

Abstract
Liver regeneration (LR) is a complex process involving intricate networks of cellular connections, cytokines, and growth factors. During the early stages of LR, hepatocytes accumulate lipids, primarily triacylglycerol, and cholesterol esters, in the lipid droplets. Although it is widely accepted that this phenomenon contributes to LR, the impact of lipid droplet deposition on LR remains a matter of debate. Some studies have suggested that lipid droplet deposition has no effect or may even be detrimental to LR. This review article focuses on transient regeneration-associated steatosis and its relationship with the liver regenerative response.
INTRODUCTION
Transient regeneration-associated steatosis (TRAS) has been observed in rodent models of partial hepatectomy (PH)–induced liver regeneration (LR), and numerous studies have attempted to establish the potential connection between TRAS and LR.1,2 However, the relationship between TRAS and LR remains a subject of controversy. Following 70% PH, mice with CAV1-/- or hepatocyte-specific knockout of AKT1/2 exhibited coexisting disorders of TRAS formation and inhibition of LR.3,4 On the other hand, mice with hepatocyte Nuclear receptor corepressor 1 (NCoR1) deficiency displayed an accelerated LR process along with enhanced TRAS formation.5 However, some studies have indicated that the deletion of fatty acid binding protein (FABP) in hepatocytes impairs TRAS formation but does not affect LR.1 Moreover, in hepatocyte peroxisome proliferator-activated receptor α (PPAR)α-deleted mice, enhanced TRAS during regeneration was accompanied by impaired LR.6 The disruption of key genes involved in lipid metabolism, transport, and regulation may influence energy metabolism during LR, and the effect of the disturbance of energy metabolism on the progress of LR may play an important role in those contradictory results. The theoretical basis of most studies is the utilization of lipid droplets (LDs) as sources of energy in the regeneration process.7,8 However, a recent study has suggested that LD formation can inhibit the expression of MIER1 by influencing protein synthesis, thereby promoting the progress of LR.2 During LR, not only do triglyceride (TG)and cholesterol levels increase in the liver but there is also a transient elevation in the levels of free fatty acids (FFAs) in the peripheral blood.1,5 However, certain studies have demonstrated a downward trend in the expression levels of genes associated with de novo synthesis of FFAs during the early stages of regeneration.9 In summary, the mechanisms underlying TRAS formation and its effects on LR remain unclear. It is only through an in-depth exploration of these intricate mechanisms that TRAS can be manipulated for the advancement of LR. This review aims to provide valuable insights into the mechanism of TRAS formation and its relationship with LR, thereby facilitating a deeper understanding of this intriguing phenomenon.
LIVER REGENERATION AND GROWTH, HEPATOCYTE PROLIFERATION, AND HYPERTROPHY
Detoxification is a critical function of the liver, whereby it transforms endogenous metabolites, such as ammonia and exogenous drugs, and toxins into less toxic or nontoxic compounds.10 However, this may result in liver injury. The liver is the “injury-privileged organ” of vertebrates. It has a strong regenerative ability after injury caused by biotic (viruses, parasites, etc.) and abiotic (toxins, drugs, and PH) factors.11 Long-term chronic injury caused by NAFLD may cause some hepatocytes to proliferate to maintain liver homeostasis. However, in cases of acute injury such as PH, NAFLD may interfere with the process of LR.12,13 The energy metabolism of hepatocytes changes during LR to meet the rapid proliferation and hypertrophy requirements.14,15 LR is a complex interactive network involving multiple cellular and molecular interactions,11 with close relationships between the liver and other organs of the body.16 The main manifestations of LR are the division and hypertrophy of mature hepatocytes or progenitor cells after the death or loss of hepatocytes, which can restore the original liver weight and function.7,17 Changes in liver size can also occur under pathological and physiological conditions, such as cachexia, severe wasting, fetal development, individual growth, and pregnancy.11,18 Although there are many similar biological metabolic processes and signaling molecule transmissions in LR and liver growth, the 2 processes are not the same. In addition, hepatocyte proliferation events, such as the transplantation of an entire liver with a relatively small mass into a recipient with a large body weight, would also result in liver growth. This process can be regarded as an acute loss of parenchyma in the liver of a recipient with a large body weight, which can be categorized as LR despite relatively small transplanted livers without significant injury, as liver resection.19
The complexity of LR arises from many factors (Figure 1). First, the source of regenerating hepatocytes was multivariate.20,21 The reentry of differentiated mature hepatocytes into the cell cycle to divide and proliferate and to promote the recovery of liver weight and function plays an important role in LR after PH.20 In cases of severe liver injury resulting in significant hepatocyte loss or chronic liver injury inhibiting hepatocyte replication, liver progenitor cells emerge as the primary cellular source of LR.22,23 Furthermore, apart from liver progenitor cells, biliary epithelial cells have also been identified as one of the sources involved in LR.24,25 Second, the LR microenvironment is complex. In addition to resident hepatocytes, cholangiocytes, sinusoidal endothelial cells, KC, and HSC within the liver, numerous platelets, neutrophils, and macrophages are involved in regulating LR through extensive and complex intercellular information communication.11,26–28 Third, changes in nutrients, the vagus nerve, and mechanical forces brought about by blood flow and the extracellular matrix are also important factors affecting LR.18,29–32 There is a close relationship between the liver and other organs16; whether changes in the transcriptional levels of the kidney and lung after PH regulate the LR process need to be further studied.33 When the liver is damaged and needs to be repaired, “hepatostat”11 in the body mobilizes multiple regenerative mechanisms to quickly restore the liver’s size, weight, and function to guarantee and maintain body homeostasis.
FIGURE 1.
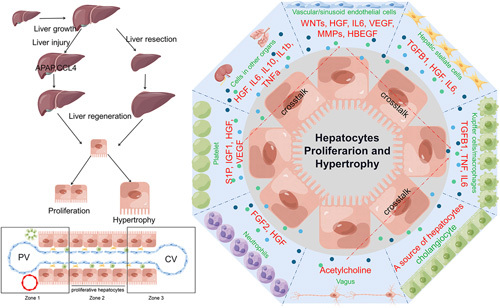
A schematic diagram of liver growth, regeneration, and regulation of intercellular crosstalk and secretion in the regenerative microenvironment. Liver growth and regeneration after injury involve the division and hypertrophy of hepatocytes. This process is not only regulated by biliary epithelial cells, sinusoidal endothelial cells, KC, and HSC but also by a large number of platelets, neutrophils, macrophages, and vagus nerves through extensive and complex intercellular communication. Significant transcriptional changes occur in other organs such as the lung and kidneys after PH. Abbreviation: PH, partial hepatectomy.
ENERGY METABOLISM OF HEPATOCYTES AND REMODELING IN LR
Hepatocytes do not continuously proliferate in adult vertebrates and play an important role in maintaining body homeostasis. The liver is responsible for the biochemical metabolism of sugars, lipids, proteins, and amino acids, and its function is conserved in mammals.34 The hepatic lobule is the basic structural and functional unit of the liver. As early as 100 years ago, Noël proposed the regionality of hepatic lobules,35 and Deane first proposed the concept of zonation.36 The application of advanced techniques has facilitated the identification of markers and metabolic functions of hepatocytes in different compartments of the hepatic lobule, as shown in Figure 2. PCK1, HAL, CPS1, CDH1, IGF1, CYP7A1, GLS2, HSD3B7, HMGCS1, HSD17B13, ASS1, and ARG1 were highly expressed in portal vein zone hepatocytes. GLUL, CYP2E1, CYP1A2, CYP3A4, OAT, IGFBP1, NT5E, ADH4, and BCHE were highly expressed in central vein zone hepatocytes. HAMP, HAMP2, IGFBP2, CYP8B1, HINT1, COX7C, APOC1, FABP1, MT2A, MT1G, and NDUFB1 were highly expressed in the hepatocytes in the middle lobule. The marker genes in the central region of the lobule have not been completely determined, and some genes in the central vein and portal vein regions extend into the hepatocytes in the middle lobule, such as CYP2E1 or ARG1.35,37 The physiological direction of blood flow and the differential expression of proteins in hepatocytes and endothelial cells at different sites determine, to some extent, the distribution of metabolic functions of hepatocytes in the hepatic lobule. Oxygen content is higher around the portal vein than around the central vein, which may indicate that periportal hepatocytes are mainly responsible for high oxygen consumption biological functions, such as protein secretion and FFA oxidative metabolism. The differential distribution of enzymes in hepatocytes further controls the spatial differential distribution of hepatocyte metabolism. Overall, hepatocytes in zone 1, in addition to being responsible for protein secretion and oxidative metabolism of FFA, perform functions such as gluconeogenesis, cholesterol biosynthesis, glutaminolysis, urea synthesis, and FFA uptake. The main functions exerted by the hepatocytes in zone 3 are glycolysis, lipid synthesis, bile acid synthesis, glutamine synthesis, and drug metabolism (high expression of CYPs). A detailed description of liver lobule zonation and metabolic heterogeneity is available in 4 review articles by Martini, Ben, Moshes, and Paris J.35,37–39
FIGURE 2.
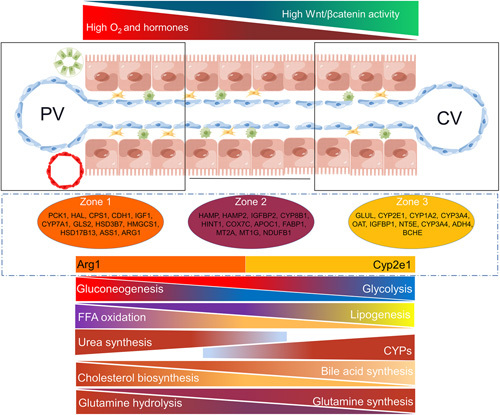
Heterogeneity of metabolism, environment, and gene expression in the hepatic lobules. The hepatic lobule has a strict spatial structure, and different regions have different environments and gene expression profiles of hepatocytes and endothelial cells. The differences in the microenvironment and gene expression together construct spatial metabolic heterogeneity at different locations in the hepatic lobule. Abbreviations: CV, central vein; PV, portal vein.
The liver is chronically exposed to damage by multiple biotic and abiotic factors, and LR is important for maintaining liver homeostasis. Two main aspects are involved in the maintenance of LR: hypertrophy and division of hepatocytes.40 However, massive loss of liver parenchyma, as described earlier, leads mature hepatocytes to reenter the cell cycle and become rapidly proliferating. Rapidly proliferating cells, such as hepatocytes during LR and cancer cells, often have a metabolic state that is distinct from that of quiescent cells.41,42 Metabolic reprogramming is one of the hallmark features of cancer, such as characteristic changes in glucose, glutamine, and lipid metabolism, and similarly, proliferating hepatocytes have metabolic reprogramming.42–44 The coordinated regulation of cellular metabolic patterning, growth, and division during tissue renewal and regeneration is a prerequisite for tissue recovery after injury, and cellular metabolism sets the stage for tissue homeostasis and regeneration.14 In terms of LR, some studies have revealed that zone 1 hepatocytes are one of the cellular sources of early regenerating liver,45,46 and lipid metabolism is the main source of energy. From recent studies, it can be inferred that after massive loss of liver parenchyma, dramatic glucose reduction may enhance lipolysis in the peripheral adipose tissue, resulting in increased plasma FFA content. FFA uptake and de novo synthesis in the liver may eventually lead to increased lipid levels in regenerating hepatocytes and serve as an energy source during LR.3–5,7,47,48 PTEN-/- mice developed hepatocyte hypertrophy and mitotic inhibition after PH, which mainly provided energy for liver hypertrophy by enhancing FFA β oxidation.7 The proliferation of hepatocytes in zone 2 plays an important role in maintaining liver homeostasis, but for zone 1 and zone 3 liver injuries caused by the 3,5-diethoxycarbonyl-1,4-dihydro-collidine (DDC) diet and CCl4 injection, the source of proliferating hepatocytes is distributed in the noninjured area.49 These findings indicate that hepatocytes with proliferative capacity are distributed in different positions within the hepatic lobules, enabling them to better respond to liver damage caused by different factors and contribute to the maintenance and restoration of liver homeostasis. While some researchers believe that the process of LR after PH is mainly derived from hepatocytes in zone 2, there is also evidence of proliferating hepatocytes in zone 1 and zone 3.50 The energy sources on which the LR process depends are not always lipids in different genetic backgrounds in mice.8 The division of hepatocytes in the regenerating liver of CDK1-/- mice is inhibited, and the original liver size is restored mainly through hepatocyte hypertrophy. The level of mitochondrial oxidative metabolism is reduced in hypertrophied hepatocytes; however, the metabolic flux of ALT is markedly increased for energy supply through pyruvate oxidative metabolism.15 Significant metabolic remodeling occurs during LR, which can meet the demand for energy and substance-synthetic substrates required for cell hypertrophy and proliferation to maintain homeostasis of liver metabolism and structural function.
POTENTIAL LINK BETWEEN LIPOLYSIS IN ADIPOSE TISSUE AND LR
When the body’s energy supply is adequate, adipocytes store FFA inside the cells as nontoxic TG, which is an important energy depot for the body. In the fasted state, FFAs are released from white adipose tissue lipolysis into the blood, where transport through serum albumin to energy-consuming tissues serves to supply energy and biosynthetic substrates. Both neutral and acidic lipolysis are involved in the lipolysis of peripheral adipose tissue. Acid lipolysis is carried out mainly by lysosomal acid lipase in lysosomes, whereas neutral lipolysis is mediated mainly by adipose triglyceride lipase (ATGL), hormone sensitive lipase (HSL), and monoacylglycerol lipase51; 90% of TG breakdown is carried out by ATGL and HSL.52 In addition, lipolysis also occurs in nonadipose tissues and is mediated by noncanonical pathways that have not yet been fully defined.51 The regulatory mechanisms of ATGL and HSL are highly complex, with multiple endocrine, autocrine, and paracrine factors, hormones, and neurotransmitters that regulate ATGL and HSL at the transcriptional and posttranscriptional levels to induce lipolysis of adipose tissue. These include catecholamines (adrenaline and noradrenaline), glucocorticoids, thyroid hormones, eicosanoids, atrial natriuretic hormone, growth hormone, IL (IL6 and IL1β), TNFα, and leptin.51 In contrast, insulin is a well-defined inhibitor of lipolysis.51 In addition, endothelin 1; sphingosine-1-phosphate, and prostaglandin E2 can participate in the regulation of lipolysis in adipose tissue to a certain extent.53–58 During LR, hepatocyte proliferation undergoes significant alterations in energy metabolism, and the demand for energy sources shifts accordingly. Cytokine, hormone, and metabolite storms occur during LR. After PH, serum leptin levels increase markedly, accompanied by a marked decrease in blood glucose.59,60 Many metabolites, such as sphingosine-1-phosphate derived from sphingolipid metabolism and the prostaglandin family of arachidonic acid metabolites (eg, prostaglandin E2), are generated in the regenerative microenvironment.61,62 Inflammatory factors and cytokines also produce changes as LR occurs and progresses, such as the IL family (IL6 and IL1β), the TNFα family, the TGFβ family, the EGF and FGF families, and others.11 Changes in mechanical forces are sensed by vascular endothelial cells, with the consequent secretion of a large number of vascular endothelial cell-derived cytokines and regulatory proteins such as endothelin 1.63 During LR, a large number of cytokines and metabolites are present in the peripheral blood, some of which are the same factors that regulate lipolysis in peripheral adipose tissue. However, whether LR regulates TG hydrolysis in peripheral adipose tissue and whether the secretory cytokine storm has an impact on the adipose tissue lipolysis process remain unclear. Herein, we delineated the cues that may regulate adipocyte lipid breakdown during LR based on literature data, as shown in Figure 3.
