

Abstract
Plant synthetic biology requires precise characterization of genetic elements to construct complex genetic circuits that can improve plant traits or confer them with new characteristics. Transcriptional reporter assays are essential to quantify the effect of gene expression regulator elements. Additionally, transcriptional reporter systems are a key tool in understanding control of gene expression in biology. In this work, we construct and characterize a dual color luciferase ratiometric reporter system that possesses several advantages over currently used reporters. It is ratiometric, thus reducing variability and increasing consistency between experiments; it is fast, as both reporters can be measured at the same time in a single reaction, and it is less expensive to perform than current dual luciferase reporter assays. We have validated our system quantifying the transcriptional capability of a panel of promoters and terminators commonly used in synthetic biology with a broad range of expression magnitudes, and in a biologically relevant system, nitrate response.
Keywords: synthetic biology, transcriptional regulation, reporter assay, luciferase, plant
Graphical Abstract
INTRODUCTION
Agronomy faces significant global challenges and addressing these will require the adoption of genetic manipulation technologies used in synthetic biology. Synthetic biology tools can aid in both understanding endogenous transcriptional regulation and designing genetic circuits to confer plants novel traits. Complex DNA constructs can be assembled from different genetic elements with modular cloning toolkits (e.g., Golden-Braid1) together with the Phytobrick standard that systematizes sequence requirements for cloning.2 In plants, the most commonly employed reporters to measure transcriptional activity are fluorescent proteins, beta-glucuronidase, and luminescent proteins.3 Variability commonly found between samples and experiments can be reduced using ratiometric assays with a second invariable reporter as an internal reference (e.g., renilla/firefly luciferase).1 However, activity of these dual reporters is measured by different methods,4,5 or in sequential reactions with distinct kinetics.6 Additionally, these assays often require sample homogenization and protein extraction, and are thus unpractical for high-throughput assays.
Here, we developed a ratiometric dual color luciferase reporter assay to quantify transcriptional activity of genetic elements in plants. This GREAT (green/red luciferase ratiometric) reporter system is based on two luciferases that process the same substrate and emit light at different wavelengths. This system is (i) ratiometric, with both reporters measured by the same method and at the same time, (ii) has a high dynamic range output that allows precise comparison of a wide range of expression magnitudes, (iii) requires minimal sample preparation time, and (iv) is less expensive than currently used dual luciferase methods.
RESULTS AND DISCUSSION
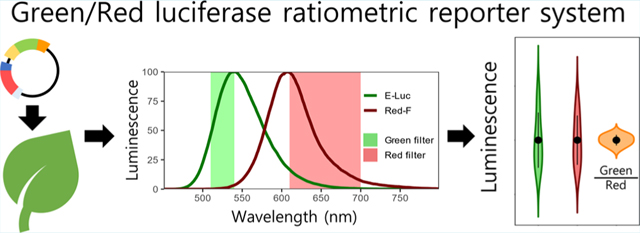
Previous characterization of luciferases with different attributes has been performed mainly in cell culture or animals.6−8 As metabolites in plant extracts can alter luciferase emission properties,9 we surveyed a panel of luciferases that emit green or red luminescence6 by transiently expressing them in Nicotiana benthamiana leaves through agroinfiltration. We extracted total protein from leaf tissues and measured luciferase emission spectra and stability 3 days postinfiltration (Materials and Methods). We selected E-Luc10 and Red-F11 as green- and red-emitting luciferases, respectively, because this pair of luciferases exhibited the most distant emission peaks (Figure 1A, 541 and 608 nm, respectively). Additionally, both luciferases showed very similar and stable reaction kinetics that allow reliable relative measurements starting 10 min after extraction and substrate mixing, with stable luminescence for over 2 h (Figure 1B). These properties allow characterization of many samples in a standard luminometer without the need for expensive injectors required for time-sensitive coelenterazine-using luciferases (e.g.,Renilla luciferase). Using a single substrate to measure luminescence from both luciferases also greatly reduces the economic cost of the assay (Promega Steady-Glo Assay used herein costs 1.12$ per reaction compared to Promega Dual Luciferase Reporter assay cost of 2.29$ per reaction). Moreover, we were able to measure stable luminescence directly from leaf discs, effectively eliminating sample preprocessing (Figure 1B, Materials and Methods). This method greatly reduces sample processing time (protein extraction takes 20 min, see Materials and Methods) and labor (using a single substrate halves luminescence measuring time compared to renilla/firefly assays), and thus, is amenable for high-throughput measurements.
