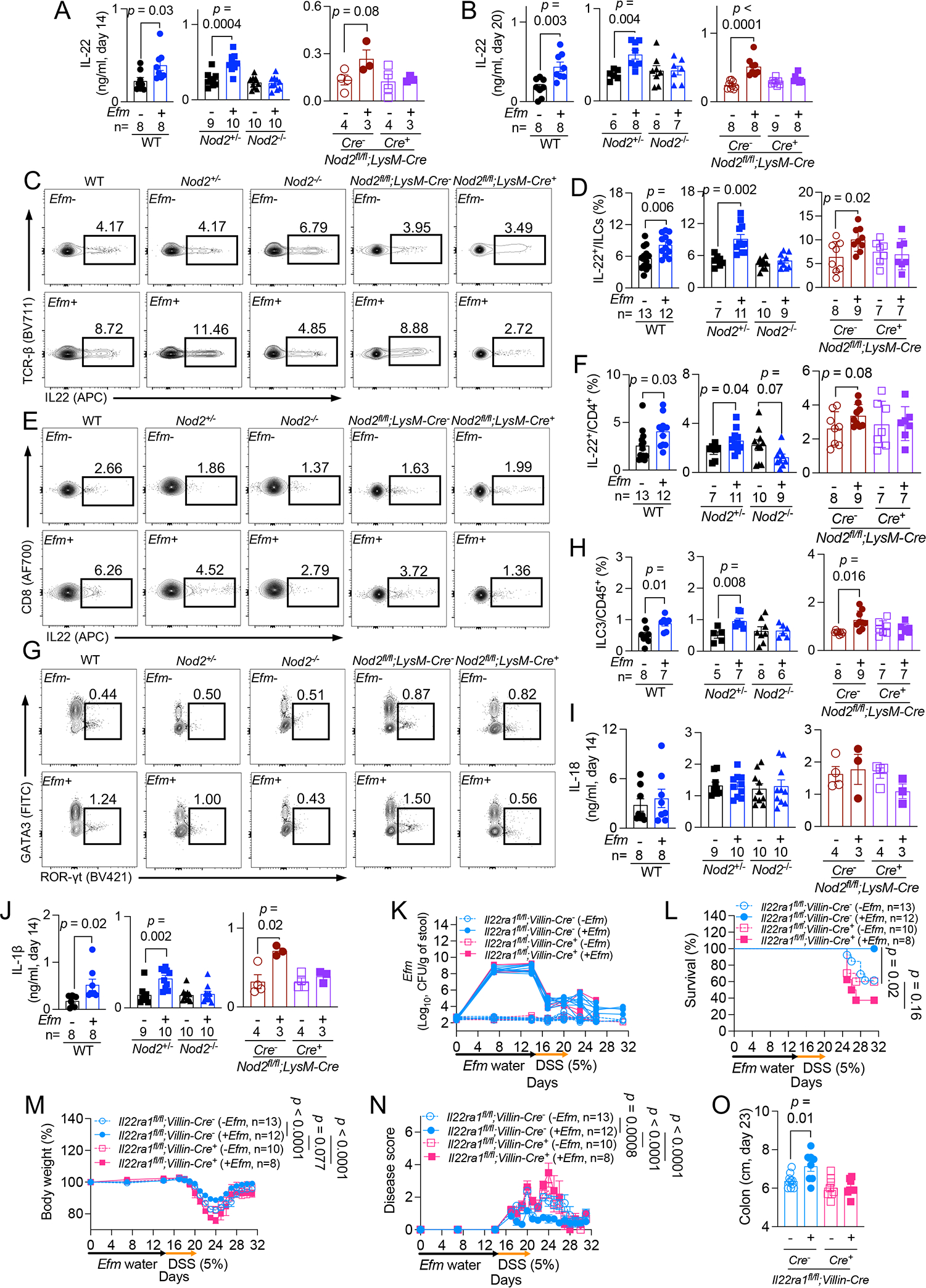
A and B) Quantification of IL-22 on days 14 (A) and 20 (B) in gut explants from WT, Nod2+/−, Nod2−/−, and Nod2fl/fl;LysM-Cre− and Cre+ mice ± Efm inoculation. C-F) Representative flow cytometry plots and quantification of proportion of colonic IL-22+ innate lymphoid cells (ILCs) (C and D) and CD4+ T cells (E and F) in indicated mice ± Efm inoculation. G and H) Representative flow cytometry plots and quantification of group 3 ILCs (ILC3s) from mice in C-F. I and J) Quantification of IL-18 (I) and IL-1β (J) in gut explants from A. K-O) DSS treatment of Il22ra1fl/fl;Villin-Cre− and Cre+ mice following administration of Efm or control examined for Efm burden in the stool (K), survival (L), changes in body weight (M), disease score (N), and colon length on day 23 (O). Data points in A, B, D, F, H-J, and O and lines in K represent individual mice. Data points in M and N represent mean ± SEM. Bars represent mean ± SEM and at least two independent experiments were performed. Indicated p values by unpaired t test, two-tailed in A, B, D, F, H, J, and O, log-rank Mantel-Cox test in L, and two-way ANOVA test in M and N. See also Figures S5 and S6.
IL-1β from intestinal myeloid cells induces IL-22 production from ILC3s79,80. Also, myeloid-derived IL-1β in response to the microbiota depends on NOD281. The secretion of IL-1β, but not IL-18, was induced in Efm-colonized WT, Nod2+/−, and Nod2fl/fl;LysM-Cre− mice on days 14 and 20 (Figures 6I, 6J, S6A, and S6B). IL-23, another cytokine that induces IL-22 from lymphoid cells82, was not detected in gut explants. Efm did not induce IL-1β in Nod2−/− and Nod2fl/fl;LysM-Cre+ mice, indicating that Nod2-dependent sensing of Efm in myeloid cells is required for the upregulated IL-1β secretion (Figures 6J and S6B). Rm 13 mice also displayed increased IL-1β and IL-22 (Figure S6C).
To confirm whether IL-22 is required for Efm-mediated protection, we generated Il22ra1fl/fl;Villin-Cre mice in which IL-22 receptor subunit α is deficient in intestinal epithelial cells and confirmed the Efm shedding in stools following inoculation in all groups (Figure 6K). Il22ra1fl/fl;Villin-Cre+ mice displayed exacerbated intestinal injury compared with Il22ra1fl/fl;Villin-Cre− mice, which did not improve with Efm colonization (Figures 6L–6O).
Based on these results in mice, we examined the correlation between Efm burden and IL-22 production in NIBD and IBD patients. We detected IL-22 protein in NIBD and IBD stool extracts (Figure S6D). Although the detection frequency of IL-22 was comparable between NIBD and IBD, IL-22 intensity was higher in IBD samples (Figures S6E and S6F) as described previously83. Among NIBD patients, Efm colonization was associated with higher IL-22 levels than the Efm-negative patients (Figure S6F). If the heightened IL-22 in IBD patients reflects unresolved inflammation, this relationship would be lost in IBD patients. Indeed, IL-22 levels were comparable between Efm-positive and -negative IBD patients (Figure S6F). Among Efm-positive individuals, we observed a correlation between IL-22 and Efm burden in NIBD patients, which was less obvious in IBD patients (Figure S6G), suggesting a conserved association between Efm colonization and IL-22 production in mice and humans.
Efm directly induces IL-1β upstream of IL-22
Efm may act through other microbiota members. However, alpha and beta diversities of the gut microbiota were similar when comparing 16S rRNA sequencing of stool from mice ± Efm colonization (Figures S6H and S6I). Analysis of composition of microbiomes (ANCOM) revealed that Enterococcus and Stenotrophomonas were the only two taxa significantly altered by Efm colonization (Figures S6J and S6K). Of note, endogenous Enterococcus was undetectable by sequencing, but readily detected by the culture-based method (Figure S6L). Given that Efm colonization marginally changes in the microbiota composition, we tested whether Efm would be sufficient for promoting the IL-22 response. Introducing Efm into germ-free (GF) mice led to stable colonization without changes in body weight and colon length (Figures S6M–S6O). IL-1β and IL-22 were increased in Efm-monocolonized mice compared with GF control mice while IL-18 was comparable (Figure S6P).
To confirm whether IL-1β is required for IL-22 production, we quantified these cytokines in gut explants from IL-1 receptor-deficient mice. Although Efm promoted IL-1β secretion in both Il1r1+/− and Il1r1−/− mice, IL-22 was not induced in Il1r1−/− mice (Figure S6Q). IL-1β can be produced following its transcription (priming) and post-translational activation by the NLRP3 inflammasome84,85. Consistent with a role for NOD2 in priming86, Efm induced Il1β and Nlrp3 expression (Figure S6R). Il18 transcript and IL-18 secretion were not affected by Efm, possibly due to NLRP3-independent regulation of IL-18 in the gut87,88. Efm increased secretion of IL-1β and IL-22 in Nlrp3+/− mice but not in Nlrp3−/− mice (Figure S6S). Thus, inflammasome priming by Efm induces Il1β expression to promote IL-22 production.
NOD2 R702W equivalent impairs Efm-mediated protection in mice
The three major NOD2 variants linked to IBD – R702W, G908R, and a frameshift deletion mutation at L1007 (L1007fs) – result in the loss of muropeptide recognition and NF-κB signaling in vitro89,90. Mice harboring a frameshift mutation equivalent to L1007fs display a compromised cytokine response during bloodstream infection by Efl42, confirming that it results in loss-of-function. The most common variant R702W is detected in up to 5% of individuals of European descent (Figure S7A) has not been studied using an in vivo model (Figure 7A). Thus, we generated mice harboring the equivalent of the human R702W variant (Q675W, Figure S7B). We confirmed the absence of other mutations in Nod2, and that NOD2 Q675W protein was produced (Figures S7C and S7D). Efm was detected in stool collected from Nod2Q675W/+ (littermate controls) and Nod2Q675W/Q675W mice following administration (Figure S7E). Nod2Q675W/+ and Nod2Q675W/Q675W mice displayed modest mortality, weight loss, or disease score following DSS treatment without Efm (Figures 7B–7D), partly due to the high levels of endogenous Enterococcus colonization compared with the other mice raised in Rm 6 (Figure S7G). Efm did not improve disease parameters in Nod2Q675W/Q675W mice, and as such, Efm colonized mice were more susceptible to DSS than similarly treated Nod2Q675W/+ mice (Figures 7B–7H, and S7F;see Table S4). Also, IL-1β and IL-22 were not induced by Efm in Nod2Q675W/Q675W mice (Figure 7I). Thus, the NOD2 R702W equivalent in mice impairs Efm-mediated protection.
Figure 7. NOD2 Q675W impairs Efm-mediated protection.
