

Abstract
Background:
Group 2 innate lymphoid cells (ILC2s) are critical mediators of type 2 respiratory inflammation, releasing IL-5 and IL-13 and promoting the pulmonary eosinophilia associated with allergen provocation. Although ILC2s have been shown to promote eosinophil activities, the role of eosinophils in group 2 innate lymphoid cell (ILC2) responses is less well defined.
Objective:
We sought to investigate the role of eosinophils in activation of ILC2s in models of allergic asthma and in vitro.
Methods:
Inducible eosinophil-deficient mice were exposed to allergic respiratory inflammation models of asthma, such as ovalbumin or house dust mite challenge, or to innate models of type 2 airway inflammation, such as inhalation of IL-33.Eosinophil-specific IL-4/13–deficient mice were used to address the specific roles for eosinophil-derived cytokines. Direct cell interactions between ILC2s and eosinophils were assessed by in vitro culture experiments.
Results:
Targeted depletion of eosinophils resulted in significant reductions of total and IL-5+ and IL-13+ lung ILC2s in all models of respiratory inflammation. This correlated with reductions in IL-13 levels and mucus in the airway. Eosinophil-derived IL-4/13 was necessary for both eosinophil and ILC2 accumulation in lung in allergen models. In vitro, eosinophils released soluble mediators that induced ILC2 proliferation and G protein–coupled receptor–dependent chemotaxis of ILC2s. Coculture of ILC2s and IL-33–activated eosinophils resulted in transcriptome changes in both ILC2s and eosinophils, suggesting potential novel reciprocal interactions.
Conclusion:
These studies demonstrate that eosinophils play a reciprocal role in ILC2 effector functions as part of both adaptive and innate type 2 pulmonary inflammatory events.
Keywords: Eosinophil, group 2 innate lymphoid cell, asthma, IL-33, IL-4, IL-13, lung, inflammation, eosinophil deficient
GRAPHICAL ABSTRACT

Allergic asthma is a chronic disease generally characterized by increased production of type 2 cytokines (eg, IL-4, IL-5, and IL-13) and accumulation of lymphocytes and eosinophils into the airway.1 Group 2 innate lymphoid cells (ILC2s) have been identified as critical upstream mediators of type 2 inflammation and the induced airway eosinophilia in translational models of allergic asthma.2–4 In humans, their numbers and activation state are increased in subgroups of people with asthma5–8 and chronic rhinosinusitis with nasal polyps.9 ILC2s are lineage-negative immune cells of lymphoid origin found constitutively in the lung and can recruit from other tissues when the lung is inflamed.10,11 ILC2s respond strongly to epithelial-derived cytokines, in particular IL-33, to produce IL-5, IL-13, and other mediators.12–14 More recently, ILC2s have been identified to have reciprocal interactions with other cells such as macrophages,15,16 mast cells,17 basophils,18 dendritic cells (DCs),19 adaptive immune cells,20 and stromal cells.21 Although ILC2s are proposed to be upstream mediators of eosinophils, little is known regarding the reciprocal interactions between eosinophils and ILC2s despite their co-occurrence in allergic diseases. Moreover, while mice deficient in ILC2s have dramatically reduced type 2 inflammation and eosinophil recruitment in models of asthma,3,22,23 mice deficient in eosinophils also fail to promote type 2 inflammation.24 Taken together, these findings suggest a reciprocal interaction between ILC2s and eosinophils that is not currently well defined.
Depletion of eosinophils by antibody25 or by genetic knockout (eg, congenitally eosinophil-deficient mouse [PHIL], ΔdblGATA-1, or MBP−/−EPX−/−)24,26–31 have identified eosinophils as essential for type 2 inflammation in both allergen-dependent models and transgenic cytokine overexpression models of severe eosinophilic asthma. In part, eosinophils mediate these effects through immune modulation of other cells, such as DCs, macrophages, and T cells. For example, eosinophils activate DCs,28 induce accumulation and polarization of alternatively activated (M2) macrophages,27,32,33 and recruit TH2 T cells to mouse lung on allergen challenge.27,34 In the absence of eosinophils, these events are highly attenuated and adoptive transfer of eosinophils back into eosinophil-deficient mice can increase the type 2 respiratory inflammation in models of allergic asthma. Specifically, eosinophils activated by IL-33 produce IL-13,27,33,35 which has been found to be a mediator required for increasing TH2 cells and M2 macrophage in lung, along with histopathologic changes in models of allergic asthma.27,32 Thus, similar functions exist between eosinophils and ILC2s in their ability to respond to and induce type 2 immune responses, suggesting a potential additional network between eosinophils and ILC2s in asthma.
The aim of these studies was to define the contribution of eosinophils to ILC2 activities in type 2 pulmonary inflammation and their potential reciprocal interactions in ex vivo assays. Eosinophil-deficient and inducible eosinophil-deficient mice underwent either allergen or IL-33 inhalation models of type 2 respiratory inflammation. We identified that IL-4 and IL-13 from eosinophils was critical for both eosinophil and ILC2 accumulation in allergic respiratory inflammation. In vitro experiments with purified eosinophils and ILC2s demonstrated a capacity for eosinophils to promote chemotaxis and proliferation through soluble mediators, while cell contact enhanced ILC2 activation. Analysis of the transcriptome revealed reciprocal changes in ILC2s and eosinophils on coculture. Overall, these in vivo translational studies of allergic asthma show that eosinophils are a critical component of the network of type 2 immune cells that promote ILC2 effector functions in the lungs. The data from our in vitro studies demonstrate that eosinophils have the capacity to directly modulate ILC2 activities, which may promote a reciprocal amplification of eosinophil and ILC2 functions in type 2 pulmonary inflammation.
METHODS
Mice
All mice were 8 to 12 weeks old and were bred on a C57BL/6J background. Wild-type (WT), IL-4−/−, and IL-4/13fl/fl mice were purchased from The Jackson Laboratory (Bar Harbor, Me). PHIL,31 inducible eosinophil-deficient mice (iPHIL),26 IL-5 transgenic NJ.1638,36 eosinophil-specfic Cre (eoCre)37 crossed to IL-4/13fl/fl to generate eoCre-4/13fl/fl, and IL-13−/−38 mice were maintained at Mayo Clinic Arizona. WT and/or Credeficient littermates were used as controls. Protocols and studies involving animals were performed in accordance with National Institutes of Health and Mayo Foundation Animal Care and Use Committee institutional guidelines and according to protocols granted by the Austrian Federal Ministry of Science and Research.
Lung inflammation protocols
Allergen-induced pulmonary inflammation included the sensitization and challenge of mice to either ovalbumin (OVA) or house dust mite (HDM) similar to previous protocols.26–28
OVA protocol.
Mice were provided intraperitoneal injections of OVA (A5503, Sigma-Aldrich, St Louis, Mo) with Imject Alum (Thermo Fisher Scientific, Waltham, Mass) on days 0 and 10 and were challenged with 25 μg/25 μL OVA in PBS intranasally or PBS alone on days 22–25 and humanely killed on day 28.
HDM protocol.
Mice were intranasally sensitized with 25 μg/25 μL of HDM extract (XPB82D3A2.5; Stallergenes Greer, Lenoir, NC) in PBS on days 0–2, and challenged intranasally with 25 μg/25 μL HDM or PBS alone on days 14–17 and humanely killed on day 20.
IL-33 inhalation protocol.
Mice were provided IL-33 500 ng/50 μL (R&D Systems, Minneapolis, Minn) in PBS by intratracheal transfer or PBS alone on days 4 and 6 and humanely killed on day 8.
Methods of eosinophil depletion.
As previously described,26 inducible eosinophil deficiency in iPHIL mice was achieved by intraperitoneal administration 15 ng/g body weight of diphtheria toxin (DT) (D0564; Sigma-Aldrich) before and throughout challenges to deplete eosinophils at the time of challenge. WT littermates were also provided DT.
Enzyme-linked immunosorbent assay
ELISA for IL-13 in bronchoalveolar lavage (BAL) was performed according to the manufacturer’s protocol (R&D Systems).
Histology
Lungs were fixed in formalin and processed as described previously.39 Hematoxylin and eosin (HE) staining, periodic acid–Schiff (PAS) staining, and immunohistochemistry for major basic protein 1 (MBP-1; clone MT2–14.7.3; Mayo Clinic Arizona) were completed as described previously.39 Images were captured with a Zeiss Axioscope at 20× magnification (Carl Zeiss, Jena, Germany). Mucus indexing was quantified as described elsewhere40 with some modification. Entire slides were digitally scanned with Aperio Slide scanner (Leica Biosystems, Deer Park, Ill), and image analysis was completed with QuPath v0.4.2 (qupath.githu-b.io). The ratio of mucin-containing area (mm2) was divided by the basement membrane (mm) for each airway. The average of the top 20 airways was expressed as the mucus index.
Lung isolation and flow cytometry
Total leukocytes from lungs were prepared using strategies described previously.27 Perfused lungs were digested with collagenase IV (Worthington Biochemical, Lakewood, NJ) for 30 minutes at 37°C and mechanically homogenized, then underwent lysis of red blood cells with water followed by several washes with fluorescence-activated cell sorting (FACS) buffer (PBS, 0.5% [wt/vol] BSA, 2 mmol EDTA). Single cell suspensions of lung leukocytes were counted by hemacytometer and labeled for detection of cell populations by flow cytometry. Cells were first labeled with viability dye (Fixable Viability Dye UV455 or Fixable Viability Dye eFluor 780; eBioscience; Thermo Fisher Scientific) followed by incubation with FcγR-specific blocking antibody (2.4G2) and then by fluorochrome-labeled antibodies. A list of the antibodies used in this study, along with more detailed methods, are available in this article’s Methods section in the Online Repository www.jacionline.org. All cells were gated on single cells, live cells, and CD45.2 positive before additional gating strategies. Eosinophils were (SSCmedium/hiGr1low/mediumCd11b+Siglec-F+F4/80low) and T cells (SSClowCD90+CD4+). Lung ILC2s were lineage (Lin) negative (CD3, CD4, CD8α, CD19, B220, CD11c, CD11b, Gr1, TER119, FεR1, NK1.1) and CD90+Sca-1+KLRG1medium positive, where KLRG1 is generally medium for lung ILC2s41–43 and high for inflammatory ILC2s.44 This population of ILC2s was also confirmed to be positive for CD127 and GATA-3. (For the gating strategy of ILC2s, see Fig E1 in the Online Repository at www.jacionline.org.) The Foxp3+/transcription factor fixation/staining kit (eBio-science) was used to measure Ki-67. Intracellular cytokines were measured after culture for 4 hours with eBioscience Cell Stimulation Cocktail, followed by fixation and permeabilization and intracellular labeling for IL-5 and IL-13. Some data were acquired on a Beckman Coulter (Fullerton, Calif) CyAn ADP flow cytometer, with the majority of data being acquired and confirmed by acquisition on a LSR II Fortessa flow cytometer (BD Biosciences, Franklin Lakes, NJ) by BD FACSDiva v8.0 software and analyzed by FlowJo v10.5 software (Treestar, Ashland, Ore).
