

Abstract
Dolastatin 16, a marine cyclic depsipeptide, was initially isolated from the sea hare Dolabella Auricularia by Pettit et al. Due to the lack of information regarding its bioactivity, target identification becomes an indispensable strategy for revealing the potential targets and mechanisms of action of Dolastatin 16. Network pharmacology was utilized to identify targets associated with the disease, gene ontology, and KEGG pathways. The results highlighted Matrix Metalloproteinase-9 (MMP9) as a potential target of Dolastatin 16 through network pharmacology analysis. This target was found to be primarily involved in the TNF signaling pathway and in foot ulceration-associated diabetic polyneuropathy. Furthermore, the binding mode and dynamic behavior of the complex were investigated through molecular docking and molecular dynamics studies. In the docking study, a native ligand (a hydroxamate inhibitor) and (R)-ND-336 were employed as ligand controls, demonstrating binding energy values of − 6.6 and − 8.9 kcal/mol, respectively. The Dolastatin 16 complex exhibited a strong affinity for MMP9, with a binding energy value of − 9.7 kcal/mol, indicating its high potential as an inhibitor. Molecular dynamics also confirmed the stability of the MMP9-Dolastatin complex throughout the simulation process. Dolastatin 16 has the potential to act as an MMP9 inhibitor, offering promise for accelerating the wound healing process in diabetic foot conditions.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40203-023-00161-5.
Keywords: Diabetic wound, Dolastatin 16, MMP9, Molecular docking, Molecular dynamics, Network pharmacology
Introduction
Natural cyclic peptides offer valuable pharmacological properties applicable to therapeutic agents. Over the past two decades, more than 40 cyclic peptide-based drugs have received clinical approval, primarily derived from natural products (Jing and Jin 2020). These compounds have demonstrated high metabolic stability, strong binding affinity, proteolytic stability, and target selectivity. Additionally, cyclic peptides possess an expanded surface area, allowing them to tightly interact with specific targets, thereby exhibiting remarkable selectivity and affinity (Nielsen et al. 2017; Zorzi et al. 2017). Notably, these substances include proline-rich marine cyclic peptides (Fang et al. 2016).
Dolastatin 16, a proline-rich cyclic depsipeptide, was first isolated from the sea hare Dolabella Auricularia in Papua New Guinea. The structure of Dolastatin 16 consists of cyclo–(Pro1–Dpv2–Pro3–Dml4–O-Lac5–Pro6–O-Hiv7–MeVal8) (Fig. 1). In (1997), Pettit et al. reported its remarkable cytotoxicity against various human cancer cell lines. However, in subsequent attempts, synthetic Dolastatin 16 failed to reproduce cancer cell growth inhibition. The result revealed that another compound, not chemically detected, was observed in the 1997 study (Pettit et al. 2014). Since then, the bioactivity of Dolastatin 16 has remained a historical question. Target identification is necessary to unravel the potential target and mechanism of action of Dolastatin 16.
Fig. 1.

Structure of Dolastatin 16, modified from (Casalme et al. 2017; Pettit et al. 1997)
In drug discovery research, target identification plays a crucial role in validating appropriate targets for drugs, leading to desired therapeutic effects. Nowadays, there are two primary approaches to drug target identification: experimental and computational studies. Experimental verification involves biological assays, proteomic analysis, and mass spectrometry (Tsuda et al. 2022). Regarding the computational approach, the network pharmacology method integrates network biology and multi-omics data to identify biomarkers and discover potential therapeutic targets. This approach combines graph theory and pathway analysis to investigate the important components within the network (Jain et al. 2018).
Khairy et al. (2023) reported that network pharmacology-based analysis was useful for predicting the anti-inflammatory targets of certain active constituents from plants. Similarly, Hu et al. (2023) successfully investigated the therapeutic targets and mechanisms of proton pump inhibitors in diabetes-associated breast cancer using network pharmacology analysis. Therefore, network pharmacology represents a valuable tool for unraveling the potential targets of drugs. With the advancement of computational technologies, this approach might also help address the complexity of tasks in the drug discovery process.
In a study by Liang et al. (2018), the target prediction of Dolastatin 16 was conducted using a combination of computational screening, molecular docking, and experimental validation. Peptidyl–prolyl cis–trans isomerase FKBP1A (FKBP12) was predicted as a potential target of Dolastatin 16 via the PharmMapper database, and the results were further verified through chemical–protein interactome (CPI) experimental studies. In the present study, however, we comprehensively investigated the potential target of Dolastatin 16 from different perspectives, employing network pharmacology, molecular docking, and molecular dynamics studies. The primary objective was to identify the most crucial therapeutic target of Dolastatin 16, as well as its mechanism of action in the disease. Molecular docking and molecular dynamics simulations were additionally performed to validate the interaction stability between Dolastatin 16 and its target.
Material and methods
Network pharmacology analysis
Target identification of dolastatin 16
The simplified molecular-input line-entry specification (SMILES) information of Dolastatin 16 was obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/), and subsequently submitted to Super-PRED (https://prediction.charite.de/), SWISS Target Prediction (http://www.swisstargetprediction.ch/), and SEA Target (https://sea.bkslab.org/) to predict the targets of Dolastatin 16. Homo sapiens was designated as the organism for data retrieval. To standardize protein IDs and eliminate duplicate proteins, UniProt ID (https://www.uniprot.org/) was used for protein ID alignment. The overall steps of this study are shown in Fig. 2.
Fig. 2.