FIGURE 3.
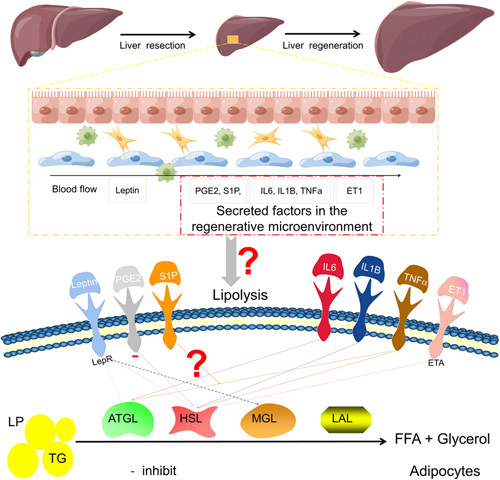
The potential link between LR and peripheral adipose tissue lipolysis. During LR, cytokines and metabolite storms are produced, such as interleukin family, TNF family, ET1, S1P, and PGE2, some of which have been shown to affect lipolysis in peripheral adipose tissue. Abbreviations: ET1, endothelin 1; LR, liver regeneration; PGE2, prostaglandin E2; SIP, sphingosine-1-phosphate.
METABOLISM OF FFA IN HEPATOCYTES
Hepatocytes are the core sites of lipid metabolism, participating in balancing and buffering lipid metabolism within the body, and therefore, often experience increased LD deposition in pathological hepatocytes. The sources of TG or FFA in hepatocytes mainly include the following: (1) excess dietary TGs are transported to the liver through chylomicrons (CM), (2) increased FFA synthesis from de novo lipogenesis, (3) excess FFA influx into hepatocytes from lipolysis within adipose tissue, (4) reduced export of lipids from the liver through VLDL, and (5) reduced oxidation of FFA.64 Dietary TGs are decomposed into monoacylglycerol and FFA by treatment with bile acid and pancreatic lipase. After being absorbed by intestinal mucosal epithelial cells, TGs are resynthesized, packaged as CM, and enter blood circulation through the lymphatic system.65–67 CM utilization is facilitated by lipoprotein lipase in the microvasculature of adipose and muscle tissues.68 CM remnants not utilized by adipose and muscle tissues are transported to the liver, where they are further metabolized.69 Hydrolysis of TG in peripheral adipose tissue proceeds through a series of lipase-mediated lipolyses, which releases FFA.51 Multiple proteins have been identified to be involved in the transport of FFA from plasma into hepatocytes, including plasma membrane FABP, CD36, caveolin-1, and very long-chain acyl CoA synthetases (ACSVL/FA transport proteins, also known as FATP/solute carrier family 27A1-6, SLC27A1-6).64,68,70 In addition to FFA released from peripheral adipose tissue, de novo synthesis of FFA in hepatocytes can also lead to an increase in TG content in hepatocytes.68,69 De novo synthesis of FFA is carried out in the cytoplasm, and its synthetic substrate acetyl-CoA is mainly in the mitochondria and cannot freely cross the mitochondrial membrane. Therefore, a citrate-pyruvate transport system in the cell promotes the transport of acetyl-CoA from the mitochondria to the cytoplasm for FFA synthesis. Acetyl-CoA is first converted to malonyl-CoA by ACC, and malonyl-CoA is then converted to palmitate by FAS.68 FFA oxidative metabolism is used to provide energy, mainly in the mitochondria, which, in turn, reduces lipid levels in the liver. Once the intracellular FFA oxidative metabolism is inhibited, it is obvious that the intracellular FFA utilization is reduced, resulting in fat accumulation, manifested as intracellular LD deposition. In addition to FFA oxidative metabolism, outward transport is the only way hepatocytes reduce intracellular lipids. Because of its hydrophobicity, microsomal triglyceride transfer protein (MTP) assists TG in the endoplasmic reticulum and Golgi apparatus with cholesterol, phospholipids, and ApoB100 together packaged into water-soluble VLDL before being outputted from the liver.64,68,70 A schematic diagram of FFA metabolism in hepatocytes is shown in Figure 4.
FIGURE 4.
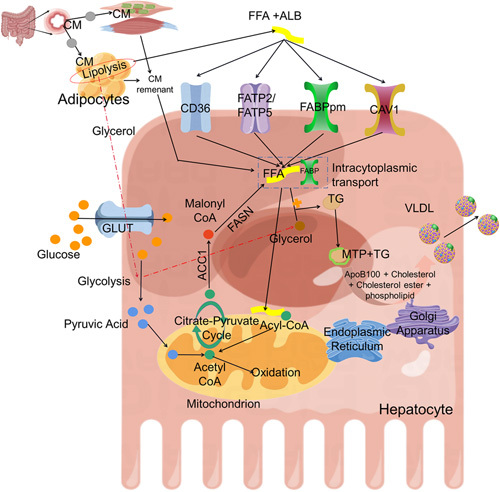
Schematic diagram of FFA metabolism in hepatocytes. Hepatocytes are important centers of lipid metabolism. First, fat in the digestive tract is absorbed into the blood in the form of chylomicrons(CM), and part of it is stored and utilized in adipose tissue and muscle tissue. CM remnants enter the liver for further metabolism. In peripheral adipose tissue, TG is hydrolyzed by lipase under the stimulation of various factors. FFAs are released and combined with ALB (albumin) to transport in the blood and then transported into the liver through the FFA transporter on the surface of the liver cell membrane. It can be converted into LD stored in cells and can also be further packaged into VLDL to secrete hepatocytes. FFA provides energy through oxidative metabolism in mitochondria. In addition to the above metabolism, cells can also use acetyl coenzyme A for the de novo synthesis of FFA. Abbreviations: ALB, albumin; CM, chylomicrons; FFA, free fatty acids; MTP, microsomal triglyceride transfer protein; TG, triglyceride.
LD DEPOSITION IN LR
During the early phase of LR, large amounts of TGs are stored in hepatocyte LD, mainly composed of TG and cholesteryl esters.48,71,72 This phenomenon has been defined by some scholars as TRAS.7 Interestingly, some investigators have revealed that there are 4 times increased levels of hepatocyte proliferation during LR after PH, accompanied by 3 times increased levels of LD accumulation.73 In comparison to standard PH (70%), extended PH (86%) has been associated with higher mortality rates and a lower rate of hepatocyte proliferation. MRI, lipid content analysis, and pathological examination have consistently demonstrated significantly higher lipid content in the liver of the extended PH group compared with the standard PH group at 48 hours after operation. This increased LD deposition is closely associated with mortality in the extended PH group.74 Metabolism is dynamically altered during LR after PH, and the division of labor among hepatocyte populations balances the proliferation and metabolic requirements of the regenerating liver.50 The change in gene expression of lipid metabolism can clarify that TRAS is a biological process with strict regulation.75 With more in-depth studies on LR, the mechanism of TRAS development and its effects on LR are gradually being recognized. However, there is no consensus regarding the relationship between TRAS and LR.1,48,76,77 According to earlier studies, there are 3 possible combinations of the interaction between TRAS and LR: (1) the formation of LD had little impact on LR; (2) the accumulation of LD promotes LR; and (3) the formation of LD impedes LR. Although studies have shown that the expression levels of genes related to de novo synthesis of FFA are inhibited during LR,9 some researchers still believe that LD deposition may also be partially derived from de novo FFA synthesis in hepatocytes.5 In addition to de novo synthesis and transport, VLDL-mediated lipid output and oxidative metabolism of hepatocytes also affect TRAS. The relationship between TRAS and LR is summarized in Supplemental Table S1, http://links.lww.com/HC9/A521. At the same time, we also described the interference of those influencing factors on lipid metabolism in the liver (Figure 5). Taken together with the relationship between TRAS and LR in previous literature, we summarize that there may be the following 5 reasons for the above 3 phenotypes: (1) impaired LR coexists with increased LD deposition: metabolic inhibition of FFA leads to increased accumulation of LD, and energy supply impairment of regenerating hepatocytes results in LR inhibition, as typically exemplified by PPAR α effects on LR; (2) reduced LD deposition inhibits LR: severe impairment of hepatic FFA transport induces reduced LD deposition in the liver, leading to insufficient supply for subsequent oxidative metabolism, inhibiting LR, such as CAV1 deficiency (partly related to the suppression of oxidative metabolism); (3) reduced LD deposition inhibits LR: impaired storage of lipids results in reduced LD that inhibits LR, as knockout of Plin2 causes increased VLDL formation and facilitates lipid transport out of hepatocytes; (4) LD deposition does not affect LR: some transporters affect lipid transport to a lesser extent, other lipid transporters compensate, and the deposited LD are sufficient to cope with the regenerative energy requirements; (5) LD deposition did not affect LR: there was no difference in the peak of fat accumulation, but the time of fatty accumulation was extended. In LR, hepatocyte hypertrophy substitutes for hepatocyte proliferation to restore liver homeostasis. For example, PTEN deficiency enhances the oxidative metabolism in hepatocytes. The relationship between TRAS and LR is intricate, and genetically engineered mice often harbor numerous associated gene expression changes that influence the relationship between TRAS and LR. Next, we describe the complex relationships in detail.
FIGURE 5.
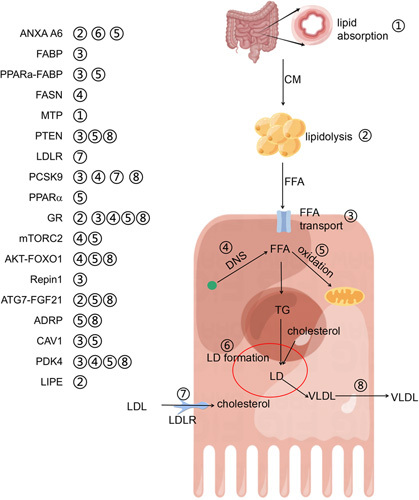
A brief summary of the regulatory mechanism of lipid droplet (LD) deposition during liver regeneration as reported in the literature. This figure depicts eight important processes of lipid metabolism: ① lipid absorption in the digestive tract; ② lipid hydrolysis in peripheral adipose tissue; ③ transport of free fatty acids into hepatocytes; ④ de novo synthesis of fatty acids; ⑤ oxidative metabolism of fatty acids; ⑥ formation of lipid droplets; ⑦ LDL transport into hepatocytes; and ⑧ VLDL assembly and transport outside hepatocytes. The left-hand side of the figure summarizes important regulatory factors and their potential involvement in lipid metabolism. The red circle represents the formation of LD, while the green dots represent acetyl-CoA. Abbreviations: CM, chylomicron; DNS, de novo synthesis; LD, liquid droplet.
LD deposition does not affect LR
Annexin A6
Annexin A6 (ANXA A6), a member of the annexin family, is involved in a variety of cellular functions, such as membrane transport, cholesterol homeostasis, and signal transduction.78–81 ANXA A6 gene knockout has no significant effect on normal growth and development in mice82 but can avoid weight gain and obesity caused by high-fat feeding.83 ANXA A6 plays an important role in energy metabolism.14 In ANXA A6-/- mice, fatal hypoglycemia occurred after PH, the survival rate was severely reduced, and LD deposition was delayed during LR but did not affect the peak of LD deposition. Although the expression of ANXA A6 by the adeno-associated virus and glucose supplementation were able to rescue death after PH, the authors did not investigate its effect on LD deposition. Interestingly, despite increased mortality, aggravation of hypoglycemia, and delayed appearance of the peak of LD deposition after PH, reentry into the hepatocyte cell cycle was not inhibited in the early phase of LR.60 The regulatory mechanism of ANXA A6 on LD deposition during LR is unclear, but previous findings seem to provide some hints. The effect of ANXA A6 on LD metabolism differs among cells. First, primary hepatocytes from ANXA A6-/- mice and cell lines in which ANXA A6 was knocked down exhibited a reduction in LD deposition after OA treatment, possibly through a mechanism involving cytoplasmic phospholipase A2α inhibition84 because cytoplasmic phospholipase A2α was involved in the formation of LD.85 Second, loss of ANXA A6 results in increased LD deposition in adipocytes, along with severely suppressed catecholamine-induced phosphorylation of HSL, impairing lipolysis in adipocytes.86 These mechanisms may play an important role in inhibiting LD deposition during LR after PH in ANXA A6-/- mice. Although there is no study on the effect of downregulation of ANXA A6 expression on FFA metabolism in hepatocytes, some researchers found that the downregulation of ANXA A6 expression in triple-negative breast cancer can increase the expression of FABP4 and also promote the uptake and oxidative metabolism of FFA in mitochondria with intracellular lipid reducing.87 The decrease in LD deposition caused by ANXA A6 deficiency may come from many aspects, such as decreased lipolysis of peripheral TG, inhibition of LD formation, and enhanced oxidative metabolism.