ILC2 isolation and culture
Naive WT mice were intratracheally administered 500 ng/50 μL of IL-33 (R&D Systems) in PBS on days 0 and 2 and humanely killed on day 4 to isolate lung single cell suspensions as described above. Cells were depleted of Lin+ cells by magnetic bead separation according to the manufacturer’s recommendations (Miltenyi Biotec, San Diego, Calif). The lineage-depleted population was sorted by flow cytometry for ILC2s (Lin−CD45+CD90.2+Sca-1+ST2+) on a BD Biosciences FACS Aria III SORP Cell Sorter. The purity of sorted ILC2s was >99%. ILC2s were cultured in complete RPMI 1640 with IL-2 (10 ng/mL), IL-7 (10 ng/mL), IL-25 (10 ng/mL), and IL-33 (5 ng/mL) (R&D Systems) to expand and maintain ILC2s for 8–10 days. Before coculture, ILC2s were rested in IL-2 (10 ng/mL) and IL-7 (10 ng/mL) for 24 hours, followed by 24 hours of IL-7 (10 ng/mL), as previously described.45 This resulted in reduced proliferation, activation (eg, IL-5 and IL-13 production), and smaller cell morphology while maintaining >95% viability. Cells were washed 3 times in complete RPMI 1640 media to remove cytokines before coculture experiments with eosinophils. More details are available in Fig E2, in Fig E3, A, and in the Methods in the Online Repository at www.jacionline.org.
Eosinophil isolation and culture
Purified blood–derived eosinophils (>97% purity) were isolated from NJ.1638 mice as described previously.27,28 In some experiments, bone marrow–derived eosinophils26 from WT mice were generated to confirm the results found with peripheral blood eosinophils. Eosinophils were either cultured in complete RPMI 1640 media for 24 hours at 2.5 × 106/mL with IL-5 media (10 ng/mL IL-5; PeproTech, London, United Kingdom) to generate IL-5–treated eosinophils (IL-5 EOS) or were cultured in IL-33 Act media (10 ng/mL IL-5 with 10 ng/mL granulocyte-macrophage colony-stimulating factor [GM-CSF; PeproTech] and 40 ng/mL IL-33 [R&D Systems]) to generate IL-33–activated eosinophils (IL-33 Act EOS; Fig E3, B). Cells were washed 3 times in complete RPMI 1640 media to remove cytokines before coculture experiments with ILC2s. For chemotaxis assays, cell-free supernatants were collected from eosinophils cultured for 48 hours in IL-5 media or IL-33 Act media. Controls included IL-5 media and IL-33 Act media that were not cultured with eosinophils. For adoptive transfer experiments, cultured eosinophils were washed and transferred intratracheally into the lungs of OVA-sensitized/challenged mice as described previously.27
Chemotaxis assay
Supernatant from IL-5 media or IL-33 Act media cultured for 48 hours with or without eosinophils was placed in the bottom of 96-well plates that contained 3 μm Transwell inserts (Corning, Corning, NY). ILC2s (1 × 105 cells) in cytokine-free media were placed into the top of 3 μm Transwell inserts in a 96-well plate in order to measure migration into the bottom well after 1 hour at 37°C. The fluid of the lower chamber was collected, and relative cell counts were measured. Different conditions with eosinophil-conditioned media described above were used to measure chemotaxis and chemokinesis. In a set of experiments, ILC2s were pretreated for 60 minutes with pertussis toxin (Invitrogen; Thermo Fisher Scientific) at increasing doses ranging from 3 to 300 ng/mL.
ILC2 and eosinophil coculture experiments
In vitro experiments included the culture of ILC2s and eosinophils either together or alone in cytokine-free media for 24 hours with different end point measurements completed. Rested ILC2s were placed at 5 × 105 cells in cytokine-free complete RPMI 1640 media per well of a 24-well Transwell plate. For noncontact in vitro assays, ILC2s were placed on the top of 0.4 μm Transwell unit (to separate the cells) to allow only soluble components to pass through to the bottom of the well. ILC2s were cultured either alone or with 2.5 × 106 IL-5 EOS or IL-33 Act EOS that had been washed of cytokines before placing in bottom of the 24-well plate for 24-hour coculture. ILC2 proliferation and production of IL-5 and IL-13 cytokines were measured by intracellular flow cytometry. Cell-free supernatants were analyzed by multiplex Luminex assay (Eve Technologies, Calgary, Alberta, Canada). Cytospins of cells were stained with HE. In a set of experiments, eosinophils and ILC2s were isolated at the end of the 24-hour coculture by flow cytometry sorting with a BD Biosciences FACS Aria III SORP Cell Sorter to obtain pure populations of ILC2s or eosinophils for bulk RNA sequencing (RNA-Seq). More information about coculture and isolation of eosinophils and ILC2s is available in Fig E3, C and D.
RNA isolation and RNA-Seq
Isolated ILC2s and eosinophils from the 24-hour coculture were processed in TRIzol, quick frozen on dry ice, and stored at −80°C until sent to Genewiz (South Plainfield, NJ) for RNA isolation and next-generation sequencing. After raw data were trimmed and processed, differential gene expression (DEG) was determined by DESeq2 with either false discovery rate (FDR) < 0.05 or P <.05 and absolute log2-fold changes of >1. Functional pathway analysis and gene set enrichment analysis were completed with online tools such as iDEP.96,13 Reactome,46 and ToppGene.47,48 Additional information on RNA-Seq and data analysis is available in the Methods in the Online Repository www.jacionline.org. Data are deposited in the Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo) database under accession number GSE220453.
Statistical analysis
Data were analyzed by GraphPad Prism 8.1 for Windows (GraphPad Software, La Jolla, Calif). Statistical analysis was performed by unpaired Student t tests or 1-way ANOVA with post hoc analysis for comparisons depending on Gaussian distribution, standard deviations by multiple comparisons for comparison to control (Dunnet), or between all groups (Tukey). Some experiments were measured with 2-way ANOVA with Tukey post hoc multiple comparison test. Error bars represent means ± SEMs. Differences between means were considered significant and labeled as follows: ****P < .0001, ***P < .001, **P < .01, *P < .05; ns, nonsignificant.
RESULTS
Eosinophil depletion at time of allergen challenge is sufficient to attenuate ILC2 accumulation and activation in lung of OVA-treated mice
We have previously shown that type 2 lung inflammation and pathologies are dependent on eosinophils in acute models of asthma with OVA sensitization/challenge,26,28,30,31,34 yet we had not measured the effects of eosinophils on ILC2 activities in this model. To test this, we used iPHIL mice that have complete and specific depletion of eosinophils by intraperitoneal administration of DT.26
OVA-sensitized WT and iPHIL mice were administered DT 4 days before and throughout the OVA or PBS (control) challenges (Fig 1, A). The complete depletion of pulmonary eosinophils in iPHIL mice was verified in BAL fluid and lung by flow cytometry (Fig 1, B and C) and by immunohistochemistry for eosinophil-specific major basic protein 1 (MBP-1) (Fig 1, M). Total lung ILC2s were significantly reduced in OVA-treated iPHIL mice and OVA-treated WT littermates (Fig 1, D) and resulted in similar reductions to that seen with congenic eosinophil-deficient PHIL mice (Fig E1, D). We confirmed that iPHIL mice are otherwise similar to WT mice in their response to OVA-induced ILC2 accumulation when eosinophilia is maintained (Fig E1, C and D), demonstrating that the reduction in ILC2s is eosinophil dependent.
FIG 1.

Eosinophil-depletion during OVA allergen challenge significantly reduced ILC2 accumulation and activation and type 2 pulmonary inflammation. (A) Scheme representing OVA experimental model with DT treatment in WT and eosinophil-inducible depletion iPHIL mice. Single cell lung suspensions were measured by flow cytometry. Total number of eosinophils in (B) BAL fluid and (C) lung. Total lung ILC2s (D) and total lung (E) IL-5+ ILC2s and (F) IL-13+ ILC2s. (G) Representative flow cytometry plots of ILC2s (CD45+Lin−CD90+Sca-1+KLRG1+) for expression of IL-5 and IL-13. Y-axis shows C90; x-axis, IL-5 or IL-13 after gating for ILC2s. Values in parentheses are cytokine-positive ILC2s out of live cells. Summary of data for percentage (H) IL-5+ ILC2s/ILC2s and (I) IL-13+ ILC2s/ILC2s in lung. (J) Total IL-13+ TH2 cells in lung. (K) BAL fluid IL-13 levels measured by ELISA. (L) Mucus index shows relative PAS-positive pixels per airway. (M) Representative images of HE-stained slides, and PAS-stained slides for mucus (purple stain) in airway. Immunohistochemistry for major basic protein 1 (MBP-1) to identify eosinophils in lung tissue. Scale bar = 100 μm. Data are shown as means 6 SEMs (n = 4–8 mice) from 2 to 3 independent experiments. Student t test (K and L) or 1-way ANOVA with Tukey multiple comparison test (B, C, D, E, G, H, I, and J). ****P < .0001, ***P < .001, **P < .01, *P < .05.
To determine if the reduced ILC2 numbers in the lungs of eosinophil-depleted mice also displayed a reduced activation state, which is commonly measured by expression of IL-5 and IL-13, we measured these cytokines in lung ILC2 by intracellular flow cytometry. Total numbers of ILC2s expressing IL-5 and IL-13 were significantly reduced in the absence of eosinophils in OVA-treated mice (Fig 1, E and F). All of the IL-5– and IL-13–positive ILC2s also expressed ST2 (see Fig E4, A, in the Online Repository at www.jacionline.org). Moreover, ILC2s had a reduced activation state: percentages of IL-5+ and IL-13+ ILC2s out of total ILC2s (ie, the ratio of active ILC2s in lung) were reduced in eosinophil-depleted mice (Fig 1, G, H, and I, and Fig E4, B and C).
TH2 cells are also important to type 2 inflammation in allergic respiratory inflammation49 and rely on eosinophils for recruitment and activation during OVA challenge.27,28,34 As anticipated, TH2 cells expressing IL-13 and IL-5 (Fig 1, J, and Fig E4, D) were reduced in OVA-treated, eosinophil-depleted mice. These mice also had reduced IL-13 levels in BAL fluid (Fig 1, K). IL-13 is critical for mucus production in models of respiratory inflammation. Accordingly, PAS of lungs showed a reduction in mucus (ie, purple staining) in the airway epithelium of OVA-treated iPHIL mice WT mice (Fig 1, L and M). Together, these results demonstrate that conditional depletion of eosinophils at the time of OVA challenge is sufficient to reduce activation and accumulation of ILC2s along with reduced TH2 cells, IL-13 levels, and mucus accumulation in lung.
ILC2s are attenuated in mice depleted of eosinophils in an HDM model of asthma
The HDM model of asthma is distinct from the OVA model, both in type of allergen and route of sensitization,50 in addition to the reported dependency of ILC2s and T-cell interactions that promote lung inflammation.20,49 WT and iPHIL mice were administered DT 4 days before HDM challenge and throughout the challenge phase (Fig 2, A). The administration of DT was sufficient to completely ablate eosinophil accumulation in airway and lung in iPHIL mice (Fig 2, B, C, and J). Depleting eosinophils during HDM allergen challenge resulted in reduced numbers of total ILC2s (Fig 2, D) and numbers of cytokine-expressing ILC2 WT littermates (Fig 2, E and F). Unlike the OVA model, the percentage or ratio of activated ILC2s was not significantly reduced (Fig E4, E–G). However, IL-13+ TH2 cells and IL-5+ TH2 cells were significantly reduced in HDM-challenged iPHIL mice (Fig 2, G, and Fig E4, H). Taken together, reduced accumulation of IL-13–producing cells in lung was evident in the reduced levels of IL-13 in BAL fluid (Fig 2, H) and reduced mucus production in the airway (Fig 2, I and J) of iPHIL mice. These results suggest airway eosinophilia promotes accumulation of activated lung ILC2s, TH2 cells, and IL-13 levels and mucus production in the HDM model of asthma.
