

Abstract
Objectives
Type I interferon (IFN) plays a role in the pathogenesis of systemic lupus erythematosus (SLE), but insufficient attention has been directed to the differences in IFN responses between ancestral populations. Here, we explored the expression of the interferon gene signatures (IGSs) in SLE patients of European ancestry (EA) and Asian ancestry (AsA).
Methods
We used gene set variation analysis with multiple IGS encompassing the response to both type 1 and type 2 IFN in isolated CD14+ monocytes, CD19+B cells, CD4+T cells and Natural Killer (NK) cells from patients with SLE stratified by self-identified ancestry. The expression of genes upstream of the IGS and influenced by lupus-associated risk alleles was also examined. Lastly, we employed machine learning (ML) models to assess the most important features classifying patients by disease activity.
Results
AsA patients with SLE exhibited greater enrichment in the IFN core and IFNA2 IGS compared with EA patients in all cell types examined and, in the presence and absence of autoantibodies. Overall, AsA patients with SLE demonstrated higher expression of genes upstream of the IGS than EA counterparts. ML with feature importance analysis indicated that IGS expression in NK cells, anti-dsDNA, complement levels and AsA status contributed to disease activity.
Conclusions
AsA patients with SLE exhibited higher IGS than EA patients in all cell types regardless of autoantibody status, with enhanced expression of genetically associated genes upstream of the IGS potentially contributing. AsA, along with the IGS in NK cells, anti-dsDNA and complement, independently influenced SLE disease activity.
Keywords: Autoantibodies; Autoimmune Diseases; Lupus Erythematosus, Systemic; Polymorphism, Genetic
WHAT IS ALREADY KNOWN ON THIS TOPIC
Type 1 interferon (IFN) plays an important role in systemic lupus erythematosus (SLE) pathogenesis.
The expression of the interferon gene signature (IGS) differs in lupus patients of different ancestries.
Regulation of IFN in patients of East Asian ancestry has not been examined.
WHAT THIS STUDY ADDS
The expression of the IGS is higher in lupus patients of Asian ancestry compared with those of European ancestry and cannot be explained by differences in autoantibody levels.
Overexpression of genetically regulated genes upstream of the IGS may contribute to the enhanced IGS in lupus patients of Asian ancestry.
The IGS along with serologic abnormalities and Asian ancestry independently contribute to disease activity in patients with lupus.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Understanding the ancestral contribution to the IFN response could contribute to more precise use of IFN antagonists, such as anifrolumab.
Better knowledge of the various contributors to disease activity could provide new insights into disease pathogenesis and the more effective use of targeted therapies.
Introduction
Systemic lupus erythematosus (SLE) is a multisystem female-dominant autoimmune disease that is observed more frequently among patients of non-European ancestry.1 2 Various autoantibodies, such as anti-dsDNA, anti-RNP (ribonucleoprotein), anti-Smith and anti-SSA, as well as type 1 interferon (IFN) contribute to disease pathogenesis.3 4
The type I IFN family includes 13 subtypes of IFNα as well as IFNβ, IFNω and IFNκ, whereas IFNγ comprises the sole member of the type II IFN family. Both type I and II IFNs activate distinct canonical signalling pathways that include the JAK kinases and the STAT family of transcription factors.5 Elevated production of IFNs in SLE results in the increased expression of downstream target genes collectively referred to as the interferon gene signature (IGS), and include groups of genes that are uniquely induced by individual IFN species, as well as a large number of genes induced by all type 1 and type 2 subtypes.1 Because it is difficult to measure the serum levels of type 1 IFN accurately, IGSs are often used as proxies to estimate IFN activity.6 However, there is no uniform IGS and many different genes have been used to create different signatures.7–11
Although the IGS is found in a majority of patients with SLE, the frequency and magnitude of expression varies among patients of different ancestral backgrounds. For example, the IGS is more frequently observed in lupus patients of African ancestry (AA) compared with those of European ancestry (EA) and is associated with the elevated production of autoantibodies to RNA-binding proteins, including anti-RNP, anti-Smith and anti-SSA (Ro).3 SLE patients of African ancestry have also been shown to harbour a higher burden of single nucleotide polymorphism (SNP) diversity regulating the IFN pathway compared with their EA counterparts.12 13 Notably, SNP associations mapping to upstream and downstream IFN molecules are also present in SLE patients of Asian ancestry (AsA), but the differential regulation of the IGS in AsA patients with SLE has not been thoroughly explored.12 14 15
This study aimed to differentiate the SLE-driven IFN response between Asian and European ancestral populations. Differences in the IFN response could affect clinical manifestations as well as response to treatment. Since the IFN response is dynamic and is dependent on context and cell type,7 we also explored the differences in the IGS among multiple cell types, including CD14+ monocytes, CD19+B cells, CD4+T cells and Natural Killer (NK) cells between AsA and EA patients with SLE.
The results indicate that AsA patients with SLE have a higher IGS than EA patients and this is manifest in all cell types. Although anti-RNP is associated with an increased IGS in AsA patients with SLE, augmented expression is also noted in AsA patients lacking detectable autoantibodies.3 16 An increased expression of genes upstream of the IGS suggests that genetic associations are more contributory to the enhanced IGS associated with SLE in AsA patients.
Materials and methods
Patient and public involvement
The patient data are from a publicly available dataset (GSE164457). Patients were recruited at UCSF as part of the CLUES database and methods of cell isolation are detailed in the original report.17 Ancestry was assessed using a structured verbal interview. Additionally, if the patient was of Asian ancestry, their ethnicity and country of origin were also assessed using the structured verbal interview. The patients and the public were not involved in the design of this report.