FABP, PPARα- FABP, and FAS in hepatocytes and MTP in the small intestine
The complete process of lipid metabolism involves the absorption of lipids in the digestive tract, lipolysis of peripheral adipose tissue, import of FFA by fatty acid transport-related proteins to hepatocytes, export of lipids from hepatocytes through VLDL, and de novo synthesis and oxidation in cells.88 Four knockout models have been used to study the effect of lipid metabolism on TRAS and regeneration, namely, hepatocyte-specific knockout FABP mice, small intestine-specific knockout MTP mice, hepatocyte PPARα and FABP double knockout, and hepatocyte-specific knockout FAS mice. However, there was no correlation between TRAS and LR.1 It is important to note, however, that TRAS mainly arises from lipolysis in peripheral adipose tissue and possibly in part from de novo synthesis, so TG only partially decreased during LR as a result of small intestine-specific knockout of MTP. Serum FFA levels were significantly higher at 6 hours. MTP in the small intestine is very important for CM-mediated lipid absorption. The absence of MTP leads to a large accumulation of TG in small intestinal mucosal cells and reduced absorption.89 There are few studies on MTP and fat metabolism in the small intestine; however, the absence of MTP in adipose tissue significantly promotes ATGL-mediated lipolysis.90 There is no clear conclusion on whether the deinhibition of ATGL by MTP deficiency exists in the small intestine at the same time, but it may also be a possible reason for the increase in serum FFA levels. FABP is a fatty acid transport-related protein in hepatocytes, and hepatocyte-specific knockout of FABP resulted in a partial decline in hepatic TG during LR. The overall trend of hepatic TG in both groups of mice was consistent with that in control mice. There are 4 mechanisms of FA transport on the hepatocyte surface: CD36, FABP, FATP, and CAV1.64 The knockout of FABP may only partially affect FA transport and LD formation. In the hepatocyte-specific knockout FAS model, although some researchers have suggested that the de novo synthesis of FFA is related to the formation of LD during LR,5 LD deposition and LR were not significantly affected. The relationship between the de novo synthesis of FFA and the formation of LD and the extent of their contribution still needs to be clarified. Finally, in the PPARα and PPARα-FABP double knockout mouse model, knockout of FABP reversed the increased LD deposition resulting from PPARα knockout, illustrating that FABP plays a role in the increased TG content during LR. Paradoxically, however, the author did not find a difference in LR, possibly because they did not dynamically analyze changes throughout the LR. Although the above 4 models have found that LR is not related to lipid deposition, the above 4 models may not truly reflect the relationship between TRAS and LR. This may be because other homofunctional proteins or processes may play a compensatory role in the case of interfering with a single factor affecting lipid metabolism.
PTEN
PTEN, a tumor proliferation suppressor with dual-specificity phosphatase activity of lipid phosphatase and protein phosphatase, is involved in the regulation of several signaling pathways and plays an important role in cell growth, cell cycle, and material metabolism.91 The lipid phosphatase properties of PTEN are mainly able to inactivate phosphatidylinositol triphosphate by converting phosphatidylinositol triphosphate dephosphorylation to phosphatidylinositol bisphosphate, which further inhibits Pl3K/AKT and the activation of downstream signaling pathways.92 PI3K/AKT is a core signaling pathway involved in cell proliferation, cell cycle regulation, and substance metabolism in cancer and LR.93,94 First, PTEN-specific deletion in adipose tissue improved glucose tolerance and increased insulin sensitivity in obese and diabetic mice.95 Second, the specific knockout of PTEN in hepatocytes leads to markedly increased lipid synthesis and lipid import in the liver, resulting in fatty liver disease and even progression to liver cancer.96–98 Third, the downregulation of PTEN expression leads to decreased assembly and secretion of VLDL in hepatocytes.99 The downregulation of PTEN has been found in several patient and animal models of diseases, such as HBV/HCV infection, metabolic syndrome, and hepatoma progression triggered by dietary unsaturated FFA.100–102 Fat accumulation in the liver occurs early in the LR process, and a decrease in PTEN expression in liver tissue occurs during this process.7 Some evidence indicates that PTEN is involved in the process of LR and the regulation of energy metabolism.7,103 In the LR model of hepatocyte-specific PTEN knockout mice, liver weight increased, liver cell area expanded, and hypertrophy was observed, but mitosis decreased. Although the deletion of PTEN in hepatocytes resulted in increased LD deposition in nonproliferating hepatocytes, the peak of lipid deposition during LR was not significantly increased but prolonged the time of lipid content elevation. Downregulation of PTEN expression induces AKT signaling pathway activation, leading to enhanced lipid oxidation, thereby mediating metabolic adjustments and hepatocyte hypertrophy during LR.7 However, a recent study found that PARK7 knockout led to an increase in PTEN expression levels, inhibiting LR and leading to enhanced TRAS, and genomic depletion or pharmacological inhibition of PTEN restored the delayed LR.103 According to the 2 studies mentioned above, it can be found that the expression level of PTEN affects the deposition of lipids during LR. Loss of PTEN expression results in a prolonged period of lipid deposition, whereas increased expression of PTEN results in increased lipid deposition. In addition, the knockout of PTEN in other cell types also affects LR. The loss of PTEN expression in bone marrow-derived cells leads to M2 differentiation, which can promote LR after PH. In vitro studies also showed that the conditioned medium of KC with PTEN deficiency could promote the proliferation of hepatocytes.104 In summary, PTEN is involved in regulating LR and lipid metabolism during LR, and PTEN expression in hepatocytes and other cells has different effects on LR.
LDL receptor (LDLR)
LDLR mainly clears cholesterol esters (CE)in the serum by endocytosis of LDL and other lipoproteins. LDLR almost disappeared in the livers of mice overexpressing proprotein convertase subtilisin/kexin type 9 (PCSK9), and the whole or hepatocyte-specific knockout of PCSK9 showed an obvious increase in LDLR expression level.105 Previous studies have shown that the expression of PCSK9 (mRNA) increases during LR after PH.106 Therefore, the expression level of LDLR may decrease during LR. In LDLR-/- mice, the LR process was impaired, which was mainly accompanied by a decrease in the expression levels of cytokines such as IL6, TNF, and HGF. LD deposition did not change significantly during LR, but there were significant differences in the lipid profiles. Compared with WT mice, in the basic-state liver of LDLR-/- mice, the CE level was higher (increased by 2.5 times), and phosphatidylethanolamine (decreased by 1.7 times) and ceramide (decreased by 2.5 times) were lower. After PH, the CE level in WT mice decreased gradually after 48 hours, but the CE level in LDLR-/- mice remained high. In addition to the difference in the changes in CE, the level of sphingolipids in LDLR-/- mice liver 3 hours after surgery was significantly higher than that in the control group, and the level of ceramide was significantly higher than that in the control group 2 days later.107 Mice with PCSK9-/- showed increased expression levels of inflammatory factors, such as IL6 and TNFα, which were reversed by small interfering RNA-targeted LDLR; thus, there may be an endogenous regulation mechanism of IL6 and TNFα by LDLR.108 Although, in the basal state, loss of PCSK9 expression levels resulted in increased cytokine expression levels, LR induced by PH was significantly suppressed in PCSK9-/- mice.105 Cholesterol can promote the inflammatory response and proliferation of hepatocytes,109 but a long-term cholesterol diet resulting in increased intracellular cholesterol content inhibits the hepatic regenerative response after PH.110 In general, the expression level of LDLR may be reduced during LR; therefore, LDLR may not be important for LR and lipid deposition. The impaired regeneration ability of hepatocytes in LDLR-deficient mice may be related to a decrease in cytokine expression and a change in lipid composition. However, the effect of lipid composition on LR remains unclear.
LD deposition is a favorable factor for LR
NCoR1
Studies on interactions between NCoR1, nuclear receptors, deacetylases, and transducing beta-like 1 have revealed that they can form a corepressor complex for the downregulation of target genes; therefore, loss of NCoR1 leads to increased expression levels of its target genes.111–113 In mice, hepatocyte-specific knockout of NCoR1 (NCoR1 hepa-/-) results in hepatic steatosis,111 and downregulation of NCoR1 expression, leading to elevated expression of metabolism-related genes, may be responsible for hepatic steatosis, such as lipogenesis and lipid oxidation genes.114 The expression of NCoR1 is suppressed after PH, and this suppression may play an important role in driving the progression of LR. In NCoR1 hepa-/- mice, NCoR1 loss accelerated LR but did not alter its outcome. This accelerated LR was accompanied by an increase in the TG content in the liver. Notably, however, basal levels of TG were higher in the livers of NCoR1hepa-/- mice than in the controls, with a similar trend in the LR process in the 2 groups. Orlistat, a FAS inhibitor, reduced and delayed the peak of TG accumulation during LR in both groups while eliminating the difference. Based on the above results, the authors affirmed that de novo synthesis of FFA in hepatocytes may be involved in LD deposition during LR.5 However, the effects of NCoR1 and orlistat on TG deposition, LR, and lipid synthesis must be assessed objectively. First, the loss expression of NCoR1 can also promote the enhancement of FFA oxidative metabolism,115 which provides energy for the early stage of LR and is important for accelerating the progress of LR.7,45 Second, the main reason why orlistat strongly inhibited the level and rate of TG accumulation during LR may be that orlistat is an inhibitor of FFA synthase and lipases. Orlistat inhibits the absorption of dietary-derived FFA116 and also reduces lipolysis in adipocytes.117 Inhibition of lipolysis in peripheral adipose tissue and de novo synthesis of FFA may result in decreased TG deposition during LR, and insufficient energy metabolism leads to obvious inhibition of LR in both groups. The effect of de novo synthesis of FFA on TRAS and LR rate remains to be further studied, but, in the case of insufficient transport, such as the absence of CAV1,3 the reduction of LD deposition during regeneration can be reversed by exogenous supplementation of glucose, which, to some extent, suggests that de novo synthesis of FFA plays a role in LD deposition and is important evidence that transport and de novo synthesis complement each other.
Glucocorticoid receptor (GR)
Glucocorticoids (GC) promote gluconeogenesis in the liver by facilitating the transport of protein and lipid metabolites in skeletal muscle and liver, respectively. Long-term administration of GC leads to severe metabolic syndrome with enhanced expression of genes involved in glycolipid metabolism after binding to GR in the liver.118 One study demonstrated that GR mRNA expression is elevated twice, 3–6 hours and 24–36 hours, after PH.119 TG accumulation and regenerative capacity are significantly inhibited following PH in mice with hepatocyte-specific knockout of GR.75 However, exogenous supplementation with GC has been found to inhibit DNA synthesis and perturb LR after PH in rats.120 These contradictory results seem to indicate that the biological effects mediated by GR expression are not only due to the direct effect of GC but also by internal regulation. Many studies have been conducted on the regulation of GR activation during lipid metabolism in the basic state. Dexamethasone is a commonly used GC. Resection of the adrenal gland in rats did not change FABP expression in the liver, but dexamethasone treatment significantly reduced the expression of FABP.121 Long-term chronic administration of dexamethasone can increase the expression level of CD36 in the liver of mice, increase the transport of FFA into the liver, and lead to fatty liver.122 Similarly, dexamethasone treatment increased the expression of FATP in the chicken liver.123 GR activation affects LD metabolism in the liver in 2 ways. The activation of GR by CORT118335 can only induce the partial effect of GC, mainly by promoting the secretion of VLDL in hepatocytes, reducing the deposition of lipids in hepatocytes, and not increasing the transfer of FFA into hepatocytes.124 GC/GR not only affects the inward and outward transport of FFA in hepatocytes but also increases the synthesis of FFA,125 lipolysis in adipose tissue,126 and mitochondrial oxidative phosphorylation.127 The effect of GR on lipid metabolism in the liver is multilevel, but there are few studies on GR and TRAS, and their relationship needs to be further explored.
Caveolin-1 (CAV1)
In many mammalian cell types, there are abundant submicroscopic plasmalemma pits, which are named caveolae. The functions of caveolae are complex, mainly involving endocytosis, signal transduction, lipid regulation, calcium signal transduction, mechanical sensation, and other processes.128 Caveolins are the main protein components of caveolae, mainly caveolin-1 (Cav-1), Cav-2, and Cav-3, which are selectively distributed and expressed in tissues,129–131 and CAV1 and CAV3 are essential for the formation of caveolae.128 Cav1 is closely related to the physiological and pathological functions of the liver.132 In all tissues, the expression of CAV1 was the lowest in the liver, but there were obvious differences in different liver cells.128,133 Three knockout mouse models were used to investigate the relationship between CAV1 and LR expression. The Jackson Laboratory produces jax CAV1-/-, k CAV1-/- mice bred from the kurzchalia laboratory, and BALB /C CAV1-/- mice produced by mating k CAV1-/- mice with BALB/C mice. The expression of CAV1 during LR is a process of change. After increasing at the early stage of LR, it becomes phosphorylated at the Y14 site and translocated to the cell membrane, returning to normal levels at approximately postoperative day 5. These results seem to reveal that CAV1 plays a role in LR. However, another study reported an increased rate of LR in jax CAV1-/- mice, with the liver-to-body weight ratio exceeding that of control mice at 80 hours after PH.134 However, in both k CAV1-/- and BALB/C CAV1-/- mice, Manuel et al found that the 3-day survival rate of CAV1-/- mice was significantly decreased after 70% PH,3 accompanied by a significantly poorer level of regenerative liver recovery.8 In addition to the different effects on the LR process, the effect of CAV1 on LD deposition during LR was also different in those studies. Using 2 different genetic backgrounds of CAV1-/- mice, Manuel et al asserted that the loss of expression of the CAV1 gene severely inhibits LD deposition during LR. However, a completely different result was reported by Rafael et al. The differing results of the 2 studies may be attributed to several reasons. First, the genetic backgrounds of the mice used by the 2 investigators were different, with the former using C57BL/6J (75% 129 Sv, 20% C57BL/6 J, and 5% SJL mixture) and BALB/C strains and the latter using CAV1 tm1mls/J strain mice. Second, the sites of gene knockdown in the 2 groups were inconsistent, with the former mainly targeting the exon 3 region and the latter mainly targeting exons 1 and 2. Third, the 3 strains of mice differ in their energy sources, and studies on different genetic backgrounds have found that JAX CAV1 mice are more reliant on carbohydrates for energy supply and rely primarily on aerobic glycolysis during LR, not lipid metabolism, unlike the reliance on lipid metabolism exhibited by the other 2 strains of mice.8,135,136 The effects of CAV1 on LR and lipid deposition may result from multifaceted causes. First, CAV1 is involved in the mitochondrial FFA oxidative metabolism.132 Second, it has recently been shown that loss of CAV1 can lead to enhanced lipid metabolism resulting from the activation of the cAMP signaling pathway by autocrine PGI2, leading to reduced LD accumulation in cells.137 CAV1 is an important fatty acid transporter involved in FFA input into hepatocytes.138 Taken together, the impact of CAV1 on LR has been well-established. The difference in outcomes resulting from different exon knockouts still needs further exploration, and the impact on the occurrence of TRAS may also be a result of their multifaceted interference.
