

Patients with deficiency of zeta-chain-associated protein kinase 70 (ZAP-70) protein generally present as combined immunodeficiency (CID) with severe recurrent infections and dermatological findings during the first years of life. They also suffer from diarrhea, mainly resulting from viral agents, lymphoproliferation, and autoimmunity (autoimmune cytopenia, bullous pemphigoid, nephrotic syndrome, and adrenal insufficiency). The most striking immunological findings are severely decreased CD3+CD8+ T-cell counts with decreased proliferation. The current definitive treatment of ZAP-70 deficiency is hematopoietic stem cell transplantation (HSCT).1
To date, 52 patients with biallelic mutations in the ZAP70 gene have been described in the literature.1,2 Herein, we report a patient with a novel missense mutation in the ZAP70 who presented with atypical skin lesions and a rapid decrease in CD8+ T-cell counts on immunological evaluations between 6 and 9 months of age. Despite undetectable ZAP-70 protein, the patient did not present severe infections in the first year of life. This description expands the spectrum of disease caused by mutations in the ZAP70, thus providing vigilance for early diagnosis and treatment.
A 20-month-old boy, born to consanguineous parents of Kurdish descent, was admitted to the hospital due to an extensive rash covering his body, starting on the third day of life. He also had continual diarrhea, with a frequency of two times per day since birth. Local treatments and antihistamines did not improve his skin symptoms. Physical examination revealed no specific features other than erythematous papular lesions on the dorsum of arms, malar rash like lesions, and livedo reticularis formation on his legs (Figure 1A). Complete blood count showed a lymphocyte count of 4700/mm3, eosinophil of 2100/mm3, hemoglobin of 10.7 g/dl, and platelet of 988,000/mm3. Immunoglobulin levels were normal except for an IgE level of 499 mg/dl. Lymphocyte immunophenotyping revealed moderate low CD3+ and CD8+ T cells, accompanied by decreased naive and increased memory CD4+ and CD8+ T cells. Levels of recent thymic emigrants were also diminished compared to healthy controls. Detailed blood test