Flow chart of the network pharmacology, molecular docking, and molecular dynamics-based study
Construction of protein–protein interactions (PPI)
All proteins resulting from the previous step were input into the STRING database (https://string-db.org/) using the "multiple protein" option. The parameter settings included Homo sapiens as the organism, "full STRING network" as the type of network, a "high confidence" score (0.700), and an FDR stringency of "medium (5 percent)". PPI data in TSV format was downloaded from the "explore" option and then analyzed using Cytoscape 3.9.1 software to visualize and construct the PPI network.
Network construction and topological analysis using cytoscape software
The TSV format of PPI data was imported into Cytoscape 3.9.1 software ((http://www.cytoscape.org/) for topological analysis. In this analysis, the term "degree" refers to the number of connections among nodes within the network. The degree value indicates the importance of core targets (Tao et al. 2020). In this study, nodes with the highest degree were determined as the most important nodes in the network and categorized as "core targets". Additionally, the accuracy of the listed core targets was evaluated based on the three databases (Super-PRED, SWISS Target, Sea Target) used in this study. The target with the highest accuracy (found in two–three databases) was selected for further analysis.
Disease analysis, gene ontology biological process, and KEGG pathway
The core targets were input into the DAVID database (https://david.ncifcrf.gov/) to obtain predictions for diseases, gene ontology, and KEGG pathway information. The parameter was set at FDR < 0.05, and the results were visualized as a bar graph.
Molecular docking studies
Protein and ligand selection
The information regarding the protein targets was sourced from the RCSB database (https://www.rcsb.org/). PDB IDs with resolutions within the range of 2.0–3.0 Å were selected. The structure of Dolastatin 16 and several protein inhibitors, which were not provided in the co-crystallized ligands, were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). The database https://cactus.nci.nih.gov/translate/ was employed to convert 2D ligand structures to their 3D structure. The protein inhibitors were used as standards for conducting and comparing the outcomes of this molecular docking simulation.
Binding pocket analysis
The analysis of binding sites was conducted based on available literature. The list of binding sites for each target is presented in Table 1.
Table 1.
The binding sites of the protein targets
| Targets | PDB ID | Binding Pockets | References |
|---|---|---|---|
| PIK3CA | 4JPS | Met772/Pro778/Ile800/Lys802/Asp810/Tyr836/Val851/Ser854/Gln859/Asp933 | (Furet et al. 2013) |
| CASP3 | 1RE1 | Glu379/Gln351/Asp345/Gly346/Ser343/Arg341/ Tyr338/Trp340/Asn342/Trp348/Phe380/Glu381/ Ser381a/Phe381b/Phe381h | (Gond et al. 2022) |
| NFKB1 | 4KIK | Leu21/Gly22/Val29/Ala42/Lys44/Val74/Met96/ Glu97/Tyr98/Cys99/Glu149/Val152/Ile165 | (Liu et al. 2013) |
| MMP9 | 1GKC (Catalytic site) | Gly186/Leu187/Leu188/Ala189/Leu222/Val223/ Glu227/Leu243/Val398/His401/His405/His411/Pro421/Tyr423/et422 | (Mathpal et al. 2022; Rowsell et al. 2002) |
| 1ITV (Hemopexin site) | Asn7/Ile8/Lys26 | (Cha et al. 2002) | |
| LYN | 2ZVA | Leu253/Ala273/Lys275/Glu290/Met294/Val303/ Ile317/Thr319/Met322/Ala323/Gly325/Leu374/ Ala384 | (Williams et al. 2009) |
| KDR | 3WZD | Gly841/Val848/Leu840/Ala866/Glu885/Val899/Val916/he918/Cys919/Lys920/Gly922/Asn923/Leu1035/Cys1045/Phe1047 | (Okamoto et al. 2014) |
| JAK2 | 6VGL | Leu855/Gly856/Ala880/Val863/Val911/Met929/ Glu930/Typ931/Leu932/Asn981/Leu983/Asp994 | (Davis et al. 2021) |
Protein and ligand preparation
The protein structures downloaded from the RCSB database underwent cleaning by eliminating water molecules and unnecessary chains through Biovia Discovery Studio Visualizer software. Additionally, the proteins were separated from previously bound ligand groups. Polar hydrogen atoms were added to both the protein structure and ligand, and the results were saved in PDB format.
Molecular docking
A molecular docking study was conducted using Dolastatin 16 as the ligand and the primary targets obtained from the network pharmacology analysis as macromolecules. However, three targets (VEGFA, ITGB1, and PRKCD) were excluded from this study due to the majority of their inhibitors being available in antibody form, and the crystal structure of PRKCD not being present in the RCSB database. The AutoDock Vina program integrated into PyRx 0.9.8 software was utilized for the molecular docking study (Dallakyan and Olson 2015; Stanzione et al. 2021; Eberhardt et al. 2021). Ligands and target proteins, prepared in the previous step, were formatted in PDB file format and subjected to AutoDock Vina. The SDF file format of Dolastatin 16 and several standard compounds were subjected to energy minimization using the Open Babel plugin in the PyRx program. These were then converted to PDBQT format (O’Boyle et al. 2011). Before simulation, the grid box was adjusted to cover binding site residues. The grid spacing was set to 0.375 Å, and the exhaustiveness was set to the default value of 8 (Rathod et al. 2022). For validation, the RMSD of native ligands in post-molecular docking states was compared to pre-docking states using the LigRMSD website (https://ligrmsd.appsbio.utalca.cl/). The docking simulation was executed through 5 iterations. The best pose was selected for further analysis and visualization using Biovia Discovery Studio Visualizer, Pymol, and Chimera.