Gene expression datasets
All datasets are summarised in online supplemental table S1.
rmdopen-2023-003475supp002.xlsx (38.5KB, xlsx)
Quality control and data normalisation
Microarrary data (GSE49454, GSE39088, Affymetrix and GSE88884; Illumina) were processed as previously described.18 Briefly, unnormalised arrays were inspected for visual artefacts or poor RNA hybridisation using QC plots. Datasets were annotated using their native chip definition files. Probes missing gene annotation data were removed. Raw data (CEL files) from the Affymetrix platform were background corrected and normalised using GC robust multiarray average or robust multichip average algorithms, whereas raw data files from the Illumina chip were read and normalised using neqc (limma R package). RNA-Seq data (GSE164457) were processed from FASTQ files as previously described.19 Principal component analysis was used to inspect the raw data files from each dataset for outliers (online supplemental figure S1). All log2-transformed data were formatted into R expression set objects (esets).
rmdopen-2023-003475supp001.pdf (2.4MB, pdf)
Differentially expressed gene (DEG) analysis
The differential gene expression between Asian and European patients with SLE in GSE164457 was carried out using the DESeq2 pipeline. P values were adjusted for multiple hypothesis testing using the Benjamini-Hochberg correction, which resulted in a false discovery rate (FDR) for each gene. A FDR <0.2 was employed to avoid falsely excluding genes of interest. A fold change >1.5 was further used to filter only the most significant DEGs.
Generation of IFN gene signatures
Multiple gene signatures for type I and type II IFN, as well as IL12 and TNF, have been previously described.7–11 Additional IFN gene signatures were generated using the Singscore R package. This method, which uses rank-based statistics to score gene impact (high vs low expression) at the single sample level, was applied to genes collated from the seven IFN modules derived from Catalina et al (IFN Core, IFNA2, IFNB1, IFNW1, IFNG, IL12 and TNF) to determine the most impactful genes contributing to the IGS in each cell type (CD14, CD19, CD4 and NK) included in GSE164457. All gene sets are listed in online supplemental tables S2–S4.
Identification of IGS upstream genes
Using all SLE-associated single nucleotide polymorphisms (SNPs) from genome-wide association studies (GWAS) and immunochip analyses, we identified the most likely genetically regulated genes in SLE patients of EA, AsA and African ancestry (AA), as previously described.15 20 From this, we selected all genes upstream of IFN production, as well as those related to IFN signalling and the IFN signalling pathways. The final list of genetically regulated IGS upstream genes can be seen in online supplemental table S4.
Gene set variation analysis (GSVA)
The R/Bioconductor package GSVA (V.1.25.0) was used as a non-parametric, unsupervised method to estimate the variation in enrichment of predefined gene sets in RNA-seq dataset samples as previously described. In brief, a matrix of log2-transformed gene expression values for each sample and predefined gene sets were used as inputs for the GSVA algorithm. Then enrichment scores (GSVA scores) for each gene set were calculated using a Kolmogorov-Smirnoff–like random walk statistic. GSVA scores for each patient and control were calculated and normalised to scores between −1 (no enrichment) and +1 (enriched). Significance of gene set enrichment between cohorts was calculated using a Welch’s t-test, and p value <0.05 was considered significant.
Machine learning (ML) and generation of complex heatmaps
K-means clustering and patient classification by SLE disease activity index (SLEDAI) was carried out in a Jupyter notebook using the sklearn package. Data were organised using the NumPy and Pandas libraries. For k-means clustering, the optimal number of clusters was determined by the elbow method (online supplemental figure S2) and heatmaps were generated using the Complex Heatmap R package.
For patient classification by SLEDAI, the data were divided into a training and testing set using a 70:30 train/test split. Collinear features were removed and signature C11 as well as the IFNA2 and IFNG modules in all four cell types were selected to train the models. We included the following clinical variables as features: sex, ancestry, age, C3 level, anti-dsDNA titre, anti-RNP titre and anti-SM titre. The autoantibodies and C3 were log-normalised. Feature importance was calculated by Gini Feature Importance score. Anti-SSA titres were excluded for negligible feature importance. Nine different ML methods were trained using 10-fold cross-validation. The Decision Tree (DTR) model was selected since it had the highest test accuracy and the highest area under the curve (AUC) score of the receiver operating characteristic (ROC) curves among all nine models. Visualisations for patient classification by SLEDAI, as well as the elbow plots, were created using the matplotlib package in Python.
Statistical analysis and data visualisation
Data processing and analysis were conducted within the R programming platform using relevant Bioconductor packages. Data visualisation and statistical analyses were conducted using GraphPad Prism V.9.4.1. Unless otherwise noted, significance for the violin plots was determined using a t-test with Welch’s correction and the significance for the bar plots was determined using a χ2 test of proportion.
Results
Contribution of ancestry to IGS expression
RNA-seq data derived from 120 patients enrolled in the CLUES (GSE164457) dataset were used to examine the enrichment of multiple IGS in a trans-ancestral patient cohort.17 In general, EA and AsA patients exhibited similar clinical characteristics, but EA patients tended to be older and AsA patients were significantly more likely to be positive for anti-SSA autoantibody and to be receiving mycophenolate (online supplemental table S5). Notably, there was no difference in mean disease activity as measured by SLEDAI between SLE patients of AsA or EA.