AKT1/2-FoxO1
AKT (protein kinase B) is a serine/threonine protein kinase that contains 3 isoforms, AKT1, AKT2, and AKT3, encoded by 3 separate genes.139 These 3 AKT isoforms exert various effects and exhibit different biological functions in different disease models. In inflammatory models, AKT1 null mice exhibit impaired growth, vascular dysfunction, and reduced leukocyte recruitment. Mice deficient in AKT2 exhibit defects in glucose homeostasis and insulin resistance. AKT3-/- mice exhibit impaired brain development.140–144 The distribution of AKT varies among organs throughout the body, as does its distribution in different cell types in the same organ. Only 2 isoforms, AKT1 and AKT2, are present in the liver, with the expression level of AKT2 being predominant, reaching approximately 85% of the total, and the expression level of AKT1 is relatively high in endothelial and HSC.139 AKT is an important kinase that mediates LR and plays various roles in different cell types in the LR microenvironment. The activation of AKT in liver parenchymal cells regulates LR after PH.145 Chronic liver injury resulting from the DDC diet is also accompanied by the activation of AKT, but predominantly AKT1.146 Activation of AKT is not always favorable for the liver, and p-AKT in endothelial cells enhances the ability of endothelial cells to activate stellate cells, promoting the progression of liver fibrosis. Targeted inhibition of AKT in endothelial cells by nanomaterial-loaded honokiol, which activates ERK1/2 and inhibits AKT, can promote LR in a state of liver fibrosis resulting from bile duct ligation and CCL4 without promoting the progression of liver fibrosis.147 Adenoviruses expressing AKT can rapidly increase the growth of the liver and, in a short time, can also lead to an obvious increase in liver weight and intracellular stored lipids and glycogen. Inhibition of AKT phosphorylation by blocking PI3K or PDK1 impairs LR.148–150 These results suggest that the effects of AKT on LR within different cells are 2-sided in different liver disease states. Although AKT1 and AKT2 share their respective functional characteristics, LR is not impaired by specific AKT1 or AKT2 knockout in hepatocytes and is perturbed in AKT1 and AKT2 double knockout mice, resulting in impaired LR. In mice with AKT1/2 hepatocyte-specific knockout, poor LR is accompanied by reduced LD formation.4 AKT is at the crossroads of exerting biological functions and has complex effects on cell function. There are many regulatory targets upstream and downstream, such as PI3K, GSK3, FOXO, and mTORC1.151 FoxO1, an important member of the FOXO family, is involved in the regulation of substance metabolism, and the selective regulation of FoxO1 function can regulate hepatocyte glycolipid metabolism.152,153 Hepatocyte-specific knockout of FoxO1 alleviated the inhibitory effect of AKT1/2 double knockout on LR while restoring the normalization of lipid deposition in the liver.4 The above results illustrate that the effect of AKT knockout on LD content during LR partially results from the attenuation of FoxO1 phosphorylation with the function of FoxO1 enhancement. However, it is strange that targeted liver FoxO1 gain-of-function mutations can induce diabetes.154 FoxO1 transcriptionally activates MTP and apolipoprotein CIII genes, promotes hepatic VLDL assembly, inhibits postsecretory catabolism, enhances LD and VLDL transport outside the liver,155 and is also able to suppress de novo synthesis of FFA in cells.156 Interestingly, the inhibition of FoxO1 can inhibit the formation of LD in adipocytes, suggesting that FoxO1 has multiple effects on lipid metabolism.157 Taken together, multiple mechanisms are involved in TRAS caused by reduced inactivation of FoxO1 resulting from AKT knockout.
Replication initiator 1 (Repin1)
Repin1 is widely distributed throughout the body but is relatively highly expressed in the liver and intra-abdominal adipose tissue158 and is closely associated with obesity, abnormal lipid metabolism, and NAFLD.159,160 In adipocytes, Repin1 regulates the expression of genes involved in adipogenesis, LD formation and fusion, and glucose and FFA transport.161 Hepatocyte-specific Repin1 deficiency results in dyslipidemia, reduced hepatic lipid deposition, and altered hepatic lipidome structure, preventing adipocyte hypertrophy induced by high-fat feeding.162 Loss of Repin1 does not affect de novo FFA synthesis but rather affects LD content in hepatocytes. In hepatocytes from Repin1-/- mice, the levels of vehicle-associated membrane protein 4 and synaptosomal-associated protein expression were severely decreased (80% and 40%, respectively), and the expression of CD36 was reduced by half. These results imply that the function of Repin1 is tightly linked to lipid metabolism in the body. Paradoxically, however, a sharp decrease in the mRNA expression level of Repin1 occurs early after PH (more than 80% reduction at 6h after surgery), but a marked increase in LD accumulation occurs early in LR.163 This trend indicates that the expression of Repin1 does not seem to have a direct effect on LD deposition during LR. Depletion of hepatocyte Repin1 delays but does not inhibit the outcome of LR and concomitantly inhibits the deposition of LD in the liver, mainly as indicated by the reduced number of LD and total area of LD.163 In summary, the relationship between Repin1 and LD deposition during LR is puzzling, and its effect on TRAS is not a simple surface relationship observed in current studies.
Autophagy related 7(ATG7)
Autophagy is a protective biological process mediated by lysosomes to degrade intracellular materials and is mainly completed by a cluster of autophagy-related proteins and classified into macroautophagy, microautophagy, and chaperone-mediated autophagy.164 Autophagy is involved in the regulation of lipid metabolism and is particularly important for energy metabolism in the liver.164,165 Pharmacological inhibition of autophagy (with 3-methyladenine) and knockdown of ATG5 expression by RNAi both promoted increased intracellular TG levels, and a conditional knockout of ATG7 in hepatocytes resulted in high levels of intracellular TG and NAFLD presentation.165–167 Paradoxically, however, another study suggested that Oil Red O staining of liver tissue at 0 hour after PH with the hepatocyte-specific knockout of ATG7 showed no significant increase in lipid content.168 Autophagy is involved in LR, inhibition of autophagy impairs LR, and the promotion of autophagy accelerates this process.169–172 After 70% portal vein ligation in rats, LR on the nonligated side was accompanied by increased expression levels of LC3B and Beclin1, and positively correlated with the expression levels of CyclinD1.173 Similarly, enhanced autophagy was observed in LR after PH.174 The simultaneous presence of enhanced autophagy and increased LD deposition during LR seems to be understandable. Poor diet and reduced blood glucose levels in rodents following PH, similar to the starvation state, induce hepatocyte autophagy and increase FFA delivered from adipose tissue lipolysis to the liver.165 ATG7 knockout, similar to ATG5 knockout, resulted in an enlarged liver in the absence of injury,168,175 but the inhibition of autophagy resulting from ATG7 knockdown in hepatocytes attenuated the accumulation of LD and inhibited LR.168 In wild-type mice, starvation176 and PH177 induce an increase in FGF21 expression levels, and FGF21 not only promotes autophagy but also increases FFA metabolism in the liver. Hepatocyte-specific knockout of ATG7 leads to impaired LR after PH and inhibits LD deposition, which may be due to many reasons. Inhibition of autophagy can inhibit the oxidation of FFA and reduce the transmission of TG by VLDL.178 Therefore, ATG7 knockout can also reduce intracellular LD consumption to a certain extent. However, in mice with a hepatocyte-specific knockout of ATG7, the expression level of liver FGF21 was significantly increased, which could compensate for the inhibitory effect of autophagy reduction to some extent.179 In addition to stimulating liver FFA oxidation, FGF21 also reduces FFA flux into the liver by increasing peripheral lipoprotein catabolism and reducing lipolysis of adipocytes.180 Eventually, in hepatic-specific knockout ATG7 mice, FGF21 may play an important role in impairing LD deposition during LR. In summary, autophagy is involved in the maintenance of organ homeostasis. The effects of autophagy on the liver differ in basal and surgical states, and the relationship between autophagy and the maintenance of liver homeostasis and lipid metabolism is complex.
mTORC2
mTOR is a serine/threonine protein kinase that recruits other proteins to form 2 different complexes: mTOR complex 1 (mTORC1) and complex2 (mTORC2). mTORC2 mainly controls cell survival and migration by phosphorylating various kinases, including AKT, PKC, and SGK1.181 As one of the important targets of mTORC2, AKT plays an important role in LR and lipid deposition during LR.4 What role does mTORC2 play in the LR? First, in C57BL/6 mice with hepatocyte-specific knockout Rictor, the slight decrease in liver weight and the decrease in liver/body weight ratio in 5–8-week-old mice were mainly due to changes in cell size rather than changes in cell proliferation. The mTOR signaling pathway is activated during LR after PH and is accompanied by AKT activation. Mice with Rictor knockout in hepatocytes showed significant inhibition of LR after PH, with increased postoperative mortality and reduced deposition of LD.145 However, another study found that a hepatocyte-specific Rictor knockout resulted in the inhibition of LR and hepatocyte proliferation, accompanied by increased intrahepatic LD deposition. The main reason for this is that the activity of PPARα in the liver is inhibited, and the oxidative metabolism of FFA is severely inhibited.182 mTORC2 and AKT1 were activated in the DDC diet-induced chronic liver injury model. Since the source of proliferating hepatocytes in LR induced by the DDC diet differs from that following PH, Rictor knockdown only reduces the proliferation of oval cells in the liver.146 LR is clearly inhibited in mice with hepatocyte deficiency of mTORC2. In terms of lipid deposition, it is surprising that researchers have obtained 2 completely opposite conclusions, which may require more objective experimental studies.
Adipose differentiation-related protein (ADRP, Plin2)
LD protein is an important protein component of LD, and in hepatocytes, there are 5 different protein types, Perilipin1(Plin1)–Plin5,183,184 and each perilipin protein has distinct roles. Adipose differentiation-related protein is the only widely expressed constitutive LD protein, and its expression level of Plin2 is correlated with the TG content and density of LD.185,186 The TG content in hepatocytes was significantly reduced in Plin2-/- mice; however, hepatic lipogenesis, VLDL secretion, and lipid uptake and utilization were not significantly different from those in WT mice,187,188 despite uncontrolled VLDL-mediated lipid efflux following PH.189 Plin2 deficiency led to lipid reduction, whereas, in Atg7-/- mice, Plin2 deficiency did not reduce lipid levels. Therefore, the authors conclude that Plin2 deficiency induces decreased LD deposition caused by autophagy in mouse hepatocytes.190 In hepatocyte-specific Plin2 knockout mice, the function of ATGL in hepatocytes is increased, thereby promoting lipolysis.191 The increased function of autophagy and ATGL may be an important reason for the reduction in lipid deposition in the hepatocytes. The above clues seem to be a reasonable explanation for the decrease in LD deposition during LR. In Plin2-/- mice, hepatocyte entry into the cell cycle and LD deposition were impaired after PH.189 Compared with WT mice, there was no difference in the expression of FFA synthesis-related and FFA transport-related genes in the hepatocytes of Plin2-/- mice. The observed LD deposition disorder was mainly due to VLDL-enhanced lipid outward transport, resulting in decreased intracellular lipid storage.189 The reason for the impaired LR may be that insufficient LD deposition decreases the FFA β oxidation energy supply. On the other hand, β oxidation of regenerated livers in Plin2-/- mice is severely inhibited.188,189 Plin2 is essential for the storage and oxidative metabolism of FFA, and its effects on TRAS are multiple.
Pyruvate dehydrogenase kinase 4 (PDK4)
PDK4 is a key factor in metabolic regulation. It inactivates the mitochondrial pyruvate dehydrogenase complex by phosphorylating pyruvate dehydrogenase, thus affecting pyruvate conversion metabolism. PDK4 coordinates glucose and fat metabolism and maintains normal blood glucose levels.192,193 The increased expression of PDK4 is an important marker for the conversion of cellular energy metabolism to FFA oxidation metabolism.194 The mRNA expression level of PDK4 increases in the early stage of LR, and the protein level of PDK4 increases significantly within 24 hours after operation.9 To a certain extent, this result also indicates that cell energy metabolism shifts to lipid metabolism during LR, which seems to indicate that it plays an important role in promoting LR. Interestingly, PDK4-/- mice showed a significant acceleration of LR after PH and increased LD deposition during LR. There are many reasons for the effect of PDK4 on LD deposition. First, the increase in the expression level of Plin2 is important for the retention of VLDL in hepatocytes and reduces the loss of VLDL caused by insufficient Plin2. Second, the expression levels of CD36 and FABP in PDK4-/- mice were increased, which enhanced the transport of FFA into hepatocytes. Third, the gene expression levels of FFA de novo synthesis decreased. Fourth, the lack of PDK4 enhances the oxidative metabolism of FFA.9 Therefore, the mechanism by which PDK4 affects TRAS is complex and requires further investigation.
PCSK9
PCSK9 is a mammalian proprotein convertase with complex biological functions.106 PCSK9 induces the degradation of LDLR, leading to the accumulation of LDL cholesterol in the blood.195–198 Therefore, affecting the expression or activity of PCSK9 leads to abnormal serum cholesterol.199–201 Systemic and hepatocyte-specific knockdown of PCSK9 reduced plasma cholesterol levels by 42% and 27%, respectively.105 However, a significantly greater proportion of hepatic steatosis and higher visceral fat content were observed in the patient population carrying R46L, a low expression mutation of PCSK9.202 PCSK9 is highly expressed in both fetal and mature livers, and its levels in the regenerating liver after PH peak at 2–3 days in rodents.105,106 LR potential is impaired in PCSK9-/- mice. Interestingly, in PCSK9-/- mice, feeding with high cholesterol markedly reduced necrosis in the liver and seemed to reduce liver damage, but the LR index was not elevated. In contrast, in WT mice, high cholesterol feeding increased the lipid content in the liver but decreased the LR index (4.8±0.7 vs. 3.6±0.5), and the extent of the decrease exceeded that in mice on a chow diet PCSK9-/- at the same time (3.7±0.5). It is difficult to understand that, although reduced expression or function of PCSK9 promotes fat accumulation in the liver, a significant reduction in lipid content occurs in the regenerated liver 72 hours after PH in PCSK9-/- mice.105 The decrease in lipid content is accompanied by the inhibition of LR. The effects of PCSK9 on lipid metabolism in hepatocytes are complex. As mentioned earlier, PCSK9 affects cholesterol metabolism by inducing LDLR degradation. Second, because PCSK9 induces CD36 degradation, in PCSK9 knockdown mice, increased CD36 expression levels promote FFA uptake by hepatocytes, ultimately leading to increased TG content in the liver.203 Third, PCSK9 promotes VLDL secretion from the liver and increases TG export outside the liver, whereas loss of PCSK9 expression reduces VLDL secretion and promotes TG accumulation within the liver.204 Fourth, PCSK9 also increases the levels of the lipid-generating enzymes FAS, stearoyl-CoA desaturase, and diacylglycerol acyltransferase 2, thereby participating in de novo FFA synthesis and leading to an increase in FFA.205 In summary, it is not difficult to determine that the effect of PCSK9 on LR and TRAS is extremely complex, and its specific mechanism requires further exploration.