Figure 1.

Characterization and validation of a ratiometric dual color luciferase reporter system. (A) Emission spectra of E-Luc and Red-F luciferases. Wavelengths transmitted by green- and red-light filters are shown. (B) Luminescence stability from protein extract or leaf discs. (C) Signal deconvolution of Red-F and E-Luc. Deconvoluted signal is compared to expected red or green luminescence in counts per second (cps). (D) Correlation of deconvoluted green and red luminescence from plants agroinfiltrated with pGREAT2. The orange line represents linear regression. (E) Dispersion of luminescence in samples from panel D. Green and red luminescence are normalized to the average luminescence. The black dot represents the average, and the vertical lines are the standard deviation. (F) Green/red luminescence from plants agroinfiltrated with pGREAT1−25 plasmids. Green/red luminescence is relative to pGREAT1 (pNOS:E-Luc:NOSt). (G) Green/red luminescence of an agroinfiltrated plasmid in which E-luc is under control of an estradiol inducible promoter (pGREAT26). Leaves were infiltrated with 20 μM β-estradiol. (H) Green/red luminescence of Arabidopsis root protoplasts transformed with a plasmid in which E-luc is under control of the Nitrate-Regulated Promoter (pGREAT27). Protoplasts were incubated with increasing nitrate concentrations. In panels B and C, the line represents average luminescence, the faded band represents a 95% confidence interval. Error bars in G and H represent the standard error of the mean. t test statistical significance in G and H is denoted as follows: *, p- val < 0.05; **, p-val < 0.01; ***, p-val < 0.001. n = 3 for each treatment/condition except in panels D and E, where n = 8 and panel C, where n = 3−6.
As there is partial signal overlap between E-Luc and Red-F, we used a previously described simple deconvolution method that allows precise adscription of luminescence signal to each luciferase (Promega Technical Manual TM062). Other deconvolution methods (e.g., simultaneous equations6) could be adapted to this reporter system. Calibration constants are calculated measuring green-, red-filtered, and total luminescence (Figure 1A). These constants only need to be calculated once if the measurement conditions that affect luminescence are kept fixed in subsequent experiments (e.g., temperature, pH). We tested signal deconvolution for each luciferase by measuring luminescence of a serial dilution of one luciferase to which we added different amounts of the other luciferase. We then compared the deconvoluted signal to expected red or green luminescence measured from extracts with only one of the luciferases. We found that when luminescence from the second luciferase is very high compared to the first one, the deconvoluted signal of the first luciferase can be overestimated (Figure 1C, leftmost panels). When the amount of the second luciferase was reduced, this overestimation was eliminated and the deconvoluted luminescence perfectly matched the expected luminescence value (Figure 1C). On the basis of these results, we selected a Red-F transcriptional unit that produced moderately low luminescence (pNOS:Red-F:NOSt) as the normalizer reporter for our ratiometric system. We recommend conducting a similar assay to the one described here and including the deconvolution deviation in final calculations when the expected measurements will vary over a wide range of magnitudes.
We then constructed a ratiometric vector including both luciferases to quantify how ratiometric normalization reduces variability when measuring transcriptional activity (pGREAT2). We agroinfiltrated several Nicotiana benthamiana leaves and, as expected, we observed a great degree of variability when measuring green luminescence in different leaves, quantified by a coefficient of variability (CV) of 50.93%. This variability was also proportional to red luminescence (CV of 45.44%), suggesting that each leaf exhibited diverse degrees of transcription/translation that affected both luciferases equally (Figure 1D). When green luminescence was normalized by red luminescence, variability was greatly reduced to a CV of 9.85% (Figure 1E).