Molecular dynamics simulation
To explore the stability of protein–ligand interactions, molecular dynamics simulations were performed using YASARA v 21.12.19.M.64 software over a period of 50.00 ns, with a timestep of 2.5 fs and the AMBER14 force field. Before simulation, parameters were configured in the md_run.mcr. The simulation was carried out at a temperature of 310 K, pH 7.4, NaCl concentration of 0.9%, and a pressure of 1 atm. The results of RMSD, RMSF, and Radius of Gyration were obtained through the execution of md_analyze.mcr. This study also conducted MM-PBSA (Molecular Mechanics-Poisson Boltzmann Surface Area) binding free energy calculations by subjecting the trajectories to md_analyzebindenergy.mcr. MM-PBSA calculations are based on the following equations:
Binding energy = EpotReceptor + EsolvReceptor + EpotLigand + EsolvLigand – EpotComplex – EsolvComplex.
A more positive binding energy value indicates greater binding stability of the protein–ligand complex (Mitra and Dash 2018). Snapshots from each time point of the simulation were presented by running md_play.mcr, and molecular visualization was performed through Chimera 1.16 and Biovia Discovery Studio Visualizer software.
Results and discussion
Results
Network pharmacology analysis
Database retrieval of Dolastatin 16 potential targets
Upon submitting the SMILES code of Dolastatin 16 to the database, it resulted in 114 targets (Super PRED), 103 targets (SWISS Target), and 7 targets (SEA Target). The complete list of all targets is provided in SI Table 1. These listed targets were compiled and combined into an Excel format, which was then imported into UniProt (https://www.uniprot.org/) to eliminate duplicate protein targets and obtain valid protein ID names.
Protein–protein interaction networks
Protein IDs resulting from the Uniprot alignment were assessed for interactions using the STRING database (https://string-db.org/). To visualize these interactions, the outcomes were imported into Cytoscape 3.9.1 software for topological analysis. The findings of this analysis are presented in Fig. 3. Circular nodes represent gene targets, with node color proportional to the number of degrees. Nodes in purple (PIK3CA, VEGFA, CASP3, NFKB1, MMP9, LYN, PRKCD, ITGB1, KDR, and JAK2) have higher degrees. The specific degree values are provided in SI Table 2.
Fig. 3.

The illustration of PPI network from all targets
To identify the potential target of Dolastatin 16, all core targets were compared to the three databases previously utilized for predicting protein targets of Dolastatin 16. A consensus was reached by considering targets detected in two to three databases. The outcome revealed that only MMP9 was detected in two databases. SI Table 3 displays the probabilities of the targets found in the databases. Subsequently, our focus shifted to MMP9 as the potential target of Dolastatin 16.
Analysis of disease, gene ontology biological process, and KEGG pathway
The list of 10 core targets yielded results for diseases, gene ontology biological processes, and KEGG pathways from the DAVID database (https://david.ncifcrf.gov/) as depicted in Fig. 4.
Fig. 4.

Enrichment analysis. a Disease prediction, b gene ontology biological process, and c KEGG pathway related to 10 core targets. The involvement of MMP9 in specific diseases, biological processes, and pathway terms is illustrated in brown color
Network construction of compound-core targets-diseases
After obtaining the results of core targets and diseases, a visualization of network construction was presented to understand the relationship between Dolastatin 16, its main targets, and associated diseases, as depicted Fig. 5
Fig. 5.

Network diagram of Dolastatin16-core targets-diseases. The color of the core target nodes transitions from yellow to purple, indicating a shift from low to high degree values in the network
Pathway mechanisms of Dolastatin 16 against MMP9 in diabetic wound healing
The pathway mechanism of NFKB signaling mediated by TNF-α was adopted and adapted from https://www.kegg.jp/pathway/hsa04668 along with information from literature (Yu et al. 2020), which is provided in the Supplementary Material. The role of Dolastatin 16 in accelerating diabetic wound healing is depicted in Fig. 6. The figure has been modified from previous studies (Nguyen et al. 2018; Chen et al. 2023). These pathway mechanisms were generated using the BioRender online tool (https://www.biorender.com).
Fig. 6.

The role of Dolastatin 16 in diabetic wound healing process
Molecular docking studies
In the docking simulation, the grid box center was adjusted to the binding site of the co-crystallized ligand (a reverse hydroxamate) within the MMP9 receptor. The specific grid box details can be found in SI Table 4. To attain the optimal pose of the model, this study conducted simulations through 5 iterations, wherein the RMSD of control docking was compared against the initial pose for each iteration. Figure 7 depicts the best model of the ligand control in the MMP9. The similarity in conformational orientation between the docked pose and the co-crystallized ligand (RMSD 0.98 Å) validates the accuracy of the docking protocol. The results of the binding affinity are presented in Fig. 8.
Fig. 7.
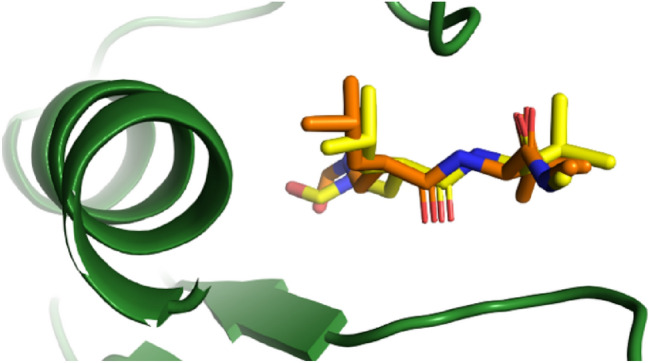
The best re-docking model with RMSD value of 0.98 Å (Original: yellow, Docked: orange)
Fig. 8.

The value of binding affinity and hydrogen bond of each target in the Dolastatin 16 complex
Molecular dynamics simulation
Upon completion of the MD simulations, we proceeded to analyze the outcomes derived from RMSD, RMSF, Radius of Gyration, and MM-PBSA binding free energy calculations. The figures depicting RMSD and MM-PBSA binding free energy are presented in Fig. 10, while the remaining results are provided in the Supplementary Information.