results and immunophenotyping are shown in Table 1. Skin prick test and specific IgE levels were all negative and biopsy from the skin lesions showed minimal vacuolar degenerations with focal parakeratosis. Immunohistochemical examination of the biopsy revealed infiltrating T lymphocytes, predominantly CD4+ T cells. Maternal engraftment was excluded by using short tandem repeat analysis in the patient. Viral serology for Cytomegalovirus (CMV), Epstein-Barr virus (EBV) were negative. Interestingly, a follow-up lymphocyte subset analysis at 9 months of age showed a severe decrease in CD8+ T cells (1.2%). The patient’s family history revealed an older sister with EBV-related B-cell lymphoproliferative disease; she died at 22 months of age, due to systemic aspergillosis leading to multiorgan failure.
FIGURE 1.
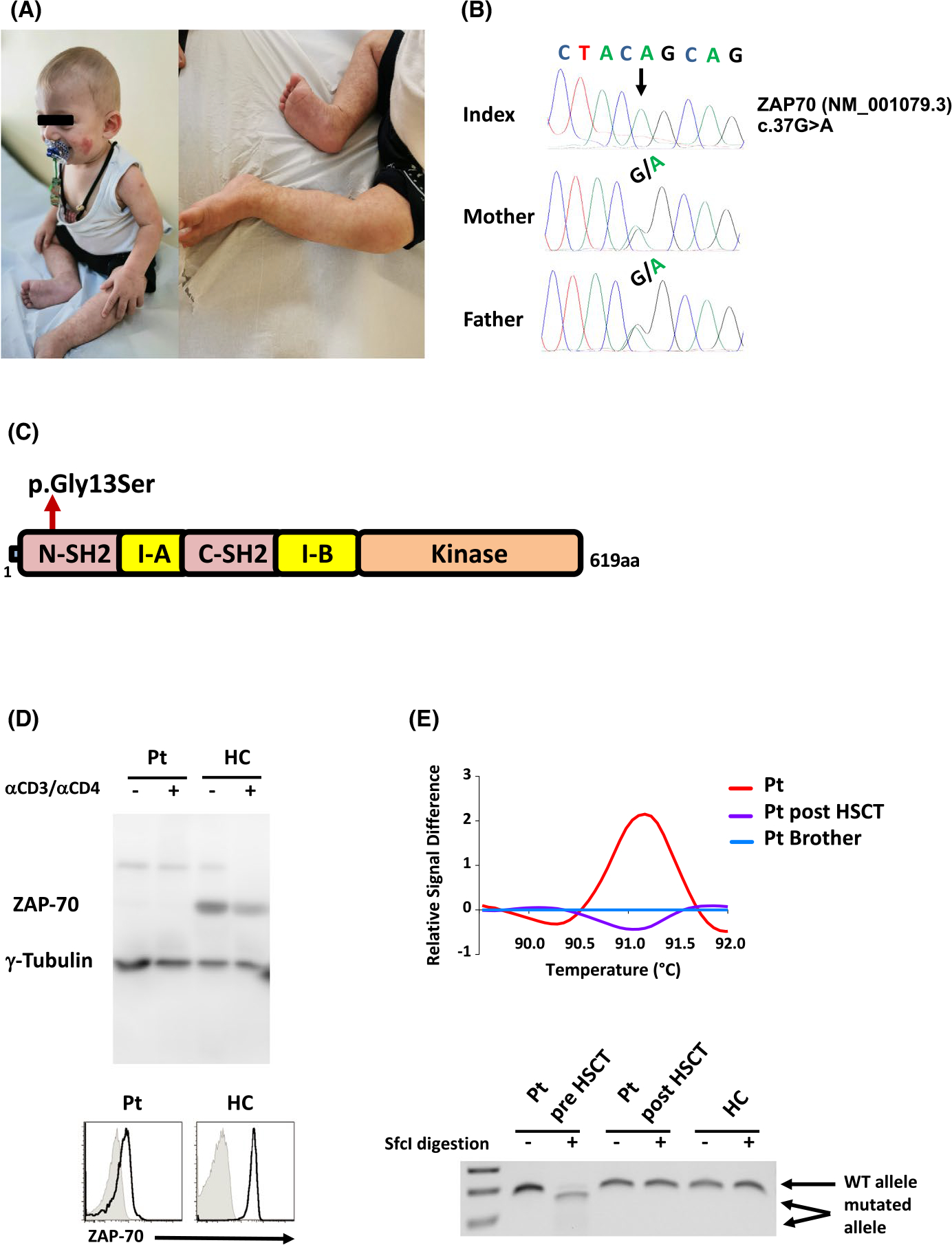
Clinical features and functional consequence of the ZAP70 mutation. (A) Cutaneous findings in the patient. A malar rash and erythematous papular lesions on the dorsum of arms, and livedo reticularis formation on the legs are shown. (B) Mutation segregation in the family depicting the 37G>A change. (C) Schematic diagram of the ZAP-70 protein showing the position of the Gly13Ser missense mutation. The domain architecture of the ZAP-70 protein consists of tandem Src homology 2 (SH2) domains, an interdomain A (I-A) between the SH2 domains, an interdomain B (I-B), and a kinase domain (D) Western blot analysis of the ZAP-70 protein in the patient (Pt) and healthy control (HC) T cells in unstimulated and anti-CD3/anti-CD4 stimulated conditions. Tubulin staining is shown as a loading control (top). ZAP-70 levels in CD4+ T cells were almost monitored by intracellular staining with a monoclonal anti-ZAP-70 Ab (solid lines) versus isotype controls (gray filled) and representative histograms are shown (bottom). (E) The PCR-amplified 169bp genomic product harboring the 37G>A mutation from the patient pre- and post-transplant, as well as from his brother can be distinguished by high-resolution melting analysis (HRM; top). Allele-specific restriction profile (SfcI) of the ZAP70 gene in the patient before (pre) and post-HSCT as well as in a HC (bottom)
TABLE 1.