FIG 2.

Eosinophil-depletion during HDM allergen challenge resulted in reduced pulmonary ILC2 accumulation and type 2 inflammation. (A) Scheme representing HDM experimental model with DT treatment in WT and iPHIL mice. Total number of eosinophils in (B) BAL fluid and (C) lung. (D) Total lung ILC2s and total lung (E) IL-5+ ILC2s and (F) IL-13+ ILC2s by intracellular flow cytometry. (G) Total IL-13+ TH2 cell in lung. (H) BAL fluid IL-13 levels measured by ELISA. (I) Mucus index shows relative PAS-positive pixels per area of airway. (J) Representative slides of lunghistologyfor HE, PAS, and immunohistochemistryfor MBP-1.Scale bar = 100 μm.Data are shown as means ± SEMs(n = 3–9mice)from 2 to 3 independent experiments by Student t test(HandI)or 1-wayANOVA with Tukey multiple comparison test (B, C, D, E, F, and G). ****P < .0001, ***P < .001, **P < .01, *P < .05.
Eosinophil-derived IL-4 and IL-13 play a role in the immune network that promotes ILC2 accumulation in OVA models of allergic respiratory inflammation
We have identified above that eosinophil ablation reduced the relative number of IL-13–expressing ILC2s and TH2 cells and level of IL-13 in the airway; however, the contribution of eosinophil-derived IL-13 remains unclear. Indeed, the role of IL-4 and IL-13 from eosinophils in airway inflammation has been controversial.22,27,39,51–54 To test the endogenous role of eosinophil-derived IL-4 and IL-13 in an OVA model of asthma, we generated mice with eosinophil-specific deletion of IL-4 and IL-13 (ie, eoCre-4/13fl/fl mice). Mice with eosinophil-specific endogenous deletion of IL-4/13 had reduced eosinophil accumulation in lung in response to allergen challenge (Fig 3, A); however, this failure to accumulate was not due to changes in peripheral levels of eosinophils (see Fig E5, A, in the Online Repository at www.jacionline.org). Similar to eosinophils, ILC2 (Fig 3, B), T-cell (Fig 3, C), and CD206+ macrophage (Fig E5, B) accumulation in lung of OVA-treated eoCre-4/13fl/fl mice was reduced as compared to littermate 4/13fl/fl mice. Similar results were found when the HDM model of asthma was used (Fig E5, C). These data demonstrate that in diverse allergic models of asthma, eosinophils and/or eosinophil-derived IL-4/13 are required to promote accumulation of type 2 immune cells in the lungs with allergen challenge.
FIG 3.
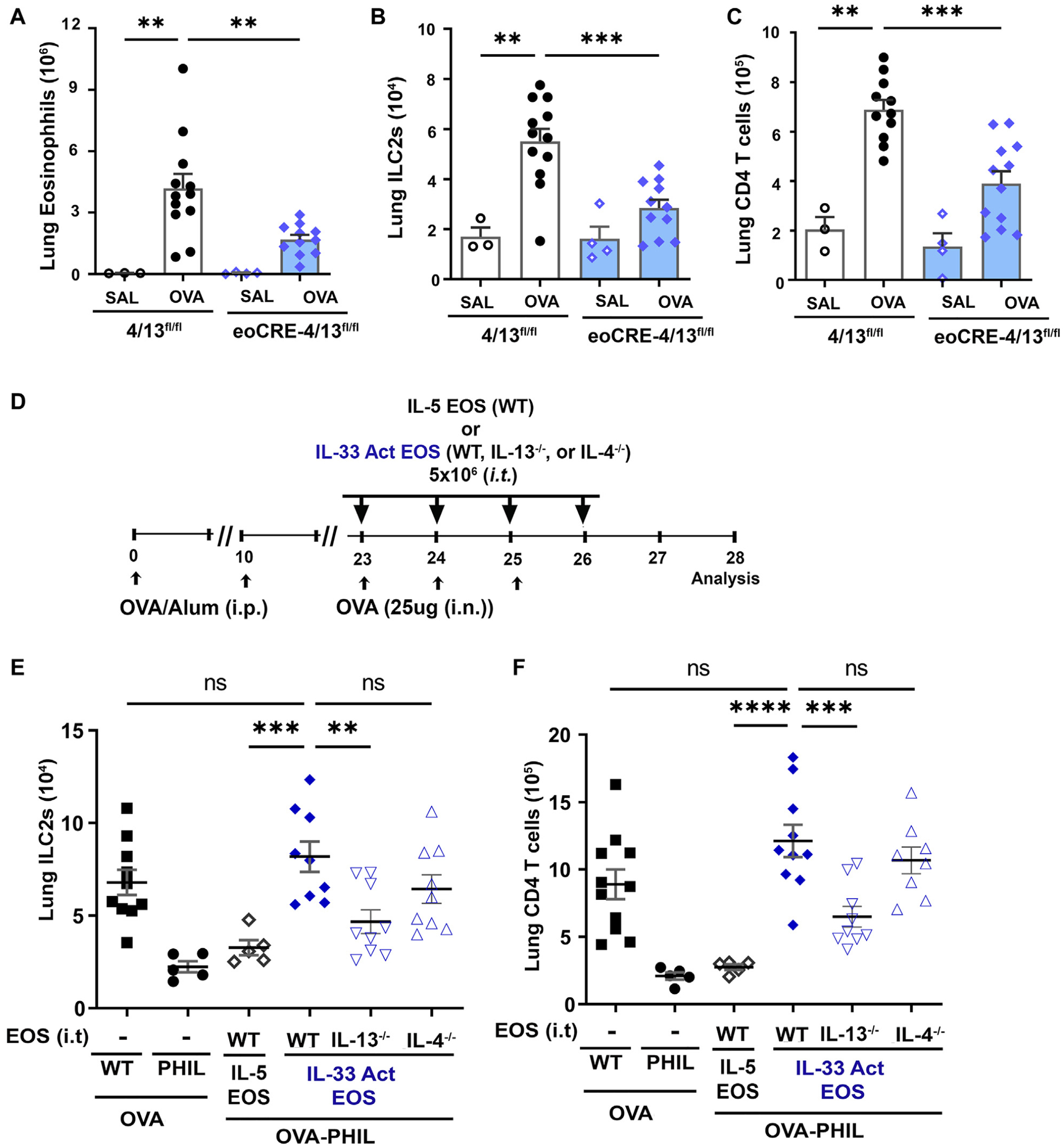
Eosinophil-derived IL-4 and IL-13 contribute to network of ILC2 and T cell accumulation in an allergic model of asthma. Mice with eosinophil-specific knockout of IL-4/13 (eoCre-4/13fl/fl) and their littermate controls (4/13fl/fl) underwent OVA sensitization/challenge. Total lung (A) eosinophils, (B) ILC2s, and (C) CD4 T cells in lung were determined by flow cytometry. (D) Scheme representing OVA experimental model with intratracheal adoptive transfer of eosinophils into eosinophil-deficient PHIL mice. WT mice and PHIL mice were provided saline as controls. Cohorts of PHIL mice received IL-5–treated WT or IL-33–activated WT, IL-13−/−, or IL-4−/− eosinophils that were adoptively transferred intratracheally during allergen challenge. Total (E) ILC2s and (F) CD4 T cells in lung were determined by flow cytometry. Data are shown as means ± SEMs (n = 3–12 mice) from 3 independent experiments by 1-way ANOVA with Tukey multiple comparison test (A, B, and C). To specifically determine the significance between adoptive transfer mice, we used 1-way ANOVA with Dunnet test and control group (WT IL-33 Act EOS) (D and E). ****P < .0001, ***P < .001, **P < .01, ns, nonsignificant.
As a result of the failure of eosinophils to accumulate/recruit into lung of eoCre-4/13fl/fl mice, we performed studies that used adoptive transfer of eosinophils directly into lung of eosinophil-deficient mice to analyze the lung-specific roles of eosinophil-derived IL-4 and IL-13. In particular, we have previously demonstrated with this model that IL-13 and not IL-4 from eosinophils was necessary for promoting TH2 cells, M2 macrophages, and lung histopathologies in eosinophil-deficient PHIL mice challenged with OVA.27 Using the same procedures27 (Fig 3, D), comparisons were made between adoptive transfer of IL-33–activated WT, IL-13–deficient, or IL-4–deficient eosinophils into lung of OVA-challenged PHIL mice. IL-5–treated eosinophils and PBS transfers were controls. As anticipated on the basis of our previous studies,27 the transfer of IL-33–activated eosinophils into lung of OVA-treated PHIL mice led to accumulation of lung ILC2s (Fig 3, E) and T cells (Fig 3, F) at levels comparable to OVA-treated WT mice. Importantly, IL-33–activated, IL-13–deficient eosinophils were unable to increase number of ILC2s and CD4 T cells in lung of mice with IL-33–activated WT or IL-4–deficient eosinophils in this adoptive transfer model. These studies demonstrate that eosinophils and/or eosinophil-derived IL-13 are required for the network of type 2 inflammation that promotes accumulation of ILC2s and CD4 T cells to lung of OVA allergen–challenged mice. Future studies will elucidate the contribution of each cell to the type 2 inflammation found in these models of allergic asthma.
Eosinophils contribute to ILC2 accumulation and activation in an innate IL-33 inhalation model of lung inflammation
The allergen models of type 2 respiratory inflammation rely on TH2 cells that have complex interactions with eosinophils and ILC2s, confounding the ability to distinguish TH2-independent roles of eosinophils with ILC2s. Therefore, we chose a T-cell–independent model of innate IL-33–induced pulmonary inflammation that induces both ILC2 and eosinophil accumulation on exposure in vivo.12,13,33 While depletion of ILC2s attenuated eosinophil responses to IL-33–induced asthma,23 the consequence of eosinophil depletion on ILC2s activities in this model remain unknown. This is important for 2 reasons in IL-33–induced lung inflammation. First, eosinophils are present in low numbers in naive lung,55 and second, eosinophils can be directly activated by IL-33.27,33 To measure the effect of eosinophil depletion on ILC2 activities, we used iPHIL mice injected with DT before inhalation of IL-33 (Fig 4, A). Pulmonary eosinophilia was ablated by DT injection (Fig 4, B and J). In the absence of eosinophils, the numbers of lung ILC2s (Fig 4, C), IL-5+ and IL-13+ ILC2s (Fig 4, D and F), and the ratio of IL-5 and IL-13 expressing (ie, activated) ILC2s (Fig 4, E and G) were significantly reduced in this IL-33–induced inflammation model. Thus, in a T-cell–independent manner, eosinophils mediate, directly or indirectly, increased ILC2 accumulation and activation state in response to IL-33–induced inflammation.
FIG 4.