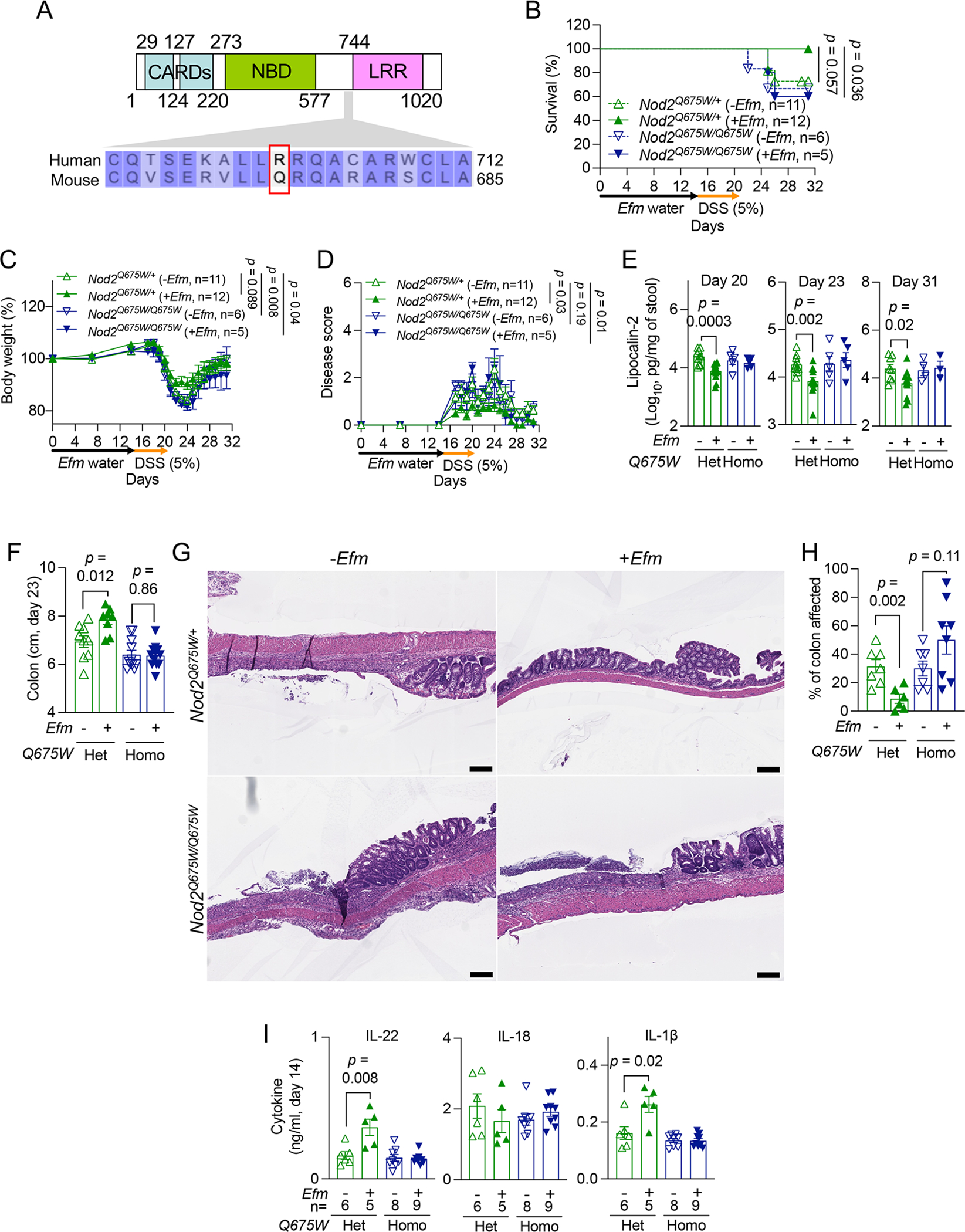
A) NOD2 protein region targeted by mutagenesis. Arginine at position 702 in human NOD2 and the corresponding glutamine at position 675 in mouse NOD2 are indicated in the red box. Numbers refer to human amino acid positions. B-F) DSS-treated Nod2Q675W/+ and Nod2Q675W/Q675W mice ± Efm inoculation were examined for survival (B), changes in body weight (C), disease score (D), fecal LCN2 (E), and colon length (F) on day 23. G and H) Representative images of H&E-stained sections of the colon (G) and quantification of the proportion of colon affected (H). I) Quantification of IL-22 (left), IL-18 (middle), and IL-1β (right) on day 14 in gut explants from Nod2Q675W/+ and Nod2Q675W/Q675W mice ± Efm inoculation. Bars, 200 μM. Data points in E, F, H, and I represent individual mice. Data points in C and D represent mean ± SEM. Bars represent mean ± SEM from at least two independent experiments. CARD, caspase recruitment domain; NBD, nucleotide-binding domain; LRR, leucine-rich repeat domain; Het, heterozygotes; Homo, homozygotes. Indicated p values by log-rank Mantel-Cox test in B, two-way ANOVA test in C and D, and unpaired t test, two-tailed in E, F, H, and I. See also Figures S7 and Table S4.
DISCUSSION
Previous studies demonstrated excessive REG3 gene expression in IBD despite that AMP secretion is generally associated with an improved mucosal barrier39. Here, we revisited this counterintuitive observation. We showed increased REG3 antimicrobial activity in IBD patients compared with controls matched for gastrointestinal symptoms, indicating that REG3 overproduction is specific to IBD flares. Because enterococci are among the most sensitive bacteria to REG3-mediated killing6,9, we focused our analysis on this group and showed their reduced presence and diversity in IBD patients. Our results in mice show that SagA-secreting enterococci such as Efm activate NOD2 in myeloid cells to induce IL-1β, leading to an increase in lymphoid cell types that produce the protective cytokine IL-22. Our findings in humans further support this mechanism by showing that IL-22 levels were associated with Efm colonization in NIBD patients, who display heightened NOD2 activity compared with IBD patients. Once the inflammation has been resolved in a non-IBD setting, enhanced IL-22 may maintain optimal REG3 expressions through STAT3 activation91, perpetuating gut homeostasis. In IBD, either depletion of NOD2 ligand due to REG3 overproduction or genetic deficiency in NOD2 function disrupts this anti-inflammatory circuit (Figure S7H). This model helps explain how IBD patients with intact NOD2 can develop disease presentations similar to those observed in individuals with NOD2 variants once inflammation is established.
Our study also reveals how endogenous Enterococcus in the mouse microbiota can influence experimental outcomes. WT B6 mice maintained in separate rooms displayed substantial differences in susceptibility to DSS due to differences in endogenous Enterococcus burden. Also, untreated Nod2+/− and Nod2−/− mice displayed 4-logs lower Enterococcus burden than with Efm inoculation (Figures S2C and S2D), and hence comparable susceptibility to intestinal injury. Additionally, not all enterococci are protective. Enterococcus isolated from mice resistant to intestinal injury (Rm 13) better-protected recipient mice compared with Enterococcus isolated from mice susceptible to DSS (Rm 6) (Figures 3T, 3U, and S2P). Therefore, it may be necessary to monitor Enterococcus colonization carefully in studies that examine NOD2 function.
Observations with mice deficient in Reg3γ or expressing human REG3A in hepatocytes indicate these AMPs suppress intestinal inflammation10,92. Although we cannot rule out differences in how these proteins are regulated in mice versus humans, these observations could be explained by a model in which REG3 proteins are protective during homeostasis and harmful during chronic inflammation. Altered availability of oxygen and nitrate in the inflamed gut favors colonization by facultative anaerobic Enterobacteriaceae species over the strictly anaerobic Firmicutes, the phylum that includes enterococci93,94. Other Firmicutes reduced in IBD patients encode DL-endopeptidases, such as Streptococcus cristatus, Lactobacillus rhamnosus, and Bacteroides caccae40. AMP dysregulation could exacerbate depletion of NOD2-activating species already vulnerable due to imbalanced redox status of the inflamed environment. REG3 overproduction may explain why individuals experiencing transient gastroenteritis recover while IBD patients remain susceptible to disease flare.
Our findings have relevance to other disease settings such as cancer. Probiotics engineered to secrete SagA were shown to improve the efficacy of immune checkpoint blockade therapy in reducing tumors in mice50. Although adverse events such as colitis hinder cancer immunotherapy strategies, our results predict that inducing NOD2 through SagA would promote checkpoint inhibitor efficacy while also reducing intestinal inflammation. However, our findings with the Nod2 Q675W knock-in mice suggest that this approach will fail in individuals homozygous for analogous loss-of-function variants of NOD2. Genetic information may be useful for matching patients with optimal microbiota-targeting therapies. Additionally, administration of live bacteria is associated with substantial safety concerns. In this context, it is notable that we achieved a similar degree of protection against intestinal injury by administering sterile supernatant collected from SagA-producing bacterial cultures. Postbiotic strategies using bacterial products may be a safe alternate to beneficially trigger NOD2.
Limitations of the study
The consequence of Enterococcus depletion on intestinal inflammation and the immune mechanism was investigated in an animal model. A long-term goal would be to test the efficacy and safety of therapies based on this mechanism. For instance, IL-1β production promotes intestinal inflammation in the IL-10-deficient setting95. Also, our proposed immune circuit induced by NOD2 in myeloid cells is not mutually exclusive with the multitude of other ways in which NOD2 can protect the intestinal barrier.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ken Cadwell (Ken.Cadwell@Pennmedicine.upenn.edu).
Materials availability
The materials in the current study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
16S rRNA sequencing and whole genome sequencing data are deposited in the NCBI Sequence Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers for the datasets are listed in the key resources table.
This study does not report original codes.
Any additional information required to reanalyze the data reported from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-Human Reg1A antibody | R&D Systems | Cat#: MAB4937; RRID: AB_2102139; Clone: 431202 |
| Mouse anti-Human Reg3A antibody | R&D Systems | Cat#: 62257S; RRID: AB_1964697; Clone: 512124 |
| Rabbit anti-REG3G antibody | Abcam | Cat#: ab233480 |
| Rabbit anti-REG4 antibody | Abcam | Cat#: ab255820; Clone: EPR22810-327 |
| Rabbit anti-SagA antibody | Dr. Hang (Scripps Research Institutes)41 | |
| Mouse anti-IL-22 antibody | R&D Systems | Cat#: MAB782; RRID: AB_2295882; Clone: 142938 |
| Mouse anti-NOD2 antibody | Invitrogen | Cat#: MA1-16611; RRID: AB_568643; Clone: 2D9 |
| Mouse anti-β-Actin antibody | Sigma | Cat#: A5441; RRID: AB_476744; Clone: AC-15 |
| IRDye 680RD Goat anti-Rabbit IgG Secondary Antibody | LI-COR | Cat# 925-68071; RRID: AB_2721181; |
| IRDye 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR | Cat#: 925-32210; RRID: AB_2687825; |
| IRDye 800CW Goat anti-Rat IgG Secondary Antibody | LI-COR | Cat#: 925-32219; RRID: AB_2721932; |
| PerCP/Cyanine5.5 anti-mouse/human CD11b Antibody | Biolegend | Cat#: 101228; RRID: AB_893232; Clone: M1/70 |
| PerCP/Cyanine5.5 anti-mouse CD11c Antibody | Biolegend | Cat#: 117328; RRID: AB_2129641; Clone: N418 |
| PerCP/Cyanine5.5 anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | Cat#: 108428; RRID: AB_893558; Clone: RB6-8C5 |
| PerCP/Cyanine5.5 anti-mouse TER-119/Erythroid Cells Antibody | Biolegend | Cat#: 116228; RRID: AB_893636; Clone: TER-119 |
| PerCP/Cyanine5.5 anti-mouse NK-1.1 Antibody | Biolegend | Cat#: 108728; RRID: AB_2132705; Clone: PK136 |
| PE anti-T-bet Antibody | Biolegend | Cat#: 644810; RRID: AB_2200542; Clone: 4B10 |
| PE- Cyanine7 anti-mouse/human CD44 antibody | Biolegend | Cat#: 103030; RRID: AB_830787; Clone: IM7 |
| APC/Cyanine7 anti-mouse CD4 Antibody | Biolegend | Cat#: 100414; RRID: AB_312699; Clone: GK1.5 |
| Brilliant Violet 570 anti-mouse CD62L Antibody | Biolegend | Cat#: 104433; RRID: AB_10900262; Clone: MEL-14 |
| Brilliant Violet 605 anti-mouse CD127 (IL-7Ra) Antibody | Biolegend | Cat#: 135041; RRID: AB_2572047; Clone: A7R34 |
| Brilliant Violet 650 anti-mouse NK-1.1 Antibody | Biolegend | Cat#: 108736; RRID: AB_2563159; Clone: PK136 |
| Alexa Fluor 700 anti-mouse CD8a Antibody | Biolegend | Cat#: 100730; RRID: AB_493703; Clone: 53-6.7 |
| Brilliant Violet 711 anti-mouse TCR β chain Antibody | Biolegend | Cat#: 109243; RRID: AB_2629564; Clone: H57-597 |
| PerCP/Cyanine5.5 anti-mouse CD19 | eBioscience | Cat#: 45-0193-82; RRID: AB_1106999; Clone: 1D3 |
| PE anti-mouse IL-22 Antibody | eBioscience | Cat#: 12-7221-82; RRID: AB_10597428; Clone: 1H8PWSR |
| APC anti-mouse FOXP3 Antibody | eBioscience | Cat#: 17-5773-82; RRID: AB_469457; Clone: FJK-16s |
| PE-CF594 anti-mouse γδ T-Cell Receptor Antibody | BD Bioscience | Cat#: 563532; RRID: AB_2661844; Clone: GL3 |
| BUV395 anti-mouse CD45 Antibody | BD Bioscience | Cat#: 564279; RRID: AB_2651134; Clone: 30-F11 |