Fig. 10.

Molecular dynamic result. a RMSD-Cα, b RMSD-Ligand movement, and c MM-PBSA binding energy calculation plot of ligands-MMP9 over the 50 ns MD simulations. The colors green, blue, and orange correspond to MMP9, (R)-ND-336, and Dolastatin 16, respectively
Discussion
This study employed a network pharmacology approach to explore potential targets for Dolastatin 16. The process involved several steps, including target identification using databases, protein–protein interaction (PPI) analysis, topological analysis using Cytoscape 3.9.1 software, and target accuracy assessment. From a total of 202 related targets, the top 10 with the highest degrees resulted from the topological analysis. These targets were then cross-referenced with target prediction databases to identify the target with the highest frequency. MMP9 emerged as the most frequent target across both the Super PRED and SWISS Target databases, thus becoming the focus of subsequent analysis.
Subsequently, the 10 core targets underwent further examination through disease analysis, gene ontology biological processes, and KEGG pathway exploration. Based on the results in Fig. 4a, Diabetic Neuropathy, encompassing conditions like Diabetic Polyneuropathies, Diabetic Mononeuropathy, Asymmetric Diabetic Proximal Motor Neuropathy, Diabetic Asymmetric Polyneuropathy, Diabetic Autonomic Neuropathy, Diabetic Amyotrophy, Symmetric Diabetic Proximal Motor Neuropathy, and Diabetic Neuralgia emerged as the most probable disease association. Figure 4b outlined the top four biological processes in which MMP9 played a role, including positive regulation of cell migration (GO:0030335), negative regulation of apoptotic process (GO:0043066), apoptotic process (GO:0006915), and positive regulation of protein phosphorylation (GO:0001934). These processes were associated with pathways like Lipid and Atherosclerosis (hsa05417), Fluid Shear Stress and Atherosclerosis (hsa05418), and TNF Signaling Pathway (hsa04668), as depicted in Fig. 4c. Although atherosclerosis and diabetes often involve chronic inflammation through the TNF signaling pathway, this study focused exclusively on the diabetic context. Figure 5 highlighted the network connections among Dolastatin 16, core targets, and diabetic polyneuropathy.
Diabetic neuropathy stands as one of the most prevalent complications of diabetes, significantly impacting sensory function. Over time, nearly 50% of individuals with diabetes develop diabetic neuropathy, with type 2 diabetes mellitus patients being most affected (6,100/100,000 persons per year) (Feldman et al. 2019). This chronic condition gives rise to neuropathic pain, painless foot ulcers, and autonomic dysfunction (Bönhof et al. 2018). Among various types of diabetic neuropathies, diabetic polyneuropathy is the most common condition, often leading to foot ulceration (De Gregorio et al. 2020). Within the diabetic population, foot disorders contribute to approximately 85% of all amputation cases (Begum et al. 2022). Without effective interventions, these cases are expected to rise in the coming decades.
The TNF signaling pathway assumes a crucial role in the pathogenesis of diabetic foot ulceration (DFU). Tumor necrosis factor alpha (TNF-α), a member of the TNF family, stands as a potent proinflammatory cytokine produced by macrophages during acute inflammation. Its presence has also been documented in patients with infected diabetic foot ulcers (Dhamodharan et al. 2019). In this pathway, the inflammatory route involves NF-κB signaling, which facilitates MMP9 activation during diabetic ulceration. The NF-κB family comprises five members: p65 (RelA), RelB, c-Rel, p105/p50, and p100/p52. Upon NF-κB pathway activation, the heterodimers RelA and p50 play a pivotal role as transcription mediators for target genes.
As indicated in Supplementary data Fig. 1, in cases of inflammation, TNF-α is released due to foot ulceration, transmitting extracellular signaling to its receptor TNFR-1 (TNF receptor-1) that features a Death Domain (DD). TNFR1 subsequently recruits TRADD (TNF receptor activated protein with a death domain), TRAF2/5 (TNF receptor-associated factor 2 and 5), and E3 ubiquitin ligase cIAP1/2 (cellular inhibitor of apoptosis 1 and 2). Additionally, it recruits RIP1 (Receptor interacting protein 1) (Hsu et al. 1995, 1996; Baker et al. 2011). Upon activation of the complete receptor complex, the NF-κB pathway becomes regulated.
Ubiquitination plays a pivotal role in NF-κB activation. It begins with K63-linked ubiquitination binding to RIP1, which is then recruited to NEMO to form the TAK1-IKK complex. The IKK complex comprises two catalytic subunits, IKKα (IKK1), IKKβ (IKK2), and IKKγ (also known as NF-κB essential modulator or NEMO). TAK1 phosphorylates and activates IKKβ. In the absence of external stimuli, NF-κB dimers remain inactive, sequestered in the cytoplasm by the protein inhibitor IκBα, which prevents their nuclear entry. To activate NF-κB signaling, IKKβ mediates IκBα phosphorylation, a crucial step in NF-κB activation. Phosphorylated IκBα associates with the βTrCP protein, initiating K48-linked ubiquitination of IκBα. This ubiquitinated form of IκBα is directed to the cytosolic proteosome and subsequently degraded. This process allows NF-κB dimers (p50 and p65) to translocate into the nucleus, where they bind to DNA, driving gene expressions such as extracellular matrix remodeling involving MMP9, MMP3, and MMP14 (Hayden and Ghosh 2008; Israël 2010; Wertz and Dixit 2010). Figure 1 (Supplementary data) illustrates Dolastatin 16 as the MMP9 inhibitor in diabetic wound healing.