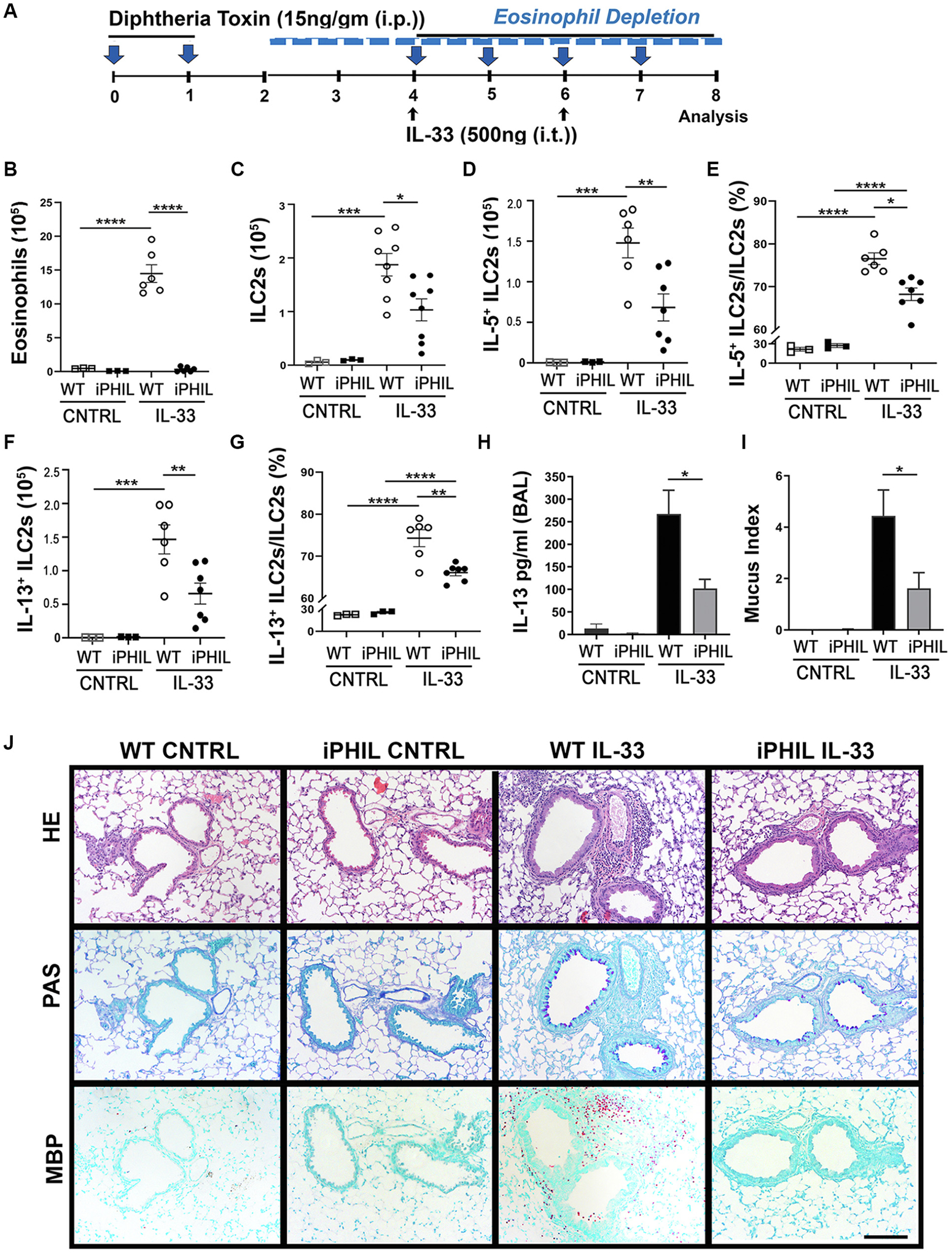
Eosinophils are required for IL-33–induced lung inflammation. (A) Scheme representing IL-33 experimental model with DT treatment in WT and iPHIL mice. Total lung (B) eosinophils and (C) ILC2s were determined by flow cytometry. Total (D) IL-5+ ILC2s and (E) percentage of IL-5+ ILC2s/ILC2s and total (F) IL-13+ ILC2s and (G) percentage of IL-131 ILC2s/ILC2s were measured by flow cytometry. (H) BAL fluid was measured for IL-13 by ELISA. (I) Mucus index shows relative PAS-positive pixels per area of airway. (J) Representative slides of lungs histology for HE, PAS, and immunohistochemistry for MBP-1. Scale bar = 100 μm. Data are shown as means ± SEMs (n = 3–8 mice) from 2 to 3 independent experiments by Student ttest (H and I) or 1-way ANOVA with Tukey multiple comparison test (B, C, D, E, F, and G). ****P < .0001, ***P < .001, **P < .01, *P < .05.
IL-33 stimulation of ILC2s and eosinophils, in addition to other innate cells, results in their production of IL-13.13 As anticipated, the combined absence of eosinophils and reduction in ILC2s in lung of iPHIL mice resulted in reduced levels of IL-13 in BAL fluid (Fig 4, H) and reduced mucus production (Fig 4, I and J). The contribution of IL-13 from each cell is difficult to measure because the standard methods of flow cytometry for intracellular staining of IL-13 (eg, phorbol 12-myristate 13-acetate [PMA]/ionomycin stimulation) results in eosinophil cell death.56 Other methods to measure IL-13 production indicate eosinophils produce less IL-13 than ILC2s, yet eosinophils are present in the airway and lung at significantly greater numbers than ILC2s (see Fig E4, A–C, and Fig E6, C, in the Online Repository). Overall, these findings demonstrate that eosinophils are required for increased IL-13 levels in the airway, and this coincides with direct or indirect roles for eosinophil-dependent activation of ILC2s and promotion of mucus production in response to IL-33–induced pulmonary inflammation.
Activated eosinophils induce chemotaxis of ILC2s through G protein–coupled receptor–dependent mechanisms
To test if eosinophils can directly influence ILC2 functions, we performed a series of in vitro studies with isolated pure populations of eosinophils and ILC2s. We first tested the ability of eosinophils to increase the migratory capacity of ILC2s in vitro. Importantly, these assays showed that conditioned media from IL-33–activated eosinophils differentially enhanced the migration of ILC2s relative to either control media (ie, no cytokines or eosinophils) or conditioned media derived from IL-5–treated eosinophils (Fig 5, A), and this was not due to chemokinesis (Fig 5, B). Pretreatment of ILC2s with pertussis toxin resulted in inhibition of migration of ILC2s, suggesting a G protein–coupled receptor (GPCR)-dependent mechanisms of chemotaxis by ILC2s (Fig 5, C). Overall, these data demonstrate a potential for activated eosinophils to promote ILC2 chemotaxis through GPCR signaling pathways.
FIG 5.

IL-33–activated eosinophils induce chemotaxis of ILC2s in a GPCR-dependent mechanism. (A) Isolated pure populations of peripheral blood eosinophils and lung-derived ILC2s were obtained to complete in vitro migration assays. Cell-free media from 48-hour cultures that included no cytokines (CN), IL-5 media, or IL-33 Act media that had been cultured with or without eosinophils were used to measure migration. After 1 hour, relative counts of ILC2s in the bottom well were counted and normalized to no cytokine in media samples. (B) Chemokinesis was tested by adding cell-free media from IL-33 Act media with eosinophils (IL-33 Act media + EOS) to either both top and bottom wells or bottom wells alone. (C) ILC2s were pretreated for 1 hour with increasing doses of PTX, then tested for response to IL-33 Act media + EOS. Migration was normalized to IL-33 Act media without EOS. Data are shown as means ± SEMs. Each data symbol represents mean value per experiment, where each experiment included >3 technical replicates. A total of 3–6 independent experiments were completed per study. We used 1-way ANOVA with Tukey multiple comparison test (A and B). To measure dose response to PTX, we used 1-way ANOVA with Dunnet test and control group IL-33 Act media + EOS 0 ng/mL PTX (C). ****P < .0001, ***P < .001. PTX, Pertussis toxin.
IL-33–activated eosinophils promote activation of ILC2s ex vivo
We have demonstrated that ILC2 activation state (eg, IL-5 and IL-13 production) was reduced in the absence of eosinophils in the in vivo studies. To better define the direct contribution of eosinophils to ILC2 activation, we completed in vitro coculture studies with isolated cell populations. In particular, we used rested ILC2s to allow for measuring the extent of activation induced by eosinophils. Rested ILC2s produce little IL-5 and IL-13 and are in a quiescent state, as reported by others.45 These rested ILC2s were either cultured alone or with IL-5–treated or IL-33–activated eosinophils that had been treated with cytokines for 24 hours and then washed of all cytokines before coculture with ILC2s. Morphologic analysis revealed that coculture of ILC2s with IL-33–activated eosinophils promoted a dramatic increase in the cytoplasmic area of rested ILC2s (Fig 6, A), which is generally indicative of an activated state for lymphocytes. The ratio of IL-13+ and IL-5+ ILC2s to total ILC2s increased on culture with IL-5–treated eosinophils that was greatly increased when cultured with IL-33–activated eosinophils (Fig 6, B). IL-13 and IL-5, in addition to GM-CSF, IL-6, and CCL5, were elevated in supernatants of the coculture of ILC2s with IL-33–activated eosinophils, while IL-4 was reduced in cells cultured alone (Fig 6, C). To determine if the activation of ILC2s by eosinophils was cell contact dependent, we completed Transwell studies (0.4 μm inserts) to physically separate eosinophils and ILC2s. We found that ILC2 and eosinophil cell contact, while not essential, was a major contributor of ILC2 activation in this in vitro system (Fig 6, D). Conversely, soluble mediators that can diffuse through the Transwell insert (ie, cell contact-independent) were sufficient to induce proliferation of ILC2s as measured by Ki-67 staining (Fig 6, E). In particular, IL-5–treated eosinophils promoted proliferation of rested ILC2s, indicating a potential for reciprocal interactions of these cells in more homeostatic environments. While eosinophils have the potential to produce IL-2 and IL-7, little IL-2 and IL-7 were detected in the culture conditions (data not shown). Taken together, these results demonstrate that IL-33–activated eosinophils induced significant cytokine production by ILC2s, and that IL-5–treated eosinophils were sufficient to promote proliferation of ILC2s.
FIG 6.

IL-33–activated eosinophils promote ILC2 activation in vitro and IL-5–treated eosinophils are sufficient to promote proliferation. Rested ILC2s were cocultured in cytokine-free media for 24 hours with no eosinophils, IL-5 EOS, or IL-33 Act EOS and then measured for morphology, cytokine expression, and proliferation at the end of 24 hours. (A) Representative HE staining of ILC2s. Scale bar = 10 μm. (B) Representative plots of intracellular IL-5 and IL-13 in ILC2s. (C) Cell-free supernatant from cocultures was measured for cytokines by multiplex assay. ILC2s were cultured alone, with IL-5 EOS, or with IL-33 Act EOS. IL-5 EOS and IL-33 Act EOS were cultured alone (no ILC2s) (right). (D) Cultures that prevented cell contact between ILC2s and eosinophils by 0.4 μm Transwell inserts (no contact) and cultures that allowed contact (contact) were measured by flow cytometry for intracellular IL-5 and IL-13 in ILC2s and (E) proliferation by Ki-67. Data are shown as means ± SEMs. One-way ANOVA with Tukey multiple comparison test (C). A representative of 3 independent experiments is shown in (D) and (E). We used 2-way ANOVA with Tukey multiple comparison test (D and E). ****P < .0001, ***P < .001, **P < .01. Specific significances are shown for ease of viewing differences between groups and conditions of “contact” and “no contact” and treatment conditions.