GSVA was applied to RNA-Seq data from each of the cell types, including CD14 monocytes, CD19 B cells, CD4 T cells and NK cells, to determine the enrichment of previously reported IGS that captured genes regulated by both type 1 and 2 IFN (IFN core) as well as signatures using genes uniquely regulated by IFN α2, β1, ω and γ, and as controls, the inflammatory cytokines IL12 and TNF (tumour necrosis factor).7 Stable k-means clustering of the GSVA scores separated the samples into four distinct patient subsets. Each subset was designated by an arbitrary colour (red, green, blue and salmon) and visualised in a complex heatmap showing patient-by-patient variation of IGS expression and the clinical parameters associated with each sample (figure 1A).
Figure 1.

Patient subsets are defined by differential IGS enrichment. (A) K-means clustering of IFN GSVA enrichment scores from 113 patients with SLE across four cell types. The complex heatmap shows patient-by-patient variation in seven IGSs along with ancestry, age, SLEDAI, medications and autoantibody titre. Hydroxychloroquine is abbreviated as HCQ and mycophenolate mofetil is abbreviated as MMF. The distribution of AsA and EA patients in each subset is also shown. (B) Violin plots of GSVA scores of the seven different interferon modules in each of the four patient subsets within monocytes. Significance measures are as follows: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 determined by Welch’s t-test. The red subset contains 32 patients, the green subset contains 26 patients, the blue subset contains 37 patients and the salmon subset contains 18 patients. (C) Bar plots showing the percentage of patients in each subset that are GSVA positive for the various IGSs within monocytes. Statistical significance is shown using a χ2 test of proportion. AsA, Asian ancestry; EA, European ancestry; GSVA, gene set variation analysis; IFN, interferon; IGS, interferon gene signature.
Lower levels of IGS enrichment were observed in the red patient subset, whereas patients in the blue subset showed consistently higher IGS expression across all cell types. Overall, higher IGS expression was also seen in the green and salmon subsets compared with red, particularly of the IFN core and IFNA2 signatures, except in B cells (salmon) and NK cells (green) (figure 1A). Patients in the red subset (lowest IGS expression) were predominantly EA (24/32, 75%) and had significantly lower disease activity as measured by SLEDAI (mean 1.75±2.23) (online supplemental table S6). This subset also contained significantly fewer autoantibody-positive patients. In contrast, the majority of the blue and salmon subsets were composed of AsA patients (24/37, 64.9% and 13/18, 72.2%, respectively) who had demonstrably higher IGS expression in at least three of the four cell types and were associated with significantly higher disease activity, especially in the salmon subset (mean SLEDAI 4.06±3.98), and more frequently expressed anti-dsDNA, anti-RNP, anti-Sm and anti-SSA (online supplemental table S6). In general, the salmon subset also displayed elevated expression of the IL12 gene signature but a significantly lower tumour necrosis factor (TNF) signature, implying a Th1 bias.
When IGS expression in each patient subset was examined, the red subset had significantly less enrichment of the IFN core and IFNA2 signatures in monocytes, but not the other IGS nor the TNF and IL-12 signatures (figure 1B,C). The salmon subset was enriched in IFNG and IL12 signatures compared with the other three patient subsets. Examination of B, T and NK cells revealed generally similar patterns of expression with significantly decreased enrichment of the IGS in the red subset (online supplemental figure S3). Additional differences were noted in the NK and B cell subsets. When subsets were analysed for the percentage of subjects with positive IGS expression (positive GSVA score) for each IGS, similar results were observed (figure 1C and online supplemental figure S3). Together, these results indicate that patients in the EA dominant red subset exhibit less enrichment of the IFN core and IFNA2 signatures, as well as lower disease activity and a decreased frequency of autoantibodies.
These findings were confirmed using a number of additional IGS that were largely designed to discriminate patients with a positive or negative IGS. For this purpose, Singscore was used to identify the most important contributors to the IFN core and IFNA2 signatures, yielding a new 14-gene IFN core signature (signature A) and a 2-gene IFNA2 signature composed of HLA-DRB5 and IFI44L (signature F) (online supplemental table S3). In addition, we identified four other high performing IGS from the literature, including those by Yiu et al (signature B), Yao et al (signature C), Catalina et al (signature D) as well as the small gene signature used in the anifrolumab clinical trial (signature E) (online supplemental table S3).8–11 GSVA using these gene signatures as input separated the patient samples into two subsets (online supplemental figure S2B); an EA dominant group with comparatively low IGS expression (red), less mean disease activity and lower frequency of autoantibodies, and an AsA dominant, high IGS subset (green) exhibiting greater mean disease activity and higher frequency of autoantibodies (figure 2). It is notable that while all six gene sets performed similarly, the two gene signatures composed of fewer genes (signatures E and F) separated patients less effectively. This was evident for a number of patients who were negative using signatures E or F, but were positive for enrichment of other IGSs. To confirm this, we examined additional patient datasets and found that up to 29% of subjects who scored negatively using IGS E were enriched in the other IGSs (online supplemental table S7).
Figure 2.

Patient subsets defined by differential interferon gene signature (IGS) enrichment using alternative gene sets. Complex heatmap of the 113 patients assessed by gene set variation analysis (GSVA) using alternative gene sets to determine IGS enrichment. The gene signatures used include (A) the IFN core signature generated by Singscore, (B) signature from Yiu et al, (C) signature from Yao et al, (D) signature from Catalina et al, (E) signature from Morand et al and (F) the IFNA2 gene signature generated by Singscore.7–10
Consistent with the results observed in figure 1, patients divided relatively evenly between the high and low IGS subsets. The high IFN expression subset consisted mainly of AsA patients (65%), whereas the low IFN expression subset was mainly EA patients (65%). The high IGS expression subset had a significantly higher mean SLEDAI score, a lower mean age and significantly more autoantibody positive patients (online supplemental table S8).