During the inflammatory phase, wound healing in diabetic individuals differs from that in normal individuals. In diabetic cases, wounds are more susceptible to bacterial infection due to impaired immune responses, leading to elevated levels of pro-inflammatory cytokines, proteases, and reactive oxygen species (ROS). This, in turn, damages structural components of the extracellular matrix (ECM) (Cho et al. 2019; Demidova-Rice et al. 2012). When combined with pro-inflammatory cytokines, ROS can exacerbate wounds by upregulating the expression of matrix metalloproteinases (MMPs), contributing to ECM and growth factor degradation (Chang 2016).
Matrix metalloproteinases (MMPs), a family of zinc-dependent endopeptidases, play a vital role in both physiological and pathophysiological tissue remodeling. MMPs contribute to various phases of wound healing by degrading almost all ECM protein components (Nagase et al. 2006; Yabluchanskiy et al. 2013). They are categorized into collagenases (e.g., MMP1, MMP8, MMP13), matrilysins (e.g., MMP7), stromelysins (e.g., MMP3, MMP10, and MMP11), gelatinases (e.g., MMP2 and MMP9), and membrane-type metalloproteinases (e.g., MMP14) (Laronha and Caldeira 2020; Rohani and Parks 2015). In diabetic foot ulcer conditions, elevated levels of MMPs with protease activity lead to protein degradation, ECM deterioration, granulation tissue remodeling, modulation of angiogenesis, and regulation of growth factor activity (Chang and Nguyen 2021; Park et al. 2014; Tardáguila-García et al. 2019).
MMP9 comprises a catalytic domain containing two zinc ions, five calcium ions, and three repeats homologous to the type II module of fibronectin. It is involved in the degradation of ECM for both physiological and pathophysiological processes related to tissue remodeling (Yabluchanskiy et al. 2013). Additionally, MMP9 is implicated in inflammatory processes such as diabetes, arthritis, and cancer (Halade et al. 2013). In diabetic foot conditions, elevated MMP9 levels are primarily responsible for delayed wound healing. Nguyen et al. reported high levels of active MMP9 in severely infected wounds based on affinity resin and proteomic analysis (Nguyen et al. 2018). This suggests that targeting MMP9 holds promise as a strategy for treating diabetic ulcers.
As illustrated in Fig. 6, when skin injury occurs, an influx of neutrophils is drawn to the injury site, releasing cytokines such as MMP9 and ROS. These factors play a role in pathogen/bacteria elimination and thrombus formation regulation. Elevated ROS levels trigger NF-κB activation, resulting in increased production and overexpression of MMP9, ultimately contributing to chronic wound development (Nguyen et al. 2018). Dolastatin 16 could potentially act as an MMP9 inhibitor, accelerating the wound healing process in diabetic foot patients.
To validate MMP9 as a potential target for Dolastatin 16, this study conducted molecular docking and molecular dynamics studies. In the molecular docking simulation, both the catalytic domain (PDB ID: 1GKC) and hemopexin domain (PDB ID: 1ITV) of MMP9 were used. In the catalytic site, the inhibitor was not directed to bind to the catalytic Zn2+ ion due to its conservation across all MMPs. Targeting non-zinc binding sites offers a promising strategy for obtaining more selective MMP9 inhibitors (Jacobsen et al. 2010). According to Fig. 8, the result indicated a low binding energy value of − 9.7 kcal/mol for the MMP9-catalytic site. Dolastatin 16's binding energy value with MMP9 was also lower than that of the native ligand reverse hydroxamate (− 6.6 kcal/mol) and (R)-ND-336 (− 8.9 kcal/mol), suggesting Dolastatin 16's potential potency compared to the inhibitors in 1GKC and (R)-ND-336. The latter is an MMP9 inhibitor designed by Nguyen et al., exhibiting greater efficacy than Becaplermin, an FDA-approved drug for diabetic foot ulcer treatment (Nguyen et al. 2018). Hence, (R)-ND-336 served as an additional ligand control in this study.
Figure 9 showcases Dolastatin 16's binding to the MMP9 site involving key residues. Four hydrogen bonds were observed in the MMP9 binding, including Leu187, Leu188, Ala189, and Pro421. Among these, the hydrogen bond of Pro421 had the closest distance (1.80 Å) to the Dolastatin 16 structure, potentially enhancing the binding affinity. Other significant residues in the MMP9 binding pocket, such as Val398, His401, His405, and Tyr423, participated in hydrophobic contacts. These results suggest that Dolastatin 16 strongly binds to MMP9 via hydrogen bonds and hydrophobic interactions. More detailed interactions of Dolastatin 16 with MMP9 residues and other protein targets are provided in SI Table 5. Another study investigating an MMP9 inhibitor via molecular docking involved (I-3, II-3)-biacetin, reported as a potent inhibitor (docking score: − 7.5 kcal/mol) targeting MMP9 through non-zinc binding interactions. This highlights the potential of targeting non-zinc binding sites in designing new MMP9 inhibitors (Nanjan et al. 2015).
Fig. 9.

Molecular docking result. a Dolastatin 16 binding within the MMP9 binding site (yellow cavity) (PDB ID: 1GKC), b 3D view of Dolastatin 16 binding to the pocket, with hydrogen bonds depicted as dashed lines, and c 2D interaction representation of Dolastatin 16 with MMP9 (PDB ID: 1GKC)
The structural behavior, molecular flexibility, and stability of the MMP9 complex with Dolastatin 16 were examined through a 50 ns MD simulation using YASARA software. This study analyzed RMSD, RMSF, Radius of Gyration, and MM-PBSA binding free energy. For clarity, only RMSD and MM-PBSA binding energy are presented in the main text, while the remaining figures are available in the Supporting Information. The changes in the Dolastatin 16 complex were evaluated and compared across different systems. Conformational changes in protein–ligand complexes during the simulation were assessed using RMSD (Dong and Wang 2023). Figure 10a displays the RMSD-Cα plots for the native protein MMP9 and the protein–ligand complexes. The complexes exhibit fluctuations in comparison to the native protein, suggesting protein intervention due to ligand interaction. However, the overall patterns remain similar throughout the simulation. In the initial 10 ns, the complexes experienced an increase to approximately 3 Å, followed by a decrease to around 2 Å during the subsequent 30 ns. The graph then gradually increases until the end of the simulation. The significant change in MMP9 structure due to ligand binding may indicate its importance for inhibition.