| BV421 anti-mouse RORgt Antibody | BD Bioscience | Cat#: 562894; RRID: AB_2687545; Clone: Q31-378 |
| BV421 anti-mouse IL-10 Antibody | BD Bioscience | Cat#: 566295; RRID: AB_2739668; Clone: JES5-16E3 |
| Alexa Fluor 488 anti-mouse GATA3 Antibody | BD Bioscience | Cat#: 560163; RRID: AB_1645302; Clone: L50-823 |
| Bacterial strains | ||
| Enterococcus faecalis OG1RF | Dr. Hang (Scripps Research Institutes), originally acquired from ATCC 47077. | N/A |
| Enterococcus faecium Com15 | Dr. Hang (Scripps Research Institutes), originally acquired Dr. Gilmore (Harvard Medical School). | N/A |
| Salmonella Typhimurium SL1344 | Gift from Dr. Littman (NYU) | N/A |
| Lactococcus lactis thyA auxotroph | Dr. Hang (Scripps Research Institutes)50, generated in collaboration with Rise Therapeutics | N/A |
| Lactococcus lactis thyA expressing wild-type SagA | Dr. Hang (Scripps Research Institutes)50, generated in collaboration with Rise Therapeutics | N/A |
| Lactococcus lactis thyA expressing catalytic mutant SagA | Dr. Hang (Scripps Research Institutes)50, generated in collaboration with Rise Therapeutics | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bacto brain heart infusion | BD | Cat#: 237500 |
| Sabouraud dextrose broth | Millipore | Cat#: S3306 |
| Nutrient Broth | Millipore | Cat#: 70122 |
| BsaHI | NEB | Cat#: R0556S |
| Dulbecco’s Modified Eagle’s Medium | Corning | Cat#: 10-017-CV |
| Fetal bovine serum | Peak Serum | Cat#: PS-FB2 |
| Penicillin-Streptomycin Solution | Corning | Cat#: 30-002-CI |
| Normicin | InvivoGen | Cat#: ant-nr |
| Zeocin | InvivoGen | Cat#: ant-zn |
| Blastocidin | InvivoGen | Cat#: ant-bl |
| Trypsin-EDTA | Gibco | Cat#: 25200056 |
| M17 Broth | BD | Cat#: 218561 |
| M17 Agar | BD | Cat#: 218571 |
| Bile esculin azide agar | Millipore | Cat#: 06105 |
| HiCrome Enterococcus faecium Agar Base | HIMEDIA | Cat#: M1580 |
| Enterococcus faecium Selective Supplement | HIMEDIA | Cat#: FD226 |
| Lactose | Sigma-Aldrich | Cat#: 17814 |
| Thymidine | Sigma-Aldrich | Cat#: T9250 |
| Phosphate-buffer saline | Corning | Cat#: 21040CV |
| Triton X-100 | Sigma-Aldrich | Cat#: T8787 |
| Glycerol | Sigma-Aldrich | Cat#: G5516 |
| Halt Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher | Cat#: 78440 |
| Bolt 4-12% Bis-Tris Plus Gels | Invitrogen | Cat#: NW04120BOX |
| Intercept (TBS) Blocking Buffer | LI-COR | Cat#: 927-60003 |
| Trypticase soy agar with 5% sheep blood | Henry Schein | Cat#: 9873908 |
| HEK-Blue detection medium | InvivoGen | Cat#: hb-det2 |
| Dextran sulfate sodium | TdB Consultancy | Cat#: DB001 |
| Ampicillin | Amercian Bioanalytical | Cat#: 69-52-3 |
| Streptomycin | Sigma-Aldrich | Cat#: S6501 |
| Low melting point agar | Promega | Cat#: V2111 |
| 10% Formalin | Thermo Fisher | Cat#: SF99-4 |
| Roswell Park Memorial Institute 1640 Medium | Gibco | Cat#: 61870127 |
| L-glutamine | Corning | Cat#: 25-005-CI |
| HEPES | Corning | Cat#: 25-060-CI |
| Minimum Essential Medium Nonessential Amino Acids | Corning | Cat#: 25-025-CI |
| Sodium pyruvate | Corning | Cat#: 25-000-CI |
| β-mercaptoethanol | Gibco | Cat#: 21985023 |
| Hank’s Balanced Salt Solution | Gibco | Cat#: 14175103 |
| DL-dithiothreitol | Sigma Aldrich | Cat#: D0632 |
| Collagenase | Sigma Aldrich | Cat#: C2139 |
| Deoxyribonuclease I | Sigma Aldrich | Cat#: DN25 |
| Percoll | Sigma-Aldrich | Cat#: P1644 |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | eBioscience | Cat#: 00-4975-93 |
| Bovine Serum Albumin | Thermo Fisher | Cat#: BP1600-100 |
| Fixation Buffer | Biolegend | Cat#: 420801 |
| Intracellular Staining Permeabilization Wash Buffer | Biolegend | Cat#: 421002 |
| Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher | Cat#: 00-5523-00 |
| Zombie UV Fixable Viability Kit | Biolegend | Cat#: 423108 |
| Critical commercial assays | ||
| 10 ml syringe | BD | Cat#: 302995 |
| Gauze pad | 4MD Medical | Cat#: PROP157025 |
| Millex-GS Syringe Filter Unit, 0.22 μm | Millipore | Cat#: SLGL0250S |
| Nalgene Rapid-Flow Sterile Disposable Filter Units with PES Membranes | Thermo Fisher | Cat#: 567-0020 |
| Human Reg3A DuoSet ELISA | R&D systems | Cat#: DY5940-05 |
| Mouse Lipocalin-2/NGAL DuoSet ELISA | R&D systems | Cat#: DY1857-05 |
| Mouse IL-22 DuoSet ELISA | R&D systems | Cat#: DY582-05 |
| Mouse IL-18 DuoSet ELISA | R&D systems | Cat#: DY7625-05 |
| Mouse IL-1 beta/IL-1 F2 DuoSet ELISA | R&D systems | Cat#: DY401-05 |
| DNeasy PowerSoil Pro Kit | Qiagen | Cat#: 47016 |
| MagMAX DNA Multi-Sample Ultra 2.0 kit | Thermo Fisher | Cat#: A36570 |
| RNeasy Mini Kit | Qiagen | Cat#: 74106 |
| RNase-Free DNase Set | Qiagen | Cat#: 79256 |
| 70 μm nylon mesh | ELKO Filtering | Cat#: 03-70/33 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat#: 4368814 |
| LightCycler 480 SYBR Green I Master | Roche | Cat#: 04707516001 |
| TOPO TA Cloning Kit | Invitrogen | Cat#: 450002 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Nod2−/− | Jackson Laboratory | Cat#: 005763 |
| Mouse Nod2fl/fl | Dr. Keestra-Gounder (University of Colorado Anschutz Medical Campus) | N/A |
| Mouse: Il22ra1fl/fl | Dr. Sergei Koralov (New York University Grossman school of medicine), originally acquired from Jackson Laboratory (Cat#: 031003) | N/A |
| Mouse: LysM-Cre | Jackson Laboratory | Cat#: 004781 |
| Mouse: Villin-Cre | Jackson Laboratory | Cat#: 004586 |
| Mouse: Il1r1 | Jackson Laboratory | Cat#: 003245 |
| Mouse Nlrp3 | Jackson Laboratory | Cat#: 021302 |
| Mouse: Nod2Q675W/Q675W | This study | N/A |
| Cell line: HEK-Blue Null2 | InvivoGen | Cat#: hkb-hnod2 |
| Cell line: HEK-Blue NOD2 | InvivoGen | Cat#: hkb-null2 |
| Deposited data | ||
| 16S rRNA sequencing data related to Figure 2 | This study | BioProject: PRJNA915760 |
| Whole genome sequencing data related to Figure S1 | This study | BioProject: PRJNA978059 |
| 16S rRNA sequencing data related to Figure S6 | This study | BioProject: PRJNA977252 |
| Software and Algorithms | ||
| Prism 9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | BD | https://www.flowjo.com |
| NCBI Gene and Protein Data Base | NCBI | http://www.ncbi.nlm.nih.gov |
| Nucleotide blast | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Blastx | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Fiji/ImageJ | NIH | https://fiji.sc |
EXPERIENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
Age- and gender-matched 7 to 10-week-old mice on the C57BL/6J (B6) background were used. All mice were bred on-site. Mice harboring gene deletions were compared to their respective littermate control mice indicated in the results section. Nod2−/−, LysM-Cre, Villin-Cre, Il1r1, and Nlrp3 mice were purchased from The Jackson Laboratory. Il22ra1fl/fl and Nod2fl/fl mice were obtained from Sergei Koralov (New York University Grossman School of Medicine) and A. Marijke Keestra-Gounder (University of Colorado Anschutz School of Medicine)96, respectively. To generate cell type-specific knock-out mice, Nod2fl/fl mice were bred with LysM-Cre and Villlin-Cre mice and Il22ra1fl/fl mice were bred with Villin-Cre mice. Mice were assigned numbers to facilitate blind data collection and distributed randomly to treatment groups. All animal studies were performed according to approved protocols by the NYU Grossman School of Medicine Animal Care and Use Committee (IACUCs).
Generation of Nod2 Q675W knock-in mice
CRISPR–Cas9 gene-targeting mixture containing sgRNA (5’-AGCGGGCACGTGCCTGACGC-3’) targeting exon 4 of B6 Nod2 and template (5’-TCACAGCAGCCTTCCTAGCAGGTCTGTTGTCCCAGCAGCATCGGGACCTGTTGGCTGCATGCCAGGTCTCCGAGAGGGTACTTCTATGGCGTCAGGCACGTGCCCGCTCGTGTCTGGCCCACAGCCT-3’) (synthesized at Integrated DNA Technologies) and Cas9 mRNA were injected into the cytoplasm of zygotes generated from B6 females impregnated by B6 males, and then the microinjected embryos were incubated in potassium-supplemented simplex optimized medium (KSOM) at 37°C for one day and subsequently transferred into pseudopregnant CD-1 female mice at the two-cell stage by the Rodent Genetic Engineering Laboratory at NYU Grossman School of Medicine. The resulting F0 chimeras were screened through genotyping PCR. Amplicons were generated using a pair of primers (Fwd 5’-CTTTTCAGCTGTGGCCGGCT-3’ and Rev 5’-TTTGCCACAGGCCCAATCGG-3’) flanking the targeting sites from tail DNA from chimeras and wild-type mice and cut by BsaHI (NEB) (Figure S7A). A 3042-bp region containing the Nod2 coding region was sequenced to verify correct gene targeting (Figure S7C). All mice used in experiments were backcrossed with B6 mice at least 3 generations.
Gnotobiotics
Previously described GF mice97 were maintained in flexible film isolators, and absence of fecal bacteria and fungi was confirmed by aerobic culture in brain heart infusion (BHI) (BD), Sabouraud dextrose (Millipore), and nutrient broth (Millipore) and qPCR for bacterial 16S and eukaryotic 18S ribosomal RNA (rRNA) genes through sampling of stool from individual cages in each isolator on a monthly basis. Mice were transferred into individually ventilated Tecniplast ISOcages for DSS treatment to maintain sterility under positive air pressure.
Cell culture
HEK-Blue Null2 (InvivoGen) and HEK-Blue NOD2 (InvivoGen) were cultured at 37°C in 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning) in the presence of 10% fetal bovine serum (FBS, Peak Serum), 100 IU/ml penicillin and 100 μg/ml streptomycin (Corning), 100 μg/ml normicin (InvivoGen), and appropriate antibiotics (100 μg/ml zeocin (InvivoGen) for HEK-Blue Null2 and 30 μg/ml blastocidin (InvivoGen) and 100 μg/ml zeocin for HEK-Blue NOD2). Cells were maintained at no greater than 70% confluency and subcultured using Trypsin-EDTA (Gibco). Cell lines were routinely cultured without antibiotics to ensure no bacterial infection and tested for mycoplasma.