LIPE in adipose tissues
LIPE, also known as hormone sensitive lipase (HSL), encodes an enzyme responsible for TG decomposition and increased production of FFA.52 As previously mentioned, we have analyzed and summarized the regulatory mechanism of lipolysis in adipose tissue and its connection to LR. During the early stages of LR, the weights of epididymal and inguinal white adipose tissues were significantly decreased, accompanied by increased phosphorylation of HSL in adipose tissues.2 These findings not only indicate the influence of HSL in peripheral adipose tissue during LR but also suggest the involvement of peripheral adipose tissue lipolysis in the redistribution of lipids induced by LR. Adipose tissue-specific knockout of LIPE significantly reduced LD deposition during LR and decreased FFA in peripheral blood.2 The liver weight-to-body weight ratio and Ki67 expression demonstrated that the knockout of HSL in adipose tissue inhibited LR.2 Most previous studies have focused on the energy supply aspect of TRAS’s effect on LR.7,8 However, through an in-depth exploration of the impact of LD on LR, Chen et al discovered that TRAS formation inhibits MIER1 translation by enhancing EIF2S1 phosphorylation, thereby initiating LR.2 Notably, a recent study indicated that embryonic stem cells have higher LD content and a lower rate of lipid hydrolysis, with enhanced LD catabolism occurring during differentiation.206 This suggests a potential role for LD in maintaining the stemness of embryonic stem cells. Does TRAS formation affect the reentry of hepatocytes into the cell cycle during LR? Interestingly, in vitro treatment of hepatocytes with palmitic acid increased EIF2S1 phosphorylation and decreased cellular protein synthesis.2 The enhanced phosphorylation of EIF2S1 caused by TRAS coincides with extensive protein translation during the early stages of regeneration. Inhibition of EIF2S1 phosphorylation in colon cancer cells reversed the inhibitory effect of 3,3’-diindolylmethane on cyclin D1 translation.207 Studies have shown increased expression of CyclinD1 protein during the early stages of LR.208 The inhibition of MIER1 translation by TRAS may partially contribute to the initiation of LR, but the process of hepatocyte reentry into the cell cycle for regeneration initiation is complex, and the mechanism of TRAS’s effect on LR requires further exploration.
LD deposition inhibits LR
PPARα
PPARs are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily, and FFA is one of their common ligands. They regulate key aspects of energy metabolism within cells. PPARs play pivotal roles in the regulation of hepatic lipid metabolism.209 In mammals, there are 3 isoforms of PPAR, alpha (α), beta/delta(β/δ), and gamma (γ), which are differentially expressed in various tissues, with PPARα being the predominant isoform in the liver.209 In rodents, sustained activation of PPARα by endogenous or exogenous compounds leads to hepatocyte swelling, inhibition of apoptosis, and promotion of hepatocyte proliferation, resulting in hepatomegaly, which can eventually progress to liver tumors. However, in humans, it does not cause hepatocarcinogenesis, mainly because of differences in the structure and function of PPARα between mouse and human.210,211 In the C57BL/6 strain genetic background, increased expression of PPARα accompanies the process of LR after PH6 and is involved in regulating LR.212,213 However, global deletion of PPARα in mice in the SV/129 genetic background did not affect the LR process after PH.214 In addition, in the C57BL/6 mice, Elizabet et al. also showed that PPARα depletion had no significant effect on LR. However, only a one-time point BrdU-positive result was given in the article, and the LR process was not analyzed comprehensively.1 Several studies have shown that both global and liver-specific knockdown of PPARα expression in the C57BL/6 mice can suppress PH-induced LR.6,215,216 The reason for the inconsistent effect of PPARα on LR across studies may lie in the fact that the effects of hepatocyte and nonhepatocyte knockout on LR are different. A recent study showed that, in bone marrow cell-specific PPARα knockout mice, knocking down PPARα in bone marrow cells led to the differentiation of macrophages into M1 type, immersed in the liver during LR, and secreted a large number of important cytokines, such as TNFα and IL6,217 thereby accelerating LR after PH. Both whole-body and hepatocyte-specific PPARα knockdown showed increased LD deposition during LR.6,216 PPARα is a key factor in controlling lipid β oxidation, and activators of PPARα can mitigate the degree of fatty liver.218 Knockdown of PPARα leads to severe inhibition of FFA β oxidation and increased lipid accumulation in hepatocytes and is an important trigger for impaired LR and increased lipid deposition during LR. PPARα in different tissues exerts different effects on LR, and the effects of PPARα in hepatocytes on LR and TRAS may largely depend on the function of FFA oxidative metabolism.
PERSPECTIVE: HOW TO GAIN A CORRECT INSIGHT INTO THE RELATIONSHIP BETWEEN LR AND LD IN FATTY LIVER AND TRAS?
To better probe LD deposition during LR, we must clarify that the relationship between LD deposition and LR is not that between fatty liver and LR. Hepatic steatosis, a pathological concept characterized by massive intracellular accumulation of neutral fat, is an important pathological change that occurs during the progression of several diseases, such as viral infection, alcohol consumption, metabolic syndrome, obesity, and diabetes.219–221 LDs are ubiquitous “organelles” that store neutral lipids in various cells, and the metabolic imbalance of LD is an important reason for their massive accumulation in cells, leading cells to steatosis.222 The LD membrane originates from the phospholipid monolayer structure in the endoplasmic reticulum, and the proteins on the surface of the LD are essential for coordinating the continued accumulation and decomposition of LD. LDs in hepatocytes are mainly composed of triacylglycerol, cholesteryl esters, or retinyl ester.223
Mitochondrial dysfunction has been found in patients and animal models of NAFLD, and endoplasmic reticulum stress further promotes hepatocyte injury and accelerates NAFLD progression.224–226 Hepatocyte injury activates cells in the regenerative microenvironment, such as stellate cells, which release inflammatory and growth factors227,228 that together orchestrate and promote hepatocyte proliferation to maintain liver structural, metabolic, and functional homeostasis. However, the regenerative response in steatotic livers after injury remains controversial in the presence of exogenous injuries, such as PH.1,229,230 In recent years, there has been a gradual consensus that severe steatosis plays a detrimental role in regeneration and function after PH and liver transplantation.231,232 Severely steatotic livers (>60% macrovesicular steatosis) are routinely excluded from transplant donor livers because they often cause severe postoperative complications, such as primary nonfunction.232 More than 20% of patients who are prepared for PH have varying degrees of steatosis, which is also a significant risk factor for increased postoperative mortality.231 In humans, the above evidence indicates that steatosis has significant adverse effects on LR and liver function. However, the effect of hepatocyte steatosis on LR has not yielded consistent results in rodent models of NAFLD. In murine NAFLD models caused by ob/ob and db/db knockouts, significant LR failure was not rescued by leptin supplementation after PH. The relationship between hepatic steatosis and LR may not be accurately revealed by ob/ob and db/db knockout–induced NAFLD models.233,234 In another model of NAFLD induced by the methionine and choline defi diet (4 wks), steatosis did not affect LR after PH,235 and in rats (5 wks of feeding), steatosis was able to inhibit LR resulting from portal vein ligation.13 Svenja et al found that a high-fat diet for 6 weeks enhanced LR induced by 70% PH.236 However, Gao et al did not find any interference with LR in rats fed a high-fat diet for 6 weeks.237 Another study indicated that LR after associating liver partition and portal vein ligation for staged hepatectomy was inhibited in a rat model of high-fat diet-induced NAFLD rat model.12 More interestingly, after PH in mice fed a high-fat diet for 9–10 weeks, LR was accelerated at 2 d and inhibited at 8 d, respectively.238 The reasons for the different results of the above studies may be complex, including animal strains, fatty liver animal model production methods, liver injury methods, and observation time. Here, we briefly summarize the information on the relationship between NAFLD and LR after PH, as shown in Supplemental Table S2. http://links.lww.com/HC9/A521.
Only by gaining the correct insight into the relationship between LR and LD in fatty liver and TRAS can LD be better used to coordinate LR. Therefore, further improvements are still needed in animal models and methods: (1) avoid changing the metabolic background of the animal liver; (2) does not affect lipid content in the basal state of the liver; (3) advanced techniques should be applied to further analyze the composition of the deposited LD; and (4) relationship between LD deposition and zonation of regeneration might be a new focus. A clear understanding of the relationship between TRAS and LR may provide a safer and more scientific direction and recognition for promoting LR. From the literature, we can find that there is a regulable space for the effect of lipid metabolism on LR. Therefore, targeting the mechanisms of TRAS formation and depletion holds promise for developing safe approaches to harness lipid metabolism to promote LR; however, some issues still need to be addressed. (1) What factors promote adipose tissue lipolysis during LR? (2) Transport of FFA depends on 4 transporters, the malfunction of which one can result in impaired LR? (3) How to maintain the storage of LD and reduce fat output in the liver? (4) How can the LR process be regulated by promoting oxidative metabolism? Ultimately, multifaceted synergistic control may be a safe and effective method to promote LR.
Supplementary Material
AUTHOR CONTRIBUTIONS
Yuelei Hu and Ruilin Wang: Data curation, formal analysis, and writing and original draft; Yuelei Hu and Juan Liu: Conceptualization; Jiahong Dong and Yunfang Wang: Conceptualization, supervision, validation, and writing and review editing.
ACKNOWLEDGMENT
We would like to express our sincere gratitude to all the members of the laboratories who have made significant contributions to both current and past research in the field of molecular target therapy. Furthermore, we extend our appreciation to the Figdraw online website (https://www.figdraw.com/static/index.html) for providing valuable picture painting materials that were instrumental in enhancing the visual presentation of our research. Specifically, we would like to acknowledge the use of material with the ID:TUUTTe5ff5, which we utilized to create, refine, and beautify the illustrations included in this manuscript.
FUNDING INFORMATION
The review was supported by the CAMS Innovation Fund for Medical Sciences (No. 2019-I2M-5-056); National Natural Science Foundation of China (No. 82090050, 82090051, 32000970, 92168207); and Beijing Hospitals Authority’ Ascent Plan (No. DFL20190901).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: ACC, acetyl-CoA carboxylase; ADRP, adipose differentiation-related protein; ALB, albumin; ANXA A6, Annexin A6; ATG5, autophagy related 5; ATG7, autophagy related 7; ATGL, adipose triglyceride lipase; CAV1, Caveolin-1; CE, cholesterol esters; CM, chylomicron; CV, central vein; DDC, 3,5-diethoxycarbonyl-1,4-dihydro-collidine; DNS: de novo synthesis; ET1, endothelin 1; FAS, fatty acid synthase; FABP, fatty acid binding protein; FATP, fatty acid transport protein; FFA, free fatty acid; GC, glucocorticoids; GR, glucocorticoid receptor; HSL, hormone sensitive lipase; LD, lipid droplet; LDLR, LDL receptor; LR, liver regeneration; MTP, microsomal triglyceride transfer protein; NCoR1, nuclear receptor corepressor 1; PARK7, Parkinson protein 7; PCSK9, proprotein convertase subtilisin/kexin type 9; PDK4, pyruvate dehydrogenase kinase 4; PGE2, prostaglandin E2; PH, partial hepatectomy; PPAR, peroxisome proliferator-activated receptor; PV, portal vein; S1P, sphingosine-1-phosphate; TRAS, transient regeneration-associated steatosis; TG, triglyceride.
Yuelei Hu and Ruilin Wang contributed equally to this work.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Yuelei Hu, Email: huyueleijlu@126.com.
Ruilin Wang, Email: wangruilin2012@126.com.
Juan Liu, Email: lja02720@btch.edu.cn.
Yunfang Wang, Email: wangyf2011126@126.com.
Jiahong Dong, Email: dongjiahong@mail.tsinghua.edu.cn.