The study also assessed the RMSD value of ligand movement to understand the ligand's displacement from the binding site during simulation. A higher RMSD value indicates a greater distance of the ligand from the binding site. As shown in Fig. 10b, the plot for the Dolastatin 16 complex remained stable at around 4 Å during the initial 35 ns of simulation, after which it increased to approximately 6 Å until the end of the simulation. This trend suggests a notable movement of the ligand from its initial binding pocket position.
To further evaluate the most favorable bindings of the complexes, binding free energy was assessed using the MM-PBSA method. The results of MM-PBSA binding energy for the two complexes are depicted over time in Fig. 10c. A more positive value indicates a stronger complex binding (Mitra and Dash 2018). The analysis revealed that the Dolastatin 16-MMP9 complex exhibited a higher average positive MM-PBSA value of 154.908 kJ/mol, suggesting a strong binding affinity. Conversely, (R)-ND-336-MMP9 showed a lower MM-PBSA value of 74.467 kJ/mol, indicating a weaker binding. Despite the significant ligand movement from the binding site (as indicated by the RMSD ligand movement value), Dolastatin 16 maintained a strong affinity with the binding pocket throughout the simulation. Detailed visualizations of the MMP9 complexed with Dolastatin 16 during the interval period are provided in Fig. 11.
Fig. 11.

a Overall structure of native MMP9 in the catalytic domain before MD simulation, b molecular interaction between Dolastatin 16 and the MMP9 binding site: 10 ns (top), 30 ns (mid), and 50 ns (bottom) during molecular dynamic simulation
To understand the intervention of MMP9 due to ligand binding, Fig. 11 presents a comparison between the structure of native MMP9 pre and post MD simulation. The figure reveals a conformational change in the C-terminus and N-terminus structure, affected by ligand binding to the S1’ pocket of MMP9. The result also aligns with the RMSD-Cα pattern, showing fluctuations in the Dolastatin 16-MMP9 complex. Moreover, the figure demonstrates that the Dolastatin 16-MMP9 complex continues to interact with the residue keys of MMP9 through hydrogen and hydrophobic interactions until the simulation is complete. The consistent contribution of hydrogen bond interactions to the binding mode indicates that Dolastatin 16 maintains strong interactions with the MMP9 protein. Hydrophobic interactions also play a crucial role in inhibition and help stabilize the ligand in the active site. The outcome of the MD simulation shows that Dolastatin 16 fits well within the MMP9 pocket throughout the simulation period. Furthermore, through MD simulation, we can conclude that the Dolastatin 16-MMP9 complex is more stable than the (R)-ND-336-MMP9 complex, suggesting greater potential as an MMP9 inhibitor in diabetic foot conditions.
Conclusion
This study successfully investigated MMP9 as a potential target of Dolastatin 16, identified through network pharmacology, molecular docking, and molecular dynamics approaches. The target was primarily involved in the TNF signaling pathway and diabetic polyneuropathy associated with foot ulceration. Dolastatin 16 may function as an MMP9 inhibitor, thereby accelerating the wound healing process in diabetic foot patients. Molecular docking and molecular dynamics studies were employed to validate MMP9 as the target of Dolastatin 16. It was reported that the Dolastatin 16-MMP9 complex exhibits a stronger binding affinity, interacting with specific residue keys within the MMP9 binding pocket. This finding was supported by molecular dynamics studies showing stable behavior of the Dolastatin 16 complex during simulation. Notably, the critical residues within the Dolastatin 16 complex during the simulation period include Leu188, Val398, His401, Pro421, and Met422, all of which are crucial for inhibiting the enzyme's catalytic activity. Through in-silico studies, this research establishes MMP9 as a potential target of Dolastatin 16. Further validation through in-vitro studies is necessary to comprehend the functionality and mechanism of action of Dolastatin 16 against MMP9. Therefore, we will conduct the experimental analysis as a research roadmap in the further study.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors extend their sincere appreciation to the Bioinformatics Research Center INBIO Indonesia for providing essential facilities for conducting the experiments.
Author contributions
All authors contributed to the study's conception and design. The first author (DL) conducted material preparation, data collection, and analysis under the supervision of the second author (DHU). DL drafted the manuscript, and DHU reviewed it.
Funding
This research did not receive specific grants from any funding agencies in the public, commercial, or nonprofit sectors.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Conflict of interest
The authors declare no competing interests regarding this research.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dewi Luthfiana, Email: dewiluthfiana7@gmail.com.
Didik Huswo Utomo, Email: didik.huswo@gmail.com.