Human stool samples and data collection
Stool samples were collected with consent from hospitalized adult non-IBD (NIBD) and IBD patients with gastrointestinal symptoms with consent between June 27, 2019 and February 26, 2020. The protocol has been approved by the New York University School of Medicine Institutional Review Board (Mucosal Immune Profiling in Patients with Inflammatory Bowel Disease; S12–01137). The clinical data of NIBD and IBD patients were collected using EPIC EHR and REDCap 9.3.6 software. A total of 112 patients were included in the analysis, of which 56% were female, median age 44.5. Patient information including age, gender, gastrointestinal symptoms, medical history related with gastrointestinal tract, and disease activity scores (CDAI or total Mayo) are shown in Tables S1 and S2. At the time of sample acquisition and processing, investigators were blinded to the patient clinical status.
METHOD DETAILS
Bacterial species
Enterococcus faecalis OG1RF (Efl), E. faecium Com15 (Efm), and Lactococcus lactis thyA auxotroph (Lls) expressing wild-type SagA (Lls-sagAWT) and catalytic mutant SagA (Lls-sagAC443A) were previously described50. Salmonella Typhimurium SL1344 (STm) was provided by Dan Littman (NYU). Efl and Efm were grown at 37°C under ambient atmosphere in autoclaved antibiotics-free BHI broth. Lls strains and STm were cultured at the above condition in M17 broth (BD) supplemented with 2% (v/v) lactose (Sigma-Aldrich) and 20 μg/ml of thymidine (Sigma-Aldrich) (LM17 broth) and Luria Bertani (LB) broth (NYU Reagent Preparation Core), respectively. Colony forming unit (CFU) of Efl, Efm, Lls strains, and STm was enumerated on bile esculin azide (BEA) agar (Millipore), HiCrome Enterococcus faecium Agar with Selective Supplement (HIMEDIA), M17 agar (BD) supplemented with 2% (v/v) lactose and 20 μg/ml of thymidine (LM17 agar), and LB agar (NYU Reagent Preparation Core), respectively.
Preparation of human stool extract, antimicrobial activity assay, and REG3A measurement
1 g of human stools from NIBD and IBD patients were homogenized in 5 ml of phosphate-buffered saline (PBS, Corning) using TissueRuptor (Qiagen). 0.5–1 ml of human stool slurries were taken for quantification of bacterial burden. The remaining stool slurries were filtered using a 10 ml syringe (BD) with gauze (4MD Medical). Human stool extracts were collected by centrifugation at 15,000 x g for 20 min at 4°C and filter-sterilized using a 10 ml syringe with Millex-GS Syringe Filter Unit, 0.22 μm (Millipore).
For quantifying the antimicrobial activity of the human stool extracts, overnight cultures of Efl, Efm, and STm were harvested, resuspended in 50 μl of PBS, and mixed with one volume of human stool extracts or PBS. The mixture of bacteria and human stool extract was incubated in a 37°C static incubator for 24 h and then plated in serial dilution on selective agars as mentioned above for enumerating Efl, Efm, and STm.
Quantification of REG3A in human stool extracts was performed using Human Reg3A DuoSet ELISA (R&D systems) according to the manufacturer’s instructions.
Western blotting
Mouse colonic tissues were processed for immunoblotting as previously described97,98. Briefly, proximal colonic tissues (2 mm) were cut open and washed with PBS, then suspended in lysis buffer (20 mM Tris-HCl (pH 7.4, NYU Reagent Preparation Core), 150 mM NaCl (NYU Reagent Preparation Core), 1% Triton X-100 (Sigma-Aldrich), 10% glycerol (Sigma-Aldrich), and 2x Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher)) and homogenized using FastPrep-24 Classic bead beating grinder and lysis system (MP Biomedicals). Tissue homogenate was then pelleted twice at 10,000 x g for 10 min at 4°C to collect the lysates. human stool extracts and mouse colonic tissue lysates were resolved on Bolt 4–12% Bis-Tris Plus Gels (Invitrogen), transferred onto polyvinylidene difluoride membranes, and blocked using Intercept (TBS) blocking buffer (LI-COR). Membranes were probed with primary antibody overnight at 4°C. The following primary antibodies were used for western blotting studies: anti-REG1A (R&D systems, MAB4937), REG3A (R&D systems, MAB5965), REG3G (Abcam, ab233480), REG4 (Abcam, ab255820), SagA41, IL-22 (R&D systems, MAB782), NOD2 (Invitrogen, MA1–16611), and β-actin (Sigma-Aldrich, A5441). After incubation with the primary antibody, the membrane was washed and probed with the secondary antibody for 1 h at room temperature (RT). As for secondary antibodies, IRDye 680RD Goat anti-Rabbit (925–68071), IRDye 800CW Goat anti-Mouse (925–32210), and IRDye 800CW Goat anti-Rat (925–32219) were purchased from LI-COR. After additional washing, the protein was then detected with Image Studio for Odyssey CLx (LI-COR). Band intensities were measured by Fiji/ImageJ99
DNA extraction,16S rRNA sequencing analysis, and detection of Efm.
DNA from human stool samples or mouse fecal samples following 2-week-administration of Efm in drinking water or control was extracted with DNeasy PowerSoil Pro kit (Qiagen) according to the manufacturer’s instruction. Bacterial 16S rRNA gene was amplified at the V4 region using primer pairs and paired-end amplicon sequencing was performed on the Illumina MiSeq system at NYU Genome Technology Core. Sequencing reads were processed using the DADA2 pipeline in the QIIME2 software package. Taxonomic assignment was performed against the Greengenes 13_8 99% OTUs full-length sequences database100. Alpha diversity analysis was done using observed OTUs101. Beta diversity was calculated using Bray-Curtis, Jaccard, unweighted UniFrac, and weighted UniFrac distance and visualized with EMPeror102. Differentially abundant bacterial species were identified by ANCOM103.
To detect Efm from stool DNA of NIBD and IBD patients, 658-bp region specific to genomic DNA of Efm was amplified from 25 ng of the stool DNA by PCR using a pair of primers (Fwd 5’-TTGAGGCAGACCAGATTGACG-3’ and Rev 5’-TATGACAGCGACTCCGATTCC-3’) as described previously104.
Enumeration of total Enterococcus and Efm in human stool samples
The human stool slurries (200 mg/ml) were plated in serial dilution on selective agars mentioned above for enumerating CFU of total Enterococcus and Efm, respectively.
Isolation and genomic analysis of Enterococcus strains from NIBD and IBD patients
A total of 201 Enterococcus strains were isolated by plating human stool slurries on bile esculin azide agar. The colonies on the plate were further purified by streaking on trypticase soy agar with 5% sheep blood (Henry Schein). Genomic DNA was extracted using the KingFisher Flex automated extraction instrument (Thermo Fisher) and the MagMAX DNA Multi-Sample Ultra 2.0 kit reagents (Applied Biosystems). Genome sequencing was performed on an Illumina NovaSeq 6000 system at the NYU Genome Technology Core, yielding 150 bp paired-end reads. The software fastp v0.20.1105 was used with default settings to remove adapters, trim low-quality bases, and remove low-quality reads. The level of within-species cross-contamination was estimated using ConFindr106 version 0.7.4; isolates with predicted contamination greater than 10% were excluded from further analysis. Taxonomic classification to the species level was performed by running GTDBTK107 version v1.5.1 (database release 202) on genome assemblies generated using Unicycler108 version v0.4.8 in conservative mode. The presence of sagA in each genome assembly was determined using BLAST109.
Filtered reads from all species were mapped to a reference assembly of Efm strain SRR24 (RefSeq accession GCF_009734005.1) using Snippy version 4.6.0 (github.com/tseemann/snippy). A single phylogenetic tree was inferred for all species based on the resulting core alignment using RAxML version 8.2.12110 using the GTRGAMMA model of evolution.
For each of the Enterococcus species identified using GTDBTK, a core alignment of the isolates belonging to that species was generated using Snippy, using one of the isolates’ assemblies as reference. A SNP matrix was calculated using snp-dists version 0.8.2 (https://github.com/tseemann/snp-dists). Strains were then defined by clustering isolates using a threshold of 100 SNPs. Specifically, the R package igraph111 was used to create a graph whose nodes are the isolates and whose edges connect any pair of isolates differing by no more than 100 SNPs in the corresponding within-species alignment. Strains were defined as the connected components of the resulting graph.
Bacterial inoculation and DSS treatment of mice
Efm drinking water was prepared as described previously67. Briefly, overnight culture of Efm was harvested and resuspended in filter-sterilized drinking water to 109 CFU/ml, herein referred as Efm water, and administered to mice for 14 days. This drinking water was then replaced with one containing 5% dextran sulfate sodium (DSS, TdB Consultancy) for 6 days and then switched to regular drinking water for the remainder of the experiment when examining intestinal injury following Efm colonization. For experiments in which Efm was administered after the initiation of intestinal injury, mice were treated with 5% DSS for 6 days and received Efm water or control water for the remainder of the experiment. Drinking water was exchanged with freshly prepared ones every 2–3 days for all types of treatment.
For Efm colonization in GF mice, overnight cultures of Efm were harvested and resuspended in PBS to 109 CFU/100 μl and were administered to GF mice by oral gavage of 100 μl Efm inoculum on day 0. The colonization of Efm was confirmed by enumerating CFU in stool pellets on the selective agar plate mentioned above for 2 weeks.
Overnight cultures of Lls strains were harvested and resuspended in PBS to 109 CFU/100 μl. Water containing ampicillin (1 mg/ml, American Bioanalytical) and streptomycin (0.5 mg/ml, Sigma-Aldrich) was filter-sterilized using Nalgene Rapid-Flow Sterile Disposable Filter Units with PES Membranes (Thermo Fisher) for the antibiotic (abx) treatment. The mice treated with abx-containing water for 7 days were given 3% DSS for 6 days. On days 8 and 14, the mice were orally administered 100 μl inoculum of Lls strains (approximately 109 CFU).
For treatment of Lls culture supernatant, overnight cultures of Lls strains were centrifuged at 6,000 x g for 15 min at 4°C to collect the culture supernatant. The supernatants and LM17 broth control were filter-sterilized using Nalgene Rapid-Flow Sterile Disposable Filter Units with PES Membranes to avoid bacterial contamination. The mice receiving the filter-sterilized supernatants of Lls strains or LM17 broth as drinking water for 7 days were given 5% DSS for 6 days.
For administration of Rm 6 and Rm 13 Enterococcus, Enterococcus strains were collected by plating stool pellets from Rm 6 or Rm 13 mice on the selective agar plate mentioned above and then harvested for Enterococcus drinking water in the same way as Efm water preparation. The Rm 6 mice receiving the Rm 6 or Rm 13 Enterococcus drinking water for 14 days were given 5% DSS for 6 days.
The mice were monitored daily for survival and weight loss. Disease score was quantified based on five parameters in which eight was the maximum score: diarrhea (0–2), bloody stool (0–1), hunched posture (0–2), mobility (0–2), and fur ruffling (0–1).
Quantification of bacterial burden and lipocalin-2 (LCN2) in murine stool samples
Stool pellets from individual mice were weighed, homogenized in PBS, and plated in serial dilution on selective agars as mentioned above for enumerating CFU of total Enterococcus, Efm, and Lls strains, respectively. The remaining stool homogenates were centrifuged at 15,000 x g for 5 min to collect clear supernatants. LCN2 in the clear supernatants was quantified using Mouse Lipocalin-2/NGAL DuoSet ELISA (R&D systems) according to the manufacturer’s instructions.