REFERENCES
- 1. Newberry EP, Kennedy SM, Xie Y, Luo J, Stanley SE, Semenkovich CF, et al. Altered hepatic triglyceride content after partial hepatectomy without impaired liver regeneration in multiple murine genetic models. Hepatology. 2008;48:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen Y, Chen L, Wu X, Zhao Y, Wang Y, Jiang D, et al. Acute liver steatosis translationally controls the epigenetic regulator MIER1 to promote liver regeneration in a study with male mice. Nat Commun. 2023;14:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fernández MA, Albor C, Ingelmo-Torres M, Nixon SJ, Ferguson C, Kurzchalia T, et al. Caveolin-1 is essential for liver regeneration. Science. 2006;313:1628–1632. [DOI] [PubMed] [Google Scholar]
- 4. Pauta M, Rotllan N, Fernández‐Hernando A, Langhi C, Ribera J, Lu M, et al. Akt-mediated foxo1 inhibition is required for liver regeneration. Hepatology. 2016;63:1660–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ou‐Yang Q, Lin XM, Zhu YJ, Zheng B, Li L, Yang YC, et al. Distinct role of nuclear receptor corepressor 1 regulated de novo fatty acids synthesis in liver regeneration and hepatocarcinogenesis in mice. Hepatology. 2018;67:1071–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xie G, Yin S, Zhang Z, Qi D, Wang X, Kim D, et al. Hepatocyte Peroxisome Proliferator-Activated Receptor α Enhances Liver Regeneration after Partial Hepatectomy in Mice. Am J Pathol. 2019;189:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kachaylo E, Tschuor C, Calo N, Borgeaud N, Ungethüm U, Limani P, et al. PTEN Down-Regulation Promotes β-Oxidation to Fuel Hypertrophic Liver Growth After Hepatectomy in Mice. Hepatology. 2017;66:908–921. [DOI] [PubMed] [Google Scholar]
- 8. Alejandro fernández-Rojo M, Restall C, Ferguson C, Martel N, Martin S, Bosch M, et al. Caveolin-1 orchestrates the balance between glucose and lipid-dependent energy metabolism: implications for liver regeneration. Hepatology. 2012;55:1574–1584. [DOI] [PubMed] [Google Scholar]
- 9. Zhao Y, Tran M, Wang L, Shin DJ, Wu J. PDK4-Deficiency Reprograms Intrahepatic Glucose and Lipid Metabolism to Facilitate Liver Regeneration in Mice. Hepatol Commun. 2020;4:504–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piñeiro-Carrero VM, Piñeiro EO. Liver. Pediatrics. 2004;113(4 Suppl):1097–1106. [PubMed] [Google Scholar]
- 11. Michalopoulos GK, Bhushan B. Liver regeneration: Biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol. 2021;18:40–55. [DOI] [PubMed] [Google Scholar]
- 12. Zhao J, Xu H, Li Y, et al. NAFLD Induction Delays Postoperative Liver Regeneration of ALPPS in Rats. Dig Dis Sci. 2019;64:456–468. [DOI] [PubMed] [Google Scholar]
- 13. Hsiao IT, Lin KJ, Chang SI, Yen TC, Chen TC, Yeh TS. Impaired liver regeneration of steatotic rats after portal vein ligation: a particular emphasis on (99m)Tc-DISIDA scintigraphy and adiponectin signaling. J Hepatol. 2010;52:540–549. [DOI] [PubMed] [Google Scholar]
- 14. Solhi R, Lotfinia M, Gramignoli R, Najimi M, Vosough M. Metabolic hallmarks of liver regeneration. Trends Endocrinol Metab. 2021;32:731–745. [DOI] [PubMed] [Google Scholar]
- 15. Caldez MJ, Van Hul N, Koh HWL, Teo XQ, Fan JJ, Tan PY, et al. Metabolic Remodeling during Liver Regeneration. Dev Cell. 2018;47:425–438.e5. [DOI] [PubMed] [Google Scholar]
- 16. Wang F, So KF, Xiao J, Wang H. Organ-organ communication: The liver’s perspective. Theranostics. 2021;11:3317–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Yang F, Li J, Wang J, Wang X, Zhang Y, et al. Mesenchymal Stem Cells Enhance Liver Regeneration via Improving Lipid Accumulation and Hippo Signaling. Stem Cells Int. 2018;2018:7652359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lorenz L, Axnick J, Buschmann T, Henning C, Urner S, Fang S, et al. Mechanosensing by β1 integrin induces angiocrine signals for liver growth and survival. Nature. 2018;562:128–132. [DOI] [PubMed] [Google Scholar]
- 19. Ishikawa J, Takeo M, Iwadate A, Koya J, Kihira M, Oshima M, et al. Mechanical homeostasis of liver sinusoid is involved in the initiation and termination of liver regeneration. Commun Biol. 2021;4:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gao C, Peng J. All routes lead to Rome: multifaceted origin of hepatocytes during liver regeneration. Cell Regen. 2021;10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017;547:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katoonizadeh A, Nevens F, Verslype C, Pirenne J, Roskams T. Liver regeneration in acute severe liver impairment: A clinicopathological correlation study. Liver Int. 2006;26:1225–1233. [DOI] [PubMed] [Google Scholar]
- 23. Shin S, Upadhyay N, Greenbaum LE, Kaestner KH. Ablation of Foxl1-Cre-labeled hepatic progenitor cells and their descendants impairs recovery of mice from liver injury. Gastroenterology. 2015;148:192–202.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi TY, Ninov N, Stainier DY, Shin D. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology. 2014;146:776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deng X, Zhang X, Li W, Feng RX, Li L, Yi GR, et al. Chronic Liver Injury Induces Conversion of Biliary Epithelial Cells into Hepatocytes. Cell Stem Cell. 2018;23:114–122.e3. [DOI] [PubMed] [Google Scholar]
- 26. Lisman T, Porte RJ. Mechanisms of platelet-mediated liver regeneration. Blood. 2016;128:625–629. [DOI] [PubMed] [Google Scholar]
- 27. Goikoetxea‐Usandizaga N, Serrano‐Maciá M, Delgado TC, Simón J, Fernández Ramos D, Barriales D, et al. Mitochondrial bioenergetics boost macrophage activation, promoting liver regeneration in metabolically compromised animals. Hepatology. 2022;75:550–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duan JL, Ruan B, Song P, Fang ZQ, Yue ZS, Liu JJ, et al. Shear stress-induced cellular senescence blunts liver regeneration through Notch-sirtuin 1-P21/P16 axis. Hepatology. 2022;75:584–599. [DOI] [PubMed] [Google Scholar]
- 29. Beppu T, Nitta H, Hayashi H, Imai K, Okabe H, Nakagawa S, et al. Effect of branched-chain amino acid supplementation on functional liver regeneration in patients undergoing portal vein embolization and sequential hepatectomy: A randomized controlled trial. J Gastroenterol. 2015;50:1197–1205. [DOI] [PubMed] [Google Scholar]
- 30. Izumi T, Imai J, Yamamoto J, Kawana Y, Endo A, Sugawara H, et al. Vagus-macrophage-hepatocyte link promotes post-injury liver regeneration and whole-body survival through hepatic FoxM1 activation. Nat Commun. 2018;9:5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawai M, Naruse K, Komatsu S, Kobayashi S, Nagino M, Nimura Y, et al. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: Clinical and experimental studies. J Hepatol. 2002;37:240–246. [DOI] [PubMed] [Google Scholar]
- 32. Song Z, Gupta K, Ng IC, Xing J, Yang YA, Yu H. Mechanosensing in liver regeneration. Semin Cell Dev Biol. 2017;71:153–167. [DOI] [PubMed] [Google Scholar]
- 33. El’chaninov AV, Fatkhudinov TK, Arutyunyan IV, Makarov AV, Usman NY, Mikhailova LP, et al. Dynamics of Expression of Cytokine Genes and Macrophage Content in the Lungs and Kidneys after Subtotal Hepatectomy in Rats. Bull Exp Biol Med. 2018;165:136–141. [DOI] [PubMed] [Google Scholar]
- 34. Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017;27:R1147–R1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martini T, Naef F, Tchorz JS. Spatiotemporal Metabolic Liver Zonation and Consequences on Pathophysiology. Annu Rev Pathol. 2023;18:439–466. [DOI] [PubMed] [Google Scholar]
- 36. Deane HW. A cytological study of the diurnal cycle of the liver of the mouse in relation to storage and secretion. Anat Rec. 1944;88:39–65. [Google Scholar]
- 37. Paris J, Henderson NC. Liver zonation, revisited. Hepatology. 2022;76:1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kietzmann T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017;11:622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ben-Moshe S, Itzkovitz S. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol. 2019;16:395–410. [DOI] [PubMed] [Google Scholar]
- 40. Miyaoka Y, Miyajima A. To divide or not to divide: Revisiting liver regeneration. Cell Div. 2013;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hall Z, Chiarugi D, Charidemou E, Leslie J, Scott E, Pellegrinet L, et al. Lipid Remodeling in Hepatocyte Proliferation and Hepatocellular Carcinoma. Hepatology. 2021;73:1028–1044. [DOI] [PubMed] [Google Scholar]
- 43. Feng J, Li J, Wu L, Yu Q, Ji J, Wu J, et al. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S, et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. 2022;12:558–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gebhardt R. Metabolic zonation of the liver: Regulation and implications for liver function. Pharmacol Ther. 1992;53:275–354. [DOI] [PubMed] [Google Scholar]
- 46. Blikkendaal-Lieftinck LF, Kooij M, Kramer MF, Den Otter W. Cell kinetics in the liver of rats under normal and abnormal dietary conditions. An autoradiographic study by means of [3H]thymidine. Exp Mol Pathol. 1977;26:184–192. [DOI] [PubMed] [Google Scholar]
- 47. Lai HS, Chen WJ, Chen KM. Energy substrate for liver regeneration after partial hepatectomy in rats: Effects of glucose vs fat. JPEN J Parenter Enteral Nutr. 1992;16:152–156. [DOI] [PubMed] [Google Scholar]
- 48. Farrell GC. Probing Prometheus: Fat fueling the fire. Hepatology. 2004;40:1252–1255. [DOI] [PubMed] [Google Scholar]
- 49. Wei Y, Wang YG, Jia Y, Li L, Yoon J, Zhang S, et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science. 2021;371:eabb1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chembazhi UV, Bangru S, Hernaez M, Kalsotra A. Cellular plasticity balances the metabolic and proliferation dynamics of a regenerating liver. Genome Res. 2021;31:576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grabner GF, Xie H, Schweiger M, Zechner R. Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nat Metab. 2021;3:1445–1465. [DOI] [PubMed] [Google Scholar]
- 52. Schweiger M, Schreiber R, Haemmerle G, Lass A, Fledelius C, Jacobsen P, et al. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J Biol Chem. 2006;281:40236–40241. [DOI] [PubMed] [Google Scholar]
- 53. Lien CC, Yin WH, Yang DM, Chen LK, Chen CW, Liu SY, et al. Endothelin-1 induces lipolysis through activation of the GC/cGMP/Ca(2+)/ERK/CaMKIII pathway in 3T3-L1 adipocytes. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867:159071. [DOI] [PubMed] [Google Scholar]
- 54. Eriksson AKS, van Harmelen V, Stenson BM, Åström G, Wåhlén K, Laurencikiene J, et al. Endothelin-1 stimulates human adipocyte lipolysis through the ET A receptor. Int J Obes (Lond). 2009;33:67–74. [DOI] [PubMed] [Google Scholar]
- 55. Jun DJ, Lee JH, Choi BH, Koh TK, Ha DC, Jeong MW, et al. Sphingosine-1-phosphate modulates both lipolysis and leptin production in differentiated rat white adipocytes. Endocrinology. 2006;147:5835–5844. [DOI] [PubMed] [Google Scholar]
- 56. Ahmadian M, Wang Y, Sul HS. Lipolysis in adipocytes. Int J Biochem Cell Biol. 2010;42:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fain JN, Leffler CW, Bahouth SW, Rice AM, Rivkees SA. Regulation of leptin release and lipolysis by PGE2 in rat adipose tissue. Prostaglandins Other Lipid Mediat. 2000;62:343–350. [DOI] [PubMed] [Google Scholar]
- 58. Chatzipanteli K, Rudolph S, Axelrod L. Coordinate control of lipolysis by prostaglandin E2 and prostacyclin in rat adipose tissue. Diabetes. 1992;41:927–935. [DOI] [PubMed] [Google Scholar]
- 59. Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, Kato A, Yoshitomi H, et al. Circulating cytokines, chemokines, and stress hormones are increased in patients with organ dysfunction following liver resection. J Surg Res. 2006;133:102–112. [DOI] [PubMed] [Google Scholar]
- 60. Alvarez‐Guaita A, Blanco‐Muñoz P, Meneses‐Salas E, Wahba M, Pollock AH, Jose J, et al. Annexin A6 Is Critical to Maintain Glucose Homeostasis and Survival During Liver Regeneration in Mice. Hepatology. 2020;72:2149–2164. [DOI] [PubMed] [Google Scholar]
- 61. Zabielski P, Baranowski M, Zendzian-Piotrowska M, Blachnio A, Gorski J. Partial hepatectomy activates production of the pro-mitotic intermediates of the sphingomyelin signal transduction pathway in the rat liver. Prostaglandins Other Lipid Mediat. 2007;83:277–284. [DOI] [PubMed] [Google Scholar]
- 62. Tsujii H, Okamoto Y, Kikuchi E, Matsumoto M, Nakano H. Prostaglandin E2 and rat liver regeneration. Gastroenterology. 1993;105:495–499. [DOI] [PubMed] [Google Scholar]
- 63. Ratti F, Pulitanò C, Catena M, Paganelli M, Aldrighetti L. Serum levels of endothelin-1 after liver resection as an early predictor of postoperative liver failure. A prospective study. Hepatol Res. 2016;46:529–540. [DOI] [PubMed] [Google Scholar]
- 64. Canbay A, Bechmann L, Gerken G. Lipid metabolism in the liver. Z Gastroenterol. 2007;45:35–41. [DOI] [PubMed] [Google Scholar]
- 65. Lowe ME. The triglyceride lipases of the pancreas. J Lipid Res. 2002;43:2007–2016. [DOI] [PubMed] [Google Scholar]
- 66. Havel RJ. Postprandial hyperlipidemia and remnant lipoproteins. Curr Opin Lipidol. 1994;5:102–109. [DOI] [PubMed] [Google Scholar]
- 67. Iqbal J, Hussain MM. Intestinal lipid absorption. Am J Physiol Endocrinol Metab. 2009;296:E1183–E1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alves-Bezerra M, Cohen DE. Triglyceride Metabolism in the Liver. Compr Physiol. 2017;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cohen DE, Fisher EA. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin Liver Dis. 2013;33:380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. 2018;75:3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. García-Arcos I, González-Kother P, Aspichueta P, Rueda Y, Ochoa B, Fresnedo O. Lipid analysis reveals quiescent and regenerating liver-specific populations of lipid droplets. Lipids. 2010;45:1101–1108. [DOI] [PubMed] [Google Scholar]
- 72. Tijburg LB, Nyathi CB, Meijer GW, Geelen MJ. Biosynthesis and secretion of triacylglycerol in rat liver after partial hepatectomy. Biochem J. 1991;277(Pt 3):723–728; (Pt 3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zou Y, Bao Q, Kumar S, Hu M, Wang GY, Dai G. Four waves of hepatocyte proliferation linked with three waves of hepatic fat accumulation during partial hepatectomy-induced liver regeneration. PLoS One. 2012;7:e30675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wirsching A, Eberhardt C, Wurnig MC, Boss A, Lesurtel M. Transient steatosis assessed by magnetic resonance imaging predicts outcome after extended hepatectomy in mice. Am J Surg. 2018;216:658–665. [DOI] [PubMed] [Google Scholar]
- 75. Shteyer E, Liao Y, Muglia LJ, Hruz PW, Rudnick DA. Disruption of hepatic adipogenesis is associated with impaired liver regeneration in mice. Hepatology. 2004;40:1322–1332. [DOI] [PubMed] [Google Scholar]
- 76. Delahunty TJ, Rubinstein D. Accumulation and release of triglycerides by rat liver following partial hepatectomy. J Lipid Res. 1970;11:536–543. [PubMed] [Google Scholar]
- 77. Iesari S, Leclercq I, Joudiou N, Komuta M, Daumerie A, Ambroise J, et al. Selective HIF stabilization alleviates hepatocellular steatosis and ballooning in a rodent model of 70% liver resection. Clin Sci (Lond). 2021;135:2285–2305. [DOI] [PubMed] [Google Scholar]