Transcriptome analysis of ILC2 and eosinophil coculture reveals a reciprocal relationship
To determine reciprocal changes between eosinophils and ILC2s, we measured transcriptome gene expression changes in cocultured ILC2s and IL-33–activated eosinophils along with controls. To do this, we isolated ILC2s and eosinophils from the 24-hour coculture by flow cytometry followed by bulk RNA-Seq on these sorted cells. The hierarchical clustering heat map of the 1000 most variable genes showed a clear difference between ILC2s alone and ILC2s cocultured with IL-33–activated eosinophils (Fig 7, A). Parametric gene set enrichment analysis comparisons showed that many of the differentially expressed genes were related to proliferation (Fig 7, B). In addition to proliferation, pathways related to IL-5, IL-3 and IL-9 signaling, MAPK signaling, and cellular metabolism were increased, suggesting an activated state for the ILC2s. Approximately 2128 upregulated and 1575 downregulated gene DEGs were identified. A multifactor analysis plot showed log fold changes and average expression of genes with FDR < 0.05 between the 2 conditions (Fig 7, C). Examples of upregulated genes in ILC2s cultured with IL-33–activated eosinophils revealed many typical genes representing activated ILC2s,10 such as the cytokines IL-5, IL-6, IL-13, and GM-CSF, chemokine receptors such as CCR1, CCR4, and CCR8, lipid chemokine receptors Grp183 and CYSLTR1/2, costimulatory and inhibitory receptors such as CTLA4, TNFRS4, and PDCD1, transcription factor IRF4, and other mediators such as remodeling enzymes like CAPN3, VEGFA, and SERPINE1 (Fig 7, D). Gene Ontology (GO; geneontology.org) pathways were enriched in cell cycle, cell differentiation, activation, adhesion, and cell communication (Fig 7, E). To better define enrichments in immune and signaling transduction pathways, the DEGs that mapped to Immune System and Signal Transduction data sets in Reactome46 (433 genes) were analyzed by ToppGene.47,48 The top upregulated functional pathways included VEGFA signaling; IL-4/IL-13 signaling; MAPK1/3 signaling; IL-3, IL-5, and GM-CSF signaling; and Rho GTPases, EGFR, ERK, and PDGF signaling; downregulated pathways were, for example, G alpha signaling and signaling by GPCR (Fig 7, F).
FIG 7.
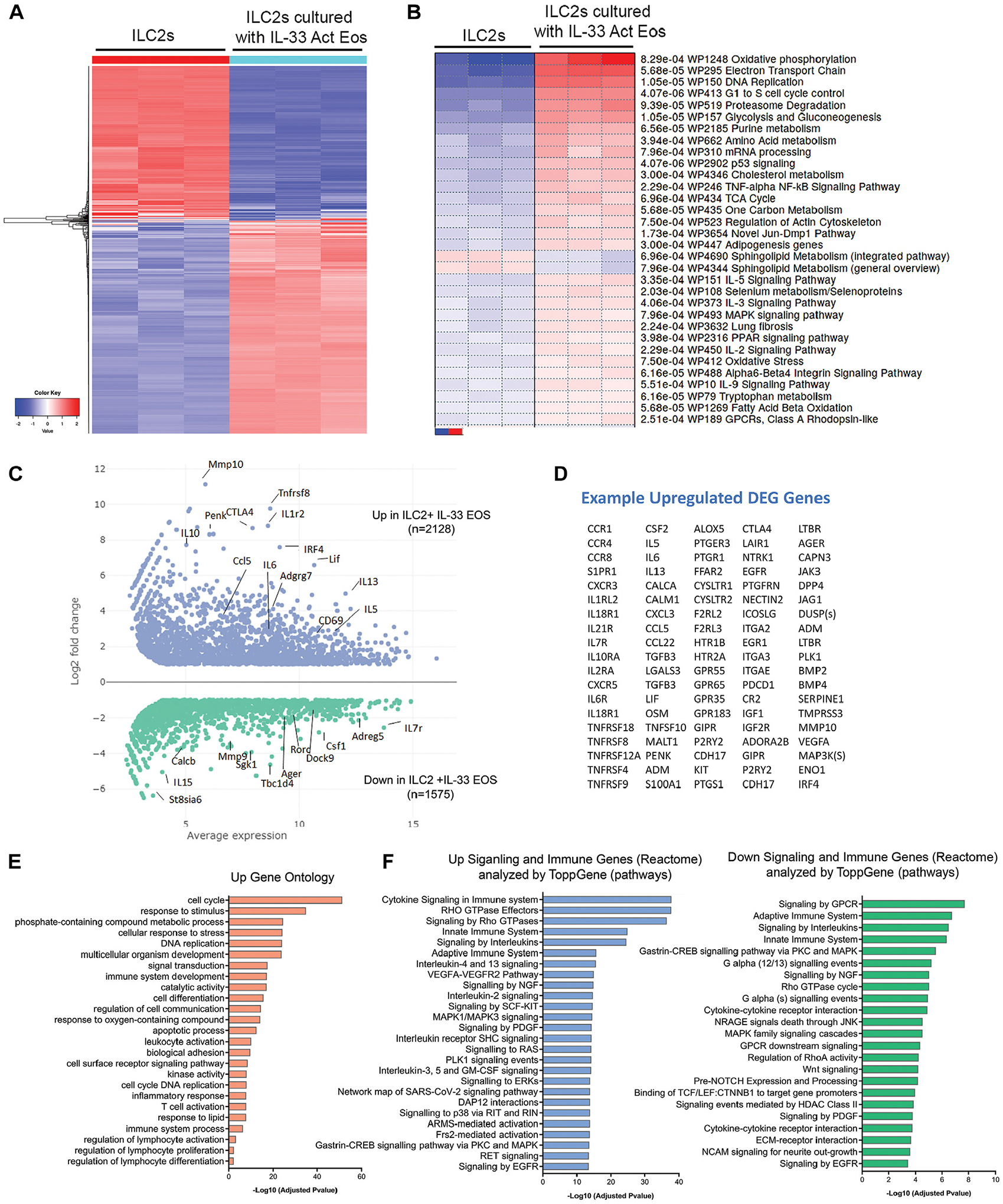
IL-33–activated eosinophils induce transcriptome changes in rested ILC2s. ILC2s and eosinophils were cultured for 24 hours as in Fig 6, then isolated for RNA-Seq. Comparisons were made between ILC2s cultured alone or with IL-33 Act EOS. (A) The 1000 most variable genes are represented in a hierarchical clustering heat map. (B) Parametric gene set enrichment (aka PGSEA) of WikiPathways. (C) MFA plot of differentially expressed genes that are FDR < 0.05 and absolute log2-fold < 1. Representative genes are labeled in MFA plot, and (D) upregulated DEG genes are shown in a table. (E) DEG genes that were FDR < 0.05 log2-fold > 1 were used to identify significant GO enrichment pathways. DEG genes that mapped to Reactome Immune System and Signal Transduction data sets were analyzed in ToppGene to identify enrichment in significant functional pathways that were both up- and downregulated between ILC2s with and without IL-33 Act EOS. MFA, Multifactor analysis.
Similar analysis was performed for eosinophils in the same coculture experiment (ie, the same batch) to allow comparison of changes in eosinophils cocultured with ILC2s. Here, IL-33–activated eosinophils with or without ILC2s were investigated. Transcriptome changes (Fig 8, A) showed that eosinophils cultured with ILC2s had increased glycolysis, prostaglandin synthesis, oxidative stress, G13 signaling, genes in the Keap1-Nrf2 pathway and reduced EGFR1 signaling, TNF-α–NF-кB signaling, and reduced integrin-mediated cell adhesion (Fig 8, B). Approximately 803 upregulated and 150 downregulated DEGs were identified (Fig 8, C). Examples of upregulated genes in IL-33–activated eosinophils cocultured with ILC2s included type 2 cytokines and chemokines such as IL-13, CCL17, CCL22, cell surface receptors CD274 and CD226, inflammation and remodeling genes such as S100A family, OSM, VEGFA, PDGF, and classical markers of upregulation in eosinophils in inflamed lung,57,58 such as CAR4, GATA-3, SELL, CD69, and FFAR2 (Fig 8, D). Enriched GO pathways included cellular activation, response to stimuli, cell communication, and collagenase activity (Fig 8, E). The top Immune System and Signal Transduction genes were enriched in functional pathways that include JAK/STAT pathways, IL-4/IL-13 pathways, signaling by GPCR, IL-6 ligand interactions, IL-10 signaling, dissolution of fibrin clotting, glycolysis, and degranulation pathways (Fig 8, F). Taken together, these transcriptome changes in ILC2s and eosinophils that occur on coculture suggest a potential for reciprocal interactions that promote changes in effector functions.
FIG 8.

ILC2s induce transcriptome changes in IL-33–activated eosinophils. ILC2s and eosinophils were cultured as in Fig 6 for 24 hours, then isolated by flow cytometry to isolate pure cell populations from culture for RNA-Seq. Comparisons were made between IL-33 Act EOS cultured with or without ILC2s. (A) Top 1000 variable genes in hierarchical clustering heat map. (B) Parametric gene set enrichment (aka PGSEA) for WikiPathways. (C) MFA plot of differentially expressed genes that are FDR < 0.05 and absolute log2-fold < 1. Representative genes are labeled in MFA plot, and (D) upregulated DEG genes are shown in a table. (E) DEG genes that were FDR < 0.05 log2-fold > 1 were used to identify significant GO-enriched pathways. DEG genes that mapped to Reactome Immune System and Signal Transduction data sets were analyzed in ToppGene to identify enrichment in significant functional pathways that were upregulated between IL-33 Act EOS with and without ILC2s. MFA, Multifactor analysis.
DISCUSSION
The presence and activation state of ILC2s are increasingly correlated with allergic diseases, and in particular asthma severity.5–8 A predominant paradigm is that ILC2s are essential upstream mediators of eosinophil activities and key inducers of type 2 inflammation in asthma. Contributing to this concept are a plethora of studies demonstrating that ILC2-deficient mice or mice deficient in ILC2 effector functions fail to recruit and activate eosinophils and fail to promote type 2 inflammation in models of lung inflammation.4,22,23 Critically, our studies suggest that ILC2 effector functions are in part dependent on eosinophils in a reciprocal network that may include other cells (eg, TH2 cells or DCs) in allergic respiratory inflammation. The implications of this are novel yet not unfamiliar, as both ILC2s10,11 and eosinophils are found in similar tissues at physiologic homeostasis and in allergic diseases.59,60 Moreover, both eosinophils and ILC2s produce similar mediators and have reciprocal interactions with innate and adaptive immune cells.
Overall, our studies demonstrated that ILC2 effector functions in models of allergic asthma and IL-33–induced pulmonary inflammation were reduced when eosinophils were ablated in mice, suggesting eosinophils, directly or indirectly, are critical for in vivo ILC2 effector functions. Using pure cell populations in vitro, we additionally discovered that eosinophils and ILC2s induced functional changes in each other that included increased cytokine production and promotion of type 2 immune responses.