Transcript analysis
Total RNA was extracted from proximal colonic tissue (2 mm) using RNeasy Mini Kit with DNase treatment (QIAGEN), and synthesis of cDNA was conducted with High-Capacity cDNA Reverse Transcription Kit (ThermoFisher) according to the manufacturer’s protocol. RT-PCR was performed using SybrGreen (Roche) on a Roch480II Lightcycler using the following primers: Reg3α, Fwd 5’-CTCAGGACATCTCGTGTCTATTCTT-3’ and Rev 5’-AGTGACCACGGTTGACAGTAGAG-3’; Reg3β, Fwd 5’-CTCTCCTGCCTGATGCTCTT-3’ and Rev 5’-GTAGGAGCCATAAGCCTGGG-3’; Reg3γ, Fwd 5’-CGTGCCTATGGCTCCTATTGCT-3’ and Rev 5’-TTCAGCGCCACTGAGCACAGAC-3’; Reg3δ, Fwd 5’-TGGAACCACAGACCTGGGCTA-3’ and Rev 5’-GAGCAGAAATGCCAGGTGTCC-3’; Reg4, Fwd 5’-CGCTGAGATGAACCCCAAG-3’ and Rev 5’-TGAGAGGGAAGTGGGAAGAG-3’; Il18, Fwd 5’-CAACCAACAAGTGATATTCTCCATG-3’ and Rev 5’-CTGACATGGCAGCCATTGT-3’; Il1β, Fwd 5’-CAACCAACAAGTGATATTCTCCATG-3’ and Rev 5’-GATCCACACTCTCCAGCTGCA-3’; Nlrp3, Fwd 5’-CTCCAACCATTCTCTGACCAG-3’ and Rev 5’-ACAGATTGAAGTAAGGCCGG-3’; Gapdh, Fwd 5’- TGGCCTTCCGTGTTCCTAC-3’ and Rev 5’- TGGCCTTCCGTGTTCCTAC-3’;. The relative expression of the respective genes to Gapdh expression was calculated using the ΔΔCT method112 and was expressed as fold change normalized to WT mice before Efm Colonization.
Histology
Quantification of colon histology data was performed blind. Colonic tissues were cut open along the length, pinned on black wax, and fixed in 10% formalin (Thermo Fisher). Tissues were embedded in 3% low melting point agar (Promega). Formalin embedding, cutting, and hematoxylin and eosin (H&E) staining were performed by the NYU Histopathology core. Sections were imaged on a Nikon Eclipse Ci microscope. Pathologic changes in colonic mucosa evaluated by Y.D. included colonic epithelium, lamina propria, muscularis mucosa, and submucosa. It was based on the state of the changes (acute or chronic) and the degree of involvement. Acute changes primarily included neutrophils in the crypt lumen, erosions, ulcers, pus, and the formation of polyps. Features of chronic changes included the following: crypt distortion (sideways crypts, branching, tortuous), crypt loss, crypt atrophy, basal plasmacytosis, and lymphoid aggregates. The involvement pattern included focal distal involvement, patchy involvement with skipping areas, diffuse involvement, and pancolonic (no skip areas) involvement. The percentage of involvement was calculated as 100 x (Length of disease involved colon / Total length of the colon).
NOD2 activity assay
Single-cell suspensions (50,000 cells/ml) of HEK-Blue Null2 or HEK-Blue NOD2 were prepared by tapping the flask and suspension with HEK-Blue detection medium (InvivoGen). Cells were aliquoted at 180 μl/well in a 96-well plate (Corning) containing 20 μl dilutes of human stool extracts in PBS. Cells were then incubated at 37°C in 5% CO2 for 24 h. To measure activity, wells were then gently pipetted up and down to mix the conditioned medium, and absorbance from the colorimetric product of the secreted alkaline phosphatase was measured at 630 nm. The NOD2 activity was calculated as fold change by normalizing to HEK-Blue Null2 cells.
Colon explant culture and cytokine detection
Colon explant culture was performed as previously described76. The proximal colon tissue (1 cm) was opened longitudinally, washed with PBS and cultured for 48 h in 1ml of complete Roswell Park Memorial Institute (RPMI) 1640 (Gibco) containing 2 mM L-glutamine (Corning), 100 IU/ml penicillin and 100 μg/ml streptomycin, 20 mM N-2-hydroxyethylpiperazine-N’-2-ethane sulfonic acid (HEPES, Corning), Minimum Essential Medium (MEM) non-essential amino acids (Corning), 1 mM sodium pyruvate (Corning), and β-mercaptoethanol (Gibco). Cytokines in supernatants were measured using the Mouse IL-22, IL-18, and IL-1 beta/IL-1 F2 DuoSet ELISA (R&D systems) according to the manufacturer’s instructions.
Isolation of LP cells
LP cells were harvested as before113. Briefly, colonic tissue including caecum was cut open and washed with PBS, and fat was removed. The tissues were incubated with 20 ml of Hank’s Balanced Salt Solution (HBSS, Gibco) with 2% HEPES, 1% sodium pyruvate, 5 mM Ethylenediaminetetraacetic acid (EDTA, NYU Reagent Preparation Core), and 1mM DL-dithiothreitol (Sigma-Aldrich) for 15 min at 37°C with 220 rpm and then with new 20 mL of HBSS with 2% HEPES, 1% sodium pyruvate, 5 mM EDTA for 10 min at 37°C with 210 rpm. The tissue bits were washed with HBSS, minced, and then enzymatically digested with collagenase (0.5 mg/ml, Sigma-Aldrich) and Deoxyribonuclease I (0.01 mg/ml, Sigma-Aldrich) for 30 min at 37°C with 200 rpm. Digested solutions were passed through 70 μm nylon mesh (ELKO Filtering) and isolated cells were resuspended in 40% Percoll (Sigma-Aldrich), layered onto 80% Percoll, and centrifuged at RT at 2,200 rpm for 22 min. Cells were recovered from the interphase and washed with RPMI containing 10% FBS (Peak Serum) (cRPMI). For the analysis of the cytokine production, LPLs were plated in cRPMI and stimulated with 1X Cell Stimulation Cocktail (plus transport inhibitors) (eBiosciecne) for 4 h at 37°C.
Flow cytometry
Surface and intracellular cytokine staining was performed per manufacturer’s instruction in PBS with 0.5% bovine serum albumin (BSA, Thermo Fisher) (BSA/PBS) for 20 min on ice. Two staining panels were prepared as described previously113. The following antibodies were used for the first panel: CD11b (101228, 1:200), CD11c (117328, 1:200), CD127 (135041, 1:33), CD4 (100414, 1:150), CD44 (103030, 1:100), CD62L (104433, 1:100), CD8 (100730, 1:100), GR1 (108428, 1:300), NK1.1 (108736, 1:150), Tbet (644810, 1:50), TCRβ (109243, 1:100), TER119 (116228, 1:200) from Biolegend, CD19 (45–0193-82, 1:100) and FOXP3 (17–5773-82, 1:100) from eBioscience, and CD45 (564279, 1:200), GATA3 (560163, 1:40), RORγt (562894, 1:100), TCRγδ (563532, 1:100) from BD Bioscience. The following antibodies were used for the second panel: CD11b (101228, 1:200), CD11c (117328, 1:200), CD127 (135041, 1:33), CD4 (100414, 1:150), CD8 (100730, 1:100), GR1 (108428, 1:300), NK1.1 (108728, 1:200), TCRβ (109243, 1:100), TER119 (116228, 1:200) from Biolegend, IL-22 (12–7221-82, 1:100) from eBioscience, and CD45 (564279, 1:200), IL-10 (566295, 1:100) and TCRγδ (563532, 1:100) from BD Bioscience. Samples were fixed with either Fixation Buffer (Biolegend) or Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher). For intracellular staining of the transcription factor, cells were permeabilized with the Foxp3/Transcription Factor Staining Buffer Set at RT for 45 min in the presence of antibodies. For intracellular staining of cytokines, cells were permeabilized with Intracellular Staining Permeabilization Wash Buffer (Biolegend) at RT for 20 min in the presence of antibodies. Zombie UV Fixable Viability Kit (Biolegend) was used to exclude dead cells. Samples were acquired on a BD LSR II (BD Biosciences) and analyzed using FlowJo software (BD).
Nod2 sequencing
Nod2 mRNA was sequenced as previously described 114. Briefly, total RNA was extracted from proximal colonic tissues (2 mm) and subjected to the synthesis of cDNA using the same kits mentioned above. A 3042-bp region containing the Nod2 coding region was amplified from cDNA by PCR using a pair of primers (Fwd 5’-ATGTGCTCACAGGAAGAGTTCC-3’ and Rev 5’-TCACAACAAGAGTCTGGCGTCCC-3’). Amplicons were cloned into pCR2.1-TOPO (Invitrogen). The plasmids were sequenced by Oxford Nanopore from SNPsaurus.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data points and bars in the figure panels represent mean values ± standard error of mean. Statistical methods in this study are described in the figure legend using GraphPad Prism 9 and Python. N represents individual patient or mouse as described in the figure legend. p < 0.05 is considered statistically significant for all assays, and individual p values are indicated in the figure panels.
Supplementary Material
Table S1. Information for IBD patients, Related to Figures 1, 2, 4, S1, and S6. M, male; F, female; CD, Crohn’s disease; UC, ulcerative colitis; CDAI, Crohn’s disease activity index; N/A, not available.
Table S3. Information for Enterococcus isolates from NIBD and IBD pateints, Related to Figure S1.
Table S4. Histological findings of colonic sections from mice receiving DSS, Related to Figures 3, 7, and S3. WT, wild type; Q675W/+, Nod2Q675W/+; Q675W/Q675W, Nod2Q675W/Q675W. WT (post) indicated mice receiving Efm after DSS treatment.
Highlights.
REG3 proteins deplete Enterococcus faecium in gut microbiota of IBD patients
E. faecium SagA inhibits intestinal inflammation in a NOD2-dependent manner
NOD2 activated myeloid cells produce IL-1β to induce IL-22 from lymphoid cells
The NOD2 R702W variant impairs E. faecium-mediated protection in mice
ACKNOWLEDGEMENT
We would like to acknowledge NYU Grossman School of Medicine Flow Cytometry and Cell Sorting, Microscopy, Genome Technology, Rodent Genetic Engineering, and Histology Cores and Clinical Microbiology Laboratory for use of their instruments and technical assistance (supported by National Institutes of Health [NIH] grants P31CA016087, S10OD01058, and S10OD018338). This work was supported in part by NIH grants DK093668 (K.C.), AI121244 (K.C.), HL123340 (K.C.), AI130945 (K.C.), AI140754 (K.C.), DK124336 (K.C.), R01AI137336 (B.S.), R01AI140754 (B.S.), AI164154 (A.M.K-G.), AI173121 (A.M.K-G.), DK122698 (F.Y.), and DK132908 (M.L.); Faculty Scholar grant from the Howard Hughes Medical Institute (K.C.), Crohn’s & Colitis Foundation (K.C.), Kenneth Rainin Foundation (K.C. and H.C.H.), Bernard Levine Postdoctoral Research Fellowship in Immunology (Y.H.C.), the Charles H. Revson Senior Fellowships in Biomedical Science (Y.H.C.), and NYU Langone Health Antimicrobial-Resistant Pathogens Program (B.S., V.J.T., and A.P.).