- 78. Reverter M, Rentero C, de Muga SV, Alvarez-Guaita A, Mulay V, Cairns R, et al. Cholesterol transport from late endosomes to the Golgi regulates t-SNARE trafficking, assembly, and function. Mol Biol Cell. 2011;22:4108–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Enrich C, Rentero C, de Muga SV, Reverter M, Mulay V, Wood P, et al. Annexin A6-Linking Ca(2+) signaling with cholesterol transport. Biochim Biophys Acta. 2011;1813:935–947. [DOI] [PubMed] [Google Scholar]
- 80. García-Melero A, Reverter M, Hoque M, Meneses-Salas E, Koese M, Conway JRW, et al. Annexin A6 and Late Endosomal Cholesterol Modulate Integrin Recycling and Cell Migration. J Biol Chem. 2016;291:1320–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Grewal T, Koese M, Rentero C, Enrich C. Annexin A6-regulator of the EGFR/Ras signalling pathway and cholesterol homeostasis. Int J Biochem Cell Biol. 2010;42:580–584. [DOI] [PubMed] [Google Scholar]
- 82. Hawkins TE, Roes J, Rees D, Monkhouse J, Moss SE. Immunological development and cardiovascular function are normal in annexin VI null mutant mice. Mol Cell Biol. 1999;19:8028–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cairns R, Fischer AW, Blanco-Munoz P, Alvarez-Guaita A, Meneses-Salas E, Egert A, et al. Altered hepatic glucose homeostasis in AnxA6-KO mice fed a high-fat diet. PLoS One. 2018;13:e0201310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cairns R, Alvarez-Guaita A, Martínez-Saludes I, Wason SJ, Hanh J, Nagarajan SR, et al. Role of hepatic Annexin A6 in fatty acid-induced lipid droplet formation. Exp Cell Res. 2017;358:397–410. [DOI] [PubMed] [Google Scholar]
- 85. Paloschi MV, Lopes JA, Boeno CN, Silva MDS, Evangelista JR, Pontes AS, et al. Cytosolic phospholipase A2-α participates in lipid body formation and PGE2 release in human neutrophils stimulated with an L-amino acid oxidase from Calloselasma rhodostoma venom. Sci Rep. 2020;10:10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Krautbauer S, Haberl EM, Eisinger K, Pohl R, Rein-Fischboeck L, Rentero C, et al. Annexin A6 regulates adipocyte lipid storage and adiponectin release. Mol Cell Endocrinol. 2017;439:419–430. [DOI] [PubMed] [Google Scholar]
- 87. Williams SD, Sakwe AM. Reduced Expression of Annexin A6 Induces Metabolic Reprogramming That Favors Rapid Fatty Acid Oxidation in Triple-Negative Breast Cancer Cells. Cancers (Basel). 2022;14:1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nguyen P, Leray V, Diez M, Serisier S, Bloc’h JL, Siliart B, et al. Liver lipid metabolism. J Anim Physiol Anim Nutr (Berl). 2008;92:272–283. [DOI] [PubMed] [Google Scholar]
- 89. Hussain MM. Intestinal lipid absorption and lipoprotein formation. Curr Opin Lipidol. 2014;25:200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rajan S, Hofer P, Christiano A, Stevenson M, Ragolia L, Villa-Cuesta E, et al. Microsomal triglyceride transfer protein regulates intracellular lipolysis in adipocytes independent of its lipid transfer activity. Metabolism. 2022;137:155331. [DOI] [PubMed] [Google Scholar]
- 91. Chen CY, Chen J, He L, Stiles BL. PTEN: Tumor Suppressor and Metabolic Regulator. Front Endocrinol (Lausanne). 2018;9:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Papa A, Pandolfi PP. The PTEN-PI3K Axis in Cancer. Biomolecules. 2019;9:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Valizadeh A, Majidinia M, Samadi-Kafil H, Yousefi M, Yousefi B. The roles of signaling pathways in liver repair and regeneration. J Cell Physiol. 2019. [DOI] [PubMed] [Google Scholar]
- 95. Kurlawalla-Martinez C, Stiles B, Wang Y, Devaskar SU, Kahn BB, Wu H. Insulin hypersensitivity and resistance to streptozotocin-induced diabetes in mice lacking PTEN in adipose tissue. Mol Cell Biol. 2005;25:2498–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Peyrou M, Bourgoin L, Poher AL, Altirriba J, Maeder C, Caillon A, et al. Hepatic PTEN deficiency improves muscle insulin sensitivity and decreases adiposity in mice. J Hepatol. 2015;62:421–429. [DOI] [PubMed] [Google Scholar]
- 97. Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci USA. 2004;101:2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhao B, Luo C, Zhang M, Xing F, Luo S, Fu S, et al. Knockdown of phosphatase and tensin homolog (PTEN) inhibits fatty acid oxidation and reduces very low density lipoprotein assembly and secretion in calf hepatocytes. J Dairy Sci. 2020;103:10728–10741. [DOI] [PubMed] [Google Scholar]
- 100. Vinciguerra M, Carrozzino F, Peyrou M, Carlone S, Montesano R, Benelli R, et al. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J Hepatol. 2009;50:1132–1141. [DOI] [PubMed] [Google Scholar]
- 101. Clément S, Peyrou M, Sanchez-Pareja A, Bourgoin L, Ramadori P, Suter D, et al. Down-regulation of phosphatase and tensin homolog by hepatitis C virus core 3a in hepatocytes triggers the formation of large lipid droplets. Hepatology. 2011;54:38–49. [DOI] [PubMed] [Google Scholar]
- 102. Peyrou M, Bourgoin L, Foti M. PTEN in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and cancer. Dig Dis. 2010;28:236–246. [DOI] [PubMed] [Google Scholar]
- 103. Qu X, Wen Y, Jiao J, Zhao J, Sun X, Wang F, et al. PARK7 deficiency inhibits fatty acid β-oxidation via PTEN to delay liver regeneration after hepatectomy. Clin Transl Med. 2022;12:e1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ma WT, Jia YJ, Liu QZ, Yang YQ, Yang JB, Zhao ZB, et al. Modulation of liver regeneration via myeloid PTEN deficiency. Cell Death Dis. 2017;8:e2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zaid A, Roubtsova A, Essalmani R, Marcinkiewicz J, Chamberland A, Hamelin J, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48:646–654. [DOI] [PubMed] [Google Scholar]
- 106. Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pauta M, Rotllan N, Vales F, Fernandez-Hernando A, Allen RM, Ford DA, et al. Impaired liver regeneration in Ldlr-/- mice is associated with an altered hepatic profile of cytokines, growth factors, and lipids. J Hepatol. 2013;59:731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Huang L, Wu H, Wu Y, Song F, Zhang L, Li Z, et al. Pcsk9 Knockout Aggravated Experimental Apical Periodontitis via LDLR. J Dent Res. 2022;101:83–92. [DOI] [PubMed] [Google Scholar]
- 109. Kaminsky-Kolesnikov Y, Rauchbach E, Abu-Halaka D, Hahn M, García-Ruiz C, Fernandez-Checa JC, et al. Cholesterol Induces Nrf-2- and HIF-1α-Dependent Hepatocyte Proliferation and Liver Regeneration to Ameliorate Bile Acid Toxicity in Mouse Models of NASH and Fibrosis. Oxid Med Cell Longev. 2020;2020:5393761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Živný P, Živná H, Palička V, Žaloudková L, Mocková P, Cermanová J, et al. Modulation of Rat Liver Regeneration after Partial Hepatectomy by Dietary Cholesterol. Acta Medica (Hradec Kralove). 2018;61:22–28. [DOI] [PubMed] [Google Scholar]
- 111. Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell. 2013;52:769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mottis A, Mouchiroud L, Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013;27:819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–123. [DOI] [PubMed] [Google Scholar]
- 114. Yamamoto H, Williams EG, Mouchiroud L, Cantó C, Fan W, Downes M, et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. 2011;147:827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yin F, Wu M, Wei X, Ren R, Liu M, Chen C, et al. Hepatic NCoR1 deletion exacerbates alcohol-induced liver injury in mice by promoting CCL2-mediated monocyte-derived macrophage infiltration. Acta Pharmacol Sin. 2022;43:2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Heck AM, Yanovski JA, Calis KA. Orlistat, a new lipase inhibitor for the management of obesity. Pharmacotherapy. 2000;20:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Grisouard J, Bouillet E, Timper K, Radimerski T, Dembinski K, Frey DM, et al. Both inflammatory and classical lipolytic pathways are involved in lipopolysaccharide-induced lipolysis in human adipocytes. Innate Immun. 2012;18:25–34. [DOI] [PubMed] [Google Scholar]
- 118. Li JX, Cummins CL. Fresh insights into glucocorticoid-induced diabetes mellitus and new therapeutic directions. Nat Rev Endocrinol. 2022;18:540–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Karabélyos C, Dobozy O, Szalai C, KlenjÁnszki K, VarjÚ K, HadhÁzy Á, et al. Elevated hepatic glucocorticoid receptor expression during liver regeneration in rats. Pathol Oncol Res. 1999;5:107–109. [DOI] [PubMed] [Google Scholar]
- 120. Tsukamoto I, Kojo S. Effect of glucocorticoid on liver regeneration after partial hepatectomy in the rat. Gut. 1989;30:387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Foucaud L, Niot I, Kanda T, Besnard P. Indirect dexamethasone down-regulation of the liver fatty acid-binding protein expression in rat liver. Biochim Biophys Acta. 1998;1391:204–212. [DOI] [PubMed] [Google Scholar]
- 122. Chen M, Bai M, YI Y, Lu S, Luo J, Li P, et al. Upregulation of hepatic CD36 via glucocorticoid receptor activation contributes to dexamethasone-induced liver lipid metabolism disorder in mice. Toxicol Lett. 2022;363:1–10. [DOI] [PubMed] [Google Scholar]
- 123. Hu X, Wang Y, Sheikhahmadi A, Li X, Buyse J, Lin H, et al. Effects of glucocorticoids on lipid metabolism and AMPK in broiler chickens’ liver. Comp Biochem Physiol B Biochem Mol Biol. 2019;232:23–30. [DOI] [PubMed] [Google Scholar]
- 124. Koorneef LL, Van Den Heuvel JK, Kroon J, et al. Selective Glucocorticoid Receptor Modulation Prevents and Reverses Nonalcoholic Fatty Liver Disease in Male Mice. Endocrinology. 2018;159:3925–3936. [DOI] [PubMed] [Google Scholar]
- 125. Yang F, Dai Y, Min C, Li X. Neonatal overfeeding induced glucocorticoid overexposure accelerates hepatic lipogenesis in male rats. Nutr Metab (Lond). 2018;15:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shen Y, Roh HC, kumari M, Rosen ED. Adipocyte glucocorticoid receptor is important in lipolysis and insulin resistance due to exogenous steroids, but not insulin resistance caused by high fat feeding. Mol Metab. 2017;6:1150–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Li R, Jia Y, Pan S, Li X, Song H, Zhao R. Glucocorticoid Receptor Mediates the Effect of High-Fat Diet on Mitochondrial Oxidative Phosphorylation in Mouse Liver. DNA Cell Biol. 2016;35:51–58. [DOI] [PubMed] [Google Scholar]
- 128. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. [DOI] [PubMed] [Google Scholar]
- 129. Kurzchalia TV, Parton RG. Membrane microdomains and caveolae. Curr Opin Cell Biol. 1999;11:424–431. [DOI] [PubMed] [Google Scholar]
- 130. Scherer PE, Lewis RY, Volonté D, Engelman JA, Galbiati F, Couet J, et al. Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. J Biol Chem. 1997;272:29337–29346. [DOI] [PubMed] [Google Scholar]
- 131. Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, et al. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996;271:15160–15165. [DOI] [PubMed] [Google Scholar]
- 132. Fernandez-Rojo MA, Ramm GA. Caveolin-1 Function in Liver Physiology and Disease. Trends Mol Med. 2016;22:889–904. [DOI] [PubMed] [Google Scholar]
- 133. Calvo M, Tebar F, Lopez-Iglesias C, Enrich C. Morphologic and functional characterization of caveolae in rat liver hepatocytes. Hepatology. 2001;33:1259–1269. [DOI] [PubMed] [Google Scholar]
- 134. Mayoral R, Fernández-Martínez A, Roy R, Boscá L, Martín-Sanz P. Dispensability and dynamics of caveolin-1 during liver regeneration and in isolated hepatic cells. Hepatology. 2007;46:813–822. [DOI] [PubMed] [Google Scholar]
- 135. Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. [DOI] [PubMed] [Google Scholar]
- 136. Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. [DOI] [PubMed] [Google Scholar]
- 137. Kuo A, Lee MY, Yang K, Gross RW, Sessa WC. Caveolin-1 regulates lipid droplet metabolism in endothelial cells via autocrine prostacyclin-stimulated, cAMP-mediated lipolysis. J Biol Chem. 2018;293:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Stremmel W, Pohl L, Ring A, Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids. 2001;36:981–989. [DOI] [PubMed] [Google Scholar]
- 139. Morales-Ruiz M, Santel A, Ribera J, Jiménez W. The Role of Akt in Chronic Liver Disease and Liver Regeneration. Semin Liver Dis. 2017;37:11–16. [DOI] [PubMed] [Google Scholar]
- 140. Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, et al. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem. 2003;278:32124–32131. [DOI] [PubMed] [Google Scholar]
- 141. Di Lorenzo A, Fernández-Hernando C, Cirino G, Sessa WC. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci U S A. 2009;106:14552–14557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A, et al. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci U S A. 2014;111:12865–12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 2001;292:1728–1731. [DOI] [PubMed] [Google Scholar]
- 144. Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Xu M, Wang H, Wang J, Burhan D, Shang R, Wang P, et al. mTORC2 Signaling Is Necessary for Timely Liver Regeneration after Partial Hepatectomy. Am J Pathol. 2020;190:817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhou Y, Xu M, Liu P, Liang B, Qian M, Wang H, et al. Mammalian Target of Rapamycin Complex 2 Signaling Is Required for Liver Regeneration in a Cholestatic Liver Injury Murine Model. Am J Pathol. 2020;190:1414–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Lao Y, Li Y, Zhang P, Shao Q, Lin W, Qiu B, et al. Targeting Endothelial Erk1/2-Akt Axis as a Regeneration Strategy to Bypass Fibrosis during Chronic Liver Injury in Mice. Mol Ther. 2018;26:2779–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hong F, Nguyen VA, Shen X, Kunos G, Gao B. Rapid activation of protein kinase B/Akt has a key role in antiapoptotic signaling during liver regeneration. Biochem Biophys Res Commun. 2000;279:974–979. [DOI] [PubMed] [Google Scholar]
- 149. Mullany LK, Nelsen CJ, Hanse EA, Goggin MM, Anttila CK, Peterson M, et al. Akt-mediated liver growth promotes induction of cyclin E through a novel translational mechanism and a p21-mediated cell cycle arrest. J Biol Chem. 2007;282:21244–21252. [DOI] [PubMed] [Google Scholar]
- 150. Haga S, Ozaki M, Inoue H, Okamoto Y, Ogawa W, Takeda K, et al. The survival pathways phosphatidylinositol-3 kinase (PI3-K)/phosphoinositide-dependent protein kinase 1 (PDK1)/Akt modulate liver regeneration through hepatocyte size rather than proliferation. Hepatology. 2009;49:204–214. [DOI] [PubMed] [Google Scholar]
- 151. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017;169:381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zhang K, Guo X, Yan H, Wu Y, Pan Q, Shen JZ, et al. Phosphorylation of Forkhead Protein FoxO1 at S253 Regulates Glucose Homeostasis in Mice. Endocrinology. 2019;160:1333–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Langlet F, Haeusler RA, Lindén D, Ericson E, Norris T, Johansson A, et al. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell. 2017;171:824–835.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380(Pt 2):297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sparks JD, Dong HH. FoxO1 and hepatic lipid metabolism. Curr Opin Lipidol. 2009;20:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281:10105–10117. [DOI] [PubMed] [Google Scholar]