Since the two subsets were largely composed of patients of different ancestries, we next sought to determine whether expression of the IGS differed between AsA and EA SLE patients. Using all six of the IGS (signatures A–F), AsA patients displayed significantly higher expression than EA patients in all cell types (p<0.01) (figure 3A and online supplemental figure S4). A significantly greater proportion of AsA patients also exhibited positive IGS expression (positive GSVA score) (figure 3B). Similar results were observed using the original seven gene modules (figure 3C and online supplemental figure S4). AsA patients demonstrated elevated IGS expression for the IFN core and IFNA2 signatures in all four cell types and were more likely to be positive for the IGS (positive GSVA score) (figure 3C,D). Additionally, AsA patients showed significant enrichment of the IFNG signature in both monocytes and T cells. We confirmed the results from GSVA with analysis of differential expression of individual genes within in each cell type. In monocytes, 12 out of the 14 total genes (85.7%) composing signature A were significantly differentially expressed in AsA SLE samples, whereas fewer genes were differentially expressed in T cells (7/14, 50%), NK cells (5/14, 35.7%) and B cells (3/14, 21.4%) (online supplemental table S9).
Figure 3.

Patients with SLE of AsA exhibit significantly greater IGS enrichment. (A) Violin plots showing GSVA scores derived using the six validation IGS modules in monocytes. Enrichment of all IGS is significantly greater in AsA compared with EA patients with SLE (p<0.001). For (A)–(D), the AsA group contains 61 patients and the EA group contains 56 patients. (B) Bar plots showing the difference in the number of IGS-positive SLE patients determined by GSVA scores using the six validation IGS modules. Significance determined by a χ2 test of proportion. (C) Violin plots of GSVA scores using the original five IGS modules (IFN core, IFNA2, IFNB1, IFNW1, IFNG) as well as IL12 and TNF in monocytes. (D) Bar plots showing the difference in the percentage of IGS-positive patients determined by GSVA scores in SLE patients of AsA and EA using the original five IFN modules as well as the IL12 and TNF modules in monocytes. AsA, Asian ancestry; EA, European ancestry; GSVA, gene set variation analysis; IFN, interferon; SLE, systemic lupus erythematosus; TNF, tumour necrosis factor.
Increased IGS in AsA patients is independent of, but augmented by, autoantibody status
Because upregulation of IGS has been reported to correlate with the presence of certain autoantibodies, especially anti-RNPs,3 we next examined the relationship between specific IGSs and autoantibody status in AsA and EA patients with SLE. We first assessed IGS enrichment in EA and AsA patients that were negative for the five measured autoantibodies (anti-SM, anti-RNP, anti-SSA, anti-dsDNA and anti-SSB). Notably, in the absence of autoantibodies, a significantly higher proportion of AsA patients with SLE were positive for the IGS in all four cell types compared with EA patients (figure 4A). In contrast, there was no difference in IGS positivity between autoantibody negative SLE patients of EA and African ancestry (AA) as previously reported (figure 4B).3 Together, these results indicate that there is enrichment of the IGS in AsA patients with SLE independent of antibody status.
Figure 4.

IGS enrichment in AsA is independent of, but augmented by, autoantibody status. (A, B) Bar plots of the percentage of AsA, AA and EA patients with SLE who are IGS positive (determined by GSVA score) and autoantibody negative in CD14, CD19, CD4, NK cells (GSE164457) and whole blood (GSE88884) using IGS signature (signature A) and IFNA2 (signature F). Statistical significance in (A) was determined using a χ2 test of proportion, whereas statistical significance in (B) was determined using a Fisher’s exact test. For (A), n=42 and for (B) n=118. (C, D) Bar plots of IGS positive patients in the presence (+) and absence (-) of anti-dsDNA antibodies (C), and in the presence (+) and absence (-) of anti-RNP antibodies (D) across all cell types. Significance is only shown within the same ancestry for clarity. For (C)–(D), n=117. For all bar plots, significance measures are as follows: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 determined using a χ2 test of proportion. AsA, Asian ancestry; EA, European ancestry; GSVA, gene set variation analysis; IGS, interferon gene signature; n.s., not significant; SLE, systemic lupus erythematosus.
The presence of autoantibodies had a variable effect on the expression of the IGS in cell types from the different ancestries, with anti-RNP and anti-Smith in general having a greater effect (figure 4D and online supplemental figure S5). The presence of anti-dsDNA was associated with a significantly increased IGS in B cells and T cells of EA patients, but only in NK cells of AsA patients, whereas anti-RNP was associated with an increased IGS in monocytes, T cells and NK cells of EA patients and monocytes, B cells and NK cells of AsA patients. Additionally, the presence of anti-SSA was associated with significantly increased IGS in B and T cells in EA patients but was not correlated with IGS in AsA (online supplemental figure S5). The presence of anti-Sm was much more impactful, as it was associated with significantly increased IGS in all cell types of EA patients and in T cells and NK cells of AsA patients (online supplemental figure S5).
AsA and EA patients differ in the expression of SLE-associated genes upstream of the IGS
Given that IGS expression by AsA and EA patients with SLE differed even in the absence of autoantibodies, it was possible that genetic factors regulating the IFN response contributed to this difference. To address this, we identified genes upstream of the IGS that are known to be regulated by SLE-associated risk variants in AsA and EA patients with lupus (online supplemental table S3). The expression of these genes was significantly enriched in AsA patients in all cell types (figure 5A), and also among SLE patients of AsA lacking autoantibodies (figure 5B and online supplemental figure S6). These results suggest that upregulation of genes known to be associated with SLE and involved in regulation of the IGS may contribute to enhanced IGS enrichment in AsA patients with SLE, even in the absence of autoantibodies.