References
- Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/J.CMET.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum F, Keni R, Ahuja TN, et al. Notch signaling: A possible therapeutic target and its role in diabetic foot ulcers. Diabetes Metab Syndr Clin Res Rev. 2022;16:102542. doi: 10.1016/J.DSX.2022.102542. [DOI] [PubMed] [Google Scholar]
- Bönhof GJ, Herder C, Strom A, et al. Emerging biomarkers, tools, and treatments for diabetic polyneuropathy. Endocr Rev. 2018;40(1):153–192. doi: 10.1210/er.2018-00107. [DOI] [PubMed] [Google Scholar]
- Casalme LO, Yamauchi A, Sato A, et al. Total synthesis and biological activity of dolastatin 16. Org Biomol Chem. 2017;15:1140–1150. doi: 10.1039/c6ob02657e. [DOI] [PubMed] [Google Scholar]
- Cha H, Kopetzki E, Huber R, et al. Structural basis of the adaptive molecular recognition by MMP9. J Mol Biol. 2002;320:1065–1079. doi: 10.1016/S0022-2836(02)00558-2. [DOI] [PubMed] [Google Scholar]
- Chang M. Restructuring of the extracellular matrix in diabetic wounds and healing: a perspective. Pharmacol Res. 2016;107:243–248. doi: 10.1016/J.PHRS.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Chang M, Nguyen TT. Strategy for treatment of infected diabetic foot ulcers. Acc Chem Res. 2021;54:1080–1093. doi: 10.1021/ACS.ACCOUNTS.0C00864. [DOI] [PubMed] [Google Scholar]
- Chen J, Qin S, Liu S, et al. Targeting matrix metalloproteases in diabetic wound healing. Front Immunol. 2023;14:1–19. doi: 10.3389/fimmu.2023.1089001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Blatchley MR, Duh EJ, Gerecht S. Acellular and cellular approaches to improve diabetic wound healing. Adv Drug Deliv Rev. 2019;146:267–288. doi: 10.1016/J.ADDR.2018.07.019. [DOI] [PubMed] [Google Scholar]
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19/COVER. [DOI] [PubMed] [Google Scholar]
- Davis RR, Li B, Yun SY, et al. Structural Insights into JAK2 Inhibition by Ruxolitinib, Fedratinib, and Derivatives Thereof. J Med Chem. 2021;64:2228–2241. doi: 10.1021/ACS.JMEDCHEM.0C01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gregorio C, Contador D, Diáz D, et al. Human adipose-derived mesenchymal stem cell-conditioned medium ameliorates polyneuropathy and foot ulceration in diabetic BKS db/db mice. Stem Cell Res Ther. 2020;11:1–21. doi: 10.1186/s13287-020-01680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidova-Rice TN, Hamblin MR, Herman IM. Acute and impaired wound healing: pathophysiology and current methods for drug delivery, part 1: normal and chronic wounds: biology, causes, and approaches to care. Adv Skin Wound Care. 2012;25:304–314. doi: 10.1097/01.ASW.0000416006.55218.D0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamodharan U, Teena R, Vimal Kumar R, et al. Circulatory levels of B-cell activating factor of the TNF family in patients with diabetic foot ulcer: Association with disease progression. Wound Repair Regen. 2019;27:442–449. doi: 10.1111/WRR.12720. [DOI] [PubMed] [Google Scholar]
- Dong J, Wang X. Identification of novel BRD4 inhibitors by pharmacophore screening, molecular docking, and molecular dynamics simulation. J Mol Struct. 2023;1274:134363. doi: 10.1016/j.molstruc.2022.134363. [DOI] [Google Scholar]
- Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J Chem Inf Model. 2021;61:3891–3898. doi: 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang WY, Dahiya R, Qin HL, et al. Natural proline-rich cyclopolypeptides from marine organisms: chemistry, synthetic methodologies, and biological status. Mar Drugs. 2016 doi: 10.3390/MD14110194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman EL, Callaghan BC, Pop-Busui R, et al. Diabetic neuropathy. Nat Rev Dis Prim. 2019;5:42. doi: 10.1038/S41572-019-0097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furet P, Guagnano V, Fairhurst RA, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorganic Med Chem Lett. 2013;23:3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Gond MK, Pandey SK, Chandra S, et al. Zinc(II) catalyzed synthesis of 2-(4-methoxyphenyl)-5-(2-pyridyl)-1,3,4-thiadiazole: Characterizations, crystal structure, DFT calculation, Hirshfeld surface analysis, and molecular docking analysis. J Mol Struct. 2022;1267:133586. doi: 10.1016/j.molstruc.2022.133586. [DOI] [Google Scholar]
- Halade GV, Jin YF, Lindsey ML. Matrix metalloproteinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther. 2013;139:32–40. doi: 10.1016/J.PHARMTHERA.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/J.CELL.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, et al. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/S1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Hu H, Wang H, Yang X, et al. Network pharmacology analysis reveals potential targets and mechanisms of proton pump inhibitors in breast cancer with diabetes. Sci Rep. 2023;131(13):1–13. doi: 10.1038/s41598-023-34524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010 doi: 10.1101/CSHPERSPECT.A000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim Biophys Acta - Mol Cell Res. 2010;1803:72–94. doi: 10.1016/j.bbamcr.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Jain B, Raj U, Varadwaj PK. Drug target interplay: A network-based analysis of human diseases and the drug targets. Curr Top Med Chem. 2018;18:1053–1061. doi: 10.2174/1568026618666180719160922. [DOI] [PubMed] [Google Scholar]
- Jing X, Jin K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med Res Rev. 2020;40:753–810. doi: 10.1002/med.21639. [DOI] [PubMed] [Google Scholar]
- Kanehisa Laboratories KEGG PATHWAY: TNF signaling pathway - Homo sapiens (human). https://www.kegg.jp/pathway/hsa04668. Accessed 27 Apr 2023