- 157. Liu L, Zheng LD, Zou P, Brooke J, Smith C, Long YC, et al. FoxO1 antagonist suppresses autophagy and lipid droplet growth in adipocytes. Cell Cycle. 2016;15:2033–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Klöting N, Wilke B, Klöting I. Triplet repeat in the Repin1 3’-untranslated region on rat chromosome 4 correlates with facets of the metabolic syndrome. Diabetes Metab Res Rev. 2007;23:406–410. [DOI] [PubMed] [Google Scholar]
- 159. Heiker JT, Klöting N. Replication initiator 1 in adipose tissue function and human obesity. Vitam Horm. 2013;91:97–105. [DOI] [PubMed] [Google Scholar]
- 160. Abshagen K, Mense L, Fischer F, Liebig M, Schaeper U, Navarro G, et al. Repin1 deficiency in liver tissue alleviates NAFLD progression in mice. J Adv Res. 2019;16:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ruschke K, Illes M, Kern M, Klöting I, Fasshauer M, Schön MR, et al. Repin1 maybe involved in the regulation of cell size and glucose transport in adipocytes. Biochem Biophys Res Commun. 2010;400:246–251. [DOI] [PubMed] [Google Scholar]
- 162. Kern M, Kosacka J, Hesselbarth N, Brückner J, Heiker JT, Flehmig G, et al. Liver-restricted Repin1 deficiency improves whole-body insulin sensitivity, alters lipid metabolism, and causes secondary changes in adipose tissue in mice. Diabetes. 2014;63:3295–3309. [DOI] [PubMed] [Google Scholar]
- 163. Abshagen K, Degenhardt B, Liebig M, Wendt A, Genz B, Schaeper U, et al. Liver-specific Repin1 deficiency impairs transient hepatic steatosis in liver regeneration. Sci Rep. 2018;8:16858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Byrnes K, Blessinger S, Bailey NT, Scaife R, Liu G, Khambu B. Therapeutic regulation of autophagy in hepatic metabolism. Acta Pharm Sin B. 2022;12:33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. [DOI] [PubMed] [Google Scholar]
- 167. Martinez-Lopez N, Singh R. Autophagy and Lipid Droplets in the Liver. Annu Rev Nutr. 2015;35:215–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Römermann D, Ansari N, Schultz‐Moreira AR, Michael A, Marhenke S, Hardtke‐Wolenski M, et al. Absence of Atg7 in the liver disturbed hepatic regeneration after liver injury. Liver Int. 2020;40:1225–1238. [DOI] [PubMed] [Google Scholar]
- 169. Xu F, Hua C, Tautenhahn HM, Dirsch O, Dahmen U. The Role of Autophagy for the Regeneration of the Aging Liver. Int J Mol Sci. 2020;21:3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Chen Y, Xu Z, Zeng Y, Liu J, Wang X, Kang Y. Altered metabolism by autophagy defection affect liver regeneration. PLoS One. 2021;16:e0250578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Lai JL, Lian YE, Wu JY, Wang YD, Bai YN. Verapamil induces autophagy to improve liver regeneration in non-alcoholic fatty liver mice. Adipocyte. 2021;10:532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhang X, Li S, Ren X, et al. TIPE1 promotes liver regeneration by enhancing ROS-FoxO1 axis mediated autophagy. FEBS J. 2022;290:1117–1133. [DOI] [PubMed] [Google Scholar]
- 173. Jia CJ, Sun H, Dai CL. Autophagy Contributes to Liver Regeneration After Portal Vein Ligation in Rats. Med Sci Monit. 2019;25:5674–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lu T, Hao J, Shen C, Gu G, Zhang J, Xu N. Partial Hepatectomy-Induced Upregulation of miR-1907 Accelerates Liver Regeneration by Activation Autophagy. Biomed Res Int. 2018;2018:3817057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ni HM, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, et al. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci. 2012;127:438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Byun S, Seok S, Kim YC, Zhang Y, Yau P, Iwamori N, et al. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat Commun. 2020;11:807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Yang C, Lu W, Lin T, You P, Ye M, Huang Y, et al. Activation of Liver FGF21 in hepatocarcinogenesis and during hepatic stress. BMC Gastroenterol. 2013;13:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Bosc C, Broin N, Fanjul M, Saland E, Farge T, Courdy C, et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat Commun. 2020;11:4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Kim J, Lee S, Lee MS. Suppressive Effect of Autocrine FGF21 on Autophagy-Deficient Hepatic Tumorigenesis. Front Oncol. 2022;12:832804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Szczepańska E, Gietka-Czernel M. FGF21: A Novel Regulator of Glucose and Lipid Metabolism and Whole-Body Energy Balance. Horm Metab Res. 2022;54:203–211. [DOI] [PubMed] [Google Scholar]
- 181. Wei X, Luo L, Chen J. Roles of mTOR Signaling in Tissue Regeneration. Cells. 2019;8:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Zhang L, Li Y, Wang Y, Qiu Y, Mou H, Deng Y, et al. mTORC2 Facilitates Liver Regeneration Through Sphingolipid-Induced PPAR-α-Fatty Acid Oxidation. Cell Mol Gastroenterol Hepatol. 2022;14:1311–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Brasaemle DL. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res. 2007;48:2547–2559. [DOI] [PubMed] [Google Scholar]
- 184. Brasaemle DL, Wolins NE. Packaging of fat: An evolving model of lipid droplet assembly and expansion. J Biol Chem. 2012;287:2273–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Brasaemle DL, Barber T, Wolins NE, Serrero G, Blanchette-Mackie EJ, Londos C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J Lipid Res. 1997;38:2249–2263. [PubMed] [Google Scholar]
- 186. Imamura M, Inoguchi T, Ikuyama S, Taniguchi S, Kobayashi K, Nakashima N, et al. ADRP stimulates lipid accumulation and lipid droplet formation in murine fibroblasts. Am J Physiol Endocrinol Metab. 2002;283:E775–E783. [DOI] [PubMed] [Google Scholar]
- 187. Chang BHJ, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, et al. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Chang BH, Li L, Saha P, Chan L. Absence of adipose differentiation related protein upregulates hepatic VLDL secretion, relieves hepatosteatosis, and improves whole body insulin resistance in leptin-deficient mice. J Lipid Res. 2010;51:2132–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Kohjima M, Tsai TH, Tackett BC, Thevananther S, Li L, Chang BHJ, et al. Delayed liver regeneration after partial hepatectomy in adipose differentiation related protein-null mice. J Hepatol. 2013;59:1246–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Tsai TH, Chen E, Li L, Saha P, Lee HJ, Huang LS, et al. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy. 2017;13:1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Griffin JD, Bejarano E, Wang XD, Greenberg AS. Integrated Action of Autophagy and Adipose Tissue Triglyceride Lipase Ameliorates Diet-Induced Hepatic Steatosis in Liver-Specific PLIN2 Knockout Mice. Cells. 2021;10:1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Xie Y, Wang H, Cheng X, Wu Y, Cao L, Wu M, et al. Farnesoid X receptor activation promotes cell proliferation via PDK4-controlled metabolic reprogramming. Sci Rep. 2016;6:18751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sun S, Liu J, Zhao M, Han Y, Chen P, Mo Q, et al. Loss of the novel mitochondrial protein FAM210B promotes metastasis via PDK4-dependent metabolic reprogramming. Cell Death Dis. 2017;8:e2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Pettersen IKN, Tusubira D, Ashrafi H, Dyrstad SE, Hansen L, Liu XZ, et al. Upregulated PDK4 expression is a sensitive marker of increased fatty acid oxidation. Mitochondrion. 2019;49:97–110. [DOI] [PubMed] [Google Scholar]
- 195. Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602–18612. [DOI] [PubMed] [Google Scholar]
- 196. Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA. 2008;105:1820–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci USA. 2004;101:7100–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, et al. NARC-1/PCSK9 and its natural mutants: Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865–48875. [DOI] [PubMed] [Google Scholar]
- 199. Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. [DOI] [PubMed] [Google Scholar]
- 200. Timms KM, Wagner S, Samuels ME, Forbey K, Goldfine H, Jammulapati S, et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet. 2004;114:349–353. [DOI] [PubMed] [Google Scholar]
- 201. Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. [DOI] [PubMed] [Google Scholar]
- 202. Baragetti A, Balzarotti G, Grigore L, Pellegatta F, Guerrini U, Pisano G, et al. PCSK9 deficiency results in increased ectopic fat accumulation in experimental models and in humans. Eur J Prev Cardiol. 2017;24:1870–1877. [DOI] [PubMed] [Google Scholar]
- 203. Demers A, Samami S, Lauzier B, Des Rosiers C, Sock ETN, Ong H, et al. PCSK9 Induces CD36 Degradation and Affects Long-Chain Fatty Acid Uptake and Triglyceride Metabolism in Adipocytes and in Mouse Liver. Arterioscler Thromb Vasc Biol. 2015;35:2517–2525. [DOI] [PubMed] [Google Scholar]
- 204. Rashid S, Kastelein JJ. PCSK9 and resistin at the crossroads of the atherogenic dyslipidemia. Expert Rev Cardiovasc Ther. 2013;11:1567–1577. [DOI] [PubMed] [Google Scholar]
- 205. Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, et al. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation. 2014;130:431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Wu Y, Chen K, Li L, Hao Z, Wang T, Liu Y, et al. Plin2-mediated lipid droplet mobilization accelerates exit from pluripotency by lipidomic remodeling and histone acetylation. Cell Death Differ. 2022;29:2316–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Zhang X, Sukamporn P, Zhang S, Min KW, Baek SJ. 3,3’-diindolylmethane downregulates cyclin D1 through triggering endoplasmic reticulum stress in colorectal cancer cells. Oncol Rep. 2017;38:569–574. [DOI] [PubMed] [Google Scholar]
- 208. Langiewicz M, Graf R, Humar B, Clavien PA. JNK1 induces hedgehog signaling from stellate cells to accelerate liver regeneration in mice. J Hepatol. 2018;69:666–675. [DOI] [PubMed] [Google Scholar]
- 209. Sinha RA, Rajak S, Singh BK, Yen PM. Hepatic Lipid Catabolism via PPARα-Lysosomal Crosstalk. Int J Mol Sci. 2020;21:2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Gonzalez FJ, Shah YM. PPARalpha: Mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2–8. [DOI] [PubMed] [Google Scholar]
- 211. Brocker CN, Yue J, Kim D, Qu A, Bonzo JA, Gonzalez FJ. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am J Physiol Gastrointest Liver Physiol. 2017;312:G283–G299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Fan S, Gao Y, Qu A, Jiang Y, Li H, Xie G, et al. YAP-TEAD mediates PPAR α-induced hepatomegaly and liver regeneration in mice. Hepatology. 2022;75:74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Yuan X, Yan S, Zhao J, Shi D, Yuan B, Dai W, et al. Lipid metabolism and peroxisome proliferator-activated receptor signaling pathways participate in late-phase liver regeneration. J Proteome Res. 2011;10:1179–1190. [DOI] [PubMed] [Google Scholar]
- 214. Rao MS, Peters JM, Gonzalez FJ, Reddy JK. Hepatic regeneration in peroxisome proliferator-activated receptor alpha-null mice after partial hepatectomy. Hepatol Res. 2002;22:52–57. [DOI] [PubMed] [Google Scholar]
- 215. Wheeler MD, Smutney OM, Check JF, Rusyn I, Schulte-Hermann R, Thurman RG. Impaired Ras membrane association and activation in PPARalpha knockout mice after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2003;284:G302–G312. [DOI] [PubMed] [Google Scholar]
- 216. Anderson SP, Yoon L, Richard EB, Dunn CS, Cattley RC, Corton JC. Delayed liver regeneration in peroxisome proliferator-activated receptor-alpha-null mice. Hepatology. 2002;36:544–554. [DOI] [PubMed] [Google Scholar]
- 217. Xie G, Song Y, Li N, Zhang Z, Wang X, Liu Y, et al. Myeloid peroxisome proliferator-activated receptor α deficiency accelerates liver regeneration via IL-6/STAT3 pathway after 2/3 partial hepatectomy in mice. Hepatobiliary Surg Nutr. 2022;11:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages(*). J Hepatol. 2020;73:757–770. [DOI] [PubMed] [Google Scholar]
- 219. Adinolfi L, Rinaldi L, Guerrera B, Restivo L, Marrone A, Giordano M, et al. NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations. Int J Mol Sci. 2016;17:803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Brar G, Tsukamoto H. Alcoholic and non-alcoholic steatohepatitis: global perspective and emerging science. J Gastroenterol. 2019;54:218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. [DOI] [PubMed] [Google Scholar]
- 222. Rakotonirina-Ricquebourg R, Costa V, Teixeira V. Hello from the other side: Membrane contact of lipid droplets with other organelles and subsequent functional implications. Prog Lipid Res. 2022;85:101141. [DOI] [PubMed] [Google Scholar]
- 223. Mashek DG. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol Metab. 2021;50:101115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:14205–14218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology. 2012;142:711–725.e6. [DOI] [PubMed] [Google Scholar]
- 226. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. [DOI] [PubMed] [Google Scholar]
- 227. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu Rev Pathol. 2016;11:451–496. [DOI] [PubMed] [Google Scholar]
- 228. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhu C, Tabas I, Schwabe RF, Pajvani UB. Maladaptive regeneration - the reawakening of developmental pathways in NASH and fibrosis. Nat Rev Gastroenterol Hepatol. 2021;18:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Picard C, Lambotte L, Starkel P, Sempoux C, Saliez A, Van Den Berge V, et al. Steatosis is not sufficient to cause an impaired regenerative response after partial hepatectomy in rats. J Hepatol. 2002;36:645–652. [DOI] [PubMed] [Google Scholar]
- 231. Veteläinen R, van Vliet A, Gouma DJ, van Gulik TM. Steatosis as a risk factor in liver surgery. Ann Surg. 2007;245:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Chu MJ, Dare AJ, Phillips AR, Bartlett AS. Donor Hepatic Steatosis and Outcome After Liver Transplantation: a Systematic Review. J Gastrointest Surg. 2015;19:1713–1724. [DOI] [PubMed] [Google Scholar]
- 233. Yamauchi H, Uetsuka K, Okada T, Nakayama H, Doi K. Impaired liver regeneration after partial hepatectomy in db/db mice. Exp Toxicol Pathol. 2003;54:281–286. [DOI] [PubMed] [Google Scholar]
- 234. Leclercq IA, Vansteenberghe M, Lebrun VB, VanHul NK, Abarca-Quinones J, Sempoux CL, et al. Defective hepatic regeneration after partial hepatectomy in leptin-deficient mice is not rescued by exogenous leptin. Lab Invest. 2006;86:1161–1171. [DOI] [PubMed] [Google Scholar]
- 235. Zhang BH, Weltman M, Farrell GC. Does steatohepatitis impair liver regeneration? A study in a dietary model of non-alcoholic steatohepatitis in rats. J Gastroenterol Hepatol. 1999;14:133–137. [DOI] [PubMed] [Google Scholar]
- 236. Sydor S, Gu Y, Schlattjan M, Bechmann LP, Rauen U, Best J, et al. Steatosis does not impair liver regeneration after partial hepatectomy. Lab Invest. 2013;93:20–30. [DOI] [PubMed] [Google Scholar]
- 237. Garnol T, Kučera O, Staňková P, Lotková H, Červinková Z. Does Simple Steatosis Affect Liver Regeneration after Partial Hepatectomy in Rats. Acta Medica (Hradec Kralove). 2016;59:35–42. [DOI] [PubMed] [Google Scholar]
- 238. Hamano M, Ezaki H, Kiso S, Furuta K, Egawa M, Kizu T, et al. Lipid overloading during liver regeneration causes delayed hepatocyte DNA replication by increasing ER stress in mice with simple hepatic steatosis. J Gastroenterol. 2014;49:305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]