Figure 5.
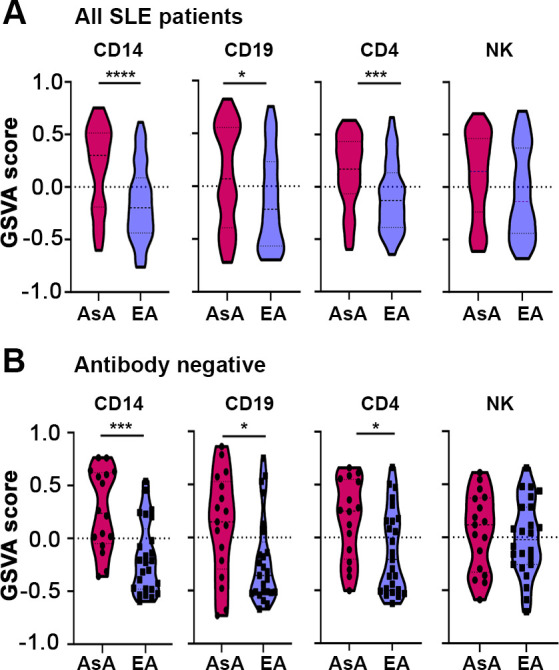
Genetically predicted genes upstream of the IGS are enriched in AsA patients with SLE. (A–B) Violin plots of GSVA scores using genetically associated upstream IGS-related genes across four cell types (A) and in patients lacking autoantibodies (B). In (A), the monocyte group consist of 61 AsA patients and 56 EA patients, the B cell group consists of 63 AsA patients and 57 EA patients, the T cell group consists of 61 AsA patients and 57 EA patients and the NK group consists of 62 AsA patients and 56 EA patients. AsA, Asian ancestry; EA, European ancestry; GSVA, gene set variation analysis; IGS, interferon gene signature; SLE, systemic lupus erythematosus.
Classification of patients by disease activity using ML
Because a number of features, including the IGS, ancestry and autoantibodies, appeared to be associated with SLEDAI scores in this cohort, we sought to determine the most important features contributing to lupus disease activity. To accomplish this, we used various ML methods to classify patients using the IGS and clinical parameters as features. We included cell type specific GSVA scores for each of the four cell types since the cell types differed in the expression of IFN (online supplemental figure S7). The patients in this dataset had a median SLEDAI of 2; therefore, we divided patients into those with a SLEDAI greater than 2 and those with a SLEDAI of less than or equal to 2. Features exhibiting collinearity were removed from the analysis.
Of the ML models, the Decision Tree (DTR) model performed best with a test accuracy of 85%, 92% precision, 73% recall and 95% specificity (figure 6A and online supplemental table S10). Gini feature importance analysis indicated that the IGS signature in NK cells displayed the highest feature importance, followed by the IFNG and IFNA2 signatures in T cells (figure 6B). Interestingly, the NK IGS captured the highest feature importance in every ML model employed, whereas the monocyte IGS C had the lowest feature importance, consistent with previous findings showing the monocyte IGS is frequently expressed by both patients with active and inactive SLE.7 Other clinical features of SLE, including C3 levels, autoantibodies and ancestry were also important features of the predictive model, although they less are impactful than the various IGS.
Figure 6.
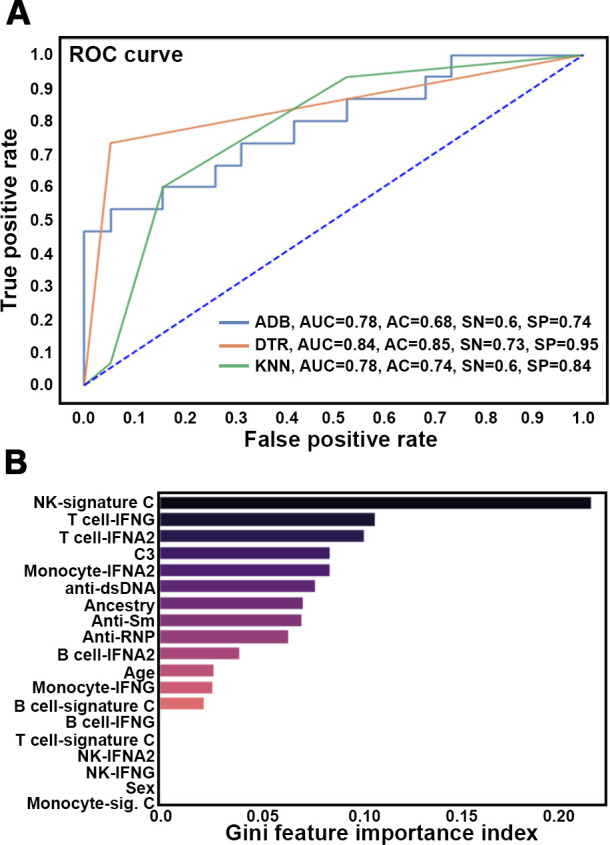
Patient classification using IGS and clinical features. (A) Receiver operating characteristic (ROC) curves for the Adaboost (ADB), Decision Tree (DTR) and K nearest neighbour (KNN) machine learning models. Values for the area under the curve (AUC), accuracy (AC), sensitivity (SN) and specificity (SP) are listed. (B) Gini Feature Importance scores (x-axis) for the indicated features (y axis) are shown.