A great deal of the work from our group has been to identify the immune-modulating roles of eosinophils in translational models of type 2 pulmonary inflammation, with a particular focus on allergen models or cytokine transgenic models of asthma.24,26–30,32,39 From these studies, using eosinophil-deficient mice (eg, PHIL, ΔdblGATA-1, or MBP−/−EPX−/−) or induced depletion of eosinophils, we have identified that eosinophils are necessary for TH2 accumulation, increased IL-13 levels in the airway, and mucus production in the lungs.
ILC2s activities have been reported to be variably intertwined with TH2 cells on the basis of the allergen model (eg, OVA and HDM).20,49 Because we have reported that eosinophils recruit and polarize TH2 cells at the time of allergen challenge,27,28,34 we wished to identify the effect of eosinophils on pulmonary ILC2s and TH2 cells in models of allergen-induced lung inflammation. We note that eosinophils are not required for production of antigen-specific T cells during sensitization, as demonstrated by eosinophil-deficient mice in allergen models.26,28 For this reason, congenic eosinophil-deficient mice and mice ablated of eosinophils only at the time of allergen challenge both produced similar results that showed impairment of ILC2 and TH2 accumulation in lung on allergen challenge with OVA. The activation state of the ILC2s (ie, IL-5 and IL-13 production) was also reduced, suggesting that ILC2s require eosinophils, whether directly or indirectly, to obtain full effector functions in the OVA model.
Unlike the OVA model, HDM-induced lung inflammation is more complex in that it has proteolytic enzymes; it has endotoxins that can activate Toll-like receptors; it may stimulate the release of IL-33; and it is delivered only through the airway by inhalation.20,61 Despite these variations, we similarly found reduced TH2 cells and reduced activated ILC2s in lung on depletion of eosinophils in the HDM model. The percentage of activated ILC2s was unchanged with or without eosinophils in the HDM model, which suggests that the allergen may have unique effects on ILC2 responses, independent of eosinophils. We found eosinophils to be a critical component of the immune network, leading to IL-13 and mucus production in the airway in both allergen models.
The role of IL-4 and IL-13 from eosinophils in allergen-induced inflammation models has been controversial.22,27,39,51–54 Eosinophils are well characterized to produce IL-4,62 and this cytokine has been demonstrated in vitro to act in an autocrine manner to enhance eosinophil migration to eotaxin.63 Peripheral eosinophil levels were unaffected in eoCre-4/13fl/fl mice, which suggests that the failure of airway eosinophilia in lung of OVA-treated eoCre-4/13fl/fl mice may have been due to impaired autocrine activation of IL-4/IL-4Rα and/or a reduction of total airway IL-13 levels. Reduced IL-13 could also result in altered M2 macrophage production of pulmonary eotaxins,64 particularly because lung M2 macrophages were reduced in eosinophil-deficient and eoCre-4/13fl/fl mice. In essence, these results may mimic some findings with patients treated with dupilumab, which blocks IL-4 and IL-13 binding to IL-4Rα, resulting in reduced tissue eosinophilia without reducing peripheral eosinophil levels.65 Consequently, we could not distinguish between reduced-airway eosinophilia or eosinophil-derived IL-4/13 as contributory to the attenuation of ILC2 or CD4 T-cell accumulation in eoCre-4/13fl/fl mice.
In order to bypass the need for eosinophil accumulation/recruitment to the lungs, we completed our adoptive transfer model, for which we had extensively characterized the role of IL-13 from IL-33–activated eosinophils as necessary for the accumulation of M2 macrophages, TH2 cells, TH2 chemokines, and IL-13 and mucus production in the airway and lung of eosinophil-deficient mice in the OVA model.27 Here we propose that the roles of IL-33–activated eosinophils and eosinophil-derived IL-13 are contributory to the overall type 2 immune network of responses in allergic asthma, including the accumulation of ILC2s in lung. Some reported roles of eosinophil-derived IL-13 may be underrepresented in the literature as a result of the methodology used to study IL-13 in eosinophils. The most common method of measuring intracellular cytokines by flow cytometry involves the use of PMA/ionomycin stimulation, which results in eosinophil lysis and degranulation.56 Moreover, single cell RNA-Seq of eosinophils derived from in situ tissues remains technologically complex.59 In agreement with this, measuring IL-13 by other means, such as immunohistochemistry, proteomics, ELISA, or RT-PCR, in patients with eosinophilic diseases35,66 and in the sputum of people with asthma,53,67 found eosinophils to be one of the significant IL-13–producing populations of cells. Additional studies to define this network of eosinophil, ILC2, and T-cell interactions, and the promotion of IL-13 and mucus production are warranted in future studies.
Eosinophils are found in low numbers as resident cells of the lungs, where they are proposed55 to perform the functions of a patrolling sentinel cell that is available to respond quickly to innate stimuli, such as IL-33. Most studies to date have used IL-33 or ST2 knockout mice, ILC2 knockout mice, ILC2 adoptive transfer, and in vitro experimentation to delineate the in vivo eosinophil and/or ILC2 roles in IL-33–induced lung inflammation.12,14,33 Most of these studies indicate that IL-33 activation of ILC2s is sufficient and essential to mediate eosinophil accumulation and type 2 pathologies. Yet other studies have shown that adoptive transfer of IL-33–activated eosinophils was able to restore pathologies in ST2-deficient mice,33 suggesting that eosinophils are a critical component of the IL-33–induced inflammation. In agreement with this, we demonstrated that depletion of eosinophils resulted in a significant reduction ILC2 accumulation, cytokine activation state of ILC2s, and levels of IL-13 and mucus in the airway of the IL-33–induced model of lung inflammation. These results demonstrate that in innate models of IL-33–induced lung inflammation, eosinophils are required for promoting ILC2 activities, IL-13, and mucus. However, all pathologies are not completely ablated, thus suggesting that some eosinophil-independent responses to IL-33 remain intact in this model.
Direct interactions between eosinophils and ILC2s has not been studied in great detail, and our findings indicate a potential for these cells to have reciprocal interactions that promote type 2 inflammation. Indeed, we found IL-33–activated eosinophils promoted chemotaxis of ILC2s through a GPCR-dependent mechanism. That IL-33 alone did not induce significant chemotaxis is in agreement with findings where IL-33 was suggested to stimulate other factors that induce ILC2 migration.68 The ligand/receptor for this eosinophil-dependent recruitment has yet to be identified, but it may involve both lipid mediators and chemokines.11 By permitting ILC2s to rest until they attained a quiescent state, we were able to measure the effect of eosinophils on inducing activation of ILC2s. Importantly, IL-5–treated eosinophils were sufficient to induce proliferation of ILC2s through a soluble mediator, suggesting a potential feed-forward mechanism between these cells, particularly in homeostatic environments. In addition to IL-5, GM-CSF is a survival cytokine for eosinophils and was highly produced by ILC2s on coculture. One may speculate that this may provide a means of increased eosinophil survival and priming by ILC2s in inflamed tissue, where IL-5Rα has been found to be downregulated on eosinophils in situ.69 Although the mechanisms are unknown, cell contact was required for increased activation of ILC2s. In humans, eosinophil and ILC2 clustering has been reported by others in situ in chronic rhinosinusitis nasal polyps.9 We propose that the large ratio of eosinophils (>5–10 times) to ILC2s in lung and airway in vivo, in addition to the ability of eosinophils to induce migration of ILC2s in vitro, suggests that cell contact is not unreasonable to occur in lung in type 2 respiratory inflammation.
Reciprocal interactions between these cells were identified by changes in transcriptome expression. Overall, both cells had increased gene expression in pathways of type 2 immune responses, extracellular matrix and remodeling factors, and costimulatory molecules. Additional research into some of these pathways may provide insight into coordinated promotion of type 2 inflammation and remodeling events. Finally, we acknowledge that this in vitro system may not represent the in vivo events that occur between multiple cells in type 2 allergic asthma. Moreover, it remains to be determined how or whether eosinophils may modulate ILC2s or TH2 cells in other allergen models or chronic models of asthma, where tissue-resident or memory ILC2s and TH2 cells may be predominant mediators of disease.23,70–72 Finally, these data lead us to question whether biologic agents targeting eosinophils for depletion59 will have beneficial additive effect on ILC2 numbers or activities. Overall, the results here provide new data to suggest additional studies in the biologic functions of eosinophils in interactions with ILC2s for future research.
The work behind this report began before the death of James J. Lee, and we would like to acknowledge his priceless contribution. We thank Tammy Brehm-Gibson for help in flow cytometry sorting and Grace Pyon for helping with scanning slides for mucus indexing.
METHODS
Lung single cell isolation details
After euthanasia, the circulatory system was perfused with PBS to remove red blood cells from lung vasculature before lung isolation for generating single cell suspensions. Lungs were diced and placed in a digestion solution comprising RPMI 1640 medium, GlutaMAX Supplement, HEPES (72400120; Gibco; Thermo Fisher Scientific), 2.0% FBS (A4766801; Gibco), 2 mol CaCl2, and 150 U/mL collagenase IV (Worthington Biochemical, Lakewood, NJ) for 30 minutes at 37°C with shaking. Digested lungs were then homogenized mechanically with frosted glass slides (12-552-5; Thermo Fisher Scientific), lysed of red blood cells with ice-cold cell-culture–grade water, and washed several times with FACS buffer (PBS, 0.5% [wt/vol] BSA, 2 mmol EDTA). Any aggregates were removed by passing the sample through a 40 μm nylon filter into a 50 mL conical tube and then pelleted, resuspended, and counted. Single cell suspensions of lung leukocytes were counted by hemacytometer with trypan blue to exclude debris and dead cells. Cells were then labeled and processed as described in text.
Antibody clones for labeling
F4/80 (BM8), Gr1 (RB6–8C5) CD3 (145–2C11), CD4 (GK1.5), CD8 (53–6.7), B220 (RA3–6B2), CD19 (1D3), CD11b (M1/70), CD11c (HL3), FεR1 (MAR-1), TER119 (Ter-119), NK1.1 (PK136), CD90.2 (53–2.1), ICOS (7E.17G9), CD127 (A7R34), CD45.2 (104), KLRG1 (2F1), CD25 (PC61.5), Ki-67 (So1A15), GATA-3 (TWAJ), CD206 (MR6F3), and IL-13 (eBio13A) were from eBioscience. Siglec-F (E50–2440), Sca-1 (E13–161.7), and IL-5 (TRFK5) were from BD Biosciences. ST2 (DJ8) was from MD Bio-products (St Paul, Minn).