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests
K.C. has received research support from Pfizer, Takeda, Pacific Biosciences, Genentech, and Abbvie. K.C. has consulted for or received an honoraria from Puretech Health, Genentech, and Abbvie. K.C. is an inventor on U.S. patent 10,722,600 and provisional patent 62/935,035 and 63/157,225. H.C.H. has received research support from Rise Therapeutics and LISCure Biosciences. U.S. patents PCT/US16/28836 (H.C.H.) and PCT/US2020/019038 (H.C.H. and M.E.G.) were obtained for the commercial use of SagA-engineered bacteria. Rise Therapeutics has licensed these patents to develop SagA-probiotics as therapeutics. J.A. reports consultancy fees, honoraria, or advisory board fees from Abbvie, Adiso, Bristol Myers Squibb, Janssen, Pfizer, Fresnius, and BioFire Diagnostics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Muniz LR, Knosp C, and Yeretssian G (2012). Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front Immunol 3, 310. 10.3389/fimmu.2012.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bevins CL, and Salzman NH (2011). Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9, 356–368. 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 3.Ramanan D, and Cadwell K (2016). Intrinsic Defense Mechanisms of the Intestinal Epithelium. Cell Host Microbe 19, 434–441. 10.1016/j.chom.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okumura R, Kurakawa T, Nakano T, Kayama H, Kinoshita M, Motooka D, Gotoh K, Kimura T, Kamiyama N, Kusu T, et al. (2016). Lypd8 promotes the segregation of flagellated microbiota and colonic epithelia. Nature 532, 117–121. 10.1038/nature17406. [DOI] [PubMed] [Google Scholar]
- 5.Lehotzky RE, Partch CL, Mukherjee S, Cash HL, Goldman WE, Gardner KH, and Hooper LV (2010). Molecular basis for peptidoglycan recognition by a bactericidal lectin. Proc Natl Acad Sci U S A 107, 7722–7727. 10.1073/pnas.0909449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cash HL, Whitham CV, Behrendt CL, and Hooper LV (2006). Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130. 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Fouts DE, Starkel P, Hartmann P, Chen P, Llorente C, DePew J, Moncera K, Ho SB, Brenner DA, et al. (2016). Intestinal REG3 Lectins Protect against Alcoholic Steatohepatitis by Reducing Mucosa-Associated Microbiota and Preventing Bacterial Translocation. Cell Host Microbe 19, 227–239. 10.1016/j.chom.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, and Hooper LV (2011). The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258. 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, DeMatteo RP, and Pamer EG (2008). Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455, 804–807. 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darnaud M, Dos Santos A, Gonzalez P, Augui S, Lacoste C, Desterke C, De Hertogh G, Valentino E, Braun E, Zheng J, et al. (2018). Enteric Delivery of Regenerating Family Member 3 alpha Alters the Intestinal Microbiota and Controls Inflammation in Mice With Colitis. Gastroenterology 154, 1009–1023 e1014. 10.1053/j.gastro.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Zhao D, Kim YH, Jeong S, Greenson JK, Chaudhry MS, Hoepting M, Anderson ER, van den Brink MR, Peled JU, Gomes AL, et al. (2018). Survival signal REG3alpha prevents crypt apoptosis to control acute gastrointestinal graft-versus-host disease. J Clin Invest 128, 4970–4979. 10.1172/JCI99261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu S, Balasubramanian I, Laubitz D, Tong K, Bandyopadhyay S, Lin X, Flores J, Singh R, Liu Y, Macazana C, et al. (2020). Paneth Cell-Derived Lysozyme Defines the Composition of Mucolytic Microbiota and the Inflammatory Tone of the Intestine. Immunity 53, 398–416 e398. 10.1016/j.immuni.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263. 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al. (2005). Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A 102, 18129–18134. 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koslowski MJ, Kubler I, Chamaillard M, Schaeffeler E, Reinisch W, Wang G, Beisner J, Teml A, Peyrin-Biroulet L, Winter S, et al. (2009). Genetic variants of Wnt transcription factor TCF-4 (TCF7L2) putative promoter region are associated with small intestinal Crohn’s disease. PLoS One 4, e4496. 10.1371/journal.pone.0004496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Courth LF, Ostaff MJ, Mailander-Sanchez D, Malek NP, Stange EF, and Wehkamp J (2015). Crohn’s disease-derived monocytes fail to induce Paneth cell defensins. Proc Natl Acad Sci U S A 112, 14000–14005. 10.1073/pnas.1510084112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, Leal T, Winter SE, Xavier RJ, and Hooper LV (2017). Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 357, 1047–1052. 10.1126/science.aal4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manichanh C, Borruel N, Casellas F, and Guarner F (2012). The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 9, 599–608. 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 19.Ni J, Wu GD, Albenberg L, and Tomov VT (2017). Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol 14, 573–584. 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong SY, and Cadwell K (2018). There was collusion: Microbes in inflammatory bowel disease. PLoS Pathog 14, e1007215. 10.1371/journal.ppat.1007215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knights D, Lassen KG, and Xavier RJ (2013). Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut 62, 1505–1510. 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho JH, and Abraham C (2007). Inflammatory bowel disease genetics: Nod2. Annu Rev Med 58, 401–416. 10.1146/annurev.med.58.061705.145024. [DOI] [PubMed] [Google Scholar]
- 24.Bonen DK, and Cho JH (2003). The genetics of inflammatory bowel disease. Gastroenterology 124, 521–536. 10.1053/gast.2003.50045. [DOI] [PubMed] [Google Scholar]
- 25.Kim YG, Kamada N, Shaw MH, Warner N, Chen GY, Franchi L, and Nunez G (2011). The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34, 769–780. 10.1016/j.immuni.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Pan Y, Yan R, Zeng B, Wang H, Zhang X, Li W, Wei H, and Liu Z (2015). Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol 16, 918–926. 10.1038/ni.3233. [DOI] [PubMed] [Google Scholar]
- 27.Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, and Kobayashi KS (2009). Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A 106, 15813–15818. 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramanan D, Tang MS, Bowcutt R, Loke P, and Cadwell K (2014). Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 41, 311–324. 10.1016/j.immuni.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchiando AM, Ramanan D, Ding Y, Gomez LE, Hubbard-Lucey VM, Maurer K, Wang C, Ziel JW, van Rooijen N, Nunez G, et al. (2013). A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe 14, 216–224. 10.1016/j.chom.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong SY, Coffre M, Ramanan D, Hines MJ, Gomez LE, Peters LA, Schadt EE, Koralov SB, and Cadwell K (2018). B Cell Defects Observed in Nod2 Knockout Mice Are a Consequence of a Dock2 Mutation Frequently Found in Inbred Strains. J Immunol 201, 1442–1451. 10.4049/jimmunol.1800014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Souza PR, Guimaraes FR, Sales-Campos H, Bonfa G, Nardini V, Chica JEL, Turato WM, Silva JS, Zamboni DS, and Cardoso CRB (2018). Absence of NOD2 receptor predisposes to intestinal inflammation by a deregulation in the immune response in hosts that are unable to control gut dysbiosis. Immunobiology 223, 577–585. 10.1016/j.imbio.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Robertson SJ, Geddes K, Maisonneuve C, Streutker CJ, and Philpott DJ (2016). Resilience of the intestinal microbiota following pathogenic bacterial infection is independent of innate immunity mediated by NOD1 or NOD2. Microbes Infect 18, 460–471. 10.1016/j.micinf.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Geddes K, Rubino S, Streutker C, Cho JH, Magalhaes JG, Le Bourhis L, Selvanantham T, Girardin SE, and Philpott DJ (2010). Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun 78, 5107–5115. 10.1128/IAI.00759-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, et al. (2013). NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 123, 700–711. 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, et al. (2004). NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 53, 1658–1664. 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bevins CL, Stange EF, and Wehkamp J (2009). Decreased Paneth cell defensin expression in ileal Crohn’s disease is independent of inflammation, but linked to the NOD2 1007fs genotype. Gut 58, 882–883; discussion 883–884. [PubMed] [Google Scholar]
- 37.VanDussen KL, Liu TC, Li D, Towfic F, Modiano N, Winter R, Haritunians T, Taylor KD, Dhall D, Targan SR, et al. (2014). Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology 146, 200–209. 10.1053/j.gastro.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Beelen Granlund A, Ostvik AE, Brenna O, Torp SH, Gustafsson BI, and Sandvik AK (2013). REG gene expression in inflamed and healthy colon mucosa explored by in situ hybridisation. Cell Tissue Res 352, 639–646. 10.1007/s00441-013-1592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogawa H, Fukushima K, Naito H, Funayama Y, Unno M, Takahashi K, Kitayama T, Matsuno S, Ohtani H, Takasawa S, et al. (2003). Increased expression of HIP/PAP and regenerating gene III in human inflammatory bowel disease and a murine bacterial reconstitution model. Inflamm Bowel Dis 9, 162–170. 10.1097/00054725-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Gao J, Zhao X, Hu S, Huang Z, Hu M, Jin S, Lu B, Sun K, Wang Z, Fu J, et al. (2022). Gut microbial DL-endopeptidase alleviates Crohn’s disease via the NOD2 pathway. Cell Host Microbe 30, 1435–1449 e1439. 10.1016/j.chom.2022.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Kim B, Wang YC, Hespen CW, Espinosa J, Salje J, Rangan KJ, Oren DA, Kang JY, Pedicord VA, and Hang HC (2019). Enterococcus faecium secreted antigen A generates muropeptides to enhance host immunity and limit bacterial pathogenesis. Elife 8. 10.7554/eLife.45343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YG, Shaw MH, Warner N, Park JH, Chen F, Ogura Y, and Nunez G (2011). Cutting edge: Crohn’s disease-associated Nod2 mutation limits production of proinflammatory cytokines to protect the host from Enterococcus faecalis-induced lethality. J Immunol 187, 2849–2852. 10.4049/jimmunol.1001854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Solache M, and Rice LB (2019). The Enterococcus: a Model of Adaptability to Its Environment. Clin Microbiol Rev 32. 10.1128/CMR.00058-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balish E, and Warner T (2002). Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol 160, 2253–2257. 10.1016/S0002-9440(10)61172-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seishima J, Iida N, Kitamura K, Yutani M, Wang Z, Seki A, Yamashita T, Sakai Y, Honda M, Yamashita T, et al. (2019). Gut-derived Enterococcus faecium from ulcerative colitis patients promotes colitis in a genetically susceptible mouse host. Genome Biol 20, 252. 10.1186/s13059-019-1879-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ben Braiek O, and Smaoui S (2019). Enterococci: Between Emerging Pathogens and Potential Probiotics. Biomed Res Int 2019, 5938210. 10.1155/2019/5938210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franz CM, Huch M, Abriouel H, Holzapfel W, and Galvez A (2011). Enterococci as probiotics and their implications in food safety. Int J Food Microbiol 151, 125–140. 10.1016/j.ijfoodmicro.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 48.Rangan KJ, Pedicord VA, Wang YC, Kim B, Lu Y, Shaham S, Mucida D, and Hang HC (2016). A secreted bacterial peptidoglycan hydrolase enhances tolerance to enteric pathogens. Science 353, 1434–1437. 10.1126/science.aaf3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedicord VA, Lockhart AAK, Rangan KJ, Craig JW, Loschko J, Rogoz A, Hang HC, and Mucida D (2016). Exploiting a host-commensal interaction to promote intestinal barrier function and enteric pathogen tolerance. Sci Immunol 1. 10.1126/sciimmunol.aai7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griffin ME, Espinosa J, Becker JL, Luo JD, Carroll TS, Jha JK, Fanger GR, and Hang HC (2021). Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 373, 1040–1046. 10.1126/science.abc9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang YC, Westcott NP, Griffin ME, and Hang HC (2019). Peptidoglycan Metabolite Photoaffinity Reporters Reveal Direct Binding to Intracellular Pattern Recognition Receptors and Arf GTPases. ACS Chem Biol 14, 405–414. 10.1021/acschembio.8b01038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, et al. (2016). Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 54, 139–148. 10.1016/j.ejca.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 53.Parikh A, Stephan AF, and Tzanakakis ES (2012). Regenerating proteins and their expression, regulation and signaling. Biomol Concepts 3, 57–70. 10.1515/bmc.2011.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang W, Wang Y, Lu Y, Zhu J, Tian X, Wu B, Du J, Cai W, and Xiao Y (2022). Reg4 protects against Salmonella infection-associated intestinal inflammation via adopting a calcium-dependent lectin-like domain. Int Immunopharmacol 113, 109310. 10.1016/j.intimp.2022.109310. [DOI] [PubMed] [Google Scholar]