Discussion
Dysregulated type I IFN signalling is a hallmark of SLE pathology.1–7 The importance of this pathway and its value as a drug target is further demonstrated by the recent approval of the type 1 IFN receptor blocking antibody, anifrolumab.21 Nonetheless, IFN signalling is complex and deconvoluting the pleiotropic biology of this pathway and its relationship with autoantibody reactivity, especially in patients of non-European descent, remains unclear. Notably, non-EA patients, especially those of AA, often present with increased autoantibody levels which may explain why these patients tend to develop more severe disease with a greater number of organ manifestations and tissue damage accrual.3 8 22
Using a diverse array of type I and II IGS, the present study demonstrates different levels of IFN involvement in AsA and EA patients with SLE. Asian ancestry was consistently linked to an elevated IGS, including increased type I IFN responsive genes as well as a difference in type II IFN expression in monocytes and T cells. Whereas there was seldom a difference noted in the enrichment of genes unique to IFNβ1 and IFNω1 between ancestries, the expression of the IFNα2 and IFN core modules was greatly enriched in AsA patients. Similar results were obtained when six additional IFN gene modules were employed. It is notable that although all modules performed in a similar manner, those with fewer genes, such as signatures E and F, were somewhat less effective in that patients identified as IGS negative with these signatures could be found to be positive when larger signatures were employed. Importantly, the GSVA was confirmed by conventional DEG analysis, supporting the conclusion that the IGS was differentially expressed between SLE patients of AsA and EA. Together, these results demonstrate that SLE patients of AsA generally express a quantitatively greater number of IFN genes that corresponds to elevated overall enrichment of IGS in these individuals compared with SLE patients of EA.
Notably, IGS enrichment in SLE patients of AsA was observed in the absence of known autoantibodies, whereas in SLE patients of EA and AA, overall IGS expression was lower in the absence of autoantibodies. Indeed, in EA and AA patients with SLE, the data indicated that autoantibodies were explanatory variables, whereas the IGS was a response variable.3 Consistent with this, in the current study, in both AsA and EA, the expression of the IGS tended to increase in association with autoantibodies, most uniformly with anti-RNP and anti-Sm. Because of the limitation in the number of patients in the dataset analysed, the independent impact of anti-RNP versus anti-SM could not be assessed. As opposed to SLE patients of AA, however, in whom the overall increased frequency of the IGS was associated with an increased frequency of autoantibodies,3 in SLE patients of AsA, except for anti-SSA, an increased frequency of autoantibodies was not observed. This suggested that features of SLE in patients of AsA other than autoantibodies contributed to their enhanced tendency to express the IGS. This possibility is supported by the observation that SLE manifests differently in AsA patients as they experience a greater prevalence of renal disease that cannot be accounted for by autoantibodies alone.22
Since autoantibodies were not the main factor in higher IGS expression in AsA patients with SLE, we examined the possibility that genes upstream of the IGS might be overexpressed in SLE patients of AsA as a possible contributor to the enhanced expression of the IGS. Indeed, Immunochip-based, GWAS and TWAS studies have revealed important ancestry-specific and trans-ancestral risk associations predisposing patients to SLE, including those involving IFN signalling and IGS expression.15 23 Examination of SNP-predicted genes upstream of the IGS revealed enrichment of these genes in monocytes, B cells and T cells in all patients as well as antibody-negative subjects, suggesting genetic factors controlling IFN responses may differ among ancestral populations of patients with SLE and may contribute to the enhanced IGS manifested by SLE patients of AsA. Many of these IGS upstream genes, including TYK2, IRF5, IRAK1, STAT1 and JAK2, were confirmed in a recent TWAS study in Asian patients with SLE.23
Notably, many of the genes we identified in our IGS upstream signature have demonstrable pathogenic roles in SLE.24–26 For example, mice lacking IRF5 were shown to be protected against SLE onset and severity. IRF7 has also been implicated as a risk allele in SLE, as the non-synonymous SNP, rs1131665, encoding a 412Q transition in IRF7 predisposes patients to develop SLE across ancestral populations.26 Numerous drugs are being developed to target these important upstream regulators of IFN including deucravicitinib (TYK2 inhibitor) and various JAK inhibitors as well as the previously mentioned anifrolumab.21 27 28 In fact, anifrolumab exhibits elevated efficacy in patients of AsA consistent with the enrichment of genetically predicted genes upstream of the IGS specifically in this patient population.21
It remains possible that other features of AsA patients contributed to the increased expression of the IGS. For example, SLE patients of AsA were also found to have an IFN gamma signature. IFNγ can induce histone acetylation of the IFN locus and thereby, result in greater expression of the IGS.29 This possibility could be explored in future studies.
There is a lack of consensus on whether expression of the IGS reflects SLE disease activity. In some studies, there is a relationship between expression of the IGS and disease activity measured in different ancestral groups with a variety of instruments (ie, SLEDAI or BILAG).30 31 In contrast, in other studies, he expression of the IGS does not reflect disease activity.7 16 32 Here, a more nuanced view emerged, with the IGS expressed in some cell populations (ie, NK cells) but not others (ie, monocytes) reflecting disease activity measured by SLEDAI. The inability of the monocyte IGS to predict disease activity is consistent with previous findings demonstrating monocytes maintain the IGS regardless of disease activity, whereas the IGS expressed by other cell types is more variable.7 This highlights the complexity of the relationship between expression of the IGS and disease activity in SLE. In unseparated blood samples, the greatest IGS is expressed in monocytes and is consistent in the current study.7 Among the lymphoid cell populations examined, IGS expression in NK and CD4 T cells was generally lower than in monocytes, as previously reported, but was significantly higher in AsA compared with EA patients with SLE.33 Furthermore, signature C in NK cells was the strongest predictor of disease activity.