Immunological evaluation of the patient before and after HSC transplantation
| Parameters | Age (6 months) | Age (9 months) | Reference values | At 10th month of HSCT | Reference values |
|---|---|---|---|---|---|
| Leukocyte count (/mm3) | 13,600 | 17,000 | 6800–17,500 | 7700 | 5500–16,500 |
| Absolute lymphocyte count (/mm3) | 4700 | 6100 | 3740–10,500 | 3100 | 2963–11,346 |
| Absolute neutrophil count (/mm3) | 5000 | 7800 | 1100–6800 | 3800 | 1200–9150 |
| Absolute monocyte count (/mm3) | 1700 (↑) | 1500 (↑) | 0–600 | 600 | 0–600 |
| Absolute eosinophil count (/mm3) | 2100 (↑) | 2330 (↑) | 0–300 | 160 | 0–300 |
| IgG (mg/dl) | 759 | 1230 (on IVIG) | 304–1231 | 1182 | 605–1430 |
| IgA (mg/dl) | 91 | 167 | 7–123 | 111 | 30–307 |
| IgM (mg/dl) | 99 | 145 | 32–263 | 67 | 66–228 |
| IgE (IU/ml) | 273 (↑) | - | 1.1–10.2 | 13.3 | 0–60 |
| Specific antibody titers | |||||
| Anti-Hbs IgG (mIU/ml) | 0.06 (↓) | - | 0–10 | - | 0–10 |
| Anti-measles IgG (IU/L) | 2.23 | - | >1.1 | - | >1.1 |
| Anti-mumps IgG (AI) | 1.0 (↓) | - | >1.1 | - | >1.1 |
| Anti-rubella IgG (IU/ml) | 0 (↓) | - | ≥15 | - | ≥15 |
| Lymphocyte subsets | |||||
| CD3+ T cell (%) | 42.3 (↓) | 46.6 (↓) | 51–85.3 | 61.3 | 51.7–84.1 |
| CD3+ T cell, count (/mm3) | 1987 (↓) | 2840 | 2340–7300 | 1890 | 1882–7372 |
| CD3+ CD4+ T (%) | 36.3 | 44.2 | 29.7–63.6 | 21.7 | 29.2–58 |
| CD3+ CD4+ T count (/mm3) | 1700 | 2696 | 1479–5050 | 672 | 1211–4696 |
| CD3+ CD8+ T (%) | 6.1 (↓) | 1.2 (↓) | 11.5–33.7 | 37.2 | 14–31 |
| CD3+ CD8+ T count (/mm3) | 287 (↓) | 73 (↓) | 584–2617 | 1147 | 567–2494 |
| CD19+ B cell (%) | 37.4 | 28.5 | 7.9–53.6 | 27.9 | 13–37 |
| CD19+ B cell, count (/mm3) | 1757 | 1738 | 450–3743 | 868 | 526–3126 |
| CD16+56+ NK cell (%) | 5 | - | 2.3–17.2 | 6.9 | 2–26 |
| CD16+56+ NK cell count (/mm3) | 235 | - | 163–894 | 214 | 105–1461 |
| Naive B cell (%) | 73 (↓) | - | 77–99 | 88.8 | 69–97 |
| NS B cell (%) | 10.7 | - | 3–13 | 4.1 | 3.6–19 |
| CS B cell (%) | 12 | - | 0.8–12 | 1.3 | 2–17 |
| CD21low CD38low activated B (%) | 2.9 | - | 0.7–6.9 | 7.4 | 0.9–9 |
| CD3+ TCRα/β cells (%) | 81.2 (↓) | - | 90–99.2 | 88.3 | 87–98 |
| CD3+ TCRγ/δ cells (%) | 9.1 | - | 2–10 | 4.8 | 2–14 |
| Recent thymic emigrants (%) | 12.8 (↓) | - | 63–90 | 54.9 | 60–83 |
| CD4+ CD45RA+ CCR7+ T cell (%) | 17.9 (↓) | - | 62–98 | 54.4 | 52–97 |
| CD4 + CD45RA- CCR7+ T cell (%) | 21.8 | - | 2.6–54 | 15.8 | 8–38 |
| CD4+ CD45RA- CCR7- T cell (%) | 52.5 (↑) | - | 0.1–13 | 25.3 | 0.3–11 |
| CD4+ CD45RA+ CCR7- T cell (%) | 7.7 | - | 0.1–76.3 | 4.5 | 0.2–55 |
| CD8+ CD45RA+ CCR7+ T cell (%) | 1.25 (↓) | - | 34–91.5 | 12.3 | 26.3–90 |
| CD8+ CD45RA- CCR7+ T cell (%) | 24.6 (↓) | - | 0.4–15.5 | 1.9 | 0.9–8.1 |
| CD8+ CD45RA- CCR7- T cell (%) | 68.3 (↓) | - | 1–68 | 42.8 | 3–40 |
| CD8+ CD45RA+ CCR7- T cell (%) | 5.8 (↑) | - | 9.6–70 | 42.9 | 13–69 |
| T-cell proliferation (anti-CD3/CD28; %) | - | Undetectable | 50–65 | 62 | 50–65 |
Note: Abnormal values are indicated in the parenthesis
Abbreviations: CS: Class switched, IVIG: Intravenous immunoglobulin, NS: Non-class switched, (-): Not done.