ILC2 isolation and culture
Naive WT C57BL/6J mice were administered 500 ng of IL-33 (R&D Systems)/50 mL PBS by intratracheal administration on days 0 and 2, and lung single cell suspensions were isolated on day 4 from humanely killed mice. Cells were labeled with PE-Cy7–labeled Lin antibodies (CD3, CD4, CD8, B220, CD19, CD11b, CD11c, Gr1, FεR1, TER119, NK1.1). These were then incubated with magnetic beads conjugated with anti–PE-Cy7 according to the manufacturer’s recommendations (Miltenyi Biotec). After being run through a magnetized LS column (Miltenyi Biotec), the negative fraction was labeled with antibodies to CD90.2, Sca-1, ST2, and CD45.2 and sorted on a BD Biosciences FACS Aria III SORP Cell Sorter. Purity of ILC2s was >99%, and generally 300,000 to 800,000 cells were obtained after sorting from 4 mice pooled together. Cells were cultured at 2 × 105/mL in complete RPMI 1640 media (comprising RPMI 1640 medium, GlutaMAX Supplement, HEPES [72400120; Gibco], 20% premium-grade FBS [A4766801; Gibco], 1× penicillin–streptomycin–glutamine [10378016; Gibco], 1× 2-mercaoptoethanol [21985023; Gibco], 1 mmol sodium pyruvate [11360070; Gibco], and 1× minimal essential amino acids [11140050; Gibco]) with cytokines for maintenance, expansion, or resting. Cells were expanded and maintained in culture similar to methods described by others,E1 with some variations. Here, cells were cultured with IL-2 (10 ng/mL), IL-7 (10 ng/mL), and IL-25 (10 ng/mL) (all R&D Systems). Cytokines and media were refreshed every 2 or 3 days. In addition, it was found that adding IL-33 (5 ng/mL; R&D Systems) once 4 days after initiation of culture aided in maintaining viability, expansion, and phenotype (eg, cell surface markers, IL-13 cytokine production, and GATA-3 expression) of ILC2s for the approximately 8–10 days of expansion. Cultures expanded more than 10-fold within the 8–10 days. As described previously,E1 before coculture with eosinophils, ILC2s were rested in IL-2 (10 ng/mL) and IL-7 (10 ng/mL) for 24 hours, followed by 24 hours of IL-7 (10 ng/mL). This resulted in reduced proliferation, activation (eg, IL-5 and IL-13 production), and smaller cell morphology while maintaining >95% viability.
Culture for RNA-Seq
ILC2s and eosinophils were cultured either together or alone in cytokine-free media for 24 hours before flow cytometry sorting for bulk RNA-Seq. Thus, no exogenous cytokines were added to the 24-hour coculture. Rested ILC2s were placed at 5 × 105 cells in cytokine-free complete RPMI 1640 media per well of a 24-well Transwell plate. ILC2s were cultured alone or with 2.5 × 106 IL-33 Act EOS that had been washed of cytokines before the 24-hour coculture. A control group included IL-33 Act EOS that was not cultured with ILC2s. Thus, ILC2s alone, ILC2s with IL-33 Act EOS, and IL-33 Act EOS alone were the groups utilized for RNA-Seq. At the end of the 24-hour coculture, eosinophils and ILC2s were isolated by flow cytometry sorting with a BD Biosciences FACS Aria III SORP Cell Sorter to obtain pure populations. Approximately 700,000 to 800,000 ILC2s and ~1.5 million eosinophils per sample were isolated after sorting. Samples were kept on ice at all times, immediately pelleted after sorting, homogenized in TRIzol, quick frozen on dry ice, and stored at −80°C until further processing.
RNA isolation and RNA-Seq
RNA Sample quality control, library preparations, and sequencing reactions were conducted at Genewiz. RNA samples were quantified with a Qubit 2.0 Fluorometer (Life Technologies; Thermo Fisher Scientific), and RNA integrity was checked with a 4200 TapeStation (Agilent Technologies, Santa Clara, Calif). Ribosomal RNA depletion was performed with the Ribozero rRNA Removal Kit (Illumina, San Diego, Calif). RNA-Seq library preparation used NEBNext Ultra RNA Library Prep Kit for Illumina, following the manufacturer’s recommendations (NEB, Ipswich, Mass). Briefly, enriched RNAs were fragmented for 15 minutes at 94°C. First-strand and second-strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at the 3ˊ ends, and universal adapter was ligated to cDNA fragments, followed by index addition and library enrichment with limited-cycle PCR. Sequencing libraries were validated with the Agilent Tapestation 4200 (Agilent Technologies) and quantified by with a Qubit 2.0 Fluorometer (Invitrogen) as well as by quantitative PCR (Applied Biosystems; Thermo Fisher Scientific).
The sequencing libraries were multiplexed and clustered on a single lane of a flow cell, then loaded on the Illumina HiSeq instrument according to the manufacturer’s instructions. The samples were sequenced using a 2 × 150 paired-end configuration. Image analysis and base calling were conducted by HiSeq Control Software. Raw sequence data (BCL files) generated from Illumina HiSeq were converted into FASTQ files and demultiplexed by Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
RNA data analysis
After investigating the quality of the raw data, the sequence read was trimmed to remove possible adapter sequences and nucleotides of poor quality by Trimmomatic v.0.36. The trimmed reads were mapped to the Mus musculus reference genome available on Ensembl using the STAR aligner v.2.5.2b. The STAR aligner is a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences. BAM files were generated as a result of this step. Unique gene hit counts were calculated by feature counts from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted.
After extraction of gene hit counts, the gene hit-count table was used for downstream analysis. Genes were processed with iDEP.96,E2 which used EdgeR and average linkage and distance correlation to obtain hierarchical clustering of the top 1000 most variable genes after normalization of genes and samples. Preranked gene set enrichment analysis (aka PGSEA) was completed within the iDEP.96 software to obtain coherently altered pathways using WikiPathways.E3 Genes underwent DESeq2 with a cutoff of threshold of FDR < 0.05 log2-fold absolute > 1 to generate multifactor analysis plots. Some genes of interest were labeled. The list of DEGs generated from this methodology in iDEP.96 were nearly identical to that generated by Genewiz. Genewiz used DESeq2 and the Wald test to generate P values and log2-fold changes. Genes with adjusted P <.05 and absolute log2-fold changes of >1 were called as DEGs for each comparison. GO biologic processes and molecular functions in particular were identified by GO Profiler.E4 As a result of the overwhelming number of enrichments in proliferation- and metabolism-related pathways, the analysis was further focused and narrowed on immune and signaling pathways. DEGs were analyzed with Reactome,E5 and 433 genes mapped to the data sets for Immune System and Signaling Transduction were identified. These were then analyzed by the ToppGene Functional Annotation tool (ToppFun)E6,E7 for enrichment in the top 50 significant functional pathways that include databases from Biosystems;E8 KEGG;E9 Biosystems: Pathway Interaction Database; Biossytems: Reactome; MSigDB C2 Biocarta v7.5.1;E10 and Panther DB.E11 Nonredundant pathways were used to generate graphs. A similar process was completed with IL-33 Act EOS compared to IL-33 Act EOS with ILC2s. Here, 125 upregulated genes were identified in Reactome Immune System and Signal Transduction and placed into ToppGene for identifying significant functional pathways.
Supplementary Material
Key messages.
Eosinophils contribute to ILC2 effector responses in models of allergic pulmonary inflammation.
Eosinophils promote ILC2 migration, proliferation, and activation in ex vivo studies with pure cell populations.
Transcriptome analysis reveals a reciprocal interaction between ILC2s and eosinophils.
Acknowledgments
Supported by the Mayo Foundation for Medical Education and Research and grants from the National Institutes of Health (HL065228, AI132840, DK121330, AI145108) and the American Lung Association. Work in R.S.’s lab is supported by the Austrian Science Fund (grants P33325 and KLI887).
Abbreviations used
- BAL
Bronchoalveolar lavage
- DC
Dendritic cell
- DEG
Differential gene expression
- DT
Diphtheria toxin
- eoCre-4/13
Eosinophil-specific Cre mice deficient in IL-4/13
- FACS
Fluorescence-activated cell sorting
- FDR
False discovery rate
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- GO
Gene Ontology
- GPCR
G protein–coupled receptor
- HDM
House dust mite
- HE
Hematoxylin and eosin
- IL-33 Act EOS
IL-33–activated eosinophils
- IL-5 EOS
IL-5–treated eosinophils
- ILC2
Group 2 innate lymphoid cell
- iPHIL
Conditional eosinophil-deficient mouse
- Lin
Lineage
- MBP-1
Major basic protein 1
- OVA
Ovalbumin
- PAS
Periodic acid–Schiff
- PHIL
Congenitally eosinophil-deficient mouse
- PMA
Phorbol 12-myristate 13-acetate
- RNA-Seq
RNA sequencing
- WT
Wild type
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Hammad H, Lambrecht BN. The basic immunology of asthma. Cell 2021;184:1469–85. [DOI] [PubMed] [Google Scholar]
- 2.Bartemes KR, Kita H. Roles of innate lymphoid cells (ILCs) in allergic diseases: the 10-year anniversary for ILC2s. J Allergy Clin Immunol 2021;147:1531–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010;464(7293):1367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doherty TA, Broide DH. Airway innate lymphoid cells in the induction and regulation of allergy. Allergol Int 2019;68:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol 2014;134:671–8.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winkler C, Hochdorfer T, Israelsson E, Hasselberg A, Cavallin A, Thorn K, et al. Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J Allergy Clin Immunol 2019;144:61–9.e7. [DOI] [PubMed] [Google Scholar]
- 7.Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O’Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol 2016;137:75–86.e8. [DOI] [PubMed] [Google Scholar]
- 8.Dhariwal J, Cameron A, Trujillo-Torralbo MB, Del Rosario A, Bakhsoliani E, Paulsen M, et al. Mucosal type 2 innate lymphoid cells are a key component of the allergic response to aeroallergens. Am J Respir Crit Care Med 2017;195:1586–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol 2016;17:636–45. [DOI] [PubMed] [Google Scholar]
- 10.Barlow JL, McKenzie ANJ. Innate lymphoid cells of the lung. Annu Rev Physiol 2019;81:429–52. [DOI] [PubMed] [Google Scholar]
- 11.Matha L, Takei F, Martinez-Gonzalez I. Tissue resident and migratory group 2 innate lymphoid cells. Front Immunol 2022;13:877005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 is more potent than IL-25 in provoking IL-13–producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol 2013;132:933–41. [DOI] [PubMed] [Google Scholar]
- 13.Drake LY, Kita H. IL-33: biological properties, functions, and roles in airway disease. Immunol Rev 2017;278:173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33–responsive lineage–CD25+CD44hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol 2012;188:1503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lei A, He Y, Yang Q, Li X, Li R. Role of myeloid cells in the regulation of group 2 innate lymphoid cell-mediated allergic inflammation. Immunology 2020;161:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi M, Samuchiwal SK, Quehenberger O, Boyce JA, Balestrieri B. Macrophages regulate lung ILC2 activation via Pla2g5-dependent mechanisms. Mucosal Immunol 2018;11:615–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimokawa C, Kanaya T, Hachisuka M, Ishiwata K, Hisaeda H, Kurashima Y, et al. Mast cells are crucial for induction of group 2 innate lymphoid cells and clearance of helminth infections. Immunity 2017;46:863–74.e4. [DOI] [PubMed] [Google Scholar]
- 18.Inclan-Rico JM, Ponessa JJ, Valero-Pacheco N, Hernandez CM, Sy CB, Lemenze AD, et al. Basophils prime group 2 innate lymphoid cells for neuropeptide-mediated inhibition. Nat Immunol 2020;21:1181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng YQ, Qin ZL, Fang SB, Xu ZB, Zhang HY, Chen D, et al. Effects of myeloid and plasmacytoid dendritic cells on ILC2s in patients with allergic rhinitis. J Allergy Clin Immunol 2020;145:855–67.e8. [DOI] [PubMed] [Google Scholar]
- 20.Li BW, de Bruijn MJ, Tindemans I, Lukkes M, KleinJan A, Hoogsteden HC, et al. T cells are necessary for ILC2 activation in house dust mite–induced allergic airway inflammation in mice. Eur J Immunol 2016;46:1392–403. [DOI] [PubMed] [Google Scholar]