Even though the IGS was the major feature associated with disease activity, other characteristics of patients with lupus also contributed. Among these were anti-dsDNA antibodies and complement levels, as reported in the literature.3 34 35 In addition, AsA was also a contributor to disease activity. Since collinear features were removed from this analysis, these results suggest that AsA, independent of the IGS and other clinical features, such as anti-dsDNA and complement levels contributed to disease activity. Assessment of the contribution of genes outside the IFN pathway in SLE patients of AsA suggested molecular processes involved in the cellular response to stress and damage, as well as altered metabolic function might contribute to this tendency.15 It is notable that anti-RNP antibodies were not found to contribute to disease activity. This is consistent with previous findings that anti-RNP was associated with lower levels of renal disease.36 This could relate to the lower likelihood that the presence of anti-RNP was associated with activation of the complement cascade, a required component of renal pathology.3
A limitation of this study is that most of the patients had inactive disease (SLEDAI <6). Despite this, we were still able to observe significant differences in disease activity between patient subsets and detect differential enrichment of multiple IGS representing type 1 and 2 IFN even among those patients with low levels of disease activity.7 16 Another limitation is the lack of additional multi-ancestral datasets for comparison and validation. In general, study location itself can impose significant challenges to diverse patient enrolment and it should be noted that even those localities capable of recruiting large cohorts of AsA patients may still skew towards genetically distinct subpopulations (ie, South Asian vs East Asian) further complicating validation efforts.31 37 Moreover, patient ancestry, ethnicity and country of origin were assessed by verbal questionnaire, raising the possibility that classification might not be completely accurate. The AsA patients included in GSE164457 were of self-identified ancestry and country of origin and did not include information on potential admixture, nor was there additional demographic data collected for the subjects of European ancestry. Finally, other demographic information, such as socioeconomic status, was not collected, so it is possible that other features of the patient groups may have contributed to the differences noted. It is notable, however, that baseline characteristics of the patient groups were similar and that medications were unlikely to play as role as the patients of AsA were more likely to be on immunosuppressives but still were enriched for IGS expression.
Overall, this study demonstrates the need for further investigation of IGS in diverse patient cohorts, especially when designing studies and clinical trials. The majority of SLE studies are conducted using EA patients,3 yet the differential IGS enrichment observed here could have a profound impact on the selection of ancestry-informed treatment options. This is exemplified by the finding that biologic drugs targeting IFN, such as anifrolumab have proven efficacy especially in AsA.21
In summary, we have employed various approaches to differentiate the IFN response between AsA and EA SLE populations, demonstrating different IFN regulatory mechanisms between ancestries, such that the IGS enrichment present in AsA individuals may be genetically motivated, whereas this response in EA is more likely to be dependent on autoantibody status.
Acknowledgments
The authors would like to thank the members of the RILITE scientific team for their careful reading of the manuscript.
Footnotes
Contributors: IR collected the data, performed the analyses and wrote the manuscript. KAO contributed data, performed analyses and wrote the manuscript. PB and EH contributed data and performed analyses. JY and MD’E collected and contributed data (CLUES dataset). ACG and PEL conceived and designed the analyses, supervised the work and wrote the manuscript. PEL is the guarantor, accpeting full responsibility for the conduct of the study.
Funding: The work presented in this manuscript was funded by a Grant from the RILITE foundation. The funder provided support in the form of salaries for authors (IR, KAO and PB). Generation of data included in GSE164457 was supported by the Centers for Disease Control (5U01DP006486) to MDE and JY.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available in a public, open access repository.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
Not applicable.
References
- 1.Ghodke-Puranik Y, Niewold TB. Genetics of the type I interferon pathway in systemic lupus erythematosus. Int J Clin Rheumtol 2013;8. 10.2217/ijr.13.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oke V, Gunnarsson I, Dorschner J, et al. High levels of circulating Interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther 2019;21:107. 10.1186/s13075-019-1878-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hubbard EL, Pisetsky DS, Lipsky PE. Anti-RNP antibodies are associated with the interferon gene signature but not decreased complement levels in SLE. Ann Rheum Dis 2022;81:632–43. 10.1136/annrheumdis-2021-221662 [DOI] [PubMed] [Google Scholar]
- 4.Iwamoto T, Dorschner J, Jolly M, et al. Associations between type I interferon and antiphospholipid antibody status differ between ancestral backgrounds. Lupus Sci Med 2018;5:e000246. 10.1136/lupus-2017-000246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazewski C, Perez RE, Fish EN, et al. Type I interferon (IFN)-regulated activation of canonical and non-canonical signaling pathways. Front Immunol 2020;11:606456. 10.3389/fimmu.2020.606456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynolds JA, Briggs TA, Rice GI, et al. Type I interferon in patients with systemic autoimmune rheumatic disease is associated with haematological abnormalities and specific autoantibody profiles. Arthritis Res Ther 2019;21:147. 10.1186/s13075-019-1929-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catalina MD, Bachali P, Geraci NS, et al. Gene expression analysis delineates the potential roles of multiple Interferons in systemic lupus erythematosus. Commun Biol 2019;2:140. 10.1038/s42003-019-0382-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catalina MD, Bachali P, Yeo AE, et al. Patient ancestry significantly contributes to molecular heterogeneity of systemic lupus erythematosus. JCI Insight 2020;5:e140380. 10.1172/jci.insight.140380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morand EF, Furie R, Tanaka Y, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med 2020;382:211–21. 10.1056/NEJMoa1912196 [DOI] [PubMed] [Google Scholar]