Next-generation sequencing revealed a novel homozygous missense mutation in the ZAP70 gene (c.37G>A), confirmed by Sanger sequencing. Both parents were heterozygous for the mutation. The other healthy brother had two WT alleles, as assessed by high-resolution melting analysis (Figure 1B and Figure S1A). The mutation led to a Gly13Ser alteration in the N-SH2 domain of ZAP-70 (Figure 1C) and was associated with a loss of protein expression analyzed by both Western blot and flow cytometry (Figure 1D). Furthermore, phytohemagglutinin (PHA) as well as anti-CD3/CD28-induced CD4+ T-cell proliferation was defective in the patient, compared to healthy family members (Figure S1B,C). Methodological details are provided in the Supplementary data.
Although the patient presented with mild clinical features, he was considered for HSCT due to the absence of ZAP-70 protein and suspected ZAP70 deficiency in the older sibling who died from severe fungal infection. At 9 months of age, he was transplanted using a bone marrow stem cell source without conditioning from a fully-matched male sibling. However, he failed to engraft. One month later, he received a second bone marrow transplantation from the same donor following a reduced toxicity conditioning regimen (treosulfan-10 mg/m2/day from days −5 to −3 and fludarabine-40 mg/m2/day from days −5 to −2) with mycophenolate mofetil and methotrexate prophylaxis for graft-versus-host disease (GVHD). Full donor engraftment was achieved, and the patient did not experience GVHD. Allele-specific DNA polymerase chain reaction analysis of the ZAP70 gene showed a normal high-resolution melting analysis and restriction profile after successful HSCT (Figure 1E). Currently, the patient is doing well, 10 months after transplantation, without any complications.
In the literature, there are three types of described mutations in ZAP70 deficiency.1 The most common is a complete loss-of-function mutation, often targeting the kinase domain. Hypomorphic mutations also exist, having been described in five patients. These patients present with less severe symptoms compared to patients with complete loss but also exhibit low levels of poorly functioning CD8+ T cells.1,3 Interestingly, Chan et al. described a compound heterozygosity for loss-and gain-of-function mutations, disrupting the autoinhibition of the protein, constituting a third form of mutations in ZAP70 deficiency. This atypical sibling pair displayed early-onset autoimmune diseases (bullous pemphigoid, colitis, hemophilia, and nephrotic syndrome) without infectious complications.4 Our patient was found to have a novel homozygous missense mutation on ZAP70, resulting in loss of protein expression and function. The mutation is located in the N-terminal SH2 domain, and to date, there are only four described patients with mutations involving this domain.1,2 Three were compound heterozygous, which included the N-SH2 and kinase domains, while the third was a single homozygous mutation found on the N-SH2 domain. Dermatitis was described in all of these patients, but apart from our report, other patients also presented respiratory and gastrointestinal infections.2,5–7
The majority of described patients with ZAP70 mutations had a clinical diagnosis of CID. Furthermore, some patients were also reported to have EBV-related lymphoproliferative disease, autoimmunity (mostly autoimmune cytopenia), hemophagocytic lymphohistiocytosis, and lymphoma.1 The cutaneous lesions in patients with ZAP-70 deficiency have consisted of an erythematous urticarial rash, bullous pemphigoid, eczema perineal ulcers, and widespread xerosis. Notably, in previously reported patients, these dermatological problems have been accompanied by more prominent clinical findings such as respiratory tract and gastrointestinal infections.1 Similar to our case, one infection-naïve patient showing eczematous skin lesions with infiltrating CD4+ T cells has been described in the literature.5 The survival and infiltration of CD4+ T cells may be attributed to an increased expression of the ZAP-70 related Syk protein tyrosine kinase, leading to an abnormal activation and differentiation of CD4+ T lymphocytes.5,8
Immunological findings of our patient were compatible with the findings of reported patients showing eosinophilia and elevated IgE levels, resembling Omenn syndrome.5,9 A unique feature of our patient was the decrease in the percentage of CD8+ T cells in the 2nd flow cytometry analysis, reflecting a loss of CD8+ T cells during the postnatal period (between 6 and 9 months of age). This phenomenon has not been described in other reported patients. However, normal to low newborn T-cell receptor excision circle levels in the same patient were reported previously, indicating that the immunological findings of disease can exacerbate over time.1 The other possibility of decrease CD8+ T cells can be explained by a potential loss of maternal T cells with age,10 but this was excluded in our patients. These data strongly suggest that serial measurements of CD8+ T cells in the setting of clinical suspicion of ZAP70 deficiency would be helpful for appropriate diagnosis.