We then used the GREAT reporter to systematically measure transcriptional activity of a set of five promoters and five terminators commonly used in plant synthetic biology. We specifically chose genetic elements with different activities to test our system over a wide range of expression levels. We Phytobrick-adapted these sequences, and built E-Luc transcriptional units with all possible combinations of promoters and terminators together with pNOS:Red-F:NOSt for ratiometric normalization, using the modular GoldenBraid system1 (pGREAT1−25, Supporting Information, Table 1). We agro-infiltrated these vectors into Nicotiana leaves and measured green/red luminescence of each plasmid, using the pGREAT1 (pNOS:E-Luc:NOSt and pNOS:Red-F:NOSt) vector as an internal normalizer, as this practice has been shown to reduce interexperiment technical variability.1 We found that our system enabled expression quantification from 0.4 to 137.2 relative luminescence units, confirming that our system can be used to reliably quantify transcriptional activity over at least 4 orders of magnitude (Figure 1F). The aforementioned characteristics of the GREAT reporter system are ideal for these types of screens by reducing the time and cost of the assay. Additionally, we used our system to measure the responsiveness of a Phytobrick-adapted β-estradiol inducible promoter (pGREAT26).12 We were able to measure a 40-fold green/red luminescence induction 24 h after treatment with 20 μM β-estradiol (Figure 1G).
We also tested if the GREAT system could be used to measure biologically relevant transcriptional responses. We cloned the Arabidopsis Nitrate-Regulated Promoter13 to control E-Luc expression (pGREAT27). We transformed Arabidopsis root protoplasts14,15 and treated them with increasing nitrate concentrations for 16 h. We observed a significant induction of green/red luminescence proportional to the nitrate concentration, validating our system (Figure 1H).
In summary, we have developed a system that possesses several advantages over other commonly used methods to measure transcriptional activity, and we have provided evidence of its potential applications. We have deposited the standardized Phytobricks and pGREAT1−27 plasmids generated herein (Supporting Information, Table 1) in the Addgene repository, including detailed sequence maps (Addgene #170873–170915). We expect that the system described in this work will help advance our understanding of transcriptional regulation and accelerate plant synthetic biology.
MATERIALS AND METHODS
Plasmid Construction.
Sequences of promoters and terminators were either PCR amplified with primers designed to remove Esp3I/BsaI/BbsI restriction sites (Supporting Information, Table 2) or synthesized de novo without these sites (Twist Biosciences) to comply with the Phytobrick standard. Q5 polymerase (New England Biolabs, cat no. M0491S) was used for PCRs, following manufacturer instructions. Because of primer length, a two-step amplification protocol was used with a single 72 °C annealing/amplification cycle. PCR fragments were gel purified (Qiagen, cat no. 28704). For GoldenGate reactions to generate Phytobricks, equimolar amounts of PCR fragments in a 2:1 ratio to 40 femtomoles of pUPD2 plasmid were used. Ten microliter reactions with 1 uL (400 units) of T4 Ligase (New England Biolabs, cat no. M0202S) and 0.5 uL (5 units) of Esp3I (New England Biolabs, cat no. R0734S) in 1× T4 Ligase Buffer were incubated for 40 cycles of 5 min 37 °C and 5 min 16 °C, with a final step of 5 min 60 °C. A 5 uL aliquot of the GoldenGate reaction was transformed into 100 uL of competent XL-1 Blue E. coli cells (UC Berkeley Macrolab) by heat shock. Transformed cells were plated on LB+Agar plates with IPTG+X-Gal selection. Three to five white colonies were selected for colony PCR, and plasmids were isolated by miniprep (Zymo Research, cat no. D4015) from positive colonies. Insert sequences were verified by Sanger Sequencing. Transcriptional units (promoter, E-luc/Red-F, terminator) were generated by a similar GoldenGate reaction, using BsaI-HFv2 (New England Biolabs, cat no. R3733S) instead of Esp3I with equimolar amounts of the three phytobricks and plasmids pDGB1 alpha1 for E-luc transcriptional units and pDGB1 alpha2 for Red-F transcriptional units as backbones. Plasmids with dual transcriptional units (pGREAT) were generated by GoldenGate reactions with Esp3I and equimolar amounts of E-luc and Red-F transcriptional units and pDGB1 omega1 as backbone. Sanger sequencing was performed to verify sequence integrity at ligation sites.
Agroinfiltration.
Agroinfiltration experiments were performed as in ref 1 with minor modifications. Briefly, 5 mL of overnight cultures of Agrobacterium tumefaciens strain GV3101+pMP90+pSOUP harboring different pGREAT plasmids were pelleted and resuspended in agroinfiltration solution (50 mM MES, pH 5.5, 100 mM MgCl2 and 200 μM acetosyringone) to an optical density of 0.1 at 600 nm. Resuspensions were incubated for 2−4 h at room temperature on a rotatory shaker at 60 rpm. Agroinfiltration were carried out through the abaxial surface of the third and fourth youngest leaf of each plant with a 1 mL needle-less syringe.