- 10.Yiu G, Rasmussen TK, Tsai BL, et al. High interferon signature leads to increased Stat1/3/5 phosphorylation in PBMCs from SLE patients by single cell mass cytometry. Front Immunol 2022;13:833636. 10.3389/fimmu.2022.833636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao Y, Richman L, Higgs BW, et al. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha Monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum 2009;60:1785–96. 10.1002/art.24557 [DOI] [PubMed] [Google Scholar]
- 12.Manry J, Laval G, Patin E, et al. Evolutionary genetic dissection of human Interferons. J Exp Med 2011;208:2747–59. 10.1084/jem.20111680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariuki SN, Franek BS, Kumar AA, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther 2010;12:R151. 10.1186/ar3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun C, Molineros JE, Looger LL, et al. High-density genotyping of immune-related Loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet 2016;48:323–30. 10.1038/ng.3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owen KA, Bell KA, Price A, et al. Molecular pathways identified from single nucleotide polymorphisms demonstrate mechanistic differences in systemic lupus eyrthematosus patients of Asian and European ancestry. Sci Rep 2023;13:5339. 10.1038/s41598-023-32569-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Northcott M, Jones S, Koelmeyer R, et al. Type 1 interferon status in systemic lupus erythematosus: a longitudinal analysis. Lupus Sci Med 2022;9:e000625. 10.1136/lupus-2021-000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andreoletti G, Lanata CM, Trupin L, et al. Transcriptomic analysis of immune cells in a multi-ethnic cohort of systemic lupus erythematosus patients identifies ethnicity- and disease-specific expression signatures. Commun Biol 2021;4:488. 10.1038/s42003-021-02000-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Labonte AC, Kegerreis B, Geraci NS, et al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS One 2018;13:e0208132. 10.1371/journal.pone.0208132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daamen AR, Bachali P, Owen KA, et al. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci Rep 2021;11:7052. 10.1038/s41598-021-86002-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Owen KA, Price A, Ainsworth H, et al. Analysis of trans-ancestral SLE risk Loci identifies unique biologic networks and drug targets in African and European Ancestries. Am J Hum Genet 2020;107:864–81. 10.1016/j.ajhg.2020.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vital EM, Merrill JT, Morand EF, et al. Anifrolumab efficacy and safety by type I interferon gene signature and clinical subgroups in patients with SLE: post hoc analysis of pooled data from two phase III trials. Ann Rheum Dis 2022;81:951–61. 10.1136/annrheumdis-2021-221425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis MJ, Jawad AS. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology (Oxford) 2017;56:i67–77. 10.1093/rheumatology/kew399 [DOI] [PubMed] [Google Scholar]
- 23.Yin X, Kim K, Suetsugu H, et al. Biological insights into systemic lupus erythematosus through an immune cell-specific transcriptome-wide association study. Ann Rheum Dis 2022;81:1273–80. 10.1136/annrheumdis-2022-222345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song S, De S, Nelson V, et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J Clin Invest 2020;130:6700–17. 10.1172/JCI120288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu Q, Zhao J, Qian X, et al. Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum 2011;63:749–54. 10.1002/art.30193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alunno A, Padjen I, Fanouriakis A, et al. Pathogenic and therapeutic relevance of JAK/STAT signaling in systemic lupus erythematosus: integration of distinct inflammatory pathways and the prospect of their inhibition with an oral agent. Cells 2019;8:898. 10.3390/cells8080898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petri M, Bruce IN, Dörner T, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 3 trial (SLE-BRAVE-II). Lancet 2023;401:1011–9. 10.1016/S0140-6736(22)02546-6 [DOI] [PubMed] [Google Scholar]
- 28.Mease PJ, Deodhar AA, van der Heijde D, et al. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann Rheum Dis 2022;81:815–22. 10.1136/annrheumdis-2021-221664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14:36–49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braunstein I, Klein R, Okawa J, et al. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br J Dermatol 2012;166:971–5. 10.1111/j.1365-2133.2012.10825.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petri M, Singh S, Tesfasyone H, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus 2009;18:980–9. 10.1177/0961203309105529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landolt-Marticorena C, Bonventi G, Lubovich A, et al. Lack of association between the interferon-alpha signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann Rheum Dis 2009;68:1440–6. 10.1136/ard.2008.093146 [DOI] [PubMed] [Google Scholar]
- 33.Nakano M, Ota M, Takeshima Y, et al. Distinct transcriptome architectures underlying lupus establishment and exacerbation. Cell 2022;185:3375–89. 10.1016/j.cell.2022.07.021 [DOI] [PubMed] [Google Scholar]
- 34.Paz E, Adawi M, Lavi I, et al. Antinuclear antibodies measured by enzyme immunoassay in patients with systemic lupus erythematosus: relation to disease activity. Rheumatol Int 2007;27:941–5. 10.1007/s00296-007-0324-7 [DOI] [PubMed] [Google Scholar]
- 35.Kim AHJ, Strand V, Sen DP, et al. Association of blood concentrations of complement split product IC3B and serum C3 with systemic lupus erythematosus disease activity. Arthritis Rheumatol 2019;71:420–30. 10.1002/art.40747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carpintero MF, Martinez L, Fernandez I, et al. Diagnosis and risk stratification in patients with anti-RNP autoimmunity. Lupus 2015;24:1057–66. 10.1177/0961203315575586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan Z, Xu S. Population genomics of East Asian ethnic groups. Hereditas 2020;157:49. 10.1186/s41065-020-00162-w [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
rmdopen-2023-003475supp002.xlsx (38.5KB, xlsx)
rmdopen-2023-003475supp001.pdf (2.4MB, pdf)
Data Availability Statement
Data are available in a public, open access repository.