- Khairy A, Ghareeb DA, Celik I, et al. Forecasting of potential anti-inflammatory targets of some immunomodulatory plants and their constituents using in vitro, molecular docking and network pharmacology-based analysis. Sci Rep. 2023;131(13):1–24. doi: 10.1038/s41598-023-36540-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laronha H, Caldeira J. Structure and function of human matrix metalloproteinases. Cells. 2020 doi: 10.3390/CELLS9051076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang TT, Zhao Q, He S, et al. Modeling analysis of potential target of dolastatin 16 by computational virtual screening. Chem Pharm Bull. 2018;66:602–607. doi: 10.1248/cpb.c17-00966. [DOI] [PubMed] [Google Scholar]
- Liu S, Misquitta YR, Olland A, et al. Crystal structure of a human IκB kinase β asymmetric dimer. J Biol Chem. 2013;288:22758–22767. doi: 10.1074/JBC.M113.482596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathpal S, Sharma P, Joshi T, et al. Identification of Zinc-binding inhibitors of Matrix Metalloproteinase-9 to prevent cancer through deep learning and molecular dynamics simulation approach. Front Mol Biosci. 2022;9:1–16. doi: 10.3389/fmolb.2022.857430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Dash R. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP bromodomain inhibitors. J Mol Graph Model. 2018;83:42–52. doi: 10.1016/j.jmgm.2018.04.014. [DOI] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/J.CARDIORES.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Nanjan P, Nambiar J, Nair BG, Banerji A. Synthesis and discovery of (I-3, II-3)-biacacetin as a novel non-zinc binding inhibitor of MMP-2 and MMP-9. Bioorganic Med Chem. 2015;23:3781–3787. doi: 10.1016/j.bmc.2015.03.084. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Ding D, Wolter WR, et al. Validation of Matrix Metalloproteinase-9 (MMP-9) as a novel target for treatment of diabetic foot ulcers in humans and discovery of a potent and selective small-molecule MMP-9 inhibitor that accelerates healing. J Med Chem. 2018;61:8825–8837. doi: 10.1021/acs.jmedchem.8b01005. [DOI] [PubMed] [Google Scholar]
- Nielsen DS, Shepherd NE, Xu W, et al. Orally absorbed cyclic peptides. Chem Rev. 2017;117:8094–8128. doi: 10.1021/ACS.CHEMREV.6B00838. [DOI] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, et al. Open Babel: An Open chemical toolbox. J Cheminform. 2011;3:1–14. doi: 10.1186/1758-2946-3-33/TABLES/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Ikemori-Kawada M, Jestel A, et al. Distinct binding mode of multikinase inhibitor lenvatinib revealed by biochemical characterization. ACS Med Chem Lett. 2014;6:89–94. doi: 10.1021/ML500394M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kim JH, Kim EY, et al. Bioreducible polymer-delivered siRNA targeting MMP-9: suppression of granulation tissue formation after bare metallic stent placement in a rat urethral model. Radiology. 2014;271:87–95. doi: 10.1148/RADIOL.13130980. [DOI] [PubMed] [Google Scholar]
- Pettit GR, Xu J, Hogan F, et al. Isolation and structure of the human cancer cell growth inhibitory cyclodepsipeptide Dolastatin 16. J Nat Prod. 1997;3864:752–754. doi: 10.1021/np9700230. [DOI] [PubMed] [Google Scholar]
- Pettit GR, Smith TH, Arce PM, et al. Antineoplastic agents 599 Total synthesis of Dolastatin 16. J Nat Prod. 2014 doi: 10.1021/np500925y. [DOI] [PubMed] [Google Scholar]
- Rathod S, Shinde K, Porlekar J, et al. Computational exploration of anti-cancer potential of flavonoids against Cyclin-Dependent Kinase 8: An in silico molecular docking and dynamic approach. ACS Omega. 2022 doi: 10.1021/ACSOMEGA.2C04837/ASSET/IMAGES/LARGE/AO2C04837_0012.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015;44–46:113–121. doi: 10.1016/J.MATBIO.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Rowsell S, Hawtin P, Minshull CA, et al. Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J Mol Biol. 2002;319:173–181. doi: 10.1016/S0022-2836(02)00262-0. [DOI] [PubMed] [Google Scholar]
- Stanzione F, Giangreco I, Cole JC. Use of molecular docking computational tools in drug discovery. Prog Med Chem. 2021;60:273–343. doi: 10.1016/BS.PMCH.2021.01.004. [DOI] [PubMed] [Google Scholar]
- Tao Q, Du J, Li X, et al. Network pharmacology and molecular docking analysis on molecular targets andmechanisms of Huashi Baidu formula in the treatment of COVID-19. Drug Dev Ind Pharm. 2020;46:1345. doi: 10.1080/03639045.2020.1788070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardáguila-García A, García-Morales E, García-Alamino JM, et al. Metalloproteinases in chronic and acute wounds: A systematic review and meta-analysis. Wound Repair Regen. 2019;27:415–420. doi: 10.1111/WRR.12717. [DOI] [PubMed] [Google Scholar]
- Tsuda T, Arai A, Kita M. Ligand-dissociation-type N, N-dimethylaminopyrene probes for in-situ site-specific protein labeling. Chem - an Asian J. 2022;17:e202200631. doi: 10.1002/asia.202200631. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010 doi: 10.1101/CSHPERSPECT.A003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams NK, Lucet IS, Peter Klinken S, et al. Crystal structures of the Lyn protein tyrosine kinase domain in its Apo- and inhibitor-bound state. J Biol Chem. 2009;284:284–291. doi: 10.1074/JBC.M807850200. [DOI] [PubMed] [Google Scholar]
- Yabluchanskiy A, Ma Y, Iyer RP, et al. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology (bethesda) 2013;28:391–403. doi: 10.1152/PHYSIOL.00029.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lin L, Zhang Z, et al. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5:209. doi: 10.1038/s41392-020-00312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: past, present and future. Curr Opin Chem Biol. 2017;38:24–29. doi: 10.1016/J.CBPA.2017.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.