Protein Extraction.
Protein extraction was performed 3 days postinfiltration. Briefly, 30 mg of leaf tissue per sample was collected into a 2 mL tube (Biospec Products, cat no. 330TX) with two 3.2 mm chrome steel beads (RPI, cat. no. 9840) and flash frozen in liquid N2 immediately. Frozen samples were ground in a Mini-beadbeater (Biospec Products, cat. no. 3110Bx) tissue homogenizer for 5 s at 25 Hz frequency, twice. Then, 100 μL of 1× Passive Lysis buffer (Promega, cat no. E1941) was added per tube, and the tube was vigorously vortexed until the ground sample was completely dissolved and no clumps were visible. Tubes were left at room temperature for 10 min, then centrifuged at 12000g to pellet cell debris, and the supernatant was transferred to a new Eppendorf tube.
Luminescence Quantification.
Promega Steady-Glo Assay (Promega, cat no. E2510) was used following manufacturer instructions with minor modifications. A 20 μL aliquot of protein extract or a 1/4 in. leaf disc produced with a leaf punch (Thomas Scientific, cat no. 1197R21) or 3 × 104 protoplasts were added to 100 μL of Steady-Glo reagent per well of Lumitrac 200 96-well plate (Greiner Bio-one, cat no. 655095). Luminescence was measured in a Tecan M-100PRO plate reader at 25 °C, using Lumi Green and Lumi Red filters and an integration time of 1 s per well. To calculate signal deconvolution coefficients, total, Lumi Green-, and Lumi Red-filtered luminescence were measured, and a Promega Chroma-Luc Technology calculator spreadsheet (Promega Technical Manual TM062) was used.
Arabidopsis Root Protoplast Transformation.
Arabidopsis root protoplasts were generated as described in ref 2, and PEG-Calcium transformations were performed as described in ref 3.
Supplementary Material
ACKNOWLEDGMENTS
M.P.L. acknowledges support of a USDA BBT EAGER award (2019-67013-29104) and a USDA NIFA award (2018-67021-27964). G.S.D. is supported by the Resnick Sustainability Institute Postdoctoral Fellowship, and G.S.D. and S.M.B. acknowledge support of a USDA BBT-EAGER award (2019-67013-29012), and partial support by an HHMI Faculty Scholar fellowship. pRedF was a gift from Koen Venken (Addgene #118057).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.1c00248.
Plasmids generated in this work (XLSX)
Primers used in this work (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acssynbio.1c00248
The authors declare no competing financial interest.
Contributor Information
Eduardo González-Grandío, University of California Berkeley, Chemical and Biomolecular Engineering, Berkeley, California 94720, United States.
Gozde S. Demirer, University of California Davis, Department of Plant Biology and Genome Center, Davis, California 95616, United States
Wenhe Ma, University of California Berkeley, Chemical and Biomolecular Engineering, Berkeley, California 94720, United States.
Siobhan Brady, University of California Davis, Department of Plant Biology and Genome Center, Davis, California 95616, United States.