- 21.Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity 2019;50:707–22.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol 2012;42:1106–16. [DOI] [PubMed] [Google Scholar]
- 23.Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT Jr, Rollins DR, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol 2015;136:59–68.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobsen EA, Lee NA, Lee JJ. Re-defining the unique roles for eosinophils in allergic respiratory inflammation. Clin Exp Allergy 2014;44:1119–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kung TT, Stelts DM, Zurcher JA, Adams GK 3rd, Egan RW, Kreutner W, et al. Involvement of IL-5 in a murine model of allergic pulmonary inflammation: pro-phylactic and therapeutic effect of an anti–IL-5 antibody. Am J Respir Cell Mol Biol 1995;13:360–5. [DOI] [PubMed] [Google Scholar]
- 26.Jacobsen EA, Lesuer WE, Willetts L, Zellner KR, Mazzolini K, Antonios N, et al. Eosinophil activities modulate the immune/inflammatory character of allergic respiratory responses in mice. Allergy 2014;69:315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobsen EA, Doyle AD, Colbert DC, Zellner KR, Protheroe CA, LeSuer WE, et al. Differential activation of airway eosinophils induces IL-13–mediated allergic Th2 pulmonary responses in mice. Allergy 2015;70:1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobsen EA, Zellner KR, Colbert D, Lee NA, Lee JJ. Eosinophils regulate dendritic cells and Th2 pulmonary immune responses following allergen provocation. J Immunol 2011;187:6059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ochkur SI, Jacobsen EA, Protheroe CA, Biechele TL, Pero RS, McGarry MP, et al. Co-expression of IL-5 and eotaxin-2 in mice creates an eosinophil-dependent model of respiratory inflammation with characteristics of severe asthma. J Immunol 2007;178:7879–89. [DOI] [PubMed] [Google Scholar]
- 30.Ochkur SI, Doyle AD, Jacobsen EA, LeSuer WE, Li W, Protheroe CA, et al. Front-line science: eosinophil-deficient MBP-1 and EPX double-knockout mice link pulmonary remodeling and airway dysfunction with type 2 inflammation. J Leukoc Biol 2017;102:589–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O’Neill KR, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science 2004;305(5691):1773–6. [DOI] [PubMed] [Google Scholar]
- 32.Doyle AD, Mukherjee M, LeSuer WE, Bittner TB, Pasha SM, Frere JJ, et al. Eosinophil-derived IL-13 promotes emphysema. Eur Respir J 2019;53(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol 2010;185:3472–80. [DOI] [PubMed] [Google Scholar]
- 34.Jacobsen EA, Ochkur SI, Pero RS, Taranova AG, Protheroe CA, Colbert DC, et al. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector T cells. J Exp Med 2008;205:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchida AM, Ro G, Qiang L, Peterson KA, Round J, Dougan M, et al. Human differentiated eosinophils release IL-13 in response to IL-33 stimulation. Front Immunol 2022;13:946643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macias MP, Fitzpatrick LA, Brenneise I, McGarry MP, Lee JJ, Lee NA. Expression of IL-5 alters bone metabolism and induces ossification of the spleen in transgenic mice. J Clin Invest 2001;107:949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doyle AD, Jacobsen EA, Ochkur SI, Willetts L, Shim K, Neely J, et al. Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J Leukoc Biol 2013;94:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, et al. Impaired development of Th2 cells in IL-13–deficient mice. Immunity 1998;9:423–32. [DOI] [PubMed] [Google Scholar]
- 39.Jacobsen EA, Ochkur SI, Doyle AD, LeSuer WE, Li W, Protheroe CA, et al. Lung pathologies in a chronic inflammation mouse model are independent of eosinophil degranulation. Am J Respir Crit Care Med 2017;195:1321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Y, Abdullah LH, Doyle SP, Nguyen K, Ribeiro CM, Vasquez PA, et al. Baseline goblet cell mucin secretion in the airways exceeds stimulated secretion over extended time periods, and is sensitive to shear stress and intracellular mucin stores. PLoS One 2015;10:e0127267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li BWS, Stadhouders R, de Bruijn MJW, Lukkes M, Beerens D, Brem MD, et al. Group 2 innate lymphoid cells exhibit a dynamic phenotype in allergic airway inflammation. Front Immunol 2017;8:1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spits H, Mjosberg J. Heterogeneity of type 2 innate lymphoid cells. Nat Rev Immunol 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Entwistle LJ, Gregory LG, Oliver RA, Branchett WJ, Puttur F, Lloyd CM. Pulmonary group 2 innate lymphoid cell phenotype is context specific: determining the effect of strain, location, and stimuli. Front Immunol 2019;10:3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. IL-25–responsive, lineage-negative KLRG1hi cells are multipotential “inflammatory” type 2 innate lymphoid cells. Nat Immunol 2015;16:161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol 2016;17:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sidiropoulos K, Viteri G, Sevilla C, Jupe S, Webber M, Orlic-Milacic M, et al. Reactome enhanced pathway visualization. Bioinformatics 2017;33:3461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37: W305–11, Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danopoulos S, Bhattacharya S, Mariani TJ, Al Alam D. Transcriptional characterisation of human lung cells identifies novel mesenchymal lineage markers. Eur Respir J 2020;55:1900746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gold MJ, Antignano F, Halim TY, Hirota JA, Blanchet MR, Zaph C, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol 2014;133:1142–8. [DOI] [PubMed] [Google Scholar]
- 50.Gregory LG, Lloyd CM. Orchestrating house dust mite–associated allergy in the lung. Trends Immunol 2011;32:402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voehringer D, Reese TA, Huang X, Shinkai K, Locksley RM. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J Exp Med 2006;203:1435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Symowski C, Voehringer D. Th2 cell–derived IL-4/IL-13 promote ILC2 accumulation in the lung by ILC2-intrinsic STAT6 signaling in mice. Eur J Immunol 2019;49:1421–32. [DOI] [PubMed] [Google Scholar]
- 53.Berry MA, Parker D, Neale N, Woodman L, Morgan A, Monk P, et al. Sputum and bronchial submucosal IL-13 expression in asthma and eosinophilic bronchitis. J Allergy Clin Immunol 2004;114:1106–9. [DOI] [PubMed] [Google Scholar]
- 54.Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol 2016;17:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chojnacki A, Wojcik K, Petri B, Aulakh G, Jacobsen EA, LeSuer WE, et al. Intra-vital imaging allows real-time characterization of tissue resident eosinophils. Commun Biol 2019;2:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fukuchi M, Miyabe Y, Furutani C, Saga T, Moritoki Y, Yamada T, et al. How to detect eosinophil ETosis (EETosis) and extracellular traps. Allergol Int 2021;70: 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johansson MW. Eosinophil activation status in separate compartments and association with asthma. Front Med (Lausanne) 2017;4:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wen T, Mingler MK, Wahl B, Khorki ME, Pabst O, Zimmermann N, et al. Carbonic anhydrase IV is expressed on IL-5–activated murine eosinophils. J Immunol 2014;192:5481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobsen EA, Jackson DJ, Heffler E, Mathur SK, Bredenoord AJ, Pavord ID, et al. Eosinophil knockout humans: uncovering the role of eosinophils through eosinophil-directed biological therapies. Annu Rev Immunol 2021;39:719–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weller PF, Spencer LA. Functions of tissue-resident eosinophils. Nat Rev Immunol 2017;17:746–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol 2018;19:375–85. [DOI] [PubMed] [Google Scholar]
- 62.Spencer LA, Melo RC, Perez SA, Bafford SP, Dvorak AM, Weller PF. Cytokine receptor–mediated trafficking of preformed IL-4 in eosinophils identifies an innate immune mechanism of cytokine secretion. Proc Natl Acad Sci U S A 2006;103:3333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heller NM, Gwinn WM, Donnelly RP, Constant SL, Keegan AD. IL-4 engagement of the type I IL-4 receptor complex enhances mouse eosinophil migration to eotaxin-1 in vitro. PLoS One 2012;7:e39673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patial S, Lewis BW, Vo T, Choudhary I, Paudel K, Mao Y, et al. Myeloid-IL4Ralpha is an indispensable link in IL-33–ILCs–IL-13–IL4Ralpha axis of eosinophil recruitment in murine lungs. Sci Rep 2021;11:15465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olaguibel JM, Sastre J, Rodriguez JM, Del Pozo V. Eosinophilia induced by blocking the IL-4/IL-13 pathway: potential mechanisms and clinical outcomes. J Investig Allergol Clin Immunol 2022;32:165–80. [DOI] [PubMed] [Google Scholar]
- 66.Schmid-Grendelmeier P, Altznauer F, Fischer B, Bizer C, Straumann A, Menz G, et al. Eosinophils express functional IL-13 in eosinophilic inflammatory diseases. J Immunol 2002;169:1021–7. [DOI] [PubMed] [Google Scholar]
- 67.Schofield JPR, Burg D, Nicholas B, Strazzeri F, Brandsma J, Staykova D, et al. Stratification of asthma phenotypes by airway proteomic signatures. J Allergy Clin Immunol 2019;144:70–82. [DOI] [PubMed] [Google Scholar]
- 68.Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol 2014;133:1184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu LY, Sedgwick JB, Bates ME, Vrtis RF, Gern JE, Kita H, et al. Decreased expression of membrane IL-5 receptor alpha on human eosinophils: I. Loss of membrane IL-5 receptor alpha on airway eosinophils and increased soluble IL-5 receptor alpha in the airway after allergen challenge. J Immunol 2002;169:6452–8. [DOI] [PubMed] [Google Scholar]
- 70.Verma M, Verma D, Alam R. Role of type-2 innate lymphoid cells (ILC2s) in type-2 asthma. Curr Opin Allergy Clin Immunol 2022;22:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobayashi T, Iijima K, Matsumoto K, Lama JK, Kita H. Lung-resident CD69+ST2+ Th2 cells mediate long-term type 2 memory to inhaled antigen in mice. J Allergy Clin Immunol 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, et al. A critical role for eosinophils in allergic airways remodeling. Science 2004;305(5691):1776–9. [DOI] [PubMed] [Google Scholar]
REFERENCES
- E1.Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol 2016;17:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E2.Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics 2018; 19:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E3.Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, et al. WikiPathways: connecting communities. Nucleic Acids Res 2021;49(D1): D613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E4.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019;47(W1):W191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E5.Sidiropoulos K, Viteri G, Sevilla C, Jupe S, Webber M, Orlic-Milacic M, et al. Reactome enhanced pathway visualization. Bioinformatics 2017;33:3461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E6.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009; 37(Web Server issue):W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E7.Danopoulos S, Bhattacharya S, Mariani TJ, Al Alam D. Transcriptional characterisation of human lung cells identifies novel mesenchymal lineage markers. Eur Respir J 2020;55:1900746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E8.Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, et al. The NCBI BioSystems database. Nucleic Acids Res 2010;38(Database issue):D492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E9.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1999;27:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E10.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E11.Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, et al. Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 2019;14:703–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E12.McGarry MP, Protheroe CA, Lee JJ. Mouse hematology: a laboratory manual. Cold Spring. Harbor (NY): Cold Spring Harbor Laboratory Press; 2010. [Google Scholar]
- E13.Jacobsen EA, Doyle AD, Colbert DC, Zellner KR, Protheroe CA, LeSuer WE, et al. Differential activation of airway eosinophils induces IL-13–mediated allergic Th2 pulmonary responses in mice. Allergy 2015;70: 1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.