- 55.Qi H, Wei J, Gao Y, Yang Y, Li Y, Zhu H, Su L, Su X, Zhang Y, and Yang R (2020). Reg4 and complement factor D prevent the overgrowth of E. coli in the mouse gut. Commun Biol 3, 483. 10.1038/s42003-020-01219-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bradley CR, and Fraise AP (1996). Heat and chemical resistance of enterococci. J Hosp Infect 34, 191–196. 10.1016/s0195-6701(96)90065-1. [DOI] [PubMed] [Google Scholar]
- 57.McHugh CP, Zhang P, Michalek S, and Eleazer PD (2004). pH required to kill Enterococcus faecalis in vitro. J Endod 30, 218–219. 10.1097/00004770-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 58.Maria R, Dutta SD, Thete SG, and AlAttas MH (2021). Evaluation of Antibacterial Properties of Organic Gutta-percha Solvents and Synthetic Solvents Against Enterococcus faecalis. J Int Soc Prev Community Dent 11, 179–183. 10.4103/jispcd.JISPCD_422_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luddin N, and Ahmed HM (2013). The antibacterial activity of sodium hypochlorite and chlorhexidine against Enterococcus faecalis: A review on agar diffusion and direct contact methods. J Conserv Dent 16, 9–16. 10.4103/0972-0707.105291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piewngam P, Zheng Y, Nguyen TH, Dickey SW, Joo HS, Villaruz AE, Glose KA, Fisher EL, Hunt RL, Li B, et al. (2018). Pathogen elimination by probiotic Bacillus via signalling interference. Nature 562, 532–537. 10.1038/s41586-018-0616-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, Lagoudas GK, Vatanen T, Fornelos N, Wilson R, et al. (2017). A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med 9, 103. 10.1186/s13073-017-0490-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. (2015). Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18, 489–500. 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. (2019). Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4, 293–305. 10.1038/s41564-018-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, and Vijay-Kumar M (2012). Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 7, e44328. 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Forster SC, Clare S, Beresford-Jones BS, Harcourt K, Notley G, Stares MD, Kumar N, Soderholm AT, Adoum A, Wong H, et al. (2022). Identification of gut microbial species linked with disease variability in a widely used mouse model of colitis. Nat Microbiol 7, 590–599. 10.1038/s41564-022-01094-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moon C, Baldridge MT, Wallace MA, Burnham D,CA, Virgin HW, and Stappenbeck TS (2015). Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521, 90–93. 10.1038/nature14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kommineni S, Bretl DJ, Lam V, Chakraborty R, Hayward M, Simpson P, Cao Y, Bousounis P, Kristich CJ, and Salzman NH (2015). Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 526, 719–722. 10.1038/nature15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Z, Downing S, and Tzanakakis ES (2019). Four Decades After the Discovery of Regenerating Islet-Derived (Reg) Proteins: Current Understanding and Challenges. Front Cell Dev Biol 7, 235. 10.3389/fcell.2019.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edwards JA, Tan N, Toussaint N, Ou P, Mueller C, Stanek A, Zinsou V, Roudnitsky S, Sagal M, Dresner L, et al. (2020). Role of regenerating islet-derived proteins in inflammatory bowel disease. World J Gastroenterol 26, 2702–2714. 10.3748/wjg.v26.i21.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Teng F, Kawalec M, Weinstock GM, Hryniewicz W, and Murray BE (2003). An Enterococcus faecium secreted antigen, SagA, exhibits broad-spectrum binding to extracellular matrix proteins and appears essential for E. faecium growth. Infect Immun 71, 5033–5041. 10.1128/IAI.71.9.5033-5041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, and Medzhitov R (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241. 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 72.Kernbauer E, Ding Y, and Cadwell K (2014). An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98. 10.1038/nature13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ferrand A, Al Nabhani Z, Tapias NS, Mas E, Hugot JP, and Barreau F (2019). NOD2 Expression in Intestinal Epithelial Cells Protects Toward the Development of Inflammation and Associated Carcinogenesis. Cell Mol Gastroenterol Hepatol 7, 357–369. 10.1016/j.jcmgh.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mukherjee T, Hovingh ES, Foerster EG, Abdel-Nour M, Philpott DJ, and Girardin SE (2019). NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys 670, 69–81. 10.1016/j.abb.2018.12.022. [DOI] [PubMed] [Google Scholar]
- 75.Trindade BC, and Chen GY (2020). NOD1 and NOD2 in inflammatory and infectious diseases. Immunol Rev 297, 139–161. 10.1111/imr.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neil JA, Matsuzawa-Ishimoto Y, Kernbauer-Holzl E, Schuster SL, Sota S, Venzon M, Dallari S, Galvao Neto A, Hine A, Hudesman D, et al. (2019). IFN-I and IL-22 mediate protective effects of intestinal viral infection. Nat Microbiol 4, 1737–1749. 10.1038/s41564-019-0470-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, and Mizoguchi A (2008). IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 118, 534–544. 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chu H, Khosravi A, Kusumawardhani IP, Kwon AH, Vasconcelos AC, Cunha LD, Mayer AE, Shen Y, Wu WL, Kambal A, et al. (2016). Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 352, 1116–1120. 10.1126/science.aad9948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu WH, Kim M, Chang LC, Assie A, Saldana-Morales FB, Zegarra-Ruiz DF, Norwood K, Samuel BS, and Diehl GE (2022). Interleukin-1beta secretion induced by mucosa-associated gut commensal bacteria promotes intestinal barrier repair. Gut Microbes 14, 2014772. 10.1080/19490976.2021.2014772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seo SU, Kuffa P, Kitamoto S, Nagao-Kitamoto H, Rousseau J, Kim YG, Nunez G, and Kamada N (2015). Intestinal macrophages arising from CCR2(+) monocytes control pathogen infection by activating innate lymphoid cells. Nat Commun 6, 8010. 10.1038/ncomms9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, Putzel GG, Kabata H, Kelsen JR, Baldassano RN, et al. (2019). Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409. 10.1038/s41586-019-1082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ebbo M, Crinier A, Vely F, and Vivier E (2017). Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol 17, 665–678. 10.1038/nri.2017.86. [DOI] [PubMed] [Google Scholar]
- 83.Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, and Mizoguchi E (2018). Clinical importance of IL-22 cascade in IBD. J Gastroenterol 53, 465–474. 10.1007/s00535-017-1401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Swanson KV, Deng M, and Ting JP (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19, 477–489. 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang Y, Xu W, and Zhou R (2021). NLRP3 inflammasome activation and cell death. Cell Mol Immunol 18, 2114–2127. 10.1038/s41423-021-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183, 787–791. 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yao X, Zhang C, Xing Y, Xue G, Zhang Q, Pan F, Wu G, Hu Y, Guo Q, Lu A, et al. (2017). Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat Commun 8, 1896. 10.1038/s41467-017-01917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu Q, and Kanneganti TD (2017). Cutting Edge: Distinct Regulatory Mechanisms Control Proinflammatory Cytokines IL-18 and IL-1beta. J Immunol 198, 4210–4215. 10.4049/jimmunol.1700352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. (2003). Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 278, 5509–5512. 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 90.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603. 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 91.Murano T, Okamoto R, Ito G, Nakata T, Hibiya S, Shimizu H, Fujii S, Kano Y, Mizutani T, Yui S, et al. (2014). Hes1 promotes the IL-22-mediated antimicrobial response by enhancing STAT3-dependent transcription in human intestinal epithelial cells. Biochem Biophys Res Commun 443, 840–846. 10.1016/j.bbrc.2013.12.061. [DOI] [PubMed] [Google Scholar]
- 92.Loonen LM, Stolte EH, Jaklofsky MT, Meijerink M, Dekker J, van Baarlen P, and Wells JM (2014). REG3gamma-deficient mice have altered mucus distribution and increased mucosal inflammatory responses to the microbiota and enteric pathogens in the ileum. Mucosal Immunol 7, 939–947. 10.1038/mi.2013.109. [DOI] [PubMed] [Google Scholar]
- 93.Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. (2013). Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711. 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee JY, Tsolis RM, and Baumler AJ (2022). The microbiome and gut homeostasis. Science 377, eabp9960. 10.1126/science.abp9960. [DOI] [PubMed] [Google Scholar]
- 95.Shouval DS, Biswas A, Kang YH, Griffith AE, Konnikova L, Mascanfroni ID, Redhu NS, Frei SM, Field M, Doty AL, et al. (2016). Interleukin 1beta Mediates Intestinal Inflammation in Mice and Patients With Interleukin 10 Receptor Deficiency. Gastroenterology 151, 1100–1104. 10.1053/j.gastro.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim D, Kim YG, Seo SU, Kim DJ, Kamada N, Prescott D, Chamaillard M, Philpott DJ, Rosenstiel P, Inohara N, and Nunez G (2016). Nod2-mediated recognition of the microbiota is critical for mucosal adjuvant activity of cholera toxin. Nat Med 22, 524–530. 10.1038/nm.4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martin PK, Marchiando A, Xu R, Rudensky E, Yeung F, Schuster SL, Kernbauer E, and Cadwell K (2018). Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol 3, 1131–1141. 10.1038/s41564-018-0229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matsuzawa-Ishimoto Y, Yao X, Koide A, Ueberheide BM, Axelrad JE, Reis BS, Parsa R, Neil JA, Devlin JC, Rudensky E, et al. (2022). The gammadelta IEL effector API5 masks genetic susceptibility to Paneth cell death. Nature 610, 547–554. 10.1038/s41586-022-05259-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, and Gregory Caporaso J (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lozupone C, Lladser ME, Knights D, Stombaugh J, and Knight R (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J 5, 169–172. 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vazquez-Baeza Y, Pirrung M, Gonzalez A, and Knight R (2013). EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2, 16. 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, and Peddada SD (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 26, 27663. 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheng S, McCleskey FK, Gress MJ, Petroziello JM, Liu R, Namdari H, Beninga K, Salmen A, and DelVecchio VG (1997). A PCR assay for identification of Enterococcus faecium. J Clin Microbiol 35, 1248–1250. 10.1128/jcm.35.5.1248-1250.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Low AJ, Koziol AG, Manninger PA, Blais B, and Carrillo CD (2019). ConFindr: rapid detection of intraspecies and cross-species contamination in bacterial whole-genome sequence data. PeerJ 7, e6995. 10.7717/peerj.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chaumeil PA, Mussig AJ, Hugenholtz P, and Parks DH (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wick RR, Judd LM, Gorrie CL, and Holt KE (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13, e1005595. 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J Mol Biol 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 110.Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Csardi G, and Nepusz T The igraph software package for complex network research.
- 112.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, and Speleman F (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, RESEARCH0034. 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dallari S, Heaney T, Rosas-Villegas A, Neil JA, Wong SY, Brown JJ, Urbanek K, Herrmann C, Depledge DP, Dermody TS, and Cadwell K (2021). Enteric viruses evoke broad host immune responses resembling those elicited by the bacterial microbiome. Cell Host Microbe 29, 1014–1029 e1018. 10.1016/j.chom.2021.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jang KK, Kaczmarek ME, Dallari S, Chen YH, Tada T, Axelrad J, Landau NR, Stapleford KA, and Cadwell K (2022). Variable susceptibility of intestinal organoid-derived monolayers to SARS-CoV-2 infection. PLoS Biol 20, e3001592. 10.1371/journal.pbio.3001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information for IBD patients, Related to Figures 1, 2, 4, S1, and S6. M, male; F, female; CD, Crohn’s disease; UC, ulcerative colitis; CDAI, Crohn’s disease activity index; N/A, not available.
Table S3. Information for Enterococcus isolates from NIBD and IBD pateints, Related to Figure S1.
Table S4. Histological findings of colonic sections from mice receiving DSS, Related to Figures 3, 7, and S3. WT, wild type; Q675W/+, Nod2Q675W/+; Q675W/Q675W, Nod2Q675W/Q675W. WT (post) indicated mice receiving Efm after DSS treatment.
Data Availability Statement
16S rRNA sequencing and whole genome sequencing data are deposited in the NCBI Sequence Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers for the datasets are listed in the key resources table.
This study does not report original codes.