Markita P. Landry, University of California Berkeley, Chemical and Biomolecular Engineering, Berkeley, California 94720, United States
REFERENCES
- (1).Vazquez-Vilar M; Quijano-Rubio A; Fernandez-Del-Carmen A; Sarrion-Perdigones A; Ochoa-Fernandez R; Ziarsolo P; Blanca J; Granell A; Orzaez D. GB3.0: A Platform for Plant Bio-Design That Connects Functional DNA Elements with Associated Biological Data. Nucleic Acids Res. 2017, 45 (4), 2196–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Patron NJ; Orzaez D; Marillonnet S; Warzecha H; Matthewman C; Youles M; Raitskin O; Leveau A; Farre G; Rogers C; Smith A; Hibberd J; Webb AAR; Locke J; Schornack S; Ajioka J; Baulcombe DC; Zipfel C; Kamoun S; Jones JDG; Kuhn H; Robatzek S; Van Esse HP; Sanders D; Oldroyd G; Martin C; Field R; O’Connor S; Fox S; Wulff B; Miller B; Breakspear A; Radhakrishnan G; Delaux P-M; Loque D; Granell A; Tissier A; Shih P; Brutnell TP; Quick WP; Rischer H; Fraser PD; Aharoni A; Raines C; South PF; Ane J-M; Hamberger BR; Langdale J; Stougaard J; Bouwmeester H; Udvardi M; Murray JAH; Ntoukakis V; Schafer P; Denby K; Edwards KJ; Osbourn A; Haseloff J. Standards for Plant Synthetic Biology: A Common Syntax for Exchange of DNA Parts. New Phytol. 2015, 208 (1), 13–19. [DOI] [PubMed] [Google Scholar]
- (3).De Ruijter NCA; Verhees J; Van Leeuwen W; Van Der Krol AR Evaluation and Comparison of the GUS, LUC and GFP Reporter System for Gene Expression Studies in Plants. Plant Biol. 2003, 5, 103–115. [Google Scholar]
- (4).Feike D; Korolev AV; Soumpourou E; Murakami E; Reid D; Breakspear A; Rogers C; Radutoiu S; Stougaard J; Harwood WA; Oldroyd GED; Miller JB Characterizing Standard Genetic Parts and Establishing Common Principles for Engineering Legume and Cereal Roots. Plant Biotechnol. J 2019, 17 (12), 2234–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Brückner K; Schäfer P; Weber E; Grützner R; Marillonnet S; Tissier A. A Library of Synthetic Transcription Activator-like Effector-Activated Promoters for Coordinated Orthogonal Gene Expression in Plants. Plant J. 2015, 82 (4), 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sarrion-Perdigones A; Chang L; Gonzalez Y; Gallego-Flores T; Young DW; Venken KJTT Examining Multiple Cellular Pathways at Once Using Multiplex Hextuple Luciferase Assaying. Nat. Commun 2019, 10 (1), No. 13651-y, DOI: 10.1038/s41467-019-13651-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Doi M; Sato M; Ohmiya Y. In Vivo Simultaneous Analysis of Gene Expression by Dual-Color Luciferases in Caenorhabditis Elegans. Int. J. Mol. Sci 2021, 22 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Branchini BR; Southworth TL; Fontaine DM; Kohrt D; Florentine CM; Grossel MJ A Firefly Luciferase Dual Color Bioluminescence Reporter Assay Using Two Substrates to Simultaneously Monitor Two Gene Expression Events. Sci. Rep 2018, 8 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Moyle RL; Birch RG Sugarcane Loading Stem Gene Promoters Drive Transgene Expression Preferentially in the Stem. Plant Mol. Biol 2013, 82 (1−2), 51–58. [DOI] [PubMed] [Google Scholar]
- (10).Nakajima Y; Yamazaki T; Nishii S; Noguchi T; Hoshino H; Niwa K; Viviani VR; Ohmiya Y. Enhanced Beetle Luciferase for High-Resolution Bioluminescence Imaging. PLoS One 2010, 5 (4), e10011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Branchini BR; Southworth TL; DeAngelis JP; Roda A; Michelini E. Luciferase from the Italian Firefly Luciola Italica: Molecular Cloning and Expression. Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol 2006, 145 (2), 159–167. [DOI] [PubMed] [Google Scholar]
- (12).Zuo J; Niu QW; Chua NH Technical Advance: An Estrogen Receptor-Based Transactivator XVE Mediates Highly Inducible Gene Expression in Transgenic Plants. Plant J. 2000, 24 (2), 265–273. [DOI] [PubMed] [Google Scholar]
- (13).Wang R; Guan P; Chen M; Xing X; Zhang Y; Crawford NM Multiple Regulatory Elements in the Arabidopsis NIA1 Promoter Act Synergistically to Form a Nitrate Enhancer. Plant Physiol. 2010, 154 (1), 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Birnbaum K; Jung JW; Wang JY; Lambert GM; Hirst JA; Galbraith DW; Benfey PN Cell Type-Specific Expression Profiling in Plants via Cell Sorting of Protoplasts from Fluorescent Reporter Lines. Nat. Methods 2005, 2 (8), 615–619. [DOI] [PubMed] [Google Scholar]
- (15).Yoo SD; Cho YH; Sheen J. Arabidopsis Mesophyll Protoplasts: A Versatile Cell System for Transient Gene Expression Analysis. Nat. Protoc 2007, 2 (7), 1565–1572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.