In conclusion, ZAP70 deficiency is characterized by a broad spectrum of clinical presentations. Even in patients with subtle clinical findings, progressive reduction in CD8+ T-cell counts should evoke the possibility of ZAP70 deficiency, improving early diagnosis and promoting a better outcome for these patients.
Supplementary Material
FUNDING INFORMATION
This work was supported by grants from the Scientific and Technological Research Council of Turkey (318S202) to S.B, and AFM Telethon to V.Z. and N.T.
Footnotes
CONFLICT OF INTEREST
None to declare.
ETHICAL APPROVAL
The study was approved by the Ethics Committee of Marmara University, School of Medicine.
CONSENT TO PARTICIPATE
Informed consent for participation was obtained from the family.
CONSENT FOR PUBLICATION
Informed publication consent was obtained from the family.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/pai.13756.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
The data generated during the study are included in this published article and its supplementary file.
REFERENCES
- 1.Sharifinejad N, Jamee M, Zaki-Dizaji M, et al. Clinical, immunological, and genetic features in 49 patients with ZAP-70 deficiency: a systematic review. Front Immunol. 2020;11:831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaman K, Abrams M, Dobbs K, et al. Novel compound heterozygous mutations in ZAP70 leading to a SCID phenotype with normal downstream in vitro signaling. J Clin Immunol. 2021;41(2):470–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picard C, Dogniaux S, Chemin K, et al. Hypomorphic mutation of ZAP70 in human results in a late onset immunodeficiency and no autoimmunity. Eur J Immunol. 2009;39(7):1966–1976. [DOI] [PubMed] [Google Scholar]
- 4.Chan AY, Punwani D, Kadlecek TA, et al. A novel human autoimmune syndrome caused by combined hypomorphic and activating mutations in ZAP-70. J Exp Med. 2016;213(2):155–16 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katamura K, Tai G, Tachibana T, et al. Existence of activated and memory CD4+ T cells in peripheral blood and their skin infiltration in CD8 deficiency. Clin Exp Immunol. 1999;115(1):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aluri J, Italia K, Gupta M, Dalvi A, Bavdekar A, Madkaikar M. Low T cell receptor excision circles (TRECs) in a case of ZAP 70 deficient severe combined immunodeficiency (SCID) with a novel mutation from India. Blood Cells Mol Dis. 2016;65:95–96. [DOI] [PubMed] [Google Scholar]
- 7.Llamas-G uillén BA, Pastor N, López-Herrera G, et al. Two novel mutations in ZAP70 gene that result in human immunodeficiency. Clin Immunol. 2017;183:278–284. [DOI] [PubMed] [Google Scholar]
- 8.Noraz N, Schwarz K, Steinberg M, et al. Alternative antigen receptor (TCR) signaling in T cells derived from ZAP-70-deficient patients expressing high levels of Syk. J Biol Chem. 2000;275(21):15832–15838. [DOI] [PubMed] [Google Scholar]
- 9.Turul T, Tezcan I, Artac H, et al. Clinical heterogeneity can hamper the diagnosis of patients with ZAP70 deficiency. Eur J Pediatr. 2009;168(1):87–93. [DOI] [PubMed] [Google Scholar]
- 10.Muller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood. 2001;98(6):1847–1851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated during the study are included in this published article and its supplementary file.