Any additional information required to reanalyze the data reported from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-Human Reg1A antibody | R&D Systems | Cat#: MAB4937; RRID: AB_2102139; Clone: 431202 |
| Mouse anti-Human Reg3A antibody | R&D Systems | Cat#: 62257S; RRID: AB_1964697; Clone: 512124 |
| Rabbit anti-REG3G antibody | Abcam | Cat#: ab233480 |
| Rabbit anti-REG4 antibody | Abcam | Cat#: ab255820; Clone: EPR22810-327 |
| Rabbit anti-SagA antibody | Dr. Hang (Scripps Research Institutes)41 | |
| Mouse anti-IL-22 antibody | R&D Systems | Cat#: MAB782; RRID: AB_2295882; Clone: 142938 |
| Mouse anti-NOD2 antibody | Invitrogen | Cat#: MA1-16611; RRID: AB_568643; Clone: 2D9 |
| Mouse anti-β-Actin antibody | Sigma | Cat#: A5441; RRID: AB_476744; Clone: AC-15 |
| IRDye 680RD Goat anti-Rabbit IgG Secondary Antibody | LI-COR | Cat# 925-68071; RRID: AB_2721181; |
| IRDye 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR | Cat#: 925-32210; RRID: AB_2687825; |
| IRDye 800CW Goat anti-Rat IgG Secondary Antibody | LI-COR | Cat#: 925-32219; RRID: AB_2721932; |
| PerCP/Cyanine5.5 anti-mouse/human CD11b Antibody | Biolegend | Cat#: 101228; RRID: AB_893232; Clone: M1/70 |
| PerCP/Cyanine5.5 anti-mouse CD11c Antibody | Biolegend | Cat#: 117328; RRID: AB_2129641; Clone: N418 |
| PerCP/Cyanine5.5 anti-mouse Ly-6G/Ly-6C (Gr-1) Antibody | Biolegend | Cat#: 108428; RRID: AB_893558; Clone: RB6-8C5 |
| PerCP/Cyanine5.5 anti-mouse TER-119/Erythroid Cells Antibody | Biolegend | Cat#: 116228; RRID: AB_893636; Clone: TER-119 |
| PerCP/Cyanine5.5 anti-mouse NK-1.1 Antibody | Biolegend | Cat#: 108728; RRID: AB_2132705; Clone: PK136 |
| PE anti-T-bet Antibody | Biolegend | Cat#: 644810; RRID: AB_2200542; Clone: 4B10 |
| PE- Cyanine7 anti-mouse/human CD44 antibody | Biolegend | Cat#: 103030; RRID: AB_830787; Clone: IM7 |
| APC/Cyanine7 anti-mouse CD4 Antibody | Biolegend | Cat#: 100414; RRID: AB_312699; Clone: GK1.5 |
| Brilliant Violet 570 anti-mouse CD62L Antibody | Biolegend | Cat#: 104433; RRID: AB_10900262; Clone: MEL-14 |
| Brilliant Violet 605 anti-mouse CD127 (IL-7Ra) Antibody | Biolegend | Cat#: 135041; RRID: AB_2572047; Clone: A7R34 |
| Brilliant Violet 650 anti-mouse NK-1.1 Antibody | Biolegend | Cat#: 108736; RRID: AB_2563159; Clone: PK136 |
| Alexa Fluor 700 anti-mouse CD8a Antibody | Biolegend | Cat#: 100730; RRID: AB_493703; Clone: 53-6.7 |
| Brilliant Violet 711 anti-mouse TCR β chain Antibody | Biolegend | Cat#: 109243; RRID: AB_2629564; Clone: H57-597 |
| PerCP/Cyanine5.5 anti-mouse CD19 | eBioscience | Cat#: 45-0193-82; RRID: AB_1106999; Clone: 1D3 |
| PE anti-mouse IL-22 Antibody | eBioscience | Cat#: 12-7221-82; RRID: AB_10597428; Clone: 1H8PWSR |
| APC anti-mouse FOXP3 Antibody | eBioscience | Cat#: 17-5773-82; RRID: AB_469457; Clone: FJK-16s |
| PE-CF594 anti-mouse γδ T-Cell Receptor Antibody | BD Bioscience | Cat#: 563532; RRID: AB_2661844; Clone: GL3 |
| BUV395 anti-mouse CD45 Antibody | BD Bioscience | Cat#: 564279; RRID: AB_2651134; Clone: 30-F11 |
| BV421 anti-mouse RORgt Antibody | BD Bioscience | Cat#: 562894; RRID: AB_2687545; Clone: Q31-378 |
| BV421 anti-mouse IL-10 Antibody | BD Bioscience | Cat#: 566295; RRID: AB_2739668; Clone: JES5-16E3 |
| Alexa Fluor 488 anti-mouse GATA3 Antibody | BD Bioscience | Cat#: 560163; RRID: AB_1645302; Clone: L50-823 |
| Bacterial strains | ||
| Enterococcus faecalis OG1RF | Dr. Hang (Scripps Research Institutes), originally acquired from ATCC 47077. | N/A |
| Enterococcus faecium Com15 | Dr. Hang (Scripps Research Institutes), originally acquired Dr. Gilmore (Harvard Medical School). | N/A |
| Salmonella Typhimurium SL1344 | Gift from Dr. Littman (NYU) | N/A |
| Lactococcus lactis thyA auxotroph | Dr. Hang (Scripps Research Institutes)50, generated in collaboration with Rise Therapeutics | N/A |
| Lactococcus lactis thyA expressing wild-type SagA | Dr. Hang (Scripps Research Institutes)50, generated in collaboration with Rise Therapeutics | N/A |
| Lactococcus lactis thyA expressing catalytic mutant SagA | Dr. Hang (Scripps Research Institutes)50, generated in collaboration with Rise Therapeutics | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bacto brain heart infusion | BD | Cat#: 237500 |
| Sabouraud dextrose broth | Millipore | Cat#: S3306 |
| Nutrient Broth | Millipore | Cat#: 70122 |
| BsaHI | NEB | Cat#: R0556S |
| Dulbecco’s Modified Eagle’s Medium | Corning | Cat#: 10-017-CV |
| Fetal bovine serum | Peak Serum | Cat#: PS-FB2 |
| Penicillin-Streptomycin Solution | Corning | Cat#: 30-002-CI |
| Normicin | InvivoGen | Cat#: ant-nr |
| Zeocin | InvivoGen | Cat#: ant-zn |
| Blastocidin | InvivoGen | Cat#: ant-bl |
| Trypsin-EDTA | Gibco | Cat#: 25200056 |
| M17 Broth | BD | Cat#: 218561 |
| M17 Agar | BD | Cat#: 218571 |
| Bile esculin azide agar | Millipore | Cat#: 06105 |
| HiCrome Enterococcus faecium Agar Base | HIMEDIA | Cat#: M1580 |
| Enterococcus faecium Selective Supplement | HIMEDIA | Cat#: FD226 |
| Lactose | Sigma-Aldrich | Cat#: 17814 |
| Thymidine | Sigma-Aldrich | Cat#: T9250 |
| Phosphate-buffer saline | Corning | Cat#: 21040CV |
| Triton X-100 | Sigma-Aldrich | Cat#: T8787 |
| Glycerol | Sigma-Aldrich | Cat#: G5516 |
| Halt Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher | Cat#: 78440 |
| Bolt 4-12% Bis-Tris Plus Gels | Invitrogen | Cat#: NW04120BOX |
| Intercept (TBS) Blocking Buffer | LI-COR | Cat#: 927-60003 |
| Trypticase soy agar with 5% sheep blood | Henry Schein | Cat#: 9873908 |
| HEK-Blue detection medium | InvivoGen | Cat#: hb-det2 |
| Dextran sulfate sodium | TdB Consultancy | Cat#: DB001 |
| Ampicillin | Amercian Bioanalytical | Cat#: 69-52-3 |
| Streptomycin | Sigma-Aldrich | Cat#: S6501 |
| Low melting point agar | Promega | Cat#: V2111 |
| 10% Formalin | Thermo Fisher | Cat#: SF99-4 |
| Roswell Park Memorial Institute 1640 Medium | Gibco | Cat#: 61870127 |
| L-glutamine | Corning | Cat#: 25-005-CI |
| HEPES | Corning | Cat#: 25-060-CI |
| Minimum Essential Medium Nonessential Amino Acids | Corning | Cat#: 25-025-CI |
| Sodium pyruvate | Corning | Cat#: 25-000-CI |
| β-mercaptoethanol | Gibco | Cat#: 21985023 |
| Hank’s Balanced Salt Solution | Gibco | Cat#: 14175103 |
| DL-dithiothreitol | Sigma Aldrich | Cat#: D0632 |
| Collagenase | Sigma Aldrich | Cat#: C2139 |
| Deoxyribonuclease I | Sigma Aldrich | Cat#: DN25 |
| Percoll | Sigma-Aldrich | Cat#: P1644 |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | eBioscience | Cat#: 00-4975-93 |
| Bovine Serum Albumin | Thermo Fisher | Cat#: BP1600-100 |
| Fixation Buffer | Biolegend | Cat#: 420801 |
| Intracellular Staining Permeabilization Wash Buffer | Biolegend | Cat#: 421002 |
| Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher | Cat#: 00-5523-00 |
| Zombie UV Fixable Viability Kit | Biolegend | Cat#: 423108 |
| Critical commercial assays | ||
| 10 ml syringe | BD | Cat#: 302995 |
| Gauze pad | 4MD Medical | Cat#: PROP157025 |
| Millex-GS Syringe Filter Unit, 0.22 μm | Millipore | Cat#: SLGL0250S |
| Nalgene Rapid-Flow Sterile Disposable Filter Units with PES Membranes | Thermo Fisher | Cat#: 567-0020 |
| Human Reg3A DuoSet ELISA | R&D systems | Cat#: DY5940-05 |
| Mouse Lipocalin-2/NGAL DuoSet ELISA | R&D systems | Cat#: DY1857-05 |
| Mouse IL-22 DuoSet ELISA | R&D systems | Cat#: DY582-05 |
| Mouse IL-18 DuoSet ELISA | R&D systems | Cat#: DY7625-05 |
| Mouse IL-1 beta/IL-1 F2 DuoSet ELISA | R&D systems | Cat#: DY401-05 |
| DNeasy PowerSoil Pro Kit | Qiagen | Cat#: 47016 |
| MagMAX DNA Multi-Sample Ultra 2.0 kit | Thermo Fisher | Cat#: A36570 |
| RNeasy Mini Kit | Qiagen | Cat#: 74106 |
| RNase-Free DNase Set | Qiagen | Cat#: 79256 |
| 70 μm nylon mesh | ELKO Filtering | Cat#: 03-70/33 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat#: 4368814 |
| LightCycler 480 SYBR Green I Master | Roche | Cat#: 04707516001 |
| TOPO TA Cloning Kit | Invitrogen | Cat#: 450002 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Nod2−/− | Jackson Laboratory | Cat#: 005763 |
| Mouse Nod2fl/fl | Dr. Keestra-Gounder (University of Colorado Anschutz Medical Campus) | N/A |
| Mouse: Il22ra1fl/fl | Dr. Sergei Koralov (New York University Grossman school of medicine), originally acquired from Jackson Laboratory (Cat#: 031003) | N/A |
| Mouse: LysM-Cre | Jackson Laboratory | Cat#: 004781 |
| Mouse: Villin-Cre | Jackson Laboratory | Cat#: 004586 |
| Mouse: Il1r1 | Jackson Laboratory | Cat#: 003245 |
| Mouse Nlrp3 | Jackson Laboratory | Cat#: 021302 |
| Mouse: Nod2Q675W/Q675W | This study | N/A |
| Cell line: HEK-Blue Null2 | InvivoGen | Cat#: hkb-hnod2 |
| Cell line: HEK-Blue NOD2 | InvivoGen | Cat#: hkb-null2 |
| Deposited data | ||
| 16S rRNA sequencing data related to Figure 2 | This study | BioProject: PRJNA915760 |
| Whole genome sequencing data related to Figure S1 | This study | BioProject: PRJNA978059 |
| 16S rRNA sequencing data related to Figure S6 | This study | BioProject: PRJNA977252 |
| Software and Algorithms | ||
| Prism 9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | BD | https://www.flowjo.com |
| NCBI Gene and Protein Data Base | NCBI | http://www.ncbi.nlm.nih.gov |
| Nucleotide blast | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Blastx | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| Fiji/ImageJ | NIH | https://fiji.sc |